

Macrophages are critical mediators of innate immunity and must be overcome for bacterial pathogens to cause disease. The Gram-positive bacterium Staphylococcus aureus produces virulence factors that impede macrophages and other immune cells. We previously determined that production of the metabolic cofactor lipoic acid by the lipoic acid synthetase, LipA, blunts macrophage activation. A ΔlipA mutant was attenuated during infection and was more readily cleared from the host.
KEYWORDS: Staphylococcus aureus, lipoic acid, macrophages, metabolism, reactive nitrogen species, reactive oxygen species, virulence
ABSTRACT
Macrophages are critical mediators of innate immunity and must be overcome for bacterial pathogens to cause disease. The Gram-positive bacterium Staphylococcus aureus produces virulence factors that impede macrophages and other immune cells. We previously determined that production of the metabolic cofactor lipoic acid by the lipoic acid synthetase, LipA, blunts macrophage activation. A ΔlipA mutant was attenuated during infection and was more readily cleared from the host. We hypothesized that bacterial lipoic acid synthesis perturbs macrophage antimicrobial functions and therefore hinders the clearance of S. aureus. Here, we found that enhanced innate immune cell activation after infection with a ΔlipA mutant was central to attenuation in vivo, whereas a growth defect imparted by the lipA mutation made a negligible contribution to overall clearance. Macrophages recruited to the site of infection with the ΔlipA mutant produced larger amounts of bactericidal reactive oxygen species (ROS) and reactive nitrogen species (RNS) than those recruited to the site of infection with the wild-type strain or the mutant strain complemented with lipA. ROS derived from the NADPH phagocyte oxidase complex and RNS derived from the inducible nitric oxide synthetase, but not mitochondrial ROS, were critical for the restriction of bacterial growth under these conditions. Despite enhanced antimicrobial immunity upon primary infection with the ΔlipA mutant, we found that the host failed to mount an improved recall response to secondary infection. Our data suggest that lipoic acid synthesis in S. aureus promotes bacterial persistence during infection through limitation of ROS and RNS generation by macrophages. Broadly, this work furthers our understanding of the intersections between bacterial metabolism and immune responses to infection.
INTRODUCTION
The innate immune system represents a fast-acting initial line of defense to fight infection. In order to withstand innate defenses, bacterial pathogens, like the Gram-positive bacterium Staphylococcus aureus, produce a wide array of virulence factors that inhibit innate immune cell recruitment and antimicrobial activity or that directly target and kill phagocytic leukocytes, thereby facilitating pathogenesis (1–4). S. aureus is a commensal bacterium that asymptomatically colonizes the anterior nares and skin of nearly one-third of the population. Upon a breach to physical barriers to infection, S. aureus can cause a wide range of diseases, including skin lesions/cellulitis, osteomyelitis, pneumonia, endocarditis, bacteremia, and sepsis. Although a recent report by the Centers for Disease Control and Prevention detailed a steady decline in the incidence of health care-associated (HA) methicillin-resistant S. aureus (MRSA) infections from 2005 to 2012, between 2013 and 2016 this decline slowed (5). Furthermore, community-associated (CA)-MRSA infection rates have decreased at a much lower rate (5). Alarmingly, the rates of HA methicillin-susceptible S. aureus (MSSA) infections have not decreased, and CA-MSSA infections are steadily rising (5). Therefore, S. aureus poses a significant threat to human health, driving the need to understand the mechanisms by which the bacterium causes disease and how the host responds to infection.
Neutrophils and macrophages are phagocytic leukocytes of the innate immune system that protect the host by killing pathogens. Upon direct recognition via pattern recognition receptors or through the assistance of opsonins, macrophages and neutrophils phagocytose invading bacteria into phagosomes, which mature to form localized sites of microbial killing (6). Processes such as phagosome acidification, the delivery of antimicrobial peptides and proteins, the influx of bactericidal molecules from neutrophil granules, and the sequestration of microbial growth-promoting metals all facilitate growth inhibition or killing of internalized bacteria (6–8).
A major antimicrobial activity of neutrophils and macrophages is a process known as respiratory burst, which generates potent reactive oxygen species (ROS) (9). The generation of ROS occurs through the NADP (NADPH) phagocyte oxidase complex that is assembled on the phagosomal membrane (10). The NADPH oxidase complex comprises two transmembrane proteins, gp91phox and p22phox, in addition to four cytosolic proteins denoted p47phox, p67phox, p40phox, and Rac1 or Rac2 (10–13). Upon assembly and activation of the NADPH oxidase complex, it transfers electrons from NADPH to molecular oxygen, releasing superoxide (O2·−) in the phagosome (12). Superoxide can undergo spontaneous dismutation into hydrogen peroxide (H2O2) or hydroxyl radicals and other ROS (7, 8, 10, 12, 14). H2O2 can then be converted into hypochlorous acid and chloramines by myeloperoxidase (7, 15). These diverse ROS lead to the oxidization of internalized bacterial DNA, the mobilization of iron from iron sulfur-containing dehydratases promoting toxic Fenton chemistry, and the oxidation of protein residues (6, 10, 16). Collectively, these damaging effects facilitate the killing of internalized bacteria. NADPH oxidase dysfunction can lead to severe health conditions, such as chronic granulomatous disease (CGD) (17, 18). Patients with CGD commonly suffer from recurrent infections with S. aureus, as well as the fungal pathogen Aspergillus, highlighting the importance of NADPH oxidase in host defense (17, 18). In addition to NADPH oxidase-derived ROS, mitochondrial respiration also generates ROS that contribute to the destruction of internalized bacteria (19). Mitochondrial ROS (mROS) are generated when electrons from the oxidative phosphorylation machinery escape and react with molecular oxygen (20, 21). Recent studies demonstrate that mROS facilitate the inhibition of internalized S. aureus (22, 23). In summary, the production of ROS by phagocytes is important for restricting S. aureus growth.
The synthesis of nitric oxide (NO·) and the subsequent formation of reactive nitrogen species (RNS) occur in macrophages and lead to the killing of internalized bacterial pathogens (10). In response to engulfed pathogens, inducible nitric oxide synthetase (iNOS) is activated and leads to the formation of NO· and citrulline from l-arginine and oxygen (24). In the presence of oxygen, NO· is converted into nitrogen dioxide, peroxynitrite (ONOO−), dinitrogen trioxide, and other nitrogen-based reactive species, collectively forming RNS (10, 25). RNS inhibit bacterial respiration, perturb DNA replication, interfere with metal centers and tyrosine residues in proteins, and alter lipid integrity (10, 26, 27). Given the similarities in the reactivity and formation of ROS and RNS, production of both species can lead to synergistic antimicrobial effects (28). In particular, ONOO− is formed from NADPH oxidase-derived O2·− and iNOS-generated NO·, establishing a link between ROS and RNS production (10, 27). S. aureus has adapted to resist the antimicrobial effects of the ROS and RNS produced by phagocytes (29–33).
In addition to killing phagocytosed bacteria, macrophages also produce and secrete a variety of cytokines and chemokines that help to regulate both the innate and the adaptive immune systems (34–39). Though they are less efficient than dendritic cells, activated macrophages are capable of presenting antigens to engage with T cells and promote their activation (34, 40). Given their potent antimicrobial activities and critical immune-signaling functions, macrophages represent a significant mediator of the immune response to infection that S. aureus must overcome in order to cause disease.
We recently determined that S. aureus synthesis of the metabolic cofactor lipoic acid via the lipoic acid synthetase (LipA) blunts optimal macrophage activation by S. aureus (41). Found in all kingdoms of life, lipoic acid is an essential organosulfur cofactor covalently linked to proteins in large multisubunit metabolic complexes, such as pyruvate dehydrogenase (PDH) (42, 43). As a cofactor, lipoic acid engages in redox coupling in oxidative and one-carbon metabolism (44). S. aureus synthesizes lipoic acid via LipA and also salvages it from the environment through the use of lipoic acid ligases (45, 46). These diverse lipoic acid synthesis and acquisition schemes contribute to the ability of S. aureus to colonize host tissues (46). Aside from its central role in metabolism, lipoic acid also has immunosuppressive properties. High concentrations of free lipoic acid reduce the respiratory burst of neutrophils and block the translocation of the transcription factor NF-κB to the nucleus (47–51). Furthermore, free lipoic acid activates the phosphoinositide 3-kinase/Akt signaling axis to limit inflammatory cytokine production (52).
We recently determined that the lipoylated E2 subunit of pyruvate dehydrogenase (E2-PDH) is released from S. aureus, where it moonlights as an immunomodulatory factor by dampening macrophage Toll-like receptor 1/2 (TLR1/2) recognition of S. aureus lipoproteins (41). Moreover, mice infected with a ΔlipA mutant of S. aureus more readily clear infection than mice infected with wild-type (WT) S. aureus (41). Because lipoic acid synthesis is required for essential metabolic processes and has inhibitory effects on immunity, the attenuation of a ΔlipA mutant in vivo may stem either from growth deficiencies associated with lipoic acid limitation or from the improved clearance of bacteria by activated macrophages. The host represents a lipoic acid-limiting environment that requires S. aureus strains with defects in de novo lipoic acid synthesis to acquire trace amounts of the cofactor via salvage in order to survive (46). Thus far, our studies have not conclusively determined if attenuation of a ΔlipA mutant in vivo is caused by a growth defect associated with the paucity of environmental free lipoic acid or enhanced innate immune responses. However, we have demonstrated that the clearance of a ΔlipA mutant correlates with the presence of greater proportions of activated macrophages during systemic infection. Upon isolation, these cells have an improved capacity to limit bacterial growth ex vivo (41).
