

Supplemental Digital Content is available in the text
Keywords: Atherosclerosis, Macrophages, Oxidized low-density lipoprotein, Mitochondria, Metabolism
Abstract
Background:
Macrophage accumulation in the vascular wall is a hallmark of atherosclerosis. Studies showed that shifting of oxidized lipids-induced inflammatory macrophages towards an anti-inflammatory phenotype by promoting oxidative metabolism attenuated atherosclerosis progression. Therefore, this study aimed to investigate whether metformin, which has ameliorated atherosclerosis in animal models and clinical trials, modulated oxidized low-density lipoprotein (Ox-LDL) induced inflammatory status in macrophages by regulating cellular oxidative metabolism.
Methods:
Murine raw264.7 macrophages were incubated with Ox-LDL (50 μg/mL) in the presence or absence of metformin (15 μmol/L) for 24 h. Real-time polymerase chain reaction was used to quantify the transcription of classically activated (M1) pro-inflammatory and alternatively activated (M2) anti-inflammatory markers and mitochondrial DNA copy numbers. Cellular reactive oxygen species (ROS) production and mitochondrial membrane potential were detected by immunofluorescence. Cellular adenosine triphosphate (ATP) synthesis, glucose uptake, and lactic acid production were measured by commercial kit and normalized to cellular lysates. Western blotting analysis was performed to detect the expression of mitochondrial fusion/fission related proteins, enzymes mediating lipid metabolism and signaling pathway of glucose transport. Differences between groups were analyzed using one-way analysis of variance.
Results:
Metformin improved Ox-LDL-impaired anti-inflammatory phenotype in raw264.7 macrophages as shown by up-regulated transcription of anti-inflammatory markers including interleukin 10 (0.76 ± 0.04 vs. 0.94 ± 0.01, P = 0.003) and Resistin-like molecule alpha (0.67 ± 0.08 vs. 1.78 ± 0.34, P = 0.030). Conversely, Ox-LDL-diminished phosphorylation of Akt was up-regulated by metformin treatment (0.47 ± 0.05 vs. 1.02 ± 0.08, P = 0.040), associated with an improvement of mitochondrial function, characterized by decreased ROS generation (2.50 ± 0.07 vs. 2.15 ± 0.04, P = 0.040), increased lipid oxidation, and elevated cellular ATP production (0.026 ± 0.001 vs. 0.035 ± 0.003, P = 0.020). Moreover, metformin-mediated Akt activation increased Akt substrate of 160 kDa (AS160) phosphorylation (0.51 ± 0.04 vs. 1.03 ± 0.03, P = 0.0041), promoted membrane translocation of glucose transporter 1, and increased glucose influx into the cells (4.78 ± 0.04 vs. 5.47 ± 0.01, P < 0.001).
Conclusion:
This study suggested that targeting macrophage metabolism with new or existing drugs had therapeutic potential for the prevention and treatment of diabetes-accelerated atherosclerosis.
Introduction
The incidence of coronary heart disease is approximately raised by two-fold in type 2 diabetes, and remains the most important cause of morbidity and mortality in diabetic patients.[1] Accelerated atherosclerosis has been observed in diabetic patients,[2] and several studies have pointed out that hyperglycemia and insulin resistance are the two most important risk factors.[3] However, clinical trials and meta-analyses showed limited benefits of intensive glucose lowering treatment on all-cause mortality and deaths due to cardiovascular causes.[4,5] Therefore, pharmacologic interventions targeting other specific mechanisms are warranted to alter the increased risk of atherosclerosis in diabetes.
Macrophages in response to the surrounding microenvironment polarize to different phenotypes. There are two main classes of macrophages: classically activated (M1) pro-inflammatory macrophages and alternatively activated (M2) anti-inflammatory macrophages.[6] Studies have revealed that imbalance in macrophage polarization was the key pathological factor for a variety of immune-related diseases such as autoimmune diseases, tumor, and atherosclerosis.[7] Both M1 and M2 macrophages have been identified in atherosclerotic plaques of human as well as mouse,[8] and increased ratio of M1 over M2 polarization in human atherosclerosis was related to plaque instability.[9] In mouse models of atherosclerosis, the approaches to increase M1 polarization accelerated plaque formation,[10,11] while increasing M2 polarization induced atherosclerosis regression.[12,13] These suggested that manipulation of macrophage polarization might have therapeutical potential in treating atherosclerosis.
Cellular metabolism had a great impact on macrophage polarization and functionality. M1 macrophages are highly glycolytic, while M2 macrophages utilize fatty acid metabolism and mitochondrial oxidative phosphorylation (OXPHOS).[9,14] Recent studies indicated that disruption of cellular energy metabolism directly altered macrophage M1/M2 fate and inflammatory functions. Shifting of macrophage metabolism towards glycolysis drove a pro-inflammatory phenotype,[15] whereas promotion of mitochondrial OXPHOS primed macrophages for alternative activation and inhibited the production of pro-inflammatory cytokine.[16] Therefore, improvement in macrophage polarization imbalance in atherosclerosis might be achieved by regulating glycolysis and mitochondrial energy metabolism.
Metformin has been used as a glucose-lowering medication in humans for more than 60 years and is now recommended as first-line treatment for type 2 diabetes.[17] Several clinical trials suggested a cardiovascular protective effect by using metformin in individuals with diabetes,[18,19] while anti-atherosclerotic mechanisms of metformin remained poorly understood. Metformin has been implicated to improve glucose and lipid metabolism by regulating mitochondrial function and adenosine monophosphate-activated protein kinase (AMPK) activity in insulin-targeted cells,[20] but the metabolic effects on atherosclerotic-related immune cells such as macrophages remained elusive. So, this study aimed to investigate the regulatory role of metformin in macrophage energy metabolism and whether this metabolic regulation mediates phenotypic conversion of macrophages in atherosclerosis or not.
Methods
Cell culture
Raw264.7 cells, a murine macrophage cell line, were obtained from Jiangsu KeyGENBioTECH Corp., Ltd (China) and grown in high glucose Dulbecco's modified Eagle medium (Gibco, Grand Island, NY, USA) supplemented with 10% (vol/vol) fetal bovine serum, 100 U/mL penicillin and 100 mg/mL streptomycin (Gibco). The cells were incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air and were grown to 60% to 70% confluence.
