

Abstract
Background:
Collagen type IV (COL4)-related nephropathy includes a variety of kidney diseases that occur with or without extra-renal manifestations caused by COL4A3-5 mutations. Previous studies revealed several novel mutations, including three COL4A3 missense mutations (G619R, G801R, and C1616Y) and the COL4A3 chr:228172489delA c.4317delA p.Thr1440ProfsX87 frameshift mutation that resulted in a truncated NC1 domain (hereafter named COL4A3 c.4317delA); however, the mutation mechanisms that lead to podocyte injury remain unclear. This study aimed to further explore the mutation mechanisms that lead to podocyte injury.
Methods:
Wild-type (WT) and four mutant COL4A3 segments were constructed into a lentiviral plasmid, then stably transfected into human podocytes. Real-time polymerase chain reaction and Western blotting were applied to detect endoplasmic reticulum stress (ERS)- and apoptosis-related mRNA and protein levels. Then, human podocytes were treated with MG132 (a proteasome inhibitor) and brefeldin A (a transport protein inhibitor). The human podocyte findings were verified by the establishment of a mus-Col4a3 knockout mouse monoclonal podocyte using clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9) technology.
Results:
Our data showed that COL4A3 mRNA was significantly overexpressed in the lentivirus stably transfected podocytes. Moreover, the COL4A3 protein level was significantly increased in all groups except the COL4A3 c.4317delA group. Compared to the other test groups, the COL4A3 c.4317delA group showed excessive ERS and apoptosis. Podocytes treated with MG132 showed remarkably increased intra-cellular expression of the COL4A3 c.4317delA mutation. MG132 intervention improved higher ERS and apoptosis levels in the COL4A3 c.4317delA group. Mouse monoclonal podocytes with COL4A3 chr:82717932insA c.4852insA p.Arg1618ThrfsX4 were successfully acquired; this NC1-truncated mutation suggested a higher level of ERS and relatively remarkable level of apoptosis compared to that of the WT group.
Conclusions:
We demonstrated that excessive ERS and ERS-induced apoptosis were involved in the podocyte injury caused by the NC1-truncated COL4A3 mutation. Furthermore, proteasome pathway intervention might become a potential treatment for collagen type IV-related nephropathy caused by a severely truncated COL4A3 mutation.
Keywords: Collagen type IV-related nephropathy, COL4A3 mutation, Podocyte injury, Proteasome pathway, MG132, Endoplasmic reticulum stress
Introduction
Collagen type IV (COL4)-related nephropathy mainly includes Alport syndrome (AS) and thin basement membrane nephropathy, which is one of the most common hereditary glomerular diseases. Pathogenic gene mutations in the type IV Collagen α3/α4/α5 chains that code for collagen type IV alpha 3/collagen type IV alpha 4/collagen type IV alpha 5 (COL4A3/COL4A4/COL4A5, COL4A3-5, respectively) cause these diseases.[1] COL4 is a basement membrane collagen that exists in a variety of tissue basement membranes in a unique heterotrimeric superhelical chain structure (α1α1α2, α3α4α5, α3α3α4, and α5α5α6).[2,3] The glomerular basement membrane (GBM) COL4 α3/α4/α5 chain is synthesized and secreted only by podocytes as the main components of a mature GBM.[4] A significant reduction or deletion of normal COL4A3-5 caused by abnormal genetic mutations leads to an unstable GBM mechanical structure, abnormal GBM substitution components (eg, COL4 α1/α2 chain), abnormal interaction with podocytes and extra-cellular matrix receptors, and eventually podocyte injury.[5] COL4 is normally glycosylated and folded in the endoplasmic reticulum, and had been a previously reported mutation in COL4 caused by endoplasmic reticulum stress (ERS)[6]; ERS is well known to be closely related to apoptosis.[7]
A number of studies have also shown that COL4A3 and COL4A4 are new pathogenic genes for familial focal segmental glomerulosclerosis (FFSGS), including previous work that found COL4A3 missense mutations in 12.5% (5/40) of FFSGS cases.[8,9] In 2018, the Alport Syndrome Classification Working Group published a new classification scheme that categorizes collagen IV α3-α5 genetic diseases as varieties of AS.[10] To date, there are more than 1000 records of COL4A3 mutation types, including research identifying new COL4A3 mutations in Chinese FFSGS and AS.[8] The molecular mechanisms of how COL4A3 mutations cause podocyte injury remain unclear; further studies are required to provide additional therapeutic options, especially for cases of severe genotype-phenotype truncating COL4 mutation.
In this study, in order to further explore the mutation mechanisms that lead to podocyte injury, we constructed an overexpressed podocyte model of four typical mutations, including three COL4A3 missense mutations, G619R, G801R, and C1616Y, and one truncated mutation, COL4A3 p. Thr1440ProfsX87.[8] We detected possible podocyte injury indicators (ERS and apoptosis), then applied a proteasome inhibitor (MG132) to reverse the decreased expression of COL4A3. In order to compensate for the disadvantage of the overexpressed cell model, a COL4A3 knockout mouse monoclonal podocyte model was established for additional verification. Our work might provide a theoretical basis for new therapeutic treatments for kidney diseases.
Methods
COL4A3 coding sequence mutations
Four COL4A3 coding sequence (CDS) mutations associated with FFSGS or autosomal recessive Alport's syndrome (ARAS) patients have been previously reported.[8] G801R and C1616Y mutations were identified in two FFSGS families, G619R and COL4A3 p. Thr1440ProfsX8 were identified from patients with ARAS. According to the Human Genome Variation Society nomenclature method,[11] the complicated frameshift mutation was named COL4A3 chr:228172489delA c.4317delA p. Thr1440ProfsX87 (the first changed amino acid was the site 1440 threonine, and a frameshift caused early termination of the 87 amino acids located after site 1440); however, in this paper, it is hereafter referred to as COL4A3 c.4317delA for ease of understanding.
