

Abstract
Organic anion transporting polypeptides (OATP)1B1 and OATP1B3 mediate hepatic uptake of many drugs including lipid-lowering statins. Current studies determined the OATP1B1/1B3-mediated drug-drug interaction (DDI) potential of mammalian target of rapamycin (mTOR) inhibitors, everolimus and sirolimus, using R-value and physiologically based pharmacokinetic (PBPK) models. Pre-incubation with everolimus and sirolimus significantly decreased OATP1B1/1B3-mediated transport even after washing, and decreased inhibition constant values up to 8.3- and 2.9-fold for OATP1B1 and both 2.7-fold for OATP1B3, respectively. R-values of everolimus, but not sirolimus, were greater than the FDA recommended cut-off value of 1.1. PBPK models predict that everolimus and sirolimus have low OATP1B1/1B3-mediated DDI potential against pravastatin. OATP1B1/1B3-mediated transport was not affected by pre-incubation with INK-128 (10 µM, 1 h), which does however abolish mTOR kinase activity. The pre-incubation effects of everolimus and sirolimus on OATP1B1/1B3-mediated transport were similar in cells prior pre-incubated with vehicle control or INK-128, suggesting that inhibition of mTOR activity is not a prerequisite for the pre-incubation effects observed for everolimus and sirolimus. Nine potential phosphorylation sites of OATP1B1 were identified by phosphoproteomics; none of these are the predicted mTOR phosphorylation sites. We report the everolimus/sirolimus-pre-incubation-induced inhibitory effects on OATP1B1/1B3 and relatively low OATP1B1/1B3-mediated DDI potential of everolimus and sirolimus.
Keywords: drug transport, drug interactions, organic anion-transporting polypeptide, transporters, hepatocytes, hepatic transport, pharmacokinetics, physiologically based pharmacokinetic modeling
INTRODUCTION
Hepatic OATP1B1 and OATP1B3 are localized to the sinusoidal membrane of human hepatocytes and mediate the uptake of many clinically important drugs (e.g., lipid-lowering statins and anti-cancer agents)1. Decreased transport activity of OATP1B1 and OATP1B3 due to co-administered OATP inhibitors (e.g., cyclosporine [Cs] A) or genetic variation has been associated with increased systemic exposure of statins, statin-related myopathy, and even rhabdomyolysis2,3. The OATP1B1 c.521 T>C single nucleotide polymorphism has reduced transport function, and is the most robust and important predictor of statin-induced myopathy3. A static model using the R-value, which represents the predicted fold change in exposure of victim drug in the presence of a perpetrator drug, has been recommended by regulatory agencies to assess the OATP1B1- and OATP1B3-mediated DDI potential of inhibitors4–6. Assessing the in vitro inhibition constant (Ki) values against OATP1B1 and OATP1B3 is a critical step in model-based prediction of the DDI potential of perpetrator drugs/compounds. Several reports indicated that pre-incubation with some OATP inhibitors, including cyclosporine A, rifampicin, and dasatinib, decreases OATP1B1- and OATP1B3-mediated transport, resulting in reduced Ki values7–9. Currently, the mechanism underlying the pre-incubation-induced reduction in OATP1B1 and OATP1B3 transport activity remains unknown. For CsA and rifampicin, the decreased Ki values against OATP1B1 and OATP1B3 determined after inhibitor pre-incubation are close to the estimated in vivo Ki values8–10. In the recently published US FDA draft guidance for OATP1B1- and OATP1B3-mediated DDI studies, a pre-incubation with the investigational compound was recommended when assessing the Ki values in vitro5.
Mammalian Target of Rapamycin (mTOR) inhibitors everolimus (EVR) and sirolimus (SIR) are immunosuppressant drugs that are frequently used in transplant patients11,12. In addition, EVR and SIR have anticancer effects13–15. EVR has been approved for the treatment of certain cancers16, and SIR has been administered to transplant patients with cancers to achieve simultaneous immunosuppressant and anti-cancer effects13–15. Cardiovascular disease resulting from dyslipidemia is a common cause of illness and death in organ transplant recipients treated long-term with immunosuppressants17; therefore, immunosuppressant drugs, including EVR and SIR, are often co-administered with lipid-lowering statins in order to reduce the risk for cardiovascular disease in transplant patients18. Severe statin-related DDI cases were reported in transplant recipients and cancer patients concurrently using EVR or SIR with atorvastatin or simvastatin19–22. Because hepatic uptake processes have been reported as the rate-determining step of atorvastatin clearance, and systemic exposure of both atorvastatin and simvastatin acid has been reported in subjects with the c.521T>C polymorphism of OATP1B13,23, OATP inhibition can be expected to play a role in the reported DDIs. The previously reported IC50 values for EVR and SIR against OATP1B1 and OATP1B3 transport function were determined without EVR or SIR pre-incubation in transporter-overexpressing stable cell lines24. Considering the recent recommendation from the US FDA draft guidance to include a pre-incubation step when determining the Ki values in vitro, a re-evaluation of the OATP-mediated DDI potential of EVR and SIR after pre-incubation is warranted.
The mTOR kinase serves as the catalytic subunit of two multi-protein complexes, mTOR complex 1 (mTOR C1) and mTOR C2, which phosphorylate distinct downstream proteins25. EVR and SIR selectively inhibit the mTOR kinase activity of mTOR C1, but not mTOR C225, while some mTOR inhibitors, such as OSI-02726 and INK-12827, inhibit both mTOR C1 and mTOR C2 activity. OATP1B1 and OATP1B3 have putative phosphorylation sites by the mTOR kinase as predicted by the prediction of PK-specific phosphorylation (PPSP) software28. Thus, it is reasonable to hypothesize that impaired mTOR C1 kinase activity after pre-incubation with EVR or SIR is involved in regulating OATP1B1- and OATP1B3-mediated transport, presumably by altering phosphorylation of the transporters. The current studies were designed to determine the OATP1B1- and OATP1B3-mediated DDI potential of EVR and SIR, following the US FDA draft guidance and using static R-value and PBPK models. The involvement of impaired mTOR C1 kinase activity in the modulation of OATP1B1- and OATP1B3-mediated transport after EVR and SIR pre-incubation was also assessed.
MATERIALS AND METHODS
Materials.
[3H]-cholecystokinin 8 (CCK-8) (specific activity 88.0 Ci/mmol), [3H]-estradiol 17 β-D-glucuronide (E217βG) (specific activity 41.4 Ci/mmol) and [3H]-estrone-3-sulfate (E1S) (specific activity 54.0 Ci/mmol) were purchased from Perkin Elmer Life Science (Waltham, MA). [3H]-rosuvastatin was purchased from American Radiolabeled Chemicals (St. Louis, MO). The radiochemical purities determined by Perkin Elmer via high-performance liquid chromatography are greater than 90%, 97%, and 97% for [3H]-CCK-8, [3H]-E1S, and [3H]-E217βG, respectively, and 99% for [3H]-rosuvastatin. Radiochemical purity was not determined in-house before use; however, the radiolabeled compounds were used within a few months of purchase, and substrate accumulation in our control cells was comparable to data published previously8,29. EVR and SIR were purchased from LC laboratories (Woburn, MA). Unlabeled rosuvastatin was purchased from Toronto Research Chemicals (North York, ON, Canada). Unlabeled CCK-8, E217βG, dimethyl sulfoxide (DMSO), Hanks’ Balanced Salt Solution (HBSS), Dulbecco’s Modified Eagle Medium (DMEM), antibiotic antimycotic solution, Triton X-100, and trypsin-EDTA solution were purchased from Sigma-Aldrich (St. Louis, MO). Poly-L-lysine was purchased from Trevigen, Inc. (Gaithersburg, MD). Fetal bovine serum (FBS) was purchased from Hyclone Laboratories (Logan, Utah). Geneticin and HEPES buffer were obtained from Gibco (Life Technologies, Grand Island, NY). Bio-Safe II liquid scintillation mixture was purchased from Research Products International (Mt. Prospect, IL). Complete™ protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). All other materials were purchased from Thermo Fisher Scientific (Waltham, MA).
Cell culture.
Human embryonic kidney (HEK) 293 stable cell lines over-expressing OATP1B1 (HEK293-OATP1B1), OATP1B3 (HEK293-OATP1B3), and the HEK293-mock cell line were kindly provided by Dr. Dietrich Keppler30,31. The HEK293 stable cell line expressing FLAG-tagged WT-OATP1B1 was published previously32. These stable cell lines were maintained in complete DMEM medium supplemented with 10% FBS (v/v), 1% antibiotic antimycotic solution, and 600 µg/ml Geneticin in a humidified atmosphere (95% O2, 5% CO2) at 37°C. HEK293-OATP1B1, HEK293-OATP1B3, and HEK293–mock cells were seeded at a density of 1.5 × 105 cells per well of a 24-well plate coated with poly-L-lysine and were allowed to grow for 48 h to confluence before the uptake studies were performed similarly to those we published previously33.
Inhibition and Uptake Studies.
The transport and inhibition studies were conducted similar to those described previously8,34. Three scenarios, pre-incubation, co-incubation, and co-incubation after a one-hour pre-incubation step (pre+co-incubation), were used to determine the effects of mTOR inhibitors EVR and SIR on OATP1B1 and OATP1B3-mediated transport. In the co-incubation scenario, without any pre-incubation, the uptake assay was performed and substrate accumulation was determined in the presence of vehicle control or inhibitors at the concentrations indicated in the figure legends. In the pre-incubation scenario, the cells were pre-incubated in complete culture medium containing either vehicle control, SIR, or EVR for the designated treatment times and at the concentrations indicated in the figure legends. After washing three times with HBSS buffer containing 10 mM HEPES (pH 7.4), substrate accumulation was determined in the absence of inhibitor. In the pre+co-incubation scenario, cells were first pre-incubated for one hour with either inhibitor or the vehicle control before the uptake assay was performed. Substrate accumulation was determined in the presence of the inhibitor or control at the same concentrations as those used in the pre- and co-incubation steps. DMSO (≤ 0.1%, v/v) was used as the vehicle control in all scenarios. CsA served as the positive control for the pre-incubation effects. To determine the long-lasting inhibitory effects of EVR on OATP1B1- and OATP1B3-mediated transport after EVR pre-incubation, HEK293-OATP1B1 and –OATP1B3 cells were pre-incubated with culture medium containing EVR (0.2 µM) or vehicle CTL for 1 h. At the end of pre-incubation, after washing three times with HBSS buffer, cells were cultured in fresh culture medium for up to 24 h.
[3H]-E217βG (1 µM, 2 min), [3H]-E1S (25 nM, 0.5 min), and [3H]-rosuvastatin (20 nM, 0.5 min) were used as probe substrates for OATP1B1 and [3H]-CCK-8 (1 µM, 3 min) was used as the probe substrate for OATP1B3 in HEK293 stable cell lines overexpressing the transporter of interest, similar to that published previously8,32,34. The probe substrate concentrations used were below the reported Km values for OATP1B1 in HEK293 cells (E217βG - 8.29 µM, E1S - 0.46 µM, and rosuvastatin - 0.8 µM; OATP1B3 [CCK-8 – 3.8 µM])35,36 to ensure that the determined IC50 value is approximately the same as the Ki value. Substrate incubation times all fell within the linear uptake ranges as published previously8,34 or determined in current studies (Supplemental Fig. S1)
At the end of the incubation with probe substrates in HBSS-HEPES buffer (pH 7.4), the uptake assay was terminated by aspirating the incubation buffer and washing the cells three times with ice-cold HBSS-HEPES buffer. Cells were lysed in 200 µL phosphate buffered saline (PBS) buffer containing 0.5% Triton-X 100. Substrate accumulation was determined by scintillation counting (LS6500 scintillation counter, Beckman Coulter, Brea, CA) and normalized to the protein concentration (~0.5 mg/mL) determined via BCA assay (Pierce Chemical, Rockford, IL). The same uptake experiment for all substrates was conducted on a blank plate without cells in order to correct the accumulation values for nonspecific binding to the assay plate. For the experiments conducted with the substrates [3H]-E217βG and [3H]-rosuvastatin, OATP1B1-mediated uptake was calculated by performing the same experiments in HEK293-Mock cells and subtracting the values from those obtained in HEK293-OATP1B1, respectively. For other substrates, pilot experiments determined negligible accumulation in the HEK293-Mock cells, and inhibition was conducted in the transporter-expressing cells.
Prediction of OATP-mediated DDIs using the static R-value model.