Cell treatment
Cells (3 × 105/mL) were plated into six-well plates overnight for attachment. Then the media were removed and cells were treated with/without Ox-LDL (50 μg/mL; Yiyuan biotechnologies, Guangzhou, China) in the presence or absence of metformin (15 μmol/L; Sigma, St Louis, MO, USA) for 24 h.
Real-time polymerase chain reaction
Total RNA was extracted from cells using RNeasy kit (Qiagen, Hilden, Germany). The complementary DNA (cDNA) was prepared using a cDNA reverse transcription kit (Takara, Dalian, China) according to the manufacturer's instructions. The expression of messenger RNA (mRNA) was measured on real-time polymerase chain reaction (PCR) system (Applied Biosystems, Carlsbad, CA, USA) using TaqMan probes. The PCR conditions were as follows: one cycle of 95°C for 10 s, followed by 40 cycles of 60°C for 30 s and 72°C for 30 s. The primers used to quantify the transcription of tumor necrosis-alpha (TNF-α), interleukin (IL)-6, IL-10, resistin-like molecule alpha (Retnla), mitochondria (mt)-ND1 and mt-ND4 are shown in Table 1. Fold changes in the expression were calculated by Ct method using β-actin as an endogenous control for mRNA expression.
Table 1.
Primer sequences of real-time polymerase chain reaction.
Western blotting analysis
Cells were washed twice with phosphate buffer saline (PBS) and then were lysed in radio-immunoprecipitation assay buffer with protease and phosphatase inhibitors on ice for immunoblotting. The mixture was collected and centrifuged at 13,000 × g for 15 min at 4°C, and the resulting supernatants were used as the cellular lysates. Protein content was detected using bicinchoninic acid protein assay kit (Thermo Fisher, Waltham, MA, USA). Aliquots (30 μg) of cell lysates were separated on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then trans-blotted onto the polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA). After being blocked with 5% bovine serum albumin (BSA), blots were incubated with various primary antibodies including rabbit anti-Akt 1/2/3 (Thermo Fisher), rabbit anti-dynamin-related protein 1 (DRP-1), optic atrophy 1 (OPA1), mitofusin 2 (MFN2), Akt substrate of 160 kDa (AS160), nicotinamide adenine dinucleotide:ubiquinone oxidoreductase core sub-unit S3 (NDUFS3; Abcam, Cambridge, UK), rabbit anti-AMPK, P-AMPK (Thr172), acetyl-CoA carboxylase (ACC), ACC phosphorylation (p-ACC; Ser79), P-Akt (Ser473), TBC1 family domain member 1 (TBC1D1) phosphorylation (p-TBC1D1; Ser660) (CST, Beverly, MA, USA), anti-P-AS160 (T642) (Biorbyt, Cambridge, UK), anti-carnitine palmitoyltransferase (CPT)-1b (SAB, Nanjing, China), and rabbit anti-β-actin (Bio-world, St. Louis Park, MN, USA) at 4°C overnight. This was followed by incubation with horseradish peroxidase-conjugated anti-rabbit secondary antibody (Bio-world) at room temperature for 70 min. The protein bands were detected by enhanced chemiluminescence using a Syngene system. Protein quantification was measured by using Image J software (National Institutes of Health, Bethesda, Maryland, USA) and normalized by β-actin.
Immunofluorescence
Raw264.7 cells were plated on the cover glass, washed with PBS, fixed in 4% paraformaldehyde at room temperature for 30 min, permeabilized with 0.5% Triton X-100, blocked with 3% BSA, and stained with anti-glucose transporter 1 (anti-GLUT1) antibody (1:500, Abcam) overnight at 4°C. The cells were re-washed and incubated with AlexaFluor 488 anti-rabbit IgG secondary antibody (Beyotime Biotechnology, China) for 30 min. The cells were washed again and stained with 4′,6-diamidino-2-phenylindole (1:5000, Thermo Fisher) for 5 min. Images were captured with an Olympus confocal microscope (FV10i; Olympus, Tokyo, Japan) and analyzed with Olympus Confocal software.
Glucose consumption
Cells were treated as above and cultured media was collected and centrifuged at 8000 × g for 5 min at room temperature to remove cell debris. The resulting supernatant was used to determine glucose concentrations by glucose uptake kit (Jiancheng Biotechnology, China) according to the manufacturer's instructions. The 10 μL of supernatant was added to 200 μL working solutions that comprised of R1 and R2 in 1:1 ratio and incubated at 37°C for 15 min. The concentration of glucose was measured by a microplate reader (Molecular Devices M3) at 450 nm absorbance. Cell glucose consumption was expressed as the concentration of glucose in the conditional media subtracted from the total glucose of media and normalized by cellular protein concentration.
Lactic acid measurement
After treatment, the cultured media was collected and then centrifuged at 8000 × g for 5 min at room temperature. The resulting supernatant was used to determine the secretion of cellular lactic acid by lactic acid detection kit (Jiancheng Biotechnology) according to the manufacturer's instructions. The concentration of lactic acid was measured by a plate reader at 530 nm absorbance and normalized by protein concentration.
Cellular reactive oxygen species detection
Macrophages were washed with PBS and loaded with dichloro-dihydro-fluorescein diacetate probe (Sigma). After incubation for 30 min, the cells were washed with serum-free media and collected in fluorescence-activated cell sorting (FACS) tubes. The cells were centrifuged at 1000 × g for 5 min at 4°C and re-suspended in ice-cold PBS. This was repeated once and the cellular reactive oxygen species (ROS) levels were determined by fluorescent microplate reader (Molecular Devices M3) with Ex 488 nm/Em 525 nm and were normalized to protein concentrations.
