

The biological production of methane is vital to the global carbon cycle and accounts for ca. 74% of total methane emissions. The organisms that facilitate this process, methanogenic archaea, belong to a large and phylogenetically diverse group that thrives in a wide range of anaerobic environments. Two main subgroups exist within methanogenic archaea: those with and those without cytochromes.
KEYWORDS: Archaea, energy conservation, hydrogenase, methanogenesis
SUMMARY
The biological production of methane is vital to the global carbon cycle and accounts for ca. 74% of total methane emissions. The organisms that facilitate this process, methanogenic archaea, belong to a large and phylogenetically diverse group that thrives in a wide range of anaerobic environments. Two main subgroups exist within methanogenic archaea: those with and those without cytochromes. Although a variety of metabolisms exist within this group, the reduction of growth substrates to methane using electrons from molecular hydrogen is, in a phylogenetic sense, the most widespread methanogenic pathway. Methanogens without cytochromes typically generate methane by the reduction of CO2 with electrons derived from H2, formate, or secondary alcohols, generating a transmembrane ion gradient for ATP production via an Na+-translocating methyltransferase (Mtr). These organisms also conserve energy with a novel flavin-based electron bifurcation mechanism, wherein the endergonic reduction of ferredoxin is facilitated by the exergonic reduction of a disulfide terminal electron acceptor coupled to either H2 or formate oxidation. Methanogens that utilize cytochromes have a broader substrate range, and can convert acetate and methylated compounds to methane, in addition to the ability to reduce CO2. Cytochrome-containing methanogens are able to supplement the ion motive force generated by Mtr with an H+-translocating electron transport system. In both groups, enzymes known as hydrogenases, which reversibly interconvert protons and electrons to molecular hydrogen, play a central role in the methanogenic process. This review discusses recent insight into methanogen metabolism and energy conservation mechanisms with a particular focus on the genus Methanosarcina.
INTRODUCTION
Methanogenic archaea (methanogens) play a critical role in the global carbon cycle by virtue of their ability to facilitate the decomposition of organic matter to methane and carbon dioxide (CO2) in anoxic environments that lack superior electron acceptors such as Fe(III) and sulfate (1). It is estimated that approximately 2% of CO2 that is fixed into biomass by photosynthetic organisms is eventually converted into methane, a process that produces around 1 billion tons of methane annually (2, 3). Approximately 60% of this methane is oxidized to CO2 by methane-consuming organisms, while ca. 40% is released into the atmosphere (2, 3). Although geological sources of methane are significant, biological production accounts for approximately 72 to 74% of global emissions (Fig. 1) (4, 5). Moreover, human activities, especially agriculture and the petrochemical industry, have significantly increased the inputs of methane into the atmosphere. As a result, atmospheric methane concentrations have more than doubled since the onset of the industrial revolution (late 1700s), and levels continue to rise (5, 6). Evidence from carbon isotope data suggests that this increase can be primarily attributed to the enhancement of biologically produced methane by human activities (6). Rising atmospheric methane concentrations are of particular concern, as methane has a global warming potential that is 28 to 34 times higher than that of CO2 over a 100-year period (5). On the other hand, biogenic methane is a clean-burning, carbon-neutral, renewable energy source that has the potential to mitigate human-induced climate change. In either case, a more in-depth understanding of the metabolism and physiology of the organisms responsible for methane production is required to effectively manage the process.
FIG 1.

Sources of atmospheric methane. The proportion of atmospheric methane emissions caused by anthropogenic activities (red), produced by biogenic processes (blue), and the overlap between the two categories (purple) is shown. Percentages of each methane source were determined from atmospheric observation-based calculations reported by Dean et al. (5). The category “other natural sources” is primarily composed of biologically produced methane (8% of total) in freshwater and marine sediments and geologically produced methane (3% of total).
Methanogens are nearly ubiquitous in anaerobic environments and have been found to thrive in environments with a wide range of temperatures, salinities, and pHs. These include marine and freshwater sediments, wetlands, geothermal systems, permafrost soils, anaerobic sewage digesters, landfills, and the intestinal tracts of ruminants, humans, and termites (4). Wetlands are by far the largest natural source of biological methane production; however, anthropogenic production of methane by methanogens remains the largest overall source (Fig. 1). This includes emissions related to agriculture (ruminant-associated methanogenesis [18% of total methane emissions] and rice paddy soils [5%]) and landfill and waste decomposition (10%) (5).
Due to their prolific production of methane and the diverse environments in which they thrive, methanogens are ideally suited for the production of biogas, a renewable alternative to fossil natural gas that is composed primarily of methane. A variety of engineered systems have been established for the conversion of organic matter into methane by methanogens (7). More than 2,000 biogas production facilities currently operate in the United States, and it is estimated that full implementation of all potential sites could provide enough energy to power more than 3.5 million homes (8). Improving the rate and efficiency of this process could increase this number even further (7). Because biogas must be purified and methane enriched to be compatible with existing gas distribution networks, a careful consideration of the physiology of methanogens will enable both an enhancement of methane production in biogas reactors and a reduction in overall methane emissions. In this review, we discuss recent insight into the metabolism and energy conservation mechanisms utilized by methanogens, with a particular focus on how H2 is used both as a substrate for methanogenesis and as an electron carrier by cytochrome-containing methanogens.
DIVERSITY OF METHANOGENS
Methanogenic archaea belong to a large and phylogenetically diverse group with various metabolic capabilities and requirements. Until recently, known methanogens were found solely within the phylum Euryarchaeota, assigned to 8 different orders based on 16S rRNA sequence similarity. These include Methanobacteriales, Methanococcales, Methanomicrobiales, Methanosarcinales, Methanopyrales, Methanocellales, Methanomassiliicoccales, and Methanonatronarchaeales (4, 9–11). Recently, three novel taxa of methanogenic archaea have been proposed based on metagenomic data of uncultured organisms, including “Candidatus Methanofastidiosa” and members of the “Candidatus Bathyarchaeota” and “Candidatus Verstraetearchaeota” phyla (12–14), with the latter two falling outside of the Euryarchaeota in the TACK superphylum (12, 14). The discovery of methanogen lineages outside of Euryarchaeota has led to the hypothesis that the last archaeal common ancestor (LACA) was potentially a methanogen (15); however, this conclusion is not supported by recent phylogenetic analyses (16). In addition to their genetic diversity, each order of methanogenic archaea has unique characteristics, including their metabolic capabilities and the environments from which they can be isolated.
Members of the Methanobacteriales order have been isolated from a wide variety of environments, including marine and freshwater sediments, permafrost, hot springs, alkaline lakes, and the gastrointestinal tracts of various animals. Accordingly, the requirements for growth in this order are quite varied. Most species are limited to utilizing CO2 and H2 for methanogenesis, but some members of this order are capable of using formate, CO, or secondary alcohols (e.g., 2-propanol and 2-butanol) as electron donors for reduction of CO2 to methane (17). While some strains are autotrophs, many are heterotrophs that require exogenous acetate, amino acids, or vitamins for growth. Lastly, one genus, Methanosphaera, cannot reduce CO2 to methane and is limited to utilizing methanol with H2 for methanogenesis (18, 19).
