

Polycythemia vera (PV) is a clonal disease characterized by overproduction of mature erythrocytes, impaired survival, and a propensity for transformation to myelofibrosis (MF) and acute myeloid leukemia (AML).1 The JAK2 V617F mutation occurs in > 95% of PV patients, leading to hyperactive JAK-STAT signaling which mediates proliferation, differentiation, and survival.1 Recurrent mutations in other genes (e.g. TET2, DNMT3A, ASXL1, IDH1/2, and EZH2) have been identified in PV as well as other myeloid malignancies.1 Mutations in a subset of these genes (e.g. TET2, DNMT3A, and ASXL1) have also been associated with clonal hematopoiesis of indeterminate potential (CHIP).2 How these mutations modulate PV disease phenotype is not well understood.
PV is typically diagnosed in older patients (≥ 65 years old, PVold) but can occur in younger patients (≤ 45 years old, PVyoung). In a prior study, PVyoung patients were reported to be predominantly female, with lower white blood cell count and JAK2 V617F mutant allele burden, and were more likely to present with splenomegaly and splanchnic vein thrombosis (SVT) than PVold patients.3 Transformation to MF/AML was similar for both groups, but the median time to transformation was longer in PVyoung.3 The reasons for these findings are unclear. Somatic mutations have been shown to accumulate as a function of age, and a recent study demonstrated that mutation order can dictate specific clinical features in MPNs.4, 5 These observations led us to hypothesize that the mutational profile of PVyoung may differ from PVold and potentially contribute to distinct clinical phenotypes observed in these cohorts.
Samples from ten PVyoung (≤ 45 years old) and nine PVold (≥ 65 years old) JAK2 V617F+ patients were identified for genomic analysis. Similar to Stein et. al, a trend towards more females and SVT events in the PVyoung group was observed (Fig. 1a).3 When the cohort was expanded with clinical data from an additional nine young and nine old non-sequenced JAK2 V617F+ patients, the PVyoung group had significantly more females than PVold, and enrichment of SVT in PVyoung approached significance (Fig. 1a & Supp. Table S1).
Figure 1. Exome sequencing suggests JAK2 V617F is only driver present in young PV.
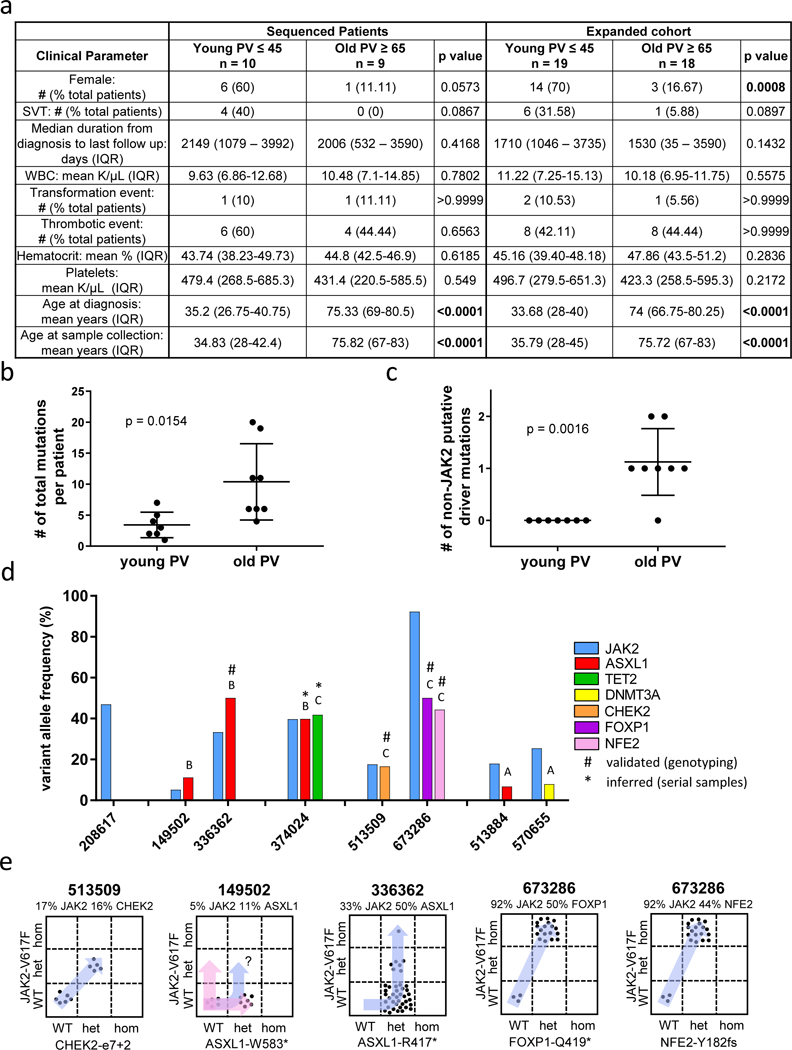
a) Clinical parameter summaries from the 19 PV patients that underwent exome sequencing (left) and an expanded cohort of 37 total patients (right). Mann-Whitney test was used to test difference between age groups for continuous variables, Fisher’s Exact test for categorical variables. “IQR” = interquartile range, “SVT” = splanchnic vein thrombosis, “WBC” = white blood cell. b) Scatter plot showing the mean +/− standard deviation of overall mutational load of young and old PV patients. c) Scatter plot showing the mean +/− standard deviation of the number of putative non-JAK2 driver mutations in young