Mitochondrial membrane potential measurement
Macrophages were washed with PBS and loaded with probe JC-1 (Sigma). After incubation for 20 min, cells were washed with serum-free media and collected in FACS tubes. The cells were centrifuged at 1000 × g for 5 min under 4°C and re-suspended in ice-cold PBS. This procedure was repeated once and green fluorescent intensity (JC-1 monomer) was determined by fluorescent microplate reader (Molecular Devices M3) with Ex 490 nm/Em 530 nm and red fluorescent intensity (JC-1 aggregates) with Ex 525 nm/Em 590 nm. Mitochondrial membrane potential was calculated as red fluorescence intensity divided by green fluorescent intensity and was normalized to protein concentrations of cellular lysates.
Cellular adenosine triphosphate measurement
Cells were washed twice with PBS and harvested in lysis buffer provided by adenosine triphosphate (ATP) detection kit (Beyotime Biotechnology) on ice. The mixture was collected and centrifuged at 12,000 × g for 5 min at 4°C, and the resulting supernatants were used to determine ATP production according to the manufacturer's instructions. The 100 μL working solutions were added to the plate, incubated at room temperature for 3 to 5 min and mixed with 10 μL of supernatant for 2 s. The relative light unit of ATP was then measured using the GloMaxTM96 luminometer by microplate reader. Cellular ATP concentrations were calculated by standard curve and normalized by protein concentrations of cellular lysates.
Statistical analysis
The data are presented as mean ± standard deviation. Differences between groups were analyzed using one-way analysis of variance by GraphPad Prism software version 8.0 (GraphPad Software Inc., San Diego, CA, USA). Statistical significance was accepted at P ≤ 0.05.
Results
Metformin improving Ox-LDL-impaired anti-inflammatory phenotype in Raw264.7 macrophages
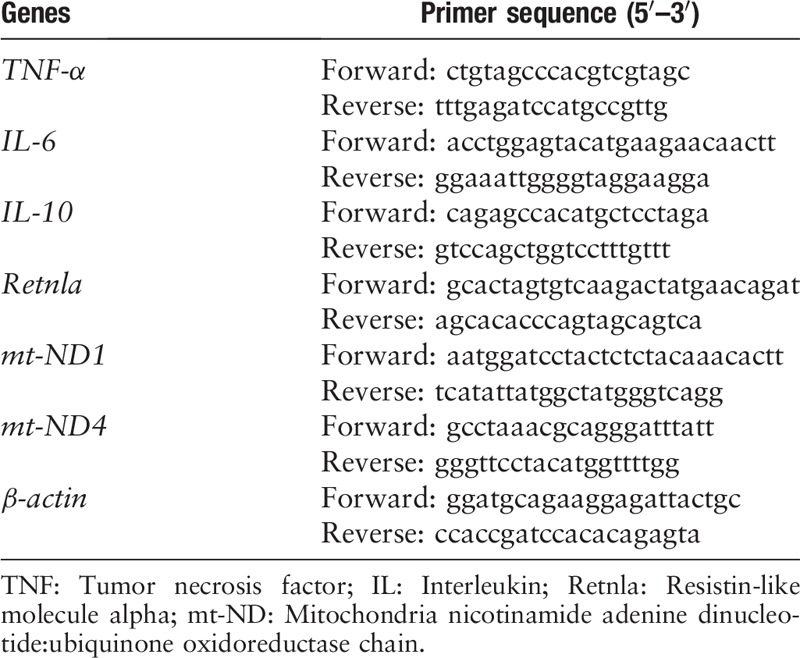
M1 macrophages have been implicated in the pathogenesis of atherosclerosis[9] and are induced by oxidatively-modified lipids and lipoproteins, the major lipid components in atherosclerotic environment.[21] So, we examined the regulatory role of metformin, with established cardiovascular protective effects in humans and animals, in macrophage polarization in Ox-LDL treated raw264.7 macrophages. We characterized the genetic signature of polarized macrophages by quantifying the transcription of M1 and M2 markers. The real time-PCR results showed that Ox-LDL produced an inflammatory phenotype in macrophages by up-regulating the transcription of M1 markers TNF-α (1.01 ± 0.01 vs. 1.69 ± 0.29, P = 0.030) and IL-6 (1.12 ± 0.17 vs. 4.90 ± 1.66, P = 0.030) [Figure 1A and 1B], as well as down-regulating M2 markers IL-10 (0.96 ± 0.05 vs. 0.76 ± 0.04, P = 0.002). Metformin treatment significantly increased the transcription of M2 markers in Ox-LDL-loaded macrophages (IL-10: 0.76 ± 0.04 vs. 0.94 ± 0.01, P < 0.001; Retnla: 0.67 ± 0.08 vs. 1.78 ± 0.34, P = 0.030) [Figure 1C and 1D], while there were no detectable changes in M1 markers (TNF-α: 1.69 ± 0.29 vs. 1.56 ± 0.22, P = 0.850; IL-6: 4.90 ± 1.66 vs. 4.00 ± 1.34, P = 0.710) [Figure 1A and 1B]. These indicated the presence of an anti-inflammatory phenotype primed by metformin in Ox-LDL-loaded macrophages.
Figure 1.
Metformin up-regulated the transcription of M2 markers in Ox-LDL loaded macrophages. Raw264.7 macrophages were incubated with/without Ox-LDL in the presence and absence of metformin for 24 h and cellular cDNA was used for real-time polymerase chain reaction of M1 and M2 markers, respectively. Transcription of M1 inflammatory markers: TNF-α (A), IL-6 (B) and M2 anti-inflammatory markers IL-10 (C) and Retnla (D) between groups. ∗P < 0.05, †P < 0.01 vs. control; ‡P < 0.01, §P < 0.05 vs. Ox-LDL. IL: Interleukin; Ox-LDL: Oxidized low-density lipoprotein; Retnla: Resistin-like molecule alpha; TNF: Tumor necrosis factor.