In this study, we sought to further examine how bacterial lipoic acid synthesis impedes infection resolution. We found that the clearance of a ΔlipA mutant during systemic infection is primarily associated with the ability of macrophages to control infection. Macrophages elicited during the course of infection with a ΔlipA mutant produced larger amounts of ROS and NO·. The ROS generated through NADPH oxidase, but not mitochondria, were necessary for macrophages from ΔlipA mutant-infected mice to restrict bacterial growth. Further, upon inhibition of iNOS, which blocks NO· production, we observed that macrophages from ΔlipA mutant-infected mice lost their heightened ability to restrict S. aureus growth. Thus, S. aureus lipoic acid synthesis compromises macrophage function through interference with the NADPH oxidase- and iNOS-derived production of ROS and RNS. Additionally, we demonstrate that the improved antimicrobial capacity of macrophages during infection with a ΔlipA mutant does not confer protection against secondary S. aureus challenge. In sum, our data suggest that lipoic acid synthesis in S. aureus blunts optimal macrophage ROS- and RNS-mediated bacterial clearance during infection, thereby aiding in the ability of the bacterium to disseminate and cause disease.
RESULTS
Macrophages are responsible for attenuation of a ΔlipA mutant during systemic infection.
To begin to determine the extent of attenuation of a ΔlipA mutant in vivo and the relative contribution of macrophages to this phenotype, we first infected mice with the WT, the ΔlipA mutant, and the ΔlipA mutant complemented with lipA (the ΔlipA + lipA strain) via intraperitoneal (i.p.) injection and enumerated the CFU at 16 h and 72 h postinfection. As early as 16 h after intraperitoneal infection, we observed a modest 5-fold decrease in the number of CFU recovered from the peritoneal lavage fluid of ΔlipA mutant-infected mice compared to the number recovered from WT strain-infected mice (Fig. 1A). This was also true in the kidneys, where we observed a 4-fold reduction in the number of CFU of the ΔlipA mutant compared to that of the WT strain (Fig. 1A). Genetic complementation of the ΔlipA mutant partially restored the number of CFU recovered to WT levels, but statistical significance (P < 0.05) was not achieved (53). At 72 h postinfection, the number of CFU recovered from the lavage fluid of mice infected with the ΔlipA mutant was significantly less than the number recovered from the lavage fluid of mice infected with the WT strain, with partial complementation occurring after infection with the ΔlipA + lipA strain (Fig. 1B). The number of CFU recovered from the kidneys of ΔlipA mutant-infected mice was greater than 100-fold less than the number recovered from the kidneys of WT strain-infected mice, an effect that was fully complemented by the ΔlipA + lipA strain (Fig. 1B). Moreover, we were unable to recover detectable numbers of CFU from a substantial proportion of the lavage fluid samples and kidneys (50% and 56%, respectively) from ΔlipA mutant-infected mice after 72 h (Fig. 1B). There were no differences noted on the basis of the sex of the mice. These data indicate that the ΔlipA mutant is attenuated in vivo and is rapidly cleared from infected mice within 72 h.
FIG 1.
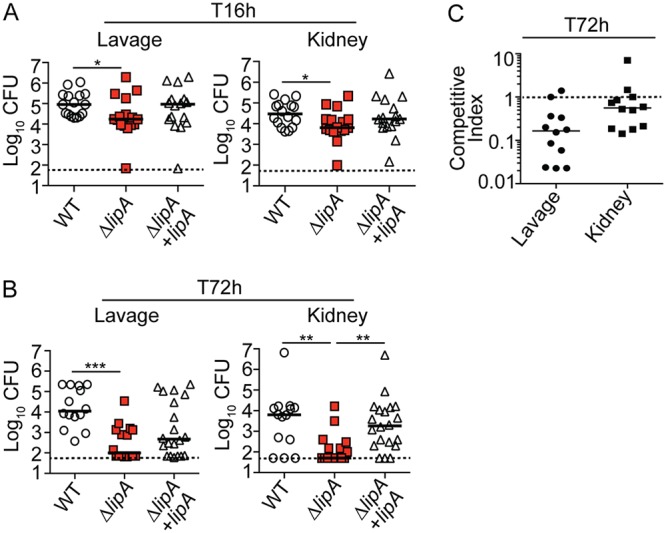
The ΔlipA mutant is attenuated during systemic infection. Bacterial burden (log10 number of CFU) in the peritoneal cavity and kidneys of mice at 16 h (T16h) after intraperitoneal infection with the WT (n = 16), ΔlipA (n = 16), or ΔlipA + lipA (n = 16) strain (A) or 72 h (T72h) after intraperitoneal infection with the WT (n = 14), ΔlipA (n = 16), or ΔlipA + lipA (n = 20) strain (B). Bars represent means. P values were determined by the Kruskal-Wallis test with Dunn’s posttest. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Dashed line, limit of detection. (C) Competitive index for the lipA::erm and WT strains after coinfection (n = 12) with a 50/50 mix of the WT and lipA::erm strains for 72 h. Dashed line, competitive index of 1.
Our prior studies indicated that (i) a ΔlipA mutant is unable to replicate in the absence of exogenous lipoic acid and (ii) activated antimicrobial macrophages are enriched at the site of infection with a ΔlipA mutant in vivo (41). Therefore, we wondered if the attenuation of the ΔlipA mutant is caused by a growth defect associated with lipoic acid limitation or if clearance is mediated by enhanced macrophage activity. We first determined if the defect of the ΔlipA mutant is rescued by WT S. aureus during mixed infection. Indeed, the attenuation of the ΔlipA mutant was largely abrogated (Fig. 1C). The ΔlipA mutant was outcompeted by the WT by approximately 8-fold in the lavage fluid and 2-fold in the kidney, whereas a 100-fold reduction in the number of CFU was seen in monoculture infections (Fig. 1C). We then depleted mice of macrophages and their precursors by administering clodronate-loaded liposomes 3 days prior to infection with 1 × 107 CFU of the WT, ΔlipA, or ΔlipA + lipA strain. Clodronate-treated mice had significantly reduced proportions of macrophages (Cd11b+ Gr1− CD11c− Ly6G− F4/80+) in the peritoneal cavity compared to nontreated mice (Fig. 2A). At 16 h postinfection, we recovered nearly identical numbers of CFU from the peritoneal cavities of clodronate-treated mice infected with the WT, ΔlipA, or ΔlipA + lipA strain (Fig. 2B). In the kidney, we recovered a wide range of numbers of CFU from clodronate-treated mice; however, there were no differences in the distribution or the average number of CFU from animals infected with the WT, ΔlipA, or ΔlipA + lipA strain. At 72 h postinfection, there were no discernible differences in the number of CFU recovered from the lavage fluid or the kidneys of S. aureus-infected clodronate-treated mice regardless of the strain used (Fig. 2C). There were no differences noted on the basis of the sex of the mice. In sum, these data suggest that in a peritonitis infection model, recruited macrophages are a primary mediator of clearance of the ΔlipA mutant during infection, arguing against a substantial growth defect imposed by lipoic acid limitation in vivo.
FIG 2.

The ΔlipA mutant is not attenuated in macrophage-depleted mice. (A) Abundance of macrophages (CD11b+ Gr1− CD11c− Ly6G− F4/80+) in the peritoneal cavity of clodronate-treated mice or nontreated mice at 72 h after intraperitoneal infection with WT S. aureus. Representative flow cytometry plots are shown along with composite data from four clodronate-treated mice and four nontreated mice. Bars represent the median. Data were analyzed using an unpaired, two-tailed Student's t test. **, P < 0.01. FSC-A, forward scatter area. (B and C) Bacterial burden (log10 number of CFU) in the peritoneal cavity and kidneys of clodronate-treated mice at 16 h after intraperitoneal infection with the WT (n = 18), ΔlipA (n = 18), or ΔlipA + lipA (n = 18) strain (B) or 72 h after intraperitoneal infection with the WT (n = 13), ΔlipA (n = 12), or ΔlipA + lipA (n = 11) strain (C). Bars represent means. Comparisons between groups in panels B and C were not statistically significant, as determined by the Kruskal-Wallis test with Dunn’s posttest.
Macrophages from ΔlipA mutant-infected mice restrict S. aureus growth and produce more ROS.