and old PV patients. Difference in mean mutation number for b and c assessed by Welch’s t-test. d) Bar graph of the variant allele frequencies of driver mutations identified in old PV. Letters above bars signify whether the mutation was predicted to be acquired before (“B”), coincident with (“C”), or after (“A”) JAK2 V617F. e) Plots of genotyping results from custom SNP assays to validate the driver mutations in four old PV patients. Each point represents the genotype of single colony. “WT” = wildtype for mutation; “het” = heterozygous for mutation; “hom” = homozygous for mutation. Variant allele frequencies from bulk cells are displayed above the plots for each patient.
Genomic DNA was isolated from either bulk peripheral blood mononuclear cells (PBMC) or granulocytes, in addition to matched normal tissue (skin or sorted CD3+ T cells) obtained from each patient. Age at sample collection was on average within 1.5 years of diagnosis (Fig. 1a). Drug treatment for patients did not significantly differ with age (Supp. Table S1). Enhanced exome capture sequencing was performed utilizing a standard xGen panel plus additional probes for recurrently mutated genes in MPN and/or AML.
Over 99% of exons sequenced had at least 20x coverage, with a mean depth of 133 reads. Of the 19 patients sequenced, three samples were removed due to sample contamination and one sample was removed due to technical artifact issues, leaving seven PVyoung and eight PVold remaining for analysis. The JAK2 V617F variant allele frequency (VAF) was not significantly different between PVold and PVyoung (p = 0.3357) (Supp. Fig. S1a). However, when clinical JAK2 V617F data was collected from 16 separate patients from the expanded cohort, a significantly higher VAF in older patients was observed (p = 0.0188) (Supp. Fig. S1b).
A total of 107 somatic mutations were identified, of which 84 were non-synonymous (Supp. Table S2). A significantly higher mutation load was observed in PVold, with a mean of 10.4 mutations versus 3.4 for PVyoung (p = 0.0154) (Fig. 1b). No large-scale chromosomal aberrations were identified (data not shown).
Secondary mutations potentially contributing to disease phenotype were identified in each cohort. Non-synonymous coding variants predicted to be deleterious to protein function through either protein truncation or having a FATHMM score > 0.5 and occurring in a gene where variants have been reported in a myeloid malignancy in multiple studies were considered. The identification of these putative secondary driver mutations revealed a striking difference in PVyoung versus PVold patients. Secondary mutations were identified in 7/8 PVold patients, namely in epigenetic regulators ASXL1, TET2, DNMT3A, transcription factors FOXP1 and NFE2, and cell cycle regulator CHEK2. In contrast, secondary driver mutations were not identified in any PVyoung patients (Fig. 1c & Table 1).
Table 1.