Two missense heterozygous mutations (G801R and C1616Y) were identified from the FFSGS family, and one homozygous G619R missense mutation was identified from a patient with ARAS. One COL4A3 c.4317delA frameshift mutation in the NC1 domain that produced the truncated COL4A3 protein was identified from the FFSGS family. Detailed information regarding these mutations is summarized in Table 1 and Figure 1A.
Table 1.
The mutation from our previous original work[8].

Figure 1.
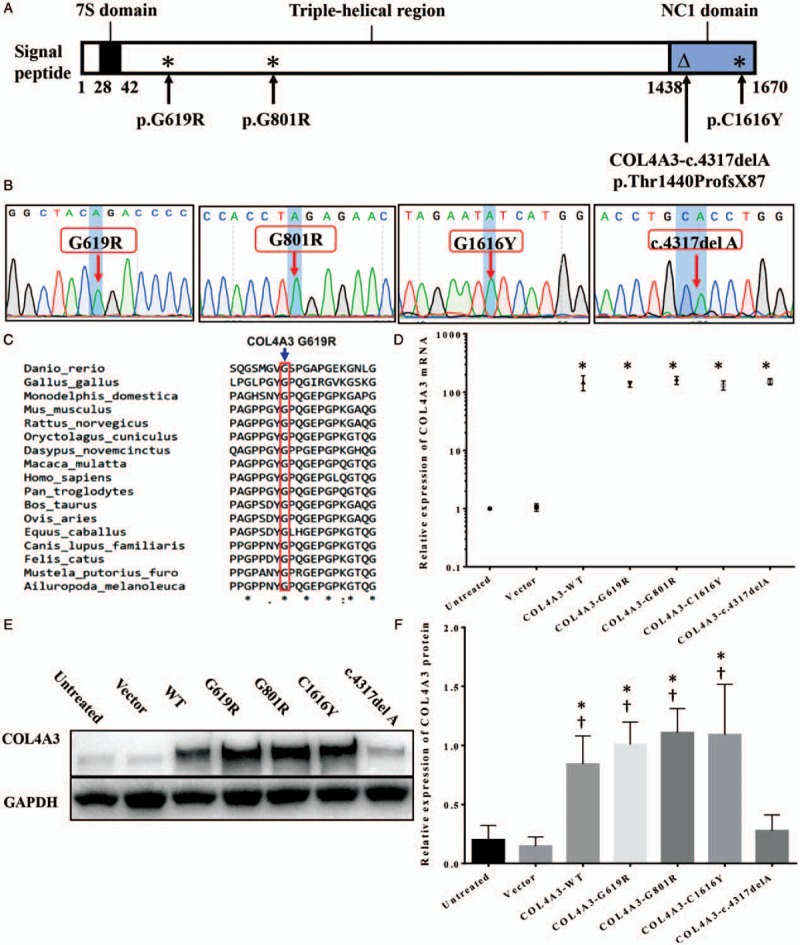
COL4A3 expression in WT and COL4A3-G619R, -G801R, -C1616Y, and c.4317delA (p.Thr1440ProfsX87) mutant transfected human podocyte cells. (A) Specific position of each COL4A3 mutation site in the COL4A3 protein that indicates the c.4317delA mutation was different from the others; it was a frameshift mutation at the DNA level that caused a truncated mutation in the protein level; (B) COL4A3 mutants constructed by lentivirus were confirmed using Sanger sequencing; (C) Evolutionary conservation analysis of the COL4A3 G619R mutation site. (D) Quantitative PCR detected COL4A3 mRNA expression levels in human podocyte cells stably transfected with different COL4A3 lentiviruses (∗compared to the empty vector group, P < 0.05); (E) Western blotting evaluated COL4A3 protein expression in human podocytes after transfection by different COL4A3 lentiviruses. The expression level of the COL4A3 c.4317delA (truncated mutation) group had an obviously lower COL4A3 protein level, unlike the WT, G619R, G801R, and C1616Y groups. (F) Three independent repetitions of COL4A3 protein expression experiment in different groups confirmed that the COL4A3 levels were differed significantly between the WT and c.4317delA groups (∗compared to the WT group, †compared to the c.4317delA group, P < 0.05). PCR: Polymerase chain reaction; WT: Wild-type.
Lentiviruses contained wild-type (WT) or mutant COL4A3 CDS
Polymerase chain reaction (PCR) amplification (KOD-Plus-Neo PCR enzyme, Toyobo, Japan) was used to acquire a full-length WT Human (Hum) COL4A3 CDS sequence, Hum-COL4A3 forward primer (5′-3′): ATGAGCGCCCGGACCG; Hum-COL4A3 reverse primer (5′-3′): TCAGTGTCTTTTCTTCATGCACACCTGAC. Four mutant COL4A3 sequences in G619R, G801R, C1616Y, and c.4317delA, respectively, were obtained by scaffold bridge PCR amplification (PrimeSTAR HS DNA Polymerase, TAKARA, Japan). Using a ClonExpress II One Step Cloning Kit (Vazyme Biotech, China), these sequences were then inserted into the multiple cloning site downstream cytomegalovirus (CMV) promoter in a pCDH-CMV-ATG2S3F-IRES-Blast lentiviral plasmid (Institute Pasteur of Shanghai, China); the CMV promoter would lead to high expression of the inserted target gene. After target gene sequence confirmation by Sanger DNA sequencing (GENEWIZ, China), the constructed plasmids were then transfected into 293T cells combined with envelope protein plasmid pCMV-VSV-G (Addgene Plasmid #8454) and packaging plasmid pCMV-dR8.2 dvpr (Addgene Plasmid #8455) to produce a lentivirus with either overexpressed WT or mutant COL4A3 (G619R, G801R, C1616Y, and c.4317delA). The successfully packaged lentivirus was stored at −70°C until use.
