

Abstract
Integrating 2D culture of adherent mammalian cells with single-cell western blotting (in situ scWB) uses microfluidic design to eliminate trypsin release of cells to suspension, prior to single-cell isolation and protein analysis. To assay HeLa cells from an attached starting state, we culture adherent cells in fibronectin-functionalized microwells formed in a thin layer of polyacrylamide gel. To integrate the culture, lysis, and assay workflow, we introduce a one-step copolymerization process that creates protein-decorated microwells. After single-cell culture, we lyse each cell in the microwell and perform western blot on each resultant lysate. We observe cell spreading after overnight microwell-based culture. scWB reports increased phosphorylation of MAP kinases (ERK1/2, p38) under hypertonic conditions. We validate the in situ scWB with slab-gel western blot, while revealing cell-to-cell heterogeneity in stress responses.
Keywords: electrophoretic separation, functional hydrogels, in situ western blot, protein phosphorylation, single-cell studies
Graphical Abstract
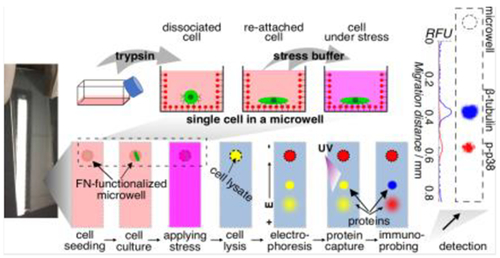
Immunoblotting single, adherent mammalian cells in 2D culture: To eliminate perturbation of cells during detachment from culture, we integrate microwell-based cell culture with single-cell immunoblotting. In a one-step process, microwells are decorated with fibronectin. After osmotic stress of HeLa cells during microwell culture, we measure phosphorylation of MAP kinases, and observe significant cell-to-cell heterogeneity in stress responses.
Introduction
Quantitative measurement of proteins with single-cell resolution on attached, adherent cells in culture eliminates biological perturbation that occurs during cell detachment from 2D culture. Cell detachment for analysis of a cell suspension can perturb membrane proteins, cytoskeletal proteins, and signaling proteins (e.g., phosphorylation state).[1] Assays performed on attached, adherent cells reduce the risk of sample-transfer loss for improved accuracy.[2] Moreover, assaying cells in culture preserves the extracellular matrix context for assessing the relationships between molecular signature, phenotype (e.g., morphology), and substrate properties (e.g., geometry or mechanical properties). These relationships are increasingly appreciated in understanding the sources of cell-to-cell heterogeneity.[3]
Immunoassays are the de facto standard for analyses of endogenous, unmodified proteins in adherent cells in culture, largely based on immunocytochemistry (ICC).[4] Nevertheless, as with any assay, ICC presents limitations. With reduced cell-suspension density, ICC profiles target proteins with single-cell resolution.[5] Target selectivity in ICC depends on the availability and specificity of immunoreagents; for detection of proteoforms (e.g., isoforms), ICC can be restricted by the availability of an isoform-specific antibody.[6] ICC offers limited throughput, with microscopy based analysis of ~100’s of cells per assay.[5b] In addition, cell aggregation and varied cell morphologies can confound cell identification.[7] As such, analytical tools that could provide selectivity for proteoform targets, increase throughput, and control cell localization for adherent cells in 2D culture would fill a gap relevant to biological inquiry.
Here we report a single-cell western blot for protein analyses of adherent cells in culture (in situ scWB). In situ scWB combines single-cell culture and western blot on one polyacrylamide (PA) gel. First, single adherent cells are seeded and cultured in individual microwells of a functionalized PA gel (in situ scWB device), thus eliminating semi-subjective cell identification in ICC.[5b, 7] Next, the cell-laden microwells are dosed with stimulants (e.g., drug, stress) by incubating the device in the stimulant solution. After dosing, each attached cell is chemically lysed in its microwell. Immediately after lysis, an electric field is applied across the device, initiating electrophoresis during which the solubilized lysate from each cell electromigrates through the PA gel and resolves based on molecular mass differences. Upon completion of electrophoresis, proteins are photo-blotted (immobilized) via UV light activation of a benzophenone methacrylamide (BPMA) incorporated into the PA gel during device fabrication.[8] Immunoprobing using fluorescently labeled immunoreagents yields high selectivity protein target detection along the electrophoresis separation lane (Figure 1a).