Greater proportions of proinflammatory macrophages are recruited to the site of infection with a ΔlipA mutant than to the site of infection with a WT strain (41). These macrophages restrict bacterial growth ex vivo (41) (Fig. 3). Our data thus far suggest that the inflammatory macrophages elicited during infection with the ΔlipA mutant are instrumental to bacterial clearance (Fig. 2B and C). Thus, we sought to determine the mechanism by which macrophages from ΔlipA mutant-infected mice restrict bacterial growth and compared it to that for macrophages from WT-infected mice. Macrophages generate ROS as one of a range of mechanisms used to inhibit the growth of phagocytosed pathogens (6). To test if ROS production by macrophages is increased in ΔlipA mutant-infected mice, we elicited macrophages to the peritoneal cavity using the WT, ΔlipA, or ΔlipA + lipA strain and reinfected the isolated macrophages ex vivo with WT S. aureus, followed by quantitation of ROS production using an indicator dye that fluoresces upon oxidation (CellROX). Upon reinfection, macrophages sourced from WT-infected mice had a net decrease in mean fluorescent intensity (MFI) (Fig. 4A). In contrast, macrophages isolated from ΔlipA mutant-infected mice had, on average, a positive fold change in MFI, suggesting that these macrophages had a higher oxidative state (Fig. 4A). This was partially complemented by the ΔlipA + lipA strain (Fig. 4A). These data suggest that macrophages from ΔlipA mutant-infected mice produce larger amounts of ROS.
FIG 3.
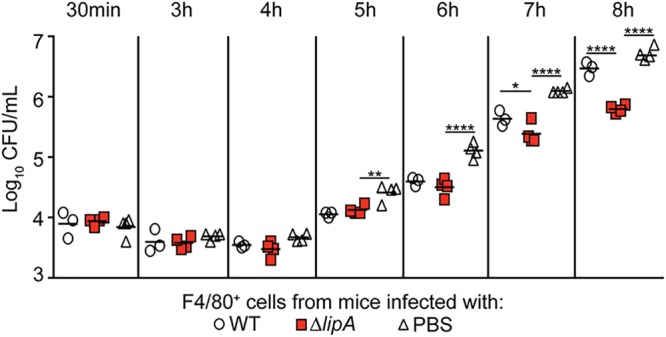
Macrophages recruited to the site of ΔlipA mutant infection restrict S. aureus outgrowth. Outgrowth (log10 number of CFU per milliliter) of WT S. aureus after infection of F4/80+ cells isolated from mice at 72 h after intraperitoneal infection with the WT strain (n = 4), ΔlipA mutant (n = 4), or PBS (n = 4). Bars represent medians. P values were determined by 2-way ANOVA with Tukey’s posttest. *, P < 0.05; **, P < 0.01; ****, P < 0.0001. The data shown are from one of at least three independent experiments.
FIG 4.

Macrophages from ΔlipA mutant-infected mice produce larger amounts of ROS that control bacterial outgrowth than macrophages from mice infected with the WT or ΔlipA + lipA strain. (A) F4/80+ macrophages were sorted from immune cells harvested from the peritoneal cavities of mice at 72 h after intraperitoneal infection with the WT (n = 19), ΔlipA (n = 20), or ΔlipA + lipA (n = 16) strain and infected ex vivo with WT S. aureus at an MOI of 0.1. Macrophages were stained with the ROS indicator CellROX deep red and analyzed by flow cytometry. Fold changes in the geometric means of CellROX fluorescence in infected cells compared to those in uninfected F4/80+ cells were quantified. **, P < 0.01 by the Kruskal-Wallis test with Dunn’s posttest. The dashed line is at a value of zero, and bars represent the median. (B) Outgrowth (log10 number of CFU per milliliter) of WT S. aureus after infection of F4/80+ cells isolated from mice at 72 h after intraperitoneal infection with the WT strain (n = 4), the ΔlipA mutant (n = 4), or PBS (n = 4) and treated with the ROS inhibitor DPI. Bars represent medians. The data shown are from one of at least three independent experiments.
To test if the increased ROS generation by macrophages isolated from ΔlipA mutant-infected mice is necessary to restrict S. aureus growth, we first inhibited ROS production using diphenyleneiodonium chloride (DPI) and compared the ability of macrophages isolated from ΔlipA mutant-infected mice to restrict growth with that of macrophages isolated from WT-infected or mock-infected (phosphate-buffered saline [PBS]-treated) mice (54). Upon inhibition of ROS production by DPI treatment, the macrophages isolated from ΔlipA mutant-infected mice no longer restricted S. aureus outgrowth, in contrast to macrophages from WT-infected or mock-infected mice (Fig. 4B). These data suggest that increased ROS production contributes to the heightened ability of macrophages isolated from ΔlipA mutant-infected mice to control bacterial outgrowth.
ΔlipA mutant-induced antibacterial ROS are generated by NADPH oxidase.
Although DPI is commonly used to inhibit the production of ROS, it can have off-target effects that lead to the inhibition of mitochondrial respiration, which interferes with mROS generation (55). Therefore, we could not use DPI to conclusively determine if the ROS derived from the mitochondria or NADPH oxidase facilitates the restriction of bacterial growth. However, recent studies have found that the delivery of mROS to the phagosome is important for the antimicrobial response to internalized S. aureus, suggesting that these species are important in bacterial clearance (22, 23). To test if macrophages from ΔlipA mutant-infected mice use mROS or NADPH oxidase-derived ROS to restrict bacterial growth, we inoculated mice with WT S. aureus or the ΔlipA mutant, isolated macrophages, and determined if inhibition of mROS with the inhibitor Necrox-5 abrogated growth restriction upon infection with WT S. aureus. Overall, mROS were found to facilitate the killing of S. aureus within macrophages, as S. aureus survival in Necrox-5-treated macrophages was greater than that in vehicle-treated macrophages (Fig. 5A). However, in the presence of Necrox-5, macrophages isolated from ΔlipA mutant-infected mice still restricted the outgrowth of S. aureus to a greater degree than macrophages from WT- or mock-infected mice (Fig. 5B and C). These data suggest that while the production of mROS contributes to the control of S. aureus within the macrophage, they are not involved in the improved restrictive capacity of macrophages isolated from ΔlipA mutant-infected mice.
FIG 5.

Macrophages isolated from ΔlipA mutant-infected mice do not use mROS to restrict bacterial outgrowth. (A) Percent survival of WT S. aureus 8 h after infecting F4/80+ cells isolated from mice treated with vehicle control (DMSO) or the mROS inhibitor Necrox-5. Error bars represent SEM (n = 8). Data were analyzed using an unpaired, two-tailed Student's t test. *, P < 0.05. (B and C) Outgrowth (log10 number of CFU per milliliter) of WT S. aureus after infection of F4/80+ cells isolated from mice at 72 h after intraperitoneal infection with the WT strain (n = 4), the ΔlipA mutant (n = 4), or PBS (n = 4) and treated with the vehicle control (DMSO) (B) or Necrox-5 (C). Bars represent medians. P values were determined by 2-way ANOVA with Tukey’s posttest. *, P < 0.05; **, P < 0.01; ****, P < 0.0001. The data shown are from one of at least two independent experiments.
Since mROS do not appear to play a role in the improved restrictive functions of macrophages sourced from ΔlipA mutant-infected mice, we wondered if NADPH oxidase-dependent ROS production might be responsible. To test this, we used a specific peptide-based inhibitor of NADPH oxidase, gp91ds-tat (56). This NADPH oxidase assembly inhibitor is a chimeric peptide containing an amino acid sequence with a high affinity for the gp91phox subunit of NADPH oxidase linked with the Tat peptide sequence from human immunodeficiency virus, which facilitates entry into cells (56, 57). We isolated macrophages from WT-, ΔlipA mutant-, and mock (PBS)-infected mice followed by reinfection with WT S. aureus ex vivo and monitored bacterial outgrowth hourly. As previously noted, macrophages isolated from ΔlipA mutant-infected mice restricted the outgrowth of S. aureus better than macrophages isolated from WT- and mock-infected mice (Fig. 6A). Treatment of macrophages sourced from ΔlipA mutant-infected mice with gp91ds-tat abrogated the growth restriction of S. aureus compared to the growth restriction obtained by gp91ds-tat treatment of macrophages isolated from WT and mock-infected mice (Fig. 6B). In sum, these data suggest that ROS generated from NADPH oxidase are necessary for the increased bacterial growth restriction imparted by macrophages isolated from ΔlipA mutant-infected mice.
FIG 6.