To determine the IC50 values, the substrate accumulation was converted to percentage of the vehicle control and plotted against the inhibitor concentrations. The IC50 values were estimated by nonlinear regression using the three-parameter model with GraphPad Prism v.7.0 (GraphPad Software, La Jolla, CA) and are summarized in Table 1. The following equation was fit to the inhibition fold change vs. CTL-concentration data:
| Eq. 1 |
Weighing was not conducted for experimental values when fitting equation 1 to the data, as the dependent variable is not wide enough, justifying not using weighing. We have tested and found that weighing does not improve the IC50 curve fitting. Based on the FDA draft guidance for in vitro DDI studies5, the R-values, which represent the predicted ratio of the victim drug AUC in the presence and absence of the investigational drug, were calculated based on Eq. 2.
| Eq. 2 |
fu,p is the unbound fraction of the inhibitor in the plasma. Iin,max is the estimated maximum plasma concentration of the inhibitor at the inlet to the liver. The Iin,max is calculated as:
| Eq. 3 |
Where Imax is the maximum plasma concentration of the inhibitor in systemic circulation, fa is the fraction absorbed, Fg is the fraction that escapes intestinal metabolism, ka is the absorption rate constant, and Qh is the hepatic blood flow rate (1500 ml/min)37. RB is the blood-to-plasma concentration ratio. faFg and ka are currently unknown values so values of 1 and 0.1/min were used as worst-case estimates, respectively, as recommended by the FDA draft guidance5. The remaining parameters were obtained from the literature and are summarized in Supplemental Table S1.
Table 1.
EVR and SIR IC50 values against OATP1B1-mediated transport of [3H]-E217βG, [3H]E1S, [3H]-rosuvastatin and OATP1B3-mediated transport of [3H]-CCK-8 in HEK293-OATP1B1 and HEK293-OATP1B3 cells, respectively. IC50 values are expressed as the mean ± SE from the model estimation (n=3 in triplicate). Determination of co- and pre+co- IC50 values and Rvalues were calculated as described in the Materials and Methods. * indicates a statistically significant difference (p<0.05 by t-test) vs. coincubation values.
| Co-Incubation |
Pre+Co-Incubation |
||||
|---|---|---|---|---|---|
| Transporter/Substrates | IC50 (μΜ) | R | IC50 (μΜ) | R | |
| Everolimus | |||||
| OATP1B1 | E217G | 0.48 ± 0.07 | 1.09 (1.06−1.15) | 0.23 ± 0.06* | 1.19 (1.16–1.26) |
| OATP1B1 | E1S | 0.61 ± 0.08 | 1.07 (1.04−1.17) | 0.16 ± 0.02* | 1.27 (1.18–1.52) |
| OATP1B1 | Rosuvastatin | 1.58 ± 0.92 | 1.03 (1.02–1.09) | 0.19 ± 0.07* | 1.23 (1.10–1.42) |
| OATP1B3 | CCK-8 | 0.51 ± 0.08 | 1.09 (1.07−1.11) | 0.19 ± 0.02* | 1.23 (1.21–1.25) |
| OATP1B1a | E1S | 4.1 ± 1.1a | 1.01 | ND | ND |
| OATP1B3a | MPA | 3.7 ± 1.3a | 1.01 | ND | ND |
| Sirolimus | |||||
| OATP1B1 | E217G | 0.64 ± 0.16 | 1.00 (1.00–1.01) | 0.22 ± 0.04 | 1.01 (1.01–1.02) |
| OATP1B1 | E1S | 0.83 ± 0.10 | 1.00 (1.00–1.01) | 0.65 ± 0.12 | 1.00 (1.00–1.01) |
| OATP1B1 | Rosuvastatin | 0.56 ± 0.30 | 1.00 (1.00–1.03) | 0.32 ± 0.11 | 1.01 (1.01–1.03) |
| OATP1B3 | CCK-8 | 0.97 ± 0.20 | 1.00 (1.00–1.01) | 0.36 ± 0.05* | 1.01 (1.00–1.01) |
| OATP1B1a | E1S | 9.8 ± 1.1a | 1.00 | ND | ND |
| OATP1B3a | MPA | 1.3 ± 1.2a | 1.00 | ND | ND |
, data from a previous publication24; MPA, mycophenolic acid 7-o-glucuronide
Pharmacokinetic Modeling and Simulations.
To evaluate the OATP1B1- and OATP1B3-mediated DDI potential of EVR and SIR against the OATP-substrate pravastatin, a PBPK modeling approach was designed utilizing the population-based Simcyp Simulator (version 17, SimCYP Ltd, Sheffield, UK). Our purpose in developing the EVR model is to assess, at the highest approved dose of EVR (10 mg), the OATP1B1- and OATP1B3-mediated DDI potential; hence, we used a clinical data from the 10-mg dose to develop the EVR model 38 and used other does (≤10 mg dose) to verify the model (reference summarized in supplemental Table S2). A fit-for-purpose PBPK perpetrator file of EVR was developed based on the parameters in Table 2.
Table 2.
Parameters values used for the EVR PBPK simulations
| EVR | Values | Reference/Comments |
|---|---|---|
| Dose (mg) | 10 | Ref.16 |
| Chemical Structure | 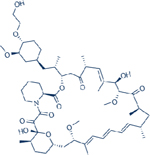 |
|
| Physicochemical properties | ||
| MW (g/mol) | 958.22 | Ref.81 |
| Log PO:W | 4.23 | Calculated using ACD software40 |
| Compound type | Neutral | Ref.82 |
| Blood-to-plasma ratio (Cb/Cp) | 3.4 at 5 ng/ml | Ref.38, see text for details |
| 4.55 at 50 ng/ml | ||
| 4.58 at 100 ng/ml | ||
| 1.9 at 500 ng/ml | ||
| 1.1 at 1 µg/ml | ||
| 0.71 at 5 µg/ml | ||
| Hematocrit value (%) | 45 | Simcyp default value |
| fu | 0.26 | Ref.42 |
| Absorption [Advanced dissolution, absorption and metabolism (ADAM) model] | ||
| fu,gut | 1 | Assumed value |
| Peff,man (x10−4 cm/s) | 6.67 | Predicted using the Simcyp mechanistic Peff (MechPeff) model Jejunum 1 regional permeability using logP input |
| Distribution (minimal PBPK with single adjusting compartment) | ||
| Vss (L/kg) | 1.55 (CV 36.0%) | Ref.83 |
| Vsac (L/kg) | 1.529 | Optimised in Simcyp Simulator |
| kin (1/h) | 0.493 | Optimised Ref.41 |
| kout (1/h) | 0.0993 | Optimised Ref.41 |
| Kp,liver | 1 | Default value |
| Elimination | ||
| CL/F (L/h) | 15.4 (CV 34.3%) | Ref.38 |
| CLR (L/h) | 0 | Ref.45 |
| Transport | ||
| Intestinal efflux intrinsic clearance | ||
| CLint,T,P-gp (µL/min) | 7 | Optimised for 10 mg daily dose at steady state47 and 10 mg weekly dose38, verified against 10 mg steady-state dose in an independent study Ref.49 |
| Interaction | ||
| Ki,OATP1B1 (µM) | 0.022 | Calibrated by CsA |
| Ki,OATP1B3 (µM) | 0.044 | Calibrated by CsA |
| CYP3A4 | ||
| Ki (µM) | 0.09 | Ref.47 |
| fu,mic | 0.14 | |
| CYP3A5 | ||
| Ki (µM) | 0.09 | Ref.47 |
| fu,mic | 0.14 | |
Po:w: neutral species octanol:buffer partition coefficient
fu,gut: unbound fraction of drug in enterocytes
Peff,man: human jejunum effective permeability
Vss: volume of distribution at steady state
Vsac: volume of distribution of single adjusting compartment
Kp,liver: liver-plasma concentration ratio
The default pravastatin (Sim-Pravastatin) and midazolam (MDZ) PBPK models within the Simcyp Simulator library were used without modification, as published previously33. Use of the pravastatin PBPK model to assess OATP1B1 and OATP1B3-mediated DDIs was validated using rifampicin as the OATP1B1 and OATP1B3 inhibitor in our previous publication33. A previously published SIR PBPK model was used in the current study without modification39.
EVR parameters are listed in Table 3. EVR is a neutral compound with a calculated n-octanol:water partition coefficient (Log PO:W) of 4.23 as estimated using Advanced Chemistry Development software (ACD/Labs, Toronto, On, Canada)40. As the experimental Log P value of EVR has not been reported, the Log P value of EVR calculated using ACD software was used in the current study, similarly to that used in a previous publication41. At the blood concentration range of 5–100 ng/ml, EVR is bound to human erythrocytes with an erythrocyte binding of approximately 85%, while at blood concentrations higher than 100 ng/ml, the blood cell uptake of EVR was concentration-dependent38. Based on data described in the EVR clinical pharmacology and biopharmaceutics review, assuming a hematocrit value of 0.45, the EVR blood-to-plasma concentration ratio (Cb/Cp) was calculated as 3.4, 4.55, 4.58, 1.9, 1.1, and 0.71 at blood concentrations of 0.005, 0.05, 0.1, 0.5, 1, and 5 mg/L. The built-in model for the concentration-dependent Cb/Cp within the Simcyp Simulator was used in the current study, employing the interpolation method. The unbound fraction of EVR in human plasma was determined to be 0.2642. The Simcyp advanced dissolution, absorption, and metabolism (ADAM) model43 was used to describe the absorption with human jejunum effective permeability (Peff,man) predicted using the mechanistic permeability (Mech Peff) model44. A liver-plasma concentration ratio (Kp,liver) value of 1 is assumed as a conservative default value for EVR since there is no reported evidence of EVR hepatic uptake. Furthermore, as the systemic concentrations of EVR are the driving concetrations for interactions with hepatic uptake transporters OATP1B1 and OATP1B3, the emphasis for this fit-for-purpose minimal PBPK model of EVR is the systemic concentrations. Sensitivity analysis has shown that the systemic concentrations of EVR were insensitive to changes to Kp,liver (Supplemental Fig. S2 A–D).
Table 3.
Effects of preincubation with EVR and SIR on transport kinetics of OATP1B1 and OATP1B3. Km and Vmax values of OATP1B1-mediated E217ßG transport and OATP1B3- mediated CCK-8 transport in vehicle control (CTL), EVR (1 μΜ, 1 h) and SIR (1 μΜ, 1 h) preincubated HEK293-OATP1B1 and -OATP1B3 cells are shown. Experiments were conducted in triplicate. Data represent mean ± SD from 3 and 5 independent experiments for OATP1B1 and OATP1B3, respectively. *, p<0.05 by one-way ANOVA followed by Dunnett’s test vs. CTL.
| Transporter | Km (µM) |
Vmax (pmol/mg P/min) |
||||
|---|---|---|---|---|---|---|
| CTL | EVR | SIR | CTL | EVR | SIR | |
| OATP1B1 | 3.3 ± 1.3 | 9.7 ± 1.8* | 9.2 ± 4.3* | 46.9 ± 11.8 | 32.3 ± 9.0* | 31.9 ± 5.1* |
| OATP1B3 | 5.8 ± 2.1 | 24.2 ± 8.7* | 22.9± 8.5* | 49.8 ± 16.6 | 39.9 ± 3.8 | 50.5 ± 5.3 |
A minimal PBPK model with a single-adjusting peripheral compartment (SAC) was used to describe the distribution of EVR based upon the ability to consider the observed concentration dependency in the blood-to-plasma ratio (Table 2). Non-linearity observed in doses higher than 10 mg was not addressed. The elimination of EVR was described using an oral clearance value of 15.4 L/h (34.3% CV) after a dose of 10 mg once per day at steady state38. Renal clearance of EVR was set to 0 based on the fact that, in humans, detection of the EVR parent drug in the urine was negligible45. It has been reported previously that transport of EVR in the basolateral-to-apical transport direction of Caco-2 monolayers was ~20 fold greater than in the apical-to-basolateral transport direction, and that the P-gp inhibitor verapamil completely inhibited the efflux of EVR 46, suggesting EVR is a substrate of P-gp. Thus, a P-gp-mediated intestinal efflux intrinsic clearance of 7 µl/min/cm2 was incorporated into the current PBPK model as the best fit value describing the reported steady state Cmax (76.7 ng/mL) of EVR following once daily 10 mg doses47 and the Cmax of EVR following weekly dose at 10 mg38. MDZ is a substrate of CYP3A48, and EVR is a inhibitor of CYP3A47. The application of the EVR PBPK model to predict internal hepatic interactions was independently verified using a clinical DDI study wherein co-administration of 10 mg EVR at steady state with a single 4 mg dose of MDZ increased the observed AUC and Cmax ratios of midazolam by 1.30 and 1.25 fold, respectively49. No sensitivity of MDZ AUC and Cmax to Kp,liver was observed over a 100 fold range; therefore, the default Kp,liver of 1 was maintained (Supplemental Fig. S2 E and F).