Human podocytes stably overexpressed WT or mutant COL4A3
Human podocytes (Icahn School of Medicine at Mount Sinai, New York, NY, USA) were cultured in 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (30 min at 56°C), 1% (vol/vol) insulin-transferrin-selenium (Thermo Fisher Scientific). To produce podocytes that stably overexpressed either WT or mutant COL4A3, the cells were infected with lentiviruses that contained WT COL4A3 or mutant COL4A3 (G619R, G801R, C1616Y, or COL4A3 c.4317delA), respectively. An empty lentiviral infection was used as a control. The cells were continuously screened with 5 ng/mL blasticidin antibiotic (Thermo Fisher Scientific), approximately 60% to 80% of the podocytes were killed between days 3 and 7. The surviving podocytes obviously multiplied after 1 week, then the stable lentivirus infection and virus replication were maintained by sustained 5 ng/mL blasticidin dosing. Until 3 to 4 weeks after transfection, the stably transfected podocytes and the untreated group were synchronously analyzed by Western blotting and real-time PCR. All transfections and experiments were performed in triplicate.
Mouse podocytes expressed NC1-truncated COL4A3
Referring to the practical protocol,[12,13] the clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9) gene-editing system was applied to establish a modified mouse podocyte that expressed NC1-truncated COL4A3. Single guide (sgRNA) was designed to target the NC1 domain in the mouse COL4A3 No. 52 exon using the http://crispr.mit.edu website (sgRNA sequence [5′-3′]: AGTTACATGTCCCTCGTCCA). The specific sgRNA was inserted downstream of the U6 promoter in the pSK-U6-gRNA plasmid. Mouse podocytes (Icahn School of Medicine at Mount Sinai) were transfected with pCDH-CMV-Cas9-GFP-Puro plasmid and pSK-U6-sgRNA using Opti-MEM medium and Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer's instructions. After 24 h, puromycin (0.5 μg/mL; Thermo Fisher Scientific) was added for up to 48 h for screening to kill most of the mouse podocytes. The remaining viable mouse podocytes were resuspended in 0.05% Trypsin-EDTA (Thermo Fisher Scientific). A single mouse podocyte was cultivated in each well of a 96-well plate using the limited dilution method and a manual cell counting instrument. After approximately 2 to 3 weeks, monoclonal mouse podocytes began to grow in the 96-well plate. Until approximately 4 weeks, confluent monoclonal mouse podocytes were digested by 0.05% Trypsin-EDTA and re-cultivated into new 24-well plates. DNA was extracted from the cultured cells from 24-well plate for Sanger DNA sequencing to confirm the mouse COL4A3 DNA modification.
MG132 and brefeldin A interference in stably transfected human podocytes
Human podocytes that stably overexpressed either WT or mutant COL4A3 were cultured on a 6-well plate. When they reached 80% confluence, 2.5 μmol/L MG132 (Sigma-Aldrich, Merck KGaA, Germany) and 10 mmol/L brefeldin A (Thermo Fisher Scientific) were added and left to stimulate the culture for 12 h. Total protein was extracted from each stable infected podocyte strain to detect the change in COL4A3 expression by Western blotting.
RNA isolation and real-time PCR
The mRNA levels of COL4A3, CHOP, and sXBP1 in human podocytes stably infected by a lentivirus containing either WT or mutant COL4A3 sequences were detected by real-time PCR. Total RNA was manually extracted using Trizol reagent (Thermo Fisher Scientific). According to the manufacturer's protocol, 0.5 mg of total RNA was reverse transcribed into cDNA utilizing the RT system (Promega, Madison, WI, USA). Real-time PCR was performed on an ABI System using SYBR Green Real-time PCR Master Mix (Vanzyme, China). The amplification primers for the target mRNA are shown in Table 2. Relative expression of the target genes was analyzed by normalization to the GAPDH housekeeping gene using the 2−ΔΔCt method.
Table 2.
Fluorescence quantitative PCR primers of human podocyte genes.
Immunoblotting
Total protein was extracted from each stable infected podocyte strain using RIPA buffer containing a protease inhibitor (Bimake, China) on a six-well plate. The target proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and detected by immunoblotting using polyvinylidene fluoride (PVDF) membranes (Merck KGaA, Germany). After blocking with 5% fat-free milk, the membranes were incubated with one of diluted anti-COL4α3 (1:2000 dilution; Novusbio, Centennial, CO, USA), -CHOP (1:2000 dilution; Cell Signaling Technology), -Grp78 (Bip) (1:2000 dilution; Cell Signaling Technology), -PERK (1:2000 dilution; Cell Signaling Technology, Danvers, MA, USA), -Grp94 (1:2000 dilution; Cell Signaling Technology), -eIF2α (1:2000 dilution; Cell Signaling Technology), -phospo-eIF2α (-p-eIF2; 1:2000 dilution; Cell Signaling Technology), -Bcl-2 (1:1000 dilution; ABclonal Technology, China), -cleaved caspase 3 (1:2000 dilution; Cell Signaling Technology), or anti-GAPDH (1:10000 dilution; Proteintech, China) primary antibodies. Then, the membranes were incubated for 1 h with 1:5000 diluted horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (Jackson Laboratory, Bar Harbor, ME, USA) and visualized using an ECL Western blotting system (Thermo Fisher Scientific).