Primary and putative secondary driver mutations in young and old PV patients
| Young PV sample |
chr | start | ref | var | type | gene | strand | mut type | aa change | Tumor VAF |
|---|---|---|---|---|---|---|---|---|---|---|
| 331060 | 9 | 5073770 | G | T | SNP | JAK2 | 1 | missense | p.V617F | 16.5 |
| 465783 | 9 | 5073770 | G | T | SNP | JAK2 | 1 | missense | p.V617F | 18.93 |
| 551599 | 9 | 5073770 | G | T | SNP | JAK2 | 1 | missense | p.V617F | 45.51 |
| 638517 | 9 | 5073770 | G | T | SNP | JAK2 | 1 | missense | p.V617F | 39.9 |
| 697576 | 9 | 5073770 | G | T | SNP | JAK2 | 1 | missense | p.V617F | 13.6 |
| 732651 | 9 | 5073770 | G | T | SNP | JAK2 | 1 | missense | p.V617F | 17.36 |
| 993645 | 9 | 5073770 | G | T | SNP | JAK2 | 1 | missense | p.V617F | 11.03 |
| Old PV sample | chr | start | ref | var | type | gene | strand | mut type | aa change |
Tumor VAF |
| 149502 | 20 | 31022264 | G | A | SNP | ASXL1 | 1 | nonsense | p.W583* | 11.13 |
| 149502 | 9 | 5073770 | G | T | SNP | JAK2 | 1 | missense | p.V617F | 5.14 |
| 208617 | 9 | 5073770 | G | T | SNP | JAK2 | 1 | missense | p.V617F | 46.87 |
| 336362 | 20 | 31021250 | C | T | SNP | ASXL1 | 1 | nonsense | p.R417* | 50 |
| 336362 | 9 | 5073770 | G | T | SNP | JAK2 | 1 | missense | p.V617F | 33.25 |
| 374024 | 4 | 106196695 | - | A | INS | TET2 | 1 | frame_shift_ins | p.T1676fs | 41.8 |
| 374024 | 9 | 5073770 | G | T | SNP | JAK2 | 1 | missense | p.V617F | 39.62 |
| 374024 | 20 | 31022441 | - | G | INS | ASXL1 | 1 | frame_shift_ins | p.G645fs | 39.78 |
| 513509 | 9 | 5073770 | G | T | SNP | JAK2 | 1 | missense | p.V617F | 17.52 |
| 513509 | 22 | 29105992 | A | C | SNP | CHEK2 | −1 | splice_site | e7+2 | 16.57 |
| 513884 | 9 | 5073770 | G | T | SNP | JAK2 | 1 | missense | p.V617F | 17.91 |
| 513884 | 20 | 31023561 | GCTGACA | - | DEL | ASXL1 | 1 | frame_shift_del | p.A1016fs | 6.71 |
| 570655 | 9 | 5073770 | G | T | SNP | JAK2 | 1 | missense | p.V617F | 25.37 |
| 570655 | 2 | 25464537 | C | T | SNP | DNMT3A | −1 | missense | p.R659H | 7.87 |
| 673286 | 9 | 5073770 | G | T | SNP | JAK2 | 1 | missense | p.V617F | 92.24 |
| 673286 | 3 | 71027072 | G | A | SNP | FOXP1 | −1 | nonsense | p.Q419* | 50 |
| 673286 | 12 | 54686735 | T | - | DEL | NFE2 | −1 | frame_shift_del | p.Y182fs | 44.33 |
chr = chromosome; ref = reference; var = variant; mut type = mutation type; aa = amino acid; VAF = variant allele frequency; INS = insertion; DEL= deletion; SNP = single nucleotide polymorphism
Mutation order in the PVold cohort was inferred from VAF or serial sampling (patient 374024 who transformed to AML). Three mutations were predicted to arise before JAK2, with four mutations occurring coincident with JAK2, and two mutations arising after JAK2 (Fig. 1d). For 4/8 PVold patients where cryopreserved PBMCs were available, mutation validation via qPCR SNP genotyping was performed on lineage negative CD34+ single cell-derived colonies (Fig. 1e). In three patients, secondary mutations were confirmed as occurring before (336362-ASXL1) or coincident (513509-CHEK2; 673286-FOXP1/NFE2) with JAK2 (Fig. 1e). For patient 149502, the identification of ASXL1 in 7/12 colonies but absence of JAK2 V617F in all colonies tested suggested that ASXL1 was acquired prior to JAK2 but could not exclude the potential existence of two independent clones (Fig. 1e). For PVold patient 374024, sequencing of serial samples associated with disease transformation revealed loss of JAK2/TET2, while ASXL1 remained present in a subclone containing new RUNX1/NRAS mutations. These findings imply acquisition of ASXL1 prior to JAK2, with subsequent AML arising from the ASXL1-only clone (Supp. Fig. S2). These data show that in PVold secondary mutations can be acquired before, coincident with, or after JAK2, with no specific predominant pattern.