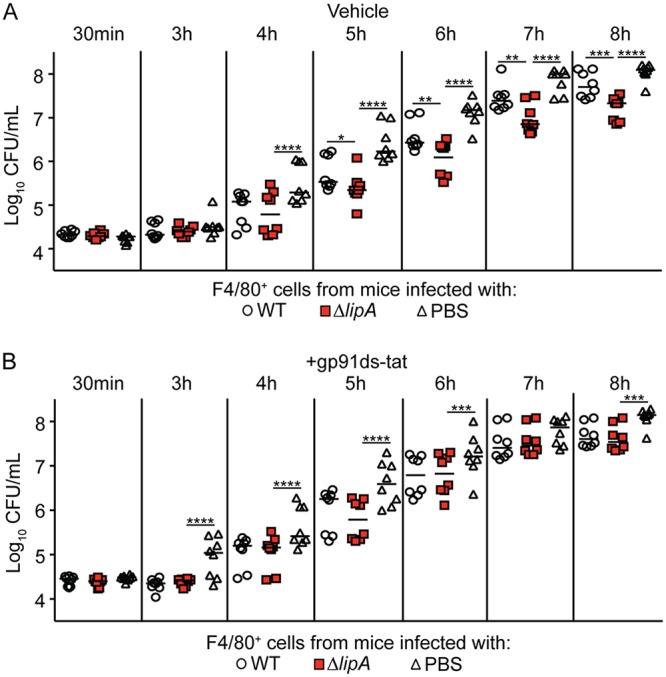
NADPH oxidase-derived ROS contributes to improved control of bacterial outgrowth by macrophages isolated from ΔlipA mutant-infected mice. The outgrowth (log10 number of CFU per milliliter) of WT S. aureus after infection of F4/80+ cells isolated from mice at 72 h after intraperitoneal infection with the WT strain (n = 8), the ΔlipA mutant (n = 8), or PBS (n = 8) amd treated with the vehicle control (water) (A) or the NADPH oxidase inhibitor gp91ds-tat (B) is shown. Bars represent medians. P values were determined by 2-way ANOVA with Tukey’s posttest. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
RNS contribute to the enhanced ability of macrophages from ΔlipA mutant-infected mice to slow bacterial outgrowth.
Besides ROS, which form in the phagosome to kill engulfed pathogens, the generation of NO· and RNS by iNOS also controls pathogen growth (10). Our data thus far suggest that the blockade of NADPH oxidase activity abrogates the restrictive capacity of macrophages from ΔlipA mutant-infected mice, implicating ROS in this process. However, antimicrobial RNS, such as ONOO−, may depend on NADPH oxidase-derived O2·− (27). Thus, macrophages treated with NADPH oxidase inhibitors may also generate fewer antimicrobial RNS. Therefore, we tested if macrophages recruited to the site of infection with the ΔlipA mutant also produce larger amounts of NO·, in addition to ROS, in which NO· and ROS together might lead to growth restriction. We stimulated macrophages isolated from WT-, ΔlipA mutant-, or ΔlipA + lipA strain-infected mice ex vivo with heat-killed S. aureus and measured the levels of NO· production by the Griess test (58). Upon restimulation, macrophages from ΔlipA mutant-infected mice produced three times more nitrite, a stable breakdown product of NO·, than macrophages from mice infected with the WT or the complemented strain (Fig. 7A). These data suggest that macrophages from ΔlipA mutant-infected mice are primed to produce larger amounts of NO· than those from WT- or ΔlipA + lipA strain-infected mice.
FIG 7.

RNS are important for restriction of bacterial growth by macrophages isolated from ΔlipA mutant-infected mice. F4/80+ macrophages were sorted from immune cells harvested from the peritoneal cavities of mice at 72 h after intraperitoneal infection with the WT (n = 20), ΔlipA (n = 19), or ΔlipA + lipA (n = 20) strain and stimulated overnight ex vivo with heat-killed WT S. aureus at an MOI of 10. The levels of nitrite, a breakdown of nitric oxide production, were measured by the Griess test (A). The fold induction of nitric oxide production was determined by comparing the levels of nitrite produced by infected cells to the levels produced by uninfected F4/80+ cells. *, P < 0.05 by the Kruskal-Wallis test with Dunn’s posttest. Bars represent the median. (B and C) Outgrowth (log10 number of CFU per milliliter) of WT S. aureus after infection of F4/80+ cells isolated from mice at 72 h after intraperitoneal infection with the WT strain (n = 8), the ΔlipA mutant (n = 8), or PBS (n = 8) and treated with the vehicle control (water) (B) or the iNOS inhibitor L-NIL (C). Bars represent medians. P values were determined by 2-way ANOVA with Tukey’s posttest. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Given the increased production of NO· in macrophages isolated from ΔlipA mutant-infected mice, we sought to test whether RNS contribute to bacterial growth restriction. We monitored the growth of WT S. aureus in macrophages isolated from WT-, ΔlipA-, or mock (PBS)-infected mice in the presence of a specific iNOS inhibitor, N6-(1-iminoethyl)-lysine hydrochloride (L-NIL), which prevents NO· production and, thus, RNS generation. We found that iNOS inhibition by L-NIL abrogated the improved growth restriction of S. aureus achieved by ΔlipA mutant-elicited macrophages compared to that achieved by macrophages isolated from WT- or mock-infected mice (Fig. 7B and C). Together, our data demonstrate that NO· production and iNOS activity are also necessary for macrophages isolated from ΔlipA mutant-infected mice to limit S. aureus growth ex vivo.
Primary infection with the ΔlipA mutant fails to protect mice from secondary S. aureus challenge.
Our current and prior studies indicate that a ΔlipA mutant not only increases macrophage ROS and RNS production to enhance bacterial killing but also leads to (i) higher neutrophil cytokine and chemokine release and (ii) elicits dendritic cells and macrophages with high surface expression of the antigen-presenting molecule major histocompatibility complex class II (MHC-II) during infection (41). Thus, we wondered if infection with a ΔlipA mutant might improve protective recall responses upon S. aureus reinfection. We immunized mice with the WT, the ΔlipA mutant, or PBS (mock infection) and allowed the infection to clear over 7 days. After 7 days, mice were rechallenged with WT S. aureus, and the numbers of CFU in the kidney were quantified over time. Mice immunized with the ΔlipA mutant had numbers of CFU in the kidneys equivalent to the numbers in the kidneys of mice immunized with the WT or mice that were not immunized at all time points (Fig. 8A). Moreover, upon extension of the immunization time course to 2 weeks, we noted that reinfection continued to yield identical numbers of CFU in the kidney regardless of whether or not the mice received primary immunization with the WT or the ΔlipA mutant (Fig. 8B). In summary, these data suggest that despite the greater host innate response elicited during infection with the ΔlipA mutant, an improved recall response does not follow.
FIG 8.

Immunization of mice with the ΔlipA mutant does not confer protection from secondary challenge. Mice were immunized by intraperitoneal injection with 1 × 108 CFU of the S. aureus WT and ΔlipA mutant or sterile PBS (mock immunized). At either 7 (A) or 14 (B) days after immunization, the mice were rechallenged via injection of 1 × 107 CFU of WT S. aureus into the retro-orbital sinus. The bacterial burden (log10 number of CFU) in the kidneys of mice was assessed 24 h (WT, n = 8; ΔlipA mutant, n = 8; PBS, n = 8), 72 h (WT, n = 8; ΔlipA mutant, n = 6; PBS, n = 8), 96 h (WT, n = 11; ΔlipA mutant, n = 12; PBS, n = 11), and 120 h (WT, n = 7; ΔlipA mutant, n = 6; PBS, n = 8) (A) or 96 h (WT, n = 11; ΔlipA mutant, n = 9; PBS, n = 8) (B) after secondary challenge. Bars represent medians. No statistical significance was achieved when comparing WT- and ΔlipA mutant-immunized animals.
DISCUSSION
In this study, we demonstrate that the synthesis of the metabolic cofactor lipoic acid by LipA is necessary for S. aureus pathogenesis. The LipA-dependent impedance of macrophage antimicrobial functions was mediated by reductions in the generation of ROS and RNS by NADPH oxidase and iNOS, respectively, leading to the improved survival of S. aureus during infection. This suppressive function was lost upon infection with a ΔlipA mutant, whose clearance was dramatically improved due to the enhanced production of ROS and RNS by macrophages. Despite the induction of a more robust antimicrobial immune response during infection with the ΔlipA mutant, the host is unable to mount a successful recall response, likely due to the rapid clearance of the microbe. Thus, this work provides new insight into our earlier observations of the immunosuppressive properties of bacterium-derived lipoic acid and emphasizes the close associations between bacterial metabolism and evasion of innate immunity.
One major outcome of this study was our determination that the attenuation of the ΔlipA mutant during peritoneal infection is mediated almost exclusively by activated macrophages. This was surprising, as we surmised that a proportion of the virulence defect of the ΔlipA mutant was derived from compromised growth due to lipoic acid limitation (41, 46). During bloodstream infection, S. aureus mutants with defects in bacterial lipoic acid synthesis and salvage lead to tissue-specific virulence defects, where infection of the kidney depends on lipoic acid salvage enzymes but infection of the heart requires LipA (46). These observations suggest that there is a varied dependency on de novo lipoic acid synthesis for S. aureus survival in different tissue sites. Although virulence defects in the heart were previously attributed to lipoic acid auxotrophy, the current study suggests the alternative possibility that the LipA-dependent reduction of antimicrobial innate immunity promotes infection persistence. Indeed, we found that LipA is not necessary for bacterial survival in the peritoneal cavity when macrophages are depleted (Fig. 2B and C). We surmise either that alternative metabolic processes take over in the absence of lipoic acid or that sufficient trace lipoic acid is available to promote survival (59–62). Incorporation of trace lipoic acid, while sufficient to maintain bacterial viability, appears to be insufficient to suppress immune responses, as described in our earlier work (41).