Since pravastatin is minimally metabolized through CYP450 enzymes50, the DDI simulation of EVR against pravastatin was performed on a fit-for-purpose basis assuming that EVR affects the disposition of pravastatin only through the inhibition of OATP1B1 and OATP1B35. Although pravastatin is also a substrate of OATP2B151, OATP1A252, and MRP253–55, inhibition of EVR and SIR on OATP2B1 and MRP2 has not been reported. OATP1A2 mRNA is expressed at a low level56 or could not be detected57,58 along the small intestine, and several laboratories were not able to determine OATP1A2 protein in intestinal samples59–61. In addition, scalable in vitro data for OATP1A2 kinetics for pravastatin are currently not available. Hence, the inhibition of OATP1A2 by EVR and SIR in the intestine is currently not specifically implemented in the pravastatin model. EVR and SIR have been reported to inhibit OATP1A224. As OATP1A2 is not specifically considered in the Simcyp pravastatin default model due to the lack of abundance and scalable in vitro kinetics data, the effects of EVR and SIR on OATP1A2 were therefore not considered when assessing the DDIs against pravastatin. After multiple doses of EVR at the highest FDA-approved 10 mg daily dose, steady state was reached at around 7 days38. As the steady-state AUC of EVR is approximately 1.5 fold higher than that of the single dose AUC38, the DDI simulation of EVR against pravastatin was performed in a 7-day trial in 100 virtual subjects (10 trials × 10 subjects) using the default Sim-Healthy volunteer data library. After multiple twice daily doses of 0.5 – 6.5 mg/m2 SIR (equivalent to ~1 – 12.5 mg) in stable renal transplant patients, steady state was achieved at around day 5–7 at all doses62. The highest recommended dose for SIR in patients at high-immunologic risk is a loading dose of up to 15 mg on day one, followed by daily maintenance doses of 5 mg11. The DDI trial design, therefore, was comprised of the highest recommended dose of SIR (15 mg) co-administered daily with 40 mg pravastatin each day for 7 days to ensure that the inhibitor reaches steady state. The DDI simulation for SIR was also performed in 100 virtual subjects (10 trials x 10 subjects) using the default Sim-Healthy volunteer data library.
CsA is a potent OATP1B1 and OATP1B3 inhibitor and was used as calibrator compound to assess OATP-mediated DDIs in the current studies, as published previously8. A sensitivity analysis was conducted to assess the impact of the in vitro determined Ki value of EVR and SIR against OATP1B1 and OATP1B3 on the estimated AUC ratio (AUCR) of pravastatin. Four potential modeling scenarios were assessed: direct use of measured in vitro pre+co-IC50 values (I); assuming saturated in vitro conditions Ki values were half of the measured pre+co-IC50 values (II) (although the experimental conditions used were already accounting for this, this was only included as it is often used as a ‘worst case’ scenario); CsA-calibrated pre+co-IC50 (III) and half of the pre+co-IC50 values (IV) according to equation 5.
| Eq. 5 |
Where in vivo Ki,OATP,CsA values are the estimated in vivo Ki values for CsA against OATP1B1 (0.019 µM) and OATP1B3 (0.032 µM)63. The in vitro Ki,OATP, EVR/SIR were IC50 values summarized in Table 1. For OATP1B1, where multiple probe substrates were used to determine the IC50 values, the lowest IC50 value was used for the simulation. The in vitro Ki,OATP,CsA are the IC50 values determined for CsA in the current studies (Supplemental Fig. S3).
Transport kinetics.
The maximal transport velocity (Vmax) and the affinity constant (Km) of OATP1B1-mediated transport of E217βG (0.1–40 µM, 2 min) and OATP1B3-mediated transport of CCK-8 (0.01–40 µM, 3 min) were determined in HEK293–1B1 and HEK293–1B3 cells, respectively, similarly to that published previously8,29. Vmax and Km values were determined in cells pre-incubated with 1 µM EVR, SIR, or vehicle control (CTL) for 1 hour and washing, or cells without pre-incubation.
The Vmax and Km values of E217βG and CCK-8 transport were estimated by fitting the Michaelis–Menten equation below to the data using GraphPad Prism v.7.0 (GraphPad Software, La Jolla, CA, USA), where v is the transport velocity of substrates and S is the substrate concentration.
Immunoprecipitation and immunoblotting
Immunoprecipitation of FLAG-OATP1B1 and immunoblotting was performed similarly to that published previously29. In brief, after immunoprecipitation with FLAG antibody (F1804, Sigma Aldrich, St. Louis, MO) and subsequent washing, denatured whole cell lysates (WCLs) (50 µg) or immunocomplex were resolved on a 10% SDS-PAGE gel (Bio-Rad, Hercules, CA). Immunoblots were probed with antibodies from Cell Signaling Technology (Danvers, MA): p70 S6 kinase (S6K) (49D7, 1:1000 dilution), phosphor-p70S6K (108D2, 1:1000), Phospho (p)-Akt (Ser473) (D9E) (1:2000 dilution), pan AKT (C67E7, 1:2000), and from Sigma-Aldrich, St. Louis, MO: β-actin (1:5000). After incubation with respective secondary antibody conjugated with horseradish peroxidase (1:5000, Santa Cruz Biotechnology, Inc., Dallas, Texas), signal was detected using Supersignal West Duro (Pierce, Rockford, IL) on a Bio-Rad ChemiDoc XRS imaging system (Bio-Rad Laboratories, Hercules, CA). Image Lab Software (Bio-Rad Laboratories, Hercules, CA) was used for densitometry. The SDS-PAGE with immunoprecipitated FLAG-OATP1B1 was stained with Coomassie Blue for subsequent phosphoproteomics analysis.
Phosphoproteomics by liquid chromatography tandem mass spectrometry (LC-MS/MS).
FLAG-OATP1B1 proteins were purified from 20 dishes (100 mm) of HEK293-FLAG1B1 cells by immunoprecipitation with FLAG antibody, as described above. Gel bands of FLAG-OATP1B1 from SDS-PAGE stained with Coomassie Blue were excised for subsequent phosphoproteomic analysis. In-gel trypsin digestion and subsequent phosphoproteomics studies by LC-MS/MS were conducted at the MS and Proteomics Resource of the WM Keck Foundation Biotechnology Resource Laboratory at Yale, USA (see Supplemental Experimental Procedures for details in MS analysis).
Statistics.
As indicated in the relevant figure legends, fold changes and associated standard errors (SEs) of substrate accumulation in treatment group(s) vs. control were estimated using generalized linear mixed models with the log link function, a fixed group effect and a random effect, while adjusting for group-specific over-dispersion. In cases of multiple comparisons, p-values were adjusted based on the Bonferroni’s method. A two-sided p-value of ≤ 0.05 defines statistical significance. The SAS software (version 9.3, Cary, NC) was used for statistical analyses.
RESULTS
Effects of EVR- and SIR-pre-incubation on OATP1B1- and OATP1B3-mediated transport.
After pre-incubation with EVR, SIR, or vehicle control and subsequent washing, OATP1B1- and 1B3-mediated transport was determined in the absence of EVR and SIR in HEK293-OATP1B1 and -OATP1B3 stable cell lines, respectively. After pre-incubation with EVR or SIR (both 0.2–5 µM) for 10 min, 30 min, and 1 h, OATP1B1-mediated [3H]-E217βG transport (1 µM, 2 min) was significantly decreased, ranging 0.72 ± 0.03 – 0.12 ± 0.02 and 0.91 ± 0.02 – 0.20 ± 0.02 fold of vehicle control (p<0.001) for EVR (Fig. 1 A) and SIR (Fig. 1 B), respectively. One hour of pre-incubation with EVR (0.1 or 0.2–10 µM) or SIR (0.1–5 µM) also significantly decreased OATP1B1-mediated transport of [3H]-E1S (25 nM, 0.5 min) (Fig. 1 C and D) and [3H]-rosuvastatin (20 nM, 0.5 min) (Fig. 1 E and F) in a concentration-dependent manner (p<0.001). After pre-incubation with EVR or SIR (both 0.2–5 µM) for 10 min, 30 min, and 1 h, OATP1B3-mediated [3H]-CCK-8 transport (1 µM, 3 min) was significantly decreased, ranging 0.81 ± 0.04 – 0.17 ± 0.03 and 0.86 ± 0.04 – 0.29 ± 0.04 fold of vehicle control (p<0.001) for EVR (Fig. 1 E) and SIR (Fig. 1 F), respectively.
Fig. 1. Effects of pre-incubation with EVR and SIR on OATP1B1- and OATP1B3-mediated transport.

Model-estimated fold change and associated SE of the accumulation of [3H]-E217βG (1 µM, 2 min) (A-B), [3H]-E1S (25 nM, 0.5 min) (C-D), [3H]-rosuvastatin (20 nM, 0.5 min) (E-F), and [3H]-CCK-8 (1 µM, 3 min) (G-H) vs. CTL in HEK293-OATP1B1 and HEK293-OATP1B3 cells after pre-incubation with 0.1 – 10 µM EVR or 0.05 – 5 µM SIR for 10 min, 30 min, and 1 hour as indicated in the legend. Substrate accumulation was determined in the absence of EVR or SIR after washing. Linear mixed effects models were fit to the data as described in the “Materials and Methods” (n=3 in triplicate). * indicates a statistically significant difference (Bonferroni-adjusted p<0.05) vs. CTL.
As the concentration-dependent pre-incubation effects of EVR and SIR on OATP1B1- and OATP1B3-mediated transport of [3H]-E217βG and [3H]-CCK-8 appear to be the greatest at one hour, a 1-h pre-incubation time point was chosen in subsequent studies to determine the effects of EVR- and SIR-pre-incubation on IC50 values of EVR and SIR against OATP1B1- and OATP1B3-mediated transport. The LDH assay showed negligible toxicity in HEK293-OATP1B1 and HEK293-OATP1B3 cell lines following one hour pre-incubation with EVR (0.001–10 µM) or SIR (0.01 – 5 µM) (Supplemental Fig. S4).
Effects of EVR- and SIR-pre-incubation on IC50 values of EVR and SIR against OATP1B1- and OATP1B3-mediated transport.
IC50 values of EVR and SIR against OATP1B1-mediated transport of [3H]-E217βG, [3H]-E1S and [3H]-rosuvastatin and OATP1B3-mediated transport of [3H]-CCK-8 was determined under co-incubation with EVR or SIR with or without 1 h pre-incubation with EVR or SIR (Fig. 2 and Table 1).
Fig. 2. Effects of pre-incubation with EVR and SIR on the IC50 values against OATP1B1 and OATP1B3.

OATP1B1- and OATP1B3-mediated transport of [3H]-E217βG and [3H]-CCK-8 is expressed as percentage of vehicle control in pre-incubation (open squares), co-incubation (closed circles), and pre+co-incubation (open circles) scenarios with 0.001 – 10 µM EVR (A-D) or 0.01 – 5 µM SIR (E-H) as inhibitor as described in the Materials and Methods. Data represent mean ± SE (n=3 in triplicate). The IC50 values were determined by fitting dose-response curves to the data by nonlinear regression analysis, and are summarized in Table 1. Solid (co-incubation) and dashed (pre+co-incubation) lines represent the fitted lines.
Pre-incubation for one hour followed by co-incubation (pre+co-incubation) with EVR significantly decreased the IC50 values of EVR against OATP1B1-mediated transport of [3H]E217βG, [3H]-E1S and [3H]-rosuvastatin by 2.1, 3.8, and 8.3 fold, respectively, and against OATP1B3-mediated transport of [3H]-CCK-8 by 2.7 fold, compared with co-incubation with EVR only (p<0.05 by t-test, n=3 in triplicate, Fig. 2 A–D, and Table 1). SIR pre+co-incubation decreased the IC50 values of SIR against OATP1B1-mediated transport of [3H]-E217βG, [3H]E1S, and [3H]-rosuvastatin by 2.9, 1.5, and 1.8 fold, respectively. However, the observed reduction in the SIR IC50 values for OATP1B1 did not reach a statistically significant level. SIR pre+co-incubation significantly decreased the IC50 of SIR against OATP1B3-mediated transport of [3H]-CCK-8 by 2.9 fold compared with co-incubation with SIR (p<0.05 by t-test, n=3 in triplicate). Pre-incubation with the positive control CsA also reduced the IC50 values against OATP1B1- and OATP1B3- mediated substrate transport (Supplemental Fig. S3 A and B).