Statistical analysis
Sequencher v 5.0 (Gene Codes Corporation, Ann Arbor, MI, USA), Snapgene v 2.3.2 (GSL Biotech LLC, Chicago, IL, USA), and DNASTAR Lasergene v 7.1 (DNASTAR, Madison, WI, USA) were applied to analyze all of the plasmids and DNA sequences. Evolutionary protein conservation analysis was performed using the ClustalW2 program (EMBL-EBI, United Kingdom). SgRNA was designed using the http://crispr.mit.edu website. Data were presented as the mean ± standard deviation. Differences between two groups were analyzed by Student's t test. Comparisons among three or more groups were analyzed by one-way analysis of variance. A P value less than 0.05 was considered statistically significant. Expression graphs were analyzed using GraphPad Prism v 5.04 software (GraphPad Software, San Diego, CA, USA) and statistical analyses were performed by using SPSS 22.0 (IBM, Armonk, New York, USA).
Results
COL4A3 overexpression in stably transfected human podocytes
All three missense point mutations, G619R, G801R, and C1616Y, were evolutionarily conserved [Figure 1C]. Since COL4A3 c.4317delA p. Thr1440ProfsX87 caused an early termination of COL4A3, it was not conserved. Analysis of the human podocytes that overexpressed either COL4A3-WT or G619R, G801R, C1616Y, or COL4A3 c.4317delA mutations [Figure 1A and 1B] showed that the CMV promoter in the lentiviral vector led to a remarkably higher expression level of COL4A3 mRNA in all cells with COL4A3-WT or the four mutations than that in the podocytes transfected with empty lentivirus (P < 0.05) [Figure 1D]; this indicated that all transfections were stable and successful. At the same time, podocytes transfected with the three COL4A3 missense point mutations (G619R, G801R, or C1616Y) showed significant overexpression of COL4A3 protein by Western blotting. However, the expression level of the COL4A3 c.4317delA group was close to that of the basal level of human podocytes in the untransfected control and empty vector transfected groups, and significantly lower than those in the cell lines with COL4A3-WT or the other three missense mutations (P < 0.05) [Figure 1E and 1F]. All groups that were stably transfected by the lentiviral vector and screened with blasticidin showed similar results over three experimental repetitions. These results suggested that the truncated COL4A3 mutation in the NC1 domain (c.4317delA) used a specific mechanism to inhibit truncated COL4A3 expression at the protein level.
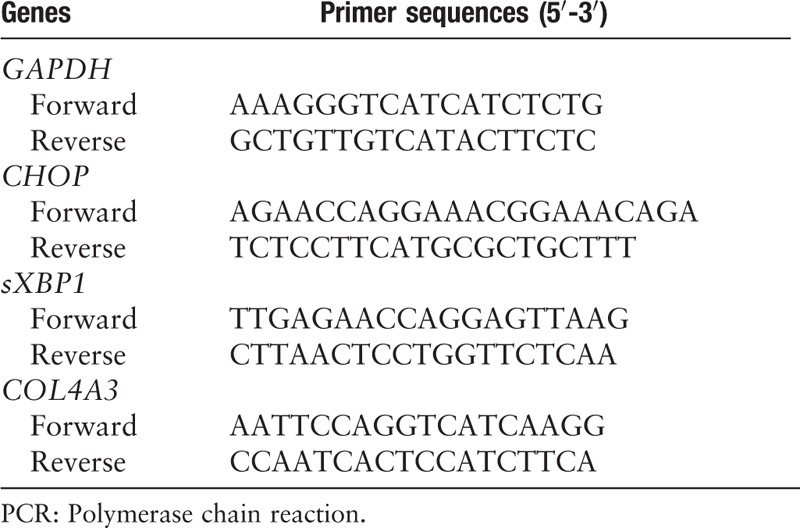
COL4A3 c.4317delA overexpression induced higher ERS- and apoptosis-related proteins in human podocytes
The previous findings demonstrated that ERS and the apoptosis pathway played a role in COL4A3 mutation-associated injury. So, we further detected ERS- and apoptosis-related protein expression in human podocytes that stably overexpressed WT or mutant COL4A3. Compared to the vector control and WT groups, both ERS- and apoptosis-related proteins in the G619R, G801R, and C1616Y groups did not differ significantly, although the mRNA levels of CHOP and sXBP-1 were significantly increased in cells with the C1616Y mutation [Figure 2]. However, the protein expression of ERS-related proteins, GRP78, GRP94, PERK, and apoptosis-related proteins, CHOP, and cleaved caspase 3, were obviously increased in the COL4A3 c.4317delA group (P < 0.05) compared to the WT group. However, apoptosis-related Bcl-2 protein expression decreased in the COL4A3 c.4317delA group compared to the WT group.
Figure 2.
Detection of ERS- and apoptosis-related proteins in human podocytes that stably overexpressed WT or mutant COL4A3. (A) mRNA detection of ERS related CHOP and sXBP-1 by real-time PCR. The results showed significantly increased ERS in the COL4A3, C1615Y, and c.4317delA groups compared to those of the WT group; (B) ERS-and apoptosis-related protein detection by Western blotting showed obviously increased ERS related proteins (GRP78, GRP94, PERK, and CHOP), increased apoptosis-related protein cleaved caspase 3, and decreased apoptosis-related Bcl-2 in the c.4317delA group compared the WT group; (C) Three independent repetitions of ERS- and apoptosis-related proteins expression detection experiments in different groups (∗compared to the COL4A3-WT group, P < 0.05). ERS: Endoplasmic reticulum stress; PCR: Polymerase chain reaction; WT: Wild-type.