Metformin improving Ox-LDL-impaired mitochondrial function
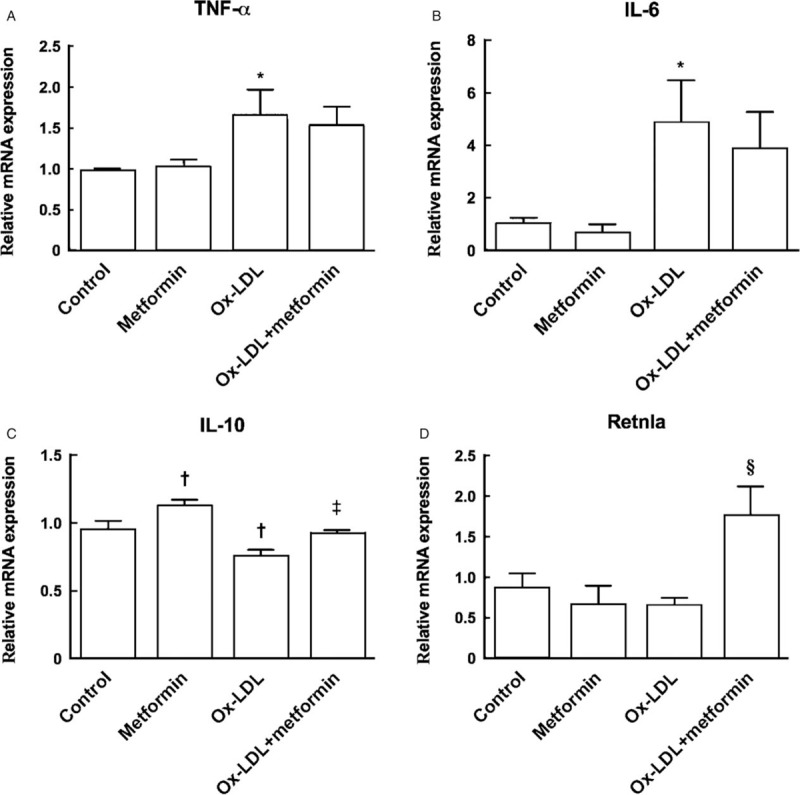
Mitochondria play a critical role in oxidative metabolism, fuel energy for polarization towards M2 macrophages, and sequential immune responses.[22] We, therefore, investigated the regulatory role of metformin in mitochondrial metabolism in Ox-LDL-treated macrophages. Our results showed that metformin significantly decreased Ox-LDL-up-regulated ROS production (2.50 ± 0.07 vs. 2.15 ± 0.04, P = 0.040) [Figure 2A], and also increased Ox-LDL-impaired mitochondrial membrane potential (0.67 ± 0.05 vs. 0.86 ± 0.05, P = 0.040) [Figure 2B], correlating with an elevated cellular ATP production in metformin-treated macrophages (0.026 ± 0.001 vs. 0.035 ± 0.003, P = 0.020) [Figure 2C]. We further tested the effects of metformin on mitochondrial DNA integrity and mitochondrial morphologies, both reported to regulate mitochondrial function.[23] The results indicated that metformin up-regulated Ox-LDL-diminished mitochondrial DNA copy number (mt-ND-4: 0.42 ± 0.16 vs. 1.04 ± 0.24, P = 0.040; mt-ND-1: 0.82 ± 0.42 vs.1.33 ± 0.40, P = 0.450) [Figure 2D], and increased the protein expression of fusion-related protein Mfn2 (0.55 ± 0.01 vs. 0.76 ± 0.01, P = 0.002),with little effects on fusion-related protein OPA1 (0.84 ± 0.03 vs. 0.77 ± 0.04, P = 0.110) and fission-related protein Drp-1 (0.77 ± 0.02 vs. 0.82 ± 0.01, P = 0.070) [Figure 2E]. All these results suggested that metformin might promote macrophage oxidative metabolism by improving Ox-LDL-impaired mitochondrial function.
Figure 2.
Metformin treatment improved mitochondrial function of macrophages after Ox-LDL incubation. (A) Metformin decreased Ox-LDL-induced ROS generation. (B) Metformin up-regulated Ox-LDL-impaired mitochondrial membrane potential. (C) Metformin increased cellular ATP production after interaction with Ox-LDL. (D) Metformin increased mt-DNA copy number in Ox-LDL-loaded macrophages. (E) Effects of metformin on protein expression of mitochondrial fusion related-proteins Mfn2, OPA1 and fission-related protein Drp-1. ∗P < 0.05, ‡P < 0.01 vs. control; †P < 0.05, §P < 0.01 vs. Ox-LDL. ATP: Adenosine triphosphate; Drp-1: Dynamin-related protein 1; MFI: Mean fluorescent intensity; Mfn2: Mitofusin 2; mt-ND: Mitochondria nicotinamide adenine dinucleotide:ubiquinone oxidoreductase chain; OPA1: Optic atrophy 1; Ox-LDL: Oxidized low-density lipoprotein; RLU: Relative light unit; ROS: Reactive oxidative species.