Prediction of OATP-mediated DDIs using static R-value and PBPK modeling.
EVR and SIR R-values determined against OATP1B1 and OATP1B3 are summarized in Table 1. For EVR, the R-values against OATP1B1 and OATP1B3 were 1.19 – 1.27 and 1.23, respectively, using the pre+co-incubation IC50 values, and were 1.03 – 1.09 and 1.09, respectively, using the co-incubation IC50 values. Using IC50 values published previously without EVR pre-incubation 24, the R values were 1.01 for both OATP1B1 and OATP1B3. For SIR, using the lowest IC50 values determined in current and previous studies 24, the R-values were all 1.
The EVR PBPK model well described the blood concentration profile of EVR administered to patients at weekly doses of 10 mg (Fig. 3 A), which is the clinical data used to develop the model 38. This model also well described other clinical data at different doses (Fig. 3 B and Supplemental Table S2 and Figs S10–S16). The EVR model was then verified by an independent clinical DDI study of EVR against MDZ49. The MDZ PBPK model well described the plasma concentration-time profile of MDZ 49 (Supplemental Fig. S5). The simulated AUC and Cmax ratios of MDZ when co-administered with 10 mg EVR were 1.15 and 1.13, respectively (Supplemental Fig. S5), comparable to the clinical data of 1.30 and 1.25, respectively49. These data suggest that the predictive performance of the EVR model can be verified by an independent clinical study. Using the CsA-calibrated pre+co-incubation IC50 values of OATP1B1 and OATP1B3, the AUC and Cmax ratios of pravastatin predicted following the 7-day trial of 10 mg daily doses of EVR and a 40 mg daily dose of pravastatin were 1.04 and 1.06, respectively (Supplemental Table S3). Using half of the CsA-calibrated pre+co-IC50 values of EVR against OATP1B1 and OATP1B3 as a worst-case scenario, the predicted pravastatin AUC and Cmax ratios were 1.10 and 1.18, respectively (Fig. 3 D and Supplemental Table S3).
Fig. 3. Simulated versus observed system concentration-time profiles of orally administered EVR, SIR, and pravastatin in healthy volunteers.
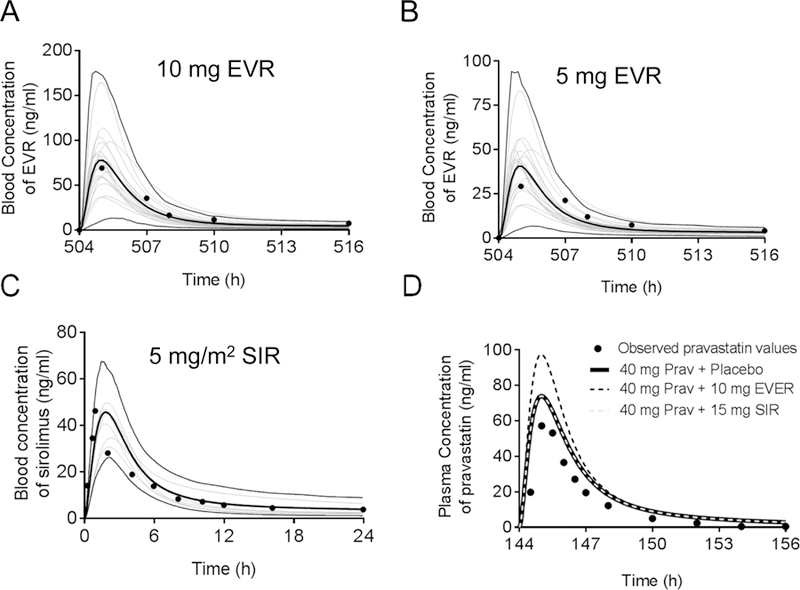
Blood concentration-time profile of EVR (A) 10 mg and (B) 5 mg weekly doses. The grey thin lines represent simulated individual trials (20) of 4 subjects using a population of 80 virtual subjects (50% female, 28–83 years) for figure A and B. The thin black lines represent the upper (95th) and lower (5th) percentiles, and the thick black line represents the simulated mean values of the healthy volunteers population (n=80). The circles denote mean values from the clinical data from the US FDA, 200938. (C) Blood concentration-time profile of a 5 mg/m2 single dose of SIR. The thin grey lines represent simulated individual trials (10) of 30 male subjects using a population of 300 virtual subjects (19–36 years). The thin black lines represent the upper (95th) and lower (5th) percentiles, and the thick black line represents the simulated mean values of the healthy volunteers population (n=300). The circles denote mean values from the clinical study by Brattstrom et al., 200064. (D) Simulated versus observed plasma concentrations of pravastatin following co-administration of a single dose pravastatin (40 mg) with placebo (black line), EVR (10 mg, black dashed line) or SIR (15 mg, grey dashed line). Lines represent the mean values of simulated virtual populations of 400 healthy volunteers (20 trials X 20 subjects, 50% female, 20–50 years). Note that the pravastatin placebo simulation and the pravastatin-SIR simulation overlap.
The SIR PBPK model 64 well described the observed blood concentration profile of SIR after a 5 mg/m2 single dose (Fig. 3 C). Under the worst-case scenario using half of the pre+co-IC50 values calibrated by CsA, the predicted AUC and Cmax ratios of pravastatin (administered at 40 mg once per day) were both 1.0, when co-administered for 7 days with the highest US FDA-approved SIR dose of 15 mg (Fig. 3 D and Supplemental Table S3).
Effects of EVR and SIR on transport kinetics of OATP1B1- and OATP1B3.
The effects of EVR and SIR pre-incubation (1 µM, 1 h) on the Vmax and Km values of OATP1B1- and OATP1B3-mediated transport were determined in HEK293–1B1 and HEK293–1B3 cells, respectively (Fig. 4 and Table 3). Pre-incubation with EVR or SIR (1 µM, 1 h) significantly increased the Km values for OATP1B1-mediated [3H]E217βG transport (3.3 ± 1.3, 9.7 ± 1.8, and 9.2 ± 4.3 µM for CTL, EVR and SIR, respectively), and significantly decreased the Vmax values (46.9 ± 11.8, 32.3 ± 9.0, and 31.9 ± 5.1 pmol/mg protein/min for CTL, EVR, and SIR, respectively). The Km values for OATP1B3-mediated [3H]CCK-8 transport were also significantly increased after pre-incubation with EVR or SIR (5.8 ± 2.1, 24.2 ± 8.7, and 22.9 ± 8.5 µM for CTL, EVR, and SIR, respectively). The Vmax values for OATP1B3 were not significantly affected by EVR or SIR pre-incubation (49.8 ± 16.6, 39.9 ± 3.8, and 50.5 ± 5.3 pmol/mg protein/min for CTL, EVR, and SIR). Without pre-incubation, co-incubation with EVR or SIR (both 5 µM) increased Km values of OATP1B1 by 6–10 fold, while it had minimal effect (1.2–1.7 fold vs. CTL) on Vmax (Supplemental Fig. S6).
Fig 4. Effects of EVR and SIR on the kinetic parameters of OATP1B1- and OATP1B3mediated transport.
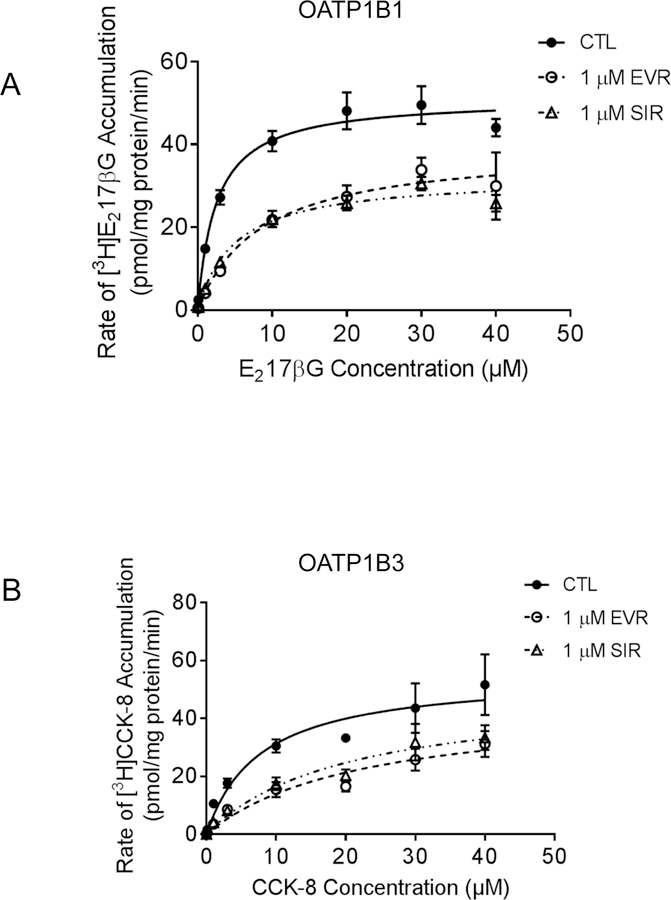
The concentration-dependent accumulation of (A) [3H]-E217βG (0.1–40 µM, 2 min) mediated by OATP1B1 and (B) [3H]-CCK-8 (0.1–40 µM, 3 min)-mediated by OATP1B3 in HEK293-OATP1B1 and –OATP1B3 cells pre-incubated with vehicle CTL, EVR (1 µM, 1 h), or SIR (1 µM, 1 h). Values of [3H]E217βG accumulation in Mock cells were subtracted from those in HEK293-OATP1B1 cells. Vmax and Km values were determined as described in the Material and Methods. Solid, dashed line, and dotted line represent the best fits of the Michaelis–Menten equation to the data of CTL (closed circles), EVR (open circles), or SIR pre-incubation (open triangles), respectively. Data represent the mean ± SD of a representative graph of three and 5 independent experiments for OATP1B1 and OATP1B3, respectively, in triplicate.
Long-lasting inhibitory effects of EVR on OATP1B1- and OATP1B3-mediated transport.
After 1 h pre-incubation with EVR (0.2 µM), OATP1B1-mediated transport of [3H]rosuvastatin was significantly decreased to 0.54 ± 0.07 fold of control (Fig 5 A). OATP1B3-mediated transport of [3H]CCK-8 was significantly decreased to 0.76 ± 0.05 fold of control (Fig 5 B). For OATP1B1-mediated transport, after washing off the EVR and culture in fresh medium, rosuvastatin accumulation was still significantly decreased even at 4 h (0.87 ± 0.03 fold of CTL, p<0.05). For OATP1B3-mediated transport, at 4, 6, and even 22 h after culturing cells in EVRfree medium after washing, [3H]CCK-8 accumulation in EVR-pre-incubated cells remained significantly decreased when compared with the cells pre-incubated with vehicle control (0.76 ± 0.04, 0.73 ± 0.04 and 0.81 ± 0.06 fold of CTL, respectively, p<0.05 at each time point).
Fig 5. Long-lasting inhibitory effects of EVR on OATP1B1- and OATP1B3-mediated transport.