All members of the Methanococcales order were isolated from marine environments and are capable of utilizing CO2 and H2 as substrates for methanogenesis. Some species are also able to use formate as an electron donor. Many members of this order are thermophilic or hyperthermophilic and were isolated from deep-sea hydrothermal vents or other marine sediments (17). As a corollary, Methanococcales species have some of the highest growth rates of all methanogens, with a rate as high as 2.4 h−1 found in Methanocaldococcus jannaschii (20, 21).
Most members of the Methanomicrobiales order are mesophilic and require near-neutral pH conditions for ideal growth. Like members of Methanococcales, all Methanomicrobiales species can reduce CO2 to methane with electrons derived from H2, and most can also use formate as an electron source. Additionally, some species are able to oxidize secondary alcohols for the reduction of CO2. Many species also require supplementation with acetate or other organic substrates, such as yeast extract or rumen fluid (17). This order includes two organisms, Methanofollis ethanolicus and Methanogenium organophilum, with the unique ability to utilize the primary alcohol, ethanol, for methanogenesis (22, 23).
The orders Methanopyrales, Methanomassiliicoccales, and Methanocellales contain few cultivated organisms, with a single species each for the first two and a single genus with three species for the last (17). Aside from that commonality, organisms from each of these orders are drastically different. Methanopyrus kandleri, isolated from hydrothermal marine sediments, grows by reduction of CO2 to methane using electrons from H2 and is the only known species capable of methanogenesis above 100°C (24, 25). The Methanomassiliicoccales order consists of one isolated species, Methanomassiliicoccus luminyensis, and at least three other “Candidatus” species identified within enrichment cultures (10, 26–28). M. luminyensis grows solely by reduction of C1 compounds with H2 and was isolated from human fecal matter (26, 29). Lastly, all three species of the Methanocellales order were isolated from the soil of rice paddies, and all utilize H2 and CO2 for methanogenesis. Two of the species can also use formate as an electron donor, and all three species require supplementation with acetate for growth (17). Sequencing of the genome of Methanocella paludicola revealed a lack of genes encoding carbon monoxide dehydrogenase/acetyl coenzyme A (acetyl-CoA) synthase (CODH/ACS); thus, this organism is unable to assimilate CO2 and requires acetate for biosynthesis (30).
Methanonatronarchaeales is the most recently recognized order of methanogens and consists of organisms isolated from sediments of hypersaline chloride-sulfate and soda lakes (11). Most organisms from this order are thermophiles and alkaliphiles, with optimal growth conditions of 50°C and a pH of 9.5 to 9.8. Additionally, all are halophiles with an optimum Na+ concentration of 4.0 M. In a mechanism that is unique among halophilic methanogens, this order overcomes osmotic stress by maintaining high intracellular potassium concentrations. Lastly, all members of this order, like those of the Methanomassiliicoccales order, generate methane by reducing methyl compounds with electrons derived from H2. However, unlike other taxa that are limited to reducing methyl compounds with H2, the Methanonatronarchaeales are also able to utilize formate as an electron source (11).
Members of the final order, Methanosarcinales, are widely distributed and can be found in environments ranging from marine and freshwater sediments to gastrointestinal tracts and anaerobic sewage digesters. Species from this order have the widest substrate range of all methanogens and can utilize CO2 plus H2, CO, acetate, or methyl compounds (e.g., methanol, methylamines, and methyl sulfides) for methanogenesis (2, 17). In addition to these substrates, a single isolate, Methermicoccus shengliensis, is able to use methoxylated aromatic compounds, which can be found in immature coal deposits, for methanogenesis via an enigmatic mechanism (31). Despite this broad substrate range, no member of the Methanosarcinales order is able to utilize formate as an electron donor, which is in stark contrast to the majority of other methanogen orders. A final difference between members of Methanosarcinales and all other orders of methanogenic archaea is that they alone contain cytochromes and the lipid-soluble electron carrier methanophenazine, the significance of which is discussed in the following sections (2).
Two putatively methanogenic taxa outside of the Euryarchaeota phylum, “Ca. Bathyarchaeota” and “Ca. Verstraetearchaeota,” were recently identified based on the presence of genes required for methanogenesis in metagenome-assembled genomes (MAGs) (12, 14). Apart from this similarity, they were obtained from quite different environments: “Ca. Bathyarchaeota” from a coal bed-associated deep aquifer and “Ca. Verstraetearchaeota” from a cellulose-degrading bioreactor. Despite the lack of a cultivated isolate from either taxon, it is thought that both reduce methyl compounds to methane based on the presence of methyl transferase genes (12, 14, 16). Additionally, a recent “Ca. Verstraetearchaeota” MAG obtained from a Yellowstone National Park hot spring appears to contain all genes required to utilize H2 and CO2 (32). While speculative, these findings suggest that the phylogenetic diversity of methanogens is significantly broader than was initially appreciated.
METABOLIC PATHWAYS FOR METHANOGENESIS
A common characteristic of all known methanogens is that they are obligate methane producers, meaning that their only system for energy conservation comes from the reduction of growth substrates to methane. As described in the previous section, nearly all methanogens are capable of reducing CO2 to methane with electrons derived from the oxidation of H2 via the CO2 reduction pathway (4, 17). This pathway differs slightly between methanogens with and without cytochromes, as is discussed in the following sections. The pathway that is utilized by cytochrome-containing methanogens is depicted in Fig. 2. In addition to this pathway, members of the Methanosarcinales order can utilize three other methanogenic pathways in order to metabolize acetate (aceticlastic pathway [Fig. 3]) or methyl compounds (methyl reduction pathway and methylotrophic pathway [Fig. 4 and 5]) (4, 33). The two pathways involving methyl compounds differ in the source of electrons used for reduction of the methyl group to methane. H2 oxidation provides electrons in the methyl reduction pathway, but in the methylotrophic pathway methyl compounds serve as both the oxidant and reductant. Whereas only species in the Methanosarcinales order are able to utilize the aceticlastic and methylotrophic pathways, three additional classes of methanogens can use the methyl reduction pathway. These include organisms from the Methanosphaera genus in the Methanobacteriales order and organisms from the Methanomassiliicoccales and Methanonatronarchaeales orders (11, 17). Based on metagenomic data, “Ca. Methanofastidiosa,” “Ca. Bathyarchaeota,” and “Ca. Verstraetearchaeota” may also rely on this pathway to reduce methyl compounds (12–14). The following sections primarily focus on the metabolic capabilities of Methanosarcina species, due to their ability to use all four methanogenic pathways.
FIG 2.

CO2 reduction pathway of Methanosarcina. In this pathway, CO2 is reduced to CH4 in a stepwise manner. For each reduction step, electrons are derived from the oxidation of H2 by the energy-converting hydrogenase (Ech), the F420-reducing hydrogenase (Frh), and the methanophenazine-reducing hydrogenase (Vht), as indicated. In the final methanogenic step, the coenzyme M (CoM)-bound methyl group is reduced by coenzyme B (CoB), thereby forming CH4 and a disulfide of the two coenzymes (CoM-CoB). Reduced forms of CoM and CoB are regenerated from the disulfide by the heterodisulfide reductase (Hdr). Methanogens that lack cytochromes do not encode Ech or Vht and instead use flavin-based electron bifurcation to couple H2 oxidation to the reduction of ferredoxin (Fd) and CoM-CoB via the MvhADG:HdrABC enzyme complex (not shown). Other abbreviations: Mch, methenyl-tetrahydrosarcinapterin (methenyl-H4SPT) cyclohydrolase; Mtr, methyl-H4SPT:CoM methyltransferase; MF, methanofuran; “red” and “ox” subscripts, reduced and oxidized states of electron carriers.