Figure 1.

In situ single-cell western blot (in situ scWB) measures protein expression in single, adherent cells in culture by integrating on-chip 2D cell culture and single-cell western blotting. a) Schematic of the in situ scWB assay for measuring osmotic stress-induced protein phosphorylation. Left: Photographs of an in situ scWB device fabricated on a standard glass microscope slide. The bottom photograph is the zoom-in of the yellow box in the top photograph. The gel on the device was stained blue for visualization. Middle: Workflow of the in situ scWB assay illustrated with one microwell from among an array of ~2000 microwells on the device. Right: A representative false-colored fluorescence micrograph from in situ scWB of stress-induced phosphorylation, and the fluorescence profile along the electrophoretic separation. β-tubulin: 50 kDa. p38: 41 kDa. b) One-step fabrication of the in situ scWB device, composed of arrays of fibronectin-functionalized microwells stippled in a thin layer of polyacrylamide (PA) gel.
Results and Discussion
To support single-cell culture in each microwell, we sought to functionalize the substrate PA gel with adhesive ligands, such as fibronectin (FN) or collagen.[9] Despite extensive use of adhesive ligand-patterned hydrogels in in vitro cell studies,[10] the geometry of the microwell (unsuitable for contact printing) and anti-fouling properties of PA gel make coating a challenge. Activating PA gel to enhance protein adhesion through chemical treatment such as sulfosuccinimidyl 6-(4’-azido-2’-nitrophenylamino)hexanoate (sulfo-SANPAH) activation can be inefficient, non-uniform, and unstable.[10a, 11] To introduce a new approach compatible with these constraints, we sought to generate FN-functionalized PA gel through FN-embedded polymerization (Figure 1b). We mixed FN in PA gel precursor, and polymerized the hydrogel using free-radical polymerization at room temperature for ~1 h. Because of the amphipathic property of FN,[12] we hypothesize that FN accumulates at the hydrophobic surface of the SU8 mold and forms an FN layer at the surface of the PA gel; a hypothesis that we investigated further.
To examine the FN distribution in the PA gel comprising the in situ scWB device, we included rhodamine-labeled FN (FN*) in the PA gel precursor and used confocal fluorescence microscopy to inspect the FN* distribution after device fabrication. Confocal microscopy reports a surface-confined layer of FN* across the device in the z-stack scan. To validate that the fluorescent layer is FN* and not simply unconjugated fluorophore, we immunoprobed the device with an AlexaFluor 647-labeled antibody against FN (anti-FN; Figure 2a). Coincident fluorescence signal from FN* and anti-FN confirms that FN* is located on the surface of the PA gel. During polymerization, hydrogen bonds are thought to form between the PA gel and FN accumulated at the gel surface,[13] thus immobilizing FN to the gel surface without covalent bonds. To form stronger covalent attachment to a PA gel, FN can be crosslinked with benzophenone of a BPMA-incorporated PA gel using UV irradiation (Supporting Information, Figure S1). The interactions between FN and the contact surfaces are hypothesized to contribute collectively to the surface accumulation of FN on the PA gel.
Figure 2.

Characterization of the fibronectin (FN) layer in microwells on the in situ scWB device. a) Representative false-color confocal fluorescence micrographs (cross section along y-z plane) indicating a thin layer of rhodamine-labeled FN (FN*) at the surface of the in situ scWB device. Microwell: diameter, 50 μm; height, 40 μm. Red: FN*. Blue: FN* probed with AlexaFluor 647-labeled antibody (anti-FN). Scale bar: 20 μm. b) Quantitated thickness of the FN* layer on in situ scWB devices of varied applied FN* concentrations (FN* conc.). c) Quantitated thickness of the FN* layer on in situ scWB devices of varied microwell diameters. n = 3. Error bar: standard deviation. d) Coefficient of variation (CV) of FN* thickness (top) and average microwell fluorescence (bottom). Straight lines are drawn to indicate the values of 0.1 (top) and 0.2 (bottom). e) Average microwell fluorescence from in situ scWB devices spanning various applied FN* concentrations. f) Linear fit to the average fluorescence intensities at each FN* concentration in e). g) Average microwell fluorescence from in situ scWB devices of various microwell diameters. Black lines in e) and g): mean value, n >1900 for each group. Error bars: standard deviation. Unless otherwise specified, the microwell diameter is 50 μm and the applied FN or FN* concentration is 10 μg ml−1.