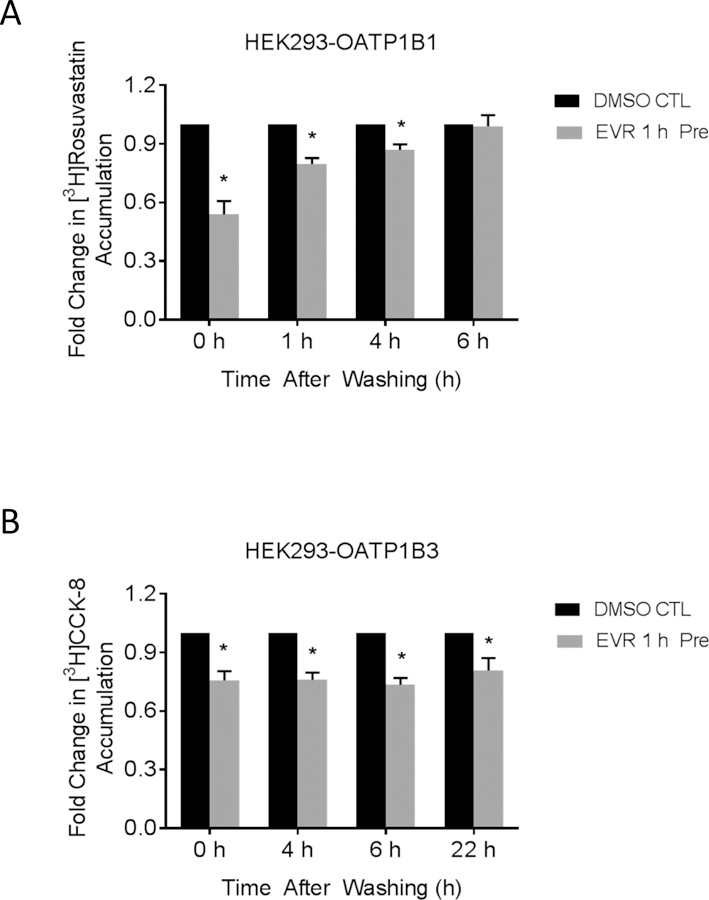
Model-estimated fold change and associated SE of the accumulation of [3H]-rosuvastatin (20 nM, 0.5 min) (A) and [3H]-CCK-8 (1 µM, 3 min) (B) vs. vehicle control (CTL). HEK293-OATP1B1 or –OATP1B3 cells were pre-incubated with CTL or 0.2 µM EVR-containing culture medium for 1 h. At the end of pre-incubation, the culture medium was removed and cells were washed three times with fresh culture medium and cultured in inhibitor-free medium for the indicated time periods. [3H]-rosuvastatin (20 nM, 0.5 min) (A) or [3H]CCK-8 (1 µM, 3 min) accumulation was determined at the indicated time points (n=3 in triplicate) in the absence of EVR after washing. Fold changes and SE were estimated using a generalized linear mixed model, as described in the “Materials and Methods”. * indicates a statistically significant difference (p<0.05) vs. CTL.
Pre-incubation effects of EVR and SIR on OATP1B1- and OATP1B3-mediated transport in cells with and without mTOR kinase activity.
We next tested whether the pre-incubation-induced inhibitory effects of EVR and SIR on OATP1B1- and OATP1B3-mediated transport are related to their mTOR C1 kinase inhibition activity. The effects of EVR and SIR on OATP1B1 and OATP1B3-mediated transport were compared in control cells with regular mTOR kinase activity and in cells in which mTOR C1 kinase activity was abolished by INK-128 prior to EVR and SIR pre-incubation. Phosphorylation of p70S6K at T389 and of AKT at S473 was used as a read out of mTOR C1 and C2 activity, respectively, similar to that published previously65–67.
In HEK293-OATP1B1 cells, compared with vehicle control treatment, pre-incubation with EVR (0.2 µM, 1 h ) or SIR (0.5 µM, 1 h ) markedly reduced the amount of phosphorylated p70S6K at T389 [P-p70s6k (T389)] without affecting protein levels of total p70S6k, as determined by immunoblotting with an antibody specific for phosphorylated p70S6k at T389 and total p70S6k, respectively (Fig. 6 A). Pre-incubation with EVR (0.2 µM, 1 h ) or SIR (0.5 µM, 1 h ) did not inhibit either the amount of phosphorylation of AKT at S473 or total AKT protein levels (Fig. 6 A). Pre-incubation with mTOR C1 and C2 dual inhibitor INK-128 (10 µM, 1 h) alone or in combination with EVR (0.2 µM) or SIR (0.5 µM) markedly reduced both phosphor-AKT (S473) and phospho-p70S6K (T389), without affecting total protein levels of either protein (Fig. 6 B). Similar effects of EVR, SIR ,and INK-128 on phosphorylated and total p70s6k and AKT were observed in HEK293-OATP1B3 cells (Supplemental Fig. S7 and S8).
Fig. 6. Effects of EVR and SIR on the phosphorylation status of mTOR downstream proteins p70s6k and AKT.
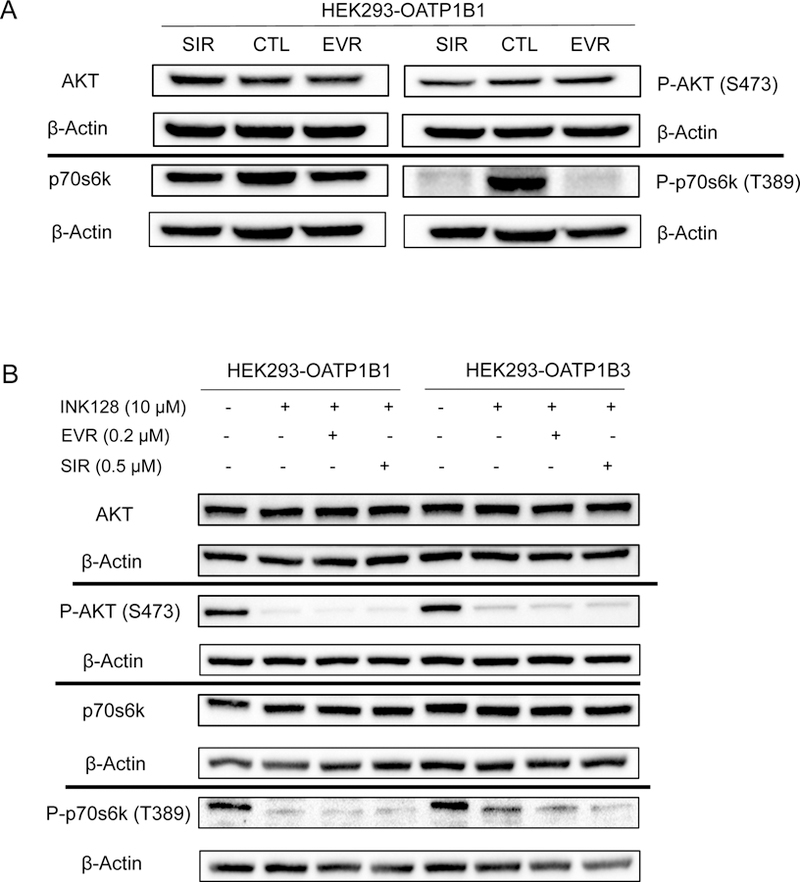
Immunoblot of phosphor-AKT (p-AKT), total AKT, phosphor-p70s6k (p-p70s6k), and total p70s6k in (A) HEK293-OATP1B1 cells after 1 h incubation with 0.2 µM EVR, 0.5 µM SIR, or vehicle control and in (B) HEK293-OATP1B1 and −1B3 cells after 1 h incubation with INK128 (10 µM) alone, or prior to incubation with EVR (0.2 µM, 1 h) or SIR (0.5 µM, 1 h) (representative figures from N=3 experiments).
In HEK293-OATP1B1 and HEK293-OATP1B3 cells, incubation with INK-128 (10 µM, 1 h) did not significantly affect OATP1B1- (Fig. 7 A) or OATP1B3-mediated transport (Fig. 7 B), compared with vehicle control pre-incubation. Pre-incubation with EVR (0.2 µM, 1 h) or SIR (0.5 µM, 1 h) alone significantly decreased OATP1B1-mediated transport to 0.67 ± 0.02 or 0.60 ± 0.05 fold of control (Fig. 7 A) and OATP1B3-mediated transport to 0.60 ± 0.03 or 0.49 ± 0.03 fold of control (Fig. 7 B), respectively. Compared with pre-incubation with EVR (0.2 µM, 1 h) or SIR (0.5 µM, 1 h) alone, there was no significant difference in accumulation of [3H]-E217βG (Fig. 7 A) or [3H]-CCK-8 (Fig. 7 B) in cells pre-incubated with INK-128 (10 µM, 1 h) prior to EVR or SIR.
Fig. 7. Effects of dual mTOR inhibitor INK-128 on pre-incubation effects of EVR and SIR.
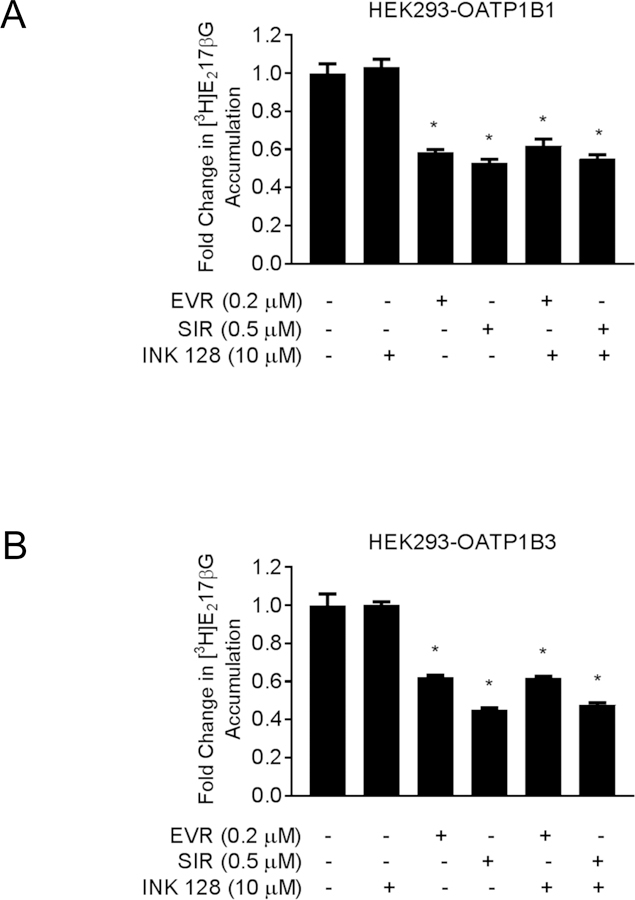
Model-estimated fold change and associated SE of the accumulation of [3H]-E217βG (1 µM, 2 min) (A) and [3H]-CCK-8 (1 µM, 3 min) (B) in HEK293-OATP1B1 and HEK293-OATP1B3 cells, respectively. The cells were pre-incubated with vehicle CTL-, 0.2 µM EVR-, or 0.5 µM SIR-containing culture medium for 1 hour either alone or in combination with 10 µM INK-128, following an earlier 1-hour treatment with 10 µM INK-128 alone. At the end of pre-incubation, the culture medium was removed and substrate accumulation was determined (n=3 in triplicate) in the absence of inhibitors after washing. Fold changes and SE were estimated using a generalized linear mixed model, as described in the “Materials and Methods”. * indicates a statistically significant difference (p<0.05) vs. CTL.
Identification of phosphorylation sites in OATP1B1.
A total of 9 potential phosphorylation sites, S293, S295, T301, T439, T441, Y442, Y590, S659 and S663, were identified on OATP1B1 from our phosphoproteomics experiments (Table 4). The measured mass for all phosphopeptides included in Table 4 had absolute mass accuracy values of less than 4 ppm, indicating accurate peptide assignments. Six phosphorylation sites (S293, T439, T441, Y442, S659, S663) are associated with phosphopeptides with high Mascot Scores of above 67, indicating that the null hypothesis that the observed match is a random event had a probability of less than 5%68. Although S295, T301, and Y590 had Mascot Scores less than 67 (62.56, 29.22, and 35.59, respectively), their peptide fragmentation pattern and identity of the phosphopeptides were manually confirmed. Six sites (S293, S295, T301, Y590, S659, S663) had PhosphoRS probability69 values near 100%, suggesting that these six sites were confidently identified as phosphorylation sites. Manual verification also confirmed that the above six sites are phosphorylation sites, as exemplified in Supplemental Fig. S9. However, localization of one phosphorylation site among the T439, T441, and Y442 residues on the same phosphopeptide cannot be verified, as these three sites exist on the same ion fragment with only one site being phosphorylated. The PhosphoRS probability value of ~33.33% for each of these three sites suggests that there was equal probability for any one of these sites to be phosphorylated. In summary, six phosphorylation sites (S293, S295, T301, Y590, S659, S663) were identified on OATP1B1 with high confidence. Three potential phosphorylation sites, T439, T441, and Y442, need to be further characterized.
Table 4.