FIG 3.

The aceticlastic pathway of methanogenesis for M. barkeri. Acetate is first converted to acetyl-CoA in an ATP-dependent manner (not shown), which is then split by acetyl-CoA decarbonylase/synthase (ACDS) into an enzyme-bound carbonyl ([CO]) and a methyl group, which gets transferred to the C1 carrier tetrahydrosarcinapterin. Oxidation of [CO] to CO2 produces reduced ferredoxin (Fdred), which is oxidized by Ech, thereby generating H2 inside the cell. H2 diffuses across the cell membrane to the Vht active site, where it is oxidized and electrons are used to reduce methanophenazine (MPred). Reduction of the methyl group bound to coenzyme M by coenzyme B produces CH4 and a disulfide of CoM and CoB, which is regenerated by reduction with electrons from MPred via the heterodisulfide reductase enzyme. Portions of the methanogenic pathway that are not required for aceticlastic methanogenesis, including the use of Frh, are shown in light gray.
FIG 4.

The methylotrophic pathway of methanogenesis for M. barkeri. In this pathway, methyl compounds undergo disproportionation, wherein the oxidation of one methyl group to CO2 provides the reducing equivalents for the reduction of three additional methyl groups to CH4. In the oxidation portion of the pathway, methyl groups are first transferred to coenzyme M by the sequential action of methyltransferases MT1 and MT2, and then transferred to tetrahydrosarcinapterin by the methyl-H4SPT:CoM methyltransferase. The methyl group is oxidized in two successive steps, first to methylene-H4SPT and then to methenyl-H4SPT, which produces two reducing equivalents in the form of reduced coenzyme F420. The third reducing equivalent comes from the oxidation of formyl-methanofuran, which produces CO2 and reduced ferredoxin. In the reduction portion of the pathway, methyl-CoM is reduced by coenzyme B, which forms CH4 and a disulfide of CoM and CoB. Regeneration of CoM and CoB requires reduction of the disulfide by the heterodisulfide reductase enzyme with electrons from reduced methanophenazine. The transfer of electrons from the oxidative to reductive portions of the pathway occurs via a hydrogen cycling mechanism in M. barkeri. In this mechanism, F420red and Fdred are oxidized by Frh and Ech, respectively, which forms H2 on the inside of the cell. The H2 then diffuses across the membrane to the Vht active site on the outside of the cell, where it is oxidized and electrons are passed to MP, thereby completing the cycle. The methyl compound R groups include –OH (methanol), –NH2 (methylamine), and –SH (methanethiol). Other examples of methyl compounds include dimethylamine, trimethylamine, and dimethyl sulfide.
FIG 5.

The methyl reductive methanogenic pathway in Methanosarcina. In this pathway, methyl groups are transferred to coenzyme M by the sequential action of methyltransferases MT1 and MT2 and then reduced to CH4 by coenzyme B, which forms a disulfide of CoM and CoB. Regeneration of CoM and CoB by the reduction of CoM-CoB occurs at the heterodisulfide reductase enzyme with electrons from reduced methanophenazine. The Vht hydrogenase generates MPred with electrons derived from the oxidation of H2. Portions of the core methanogenic pathway that are not used by the methyl reduction pathway, including Ech and Frh, are shown in light gray.
The CO2 Reduction Pathway
The CO2 reduction pathway is typified by the stepwise reduction of CO2 to methane with electrons derived from the oxidation of H2, which is mediated by a group of enzymes known as hydrogenases (Fig. 2). The overall standard free energy change (ΔG°′) for this process is −131 kJ per mole. However, when accounting for the low partial pressure of H2 in natural environments, this value drops to between −17 and −40 kJ per mole. Therefore, less than 1 mole of ATP can be synthesized from ADP and Pi (within the cell ΔG°′ = +50 kJ per mole) for each mole of methane that is produced (2). The initial reduction of CO2 to a formyl group attached to the C1 carrier molecule methanofuran (MF) requires energy input in the form of a low-redox-potential ferredoxin (Fd; E°′ ≈ −500 mV) (34–36). Methanogens have several strategies for generating reduced Fd (Fdred), which are discussed in the “Energy Conservation Mechanisms of Methanogens” section below. In one example, some species of Methanosarcina generate Fdred by the membrane-bound, proton-translocating energy-converting hydrogenase (Ech), wherein the energetically unfavorable reduction of Fd by H2 oxidation is made possible by coupling the reaction to proton translocation from the outside to the inside of the cell (2, 37, 38). The formyl group is subsequently transferred from MF to another C1 carrier molecule, tetrahydromethanopterin (H4MPT; alternatively, tetrahydrosarcinapterin [H4SPT] in species of Methanosarcina) and then converted to methenyl-H4MPT via a condensation reaction catalyzed by the cyclohydrolase enzyme, methenyl-H4SPT cyclohydrolase (Mch) (39–41). Two reduction steps follow (methenyl-H4MPT to methylene-H4MPT and methylene-H4MPT to methyl-H4MPT), in which the electron donor, reduced coenzyme F420 (F420red), is supplied by the F420-reducing hydrogenase (Frh) (42–44). After the exergonic transfer of the methyl group to the thiol of coenzyme M (CoM) by the membrane-bound methyl-H4MPT:CoM methyltransferase enzyme (Mtr) (45), the final reduction step occurs with the thiol group of coenzyme B (CoB) serving as the electron donor, resulting in production of methane and a mixed disulfide of CoM and CoB (CoM-CoB) (39, 46, 47). Continued methanogenesis relies on regeneration of CoM and CoB thiols by reducing the CoM-CoB disulfide, which is facilitated by the enzyme heterodisulfide reductase (Hdr). Non-cytochrome-containing methanogens rely on a soluble, electron-bifurcating Hdr enzyme for this reaction, as discussed below. In Methanosarcina species, the membrane-bound and cytochrome-containing HdrED catalyzes this reaction with electrons ultimately derived from H2 by way of the methanophenazine-reducing hydrogenase (Vht) and the membrane-soluble electron carrier, methanophenazine (MP) (48). Thus, the reduction of CO2 to methane with electrons derived from H2 in Methanosarcina species requires three different types of hydrogenases.
The Aceticlastic Pathway
It has been estimated that most biologically produced methane (ca. 2/3 globally) comes from the methyl group of acetate by the processes of syntrophic acetate oxidation or aceticlastic methanogenesis (49–51). The former process is facilitated by interaction between an acetate-oxidizing bacterium and an H2-consuming, CO2-reducing methanogen. Oxidation of acetate to CO2 and H2 is very energetically unfavorable (ΔG°′ = +95 kJ per mole), unless the end products are rapidly consumed by the methanogen. Overall, coupled acetate oxidation and CO2 reduction is energetically favorable (ΔG°′ = −36.0 kJ per mole), although the small amount of available energy is presumably split between the two organisms (51, 52). Syntrophic conversion of acetate to methane was originally identified in a thermophilic coculture containing a rod-shaped, acetate-oxidizing bacterium and a Methanothermobacter-like methanogen (51, 53). Subsequently, several other acetate-oxidizing bacteria, such as Clostridium ultunense strain BS, Thermacetogenium phaeum strain PB, and Thermotoga lettingae strain TMO, were identified in coculture with CO2-reducing methanogens (Methanoculleus sp. strain MAB1, Methanothermobacter thermautotrophicus strain TM, and M. thermautotrophicus strain ΔH, respectively) (54–56).