The germline JAK2 46/1 haplotype is known to predispose to JAK2 V617F+ MPN.6 Consistent with a previous report, 73.3% and 13.3% of patients were 46/1 heterozygous or homozygous, respectively, with no significant difference in distribution by age (Supp. Fig. S3a).6 When additional germline variants in genes known to potentially predispose to hematologic malignancies were identified (JAK2, MECOM, TET2, RUNX1, TP53, ATM, GATA2),7–9 however, PVyoung averaged nearly twice the number of variants per patient as PVold, although the difference was not significant (p = 0.2918) (Supp. Fig. 3b & Supp. Table S3).
The observed single driver landscape in PVyoung suggests non-genomic factors may contribute to the clinically distinct phenotype. Inflammatory cytokines are overproduced in PV and increase with normal aging.10, 11 The profile of ten selected cytokines was examined in the plasma of 15 PVyoung and 13 PVold patients in addition to 17 age-matched normal donors. No significant differences in cytokine concentrations between young and old normal plasma were identified, although this may have been related to small sample size (Supp. Fig. S4a). Compared to age-matched normals, the same cytokines were significantly increased in PVyoung and PVold plasma, namely IFN-γ, IL-2Rα, IL-6, IP-10, MIP-1α, and TNF (Supp. Fig. S4b & S4c). This abnormal cytokine profile was accentuated with age, as the fold changes of IFN-γ, IL-2Rα, IL-6, IP-10, TNF and VEGF (relative to age-matched normals) were significantly higher in old versus young patients (Supp. Fig. S4d). These data indicate that overproduction of inflammatory cytokines is a hallmark of PV that is further exacerbated by older age.
With recent research beginning to better characterize young MPN patients,12, 13 this study is the first to directly compare the mutation profile of young versus old PV patients. The finding that several secondary mutations identified in PVold can also be found with CHIP raises the possibility that these mutations were related merely to aging and occurred independent of JAK2 and the patient’s underlying PV. However, in 3/4 patients with genotyping validation, the presence of mutations within the same clone as JAK2 was confirmed. If these secondary mutations were entirely due to aging, one might expect that they would predominantly occur prior to JAK2 V617F. However, a minority of the observed non-JAK2 drivers (3/9) were likely acquired before JAK2. While these secondary mutations may nonetheless have been acquired incidentally, it remains plausible that they may contribute to the differences in clinical phenotype observed in old versus young PV patients.
The high prevalence of secondary driver mutations in PVold is consistent with the previously reported shorter median time to MF/AML transformation in PVold compared to PVyoung.3 These findings are also consistent with our prior study in primary myelofibrosis demonstrating accrual of disease-modifying mutations in conjunction with disease transformation.14 In a study focused on mutation order in MPNs, the transcriptional effects of JAK2 V617F were different depending on whether it occurred before or after TET2. In “JAK2-first” patients, single JAK2 mutant clones were larger than non-mutant and double mutant clones, suggesting that the addition of a second mutation can limit JAK2-driven HSPC proliferation.4 It is likely in PVyoung that the effects of JAK2 V617F in a single mutant landscape are different than when it is accompanied by a second mutation, and may contribute to distinct clinical phenotypes.
In conclusion, our data indicates that younger individuals develop PV in the setting of a single somatic JAK2 V617F mutation, whereas in older individuals JAK2 is predominantly accompanied by a secondary driver mutation that might not be entirely explained by age-induced mutation accumulation and which may impact PV disease presentation. Larger scale studies will be needed to verify these observations. In addition, the role of inflammatory cytokines, and the potential contribution of epigenetic15 and gender-related factors3, 15 in PV clinical phenotype remains to be further elucidated.
Supplementary Material
Acknowledgements
This work was supported by NIH grants K08HL106576 (Oh) and T32HL007088 (Fowles). This work was also supported by a grant from the Longer Life Foundation (Oh). Additional support was provided by the Washington University Institute of Clinical and Translational Sciences grant UL1TR000448 from the National Center for Advancing Translational Sciences of NIH. Support for patient sample collection and processing was provided by NIH grant P01CA101937. Exome sequencing was performed at the McDonnell Genome Institute. Technical support was provided by the Alvin J. Siteman Cancer Center Tissue Procurement Core Facility, Flow Cytometry Core, and Immunomonitoring Laboratory, which are supported by NCI Cancer Center Support Grant P30CA91842. The Immunomonitoring Laboratory is also supported by the Bursky Center for Human Immunology and Immunotherapy Programs. The authors thank D. Bender for assistance with multiplex cytokine measurement and C. Miller for assistance with sequencing analysis.