Our studies indicate that macrophages from ΔlipA mutant-infected mice are primed to produce larger amounts of ROS and NO·. This improved antimicrobial activity occurred only after infection of mice with the ΔlipA mutant and was not observed after infection of resting macrophages in culture (41). This observation suggests that the effects of the ΔlipA mutant on immune cell activation require additional temporal cues in vivo in order to fully prime the cells for enhanced antimicrobial activity. In our experiments, the ability of ΔlipA mutant-recruited macrophages to maintain improved ROS- and RNS-dependent antimicrobial activity extended beyond 24 h after isolation, which may suggest more than transient polarization of these cells. Inhibition of NADPH oxidase activity by gp91ds-tat and iNOS activity by L-NIL abolished the greater restrictive capacity of macrophages from ΔlipA mutant-infected mice. Because both inhibitors abrogated bacterial growth defects when used separately, we assume that both NADPH oxidase-derived ROS and iNOS-derived NO· are required for S. aureus growth restriction. It is known that NO· alone is not a highly reactive molecule and has an expansive range of cellular functions (27). Rather, NO· becomes highly reactive when it is converted to oxidative products like ONOO− from superoxide (27). The resulting reactive ONOO− leads to microbial destruction by reacting with iron/sulfur metal centers, causing tyrosine nitration, triggering lipid peroxidation, and damaging DNA (10). As such, elimination of either ROS production or NO· production has the potential to compromise the generation of potent antimicrobials, such as ONOO−. We propose a mechanism, based on our data, where the immunosuppressive effects of lipoic acid synthesis by S. aureus lead to the reduced generation of the antimicrobial RNS derived from superoxide and NO·. We are currently pursuing avenues to measure RNS production using boronate-containing fluorescent probes in macrophages isolated from WT- and ΔlipA mutant-infected animals, although this may be difficult due to the short half-life of species such as ONOO− (63, 64).
We previously determined that S. aureus releases lipoyl-containing E2-PDH, which has potent immunosuppressive properties that inhibit macrophage activation in a TLR1/2-dependent manner (41). This immunosuppressive response necessitates LipA-dependent lipoic acid synthesis in order to generate sufficient lipoyl-E2-PDH for release. At present, we do not yet know the molecular mechanism responsible for lipoyl-E2-PDH-mediated suppression, but we do know that TLR1/2 activation is reduced in the presence of lipoyl-E2-PDH (41). TLR2-mediated induction of ROS production by microbial pathogens is common. Recognition of Mycobacterium tuberculosis by TLR2 leads to a direct interaction between TLR2 and NADPH oxidase to ultimately stimulate ROS production (65, 66). Similarly, activation of TLR2 by heat-killed Listeria monocytogenes induces an autophagy-associated protein, Rubicon, which interacts with the p22phox subunit of NADPH oxidase to aid in its phagosomal trafficking and subsequent superoxide production (67). Furthermore, TLR2 is necessary for efficient NADPH oxidase-dependent killing of S. aureus in murine neutrophils (68). Although this study does not assign a direct role for lipoyl-E2-PDH in TLR2-based immune suppression in vivo, our data align with those of these prior studies and suggest that the LipA-dependent inhibition of TLR2 signaling, perhaps by lipoyl-E2-PDH, prevents macrophage priming and induction of NADPH oxidase. Intriguingly, despite evidence for a role of mROS in the antimicrobial activity of macrophages against S. aureus (Fig. 5A) (22, 23), these species do not appear to appreciably contribute to the enhanced restrictive function of macrophages from ΔlipA mutant-infected mice.
In order to activate NADPH oxidase, Akt (protein kinase B) must phosphorylate p47phox (69). Intriguingly, high concentrations of free lipoic acid can activate Akt (52). Because ΔlipA mutants cannot synthesize lipoic acid, one might assume lower Akt activation and NADPH oxidase activity during infection. Our data do not support this assumption, as we saw larger amounts of NADPH oxidase-derived ROS in macrophages from ΔlipA mutant-infected mice than in macrophages from mice infected with the WT or the complemented strain. We know that S. aureus-derived lipoic acid is bound to proteins; thus, we believe that the effects on Akt likely are not manifested in our infection system. Finally, free lipoic acid is known to have antioxidant activity that diminishes the respiratory burst of phagocytes (49). Therefore, mice infected with a ΔlipA mutant may also be less likely to detoxify macrophage ROS or RNS. The detailed cellular mechanism by which S. aureus lipoic acid synthesis interferes with ROS and RNS generation in macrophages during infection is under investigation.
Macrophages play a major role in the activation of the adaptive immune system through the production of cytokines and chemokines that regulate T cell function and facilitate dendritic cell maturation, leading to antigen presentation and induction of adaptive immunity (34, 40). Despite the improved activation of innate immunity and microbial clearance after infection with a ΔlipA mutant, we found that prior infection failed to protect the host from secondary challenge. We suspect that the explanation for such poor immunity to infection centers on the fact that S. aureus is destroyed too rapidly after infection with a ΔlipA mutant, a consequence of improved macrophage activation. The robust and destructive oxidative burst of macrophages interferes with optimal antigen presentation due to rapid phagolysosome fusion and destruction of antigenic peptides, lowering their ability to optimally present antigen (40). This contrasts with dendritic cells, which have different phagosomal environments, are less destructive, and can conserve antigenic information from engulfed pathogens, thereby promoting efficient antigen presentation (40). The notion that the increased antigen-destructive functions imparted by enhanced macrophage activation block the development of adaptive immunity resembles observations from studies on early antibiotic intervention in Salmonella and Chlamydia, wherein the shortened duration of antigen presentation leads to poor protective memory (70). Our data also demonstrate that NO· production is increased upon infection with a ΔlipA mutant, which is in part responsible for the antimicrobial function of recruited macrophages. Beyond its importance in the generation of RNS, NO· has diverse signaling functions that can dramatically affect adaptive immune responses, such as inhibition of T cell proliferation (71–73). Thus, it is possible that the larger amount of NO· produced by macrophages during infection with a ΔlipA mutant may interfere with optimal T cell proliferation. Whatever the ultimate mechanism, what seems clear is that improved activation of innate immune responses is unable to facilitate acquired immunity to S. aureus infection in mice.
Altogether, this work has broadened our understanding of how S. aureus lipoic acid synthesis is intimately connected with the ability to cause disease. Our studies demonstrate that lipoic acid synthesis contributes to S. aureus blunting of the antimicrobial ROS and RNS responses of macrophages during infection. Additionally, increased macrophage responses do not necessarily translate to protection against reinfection. This study adds to the growing body of work to highlight the crucial roles for bacterial metabolism in evasion of host immunity.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The wild-type Staphylococcus aureus strain used in this study was the pulsed-field gel electrophoresis type USA300 strain LAC (LAC) or a derivative of LAC cured of its erythromycin resistance plasmid (AH1263, WT). The ΔlipA mutant and its complemented strain, the ΔlipA + lipA strain, were generated and validated in a previous study (46). All S. aureus strains were grown in tryptic soy broth (TSB; Criterion), which allows for normal growth of the ΔlipA mutant (41). Unless otherwise noted, all bacteria were grown at 37°C in a shaking incubator at 200 rpm in tubes kept at a 45° angle. When solid medium was required, agar (Amresco) was supplemented into TSB at 1.5%.
Murine infection models.
For intraperitoneal infection with S. aureus, the WT, ΔlipA, ΔlipA + lipA, or lipA::erm strain was inoculated in TSB with shaking at 37°C and grown overnight. Overnight cultures were diluted 1:100 in 15 ml TSB and incubated at 200 rpm at 37°C for 3 h. The cultures were then centrifuged for 5 min at 4,200 rpm at 4°C, and the resulting cell pellets were washed 3 times in 1× sterile phosphate-buffered saline (PBS; Corning). Bacterial suspensions were then normalized to an optical density (OD) at 600 nm (OD600) of 1.1 to 1.2 (∼1 × 109 CFU/ml) prior to intraperitoneal infection. The infectious dose was assessed by serially diluting the inoculum on TSA plates. Six- to 8-week-old male and female Swiss Webster mice (Envigo) were injected intraperitoneally (i.p.) with 100 μl of PBS containing 1 × 108 CFU of S. aureus or 100 μl of sterile PBS for mock infections. For competitive index experiments, mice were infected with 200 μl of a 1:1 mixture of the OD-normalized WT and lipA::erm strains (1 × 108 CFU per strain). The mice were monitored daily until 16 or 72 h postinfection, at which point they were euthanized by CO2 narcosis, followed by lavage of the peritoneal cavity with 7 ml PBS and aseptic isolation of the kidneys. The kidneys were homogenized in 5 ml sterile PBS, and lavage fluid and kidney homogenates were serially diluted on TSA or TSA-erythromycin plates, followed by incubation at 37°C overnight to enumerate the CFU.
To deplete the mice of macrophages, dichloromethane bisphosphonate (clodronate) loaded into liposomes (Liposoma) was administered (1 ml/100 g [∼150 μl/mouse]) by i.p. injection. After 3 days, the bacteria were cultured as described above and normalized to an OD600 of 0.32 to 0.33 (∼1 × 108 CFU/ml). The mice were then injected i.p. with 100 μl of PBS containing 1 × 107 CFU of S. aureus. The mice were monitored until 16 h or 72 h postinfection, followed by enumeration of the bacterial loads from the lavage fluid and kidneys as described above.