Aceticlastic methanogenesis, which involves the dismutation of acetate to CO2 and methane by a single organism, has been observed only in two genera from the Methanosarcinales order, Methanosarcina and Methanothrix (formerly referred to as Methanosaeta [57]). Under standard conditions, aceticlastic methanogenesis has the lowest free energy of the known methane-producing pathways (ΔG°′ = −36 kJ per mole) (58–60). In the first step of this pathway, acetate is converted into acetyl-CoA in an ATP-dependent manner. The acetyl group is then split into an enzyme-bound carbonyl group and a methyl group by the acetyl-CoA decarbonylase/synthase (ACDS) enzyme complex (Fig. 3) (61, 62). Oxidation of the carbonyl to CO2 results in Fdred, which is used to reduce the methyl group to methane. After the initial cleavage of acetate, the methyl group is transferred to H4SPT, and the same reduction process involving CoM, CoB, Hdr, and MPred as described above for the CO2 reduction pathway occurs (60). In Methanosarcina species, there are two different mechanisms for transferring electrons from the oxidative to the reductive branches of the pathway. Species with active hydrogenases, such as Methanosarcina barkeri, use a hydrogen cycling mechanism, whereas species that are hydrogenase deficient, such as Methanosarcina acetivorans, have an H2-independent electron transport system. In the H2 cycling mechanism, the Ech hydrogenase proceeds in the direction opposite that described for the CO2 reduction pathway by oxidizing Fdred, which generates H2 and allows for proton translocation across the membrane, thereby contributing to the proton motive force (63). H2 then diffuses across the membrane and is oxidized by the Vht hydrogenase, where the electrons are used to reduce CoM-CoB via MPred and Hdr (Fig. 3) (37, 64, 65). Species that utilize the H2-independent electron transport system do not have Ech and instead use an Rnf complex to catalyze the transfer of electrons from Fdred to MP (66, 67). Like Ech, Rnf translocates ions across the membrane due to the exergonic nature of MP reduction with Fdred (ΔG°′ = −68 kJ per mole). However, whereas Ech translocates protons, Rnf conserves energy by translocating sodium ions (68). Despite these differences, M. barkeri and M. acetivorans have similar growth rates and yields on acetate, indicating that the two electron transport systems conserve approximately equivalent amounts of energy (69).
The Methylotrophic Pathway
Only members of the Methanosarcinales order, excluding Methanothrix species, are capable of utilizing the methylotrophic pathway for methanogenesis (17). In this pathway, methyl compounds such as methanol, methylamines, and methyl sulfides serve as the source of carbon and energy. Under standard conditions, substantially more energy is available from methylotrophic methanogenesis (ΔG°′ = −95 and −90 kJ per mole of methane from methanol and methylamine, respectively) than from aceticlastic methanogenesis (52, 59, 70). To serve as both the reductant and oxidant, methyl compounds are disproportionated such that the oxidation of one methyl group to CO2 provides the reducing equivalents needed for the reduction of three methyl groups to methane (46). At the outset, methyl groups are transferred to CoM by the sequential action of two methyltransferases (MT1 and MT2). Methanosarcina species encode multiple MT1 and MT2 isozymes that are specific for different methyl group-containing substrates (71). From the methyl-CoM level, methyl groups are either oxidized to CO2 in a reversal of the CO2 reduction pathway or reduced to methane by CoB in a manner that is identical in all four methanogenic pathways (Fig. 4). The oxidative portion of the pathway generates F420red and Fdred, which are used in the reductive branch of the pathway (72). As with aceticlastic methanogenesis, two different mechanisms are used to transfer electrons from the oxidative to the reductive portions of the pathway, depending on hydrogenase availability (33, 73). In hydrogenase-proficient species, such as M. barkeri, a hydrogen cycling mechanism is utilized in which F420red and Fdred are oxidized by the Frh and Ech hydrogenases, respectively, thereby generating H2 inside the cell. The H2 then diffuses across the cell membrane to the Vht hydrogenase active site, where it is oxidized and electrons are passed to MP to be used for the reduction of CoM-CoB (Fig. 4) (65). In hydrogenase-deficient species, such as M. acetivorans, two different membrane-bound enzyme complexes are utilized. The first, Rnf, transfers electrons from Fdred to MP and functions as described for the aceticlastic pathway (66–68). The second enzyme complex, F420 dehydrogenase (Fpo), catalyzes the transfer of electrons from F420red to MP (48). Fpo is closely related to NADH dehydrogenases found in bacteria and eukaryotes, and it similarly contributes to proton motive force by moving protons across the cell membrane (74, 75). It should be noted that M. barkeri is capable of both H2-dependent and H2-independent electron transport; however, mutant strains that lack Frh, and are thus limited to utilizing Fpo for F420:MP oxidoreductase activity, grow far slower than the wild-type strain (42).
The Methyl Reduction Pathway
In addition to the Methanosarcinales order, only three other classes of methanogens are able to utilize the methyl reduction pathway, which includes a single genus (Methanosphaera) of the Methanobacteriales order, the Methanomassiliicoccales order, and the Methanonatronarchaeales order. Based on metagenomic data, newly discovered methanogens “Ca. Methanofastidiosa,” “Ca. Bathyarchaeota,” and “Ca. Verstraetearchaeota” are also likely to rely on this pathway (12–14). In the methyl reduction pathway, methyl compounds are reduced to methane with electrons derived from H2 (76). The amount of energy made available by this reaction is fairly high under standard conditions (ΔG°′ = −95 kJ per mole), but is likely to be much lower under the H2-limiting conditions found in natural environments (2). Additionally, a strain of Methanosphaera that was isolated from a kangaroo forestomach (sp. strain WGK6) is uniquely able to reduce methanol with electrons derived from ethanol oxidation (77). As many organisms are able to reduce CO2 with electrons derived from primary and secondary alcohols, it would not be surprising for future studies to identify organisms that can couple the oxidation of a variety of substrates to methyl reduction.
In Methanosarcina species, which are unable to utilize alcohols (other than methanol) as a substrate for methanogenesis, the transfer of a methyl group to CoM, reduction to methane by CoB, reduction of CoM-CoB by Hdr, and H2:MP oxidoreductase activity of Vht all occur as described for the CO2 reduction, aceticlastic, and methylotrophic pathways (Fig. 5). The model for the methyl reduction pathway predicts that Ech and Frh should not be required for methanogenesis, which has been confirmed by mutational studies (37, 42). However, despite the ability to produce methane, the strain lacking Ech was unable to grow via this pathway unless supplemented with acetate or pyruvate, indicating that Ech has a biosynthetic role during growth with methyl compounds and H2. It is presumed that Ech is required for the H2-dependent production of Fdred that is required for acetyl-CoA and pyruvate synthesis (37).
