

Abstract
The selective functionalization of one C–H bond over others in nearly identical steric and electronic environments can facilitate the construction of complex molecules. We report site-selective functionalizations of C–H bonds, differentiated solely by remote substituents, catalyzed by artificial metalloenzymes (ArM’s) that are generated from the combination of an evolvable P450 scaffold and a noble metal-porphyrin cofactor. The generated systems catalyze the insertion of carbenes into the C–H bonds of a range of phthalan derivatives containing substituents that render the two methylene positions in each phthalan inequivalent. These reactions occur with site-selectivities up to 17.8:1 and, in most cases, with pairs of enzyme mutants that preferentially form each of the two constitutional isomers. This study demonstrates the potential of abiotic reactions catalyzed by metalloenzymes to functionalize C-H bonds with site-selectivity that is difficult to achieve with small-molecule catalysts.
Keywords: Artificial Metalloenzymes, C–H bond functionalization, site-selectivity, Iridium-Porphyrin, P450s
Graphical Abstract
Site-selective functionalizations of C–H bonds that are differentiated solely by remote substituents are obtained with nearly 10:1 ratio between constitutional isomers when catalyzed by two distinct mutants of an artificial metalloenzyme generated from an evolvable P450 scaffold and a noble metal-porphyrin cofactor.
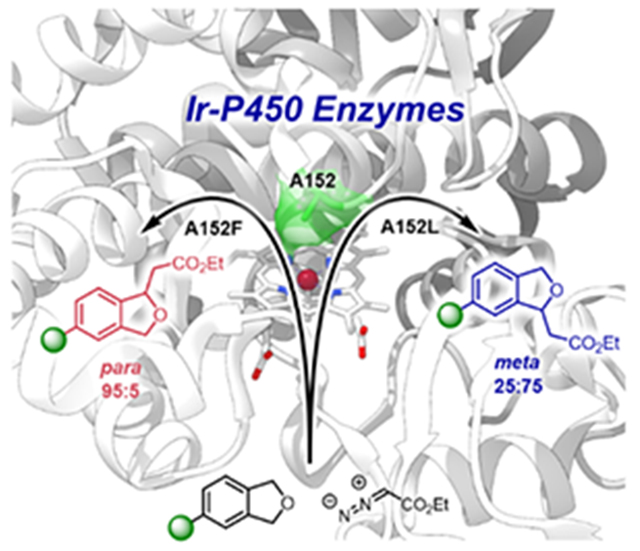
Introduction
C–H bond functionalization has changed the logic of chemical synthesis by enabling the direct addition of functional groups to a wide range of available starting materials.[1] Small-molecule, transition-metal catalysts can lead to site-selective C–H bond functionalization,[2] but many of the strategies for achieving selectivity rely on localized steric effects, directing groups, or both. Thus, it is challenging for small-molecule catalysts to distinguish between C–H bonds with similar local steric and electronic properties rendered inequivalent by the lack of symmetry of a molecule. However, enzymes, such as cytochromes P450 (CYPs), catalyze the oxidation of C–H bonds that are nearly equivalent, with high site-selectivity.[3] Such reactions often occur within the biosynthetic pathways of complex natural products, such as terpenes, alkaloids or steroids, all of which contain an abundance of similar C–H bonds.[4]
Although they can react with high selectivity, natural enzymes install a limited range of functional groups during biosynthesis. One strategy to expand the scope of reactions catalyzed by enzymes and improve the efficiency by which these reactions can occur is to conduct directed evolution. Directed evolution has proven to be a powerful methodology that allows chemists to extend the fitness of biocatalysts toward unnatural reactions,[5] Through directed evolution, in which many rounds of iterative mutagenesis are conducted, hemoproteins have been repurposed to catalyze a range of unnatural group transfer reactions to C-C multiple bonds and C-H bonds with high activity and selectivity.[6]
A complementary strategy to expand the scope of known abiological transformations is to construct artificial metalloenzymes (ArMs). These systems incorporate unnatural cofactors into evolvable protein scaffolds,[7] and the resulting ArMs are capable of catalyzing abiological reactions, often with high selectivity. Several ArMs have been constructed by exploiting the biotin-streptavidin technology,[8] unnatural amino acid anchoring,[9] and the formal replacement of the native heme cofactor with synthetic porphyrin cofactors containing noble metals.[10]
Our laboratory reported the first example of enzymatic carbene insertions into (sp3) C–H bonds, including intermolecular functionalizations. These reactions were catalyzed by artificial metalloenzymes containing an Ir(Me)–MPIX cofactor,[11] and such reactivity provides a platform to evaluate whether Ir(Me)-ArMs could be evolved to catalyze site-selective reactions of C–H bonds that are nearly equivalent and are known to yield a mixture of products with small-molecule catalysts.[12] Phthalan derivatives were chosen as model substrates because they have two sites that contain (sp3) C–H bonds with similar steric and electronic environments. Unsubstituted phthalan undergoes reaction with carbene precursors catalyzed by a P450 containing Ir(Me)–MPIX (Scheme 1).[11]
Scheme 1.