To evaluate the tunability of the one-step fabrication approach for diverse cell culture needs, we fabricated in situ scWB devices using a range of applied FN* concentrations (1–100 μg ml−1, determined based on previous work[14]) and microwell diameters (50–100 μm). Using confocal imaging, we observe FN* localized to the surface of all devices (n = 4). Moreover, the average FN* layer thickness (h) is in the same range (~20 μm) between devices fabricated with a variety of applied FN* concentrations (Figure 2b, Table S1) and microwell diameters (Figure 2c, Table S1). The microwell-to-microwell variation (CV) of the FN* layer thickness on a device is <10% for all of the applied FN* concentrations and microwell diameters (Figure 2d, top; Table S1; n = 3 representative locations across each device).
To assess the FN layer uniformity within the microwells of an in situ scWB device, we used a fluorescence scanner to image each microwell (n = ~2,000 microwells). As the applied FN* concentration increases from 1 to 100 μg ml−1, the average fluorescence intensity from a microwell (Imicrowell) increases accordingly by 2 orders of magnitude (Figure 2e, Table S2). The relationship between applied and incorporated FN* suggests a degree of controllability in the incorporated FN concentration of the FN layer (Figure 2f), and further allows calibration for design of substrates with tunable ligand densities. For each applied FN* concentration (10 μg ml−1), Imicrowell remains comparable (~1.5 fold) for devices with different microwell diameters (Figure 2g, Table S2). Finally, the CV (n = ~2,000) of the microwell fluorescence across an entire device is ≦20% for all the examined devices (Figure 2d, bottom; Table S2), which is within the acceptable CV range for this assay.[15] Compared to the observed variation in the thickness of the FN* layer (CV <10%), the larger variance of the microwell FN* fluorescence across each device likely stems from a nonideal, partial transfer of the pattern during the peel-off process. The partial pattern transfer arises from: 1) the compliance mismatch between the PA gel and the SU8 pillar that causes a partial detachment of FN* from the SU8 surface and 2) the peel-off direction that largely determines the resultant pattern of debonding defects.[16] While acceptable with the current process, process enhancements are under study, including: i) applying a uniform hydrophobic treatment to the SU8 substrate to reduce adhesion to generate interfacial crack growth and ii) developing a controlled peel-off strategy to mitigate inadvertent creation of nonuniform defect patterns across the device.
After establishing the capability to present FN on the surface of microwells cast in PA gel, we next sought to evaluate the short-term cell-culture capability of the in situ scWB device. The short-term on-chip culture should be longer than the adhesion recovery time between the dissociated cell and the microwell to allow for relevant protein measurement, and be shorter than the doubling time to maintain the singe-cell occupancy in each microwell. To accomplish this, we cultured HeLa cells overnight in each microwell (50 μm in diameter) and examined, first, cell viability and, next, cell spreading. We scrutinized viability using a calcein AM / ethidium homodimer-1 staining kit. We quantified cell spreading by measuring projected area and circularity of the calcein AM-stained cells.