Site of phosphorylation identified in OATP1B1 by LC-MS/MS. Mascot score, phosphoRS probability, ion mass and peptide mass accuracy were shown for representative phosphopeptides. Results from two independent experiments (n=2) are shown.
| Phosphorylation Site | Mascot Scorea | PhosphoRS Probabilityb | Ion Mass (Da) | Peptide Mass Accuracy (ppm)c |
|---|---|---|---|---|
| S293 | 147.53 | 99.95% | 2933.4184 | 1.8 |
| S295 | 62.56 | 99.99% | 3013.3798 | 0.1 |
| T301 | 29.22 | 99.33% | 2933.4131 | 1.5 |
| T439 | 81.98 | 33.33% | 2114.9113 | 1.0 |
| T441 | 81.98 | 33.33% | 2114.9113 | 1.0 |
| Y442 | 81.98 | 33.33% | 2114.9113 | 1.0 |
| Y590 | 36.59 | 99.99% | 2229.1441 | −3.5 |
| S659 | 105.34 | 100.00% | 2424.9364 | −1.2 |
| S663 | 105.34 | 100.00% | 2424.9364 | −1.2 |
The Mascot score is expressed as −10*LOG10(P), where P is the absolute probability that the observed match is a random event. Mascot scores greater than 67 are significant (p<0.05), suggesting that the null hypothesis that observed match is a random event had a probability of less than 5%84.
phosphoRS probability
Peptide mass accuracy= 106
(Mi-Ma)/Ma in ppm, where Mi and Ma are the observed and calculated mass, respectively, in Da85. The absolute mass accuracy value of less than 4 indicates accurate peptide assignments
DISCUSSION
The current study reports the rapid reduction of OATP1B1- and OATP1B3-mediated transport following pre-incubation with mTOR inhibitors EVR and SIR. The OATP-mediated DDI potential of EVR and SIR was then assessed using the static R-value model published in the US FDA draft guidance for in vitro OATP-mediated DDI studies in combination with PBPK modeling. The potential role of inhibition of mTOR C1 kinase in the pre-incubation effects was also evaluated.
Pre-incubation with 0.2–5 µM EVR rapidly and significantly decreased OATP1B1- and OATP1B3-mediated transport as early as ten minutes after beginning the incubation (Fig. 1). Pre-incubation with EVR for one hour significantly decreased the IC50 values against OATP1B1 and OATP1B3 and led to increased R-values for OATP-mediated DDI prediction (Table 1). Coincubation of EVR with three OATP1B1 substrates (E217βG, E1S, or rosuvastatin) or the specific OATP1B3 substrate CCK-8 yielded IC50 values ranging from 0.48 ± 0.07 (E217βG) to 1.58 ± 0.92 (rosuvastatin) for OATP1B1 and 0.51 ± 0.08 µM for OATP1B3 (Table 1). Pre-incubation followed by co-incubation with EVR reduced the IC50 values against OATP1B1 2.1, 3.8, and 8.3 fold for E217βG, E1S, and rosuvastatin, respectively, and ~2.7 fold for OATP1B3 (Table 1). Using the pre+co-incubation IC50 values recommended by the US FDA, R-values of EVR against OATP1B1 and OATP1B3 were all above the US FDA recommended cut-off value of 1.1, ranging from 1.19 (E217βG) −1.27 (E1S). Hence, the static R-value model predicts that EVR has potential to cause OATP1B1- and OATP1B3-mediated DDIs. We further conducted PBPK modeling to assess the OATP1B1- and OATP1B3-mediated DDI potential of EVR. A previously reported single dose clinical DDI study indicated that co-administration of 2 mg EVR did not influence the pharmacokinetics of pravastatin to a clinically relevant extent in healthy volunteers (Zortress, 2010). Consistent with the findings of this clinical study, a DDI simulation of 2 mg EVR with single dose pravastatin (20 mg) yielded an AUC and Cmax ratio of 1.0 (data not shown). The highest approved dose of EVR is 10 mg in cancer patients; however, the effect of 10 mg EVR on the pharmacokinetics of OATP drug substrates (e.g., pravastatin) has not been reported in a clinical study. Our simulation results indicate that even using the lowest worst-case scenario IC50 values, which are the half of the pre+co-incubation IC50 values after calibration with CsA (scenario 4), the AUC and Cmax ratios of pravastatin when co-administered with EVR were 1.10 and 1.18, respectively (Fig. 3 D), suggesting that EVR has low potential to cause OATP-mediated DDIs. However, in transplant recipients who received EVR treatment (2.5 ± 1.0 mg per day) for at least three months prior to a one-month concomitant treatment with EVR and rosuvastatin, the rosuvastatin AUC and Cmax was ~2.8 and ~2.5 fold higher than the literature data in healthy volunteers 70,71. Currently, the mechanism underlying the discrepancy between the predicted DDI potential in the current study and the observed clinical DDI of EVR against rosuvastatin70 remains unknown. Here, we reported that after EVR pre-incubation and washing, the inhibitory effects of EVR on OATP1B1 and OATP1B3 last up to 4 and 22 hs. Such longlasting inhibition characteristics of EVR may contribute to underestimated DDI prediction.
Pre-incubation with SIR also rapidly and significantly decreased OATP1B1- and OATP1B3-mediated transport (Fig. 1) and reduced the IC50 against OATP1B1 ~2.9, 1.3, and 1.8 fold for E217βG, E1S and rosuvastatin, respectively, and ~2.7 fold for OATP1B3 (Fig. 2 and Table 1). However, the static R-value and PBPK models both predict no change in the victim drug AUC, suggesting that the OATP1B1- and OATP1B3-mediated DDI potential for SIR is low. Cases of severe rhabdomyolysis, and even death, have been reported in solid-organ transplant patients concurrently administered SIR with simvastatin or fluvastatin 22,72,73. Both simvastatin and fluvastatin are extensively metabolized by CYP3A74,75, and SIR has been reported as a CYP3A inhibitor76. Though the OATP1B1 c. 521 T>C polymorphism is associated with increased systemic concentrations of simvastatin and simvastatin-induced toxicity, in the reported DDIs involving SIR and simvastatin, potential involvement of metabolic inhibition is also possible. To the best of our knowledge, neither adverse events nor clinical studies of SIR against metabolically stable statins have been reported to date. Alternatively, in the case report published by Hong et al., the transplant patient in question was initially treated with CsA as the main immunosuppressant and was switched due to the combined immunosuppressant and anticancer benefits of sirolimus therapy22. A few weeks after changing the immunosuppressive agent, the patient was admitted to the hospital with severe adverse effects, including rhabdomyolysis. Considering that CsA has been reported to inhibit OATP in a long-lasting manner even after removal of the drug77, it can be assumed that CsA also plays a potential role in this DDI. Based on the DDI prediction from the current study, it is unlikely that SIR alone will cause OATP1B1- and OATP1B3-mediated DDIs when co-administered with metabolically stable statins.
In an effort to elucidate the potential mechanism underlying the pre-incubation-induced reduction of OATP1B1- and OATP1B3-mediated transport, pre-incubation with EVR or SIR increased the Km value (~3–4 folds), with only a slight decrease in or no effect on the Vmax values of OATP1B1- and OATP1B3, suggesting that pre-incubation with EVR and SIR primarily decreased the affinity of substrates to OATP1B1 and OATP1B3 (Fig. 4 and Table 3). These results were similar to a previous report that CsA-pre-incubation markedly increased the Km values, rather than the Vmax for OATP1B177. Without pre-incubation, 5 µM EVR or SIR increased apparent Km values of OATP1B1 by 6–10 fold, while it had minimal effect (1.2–1.7 fold vs. CTL) on Vmax values (Supplemental Fig. S6), suggesting that the inhibition of OATP1B1 and OATP1B3 by EVR and SIR following co-incubation most likely occurs in a competitive manner.
In our previous study, pre-incubation with the protein kinase C activator PMA caused a rapid reduction of OATP1B3-mediated transport in association with increased phosphorylation of OATP1B3 34, suggesting that altered phosphorylation by kinase modulators is likely involved in the rapid reduction of OATP1B1- and OATP1B3-mediated transport after pre-incubation. Among the mTOR phosphorylation sites predicted by the PPSP software, after excluding the sites located in the transmembrane domain predicted by TopPred software78, four amino acids on OATP1B1 (T10, T186, S194, S682) and five amino acids on OATP1B3 (T10, T186, S194, T656, S683) were predicted sites that can be phosphorylated by the mTOR kinase. We tested whether inhibition of mTOR kinase activity is a prerequisite for EVR and SIR to exert the preincubation-induced inhibitory effects on OATP1B1- and OATP1B3-mediated transport. INK-128 is a dual inhibitor of mTOR C1 and mTOR C279. In the scenario when both mTOR C1 and C2 kinase activity is inhibited by INK-128 (Fig. 6), EVR and SIR can still exert the preincubation-induced inhibitory effects toward OATP1B1- and OATP1B3-mediated transport (Fig. 7 A and B). These data suggest that the inhibition of mTOR kinase activity is not involved in the inhibitory effects by EVR and SIR preincubation. Among all potential phosphorylation sites identified on OATP1B1 (Table 4), none of these sites was a putative phosphorylation site by the mTOR kinase. This finding may explain why altered mTOR kinase activity is not involved in regulating OATP1B1 and OATP1B3 transport function. The exact mechanism underlying the pre-incubation effects of EVR on OATP1B1- and OATP1B3-mediated transport remains unknown. Several hypotheses, such as trans-inhibition, where inhibition of OATP1B1 and OATP1B3 activity occurs from inside of the cells once the inhibitor enters the cells, and altered post-translational modification (e.g., phosphorylation), have been proposed to explain the pre-incubation-induced reduction of OATP1B1- and 1B3-mediated transport80. However, experimental data proving these hypotheses are not available. Mechanistic studies elucidating the pre-incubation-induced reduction in OATP1B1- and OATP1B3-mediated transport will expand our understanding of OATP-mediated DDIs, and warrant further investigation.
In conclusion, the current study reports that pre-incubation with mTOR inhibitors EVR and SIR potentiates the inhibitory effect toward OATP1B1 and OATP1B3 independent of their mTOR inhibition activity, and that these two clinically important immunosuppressants have low potential to cause OATP1B1- and OATP1B3-mediated DDIs.
Supplementary Material
Acknowledgements
We thank Dr. Dietrich Keppler for providing the HEK293-OATP1B1, HEK293-OATP1B3 and HEK293-Mock stable cell lines. We thank Wendy Wang for preparing the samples for LC-MS/MS data collection.
This research was supported by NIH R01 GM094268 [W. Y]. The Q-Exactive Plus mass spectrometer was funded in part by NIH SIG from the Office of The Director, National Institute of Health under Award Number (S10OD018034). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Alexandra Crowe is an American Foundation of Pharmaceutical Education Fellow.
Abbreviations:
- AUC
area under the plasma concentration-time curve
- CsA
cyclosporine A
- DDI
drug-drug interaction
- DMEM
Dulbecco’s Modified Eagle’s Medium
- DMSO
dimethyl sulfoxide
- E1S
estrone sulfate
- EVR
everolimus
- FBS
fetal bovine serum
- HBSS
Hanks’ Balanced Salt Solution
- IC50
inhibitor concentration producing 50% inhibition
- Ki
inhibition constant
- mTOR
mammalian target of rapamycin
- OATP
organic anion-transporting polypeptide
- PBPK
physiologically based pharmacokinetic modeling
- SIR
sirolimus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This article contains supplementary material available via the internet at http://wileylibrary.com.
The American Association of Pharmaceutical Scientists (AAPS)/International Transport Consortium (ITC) Joint Workshop on Drug Transporters, Baltimore, MD, April 20–22, 2015, Farasyn, T., Pahwa, S., Ding, K., Alam, K., and Yue, W., Rapid Down-Regulation of Organic Anion Transporting Polypeptide (OATP) 1B1 and 1B3 Transport Function by Mammalian Target of Rapamycin (mTOR) Inhibitors.
Farasyn, T., Alam, K., Crowe, A., Neuhoff, S., Hatley, O., Wang, X., Zhang, P., Ding, K., Li, L. and Yue, W., Down-Regulation of OATP1B1-Mediated Transport by Mammalian Target of Rapamycin (mTOR) Inhibitors Everolimus and Sirolimus: Potential Mechanism and Implication in OATP-Mediated Drug-Drug Interactions, Experimental Biology Meeting, Chicago, IL, April, 2017
Farasyn, T., Alam, K., Crowe, A., Neuhoff, S., Hatley, O., Wang, X., Zhang, P., Ding, K., Li, L. and Yue, W., Assessing the OATP1B1- and OATP1B3-mediated drug-drug interaction potential of mammalian target of rapamycin (mTOR) inhibitor everolimus, 21st North American International Society for the Study of Xenobiotics (ISSX), Providence, RI, Sept., 2017
Contributor Information
Taleah Farasyn, Department of Pharmaceutical Sciences, University of Oklahoma Health Sciences Center, Oklahoma City, OK US.