Metformin increasing Ox-LDL-impaired lipid and glucose metabolism in raw264.7 cells
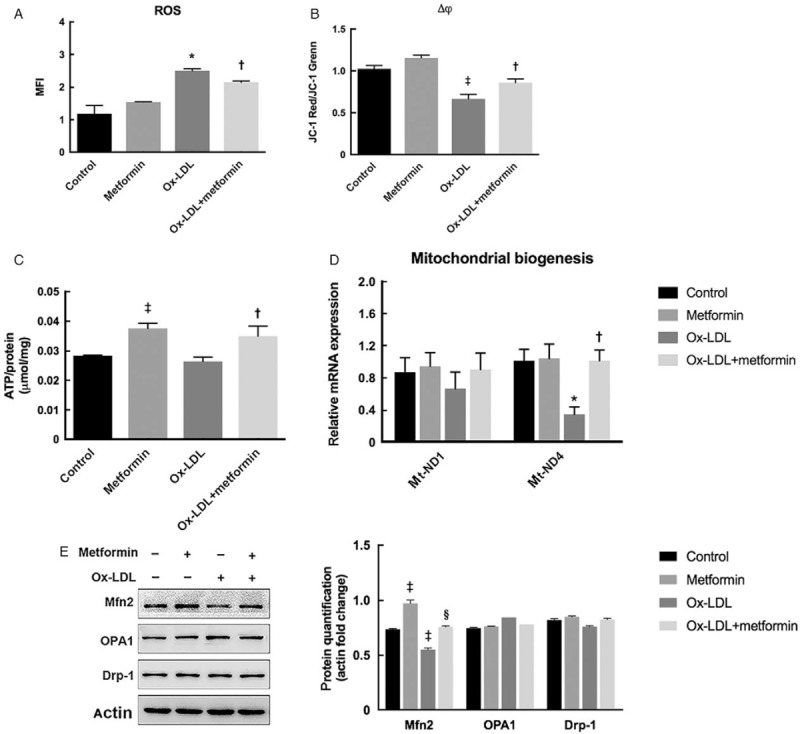
We then explored the characteristics of lipid and glucose metabolism in Ox-LDL-loaded macrophages after treatment with metformin. Our results showed that metformin significantly up-regulated Ox-LDL-impaired p-ACC (0.44 ± 0.02 vs. 0.78 ± 0.03, P = 0.002) and CPT-1b expression (0.61 ± 0.00 vs. 0.95 ± 0.01, P < 0.001), which subsequently mediated lipid synthesis inhibition and fatty acid oxidation, respectively [Figure 3A]. However, there were no detectable changes of NDUFS3 expression between groups (P = 0.230), the core sub-unit of mitochondrial complex I, which was considered as the main respiratory-chain target of metformin.[20] This indicated that metformin might improve oxidative metabolism independent of mitochondrial respiratory chain complexes regulation. With respect to glucose metabolism, we found that glucose consumption was decreased after Ox-LDL loading (5.77 ± 0.04 vs. 6.39 ± 0.03, P < 0.001), which was reversed by metformin treatment (4.78 ± 0.04 vs. 5.47 ± 0.01, P < 0.001) [Figure 3B]. In contrast, metformin significantly down-regulated Ox-LDL-induced lactic acid production in raw264.7 macrophages (8.61 ± 0.67 vs. 6.42 ± 0.06, P = 0.002) [Figure 3C]. Moreover, a decreased ratio of lactic output by glucose input in the presence of metformin was observed in Ox-LDL-treated macrophages (1.92 ± 0.15 vs. 1.18 ± 0.01, P < 0.001) [Figure 3D], implying that metformin mediated-increased glucose flux might enter into oxidative metabolic pathway instead of glycolysis in Ox-LDL-loaded macrophages.
Figure 3.
Metformin improved lipid and glucose oxidation in Ox-LDL-loaded macrophages. (A) Effects of metformin on protein expression of enzymes mediating lipid synthesis and oxidation. (B) Metformin increased Ox-LDL-impaired glucose consumption. Glucose consumption was normalized by protein concentrations of cellular lysates. (C) Metformin decreased lactic acid production in Ox-LDL-loaded macrophages. Lactic acid levels were normalized by protein concentrations of cellular lysates. (D) Metformin down-regulated the ratio of lactic acid output by glucose input in the presence of Ox-LDL. ∗P < 0.05, †P < 0.01, §P < 0.001 vs. control; ‡P < 0.01, ||P < 0.001 vs. Ox-LDL. CPT-1b: Carnitine palmitoyltransferase 1b; NDUFS3: Nicotinamide adenine dinucleotide:ubiquinone oxidoreductase core sub-unit S3; Ox-LDL: Oxidized low-density lipoprotein; p-ACC: Acetyl-CoA carboxylase phosphorylation.
Metformin-mediated Akt activation increased GLUT1 transport in Ox-LDL loaded macrophages
Two paralogue Rab GTPase activating proteins AS160 (also known as TBC1D4) and TBC1D1 have been implicated in the regulation of glut traffic to plasma membrane in insulin-targeted cells through phosphorylation by Akt and AMPK, respectively.[24] However, few studies have evaluated the possible role of AS160 and TBC1D1 in noninsulin-stimulated glucose transport. As we observed the increased membrane translocation of GLUT1, the primary glucose transporter in murine macrophages[25] by metformin treatment [Figure 4A, see white arrow], the role of metformin in AS160 and TBC1D1 regulation, as well as the upper-stream kinases Akt and AMPK were examined in Ox-LDL-loaded macrophages. Western blotting analysis showed that in the absence of Ox-LDL, metformin treatment up-regulated AMPK and TBC1D1 phosphorylation (p-AMPK: 0.59 ± 0.02 vs. 0.93 ± 0.13, P = 0.030; p-TBC1D1: 0.63 ± 0.06 vs. 0.96 ± 0.04, P = 0.020), despite little change of Akt and AS160 phosphorylation (p-Akt: 0.82 ± 0.06 vs. 0.60 ± 0.06, P = 0.060; p-AS160: 1.17 ± 0.23 vs. 0.61 ± 0.04, P = 0.080). However, after Ox-LDL loading, metformin improved Ox-LDL-diminished p-Akt, specifically Ser473, and phosphorylation of AS160 (p-Akt: 0.47 ± 0.05 vs.1.02 ± 0.08, P = 0.040; p-AS160: 0.51 ± 0.04 vs. 1.03 ± 0.03, P = 0.004), while AMPK and TBC1D1 seemed to be unaffected (p-AMPK: 0.68 ± 0.06 vs. 0.50 ± 0.07, P = 0.230; p-TBC1D1: 0.56 ± 0.07 vs. 0.71 ± 0.07, P = 0.180) [Figure 4B and 4C]. Altogether, these data implied that metformin mediated-activation of Akt-AS160 phosphorylation, instead of AMPK-TBC1D1, might play an important role in promoting glucose uptake in Ox-LDL-loaded macrophages [Supplementary Figure S1].
Figure 4.