The viability assay reports no significant difference in cell viability before and after on-chip culture (Welch’s t-test: p = 0.86, n = 3 replicates, ~1000 cells per assay), which indicates no detectable cytotoxicity for the short-term culture (Supporting Information, Figure S2). The viability results also agree with other PA gel-based in vitro studies.[9a, 17] Further, the cells cultured on unmodified devices also exhibit sustained viability after on-chip culture (Welch’s t-test: p = 0.65, n = 3 replicates, ~1000 cells per assay; Supporting Information, Figure S2). Hence, we sought more precise metrics to assess the cell status in short-term culture. Cell spreading is considered a strong indicator of robust adhesion between an adherent cell and its substrate, and robust adhesion is crucial for cell growth and proliferation.[18] In the presence of adhesive ligands, healthy cells with intact ligand receptors can effectively adhere to the substrate, and the spreading level is correlated with the ligand density.[19]
We scrutinized cell spreading after overnight culture, across a range of applied FN concentrations (0–100 μg ml−1). We observe that HeLa cells exhibit an increased polarity as applied FN concentration increases (Figure 3a), as corroborated by the changes in projected area and circularity. The average projected area increases (Spearman’s ρ = 0.8, p = 0.33) with the increase of applied FN concentration (Figure 3b). The average circularity decreases monotonically (Spearman’s ρ = −1, p = 0.08) with the increase of applied FN concentration (Figure 3c). Compared to the cells cultured on unmodified devices, those cultured on FN-modified devices start to exhibit a significant difference (p <0.001, n >100 cells) in circularity when applied FN concentrations are equal or greater than 1 μg ml−1 (Mann-Whitney test, Table S3). A similar trend is observed with projected area, but the onset FN concentration for cells to exhibit significant difference (p <0.001, n >100 cells) is 10 μg ml−1 (Mann-Whitney test, Table S3). The smaller onset value of the applied FN concentration in circularity (1 μg ml−1) than in projected area (10 μg ml−1) suggests the sensitivity of the metric ‘circularity’ in assessing cells’ morphology changes. We also observe a larger mean projected area (495 μm2) and a smaller mean circularity (~0.52) on FN-coated glass (n = 75) than on in situ scWB devices (282 μm2, ~0.72). We attribute the observation to microwell confinement (~50 μm in diameter) on cell spreading.[20] Cells spread on the in situ scWB device, and the spreading level is related to the applied FN concentration, indicating suitability of the in situ scWB device for adherent cell culture. For considerations of the device throughput and the reagent cost, we chose to fabricate microwells of 50 μm in diameter, with the applied FN concentration of 10 μg ml−1 for downstream analytical experiments.
Figure 3.

HeLa cells are viable and spread in FN-coated microwells (50 μm in diameter) on the in situ scWB device. a) Representative micrographs of the overnight cultured HeLa cells in FN-coated microwells molded in PA gel. Top: phase contrast micrographs. Bottom: false-color fluorescence micrographs. Cells were stained with calcein AM for morphology characterization. Scale bar: 10 μm. b) Comparison of the cell projected area on varied applied FN concentrations. c) Comparison of the cell circularity on varied applied FN concentrations. Boxes represent the first and third quartiles of analyte distributions. Black lines indicate the median values. Asterisks mark the mean values. Whiskers are 1.5 fold of interquartile range. Circles are outliers. Mann–Whitney significance levels: n.s., p >0.05; ***, p <0.001. n >75 for each group.
Having established the upstream cell preparation capabilities of the in situ scWB, we next sought to assess the downstream analytical capability by applying the western blotting function to measure osmotic stress-induced MAP kinase phosphorylation in single cells. Phosphorylation is dynamic.[21] Moreover, the kinases (e.g., ERK1/2 (ERK), p38) responsive to osmotic stress can be triggered by a multitude of stimuli (i.e., temperature, chemical, and mechanical perturbations).[22] Sample preparation such as trypsinization and centrifugation is thought to introduce artefacts. Consequently, we sought to design an integrated microfluidic device to circumvent such sample processing prior to protein analysis via in situ scWB.
Using the in situ scWB, we performed on-chip overnight cell culture, stimulation, and western blot analysis of single adherent HeLa cells. We investigated hyper-osmotic stress-induced phosphorylation with single-cell resolution, as our slab gel western blot analysis of pooled cells reports negligible phosphorylation of both ERK and p38 under hypo-osmotic versus hyper-osmotic stress (Figure S3). The conditions of isotonic (60 min, 300 mOsm) and hypertonic (60 min, 500 mOsm) stimulation were determined from conventional western blot analysis (Supporting Information, Figure S3). For phosphorylated-ERK (p-ERK), we observe 21.8% of the hypertonic cells having an abundance larger than 3x the standard deviation of the average isotonic abundance (nhyper = 229, niso = 181) (Figure 4a). In phosphorylated-p38 (p-p38), nearly 24.5% of the hypertonic cells have an abundance larger than 3x the standard deviation of the average isotonic abundance (nhyper = 155, niso = 157) (Figure 4a). The overall increased abundance level for both p-ERK and p-p38 under the hypertonic condition indicates apparent hyper-osmotic responses in cell populations.
Figure 4.