ENERGY CONSERVATION MECHANISMS OF METHANOGENS
Methanogens, like all living organisms, use energy derived from metabolic processes to drive growth and cellular maintenance. However, they are unable to generate net ATP via substrate-level phosphorylation and instead require ion gradient-dependent ATP generation via an ATP synthase as their principal energy conservation mechanism (70, 76). Due to the limited amount of energy available from methanogenic substrates, very few steps in the four methanogenic pathways are sufficiently exergonic to translocate ions across the cell membrane (2). Further, as mentioned above, there is a significant divide in energy conservation systems between methanogens that contain cytochromes and those that do not (2). Most methanogenic species belong to the second group, but cytochromes are ubiquitous within the order Methanosarcinales. While the utilization of cytochromes enables a unique energy conservation system, both groups share energy conservation mechanisms that are common to all methanogens (2, 70).
Establishing a Primary Na+ Ion Gradient with Mtr
A characteristic feature of all characterized methanogens is the dependence on sodium ions (Na+) for growth and methanogenesis (2, 78, 79). This requirement is likely due to the energy conservation mechanism employed by the methyl-H4MPT:CoM methyltransferase (Mtr) (45, 80, 81). The transfer of a methyl group from H4MPT to CoM is exergonic (ΔG°′ = −29 kJ per mole), thereby allowing for the translocation of approximately 2 Na+ ions across the membrane and contributing to ion motive force (58). All methanogens that are able to utilize the CO2 reduction or aceticlastic pathway (Fig. 2 and 3) have this mechanism for energy conservation. During methylotrophic methanogenesis, methyl transfer also occurs in the opposite direction (Fig. 4), such that consumption of the Na+ ion motive force is required to facilitate this endergonic reaction (70, 82).
Electron Bifurcation and Methanogenesis as a Cycle
All methanogenic pathways generate a CoM-CoB disulfide in the terminal methane-generating step. Reduction of CoM-CoB is another energy-conserving reaction common to all methanogens; however, different mechanisms for this process have evolved in methanogens with cytochromes versus those without cytochromes. Those without cytochromes utilize an energy conservation mechanism that was only recently discovered: flavin-based electron bifurcation (FBEB) (2, 83). With this mechanism, the endergonic reduction of ferredoxin (Fd; E°’≈ −500 mV) by H2 oxidation (E°′ = −414 mV) is coupled to the exergonic reduction of CoM-CoB (E°′ = −140 mV) by H2 oxidation. Thus, the overall reaction (Fdox + CoM-CoB + 2H2 → Fdred + CoM + CoB + 2H+) is energetically favorable (ΔG°′ = −50 kJ per mole) (83). The FBEB mechanism is facilitated by the MvhADG:HdrABC enzyme complex, wherein MvhADG catalyzes H2 oxidation and HdrABC catalyzes CoM-CoB and Fd reduction (83–85). The HdrABC enzyme is a class of heterodisulfide reductase that is distinct from the HdrED enzyme found in Methanosarcina species and is neither cytochrome containing nor membrane bound (86, 87). Some cytochrome-deficient methanogens are also able to couple Fd and CoM-CoB reduction to formate oxidation. In these organisms, HdrABC also forms a complex with a formate dehydrogenase (Fdh) (88, 89). The Fdred produced by FBEB is required for the initial step in the CO2 reduction pathway, the reduction of CO2 to formyl-MF (Fig. 2), which has a reduction potential of ≈−500 mV (83). Thus, in cytochrome-deficient methanogens the first and final steps of the methanogenic pathway are energetically linked via FBEB, leaving the Na+-dependent Mtr reaction as the only mechanism for generating an ion gradient for ATP synthesis.
Coupling of the first and last methanogenic steps by FBEB confirms the cyclical nature of methanogenesis, as proposed 30 years ago by Rouvière and Wolfe (90). Recent elucidation of anaplerotic mechanisms required to replenish cycle intermediates that are withdrawn for biosynthesis further supports methanogenesis as a cycle (91). The cyclization of methanogenesis by way of FBEB is now referred to as the Wolfe cycle to honor the influence of Ralph S. Wolfe on the field of methanogenic biochemistry (92).
Some methanogens with cytochromes also encode one or more putative electron bifurcating heterodisulfide reductases that are homologous to HdrABC (93). In Methanosarcina species, the HdrA2 homolog is comprised of a domain similar to MvhD (as found in the MvhADG:HdrABC enzyme complex of cytochrome-deficient methanogens) fused to a C-terminal domain with homology to HdrA (85). In vitro data suggest that the HdrA2B2C2 complex from M. acetivorans catalyzes the endergonic reduction of Fd (E°′ ≈ −500 mV) by oxidation of F420red (E°′ = −360 mV) coupled to the exergonic reduction of CoM-CoB (E°′ = −140 mV) by F420red. The reaction (Fdox + CoM-CoB + 2 F420red → Fdred + CoM + CoB + 2 F420ox) is energetically favorable, with a free energy of ≈−30 kJ per mole (94). Although biochemically characterized, the in vivo role for this FBEB mechanism in Methanosarcina species has not been fully established. Indeed, a modified strain of M. acetivorans that does not produce HdrA2B2C2 is unaffected during growth with most substrates, with the exception of slower growth with acetate (87). As F420 is not required for the aceticlastic pathway, it is not clear how this form of FBEB integrates into the energy conservation network of methanogens with cytochromes.
Electron Transport in Methanogens with Cytochromes
In contrast to methanogens without cytochromes, which can establish a transmembrane Na+ gradient only via Mtr, methanogens with cytochromes have multiple mechanisms for establishing an ion gradient via membrane-bound electron transport systems (2, 70). In each, the CoM-CoB disulfide serves as the terminal electron acceptor with translocation of H+ or Na+ ions across the membrane during electron transfer from a variety of donors (48). The electron transport mechanisms depend on the electron source (F420red, Fdred, or H2) and on whether the organism can utilize H2 as a substrate (Fig. 6 and 7). Production of simultaneous H+ and Na+ electrochemical ion gradients is a unique characteristic of these methanogens, and efficient energy conservation requires that the electrical potential from both ions be used for ATP generation (95). Recent evidence suggests that the A1AO ATP synthase from M. acetivorans can translocate both H+ and Na+ ions for ATP synthesis, a unique property that is potentially shared by other Methanosarcina species (96). Nevertheless, these organisms also encode multiple Na+/H+ antiporters, such as Mrp, to convert one ion gradient into the other or to optimize the ion ratio for the ATP synthase (70, 97). Significantly, the additional ion translocation sites within methanogens with cytochromes allow growth yields that are more than double than that of methanogens without cytochromes. Accordingly, the ATP yield per mole of methane generated during growth with CO2 and H2 is estimated to be 1.5 in methanogens with cytochromes and 0.5 in methanogens without cytochromes, which enables a growth yield of up to 7 g of cells per mole of methane in the former versus 3 g per mole of methane in the latter (2).
FIG 6.