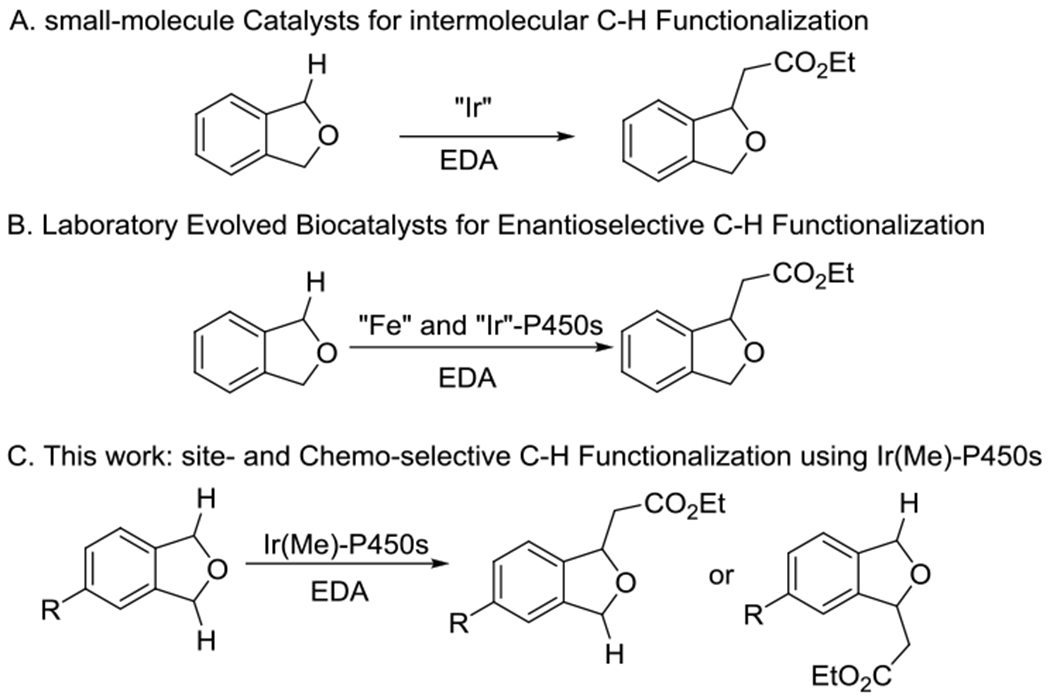
Intermolecular insertion of acceptor-only carbenes into (sp3)C–H bonds with small-molecule catalysts and biocatalysts.
Here we report that 4-substituted phthalans react to give products that are distinguished only by the position of a remote substituent and react with pairs of Ir(Me)–CYP119 mutants to give the opposite constitutional isomer as the major product for most of the derivatives. We also show that the reactions occur with high chemoselectivity for C–H bonds over typically-reactive functional groups, such as N–H bonds, and in select cases with high enantioselectivity.
Results and Discussion
Reactivity of the free Ir(Me)-MPIX cofactor and engineered Ir(Me)–hemoproteins.
To determine the role of the protein scaffold in controlling site-selectivity, we first assessed the reactivity and site-selectivity of the free Ir(Me)-MPIX cofactor for the insertion of the carbene from ethyl 2-diazoacetate (EDA) into the C–H bond of a 4-substituted phthalan. To do so, we studied the reaction of 4-bromophthalan (1a) with EDA catalyzed by the free cofactor Ir(Me)–MPIX in NaPi buffer (100 mM, pH = 6.0) . Without the protein, the free cofactor reaction gave a mixture of products resulting from a single and double insertion of the carbene into the (sp3) C–H bonds of 1a. Products from a single insertion into the C–H bonds located meta and para to the bromine atom were observed in a 1:1 ratio (Scheme 2). Selective carbene insertion into (sp3) C–H bonds catalyzed by transition metal catalysts typically utilize a donor-acceptor carbene precursor.[13] There are only a few small molecule catalysts that can handle acceptor-only carbene precursors and they require the substrate as solvent or in excess (Scheme 1A).[12, 14] We reasoned that the pre-organization of substrates within the active site of a protein would afford the opportunity to catalyze the site-selective intermolecular C–H functionalization’s using the substrates as the limiting reagents.
Scheme 2.

Site-selective intermolecular C–H insertion catalyzed by Ir(Me)-MPIX. Yields refers to the formation of all the carbene insertion products and were determined by GC with dodecane as the internal stander.
To test the capability of engineered Ir(Me)–hemoproteins to differentiate nearly identical C–H bonds and to yield products from insertion of a carbene into just one C–H bond, we conducted reactions of 4-bromophthalan 1a and EDA with previously-engineered myoglobin (Myo) mutants. Evaluation of site-selectivity from over one-hundred Ir(Me)-Myo mutants,[10b, 10d] containing mutations over six active-site residues, from clarified lysate and purified protein reaction screening did not uncover any notable site-selectivity beyond that of the free cofactor.
On the basis of our previous studies on artificial metalloenzymes generated from artificial cofactors and a P450 from the thermophile Sulfolobus solfataricus, CYP119, we investigated whether selectivity could be obtained from this scaffold possessing a more enclosed active site. We previously showed that the melting temperature of CYP119 is higher (69° C) than that for the analogous construct from the more commonly studied P450-BM3 (45° C),[11] and the carbene insertions into C-H bonds do not require the reductase domain contained within BM3.
We initially screened selectivity with T213G, C317G, a double mutant that reduced both the size and hydrogen-bonding ability of the amino acid residue at position 213 and eliminated the native axial heme-ligating cysteine at position 317. In contrast to reactions of the free cofactor, the reactions catalyzed by the double mutant CYP119 enzyme gave only the single carbene insertion products (Scheme 2). Although the site-selectivity of the initial experiment was negligible, the improved chemo-selectivity for a single insertion product is clearly imparted by the protein environment.