Phosphorylation of ERK1/2 (ERK) and p38 induced by osmotic stress in single HeLa cells is measured using in situ scWB, but not detected in scWB with ex situ 2D culture, stimulation, and trypsin release to cell suspension. Microwell: 50 μm in diameter. FN concentration: 10 μg ml−1. a) Scatter plots of the protein abundance of single cells from in situ scWB. Insets: zoom-in on y axis. b) Scatter plots of the protein abundance of single cells from scWB of trypsinized cells from conventional 2D cell culture with stimulation (normal scWB). c) Representative false-color fluorescence micrographs of immunoprobed targets in a single cell under iso- and hyper-osmotic conditions. ERK: 42, 44 kDa. Scale bar: 100 μm. d) Box plots that indicate the distribution of the normalized abundance of phosphorylated targets (p-ERK, p-p38) under different osmotic conditions. Boxes represent the first and third quartiles of analyte distributions. Black lines indicate the median values. Whiskers are 1.5 fold of interquartile range. Mann–Whitney significance levels: ***, p <0.001. n >150 for each group.
By contrast, using a version of the scWB with ex situ 2D culture, stimulation, and trypsin release to cell suspension, we observe 2.7% of the hypertonic cells having an abundance larger than 3x the standard deviation of the average isotonic abundance in p-ERK (nhyper = 187, niso = 148), and 1.5% of the hypertonic cells having an abundance larger than 3x the standard deviation of the average isotonic abundance in p-p38 (nhyper = 269, niso = 226) (Figure 4b, Figure S4). The negligible changes in p-ERK and p-p38 abundance observed in a version of the scWB with ex situ 2D culture, stimulation, and trypsin release to cell suspension are attributed to the unintended phosphorylation from trypsinization and time-in-suspension during sample preparation processes (Supporting Information, Figure S5).
Next, across a population of HeLa cells, we sought to assess any shift in mean expression level of p-ERK or p-p38 owing to osmotic stress. To correct for biological heterogeneity unrelated to the stress conditions (e.g., cell cycle, cell size), we normalized p-ERK and p-p38 expression by β-tubulin expression from the same cell and for each cell. Expression of β-tubulin by in situ scWB remains nearly constant across osmotic conditions, with 1.7% of the hypertonic cells seeing β-tubulin expression in 3x excess of the average isotonic abundance (nhyper = 229, niso = 181; Figure 4a, 4c, Figure S6). By in situ scWB, ERK and p38 show significant increases in phosphorylation under hypertonic stress (Mann-Whitney test, pp-ERK = 1.5 × 10−47, pp-p38 = 1.8 × 10−47, n >150 cells). The median phosphorylation level before versus after stress shows increases of ~5.6x (ERK) and ~5.8x (p38) (Figure 4d). The in situ scWB observations are consistent with reported population-averaged hyper-osmotic responses.[23]
Not detectable with population-averaged slab-gel western blot analysis, the in situ scWB reports larger cell-to-cell variation in phosphorylation level under hypertonic conditions for both ERK and p38. Single-cell resolution expression of ERK shows a ~6x increase (0.104 vs 0.590) in interquartile range (IQR), while p38 shows a ~3x increase (0.073 vs 0.213) in IQR (Figure 4d). The increases suggest co-existence of hyper-responders and non-responders within each cell population, thus indicating the differential cellular responses under the same osmotic stress. Such heterogeneity in kinase phosphorylation was also reported for cells stimulated with growth factors.[5]
The differential cellular response in kinase phosphorylation may be at least partially attributable to cell-cycle stage. Hyper-osmotic stress induces DNA damage to which cellular response is known to vary with stage of the cell cycle.[24] Consequently, similar dependencies in MAPK activity are anticipated. In one possible implication, the efficacy of some anti-cancer drugs depends on cell-cycle stage.[25] Thus, knowledge of osmotic stress-induced heterogeneous phosphorylation could inform cancer subclassification and bolster therapeutic efficacy.
Hyper-osmotic stress-induced protein expression, including secretion of cytokines, is reported across cell lines as well as in primary immune cells.[26] Known to play critical roles in physiology, hyper-osmotic stress-induced protein expression is involved in achieving homeostasis under stress and in immune response. Scrutinizing population-level findings with single-cell resolution aims to inform understanding of mechanistic differences in stress responses. This understanding would underpin efforts to identify, sort, and study presently unknown sub-populations of clinically relevant cells as a component of precision medicine.