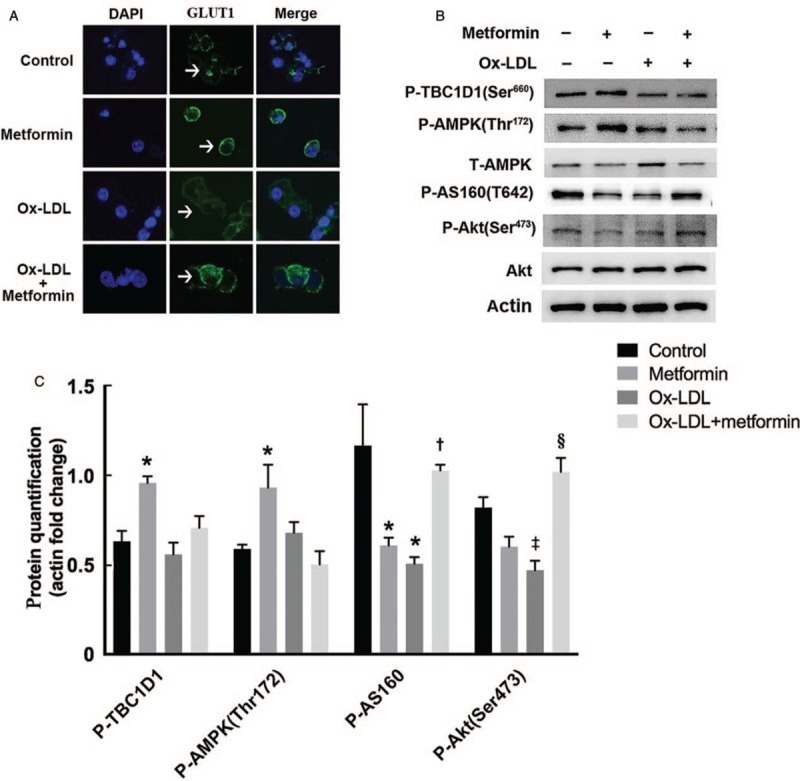
Metformin promoted Ox-LDL-impaired phosphorylation of Akt-AS160 pathway to increase GLUT1 transport. (A) GLUT1 expressions in macrophages were detected by immunofluorescence staining (green). Nuclei were stained with DAPI (blue) (original magnification ×600). (B) Metformin improved Akt-AS160 phosphorylation in the presence of Ox-LDL, while up-regulated AMPK-TBC1D1 phosphorylation in the absence of Ox-LDL. (C) Protein quantification of AMPK-TBC1D1 and Akt-AS160 pathway. Normalized by actin. ∗P < 0.05, ‡P < 0.01 vs. control; †P < 0.05, §P < 0.01 vs. Ox-LDL. AMPK: Adenosine monophosphate-activated protein kinase; AS160: Akt substrate of 160 kDa; DAPI: 4′,6-diamidino-2-phenylindole; GLUT1: Glucose transporter 1; Ox-LDL: Oxidized low-density lipoprotein; p-TBC1D1: TBC1 family domain member 1 phosphorylation.
Discussion
In this study, metformin primed Ox-LDL-induced inflammatory macrophages towards an anti-inflammatory phenotype, and cellular metabolic changes were considered as the underlying key mechanisms. Metformin improved Ox-LDL-impaired lipid oxidation and GLUT1 mediated glucose transport, both coupled with an up-regulation of mitochondrial oxidative metabolism. These findings provided new evidence supporting the protective role of metformin in atherosclerosis by regulating cellular metabolism in macrophages.
Metformin, a widely used anti-diabetic drug, has been reported to reduce atherosclerosis in humans and animal models.[26] Previous studies showed that improved mitochondrial dysfunction in endothelial cells might be the underlying mechanism of cardiovascular action of metformin.[27,28] However, few studies have elucidated the regulatory role of metformin in macrophage mitochondrial function in atherosclerosis. Our results showed that Ox-LDL-induced ROS production and mitochondrial DNA damage, both implicated in the contribution of atherosclerosis,[29,30] were ameliorated by metformin treatment. Mitochondria are a major source of cellular ROS,[31] but the exact relationship between mtDNA damage and ROS is unclear. ROS could damage mitochondrial DNA[33] and reduced ROS has been observed by increased mtDNA integrity in the cells.[34] However, Yu et al[30] found that increasing mtDNA copy number increased the abundance of mitochondrial respiratory chain complexes I, resulting in improved mitochondrial respiration independent of changes in ROS both in vivo and in vitro. Though we did not observe any up-regulation of NDUFS3, the key component of mitochondrial respiratory chain complexes I, in metformin-treated macrophages, the increased lipid oxidation, and elevated cellular ATP production implied an overall improvement of mitochondrial respiratory efficiency by metformin treatment. And the causative link between decreased ROS and increased mtDNA copy number mediated by metformin in the regulation of macrophage mitochondrial function needed further investigations.
An imbalance in the fusion/fission dynamics dramatically changed the overall mitochondrial morphology,[32] consequently altering the mitochondrial function. Mitochondrial fission is the division of a mitochondrion within a cell to form two or more separate mitochondrial compartments that are controlled by DRP1/dynamin-like protein 1/dynamin-1 and fission 1.[33] Whereas mitochondrial fusion merges two or more mitochondria within a cell to form a single compartment, which is regulated by at least three proteins: OPA1, MFN1, and MFN2[34] in mammals. Wang et al[28] demonstrated that metformin-mediated AMPK activation down-regulated hyperglycemia-induced Drp-1 expression, resulting in increased mitochondrial fusion in vascular endothelial cells and attenuation of diabetes-accelerated atherosclerosis in mouse models. However, our results showed that metformin increased Ox-LDL impaired-mitochondrial fusion in macrophages by specifically up-regulating Mfn2, with no detectable changes in AMPK phosphorylation and Drp-1 expression. This discrepancy in observation might be related with the treated cell types and extra-cellular stimuli and required further studies.
A study showed that GLUT1 distribution could be regulated by kinase-mediated phosphorylation of AS160,[35] raising the possibility that metformin might regulate GLUT1 mobilization through kinase-mediated mechanisms. Our results showed that metformin up-regulated Ox-LDL-impaired p-(Ser473) Akt expression and downstream AS160 phosphorylation. Moreover, decreased lactic acid production over increased glucose uptake was observed in metformin-treated macrophages, suggesting an increased coupling of glucose metabolism to OXPHOS.[36] Whether Akt was the target for metformin-mediated glucose oxidation [Supplementary Figure S1] requires more rigorous tests.