Directed evolution of Ir(Me)–CYP119.
To create catalysts for site-selective C–H functionalization of phthalan derivatives, we studied the reaction of 4-bromophthalan 1a with EDA catalyzed by a mutant library of CYP119 proteins.[11] This library was constructed from the double mutant scaffold containing T213G and C317G mutations followed by the introduction of additional mutations at amino acids proximal to the active site (Figure 1). The mutant library was evaluated by screening in 96-well plates. Reactions were conducted by generating a cell lysate solution of Ir(Me)-MPIX-CYP119 by reconstituting each apo-protein mutant variant with a stock solution of Ir(Me)–MPIX cofactor, followed by the addition of phthalan and carbene precursor catalyst stock solutions to the 96-well plates. The ratios of isomeric products from the insertion process were measured by high-throughput GC sampling from 96-well plates.
Figure 1.

Structure of WT Fe-CYP119 (image prepared in Chimera from PDB 1IO7).[16] Red label: mutant positions that were generated by site-directed mutagenesis (A152, T213, V254, C317); Orange label: additional positions generated by error-prone PCR for para selectivity (D177 and H340) and meta selectivity (A247).
Results from reaction screening indicated that some members of the CYP119 mutant library catalyzed reactions at one of the two methylene positions of the substituted phthalan with measurable selectivity. For example, the mutant of CYP119 PG1(para generation 1: C317G, T213G, V254L, A152F) preferentially formed the product from insertion into the benzylic C–H bond para to the bromine atom over that from insertion meta to the bromine atom with a 2.8:1 ratio and 720 TON. In contrast, the mutant of CYP119 MG1(meta generation 1: C317G, T213G, V254A, A152L) preferentially formed the product from insertion into the benzylic C–H bond meta to the bromine atom with a 1:2.7 ratio and 690 TON (Scheme 2 bar 3 and 4).
Reactions conducted at catalyst loadings lower than 0.05 mol % led to lower conversion, but they also led to lower site-selectivity and gave a mixture of products resulting from a single and double insertion of the carbene into the (sp3) C–H bonds of 1a. We assumed the lower chemo- and site-selectivities resulted from an instability of the Ir(Me)-CYP119 construct to the reaction conditions, leading to the generation of free cofactor. This decomposition presumably also influenced reactions at higher catalyst loadings. Because the proteins at this point of the evolution were generated by mutagenesis in the active site, we created a new library of mutants based on PG1 and MG1 by error-prone PCR that would contain mutations distal to the active site that could stabilize the Ir(Me)-P450 construct.
Indeed, three rounds of error-prone PCR led to enzymes that catalyzed the carbene transfer with greater site-selectivity, in many cases reaching differences in selectivity greater than 10:1 between pairs of mutants reacting at the two methylene positions of 1a and with higher turnovers. Eleven mutants were identified that catalyzed the reaction to form predominantly the para isomer, and 10 mutants were identified that preferentially formed the meta isomer. These 21 mutants that reacted with selectivity higher than those of the parent mutants were tested for activity across a series of 4-substiuted phthalan derivatives, with which small transition metal complexes,[12] including the free Ir(Me)-MPIX cofactor, reacted to give insertion products with low selectivity (Scheme S3).
Site-Selective Ir(Me)-CYP119 catalyzed carbene insertions into phthalan derivatives.
Two of the 21 mutant variants catalyzed the insertion reaction of EDA into a series of 4-substituted phthalan derivatives, having various functional groups, with good TONs and measurable chemo- and site-selectivity for either para or meta isomers. The reaction of 4-bromophthalan 1a catalyzed by the mutant PG2(PG1+D177N, H340L) gave a 3.2:1 ratio of constitutional isomers in favor of the para isomer with 1160 TON, while the same reaction catalyzed by the mutant MG2(MG1+A247S) gave a 1:3.4 ratio of isomers in favor of the meta isomer with 1456 TON (Scheme 3). Similar results were observed for reactions of 4-chlorophthalan 1b. The reaction catalyzed by the mutant PG2 formed the product from insertion into the benzylic C–H bond para to the chloro-substituent as the major product with a 3.5:1 ratio of constitutional isomers and 2286 TON, while the reaction catalyzed by the mutant MG2 yielded the meta insertion product in a 1:3.1 ratio and with 1380 TON.
Scheme 3.