For the S. aureus rechallenge model, 6- to 8-week-old male and female Swiss Webster mice were immunized by i.p. injection of 100 μl of PBS containing 1 × 108 CFU of WT S. aureus or S. aureus ΔlipA or sterile PBS. At 7 or 14 days after immunization, the mice were anesthetized with 2,2,2-tribromoethanol (Avertin; 250 mg/kg of body weight; Sigma) via i.p. injection, followed by inoculation with 100 μl PBS containing 1 × 107 CFU of S. aureus directly into the bloodstream via the retro-orbital sinus. The mice were monitored daily and then euthanized at either 24, 72, 96, or 120 h postinfection. The kidneys were isolated to enumerate the bacterial loads as described above.
S. aureus outgrowth in F4/80+ cells.
The outgrowth of S. aureus after phagocytosis by F4/80+ cells was performed as previously described (41). Briefly, at 3 days after i.p. infection with WT S. aureus, S. aureus ΔlipA, or sterile PBS, peritoneal cells were isolated by lavage with 7 ml of 1× sterile PBS. Recovered cells were pelleted by centrifugation at 1,500 rpm at 4°C for 5 min, followed by decanting the supernatant and resuspending the pellet in complete RPMI cell culture medium (RPMI plus 10% heat-inactivated fetal bovine serum [FBS]; Corning) supplemented with 100 μg/ml penicillin-streptomycin (Corning) and 50 μg/ml gentamicin (Amresco) and incubated on ice for 30 min to 1 h. After incubation, the cells were washed 3 times in 3 ml complete RPMI medium without antibiotics and incubated for 30 min in the antibiotic-free medium. The cells were then washed once and resuspended in fluorescence-activated cell sorting (FACS) wash buffer (1× PBS plus 2% heat-inactivated FBS) for magnetic bead sorting. The F4/80+ cells were sorted from total peritoneal cells using a BD IMag cell separation system (BD Biosciences) after incubation with anti-CD16/CD32 (clone 93; BioLegend) and biotinylated anti-F4/80 antibody (clone BM8; BioLegend). Magnetic bead cell sorting produced F4/80+ cells that were >96% pure (41). The sorted cells were stored in FACS wash buffer overnight at 4°C. On the following day, an overnight culture of WT LAC grown in TSB was washed 3 times in sterile 1× PBS, normalized to an OD600 of ∼0.32 (∼1.0 × 108 CFU/ml), and opsonized by incubation with 10% mouse serum for 30 min at 37°C, followed by washing 3 times in 1 ml sterile 1× PBS. The sorted F4/80+ cells were pelleted by centrifugation at 1,500 rpm at 4°C for 5 min, resuspended in fresh complete RPMI medium, and counted. The opsonized bacteria were then used to infect 5 × 105 F4/80+ cells at a multiplicity of infection (MOI) of 0.1 (unless stated otherwise) for 30 min at 37°C in sterile 1.5-ml microcentrifuge tubes placed on a rotisserie. Following the 30-min infection, samples were centrifuged at 1,500 rpm at room temperature in a benchtop centrifuge (Eppendorf) for 5 min and washed 3 times with 1 ml sterile 1× PBS. Samples were then resuspended in 1 ml complete RPMI medium and placed at 37°C on a rotisserie. In certain experiments, ROS production was blocked by the addition of 2 μM diphenyleneiodonium chloride (DPI; Sigma), mROS production was blocked by the addition of 10 μM Necrox-5 (Cayman Chemical), iNOS production was blocked by the addition of 500 μM N6-(1-iminoethyl)-lysine hydrochloride (L-NIL; Tocris), or vehicle controls (dimethyl sulfoxide [DMSO] for Necrox-5 and H2O for L-NIL) were added to the culture medium. The S. aureus CFU were enumerated hourly by removing 100-μl aliquots and lysing the bacteria with 0.1% saponin (Sigma) for 20 min on ice, followed by serial dilution and plating of the dilutions on TSA plates supplemented with 50 μg/ml of kanamycin (Amresco) and neomycin (Amresco).
For experiments where NADPH oxidase activity was blocked, the following modifications were performed. Prior to infection, F4/80+ cells were treated with 50 μM gp91ds-tat (Anaspec) or the vehicle control (H2O) for 1 h in RPMI serum-free medium to allow for entry of the inhibitor into the cells. The treated cells were then centrifuged at 1,500 rpm at room temperature in a benchtop centrifuge for 5 min and washed 3 times in sterile 1× PBS. The washed cells were resuspended in complete RPMI medium containing opsonized bacteria at an MOI of 1 and allowed to become infected for 30 min at 37°C under constant rotation. The remainder of the experiment was conducted as described above.
Assessment of ROS production.
Three days after i.p. infection with the WT, ΔlipA, or ΔlipA + lipA strain, peritoneal cells were isolated by lavage of the peritoneal cavity followed by magnetic sorting of F4/80+ macrophages as detailed above. To measure the changes in the oxidative state of isolated peritoneal F4/80+ macrophages (ROS production), 5 × 105 F4/80+ cells were infected with WT S. aureus at an MOI of 0.1 for 30 min under constant rotation at 37°C. The cells were washed free of bacteria, followed by incubation in medium containing 1.25 μM CellROX deep red (Thermo Fisher Scientific) for 1 h under constant rotation at 37°C. The cells were then washed 3 times with 200 μl of sterile 1× PBS. Fluorescence was measured using an LSRFortessa flow cytometer (BD Biosciences), and the data were analyzed using FlowJo software (FlowJo, LLC). The fold changes in the geometric mean of the CellROX fluorescence in infected cells compared to that in uninfected F4/80+ cells were assessed.
Griess test for NO· production.
Three days after i.p. infection with the WT, ΔlipA, or ΔlipA + lipA strain, peritoneal cells were isolated by lavage of the peritoneal cavity, followed by magnetic sorting of F4/80+ macrophages as described above. Immediately after sorting, isolated F4/80+ macrophages were counted and 100,000 cells were plated in a 96-well plate (Corning) in 100 μl complete RPMI medium. Heat-killed WT S. aureus was prepared by incubating a 1× PBS-washed overnight culture grown in TSB for 1 h at 60°C. The sterility of the heat-killed bacteria was confirmed by enumeration of the CFU on a TSA plate. Sorted F4/80+ macrophages were then stimulated with the heat-killed bacteria at an MOI of 10 or left unstimulated. After overnight incubation at 37°C in 5% CO2, 50 μl of supernatant was removed and a Griess test was performed to measure nitrite, which is a breakdown product of nitric oxide. Fifty microliters of 1% sulfanilic acid (Sigma) in 5% phosphoric acid (Fischer Scientific) was added to the supernatant samples, followed by incubation for 5 min, addition of 50 μl of 0.1% N-alpha-naphthyl-ethylenediamine (Sigma) in sterile water, and incubation for an additional 5 min. To quantify the amount of nitrite present in the cell culture medium, 100 mM sodium nitrate (Sigma) was diluted 1:1 and used to make a standard curve. After incubation, the sample absorbance was measured at OD550 using an ELx800 microplate reader (BioTek). The fold increase in the nitrite (nitric oxide) levels of F4/80+ macrophages was determined by comparing the nitrite production in heat-killed S. aureus-stimulated cells with that in nonstimulated cells isolated from the same original infection condition (infection with the WT, ΔlipA, or ΔlipA + lipA strain).
Assessment of macrophage depletion in clodronate-treated mice.
Mice were administered clodronate-containing liposomes (1 ml/100 g [∼150 μl/mouse]) by i.p. injection for 3 days to allow for depletion or were left untreated. The mice were then infected with 100 μl PBS containing 1 × 107 CFU of WT S. aureus by i.p. injection. After 3 days, the mice were euthanized and a lavage of the peritoneal cavity was performed. Isolated cells were pelleted by centrifugation at 1,500 rpm at 4°C for 5 min, the supernatant was decanted, and red blood cells were lysed with ACK lysing buffer (Lonza) by resuspending the pellet in 1 ml of lysing buffer and incubating for 2 min at room temperature. Cell lysis was stopped by adding 9 ml of 1× sterile PBS, mixing gently, and pelleting by centrifugation at 1,500 rpm at 4°C for 5 min. The remaining cells were suspended in 1× sterile PBS and kept on ice while they were counted. One million to 2 million cells were incubated with 50 μl of FACS wash buffer (1× PBS, 2% heat-inactivated FBS, 0.05% [wt/vol] sodium azide) containing 0.2 μg/ml of anti-CD16/CD32 (clone 93) (BioLegend) for 30 min on ice to block Fc receptors, followed by washing with FACS wash buffer and surface staining with anti-CD11b-peridinin chlorophyll protein-Cy5.5 (clone M1/70; BioLegend), anti-F4/80-phycoerythrin (PE)-Cy7 (clone BM8; BioLegend), and anti-Ly6G-PE (clone 1A8; BioLegend) for 30 min on ice. Stained cells were washed twice with FACS wash buffer and fixed using FACS fixing buffer (1× PBS, 2% heat-inactivated FBS, 2% paraformaldehyde, 0.05% [wt/vol] sodium azide). Data were collected on an LSRFortessa flow cytometer (BD Biosciences), and macrophage populations were subsequently analyzed using FlowJo software (FlowJo, LLC).