Alexandra Crowe, Department of Pharmaceutical Sciences, University of Oklahoma Health Sciences Center, Oklahoma City, OK US.
Oliver Hatley, Certara UK Ltd, Simcyp Division, Level 2-Acero, 1 Concourse Way, Sheffield, S1 2BJ, United Kingdom.
Sibylle Neuhoff, Certara UK Ltd, Simcyp Division, Level 2-Acero, 1 Concourse Way, Sheffield, S1 2BJ, United Kingdom.
Khondoker Alam, Department of Pharmaceutical Sciences, University of Oklahoma Health Sciences Center, Oklahoma City, OK US.
Jean Kanyo, Yale MS & Proteomics Resource, Yale University, New Haven, CT, US.
TuKiet Lam, Department of Molecular Biophysics and Biochemistry, Yale MS & Proteomics Resource, Yale University, New Haven, CT, US.
Kai Ding, Department of Biostatistics and Epidemiology, University of Oklahoma Health Sciences Center, Oklahoma City, OK US.
Wei Yue, Department of Pharmaceutical Sciences, University of Oklahoma Health Sciences Center, Oklahoma City, OK US.
References
- 1.König J 2010. Uptake transporters of the human OATP family: molecular characteristics, substrates, their role in drug-drug interactions, and functional consequences of polymorphisms. Handbook of experimental pharmacology (201):1–28. [DOI] [PubMed]
- 2.Shitara Y, Itoh T, Sato H, Li AP, Sugiyama Y 2003. Inhibition of transporter-mediated hepatic uptake as a mechanism for drug-drug interaction between cerivastatin and cyclosporin A. The Journal of pharmacology and experimental therapeutics 304(2):610–616. [DOI] [PubMed] [Google Scholar]
- 3.Search Collaborative Group, Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, Gut I, Lathrop M, Collins R 2008. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N Engl J Med 359(8):789–799. [DOI] [PubMed] [Google Scholar]
- 4.Vaidyanathan J, Yoshida K, Arya V, Zhang L 2016. Comparing Various In Vitro Prediction Criteria to Assess the Potential of a New Molecular Entity to Inhibit Organic Anion Transporting Polypeptide 1B1. J Clin Pharmacol 56 Suppl 7:S59–72. [DOI] [PubMed] [Google Scholar]
- 5.US FDA. 2017. In Vitro Metabolism and Transporter Mediated Drug-Drug Interaction Studies Guidance for Industry. In (CDER) USDoHaHSCfDEaR, editor, ed., Silver Spring, MD: Office of Communications, Division of Drug Information Center for Drug Evaluation and Research Food and Drug Administration. [Google Scholar]
- 6.European Medicines Agency. 2012. Guideline on the investigation of drug interactions ed., London, United Kingdom: Committee for Human Medicinal Products (CHMP),. [Google Scholar]
- 7.Izumi S, Nozaki Y, Maeda K, Komori T, Takenaka O, Kusuhara H, Sugiyama Y 2015. Investigation of the impact of substrate selection on in vitro organic anion transporting polypeptide 1B1 inhibition profiles for the prediction of drug-drug interactions. Drug Metab Dispos 43(2):235–247. [DOI] [PubMed] [Google Scholar]
- 8.Pahwa S, Alam K, Crowe A, Farasyn T, Neuhoff S, Hatley O, Ding K, Yue W 2017. Pretreatment With Rifampicin and Tyrosine Kinase Inhibitor Dasatinib Potentiates the Inhibitory Effects Toward OATP1B1- and OATP1B3-Mediated Transport. J Pharm Sci 106(8):2123–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gertz M, Cartwright CM, Hobbs MJ, Kenworthy KE, Rowland M, Houston JB, Galetin A 2013. Cyclosporine inhibition of hepatic and intestinal CYP3A4, uptake and efflux transporters: application of PBPK modeling in the assessment of drug-drug interaction potential. Pharm Res 30(3):761–780. [DOI] [PubMed] [Google Scholar]
- 10.Yoshikado T, Yoshida K, Kotani N, Nakada T, Asaumi R, Toshimoto K, Maeda K, Kusuhara H, Sugiyama Y 2016. Quantitative Analyses of Hepatic OATP-Mediated Interactions Between Statins and Inhibitors Using PBPK Modeling With a Parameter Optimization Method. Clin Pharmacol Ther 100(5):513–523. [DOI] [PubMed] [Google Scholar]
- 11.Wyeth-Ayerst Pharmaceuticals Inc. 2000. Rapamune (sirolimus) Package Insert ed., Philadelphia, PA. [Google Scholar]
- 12.Novartis. 2010. Zortress® (everolimus) tablets prescribing information ed., East Hanover, NJ. [Google Scholar]
- 13.Benjamin D, Colombi M, Moroni C, Hall MN 2011. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nature reviews Drug discovery 10(11):868–880. [DOI] [PubMed] [Google Scholar]
- 14.Faivre S, Kroemer G, Raymond E 2006. Current development of mTOR inhibitors as anticancer agents. Nature reviews Drug discovery 5(8):671–688. [DOI] [PubMed] [Google Scholar]
- 15.Zaytseva YY, Valentino JD, Gulhati P, Evers BM 2012. mTOR inhibitors in cancer therapy. Cancer letters 319(1):1–7. [DOI] [PubMed] [Google Scholar]
- 16.Novartis. 2009. Afinitor® (everolimus) tablets prescribing information ed., East Hanover, NJ. [Google Scholar]
- 17.Aakhus S, Dahl K, Widerøe TE 1999. Cardiovascular morbidity and risk factors in renal transplant patients. Nephrology Dialysis Transplantation 14:648–654. [DOI] [PubMed] [Google Scholar]
- 18.Ritz E, Wanner C 2008. Statin use prolongs patient survival after renal transplantation. J Am Soc Nephrol 19(11):2037–2040. [DOI] [PubMed] [Google Scholar]
- 19.Milowsky MI, Iyer G, Regazzi AM, Al-Ahmadie H, Gerst SR, Ostrovnaya I, Gellert LL, Kaplan R, Garcia-Grossman IR, Pendse D, Balar AV, Flaherty AM, Trout A, Solit DB, Bajorin DF 2013. Phase II study of everolimus in metastatic urothelial cancer. BJU Int 112(4):462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donders F, Kuypers D, Wolter P, Neven P 2014. Everolimus in acute kidney injury in a patient with breast cancer: a case report. J Med Case Rep 8:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barshes NR, Goodpastor SE, Goss JA 2003. Sirolimus-atorvastatin drug interaction in the pancreatic islet transplant recipient. Transplantation 76(11):1649–1650. [DOI] [PubMed] [Google Scholar]
- 22.Hong YA, Kim HD, Jo K, Park YK, Lee J, Sun IO, Chung BH, Park CW, Yang CW, Choi BS 2014. Severe rhabdomyolysis associated with concurrent use of simvastatin and sirolimus after cisplatin-based chemotherapy in a kidney transplant recipient. Experimental and clinical transplantation : official journal of the Middle East Society for Organ Transplantation 12(2):152–155. [DOI] [PubMed] [Google Scholar]
- 23.Pasanen MK, Fredrikson H, Neuvonen PJ, Niemi M 2007. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clinical pharmacology and therapeutics 82(6):726–733. [DOI] [PubMed] [Google Scholar]
- 24.Picard N, Levoir L, Lamoureux F, Yee SW, Giacomini KM, Marquet P 2011. Interaction of sirolimus and everolimus with hepatic and intestinal organic anion-transporting polypeptide transporters. Xenobiotica; the fate of foreign compounds in biological systems 41(9):752–757. [DOI] [PubMed] [Google Scholar]
- 25.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN 2002. Two TOR Complexes, Only One of which Is Rapamycin Sensitive, Have Distinct Roles in Cell Growth Control. Molecular cell 10:457–468. [DOI] [PubMed] [Google Scholar]
- 26.Bhagwat SV, Gokhale PC, Crew AP, Cooke A, Yao Y, Mantis C, Kahler J, Workman J, Bittner M, Dudkin L, Epstein DM, Gibson NW, Wild R, Arnold LD, Houghton PJ, Pachter JA 2011. Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: distinct from rapamycin. Mol Cancer Ther 10(8):1394–1406. [DOI] [PubMed] [Google Scholar]
- 27.Lou HZ, Weng XC, Pan HM, Pan Q, Sun P, Liu LL, Chen B 2014. The novel mTORC½ dual inhibitor INK-128 suppresses survival and proliferation of primary and transformed human pancreatic cancer cells. Biochem Biophys Res Commun 450(2):973–978. [DOI] [PubMed] [Google Scholar]
- 28.Xue Y, Li A, Wang L, Feng H, Yao X 2006. PPSP: prediction of PK-specific phosphorylation site with Bayesian decision theory. BMC Bioinformatics 7:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alam K, Farasyn T, Crowe A, Ding K, Yue W 2017. Treatment with proteasome inhibitor bortezomib decreases organic anion transporting polypeptide (OATP) 1B3-mediated transport in a substrate-dependent manner. PLoS One 12(11):e0186924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.König J, Cui Y, Nies AT, Keppler D 2000. Localization and genomic organization of a new hepatocellular organic anion transporting polypeptide. J Biol Chem 275(30):23161–23168. [DOI] [PubMed] [Google Scholar]
- 31.Konig J, Cui Y, Nies AT, Keppler D 2000. A novel human organic anion transporting polypeptide localized to the basolateral hepatocyte membrane. Am J Physiol Gastrointest Liver Physiol 278(1):G156–164. [DOI] [PubMed] [Google Scholar]
- 32.Alam K, Pahwa S, Wang X, Zhang P, Ding K, Abuznait AH, Li L, Yue W 2016. Downregulation of Organic Anion Transporting Polypeptide (OATP) 1B1 Transport Function by Lysosomotropic Drug Chloroquine: Implication in OATP-Mediated Drug-Drug Interactions. Molecular Pharmaceutics 13(3):839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pahwa S, Alam K, Crowe A, Farasyn T, Neuhoff S, Hatley O, Ding K, Yue W 2017. Pretreatment With Rifampicin and Tyrosine Kinase Inhibitor Dasatinib Potentiates the Inhibitory Effects Toward OATP1B1- and OATP1B3-Mediated Transport. J Pharm Sci 106:2123–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Powell J, Farasyn T, Kock K, Meng X, Pahwa S, Brouwer KL, Yue W 2014. Novel mechanism of impaired function of organic anion-transporting polypeptide 1B3 in human hepatocytes: post-translational regulation of OATP1B3 by protein kinase C activation. Drug Metab Dispos 42(11):1964–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirano M, Maeda K, Shitara Y, Sugiyama Y 2004. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J Pharmacol Exp Ther 311(1):139–146. [DOI] [PubMed] [Google Scholar]
- 36.Kitamura S, Maeda K, Wang Y, Sugiyama Y 2008. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab Dispos 36(10):2014–2023. [DOI] [PubMed] [Google Scholar]
- 37.Bradley SE, Ingelfinger FJ, et al. 1945. The estimation of hepatic blood flow in man. The Journal of clinical investigation 24:890–897. [PubMed] [Google Scholar]
- 38.US FDA. 2009. Everolimus Clinical Pharmacology and Biopharmaceutics Review. In Research CfDEa, editor, ed.: Center for Drug Evaluation and Research Office of Clinical Pharmacology. [Google Scholar]
- 39.Emoto C, Fukuda T, Cox S, Christians U, Vinks AA 2013. Development of a Physiologically-Based Pharmacokinetic Model for Sirolimus: Predicting Bioavailability Based on Intestinal CYP3A Content. CPT Pharmacometrics Syst Pharmacol 2:e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.ACD/Structure Elucidator. 2017. version 15.01 ed., Toronto, On, Canada: Advanced Chemistry Development, Inc. [Google Scholar]
- 41.Mano Y, Sugiyama Y, Ito K 2015. Use of a Physiologically Based Pharmacokinetic Model for Quantitative Prediction of Drug-Drug Interactions via CYP3A4 and Estimation of the Intestinal Availability of CYP3A4 Substrates. J Pharm Sci 104(9):3183–3193. [DOI] [PubMed] [Google Scholar]