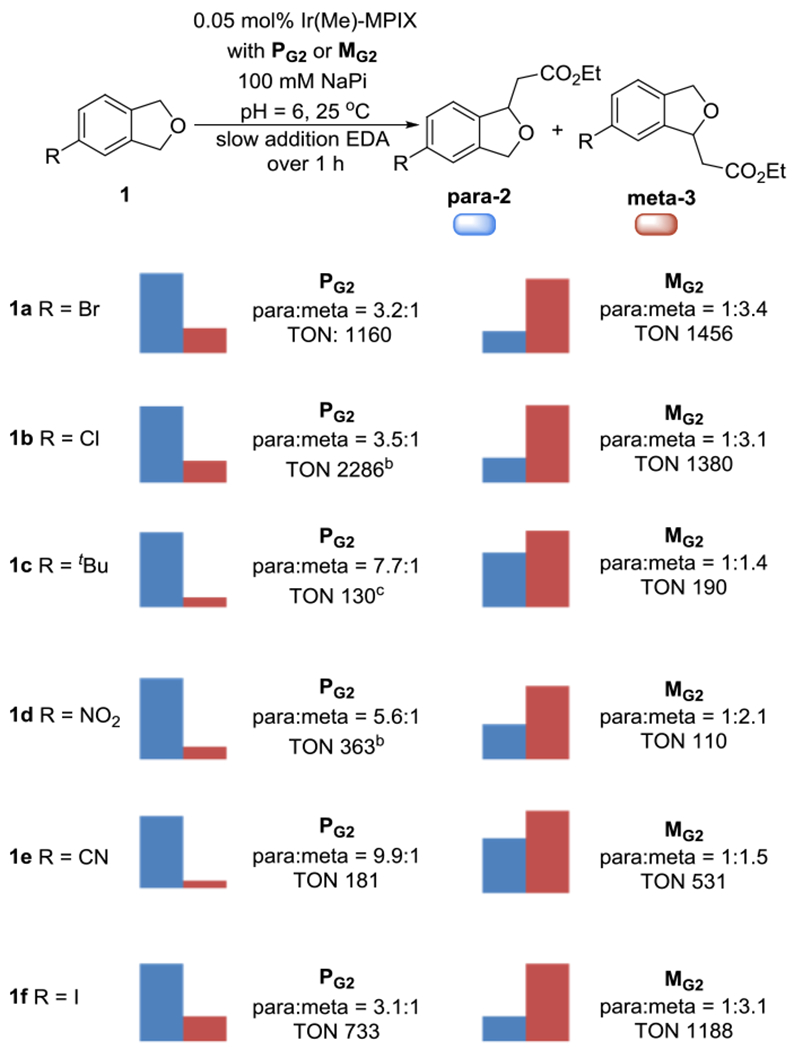
Scope of substrates that CYP119 mutants reverse site-selectivity[a] [a] TONs refers to the formation of both meta and para benzylic C–H bond insertion products and was determined by GC with dodecane as the internal stander. [b] 0.02 mol% Ir(Me)-MPIX CYP119 was used. [c] 0.1 mol% Ir(Me)-MPIX CYP119 was used.
Phthalan 1c contains a 4-tBu substituent, which is larger than the halides of 1a and 1b. However, the site-selectivity results of reactions of this sterically distinct substrate with CYP119 mutants are similar to those of reactions of 1a and 1b. The product from insertion into the benzylic C–H bond para to the tBu-substituent was favored by the mutant PG2 in a ratio of 7.7:1 and accomplished with 130 TON. Although the selectivity for the meta benzylic C–H bond of 1c from reactions catalyzed by the mutant MG2 was lower than that for reaction of 1a and 1b, the selectivity, nevertheless, was reversed to 1:1.4 in favor of the meta isomer with up to 190 TON. Thus, we were able, again, to identify a pair of mutants (PG2 and MG2) that react with nearly a >10:1 reversal of site selectivity between mutants for the two types of C–H bonds present in 1c.
Nitro-substituted reactants rarely undergo C–H bond functionalization, and the 4-nitro-substituted phthalan 1d previously did not react with EDA when catalyzed by a small-molecule iridium species.[12] However, the reaction of this substrate with EDA catalyzed by an iridium-containing ArMs formed the product from insertion into the C–H bond. Although the turnover numbers from this reaction were lower than those from reactions of the other substrates, the reaction of 4-nitrophthalan 1d catalyzed by the mutant PG2 formed the product from reaction of the benzylic C–H bond para to the nitro-substituent as the major isomer with a 5.6:1 ratio over that from reaction at the meta C–H bond (363 TON). The same reaction catalyzed by the mutant MG2 preferentially formed the product from reaction at the meta C–H bond with a 1:2.1 ratio of constitutional isomers. A similar preference for reaction with the C–H bond para to the substituent was observed from the reaction of 4-cyanophthalan 1e catalyzed by PG2 with a 9.9:1 ratio over the reaction at the methylene C–H bond meta to the cyano-substituent (181 TON). The selectivity was reversed to 1:1.5 in favor of reaction at the benzylic C–H bond meta to the cyano-substituent for the reaction catalyzed by the mutant MG2 (531 TON), again demonstrating a 10:1 reversal of site-selectivity for a pair of mutant ArMs. Reaction of the 4-iodophthalan 1f also occurred with good selectivity for insertion at the para position (3.1:1, 733 TON) when catalyzed by the mutant PG2, whereas the same selectivity in favor of reaction at the meta position (1:3.1) was observed for the reaction catalyzed by mutant MG2 (1188 TON).
Substrates for which the mutant enzymes selectively react at the para C–H bond.
In addition to reacting site-selectively for one C–H bond over another similar C–H bond, the ArMs we report prefer react with carbene insertion into benzylicic C–H bonds over other potential positions. The reaction of amide-substituted phthalan 4a catalyzed by PG2 formed the product from insertion of the carbene into the C–H bond with 17.8:1 selectivity for the benzylic C–H bond para to the amide-substituent and 470 TON. No product from insertion of the carbene into the N–H bond was observed from reactions catalyzed by any of the Ir(Me)-CYP119 constructs.
Likewise, the products of the reactions of EDA with amine substituted phthalan 4b catalyzed by the free Ir(Me)-porphyrin and artificial enzyme were distinct. The reaction of EDA with 4b catalyzed by Ir(Me)-MPIX gave the single and double carbene insertion products from addition to the C-H bond alpha to nitrogen in the pyrrolidine. However, the reaction catalyzed by the mutant PG2 occurred with high chemo- and site-selectivity for the C–H bond para to the amine-substituent (13.6:1 para selectivity and 55 TON).