The H2-independent electron transport system of Methanosarcina. Electrons enter the transport system as either reduced coenzyme F420 or reduced ferredoxin and are used for the reduction of the terminal electron acceptor, CoM-CoB. Reduction of CoM-CoB by the heterodisulfide reductase regenerates CoM and CoB for continued methanogenesis. F420 dehydrogenase (Fpo) catalyzes the exergonic transfer of electrons from F420red to MP with concomitant translocation of 2 H+ ions to the outside of the cell. Similarly, the Rnf enzyme complex facilitates the exergonic transfer of electrons from Fdred to MP, but instead translocates 3 Na+ ions outside of the cell. In both pathways, an additional 2 H+ ions are consumed from the cytoplasm during MP reduction and released outside of the cell upon MP oxidation. Thus, the transport of 2 electrons results in 4 H+ ions translocated for the F420red:CoM-CoB pathway and 3 Na+ plus 2 H+ ions translocated for the Fdred:CoM-CoB pathway (indicated in red). Brown lines trace the putative pathways of electron transport. Enzyme subunits are identified by letters, with the exception of the cytochrome c subunit (Cyt c) and a membrane-integral subunit with unknown function (*) that are cotranscribed with the rest of the rnf operon.
FIG 7.

The H2-dependent electron transport system of Methanosarcina. Electrons enter the transport system as either externally provided H2 or internally generated Fdred or F420red. All pathways involve the oxidation of H2 by Vht, and the transport of electrons by MP to the heterodisulfide reductase for reduction of the terminal electron acceptor, CoM-CoB. Regeneration of CoM and CoB enables continued methanogenesis. Frh and Ech generate H2 with electrons from F420red and Fdred, respectively. The internally produced H2 diffuses across the membrane (dashed line) and is oxidized by Vht, thereby transferring 2 H+ ions from the inside to the outside of the cell. The exergonic reaction catalyzed by Ech facilitates additional proton translocation. For each pathway, 2 H+ ions are consumed from the cytoplasm during MP reduction and released outside of the cell upon MP oxidation. Thus, the transport of 2 electrons results in 4 H+ ions translocated for the F420red:CoM-CoB pathway and 5 H+ ions translocated for the Fdred:CoM-CoB pathway (indicated in red). Brown lines trace the putative pathways of electron transport. Enzyme subunits are identified by letters.
All characterized Methanosarcina species have an H2-independent F420red:CoM-CoB electron transport pathway (60, 70). During methylotrophic methanogenesis, F420red (E°′ = −360 mV) is produced during the oxidation of methyl groups to CO2, and CoM-CoB (E°′ = −143 mV) is produced during the reduction of methyl groups to methane (Fig. 4) (98). The F420red:CoM-CoB electron transport pathway allows for the transfer of reducing equivalents between these oxidized and reduced electron carriers while also conserving energy in the form of an electrochemical H+ gradient. The key players in this electron transport chain are the 14-subunit F420 dehydrogenase (FpoABCDFHIJJKLMNO) and the membrane-bound, cytochrome-containing heterodisulfide reductase (HdrED). The two enzymes are linked by the membrane-soluble electron carrier methanophenazine (E°′ = −165 mV [Fig. 6]) (98–102). The initial reduction of MP with electrons from F420red is exergonic (ΔG°′ = −37.6 kJ per mole), which allows 2 H+ to be translocated to the outside of the cell by Fpo (75). Analysis of the amino acid sequence of Fpo from Methanosarcina species indicates a high level of similarity to the NADH:quinone oxidoreductases (complex I) of bacteria and eukaryotes, which are also H+-translocating enzyme complexes (60). Thus, it seems likely that the two enzymes share energy conservation mechanisms (75, 103). A key difference between the two is the module for electron input. Fpo lacks a homolog to the NADH-oxidizing module of complex I (NuoEFG) and instead contains an F420red-oxidizing subunit, FpoF (75). An additional 2 H+ ions are moved across the membrane during the transfer of electrons from MPred to CoM-CoB via HdrED, for a total of 4 H+ ions translocated for every 2 electrons transported by the F420red:CoM-CoB system. The HdrE subunit is a membrane-bound, heme b-containing cytochrome, and it has been suggested that protons are translocated by a mechanism similar to that of the bacterial quinone loop (the MP loop) (104, 105). Reduction of CoM-CoB occurs at the active site of HdrD with electrons obtained from HdrE, thereby completing the electron transport pathway and regenerating the free CoM and CoB thiols for further rounds of methanogenesis (106).
Some species of Methanosarcina, such as M. acetivorans, also have an H2-independent Fdred:CoM-CoB electron transport pathway. With this system, Fdred generated by the oxidation of an enzyme-bound carbonyl group to CO2 in the aceticlastic pathway (Fig. 3) or by the oxidation of formyl-MF to CO2 in the methylotrophic pathway (Fig. 4) is used to reduce the terminal electron acceptor, CoM-CoB. In a mechanism similar to the F420red:CoM-CoB pathway, electrons from the reduced electron carrier (Fdred) are transferred to MP, which is used by HdrED to reduce CoM-CoB. The exergonic transport of electrons from Fdred (E°′ = −500 mV) to MPox (E°′ = −165 mV; ΔG°′ = −65 kJ per mole) is facilitated by the membrane-bound, Na+-translocating enzyme complex, Rnf (Fig. 6) (66–68, 107). It is estimated that 3 Na+ ions are translocated for every 2 electrons transported by Rnf (68, 108). Combining this value with protons translocated by the HdrED-mediated MP loop brings the electrochemical total to 3 Na+ ions 2 H+ ions moved to the outside of the cell for every 2 electrons that pass through the Fdred:CoM-CoB electron transport pathway. The Rnf complex has been identified in a wide array of bacterial species and typically catalyzes Fdred:NAD+ oxidoreductase activity with concomitant Na+ translocation (109). However, it has been shown that Rnf from Methanosarcina does not interact with NADH/NAD+ (66, 68). Additionally, the gene cluster encoding Rnf (rnfCDGEAB) differs from all known bacterial versions by encoding a multiheme c-type cytochrome upstream of rnfC and a gene downstream of rnfB that is predicted to integrate into the membrane. Both genes are cotranscribed with the rest of the rnf operon, and it is thought that the cytochrome c subunit is responsible for transferring electrons to MP (67).
Methanosarcina species that are able to metabolize H2, such as M. barkeri, have an H2-dependent electron transport system, wherein H2 can serve as the sole source of electrons (H2:CoM-CoB pathway) or as an intermediate (Fdred:CoM-CoB and F420red:CoM-CoB pathways). In all three, the transport of electrons from H2 to CoM-CoB (i.e., the H2:CoM-CoB pathway) occurs by the same mechanism; only the source of H2 varies between the pathways (Fig. 7). Electron transport is initiated by the membrane-bound Vht, which oxidizes H2 at an externally located active site. Electrons are then transferred to MP by way of the cytochrome b-containing subunit (VhtC) (48, 74, 110). HdrED facilitates the transfer of electrons from MPred to CoM-CoB, as described above for H2-independent electron transport systems. Oxidation of H2 by Vht produces 2 external H+ ions, which, when combined with translocated protons from the MP loop, results in a total proton motive force of 4 H+ ions per 2 electrons transported (74).
