

Abstract
Supplemental Digital Content is available in the text
To the Editor: Multiple acyl-CoA dehydrogenation deficiency is an autosomal recessive lipid storage myopathy (LSM).[1] The clinical feature is heterogeneous and has been classified into three classes: a neonatal-onset form with congenital anomalies (type I), a neonatal-onset form without congenital anomalies (type II), and a late-onset form (type III).[2] Over the past 20 years, an increasing number of cases with LSM have been reported.[3,4] In this report, we described a patient with LSM harboring compound heterozygous mutations in ETFDH gene, including a novel splice mutation in exon 6 (c.684+1G>T) and a novel point mutation in exon 10 (c.1204A>T).
The patient was a 17-year-old female who complained of episodic vomiting, exercise intolerance, and muscle weakness. Her symptoms started at 13 years and the initial symptom was vomiting after a meal. At the beginning, muscle fatigue and exercise intolerance after intense exercise only affected her lower limbs. The symptoms progressed slowly in the past 4 years. Her upper limbs, as well as neck muscles were involved now. The patient had no complaint about muscle stiffness, myalgia, or numbness. Physical examination indicated that she had no obviously muscle atrophy. The muscle strength was 4 to 5 (Medical Research Council Scale) in proximal lower limbs, 4/5 in proximal upper limbs, and almost normal in distal limbs. Deep tendon reflexes were diminished and sensory examination was intact. No abnormality was evident in the serum blood, except for mild elevated creatinine kinase level (312 IU/L, normal 26–192 IU/L) and slightly increased lactate dehydrogenase level (112 IU/L, normal 8–46 IU/L). Echocardiography examinations showed the four cardiac chambers were normal in size and function. Muscle strength improved after treatment with riboflavin.
On muscle magnetic resonance imaging (MRI), T1 [Supplementary Figure 1A upper panel] and T2 [Supplementary Figure 1B upper panel] showed slightly high signal in the posterior thigh muscle group. Fat suppression [Supplementary Figure 1C upper panel] and diffusion weighted imaging [Supplementary Figure 1D upper panel] sequence revealed increased signal in semitendinosus and semimembranosus [Supplementary Figure 1C, D upper panel]. In addition, the result changed to normal after treatment with riboflavin for 5 months [Supplementary Figure 1A–D lower panel]. The MRI changes in this patient was consistent with other cases preciously described,[5] but it seems that the involvement of the thigh was mild, because there is no obviously atrophy or fat degeneration. After treatment with riboflavin, the high signal on the semitendinosus and semimembranosus disappeared 5 months later. This fact indicated that the MRI might be used to monitor the efficacy of the treatment.
Hematoxylin and eosin staining revealed mildly increased variation in fiber size, increased numbers of basophilia fibers with cytoplasmic or subsarcolemmal vacuoles [Figure 1A]. The oil red O staining showed diffuse lipid droplets accumulation, predominantly in type I fibers, suggesting LSM [Figure 1B].
Figure 1.
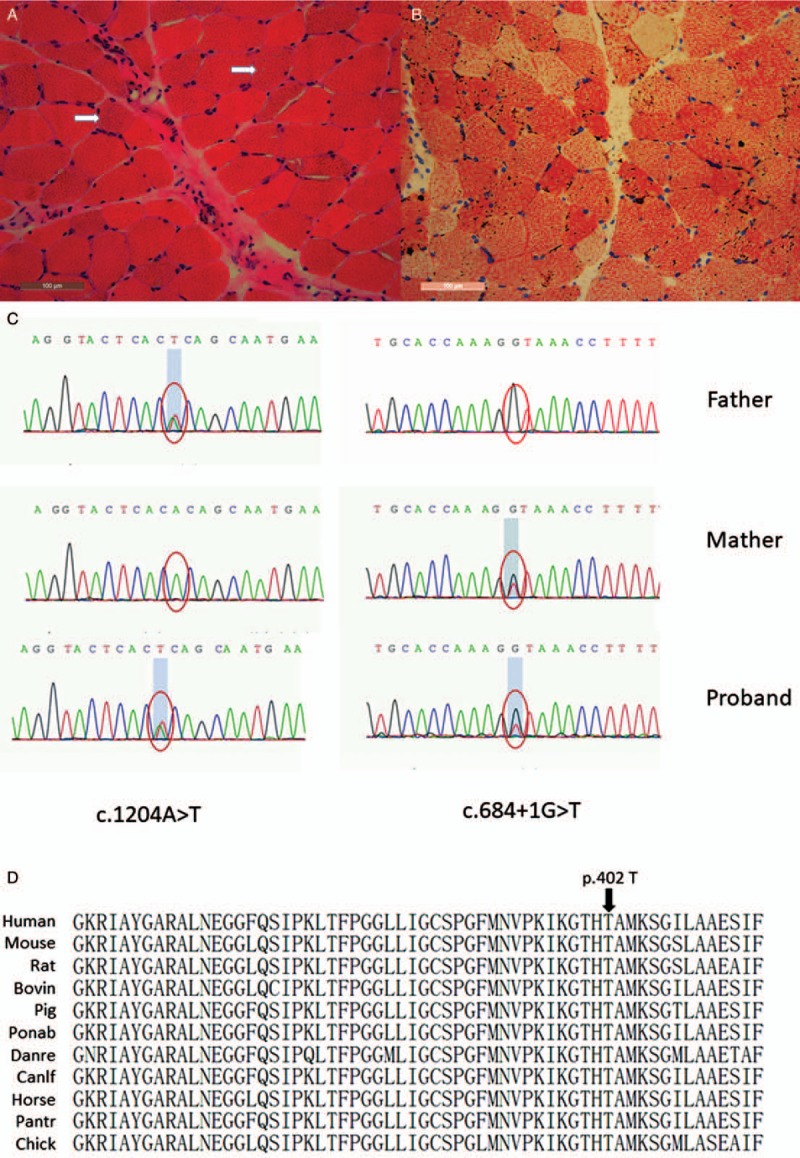
Muscle pathology of the patient with lipid storage myopathy. (A) Hematoxylin and eosin staining showed increased variation in fiber size, increased numbers of basophilia fibers with cytoplasmic or subsarcolemmal vacuoles (arrows, original magnification ×200). (B) Oil red O staining showed lipid droplets filled type I muscle fibers (original magnification ×200). Mutation analysis of ETFDH gene. (C) Two novel mutations of ETFDH gene from the proband and the proband's parents (c.684+1G>T and c.1204A>T, pThr402Ser). (D) A homology search in different species demonstrates that the threoine at 402 residue is highly evolutionarily conserved in different species.