The reactions of EDA with ester substituted phthalan 4c and ether substituted phthalan 4d catalyzed by PG2 also occurred with higher site-selectivity compared to the reactions catalyzed by free-cofactor. The reaction catalyzed by Ir(Me)-MPIX occurred with low TONs and almost no selectivity for the methylene units para and meta to the ester- and ether-substituents (Scheme S3). However, selectivity of 8.3:1 was observed for reaction at the methylene position para to the ester-substituent of 4c when catalyzed by the mutant PG2 (268 TON). The reaction of 4d occurred with 1796 TON and 6.4:1 selectivity for the methylene para to the methoxy-substituent. These reactions in Scheme 4, combined with the reactions of cyano- and nitro-substituted phthalans in Scheme 3, illustrate the potential of ArMs to enhance para-selectivity regardless of electron-donating properties of the substituents.
Scheme 4.

Scope of substrates for which mutant enzymes are selective for reaction at the para C–H Bond. TONs refers to the formation of both meta and para C–H bond insertion products and were determined by GC with dodecane as the internal stander.
Enantioselective intermolecular C–H insertion catalyzed by Ir(Me)-CYP119.
Our results show that the environment within the ArMs can control the number of insertion events, site-selectivity of the atom transfer position, and tolerate several competitive functional groups. We have shown previously that C–H insertion reactions catalyzed by Ir(Me)-P450s can occur in an enantioselective fashion.[11] However, because the current work is focused on assessing the ability of ArMs to catalyze reactions with site-selectively, we did not perform extensive evolution to enhance enantioselectivity. Nevertheless, some of the reactions did occur with substantial enantioselectivity when catalyzed by members of the mutant library. For example, the reactions of EDA with 4-fluorophthalan (7a) occurred to give the major constitutional isomer in 97:3 er and the minor isomer in 92:8 er when catalyzed by MG1+P252S (Scheme 5). The ratio of constitutional isomers was 1:1.8 in favor of the meta isomer. Although this site-selectivity is lower than that of reactions of other substituted phthalans, it does show that the enzyme can react with significant site-selectivity and enantioselectively for two C–H bonds that differ only by a remote fluorine versus hydrogen on the aryl ring. A fluorine atom has the closest size to hydrogen of any atom, and has the closest Hammett parameter to hydrogen of nearly any substituent.[15] In comparison with our prior results from the reaction of phthalan and EDA catalyzed by an Ir(Me)-P450s (84:16 er, 324 TON),[11] an increase in enantioselectivity and reactivity was observed from the same reaction catalyzed by the mutant MG1+P252S with 90:10 er and 1520 TON.[6a]
Scheme 5.
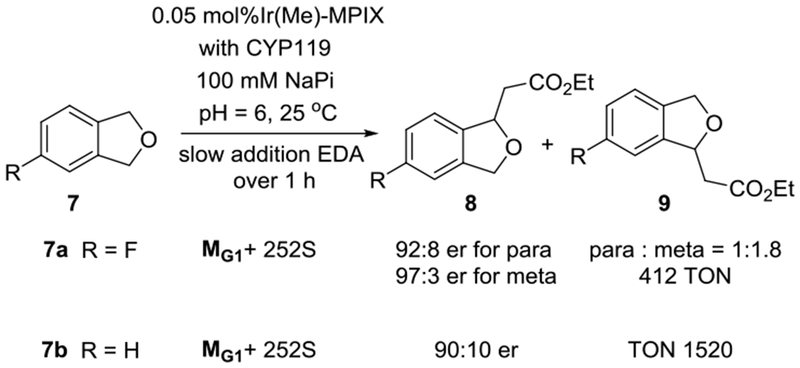
Enantioselective Intermolecular C–H Insertion Catalyzed by Ir(Me)-CYP119. TONs refer to the formation of both meta and para C–H bond insertion products with dodecane as the internal stander. The er values were determined by HPLC with a chiral column.
Scale up of the reactions with 1a and 1b.
The feasibility of this reaction for synthetic scale was assessed by conducting the reactions of 1a and 1b on a 0.3 mmol scale (Scheme 6). Similar isolated yields and selectivity were obtained with the same substrate on a smaller scale.
Scheme 6.

Scale up the reactions Catalyzed by mutant PG2.
Stability of a series of Ir(Me)-P450 mutants
The majority of mutations (A152, T213, V254 and C317) were located within the active site; however, some mutations, especially those generated by error-prone PCR (D177, H340 and A247), are located far from the active site in both PG2 and MG2 (Figure 1). As noted previously in this paper, these additional mutations appear to enhance the stability of the Ir(Me)-P450 construct because they have a larger influence on turnover number than on site-selectivity (Scheme 3 and 4). To assess this line of reasoning, we evaluated the stability of the Ir(Me)-CYP119 construct to temperature and reaction components.
To assist our evaluation of the effect of the mutations distal to the active site, we generated two mutants, Mmax (MG2 + D177N, H340L) and Pmax (PG2 + A247S), which contain all of the mutations peripheral to the active site in either MG2 or PG2. Like PG1, Pmax catalyzed the reaction of 1a with a similar site selectivity ratio of 3:1 in favor of the para isomer, but the turnover number of the reaction catalyzed by Pmax (1450 TON) was twice as high as that of the reaction catalyzed by PG1 (720 TON) (Figure 2). Likewise, the site selectivity of the reaction catalyzed by Mmax was similar to that of the reaction catalyzed by MG1 (1:2.7 for MG1 vs. 1:3.5 for Mmax), but the TON of the reaction catalyzed by Mmax (1834 TON) was more than three times higher than that of the parent MG1 mutant (580 TON) (Figure 2). These results further support the conclusion that the mutations resulting from error-prone PCR (177N, 340L and 247S) stabilize the Ir(Me)-P450 construct during the course of the reaction.