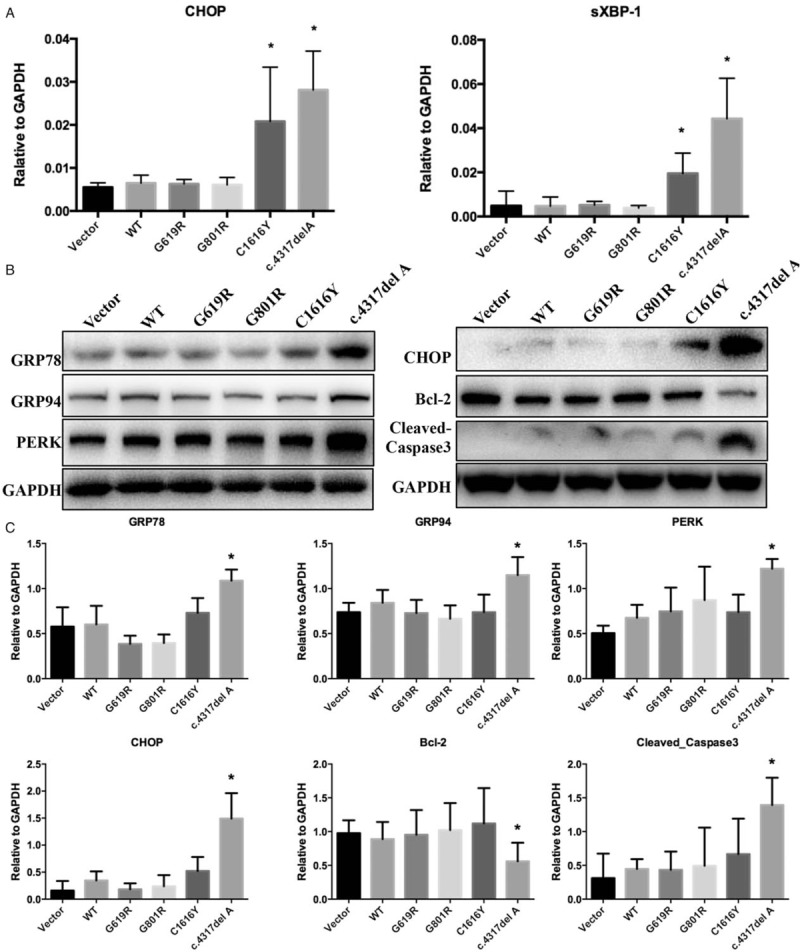
MG132 remarkably reversed lower basal intra-cellular COL4A3 expression and improved higher ERS and apoptosis in COL4A3 c.4317delA stably transfected human podocytes
We further explored the effect of proteasome degradation and endoplasmic reticulum transport pathways in human podocytes that stably overexpressed WT or mutant COL4A3 using a proteasome inhibitor (MG132), and an endoplasmic reticulum transport inhibitor (brefeldin A), respectively. The results showed that COL4A3 protein expression was slightly elevated in WT, G618R, G801R, G1616Y, and c.4317delA groups by brefeldin A intervention, while the WT group showed significantly higher expression. When COL4A3 was synthesized in the endoplasmic reticulum, the matured COL4A3 should have been transported to the Golgi apparatus for further assembly with COL4A4 and COL4A5; as this is the specific site where brefeldin A would interfere, this is where the effect was observed. Interestingly, the truncated protein (COL4A3 c.4317delA group) level was significantly increased by MG132 treatment. However, WT, and G618R, G801R, and G1616Y mutation groups showed no obvious effect when treated with MG132. Therefore, the protein degradation pathway may have played a role in inhibiting truncated COL4A3 mutation expression in the NC1 domain (c.4317delA) at the protein level. Further analysis showed MG132 intervention significantly decreased GRP78, CHOP, and cleaved caspase 3 protein levels, and increased the Bcl-2 protein level in the COL4A3 c.4317delA group [Figure 3C and 3D].
Figure 3.
The proteasome pathway and endoplasmic reticulum transport in human podocytes stably overexpressed wild-type or mutant COL4A3. (A) Protein expression in each group (COL4A3-WT and four mutations) after drug intervention (left lane-control; middle lane-brefeldin A 10 mmol/L, 12 h; right lane-MG132 2.5 μmol/L, 12 h). (B) Three independent repetitions of COL4A3 protein expression in different groups (∗compared to the control group, P < 0.05). COL4A3 protein expression is slightly elevated in G618R, G801R, and G1616Y groups by brefeldin A intervention. The WT group showed significantly higher expression. The COL4A3 c.4317delA group showed markedly different conditions; MG132 intervention produced significantly higher COL4A3 expression (P < 0.05) that appeared to reverse the lower COL4A3 expression of the untreated or brefeldin A treated groups. (C) ERS and apoptosis-related protein detection in the COL4A3 c.4317delA group after MG132 intervention (left two lanes were duplicates – control; right two lanes were duplicates – MG132 2.5 μmol/L, 12 h). (D) MG132 intervention significantly decreased ERS related proteins (GRP78 and CHOP) and apoptosis-related protein cleaved caspase 3, and increased Bcl-2 protein expression in the COL4A3 c.4317delA group (∗compared to the control group, P < 0.05). CHOP: C/EBP homologous protein; ERS: Endoplasmic reticulum stress; WT: Wild-type.
Mus COL4A3 knockout mouse monoclonal cell line showed ERS and apoptosis upregulation
The overexpressed podocyte model was not the best cell model to study mutated protein function, owing to the fact that the basal normal gene expression and strong CMV promoter produced excessive target protein levels beyond that of the normal physiological need. To establish a better cell model, we tried to construct a new COL4A3 podocyte model by applying the CRISPR/Cas9 system as described above.