Footnotes
Disclosure of Conflicts of Interest
The authors report no conflicts of interest.
Supplementary Information can be found online at the Leukemia website.
References
- 1.Tefferi A, Pardanani A. Myeloproliferative Neoplasms: A Contemporary Review. JAMA Oncol 2015. April; 1(1): 97–105. [DOI] [PubMed] [Google Scholar]
- 2.Link DC, Walter MJ. ‘CHIP’ping away at clonal hematopoiesis. Leukemia 2016. August; 30(8): 1633–1635. [DOI] [PubMed] [Google Scholar]
- 3.Stein BL, Saraf S, Sobol U, Halpern A, Shammo J, Rondelli D, et al. Age-related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk Lymphoma 2013. September; 54(9): 1989–1995. [DOI] [PubMed] [Google Scholar]
- 4.Ortmann CA, Kent DG, Nangalia J, Silber Y, Wedge DC, Grinfeld J, et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med 2015. February 12; 372(7): 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012. July 20; 150(2): 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trifa AP, Banescu C, Tevet M, Bojan A, Dima D, Urian L, et al. TERT rs2736100 A>C SNP and JAK2 46/1 haplotype significantly contribute to the occurrence of JAK2 V617F and CALR mutated myeloproliferative neoplasms - a multicentric study on 529 patients. Br J Haematol 2016. July; 174(2): 218–226. [DOI] [PubMed] [Google Scholar]
- 7.Hinds DA, Barnholt KE, Mesa RA, Kiefer AK, Do CB, Eriksson N, et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 2016. August 25; 128(8): 1121–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bougeard G, Baert-Desurmont S, Tournier I, Vasseur S, Martin C, Brugieres L, et al. Impact of the MDM2 SNP309 and p53 Arg72Pro polymorphism on age of tumour onset in Li-Fraumeni syndrome. J Med Genet 2006. June; 43(6): 531–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Godley LA. Inherited predisposition to acute myeloid leukemia. Semin Hematol 2014. October; 51(4): 306–321. [DOI] [PubMed] [Google Scholar]
- 10.Vaidya R, Gangat N, Jimma T, Finke CM, Lasho TL, Pardanani A, et al. Plasma cytokines in polycythemia vera: phenotypic correlates, prognostic relevance, and comparison with myelofibrosis. Am J Hematol 2012. November; 87(11): 1003–1005. [DOI] [PubMed] [Google Scholar]
- 11.Alvarez-Rodriguez L, Lopez-Hoyos M, Munoz-Cacho P, Martinez-Taboada VM. Aging is associated with circulating cytokine dysregulation. Cell Immunol 2012; 273(2): 124–132. [DOI] [PubMed] [Google Scholar]
- 12.Boddu P, Masarova L, Verstovsek S, Strati P, Kantarjian H, Cortes J, et al. Patient characteristics and outcomes in adolescents and young adults with classical Philadelphia chromosome-negative myeloproliferative neoplasms. Ann Hematol 2018. January; 97(1): 109–121. [DOI] [PubMed] [Google Scholar]
- 13.Szuber N, Vallapureddy RR, Penna D, Lasho TL, Finke C, Hanson CA, et al. Myeloproliferative neoplasms in the young: Mayo Clinic experience with 361 patients age 40 years or younger. Am J Hematol 2018. August 29. [DOI] [PubMed] [Google Scholar]
- 14.Engle EK, Fisher DA, Miller CA, McLellan MD, Fulton RS, Moore DM, et al. Clonal evolution revealed by whole genome sequencing in a case of primary myelofibrosis transformed to secondary acute myeloid leukemia. Leukemia 2015. April; 29(4): 869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mascarenhas J, Roper N, Chaurasia P, Hoffman R. Epigenetic abnormalities in myeloproliferative neoplasms: a target for novel therapeutic strategies. Clin Epigenetics 2011. August; 2(2): 197–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.