Figure 2.
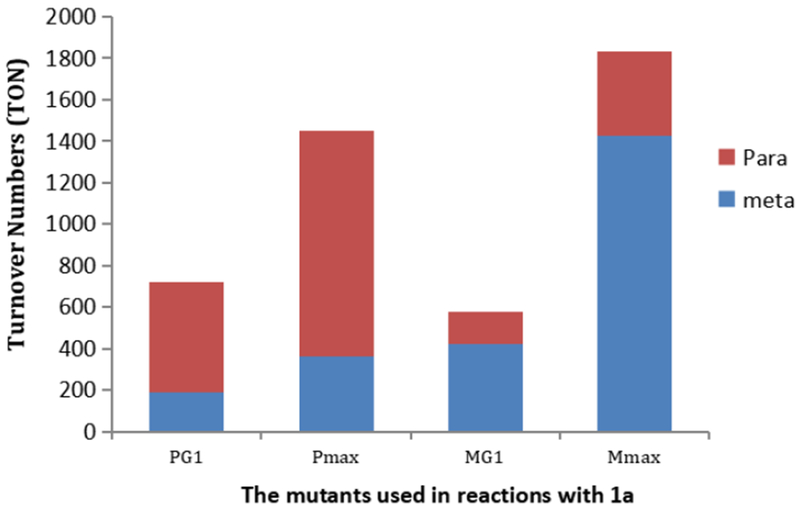
Activity studies of different mutants with slow addition of EDA with 1a. TONs refers to the formation of both meta and para C–H bond insertion products with dodecane as the internal stander.
Recently it was reported that modification of the protein scaffold during cyclopropanation of alkenes catalyzed by repurposed heme enzymes and ArMs can occur by carbene transfer into the amino acid residues of the host protein.[10c,17a,b] Because Ir(Me)-MPIX is an extremely active catalyst for carbene transfers, we suspected that this modification could limit the TON of the Ir(Me)-P450 construct and that these distal mutations might be retarding this modification. However, masses corresponding to addition of EDA to the protein or addition of the methyl group of the Ir(Me) cofactor to the protein were not observed from analysis of intact proteins by ESI-MS after addition of EDA to the six mutants of Ir(Me)-PIX CYP119 that gave the highest selectivities or addition of EDA and 4-bromophthalan 1a to the same six mutants.[17c] Instead, the carbene undergoes rapid formation of fumarate and maleate (Figure S79). Perhaps the high rate of dimerization prevents reaction of the carbene unit with amino acid residues of the protein scaffold.
Prior studies on repurposed and artificial metalloenzymes for carbene transfer showed that the CD spectrum changed upon exposure to EDA.[10c,17a] Thus, we measured the CD spectra of PG1, PG2, Pmax, MG1, MG2 and MMax in the apo- and in the holo-Ir(Me)-CYP119 forms before and after exposure to EDA. All of these CD spectra are indistinguishable from each other. No notable differences between the CD spectra of the apo- and holo-CYP119 form were observed, or between the CD spectra of these two proteins that of the WT-CYP119 protein. The α-helicity ([θ]222/[θ]208) ratio was within 1.00 and 0.96 for all of the Ir(Me)-P450 constructs, and these values are similar to those reported for the WT-CYP119 protein (Figures S60–S71).[18]
The melting temperatures (Tm) of a series of mutants were measured by differential scanning calorimetry. No significant differences among the Tm values of the mutants were observed; the average Tm value was 67.5°C with a standard deviation of 0.2°C among all the mutants (Table S2 and Figures S84–S89). The reported Tm value of WT-CYP119 is 91.9 °C. It is not surprising that the Tm values of the mutants of Ir(Me)-MPIX CYP119 are lower than that of WT-CYP119, given the mutations and exchange of an axially-ligated Fe-heme cofactor for a non-ligated artificial Ir(Me)-MPIX cofactor.[19] However, the similarity of the Tm values of the mutants indicate that changes in the active site or in the periphery of the protein have little effect on the folding and thermal stability of the Ir(Me)-PIX CYP119 construct. Therefore, the higher TON of the systems containing peripheral mutations seems to result from greater stability of the Ir(Me)-P450 constructs during catalysis.