Ethics statement.
All experiments were performed following the ethical standards of the Institutional Biosafety Committee and the Institutional Animal Care and Use Committee (IACUC) at Loyola University Chicago, Health Sciences Division. The institution is approved by the Public Health Service (PHS; approval number A3117-01 through 28 February 2022), is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (accreditation number 000180, certification dated 17 November 2016), and is registered/licensed by the U.S. Department of Agriculture (USDA; registration/license number 33-R-0024 through 24 August 2020). All animal experiments were performed in animal biosafety level 2 facilities with IACUC-approved protocols (IACUC number 2017028) under the guidance of the Office of Laboratory Animal Welfare following USDA and PHS policy on the humane care and use of laboratory animals guidelines.
Statistical analyses.
All experiments were repeated at least three independent times. Statistical analysis was conducted using Prism software (GraphPad), and the specific tests used are indicated in the figure legends. Statistical significance was defined as a P value of <0.05. The number of animals per treatment group is indicated by n in the figure legends. Unless otherwise noted, we used a Kruskal-Wallis test with Dunn’s posttest. For S. aureus ex vivo survival/growth assays with F4/80+ sorted cells, a 2-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test was used.
ACKNOWLEDGMENTS
We thank the members of the F. Alonzo laboratory for helpful discussions.
This work was supported by grants NIH R01 AI120994 to F.A. and AHA 17PRE33660173 to J.P.G.
REFERENCES
- 1.Spaan AN, Surewaard BG, Nijland R, van Strijp JA. 2013. Neutrophils versus Staphylococcus aureus: a biological tug of war. Annu Rev Microbiol 67:629–650. doi: 10.1146/annurev-micro-092412-155746. [DOI] [PubMed] [Google Scholar]
- 2.Rooijakkers SHM, van Kessel KPM, van Strijp J. 2005. Staphylococcal innate immune evasion. Trends Microbiol 13:596–601. doi: 10.1016/j.tim.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Foster TJ. 2005. Immune evasion by staphylococci. Nat Rev Microbiol 3:948–958. doi: 10.1038/nrmicro1289. [DOI] [PubMed] [Google Scholar]
- 4.Thammavongsa V, Kim HK, Missiakas D, Schneewind O. 2015. Staphylococcal manipulation of host immune responses. Nat Rev Microbiol 13:529–543. doi: 10.1038/nrmicro3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kourtis AP, Hatfield K, Baggs J, Mu Y, See I, Epson E, Nadle J, Kainer MA, Dumyati G, Petit S, Ray SM, Ham D, Capers C, Ewing H, Coffin N, McDonald LC, Jernigan J, Cardo D. 2019. Vital signs: epidemiology, and recent trends in methicillin-resistant and in methicillin-susceptible Staphylococcus aureus bloodstream infections—United States. MMWR Morb Mortal Wkly Rep 68:214–219. doi: 10.15585/mmwr.mm6809e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flannagan RS, Cosío G, Grinstein S. 2009. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol 7:355–366. doi: 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- 7.Hampton MB, Kettle AJ, Winterbourn CC. 1998. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood 92:3007–3017. [PubMed] [Google Scholar]
- 8.Winterbourn CC, Kettle AJ. 2013. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid Redox Signal 18:642–660. doi: 10.1089/ars.2012.4827. [DOI] [PubMed] [Google Scholar]
- 9.Sbarra AJ, Karnovsky ML. 1959. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J Biol Chem 234:1355–1362. [PubMed] [Google Scholar]
- 10.Fang FC. 2004. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol 2:820–832. doi: 10.1038/nrmicro1004. [DOI] [PubMed] [Google Scholar]
- 11.Vignais PV. 2002. The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci 59:1428–1459. doi: 10.1007/s00018-002-8520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Babior BM, Lambeth JD, Nauseef W. 2002. The neutrophil NADPH oxidase. Arch Biochem Biophys 397:342–344. doi: 10.1006/abbi.2001.2642. [DOI] [PubMed] [Google Scholar]
- 13.El-Benna J, Dang PM, Gougerot-Pocidalo M, Marie J, Braut-Boucher F. 2009. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp Mol Med 41:217–225. doi: 10.3858/emm.2009.41.4.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winterbourn CC. 2008. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol 4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 15.Klebanoff SJ. 2005. Myeloperoxidase: friend and foe. J Leukoc Biol 77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 16.Slauch JM. 2011. How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol Microbiol 80:580–583. doi: 10.1111/j.1365-2958.2011.07612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winkelstein JA, Marino MC, Johnston RJ, Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie P, Buckley RH, Foster CB, Chanock SJ, Dickler H. 2000. Chronic granulomatous disease: report on a national registry of 368 patients. Medicine 79:155–169. doi: 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Buvelot H, Posfay-Barbe KM, Linder P, Schrenzel J, Krause K. 2017. Staphylococcus aureus, phagocyte NADPH oxidase and chronic granulomatous disease. FEMS Microbiol Rev 41:139–157. doi: 10.1093/femsre/fuw042. [DOI] [PubMed] [Google Scholar]
- 19.West PA, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. 2011. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy MP. 2009. How mitochondria produce reactive oxygen species. Biochem J 417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koopman WJ, Nijtmans LG, Dieteren CE, Roestenberg P, Valsecchi F, Smeitink JA, Willems PH. 2010. Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxid Redox Signal 12:1431–1470. doi: 10.1089/ars.2009.2743. [DOI] [PubMed] [Google Scholar]
- 22.Abuaita BH, Schultz TL, O'Riordan MX. 2018. Mitochondria-derived vesicles deliver antimicrobial reactive oxygen species to control phagosome-localized Staphylococcus aureus. Cell Host Microbe 24:625–636. doi: 10.1016/j.chom.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen TS, Boland BB, Boland ML, Takahashi V, Tovchigrechko A, Lee Y, Wilde AD, Mazaitis MJ, Jones-Nelson O, Tkaczyk C, Raja R, Stover CK, Sellman BR. 2018. S. aureus evades macrophage killing through NLRP3-dependent effects on mitochondrial trafficking. Cell Rep 22:2431–2441. doi: 10.1016/j.celrep.2018.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stuehr DJ. 1999. Mammalian nitric oxide synthases. Biochim Biophys Acta 1411:217–230. doi: 10.1016/s0005-2728(99)00016-x. [DOI] [PubMed] [Google Scholar]
- 25.Dedon PC, Tannenbaum SR. 2004. Reactive nitrogen species in the chemical biology of inflammation. Arch Biochem Biophys 423:12–22. doi: 10.1016/j.abb.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 26.Wink DA, Mitchell JB. 1998. Chemical biology of nitric oxide: insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic Biol Med 25:434–456. doi: 10.1016/S0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 27.Pacher P, Beckman JS, Liaudet L. 2007. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pacelli R, Wink DA, Cook JA, Krishna MC, DeGraff W, Friedman N, Tsokos M, Samuni A, Mitchell JB. 1995. Nitric oxide potentiates hydrogen peroxide-induced killing of Escherichia coli. J Exp Med 182:1469–1479. doi: 10.1084/jem.182.5.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clements MO, Watson SP, Foster SJ. 1999. Characterization of the major superoxide dismutase of Staphylococcus aureus and its role in starvation survival, stress resistance, and pathogenicity. J Bacteriol 181:3898–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karavolos MH, Horsburgh MJ, Ingham E, Foster SJ. 2003. Role and regulation of the superoxide dismutases of Staphylococcus aureus. Microbiology 149:2749–2758. doi: 10.1099/mic.0.26353-0. [DOI] [PubMed] [Google Scholar]
- 31.Hochgräfe F, Wolf C, Fuchs S, Liebeke M, Lalk M, Engelmann S, Hecker M. 2008. Nitric oxide stress induces different responses but mediates comparable protein thiol protection in Bacillus subtilis and Staphylococcus aureus. J Bacteriol 190:4997–5008. doi: 10.1128/JB.01846-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vitko NP, Spahich NA, Richardson AR. 2015. Glycolytic dependency of high-level nitric oxide resistance and virulence in Staphylococcus aureus. mBio 6:e00045-15. doi: 10.1128/mBio.00045-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richardson AR, Libby SJ, Fang FC. 2008. A nitric oxide-inducible lactate dehydrogenase enables Staphylococcus aureus to resist innate immunity. Science 319:1672–1676. doi: 10.1126/science.1155207. [DOI] [PubMed] [Google Scholar]