The only electron source for the CO2 reduction (Fig. 2) and methyl reduction (Fig. 5) methanogenic pathways is external H2. During growth via these pathways, M. barkeri uses the H2:CoM-CoB electron transport pathway for energy conservation (111). However, metabolism of acetate and methyl compounds via the aceticlastic and methylotrophic pathways produces internal electron sources (Fdred and F420red) as described above. The transport of electrons from Fdred and F420red to CoM-CoB via the H2-dependent electron transport pathways involves a hydrogen cycling mechanism (Fig. 7) (37, 64, 65). In the first part of this mechanism, Fdred and F420red are oxidized by the Ech and Frh hydrogenases, respectively, and electrons are used to generate H2 on the inside of the cell with concomitant consumption of cytoplasmic protons. The H2 then diffuses across the membrane to the Vht active site, where it is oxidized releasing extracellular protons and feeding electrons to CoM-CoB via the H2:CoM-CoB electron transport pathway. The hydrogen cycling mechanism was confirmed in a series of hydrogenase mutants, wherein removal of Vht caused rapid accumulation of H2, diminished methanogenesis, and cell death. However, when both Vht and Frh were removed this effect was abrogated, indicating that Vht is required to capture H2 produced by Frh in order to maintain redox balance (65). The production of H2 inside the cell consumes 2 internal H+ ions, and oxidation by Vht produces 2 external H+ ions. Combination with protons translocated via the MP loop yields a total of 4 H+ moved across the membrane for 2 electrons that pass through the electron transport chain (42, 65). Additional energy is conserved by the proton-pumping Ech hydrogenase during the generation of H2 from Fdred, as this exergonic reaction enables auxiliary proton translocation (63). Thus, a similar amount of energy is conserved as an ion motive force by the H2-dependent and H2-independent electron transport systems (69). As highlighted by the use of H2 as an electron source and as an electron carrier, this molecule serves as an important metabolite, and the hydrogenase enzymes that enable its use are an essential component of methanogenic energy conservation systems.
HYDROGENASES OF METHANOGENS
Organisms from all three domains of life rely on the activity of hydrogenases to consume H2 as a substrate or to produce H2 from the reduction of protons. Hydrogenases belong to a large and diverse group of enzymes and are classified into groups based on their cognate electron-carrying redox partners (NAD+, Fd, F420, MP, etc.), active-site cofactors ([NiFe], [FeFe], or [Fe]), location within the cell (membrane bound or cytoplasm), or ability to conserve energy by ion translocation (112, 113). Methanogens use five different types of hydrogenase. Four of these belong to the [NiFe] group of hydrogenases, as classified by the active site transition-metals used for catalysis (112). The core of all [NiFe] hydrogenases consists of a heterodimer containing a “large” and a “small” subunit. The large subunit contains the [NiFe] active site, and the small subunit typically contains three linearly arranged Fe-S clusters of the cubane [4Fe4S]-type, which facilitate electron transport between the active site and cognate redox partner (113). Despite different mechanisms for energy conservation between methanogens with and without cytochromes, two types of [NiFe] hydrogenases are present in both methanogen classes. These include the membrane-bound energy-converting hydrogenases, such as the Ech hydrogenase found in M. barkeri, and a cytoplasmic F420-reducing hydrogenase, such as Frh. A third type of hydrogenase, exemplified by Vht in M. barkeri, contains a cytochrome and is found only in species of Methanosarcina (3). The fourth type of [NiFe] hydrogenase is Mvh, which forms a complex with the electron-bifurcating HdrABC and which is found solely in noncytochrome methanogens. Finally, a unique hydrogenase that is also found only in cytochrome-deficient methanogens is the nickel-free [Fe]-hydrogenase, which contains a unique single-Fe cofactor that is not found in any other enzyme, including the [Ni/Fe] and [Fe/Fe] hydrogenases (3, 85, 114, 115). Under nickel-limiting conditions, the [Fe]-hydrogenase replaces the F420-reducing [NiFe] hydrogenase activity required for the reduction of methenyl-H4MPT to methylene-H4MPT (Fig. 2) (116, 117). Under these growth conditions, the [Fe]-hydrogenase is also responsible for synthesis of reduced F420, which is required for numerous metabolic processes, by coupling the F420-dependent and H2-dependent methylene dehydrogenase activities.
The Energy-Converting Hydrogenase of Methanosarcina barkeri
The membrane-bound, energy-converting [NiFe] hydrogenases are evolutionarily distinct from other hydrogenases based on amino acid sequence alignments (113, 118). Aside from the conserved residues required to coordinate the [NiFe] active site and Fe-S clusters, this type of hydrogenase has very little sequence similarity to other [NiFe] hydrogenases (38). The core of all energy-converting hydrogenases contains at least six subunits; two membrane-bound hydrophobic subunits, two soluble hydrophilic subunits, and the large and small hydrogenase subunits. These six subunits, as typified by Ech from M. barkeri (Fig. 8), are highly similar to the subunits that form the catalytic core of the ion-translocating NADH:quinone oxidoreductase (complex I) of mitochondria and bacteria (38, 119). In fact, energy-converting hydrogenases are predicted to be ancestral to complex I (38).
FIG 8.
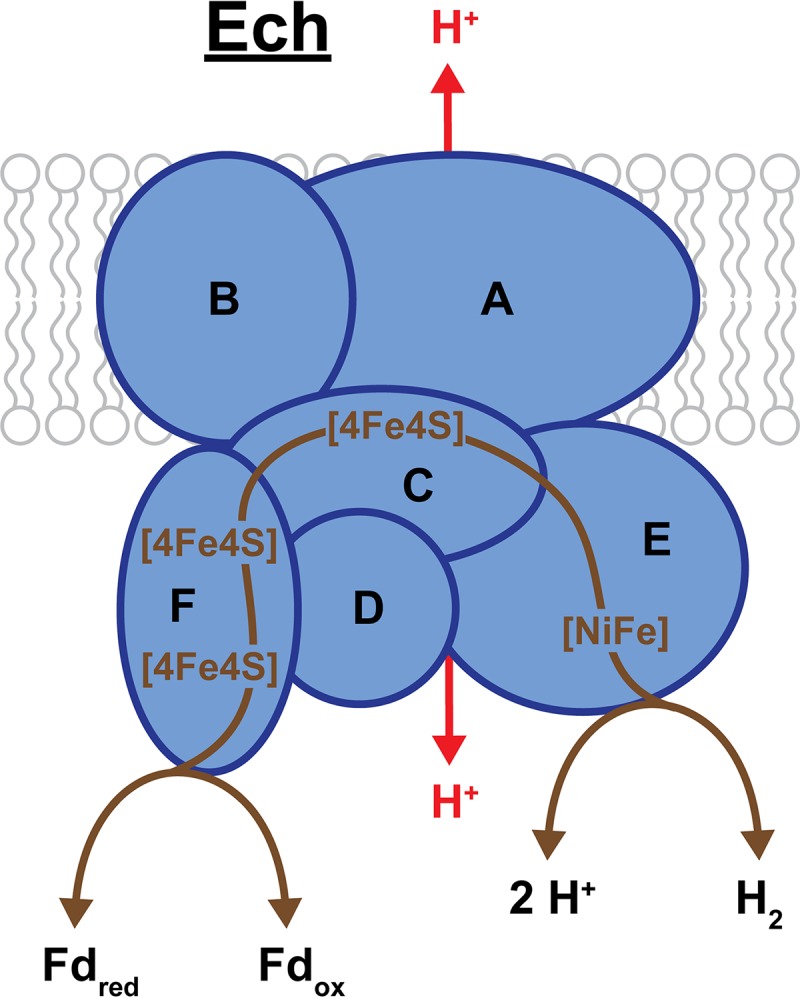
Ech of M. barkeri. Ech consists of 6 subunits (EchABCDEF), of which EchA and EchB are membrane bound and EchA is the likely location for H+ translocation (shown in red). The remaining 4 subunits are located in the cytoplasm and facilitate the transfer of electrons between Fd and H2/H+ by way of 3 cubane Fe-S clusters ([4Fe4S]). EchE contains the bimetallic [NiFe] active site for H2 formation/oxidation. Bidirectional arrows indicate Ech is a reversible enzyme, and brown lines trace the putative pathway of electron transport. In one direction, the exothermic oxidation of Fdred allows for internal H2 production and translocation of H2 to the outside of the cell.