Targeted next-generation sequencing identified two novel heterozygous mutations: a splicing mutation (c.684+1G>T) in exon 6 and a missense (c.1204A>T) in exon 10, which caused substitution of threonine with serine at 402 residue (p.Thr402Ser) [Figure 1C]. Sanger sequencing confirmed the two mutations in the proband. Family study showed the proband's mother had a G-to-T transversion (c.684+1G>T) in exon 6, while her father had an A-to-T transition (c.1204A>T) in exon 10. These results were consistent with autosomal recessive mode of Mendelian inheritance. The splicing mutation (c.684+1G>T) and missense mutation (c.1204A>T) were not found in 200 healthy Chinese controls, 1000 genome database, and ExAC database. A homology search in different species demonstrates that the threonine at 402 residue is highly evolutionarily conserved [Figure 1D]. The mutation is predicted to be disease causing by MutationTaster with probability of 0.999997. The splicing mutation (c.684+1G>T in exon 6) will result in production of a nonfunctional protein, and is generally rated disease causing. The missense mutation (c.1204A>T) caused a substitution of the conserved amino acid threonine to serine. This site was highly conserved between species, and the mutation was predicted to be disease causing by MutationTaster. This suggests that the mutation might affect the protein function. Therefore, the two mutations here are probably responsible for the development of LSM in this patient.
In conclusion, two novel mutations (c.1204A>T; p.Thr402Ser, and c.684+1G>T) were identified in this Chinese female patient with LSM. This expands the known mutational spectrum of ETFDH.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given her consent for her images and other clinical information to be reported in the journal. The patient understand that her name and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Funding
This work was supported by grants from The Young Scientists Fund of the National Natural Science Foundation of China (No. 81601093), and The Young Scientists Fund of the First Affiliated Hospital of Zhengzhou University (2015).
Conflicts of interest
None.
Supplementary Material
Footnotes
How to cite this article: Xu HL, Lian YJ, Chen X, Zhang L, Cheng X. Two novel ETFDH mutations in a patient with lipid storage myopathy. Chin Med J 2019;132:1876–1878. doi: 10.1097/CM9.0000000000000310
References
- 1.Missaglia S, Tavian D, Moro L, Angelini C. Characterization of two ETFDH mutations in a novel case of riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Lipids Health Dis 2018; 17:254.doi: 10.1186/s12944-018-0903-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grünert SC. Clinical and genetical heterogeneity of late-onset multiple acyl-coenzyme A dehydrogenase deficiency. Orphanet J Rare Dis 2014; 9:117.doi: 10.1186/s13023-014-0117-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angelini C, Tavian D, Missaglia S. Heterogeneous phenotypes in lipid storage myopathy due to ETFDH gene mutations. JIMD Rep 2018; 38:33–40. doi: 10.1007/8904_2017_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lan MY, Fu MH, Liu YF, Huang CC, Chang YY, Liu JS, et al. High frequency of ETFDH c.250G>A mutation in Taiwanese patients with late-onset lipid storage myopathy. Clin Genet 2010; 78:565–569. doi: 10.1111/j.1399-0004.2010.01421.x. [DOI] [PubMed] [Google Scholar]
- 5.Liu XY, Jin M, Wang ZQ, Wang DN, He JJ, Lin MT, et al. Skeletal muscle magnetic resonance imaging of the lower limbs in late-onset lipid storage myopathy with electron transfer flavoprotein dehydrogenase gene mutations. Chin Med J 2016; 129:1425–1431. doi: 10.4103/0366-6999.183423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.