First, we successfully established a COL4A3 knockout mouse monoclonal cell line, which was confirmed as COL4A3 chr:82717932insA c.4852insA p.Arg1618ThrfsX4 (the first changed amino acid was a site 1618 Arginine and caused early termination of the fourth amino acid after site 1618, hereafter referred to as COL4A3 c.4852insA) by Sanger sequencing [Figure 4A and 4B]. We chose this cell model because it is also frameshift mutation in the NC1 domain of COL4A3 that produced the truncated COL4A3 protein. The experiment produced two monoclonal mouse podocytes with the same mutation site from two independent transient transfection experiments (named Clone 1 and Clone 2). Compared to those of the WT group, the protein expression levels of PERK, GRP94, CHOP, and phospho-eIF2α increased obviously (eIF2α was unchanged), together with increased expression of cleaved-caspase 3, and downregulation of Bcl-2. These findings suggested a higher level of ERS and relatively remarkable level of apoptosis [Figure 4C].
Figure 4.
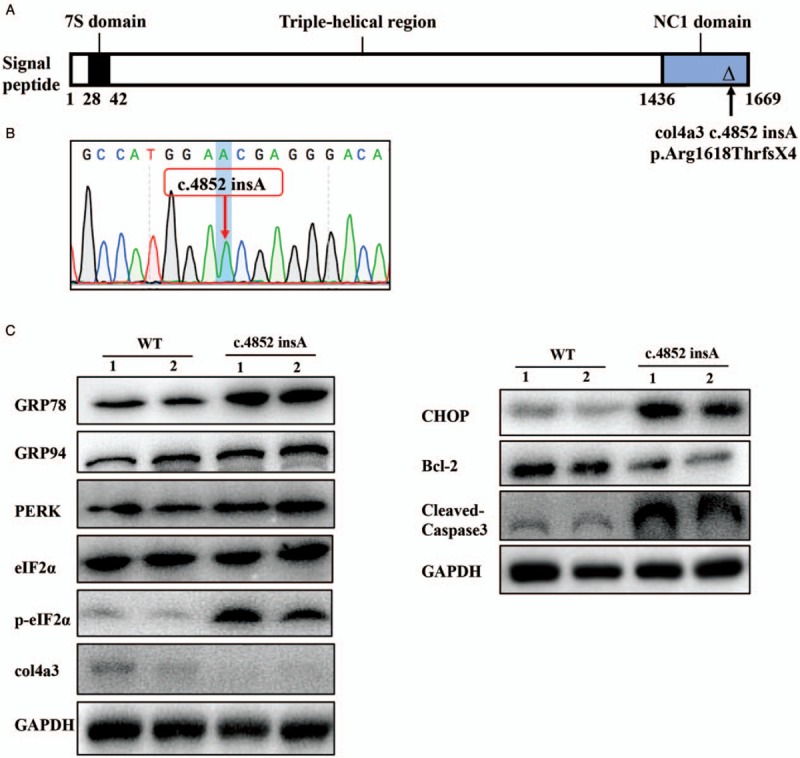
Detection of ERS- and apoptosis-related proteins in the mus COL4A3 knockout mouse monoclonal cell line. (A) Schematic diagram of the specific position of each COL4A3 mutation site in the COL4A3 protein. (B) Sequence analysis showed the generation of a knockout COL4A3 c.4852 insA p.Arg1618ThrfsX4 genotype in mouse podocytes using CRISPR/Cas9 technology. (C) Western blotting detection of mouse podocyte mutants (COL4A3 c.4852 insA p.Arg1618ThrfsX4); Clone 1 showed no obvious COL4A3 protein expression. Basal detectable COL4A3 protein expression could be observed in the WT group. This truncated mutation group presented increased ERS-related proteins (GRP78, GRP94, PERK, p-eIF2α, and CHOP), but eIF2α was unchanged. Apoptosis-related protein expression (CHOP and cleaved caspase-3) was upregulated, but Bcl-2 was downregulated. Lane 1 and 2 were duplicates; similar results were observed for the same mutation in clone 2. ERS: Endoplasmic reticulum stress; PERK: PKR-like endoplasmic reticulum kinase; p-eIF2α: Phosphorylated eukaryotic translation initiation factor 2α; WT: Wild-type.
Discussion
Podocytes are non-renewable and susceptible to damage from a variety of factors, including immune system disorders, genetic defects (gene mutation), and metabolic disorders.[14] Persistent or further aggravated injury can cause an imbalance in the stress and repair mechanisms, resulting in the activation of apoptosis-related genes that are related to the pathology and prognosis of glomerular diseases.[15] Podocyte injury is commonly observed in COL4-related nephropathy caused by COL4A mutations, and genotype-phenotype correlation in COL4-related nephropathy is relatively well documented.[16] Missense mutations usually exhibit milder phenotypic changes compared to those of truncating mutations; however, the underlying mechanism was unclear.
COL4A3 is normally glycosylated and folded in the endoplasmic reticulum before being transported to the Golgi apparatus, and subsequently, outside of the podocyte. It has been reported that COL4A3 missense mutations have been scattered along the entire collagen domain.[17] Some COL4A3 missense mutants, such as G1334E,[6] G871C, and G484R[18] might not be transported outside the podocyte, but instead, accumulate in the ER and cause an unfolded protein response activation. However, no significant ERS was observed in our three overexpressed missense point mutations. Our results could be explained by the fact that three novel missense mutations (G619R, G801R, and C1616Y) were used. Additionally, the different cell overexpression models may have also contributed to the difference; previous studies all used a transiently transfected cell model,[6,18] but ours used a stably transfected lentivirus system.