Conclusion
In summary, we introduce, validate, and apply a single-cell western blot assay to analyze hundreds of individual adherent cells in culture (in situ scWB). The integration of 2D cell culture and protein analysis minimizes the perturbations from sample preparation, and hence allows measurement of challenging protein targets, including membrane receptors, cytoskeletal proteins, focal adhesion complex, and signaling proteins as demonstrated here. The high-throughput microwell array of the in situ scWB device can interrogate hundreds of individual cells per assay, which is critical for statistical analysis of single-cell results. More importantly, the cell-culture substrate (PA gel) of the in situ scWB device can be independently tuned in geometry, ligand density, and stiffness across the physiological range, hence allowing multidimensional measurements to map the proteomic signature of each cell to culture context. As such, we envision that in situ scWB will open up new possibilities for single-cell studies.
Experimental Section
Experimental Details, including the reagents, device fabrication, cell culture, cell viability and spreading assessment, osmotic stress protocols, in situ scWB procedures, imaging and data analysis are provided in the Supporting Information.
Supplementary Material
Acknowledgements
This work was supported by the Chan Zuckerberg Biohub and the United States National Institutes of Health (R01CA203018 to A.E.H.). I.N. was supported by JSPS KAKENHI Grant (JP15KK0297). We thank the Biomolecular Nanotechnology Center of QB3 and CRL Molecular Imaging Center (supported by Helen Wills Neuroscience Institute) at University of California, Berkeley for instrumentation support. We appreciate helpful discussions with the Herr lab members throughout this project.
Footnotes
Supporting information for this article is available at: http://dx.doi.org/10.1002/anie.2019xxxxx.
Contributor Information
Dr. Yizhe Zhang, Department of Bioengineering, University of California, Berkeley, Berkeley, CA, 94720, USA
Dr. Isao Naguro, Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan
Prof. Amy E. Herr, Department of Bioengineering, University of California, Berkeley, Berkeley, CA, 94720, USA
References
- [1].a) Tsuji K, Ojima M, Otabe K, Horie M, Koga H, Sekiya I, Muneta T, Cell Transplant. 2017, 26, 1089–1102; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ren X-D, Wang R, Li Q, Kahek LAF, Kaibuchi K, Clark RAF, J. Cell Sci. 2004, 117, 3511–3518; [DOI] [PubMed] [Google Scholar]; c) Akiyama SK, Yamada KM, J. Biol. Chem 1985, 260, 4492–4500. [PubMed] [Google Scholar]
- [2].Li S, Plouffe BD, Belov AM, Ray S, Wang X, Murthy SK, Karger BL, Ivanov AR, Molecular & Cellular Proteomics 2015, 14: 10.1074/mcp.M114.045724, 1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Kiss M, Gassen SV, Movahedi K, Saeys Y, Laoui D, Cell. Immunol 2018, 330, 188–201; [DOI] [PubMed] [Google Scholar]; b) Eun K, Ham SW, Kim H, BMB Rep. 2017, 50 (3), 117–125; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dingal PCDP, Bradshaw AM, Cho S, Raab M, Buxboim A, Swift J, Discher DE, Nat. Mater 2015, 14, 951–960; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Marklein RA, Lam J, Guvendiren M, Sung KE, Bauer SR, Trends Biotechnol. 2018, 36, 105–118; [DOI] [PubMed] [Google Scholar]; e) Ribeiro AJS, Ang Y-S, Fu J-D, Rivas RN, Mohamed TMA, Higgs GC, Srivastava D, Pruitt BL, Proc. Natl. Acad. Sci. USA 2015, 112, 12705–12710; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Tseng P, Carlo DD, Adv. Mater 2014, 26, 1242–1247; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Charrier EE, Pogoda K, Wells RG, Janmey PA, Nat. Commun 2018, 9: 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Larsson L-I, Immunocytochemistry: Theory and Practice 1988, CRC Press, Boca Raton. [Google Scholar]