- 42.Kovarik JM, Kahan BD, Kaplan B, Lorber M, Winkler M, Rouilly M, Gerbeau C, Cambon N, Boger R, Rordorf C, Everolimus Phase 2 Study G 2001. Longitudinal assessment of everolimus in de novo renal transplant recipients over the first post-transplant year: pharmacokinetics, exposure-response relationships, and influence on cyclosporine. Clinical pharmacology and therapeutics 69(1):48–56. [DOI] [PubMed] [Google Scholar]
- 43.Jamei M, Turner D, Yang J, Neuhoff S, Polak S, Rostami-Hodjegan A, Tucker G 2009. Population-based mechanistic prediction of oral drug absorption. AAPS J 11(2):225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pade D, Jamei M, Rostami-Hodjegan A, Turner DB 2017. Application of the MechPeff model to predict passive effective intestinal permeability in the different regions of the rodent small intestine and colon. Biopharmaceutics & drug disposition 38(2):94–114. [DOI] [PubMed] [Google Scholar]
- 45.European Medicines Agency. 2014. Afinitor (everolimus) Summary of Product Characteristics ed., London, United Kingdom. [Google Scholar]
- 46.Crowe A, Lemaire M 1998. In vitro and in situ absorption of SDZ-RAD using a human intestinal cell line (Caco-2) and a single pass perfusion model in rats: comparison with rapamycin. Pharm Res 15:1666–1672. [DOI] [PubMed] [Google Scholar]
- 47.Kenny JR, Mukadam S, Zhang C, Tay S, Collins C, Galetin A, Khojasteh SC 2012. Drug-drug interaction potential of marketed oncology drugs: in vitro assessment of time-dependent cytochrome P450 inhibition, reactive metabolite formation and drug-drug interaction prediction. Pharm Res 29(7):1960–1976. [DOI] [PubMed] [Google Scholar]
- 48.Patki KC, Von Moltke LL, Greenblatt DJ 2003. In vitro metabolism of midazolam, triazolam, nifedipine, and testosterone by human liver microsomes and recombinant cytochromes p450: role of cyp3a4 and cyp3a5. Drug Metab Dispos 31(7):938–944. [DOI] [PubMed] [Google Scholar]
- 49.Urva S, Bouillaud E, Delaney R, Jappe A, Cheung W 2013. A phase I study evaluating the effect of everolimus on the pharmacokinetics of midazolam in healthy subjects. J Clin Pharmacol 53(4):444–450. [DOI] [PubMed] [Google Scholar]
- 50.Jacobsen W, Kirchner G, Hallensleben K, Mancinelli L, Deters M, Hackbarth I, Benet LZ, Sewing KF, Christians U 1999. Comparison of cytochrome P-450-dependent metabolism and drug interactions of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors lovastatin and pravastatin in the liver. Drug Metab Dispos 27(2):173–179. [PubMed] [Google Scholar]
- 51.Nozawa T, Imai K, Nezu J, Tsuji A, Tamai I 2004. Functional characterization of pH-sensitive organic anion transporting polypeptide OATP-B in human. J Pharmacol Exp Ther 308(2):438–445. [DOI] [PubMed] [Google Scholar]
- 52.Shirasaka Y, Suzuki K, Nakanishi T, Tamai I 2010. Intestinal absorption of HMG-CoA reductase inhibitor pravastatin mediated by organic anion transporting polypeptide. Pharm Res 27(10):2141–2149. [DOI] [PubMed] [Google Scholar]
- 53.Watanabe T, Kusuhara H, Maeda K, Shitara Y, Sugiyama Y 2009. Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J Pharmacol Exp Ther 328(2):652–662. [DOI] [PubMed] [Google Scholar]
- 54.Shirasaka Y, Suzuki K, Nakanishi T, Tamai I 2011. Differential effect of grapefruit juice on intestinal absorption of statins due to inhibition of organic anion transporting polypeptide and/or P-glycoprotein. J Pharm Sci 100(9):3843–3853. [DOI] [PubMed] [Google Scholar]
- 55.Shitara Y, Sugiyama Y 2006. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: drug-drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol Ther 112(1):71–105. [DOI] [PubMed] [Google Scholar]
- 56.Wojtal KA, Eloranta JJ, Hruz P, Gutmann H, Drewe J, Staumann A, Beglinger C, Fried M, Kullak-Ublick GA, Vavricka SR 2009. Changes in mRNA expression levels of solute carrier transporters in inflammatory bowel disease patients. Drug Metab Dispos 37(9):1871–1877. [DOI] [PubMed] [Google Scholar]
- 57.Meier Y, Eloranta JJ, Darimont J, Ismair MG, Hiller C, Fried M, Kullak-Ublick GA, Vavricka SR 2007. Regional distribution of solute carrier mRNA expression along the human intestinal tract. Drug Metab Dispos 35(4):590–594. [DOI] [PubMed] [Google Scholar]
- 58.Hilgendorf C, Ahlin G, Seithel A, Artursson P, Ungell AL, Karlsson J 2007. Expression of thirty-six drug transporter genes in human intestine, liver, kidney, and organotypic cell lines. Drug Metab Dispos 35(8):1333–1340. [DOI] [PubMed] [Google Scholar]
- 59.Groer C, Bruck S, Lai Y, Paulick A, Busemann A, Heidecke CD, Siegmund W, Oswald S 2013. LC-MS/MS-based quantification of clinically relevant intestinal uptake and efflux transporter proteins. J Pharm Biomed Anal 85:253–261. [DOI] [PubMed] [Google Scholar]
- 60.Miyauchi E, Tachikawa M, Decleves X, Uchida Y, Bouillot JL, Poitou C, Oppert JM, Mouly S, Bergmann JF, Terasaki T, Scherrmann JM, Lloret-Linares C 2016. Quantitative Atlas of Cytochrome P450, UDP-Glucuronosyltransferase, and Transporter Proteins in Jejunum of Morbidly Obese Subjects. Mol Pharm 13(8):2631–2640. [DOI] [PubMed] [Google Scholar]
- 61.Drozdzik M, Groer C, Penski J, Lapczuk J, Ostrowski M, Lai Y, Prasad B, Unadkat JD, Siegmund W, Oswald S 2014. Protein abundance of clinically relevant multidrug transporters along the entire length of the human intestine. Mol Pharm 11(10):3547–3555. [DOI] [PubMed] [Google Scholar]
- 62.Zimmerman JJ, Kahan BD 1997. Pharmacokinetics of Sirolimus in Stable Renal Transplant Patients after Multiple Dose Administration. Journal of Clinical Pharmacology 37:405–415. [DOI] [PubMed] [Google Scholar]
- 63.Yoshikado T, Yoshida K, Kotani N, Nakada T, Asaumi R, Toshimoto K, Maeda K, Kusuhara H, Sugiyama Y 2016. Quantitative Analyses of Hepatic OATP-Mediated Interactions Between Statins and Inhibitors Using PBPK Modeling With a Parameter Optimization Method. Clinical pharmacology and therapeutics [DOI] [PubMed]
- 64.Brattstrom C, Sawe J, Jansson B, Lonnebo A, Nordin J, Zimmerman JJ, Burke JT, Groth CG 2000. Pharmacokinetics and Safety of Single Oral Doses of Sirolimus (Rapamycin) in Healthy Male Volunteers. Therapeutic Drug Monitoring 22:537–544. [DOI] [PubMed] [Google Scholar]
- 65.Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, Hara K, Tanaka N, Avruch J, Yonezawa K 2003. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem 278(18):15461–15464. [DOI] [PubMed] [Google Scholar]
- 66.Moschella PC, McKillop J, Pleasant DL, Harston RK, Balasubramanian S, Kuppuswamy D 2013. mTOR complex 2 mediates Akt phosphorylation that requires PKCepsilon in adult cardiac muscle cells. Cell Signal 25(9):1904–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Breuleux M, Klopfenstein M, Stephan C, Doughty CA, Barys L, Maira SM, Kwiatkowski D, Lane HA 2009. Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Mol Cancer Ther 8(4):742–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Matrix Science Inc. 2016. Scoring: Mascot Probability Based Scoring. Mascot database help search, ed. [Google Scholar]
- 69.Taus T, Kocher T, Pichler P, Paschke C, Schmidt A, Henrich C, Mechtler K 2011. Universal and confident phosphorylation site localization using phosphoRS. J Proteome Res 10(12):5354–5362. [DOI] [PubMed] [Google Scholar]
- 70.Robertsen I, Asberg A, Granseth T, Vethe NT, Akhlaghi F, Ghareeb M, Molden E, Reier-Nilsen M, Holdaas H, Midtvedt K 2014. More potent lipid-lowering effect by rosuvastatin compared with fluvastatin in everolimus-treated renal transplant recipients. Transplantation 97(12):1266–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Martin PD, Warwick MJ, Dane AL, Cantarini MV 2003. A Double-Blind, Randomized, Incomplete Crossover Trial to Assess the Dose Proportionality of Rosuvastatin in HealthyVolunteers. Clinical Therapeutics 23(8):2215–2224. [DOI] [PubMed] [Google Scholar]
- 72.Dopazo C, Bilbao I, Lázaro JL, Sapisochin G, Caralt M, Blanco L, Castells L, Charco R 2009. Severe rhabdomyolysis and acute renal failure secondary to concomitant use of simvastatin with rapamycin plus tacrolimus in liver transplant patient. Transplantation proceedings 41(3):1021–1024. [DOI] [PubMed] [Google Scholar]
- 73.Basic-Jukic N, Kes P, Bubic-Filipi L, Vranjican Z 2010. Rhabdomyolysis and acute kidney injury secondary to concomitant use of fluvastatin and rapamycin in a renal transplant recipient. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 25(6):7. [DOI] [PubMed] [Google Scholar]
- 74.Ishigam M, Uchiyama M, Kondo T, Iwabuchi H, Inoue S, Takasaki W, Ikeda T, Komai T, Ito K, Sugiyama Y 2001. Inhibition of in vitro metabolism of simvastatin by itraconazole in humans and prediction of in vivo drug-drug interactions. Pharm Res 18(5):622–631. [DOI] [PubMed] [Google Scholar]
- 75.Fischer V, Johanson L, Heitz F, Tullman R, Graham E, Baldeck JP, Robinson WT 1999. The 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor fluvastatin: effect on human cytochrome P-450 and implications for metabolic drug interactions. Drug Metab Dispos 27(3):410–416. [PubMed] [Google Scholar]
- 76.Lampen A, Christians U, Bader A, Hackbarth I, Sewing KF 1996. Drug interactions and interindividual variability of ciclosporin metabolism in the small intestine. Pharmacology 52(3):159–168. [DOI] [PubMed] [Google Scholar]
- 77.Shitara Y, Takeuchi K, Nagamatsu Y, Wada S, Sugiyama Y, Horie T 2012. Long-lasting Inhibitory Effects of Cyclosporin A, but Not Tacrolimus, on OATP1B1- and OATP1B3-mediated Uptake. Drug Metab Pharmacokinet 27(4):368–378. [DOI] [PubMed] [Google Scholar]
- 78.Gv Heijne 1992. Membrane Protein Structure Prediction Hydrophobicity Analysis and the Positive-inside Rule. Journal of Molecular Biology 225:487–494. [DOI] [PubMed] [Google Scholar]
- 79.Liu Q, Thoreen C, Wang J, Sabatini D, Gray NS 2009. mTOR Mediated Anti-Cancer Drug Discovery. Drug Discov Today Ther Strateg 6(2):47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shitara Y, Sugiyama Y 2017. Preincubation-dependent and long-lasting inhibition of organic anion transporting polypeptide (OATP) and its impact on drug-drug interactions. Pharmacol Ther 177:67–80. [DOI] [PubMed] [Google Scholar]
- 81.Afinitor. 2009. Package insert ed., Novartis, East Hanover, NJ. [Google Scholar]
- 82.Hsieh Y, Galviz G, Long BJ 2009. Ultra-performance hydrophilic interaction liquid chromatography/tandem mass spectrometry for the determination of everolimus in mouse plasma. Rapid Commun Mass Spectrom 23(10):1461–1466. [DOI] [PubMed] [Google Scholar]
- 83.Kovarik JM, Hsu CH, McMahon L, Berthier S, Rordorf C 2001. Population pharmacokinetics of everolimus in de novo renal transplant patients: impact of ethnicity and comedications. Clinical pharmacology and therapeutics 70(3):247–254. [DOI] [PubMed] [Google Scholar]
- 84.Matrix Science Inc. 2016. Scoring: Mascot Probability Based Scoring. In search Mdh, editor, ed. [Google Scholar]
- 85.Brenton AG, Godfrey AR 2010. Accurate mass measurement: terminology and treatment of data. J Am Soc Mass Spectrom 21(11):1821–1835. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.