- 34.Mosser DM, Edwards JP. 2010. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 10:460. doi: 10.1038/nri2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernando MR, Reyes JL, Iannuzzi J, Leung G, McKay DM. 2014. The pro-inflammatory cytokine, interleukin-6, enhances the polarization of alternatively activated macrophages. PLoS One 9:e94188. doi: 10.1371/journal.pone.0094188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duque GA, Descoteaux A. 2014. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol 5:491. doi: 10.3389/fimmu.2014.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. 2008. Growth factors and cytokines in wound healing. Wound Repair Regen 16:585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 38.Serhan CN, Savill J. 2005. Resolution of inflammation: the beginning programs the end. Nat Immunol 6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 39.Gordon S. 2003. Alternative activation of macrophages. Nat Rev Immunol 3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 40.Savina A, Amigorena S. 2007. Phagocytosis and antigen presentation in dendritic cells. Immunol Rev 219:143–156. doi: 10.1111/j.1600-065X.2007.00552.x. [DOI] [PubMed] [Google Scholar]
- 41.Grayczyk JP, Harvey CJ, Laczkovich I, Alonzo F. 2017. A lipoylated metabolic protein released by Staphylococcus aureus suppresses macrophage activation. Cell Host Microbe 22:678–687.e9. doi: 10.1016/j.chom.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cronan JE. 2016. Assembly of lipoic acid on its cognate enzymes: an extraordinary and essential biosynthetic pathway. Microbiol Mol Biol Rev 80:429–450. doi: 10.1128/MMBR.00073-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spalding MD, Prigge ST. 2010. Lipoic acid metabolism in microbial pathogens. Microbiol Mol Biol Rev 74:200–228. doi: 10.1128/MMBR.00008-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reed LJ. 1998. From lipoic acid to multi‐enzyme complexes. Protein Sci 7:220–224. doi: 10.1002/pro.5560070125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laczkovich I, Teoh WP, Flury S, Grayczyk JP, Zorzoli A, Alonzo F. 2018. Increased flexibility in the use of exogenous lipoic acid by Staphylococcus aureus. Mol Microbiol 109:150–168. doi: 10.1111/mmi.13970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zorzoli A, Grayczyk JP, Alonzo F. 2016. Staphylococcus aureus tissue infection during sepsis is supported by differential use of bacterial or host-derived lipoic acid. PLoS Pathog 12:e1005933. doi: 10.1371/journal.ppat.1005933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang WJ, Frei B. 2001. α-Lipoic acid inhibits TNF-α-induced NF-κB activation and adhesion molecule expression in human aortic endothelial cells. FASEB J 15:2423–2432. doi: 10.1096/fj.01-0260com. [DOI] [PubMed] [Google Scholar]
- 48.Lee HA, Hughes DA. 2002. Alpha-lipoic acid modulates NF-κB activity in human monocytic cells by direct interaction with DNA. Exp Gerontol 37:401–410. [DOI] [PubMed] [Google Scholar]
- 49.Packer L. 1998. α-Lipoic acid: a metabolic antioxidant which regulates NF-κB signal transduction and protects against oxidative injury. Drug Metab Rev 30:245–275. doi: 10.3109/03602539808996311. [DOI] [PubMed] [Google Scholar]
- 50.Kim HS, Kim HJ, Park KG, Kim YN, Kwon TK, Park JY, Lee KU, Kim JG, Lee IK. 2007. α-Lipoic acid inhibits matrix metalloproteinase-9 expression by inhibiting NF-κB transcriptional activity. Exp Mol Med 39:106–113. doi: 10.1038/emm.2007.12. [DOI] [PubMed] [Google Scholar]
- 51.Packer L, Witt EH, Tritschler HJ. 1995. Alpha-lipoic acid as a biological antioxidant. Free Radic Biol Med 19:227–250. [DOI] [PubMed] [Google Scholar]
- 52.Zhang WJ, Wei H, Hagen T, Frei B. 2007. a-Lipoic acid attenuates LPS-induced inflammatory responses by activating the phosphoinositide 3-kinase/Akt signaling pathway. Proc Natl Acad Sci U S A 104:4077–4082. doi: 10.1073/pnas.0700305104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen J, Yoong P, Ram G, Torres VJ, Novick RP. 2014. Single-copy vectors for integration at the SaPI1 attachment site for Staphylococcus aureus. Plasmid 76:1–7. doi: 10.1016/j.plasmid.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paik W, Alonzo F, Knight KL. 2019. Probiotic exopolysaccharide protects against systemic Staphylococcus aureus infection, inducing dual-functioning macrophages that restrict bacterial growth and limit inflammation. Infect Immun 87:11–21. doi: 10.1128/IAI.00791-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Y, Trush MA. 1998. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun 253:295–299. doi: 10.1006/bbrc.1998.9729. [DOI] [PubMed] [Google Scholar]
- 56.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. 2001. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2– and systolic blood pressure in mice. Circ Res 89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 57.Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. 1994. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci U S A 91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bryan NS, Grisham MB. 2007. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic Biol Med 43:645–657. doi: 10.1016/j.freeradbiomed.2007.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wilde AD, Snyder DJ, Putnam NE, Valentino MD, Hammer ND, Lonergan ZR, Hinger SA, Aysanoa EE, Blanchard C, Dunman PM, Wasserman GA, Chen J, Shopsin B, Gilmore MS, Skaar EP, Cassat JE. 2015. Bacterial hypoxic responses revealed as critical determinants of the host-pathogen outcome by TnSeq analysis of Staphylococcus aureus invasive infection. PLoS Pathog 11:e1005341. doi: 10.1371/journal.ppat.1005341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Balasubramanian D, Harper L, Shopsin B, Torres VJ. 2017. Staphylococcus aureus pathogenesis in diverse host environments. Pathog Dis 75:ftx005. doi: 10.1093/femspd/ftx005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu Y, Nandakumar R, Sadykov MR, Madayiputhiya N, Luong TT, Gaupp R, Lee CY, Somerville GA. 2011. RpiR homologues may link Staphylococcus RNAIII synthesis and pentose phosphate pathway regulation. J Bacteriol 193:6187–6196. doi: 10.1128/JB.05930-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vitko NP, Grosser MR, Khatri D, Lance TR, Richardson AR. 2016. Expanded glucose import capability affords Staphylococcus aureus optimized glycolytic flux during infection. mBio 7:e00296-16. doi: 10.1128/mBio.00296-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen X, Chen H, Deng R, Shen J. 2014. Pros and cons of current approaches for detecting peroxynitrite and their applications. Biomed J 37:120–126. doi: 10.4103/2319-4170.134084. [DOI] [PubMed] [Google Scholar]
- 64.Sedgwick AC, Sun X, Kim G, Yoon J, Bull SD, James TD. 2016. Boronate based fluorescence (ESIPT) probe for peroxynitrite. Chem Commun (Camb) 52:12350–12352. doi: 10.1039/C6CC06829D. [DOI] [PubMed] [Google Scholar]
- 65.Schuett J, Schuett H, Oberoi R, Koch A, Pretzer S, Luchtefeld M, Schieffer B, Grote K. 2017. NADPH oxidase NOX2 mediates TLR2/6-dependent release of GM-CSF from endothelial cells. FASEB J 31:2612–2624. doi: 10.1096/fj.201600729R. [DOI] [PubMed] [Google Scholar]
- 66.Yang C, Shin D, Kim K, Lee Z, Lee C, Park SG, Bae YS, Jo E. 2009. NADPH oxidase 2 interaction with TLR2 is required for efficient innate immune responses to mycobacteria via cathelicidin expression. J Immunol 182:3696–3705. doi: 10.4049/jimmunol.0802217. [DOI] [PubMed] [Google Scholar]
- 67.Yang C, Lee J, Lee J, Lee K, Rodgers M, Min C, Kim H, Kim C, Oh B, Zandi E, Yue Z, Kramnik I, Liang C, Jung J. 2012. Autophagy protein Rubicon mediates phagocytic NADPH oxidase activation in response to microbial infection or TLR stimulation. Cell Host Microbe 11:264–276. doi: 10.1016/j.chom.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jann NJ, Schmaler M, Ferracin F, Landmann R. 2011. TLR2 enhances NADPH oxidase activity and killing of Staphylococcus aureus by PMN. Immunol Lett 135:17–23. doi: 10.1016/j.imlet.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 69.Hoyal CR, Gutierrez A, Young BM, Catz SD, Lin J-H, Tsichlis PN, Babior BM. 2003. Modulation of p47PHOX activity by site-specific phosphorylation: Akt-dependent activation of the NADPH oxidase. Proc Natl Acad Sci U S A 100:5130–5135. doi: 10.1073/pnas.1031526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benoun JM, Labuda JC, McSorley SJ. 2016. Collateral damage: detrimental effect of antibiotics on the development of protective immune memory. mBio 7:e01520-16. doi: 10.1128/mBio.01520-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bingisser RM, Tilbrook PA, Holt PG, Kees UR. 1998. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol 160:5729–5734. [PubMed] [Google Scholar]
- 72.Nabeshima S, Nomoto M, Matsuzaki G, Kishihara K, Taniguchi H, Yoshida SI, Nomoto K. 1999. T-cell hyporesponsiveness induced by activated macrophages through nitric oxide production in mice infected with Mycobacterium tuberculosis. Infect Immun 67:3221–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bogdan C. 2015. Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol 36:161–178. doi: 10.1016/j.it.2015.01.003. [DOI] [PubMed] [Google Scholar]