- [5].a) Caunt CJ, McArdle CA, J. Cell Sci. 2010, 123, 4310–4320; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ng AHC, Chamberlain MD, Situ H, Lee V, Wheeler AR, Nat. Commun 2015, 6: 7513; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Blazek M, Santisteban TS, Zengerle R, Meier M, Lab Chip 2015, 15, 726–734. [DOI] [PubMed] [Google Scholar]
- [6].a) Schnell U, Dijk F, Sjollema KA, Giepmans BNG, Nat. Methods 2012, 9, 152–158; [DOI] [PubMed] [Google Scholar]; b) Stadler C, Rexhepaj E, Singan VR, Murphy RF, Pepperkok R, Uhlén M, Simpson JC, Lundberg E, Nat. Methods 2013, 10, 315–323. [DOI] [PubMed] [Google Scholar]
- [7].Chalfoun J, Majurski M, Dima A, Stuelten C, Peskin A, Brady M, BMC Bioinformatics 2014, 15, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hughes AJ, Spelke DP, Xu Z, Kang C-C, Schaffer DV, Herr AE, Nat. Methods 2014, 11, 749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Thiele J, Ma Y, Bruekers SMC, Ma S, Huck WTS, Adv. Mater 2014, 26, 125–148; [DOI] [PubMed] [Google Scholar]; b) Caliari SR, Burdick JA, Nat. Methods 2016, 13, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Pelham RJ Jr., Wang Y-L, Proc. Natl. Acad. Sci. USA 1997, 94, 13661–13665; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Engler AJ, Sen S, Sweeney HL, Discher DE, Cell 2006, 126, 677–689; [DOI] [PubMed] [Google Scholar]; c) Prager-khoutorsky M, Lichtenstein A, Krishnan R, Rajendran K, Mayo A, Kam Z, Geiger B, Bershadsky AD, Nat. Cell Biol 2011, 13, 1457–1465; [DOI] [PubMed] [Google Scholar]; d) Rape AD, Zibinsky M, Murthy N, Kumar S, Nat. Commun 2015, 6: 8129; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Sarker B, Walter C, Pathak A, ACS Biomater. Sci. Eng 2018, 4, 2340–2349. [DOI] [PubMed] [Google Scholar]
- [11].a) Tse JR, Engler AJ, Curr. Protoc. Cell Biol 2010, 47: 10.16.1–10.16.16; [DOI] [PubMed] [Google Scholar]; b) Rowlands AS, George PA, Cooper-White JJ, Am. J. Physiol. Cell Physiol. 2008, 295, C1037–C1044; [DOI] [PubMed] [Google Scholar]; c) Winer JP, Janmey PA, McCormick ME, Funaki M, Tissue Eng. Part A 2009, 15, 147–154. [DOI] [PubMed] [Google Scholar]
- [12].a) Morgenthaler J-J, FEBS Lett. 1982, 150, 81–84; [Google Scholar]; b) Clark DC, Mackie AR, Wilde PJ, Wilson DR, Faraday Discuss. 1994, 98, 253–262; [Google Scholar]; c) Bernard A, Bosshard HR, Eur. J. Biochem 1995, 230, 416–423; [DOI] [PubMed] [Google Scholar]; d) Bernard A, Delamarche E, Schmid H, Michel B, Bosshard HR, Biebuyck H, Langmuir 1998, 14, 2225–2229; [Google Scholar]; e) Mackie AR, Gunning AP, Wilde PJ, Morris VJ, J. Colloid Interface Sci. 1999, 210, 157–166; [DOI] [PubMed] [Google Scholar]; f) Deng Y, Zhu X-Y, Kienlen T, Guo A, J. Am. Chem. Soc 2006, 128, 2768–2769; [DOI] [PubMed] [Google Scholar]; g) Meyer EE, Rosenberg KJ, Israelachvili J, Proc. Natl. Acad. Sci. USA 2006, 103, 15739–15746; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Ruiz SA, Chen CS, Soft Matter 2007, 3, 168–177. [DOI] [PubMed] [Google Scholar]
- [13].Liu L, Cooke PH, Coffin DR, Fishman ML, Hicks KB, J. Appl. Polym. Sci 2004, 92, 1893–1901. [Google Scholar]