Notably, our studies showed that metformin treatment down-regulated the protein expression of total ACC in raw264.7 macrophages, so did Ox-LDL treatment [Figure 3A] as well as other stress,[37] indicating that ACC might be sensitive to environmental cues, which might explain why there was no overall increase of p-ACC expression, while metformin significantly increased p-AMPK [Figure 4B]. Indeed, metformin obviously up-regulated the ratio of p-ACC/ACC compared to that of control [Supplementary Figure S2], consistent with the previous finding that activated AMPK phosphorylates ACC.[38] Of note, our experiments showed that metformin slightly increased ROS production, along with an increase of ATP production, in metformin-treated macrophages, suggesting that metformin induced a stress that contributed to the reduction of ACC expression, which warrants further experiments and clarification.
Multiple studies have shown that metformin provided cardiovascular protection in clinical trials and animal models of atherosclerosis, and macrophages have been widely recognized as an important player in atherosclerosis. However, the exact mechanism that metformin ameliorate atherosclerosis pathogenesis was not clear. Previous studies confirmed that metformin inhibited inflammatory phenotype in Ox-LDL-loaded macrophages in vitro,[39] and here we investigated whether metformin modulated inflammantory status of Ox-LDL loaded macrophages by regulating cellular oxidative metabolism, particularly mitochondrial function. However, the role of metformin and the inflammation of macrophage in atherosclerosis were complex. For example, anti-inflammatory cytokine IL-10 attenuated atherosclerosis in mouse models,[40,41] but promoted foam cell formation, an important contributor to atherosclerosis in vitro.[42] In the context of knowing the beneficial role of metformin in vivo (both in animals and in clinic), and the complication of its in vivo effect mechanistically, the current study was set to assess whether, and how, metformin directly acts on macrophage, as a piece of information adding to the picture of metformin pharmacology that is still puzzling the research community. The key findings of our studies were that metformin improved Ox-LDL-impaired mitochondrial function and cellular metabolism, implying the therapeutic potential of targeting macrophage metabolism in treating atherosclerosis. However, further studies are required to conclude whether or not these findings are physiologically significant.
In conclusion, this study shows that metformin regulated macrophage cellular metabolism in a way that is intimately linked to the cell's inflammatory phenotype. Metabolic reprogramming of Ox-LDL loaded macrophages towards alternatively activated M2 cells by metformin treatment provides a new therapeutic target for the treatment of atherosclerosis.
Conflicts of interest
None.
Supplementary Material
Footnotes
How to cite this article: He X, Wang L, Chen XF, Liang Q, Wang WQ, Lin AQ, Yi L, Wang Y, Gao Q. Metformin improved oxidized low-density lipoprotein-impaired mitochondrial function and increased glucose uptake involving Akt-AS160 pathway in raw264.7 macrophages. Chin Med J 2019;132:1713–1722. doi: 10.1097/CM9.0000000000000333
References
- 1.Sarwar N, Gao P, Kondapally Seshasai SR, Gobin R, Kaptoge S, Di Angelantonio E, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet 2010; 375:2215–2222. doi: 10.1016/S0140-6736(10)60484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Virmani R, Burke AP, Kolodgie F. Morphological characteristics of coronary atherosclerosis in diabetes mellitus. Can J Cardiol 2006; 22:81B–84B. 10.1016/s0828-282x(06)70991-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bornfeldt KE, Tabas I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab 2011; 14:575–585. doi: 10.1016/j.cmet.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boussageon R, Bejan-Angoulvant T, Saadatian-Elahi M, Lafont S, Bergeonneau C, Kassaï B, et al. Effect of intensive glucose lowering treatment on all cause mortality, cardiovascular death, and microvascular events in type 2 diabetes: meta-analysis of randomised controlled trials. BMJ 2011; 343:d4169–d14169. doi: 10.1136/bmj.d4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayward RA, Reaven PD, Wiitala WL, Bahn GD, Reda DJ, Ge L, et al. Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015; 372:2197–2206. doi: 10.1056/NEJMoa1414266. [DOI] [PubMed] [Google Scholar]
- 6.Biswas SK, Chittezhath M, Shalova IN, Lim J-Y. Macrophage polarization and plasticity in health and disease. Immunol Res 2012; 53:11–24. doi: 10.1007/s12026-012-8291-9. [DOI] [PubMed] [Google Scholar]
- 7.Bashir S, Sharma Y, Elahi A, Khan F. Macrophage polarization: the link between inflammation and related diseases. Inflamm Res 2016; 65:1–11. doi: 10.1007/s00011-015-0874-1. [DOI] [PubMed] [Google Scholar]
- 8.Chinetti-Gbaguidi G, Baron M, Bouhlel MA, Vanhoutte J, Copin C, Sebti Y, et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPAR gamma and LXR alpha pathways. Circ Res 2011; 108:985–995. doi: 10.1161/CIRCRESAHA.110.233775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stoger JL, Gijbels MJJ, van der Velden S, Manca M, van der Loos CM, Biessen EAL, et al. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis 2012; 225:461–468. doi: 10.1016/j.atherosclerosis.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 10.Buono C, Come CE, Stavrakis G, Maguire GF, Connelly PW, Lichtman AH. Influence of interferon-gamma on the extent and phenotype of diet-induced atherosclerosis in the LDLR-deficient mouse. Arterioscler Thromb Vasc Biol 2003; 23:454–460. doi: 10.1161/01.ATV.0000059419.11002.6E. [DOI] [PubMed] [Google Scholar]