To probe the stability of CYP119s toward the reaction components (substrate, product, solvent, etc.), Ir(Me)-CYP119 mutants were treated with components of the reaction mixture for one hour prior to initiating reactions (Figures S55–59). This pretreatment of Ir(Me)-CYP119s with the reaction products influenced the selectivity of the reaction. For example, reactions of 4-bromophthalan 1a with EDA conducted after treating PG1 or MG1 with 4-chloropthalan-product 2b for one hour occurred with negligible site-selectivity, whereas the same reaction conducted after treating Mmax or Pmax with 2b for one hour occurred with site-selectivities of 1:2 favoring the meta isomer and 1.8:1 favoring the para isomer, respectively (Figures S58–59). These results imply that the peripheral mutations stabilize the enzyme toward the C–H insertion products. This trend in effect of product on selectivity is similar to the trend in stability of the protein determined independently by CD (Figures S79–S82). Although difficult to correlate quantitatively, we found that the concentrations of the proteins containing peripheral mutations (Pmax and Mmax) were higher than those of the parent mutants PG1 and MG1 after exposure of equal concentrations of the Ir(Me)-P450 proteins to acetate-substituted product 6c.[20]
Conclusion
In summary, artificial metalloenzymes containing a noble metal porphyrin (Ir(Me)-CYP119) are capable of catalyzing the intermolecular insertion of acceptor-only carbenes into nearly equivalent C–H bonds with up to 17.8:1 site-selectively for a series of 4-substitued phthalans. Through laboratory evolution a nearly 10:1 ratio between either the meta or para constitutional isomers can be obtained using two distinct Ir(Me)-CYP119 mutants. Although not the primary focus of this study, enantioselectivity of 97:3 can be obtained with the current mutant library for the insertion of EDA into 4-fluorophthalan. The insertion reactions catalyzed by Ir(Me)-P450 tolerated a series of functional groups that have not been tolerated by small molecule catalysts for analogous insertions of acceptor-only carbenes. Furthermore, unlike other ArMs or repurposed heme enzymes, the Ir(Me)-P450s are less susceptible to deactivation from EDA insertion into the protein framework. In general, this work highlights how the combination of a highly-active transition metal active site with a readily-evolvable protein framework can lead to regioselectivities for abiotic reactions that would be difficult to achieve with small-molecule catalysts.
Supplementary Material
Acknowledgements
This work was supported by the Director, Office of Science, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Y. G. thanks a joint postdoc fellowship from Pharmaron and Shanghai Institute of Organic Chemistry (SIOC). S.N.N thanks the NIH (F32-GM126652) and the Burroughs Welcome fund (PDEB). Z. L thanks to National Science Scholarship from the Singapore Agency for Science, Technology and Research. We thank the Molecular Foundry for their assistance in DSC measurements. Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Yamaguchi J, Yamaguchi AD, Itami K, Angew. Chem. Int. Ed 2012, 51, 8960–9009 [DOI] [PubMed] [Google Scholar]; b) Brückl T, Baxter RD, Ishihara Y, Baran PS, Acc. Chem. Res 2012, 45, 826–839 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hartwig JF, J. Am. Chem. Soc 2016, 138, 2–24 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Das S, Incarvito CD, Crabtree RH, Brudvig GW, Science 2006, 312, 1941. [DOI] [PubMed] [Google Scholar]
- [2].a) Engle KM, Mei T-S, Wasa M, Yu J-Q, Acc. Chem. Res 2012, 45, 788–802 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liao K, Pickel TC, Boyarskikh V, Bacsa J, Musaev DG, Davies HML, Nature 2017, 551, 609–613 [DOI] [PubMed] [Google Scholar]; c) Anding BJ, Dairo TO, Woo LK, Organometallics 2017, 36, 1842–1847 [Google Scholar]; d) Che C-M, Lo VK-Y, Zhou C-Y, Huang J-S, Chem. Soc. Rev 2011, 40, 1950–1975 [DOI] [PubMed] [Google Scholar]; e) Metrano AJ, Miller SJ, Acc. Chem. Res 2019, 52, 199–215 [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Liao K, Negretti S, Musaev DG, Bacsa J, Davies HML, Nature 2016, 533, 230. [DOI] [PubMed] [Google Scholar]; g) Liao K, Liu W, Niemeyer ZL, Ren Z, Bacsa J, Musaev DG, Sigman MS, Davies HML, ACS Catal 2018, 8, 678–682. [Google Scholar]
- [3].a) Narayan ARH, Jiménez-Osés G, Liu P, Negretti S, Zhao W, Gilbert MM, Ramabhadran RO, Yang Y-F, Furan LR, Li Z, Podust LM, Montgomery J, Houk KN, Sherman DH, Nat. Chem 2015, 7, 653–660 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Negretti S, Narayan ARH, Chiou KC, Kells PM, Stachowski JL, Hansen DA, Podust LM, Montgomery J, Sherman DH, J. Am. Chem. Soc 2014, 136, 4901–4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Isin EM, Guengerich FP, Biochim. Biophys. Acta 2007, 1770, 314–329. [DOI] [PubMed] [Google Scholar]