- [14].Su EJ, Herr AE, Lab Chip 2017, 17, 4312–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Jelliffe RW, Schumitzky A, Bayard D, Fu X, Neely M, Ther Drug Monit. 2015, 37(3), 389–394; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Reed GF, Lynn F, Meade BD, Clin. Diagn. Lab. Immunol 2002, 9, 1235–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tang X, Ali MY, Saif MTA, Soft Matter 2012, 8, 7197–7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Hynd MR, Frampton JP, Dowell-Mesfin N, Turner JN, Shain W, J. Neurosci. Methods 2007, 162, 255–263; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wen JH, Vincent LG, Fuhrmann A, Choi YS, Hribar KC, Taylor-Weiner H, Chen S, Engler AJ, Nat. Mater 2014, 13, 979–987; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Damljanovic V, Lagerholm BC, Jacobson K, BioTechniques 2005, 39, 847–851. [DOI] [PubMed] [Google Scholar]
- [18].a) Streuli CH, Gilmore AP, J. Mammary Gland Biol. Neoplasia 1999, 4, 183–191; [DOI] [PubMed] [Google Scholar]; b) Puthalakath H, Villunger A, O’Reilly LA, Beaumont JG, Coultas L, Cheney RE, Huang DCS, Strasser A, Science 2001, 293, 1829–1832; [DOI] [PubMed] [Google Scholar]; c) Mehlen P, Puisieux A, Nat. Rev. Cancer 2006, 6, 449–458; [DOI] [PubMed] [Google Scholar]; d) Khalili AA, Ahmad MR, Int. J. Mol. Sci 2015, 16, 18149–18184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Ingber DE, Proc. Natl. Acad. Sci. USA 1990, 87, 3579–3583; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gaudet C, Marganski WA, Kim S, Brown CT, Gunderia V, Dembo M, Wong JY, Biophys. J 2003, 85, 3329–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Engler A, Bacakova L, Newman C, Hategan A, Griffin M, Discher D, Biophys. J 2004, 86, 617–628; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Reinhart-King CA, Dembo M, Hammer DA, Biophys. J 2005, 89, 676–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Ochsner M, Textor M, Vogel V, Smith ML, PLoS One 2010, 5, e9445; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Charnley M, Textor M, Khademhosseini A, Lutolf MP, Integr. Biol 2009, 1, 625–634. [DOI] [PubMed] [Google Scholar]
- [21].a) Schmelzle K, White FM, Curr. Opin. Biotechnol 2006, 17, 406–414; [DOI] [PubMed] [Google Scholar]; b) Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M, Cell 2006, 127, 635–648; [DOI] [PubMed] [Google Scholar]; c) Needham EJ, Parker BL, Burykin T, James DE, Humphrey SJ, Sci. Signal 2019, 12, eaau8645. [DOI] [PubMed] [Google Scholar]
- [22].a) de Nadal E, Ammerer G, Posas F, Nat. Rev. Genet 2011, 12, 833–845; [DOI] [PubMed] [Google Scholar]; b) Zhou X, Naguro I, Ichijo H, Watanabe K, Biochim. Biophys. Acta 2016, 1860, 2037–2052; [DOI] [PubMed] [Google Scholar]; c) Karin M, Ann. N. Y. Acad. Sci 1998, 851, 139–146; [DOI] [PubMed] [Google Scholar]; d) Cuadrado A, Nebreda AR, Biochem. J 2010, 429, 403–417. [DOI] [PubMed] [Google Scholar]
- [23].a) Nielsen M-B, Christensen ST, Hoffmann EK, Am. J. Physiol. Cell Physiol. 2008, 294, C1046–C1055; [DOI] [PubMed] [Google Scholar]; b) Li D-Q, Luo L, Chen Z, Kim H-S, Song XJ, Pflugfelder SC, Exp. Eye Res. 2006, 82, 588–596; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Maruyama T, Kadowaki H, Okamoto N, Nagai A, Naguro I, Matsuzawa A, Shibuya H, Tanaka K, Murata S, Takeda K, Nishitoh H, Ichijo H, EMBO J 2010, 29, 2501–2514; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ, J. Biol. Chem 1995, 270, 7420–7426. [DOI] [PubMed] [Google Scholar]
- [24].a) Dmitrieva NI, Burg MB, Ferraris JD, Am. J. Physiol. Renal Physiol 2005, 289, F2–F7; [DOI] [PubMed] [Google Scholar]; b) Hustedt N, Durocher D, Nat. Cell Biol 2017, 19, 1–9. [DOI] [PubMed] [Google Scholar]
- [25].Otto T, Sicinski P, Nat. Rev. Cancer 2017, 17, 93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Burg MB, Ferraris JD, Dmitrieva NI, Physiol. Rev 2007, 87, 1441–1474; [DOI] [PubMed] [Google Scholar]; b) Aramburu J, Rodriguez C, Front Immunol. 2019, 10: 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.