- 11.Whitman SC, Ravisankar P, Elam H, Daugherty A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E-/- mice. Am J Pathol 2000; 157:1819–1824. doi: 10.1016/S0002-9440(10)64820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feig JE, Parathath S, Rong JX, Mick SL, Vengrenyuk Y, Grauer L, et al. Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation 2011; 123:989–998. doi: 10.1161/CIRCULATIONAHA.110.984146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feig JE, Rong JX, Shamir R, Sanson M, Vengrenyuk Y, Liu J, et al. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc Natl Acad Sci U S A 2011; 108:7166–7171. doi: 10.1073/pnas.1016086108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jha AK, Huang SC-C, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015; 42:419–430. doi: 10.1016/j.immuni.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem 2014; 289:7884–7896. doi: 10.1074/jbc.M113.522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1 beta attenuate macrophage-mediated inflammation. Cell Metab 2006; 4:13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.American Diabetes Association Pharmacologic approaches to glycemic, treatment. Diabetes Care 2017; 40:S64–S74. doi: 10.2337/dc17-S011. [DOI] [PubMed] [Google Scholar]
- 18.Griffin SJ, Leaver JK, Irving GJ. Impact of metformin on cardiovascular disease: a meta-analysis of randomised trials among people with type 2 diabetes. Diabetologia 2017; 60:1620–1629. doi: 10.1007/s00125-017-4337-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ismail-Beigi F, Moghissi E, Kosiborod M, Inzucchi SE. Shifting paradigms in the medical management of type 2 diabetes: reflections on recent cardiovascular outcome trials. J Gen Intern Med 2017; 32:1044–1051. doi: 10.1007/s11606-017-4061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia 2017; 60:1577–1585. doi: 10.1007/s00125-017-4342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hundal RS, Gómez-Muñoz A, Kong JY, Salh BS, Marotta A, Duronio V, et al. Oxidized low density lipoprotein inhibits macrophage apoptosis by blocking ceramide generation, thereby maintaining protein kinase b activation, Bcl-X L levels. J Biol Chem 2003; 278:24399–24408. doi: 10.1074/jbc.M209179200. [DOI] [PubMed] [Google Scholar]
- 22.Yates C, Tan M, You Z, Kim J-H, Phillips JB, Angajala A, et al. Diverse roles of mitochondria in immune responses: novel insights into immuno-metabolism. Front Immunol 2018; 9:1605.doi: 10.3389/fimmu.2018.01605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kosmopoulos M, Philippou A, Tsigkou V, Simantiris S, Theodosiadis D, Papavassiliou AG, et al. Mitochondria and cardiovascular diseases—from pathophysiology to treatment. Ann Transl Med 2018; 6:256.doi: 10.21037/atm.2018.06.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cartee GD. Roles of TBC1D1 and TBC1D4 in insulin- and exercise-stimulated glucose transport of skeletal muscle. Diabetologia 2015; 58:19–30. doi: 10.1007/s00125-014-3395-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buller CL, Loberg RD, Fan M-H, Zhu Q, Park JL, Vesely E, et al. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am J Physiol Cell Physiol 2008; 295:C836–C843. doi: 10.1152/ajpcell.00554.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou J, Massey S, Story D, Li L. Metformin: an old drug with new applications. Int J Mol Sci 2018; 19:2863.doi: 10.3390/ijms19102863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Batchuluun B, Inoguchi T, Sonoda N, Sasaki S, Inoue T, Fujimura Y, et al. Metformin and liraglutide ameliorate high glucose-induced oxidative stress via inhibition of PKC-NAD(P)H oxidase pathway in human aortic endothelial cells. Atherosclerosis 2014; 232:156–164. doi: 10.1016/j.atherosclerosis.2013.10.025. [DOI] [PubMed] [Google Scholar]
- 28.Wang Q, Zhang M, Torres G, Wu S, Ouyang C, Xie Z, et al. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes 2017; 66:193–205. doi: 10.2337/db16-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Wang W, Wang N, Tall AR, Tabas I. Mitochondrial oxidative stress promotes atherosclerosis and neutrophil extracellular traps in aged mice. Arterioscler Thromb Vasc Biol 2017; 37:e99–e107. doi: 10.1161/ATVBAHA.117.309580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu EPK, Reinhold J, Yu H, Starks L, Uryga AK, Foote K, et al. Mitochondrial respiration is reduced in atherosclerosis, promoting necrotic core formation and reducing relative fibrous cap thickness. Arterioscler Thromb Vasc Biol 2017; 37:2322–2332. doi: 10.1161/ATVBAHA.117.310042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 2003; 552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer JN, Leuthner TC, Luz AL. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017; 391:42–53. doi: 10.1016/j.tox.2017.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol 2003; 23:5409–5420. 10.1128/mcb.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A 2004; 101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mendes AI, Matos P, Moniz S, Jordan P. Protein kinase WNK1 promotes cell surface expression of glucose transporter GLUT1 by regulating a Tre-2/USP6-BUB2-Cdc16 domain family member 4 (TBC1D4)-Rab8A complex. J Biol Chem 2010; 285:39117–39126. doi: 10.1074/jbc.M110.159418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev 2001; 15:1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye J, Zhu N, Sun R, Liao W, Fan S, Shi F, et al. Metformin inhibits chemokine expression through the AMPK/NF-kappaB signaling pathway. J Interferon Cytokine Res 2018; 38:363–369. doi: 10.1089/jir.2018.0061. [DOI] [PubMed] [Google Scholar]
- 38.Munday MR. Regulation of mammalian acetyl-CoA carboxylase. Biochem Soc Trans 2002; 30:1059–1064. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 39.Arai M, Uchiba M, Komura H, Mizuochi Y, Harada N, Okajima K. Metformin, an antidiabetic agent, suppresses the production of tumor necrosis factor and tissue factor by inhibiting early growth response factor-1 expression in human monocytes in vitro. J Pharmacol Exp Ther 2010; 334:206–213. doi: 10.1124/jpet.109.164970. [DOI] [PubMed] [Google Scholar]
- 40.Caligiuri G, Rudling M, Ollivier V, Jacob M-P, Michel J-B, Hansson GK, et al. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol Med 2003; 9:10–17. [PMC free article] [PubMed] [Google Scholar]
- 41.Han X, Kitamoto S, Wang H, Boisvert WA. Interleukin-10 overexpression in macrophages suppresses atherosclerosis in hyperlipidemic mice. FASEB J Off Publ Fed Am Soc Exp Biol 2010; 24:2869–2880. doi: 10.1096/fj.09-148155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oh J, Riek AE, Weng S, Petty M, Kim D, Colonna M, et al. Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. J Biol Chem 2012; 287:11629–11641. doi: 10.1074/jbc.M111.338673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.