Unlike the missense mutation, the truncated mutation located in the NC1 domain (COL4A3 c.4317delA) could not be overexpressed by stably transfected lentivirus vector podocytes, and induced excessive ERS and apoptosis. Using the Cas9 modified knockout mouse podocyte model, we verified the same phenomenon; the NC1-truncated COL4A3 mutation caused higher levels of ERS and apoptosis. We speculated that among the four mutations found in a previous study,[8] the NC1-truncated mutation may beget severe podocyte injuries including induced ERS and increased apoptosis, whereas the three missense mutations may cause pathology mainly by changing GBM composition. This phenomenon requires further clarification.
In addition, we found that the NC1-truncated mutation could not be overexpressed in human podocytes by the stably transfected lentivirus vector. The MG132 proteasome inhibitor significantly elevated the mutated protein expression, whereas the transport protein inhibitor (brefeldin A) had little effect. These data indicated that the truncated COL4A3 protein was mainly degraded by the proteasome pathway, and moreover, caused excessive podocyte ERS and apoptosis.
MG132 is a peptide aldehyde that covalently binds to the active site of the 20S proteasome and effectively blocks the proteolytic activity of the 26S proteasome complex; it has been used in the study of multiple diseases.[19] MG132 reversed mutated COL4A3 expression and improved the higher ERS- and apoptosis status in the specific truncated COL4A3 C.4317delA mutation. Our results showed a potential therapeutic effect of MG132 that might function through regulating podocyte apoptosis; previously reported MG132 protection occurred by blocking HeLa cell apoptosis induced by severe DNA damage,[20] and also against deltamethrin induced apoptosis in rat hippocampus.[21] However, further experiments in podocytes, and validation in animal models are required.
This study performed COL4A3 gene editing using CRISPR/Cas9 technology; we established a COL4A3 knockout mouse monoclonal cell line (COL4A3 c.4852 insA p.Arg1618ThrfsX4) for result verification. Other studies using CRISPR/Cas9 editing have recently been published in several important studies in the field of kidney disease[22,23]: Using a CRISPR/Cas9-mediated gene knockout of GLA in HEK-293T cells provided a successful in vitro Fabry disease model,[24] and the CRISPR/Cas9 knockout of podocalyxin in podocyte-like cells[25] and conditional CRISPR/Cas9 in Drosophila garland cell nephrocytes found that reactive oxygen species formation might be a pathological mechanism of COQ2-nephropathy.[26] The gradual improvement of Cas9 technology could be quite useful in the advanced kidney research field. Compared to the WT group, our mutant mouse podocyte model showed excessive ERS and apoptosis-related podocyte injury. The results were consistent with our overexpression of truncated COL4A3 in the lentivirus transfected human podocyte model.
In summary, ERS and ERS-induced apoptosis activation were involved in podocyte injury caused by the COL4A3 truncated mutation that caused premature protein translation termination, and were not associated with three novel missense mutations (G619R, G801R, and C1616Y). These findings may partially explain the genotype-phenotype correlation in COL4-related nephropathy. Moreover, the proteasome pathway contributed to decreased COL4A3 expression caused by the severe NC1-truncated mutation, improved the higher ERS and apoptosis-related protein expression, and might be a potential avenue for treatment. In addition, CRISPR/Cas9 technology was used to successfully construct a COL4A3 knockout mouse podocyte model expressed COL4A3 truncated protein, which verified the excessive ERS and apoptosis. The results of this study require further validation in animal models.
Acknowledgements
The authors thank Prof. Ci-Jiang He (Division of Nephrology, Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, USA) for providing the human and mouse podocyte cell lines, and Prof. Lu-Bin Jiang (Unit of Human Parasite Molecular and Cell Biology, Institute Pasteur of Shanghai, China) for providing the plasmids for the experiment.
Funding
This work was supported by grants from the National Key Research and Development Program of China (No. 2016YFC0904100), National Natural Science Foundation of China (Nos. 81870460, 81570598, and 81370015), Science and Technology Innovation Action Plan of Shanghai Science and Technology Committee (No. 17441902200), Shanghai Municipal Education Commission, Gaofeng Clinical Medicine Grant (No. 20152207), Shanghai Jiao Tong University School of Medicine, Multi-Center Clinical Research Project (No. DLY201510), and the Shanghai Health and Family Planning Committee Hundred Talents Program (No. 2018BR37).
Conflicts of interest
None.