- [5].a) Arnold FH, Angew. Chem. Int. Ed 2018, 57, 4143–4148 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Packer MS, Liu DR, Nat. Rev. Genet 2015, 16, 379. [DOI] [PubMed] [Google Scholar]; c) Sun Z, Wikmark Y, Bäckvall J-E, Reetz MT, Chem. Eur. J 2016, 22, 5046–5054. [DOI] [PubMed] [Google Scholar]
- [6].a) Zhang RK, Chen K, Huang X, Wohlschlager L, Renata H, Arnold FH, Nature 2019, 565, 67–72 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hammer SC, Knight AM, Arnold FH, Curr. Opin. Green Sust. Chem 2017, 7, 23–30 [Google Scholar]; c) Brandenberg OF, Fasan R, Arnold FH, Curr. Opin. Biotechnol 2017, 47, 102–111 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cho I, Jia Z-J, Arnold FH, Science 2019, 364, 575. [DOI] [PubMed] [Google Scholar]
- [7].a) Schwizer F, Okamoto Y, Heinisch T, Gu Y, Pellizzoni MM, Lebrun V, Reuter R, Köhler V, Lewis JC, Ward TR, Chem. Rev 2018, 118, 142–231 [DOI] [PubMed] [Google Scholar]; b) Ohashi M, Koshiyama T, Ueno T, Yanase M, Fujii H, Watanabe Y, Angew. Chem. Int. Ed 2003, 42, 1005–1008 [DOI] [PubMed] [Google Scholar]; c) Carey JR, Ma SK, Pfister TD, Garner DK, Kim HK, Abramite JA, Wang Z, Guo Z, Lu Y, J. Am. Chem. Soc 2004, 126, 10812–10813 [DOI] [PubMed] [Google Scholar]; d) Oohora K, Meichin H, Zhao L, Wolf MW, Nakayama A, Hasegawa J.-y., Lehnert N, Hayashi T, J. Am. Chem. Soc 2017, 139, 17265–17268. [DOI] [PubMed] [Google Scholar]
- [8].a) Heinisch T, Ward TR, Acc. Chem. Res. 2016, 49, 1711–1721 [DOI] [PubMed] [Google Scholar]; b) Liang AD, Serrano-Plana J, Peterson RL, Ward TR, Acc. Chem. Res 2019, 52, 585–595 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Reetz MT, Acc. Chem. Res. 2019, 52, 336–344. [DOI] [PubMed] [Google Scholar]
- [9].a) Lewis JC, Curr. Opin. Chem. Biol 2015, 25, 27–35 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lewis JC, Acc. Chem. Res 2019, 52, 576–584 [DOI] [PubMed] [Google Scholar]; c) Mirts EN, Bhagi-Damodaran A, Lu Y, Acc. Chem. Res 2019, 52, 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Natoli SN, Hartwig JF, Acc. Chem. Res 2019, 52, 326–335 [DOI] [PubMed] [Google Scholar]; b) Key HM, Dydio P, Clark DS, Hartwig JF, Nature 2016, 534, 534–537 [DOI] [PubMed] [Google Scholar]; c) Wolf MW, Vargas DA, Lehnert N, Inorg. Chem 2017, 56, 5623–5635 [DOI] [PubMed] [Google Scholar]; d) Sreenilayam G, Moore EJ, Steck V, Fasan R, Adv. Synth. Catal 2017, 359, 2076–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Key HM, Dydio P, Liu Z, Rha JYE, Nazarenko A, Seyedkazemi V, Clark DS, Hartwig JF, ACS Cent. Sci 2017, 3, 302–308 [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Dydio P, Key HM, Hayashi H, Clark DS, Hartwig JF, J. Am. Chem. Soc 2017, 139, 1750–1753. [DOI] [PubMed] [Google Scholar]
- [11].Dydio P, Key HM, Nazarenko A, Rha JY-E, Seyedkazemi V, Clark DS, Hartwig JF, Science 2016, 354, 102–106. [DOI] [PubMed] [Google Scholar]
- [12].a) For a small-molecule, iridium catalyst that gives a mixture of products, see: Weldy NM, Schafer AG, Owens CP, Herting CJ, Varela-Alvarez A, Chen S, Niemeyer Z, Musaev DG, Sigman MS, Davies HML, Blakey SB, Chem. Sci 2016, 7, 3142–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) For an engineered P411 that reacts selectively at sterically and electronically distinct positions of pyrroles, see: Brandenberg OF, Chen K, Arnold FH, J. Am. Chem. Soc 2019, 141, 8989–8995. [DOI] [PubMed] [Google Scholar]
- [13].Doyle MP, Duffy R, Ratnikov M, Zhou L, Chem. Rev 2010, 110, 704–724. [DOI] [PubMed] [Google Scholar]
- [14].Díaz-Requejo MM, Belderraín TR, Nicasio MC, Trofimenko S, Pérez PJ, J. Am. Chem. Soc 2002, 124, 896–897. [DOI] [PubMed] [Google Scholar]
- [15].Hansch C, Leo A, Taft RW, Chem. Rev 1991, 91, 165–195. [Google Scholar]
- [16].Park S-Y, Yamane K, Adachi S.-i., Shiro Y, Weiss KE, Sligar SG, Acta Crystallogr., Sect D: Biol. Crystallogr 2000, 56, 1173–1175. [DOI] [PubMed] [Google Scholar]
- [17].a) Renata H, Lewis RD, Sweredoski MJ, Moradian A, Hess S, Wang ZJ, Arnold FH, J. Am. Chem. Soc 2016, 138, 12527–12533. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) For observation of carbene insertion only after digestion of the protein and potential activation of the protein by carbene insertion, see: Yang H, Swartz AM, Park HJ, Srivastava P, Ellis-Guardiola K, Upp DM, Lee G, Belsare K, Gu Y, Zhang C, Moellering RE, Lewis JC, Nat. Chem 2018, 10, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) We cannot rule out transfer of the carbene to the porphyrin unit. We have been unable to isolate the metal-porphyrin cofactor after the reaction for analysis by mass spectroscopy.
- [18].Banerjee R, Sheet T, Proteins 2017, 85, 1975–1982 [DOI] [PubMed] [Google Scholar]
- [19].Park S-Y, Yamane K, Adachi S.-i., Shiro Y, Weiss KE, Maves SA, Sligar SG, J. Biol. Inorg. Chem 2002, 91, 491–501; b) [DOI] [PubMed] [Google Scholar]
- [20].6c was chosen because it has the weakest absorption at 280nm and high solubility compared to other 284-pthalan products.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.