Footnotes
How to cite this article: Zhang HD, Huang JN, Liu YZ, Ren H, Xie JY, Chen N. Endoplasmic reticulum stress and proteasome pathway involvement in human podocyte injury with a truncated COL4A3 mutation. Chin Med J 2019;132:1823–1832. doi: 10.1097/CM9.0000000000000294
References
- 1.Pescucci C, Mari F, Longo I, Vogiatzi P, Caselli R, Scala E, et al. Autosomal-dominant Alport syndrome: natural history of a disease due to COL4A3 or COL4A4 gene. Kidney Int 2004; 65:1598–1603. doi: 10.1111/j.1523-1755.2004. 00560.x. [DOI] [PubMed] [Google Scholar]
- 2.Van Agtmael T, Bruckner-Tuderman L. Basement membranes and human disease. Cell Tiss Res 2010; 339:167–188. doi: 10.1007/s00441-009-0866-y. [DOI] [PubMed] [Google Scholar]
- 3.Halfter W, Monnier C, Muller D, Oertle P, Uechi G, Balasubramani M, et al. The bi-functional organization of human basement membranes. PloS One 2013; 8:e67660.doi: 10.1371/journal.pone.0067660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abrahamson DR. Role of the podocyte (and glomerular endothelium) in building the GBM. Semin Nephrol 2012; 32:342–349. doi: 10.1016/j.semnephrol.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gbadegesin RA, Hall G, Adeyemo A, Hanke N, Tossidou I, Burchette J, et al. Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J Am Soc Nephrol 2014; 25:1991–2002. doi: 10.1681/ASN.2013090976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pieri M, Stefanou C, Zaravinos A, Erguler K, Stylianou K, Lapathitis G, et al. Evidence for activation of the unfolded protein response in collagen IV nephropathies. J Am Soc Nephrol 2014; 25:260–275. doi: 10.1681/ASN.2012121217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song S, Tan J, Miao Y, Li M, Zhang Q. Crosstalk of autophagy and apoptosis: involvement of the dual role of autophagy under ER stress. J Cell Physiol 2017; 232:2977–2984. doi: 10.1002/jcp.25785. [DOI] [PubMed] [Google Scholar]
- 8.Xie J, Wu X, Ren H, Wang W, Wang Z, Pan X, et al. COL4A3 mutations cause focal segmental glomerulosclerosis. J Mol Cell Biol 2014; 6:498–505. doi: 10.1093/jmcb/mju040. [DOI] [PubMed] [Google Scholar]
- 9.Xie J, Hao X, Azeloglu EU, Ren H, Wang Z, Ma J, et al. Novel mutations in the inverted formin 2 gene of Chinese families contribute to focal segmental glomerulosclerosis. Kidney Int 2015; 88:593–604. doi: 10.1038/ki.2015.106. [DOI] [PubMed] [Google Scholar]
- 10.Kashtan CE, Ding J, Garosi G, Heidet L, Massella L, Nakanishi K, et al. Alport syndrome: a unified classification of genetic disorders of collagen IV alpha345: a position paper of the Alport Syndrome Classification Working Group. Kidney Int 2018; 93:1045–1051. doi: 10.1016/j.kint.2017.12.018. [DOI] [PubMed] [Google Scholar]
- 11.Horaitis O, Cotton RG. The challenge of documenting mutation across the genome: the human genome variation society approach. Hum Mutat 2004; 23:447–452. doi: 10.1002/humu.20038. [DOI] [PubMed] [Google Scholar]
- 12.Moyer TC, Holland AJ. Generation of a conditional analog-sensitive kinase in human cells using CRISPR/Cas9-mediated genome engineering. Methods Cell Biol 2015; 129:19–36. doi: 10.1016/bs.mcb.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013; 8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reiser J, Altintas MM. Podocytes. F1000Res 2016; 5:114.doi: 10.12688/f1000research.7255.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shankland SJ. The podocyte's response to injury: role in proteinuria and glomerulosclerosis. Kidney Int 2006; 69:2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- 16.Nozu K, Nakanishi K, Abe Y, Udagawa T, Okada S, Okamoto T, et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin Exp Nephrol 2019; 23:158–168. doi: 10.1007/s10157-018-1629-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storey H, Savige J, Sivakumar V, Abbs S, Flinter FA. COL4A3/COL4A4 mutations and features in individuals with autosomal recessive Alport syndrome. J Am Soc Nephrol 2013; 24:1945–1954. doi: 10.1681/ASN.2012100985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papazachariou L, Demosthenous P, Pieri M, Papagregoriou G, Savva I, Stavrou C, et al. Frequency of COL4A3/COL4A4 mutations amongst families segregating glomerular microscopic hematuria and evidence for activation of the unfolded protein response. Focal and segmental glomerulosclerosis is a frequent development during ageing. PloS One 2014; 9:e115015.doi: 10.1371/journal.pone.0115015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rivas A, Vidal RL, Hetz C. Targeting the unfolded protein response for disease intervention. Expert Opin Ther Targets 2015; 19:1203–1218. doi: 10.1517/14728222.2015.1053869. [DOI] [PubMed] [Google Scholar]
- 20.Zhang L, Hu JJ, Gong F. MG132 inhibition of proteasome blocks apoptosis induced by severe DNA damage. Cell Cycle 2011; 10:3515–3518. doi: 10.4161/cc.10.20.17789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang X, Liang Y, Qing Y, Chen D, Shi N. Proteasome inhibition by MG-132 protects against deltamethrin-induced apoptosis in rat hippocampus. Life Sci 2019; 220:76–83. doi: 10.1016/j.lfs.2019.01.041. [DOI] [PubMed] [Google Scholar]
- 22.Higashijima Y, Hirano S, Nangaku M, Nureki O. Applications of the CRISPR-Cas9 system in kidney research. Kidney Int 2017; 92:324–335. doi: 10.1016/j.kint.2017.01.037. [DOI] [PubMed] [Google Scholar]
- 23.Miyagi A, Lu A, Humphreys BD. Gene editing: powerful new tools for nephrology research and therapy. J Am Soc Nephrol 2016; 27:2940–2947. doi: 10.1681/ASN.2016020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song HY, Chiang HC, Tseng WL, Wu P, Chien CS, Leu HB, et al. Using CRISPR/Cas9-mediated GLA gene knockout as an in vitro drug screening model for Fabry disease. Int J Mol Sci 2016; 17:2089.doi: 10.3390/ijms17122089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Freedman BS, Brooks CR, Lam AQ, Fu H, Morizane R, Agrawal V, et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat Commun 2015; 6:8715.doi: 10.1038/ncomms9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hermle T, Braun DA, Helmstadter M, Huber TB, Hildebrandt F. Modeling monogenic human nephrotic syndrome in the Drosophila Garland cell nephrocyte. J Am Soc Nephrol 2017; 28:1521–1533. doi: 10.1681/ASN.2016050517. [DOI] [PMC free article] [PubMed] [Google Scholar]