All Methanosarcina species that are able to reduce CO2 with H2 encode an energy-converting hydrogenase. Initial identification of this enzyme in M. barkeri indicated that it was highly similar to Escherichia coli-3-type hydrogenase, and it was thereby designated Ech (64, 120). Further elucidation of the coupled hydrogenase and ion-translocating activities of this enzyme led to the more accurate “energy-converting hydrogenase” designation for Ech (118). This 6-subunit enzyme (EchABCDEF) drives the endergonic reduction of Fd (E°′ ≈ −500 mV) with electrons from H2 oxidation (E°′ = −414 mV) by utilizing the proton motive force (Fig. 8) (38, 64, 121). Ech is reversible, such that oxidation of Fdred leads to both the generation of H2 and the translocation of H+ outside of the cell (63, 122, 123). EchA and EchB are the membrane-bound subunits, with EchA being the most probable location for H+ translocation based on sequence similarity to ion-translocating proteins in other organisms. The large and small hydrogenase subunits, EchE and EchC, are soluble and located within the cytoplasm. EchE contains the [NiFe] active site, and EchC contains only one [4Fe4S] cluster, which distinguishes it from the small subunit of other [NiFe] hydrogenases that typically have three clusters. However, another subunit, EchF, contains two additional [4Fe4S] clusters and is the location of Fd oxidation/reduction (3, 38, 124). Studies of an M. barkeri mutant lacking Ech indicate that this hydrogenase is required for production of methane via both the CO2 reduction and aceticlastic pathways (Fig. 2 and 3) (37, 111). Additionally, this mutant strain requires supplementation with biosynthetic precursors during growth via the methyl reduction pathway (Fig. 5), showing that Ech is required to provide the Fdred needed for pyruvate and acetyl-CoA synthesis (37).
The Coenzyme F420-Reducing Hydrogenase
The F420-reducing hydrogenase (Frh) has a critical role in the CO2 reduction pathway of methanogenesis (Fig. 2) by providing F420red required for two reduction steps. Thus, all methanogens that are able to reduce CO2 with H2-derived electrons require an Frh hydrogenase, and this enzyme has been purified from methanogens both with and without cytochromes (125–129). Frh is a reversible enzyme that catalyzes the reduction of F420 with electron obtained by the oxidation of H2 (Fig. 9). Under standard conditions this reaction has a free energy change of ΔG°′ = −11 kJ per mole. However, under in vivo conditions there is very little free energy change, and Frh activity is not directly coupled to energy conservation (3). Indeed, the redox state of F420 has been found to be in rapid equilibrium with the external concentration (partial pressure) of H2 (65, 130).
FIG 9.
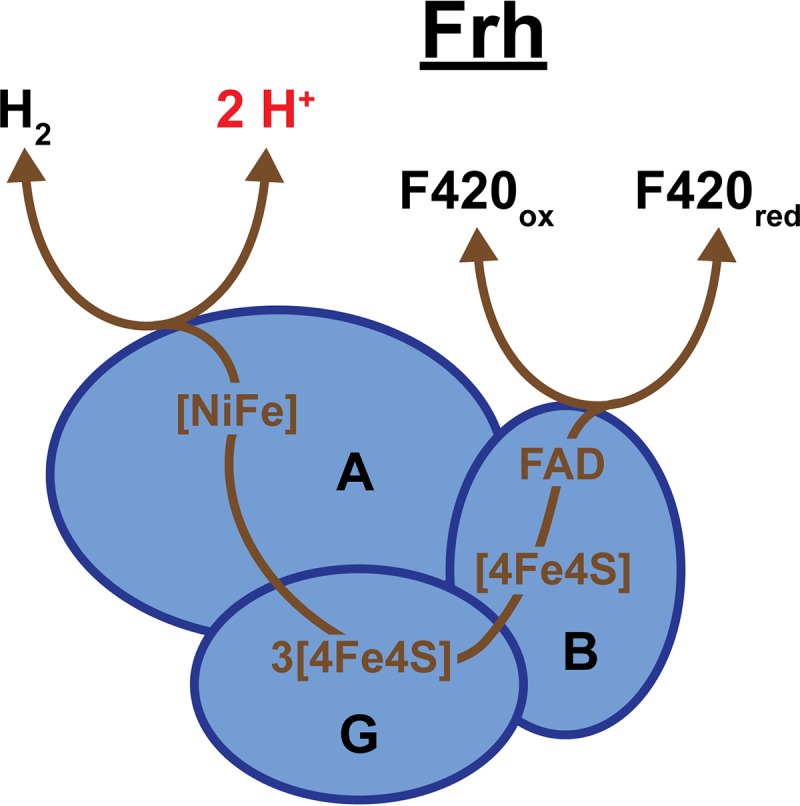
Frh. Frh is a heterotrimeric enzyme, consisting of a large subunit (FrhA), small subunit (FrhG), and a flavin adenine dinucleotide (FAD)-containing subunit (FrhB). FrhA contains the [NiFe] active site for formation or oxidation of H2, and FrhB has an FAD-containing active site for coenzyme F420 oxidation or reduction. Electrons are transmitted between active sites by four cubane Fe-S clusters ([4Fe4S]) located in FrhG and FrhB. Bidirectional arrows indicate Frh is a reversible enzyme, and brown lines trace the putative pathway of electron transport.
Initial purification and characterization of Frh indicated that the core of this hydrogenase consists of three subunits (FrhAGB) that form a membrane-associated heterotrimer (129, 131, 132). However, Frh was later found to be located within the cytoplasm and not membrane bound, as none of the subunits contain transmembrane helices. Frh, as purified from Methanothermobacter, forms a complex consisting of at least 8 α/β/γ heterotrimers (129, 131). Recently, the structure of Frh from Methanothermobacter marburgensis was determined by cryo-electron microscopy, which suggested that the enzyme is made up of 12 copies of the α/β/γ heterotrimer (133). The structure determination also verified the locations of key components for electron transport within this enzyme: the [NiFe] active site, [4Fe4S] clusters, and flavin adenine dinucleotide (FAD) (Fig. 9). FrhA is the large hydrogenase subunit and contains the bimetallic [NiFe] active site for H2 oxidation or formation. FrhB facilitates F420 redox reactions with an FAD-containing active site. Transport of electrons between the [NiFe] and FAD active sites occurs via four [4Fe4S] clusters. Three of these clusters are located in the small hydrogenase subunit, FrhG, and the fourth is located within FrhB (3, 133).
Whereas methanogens without cytochromes use Frh solely to produce F420red from H2 for two sequential reduction steps in the CO2 reduction pathway, methanogens with cytochromes are also able to take advantage of the reversibility of Frh during methylotrophic methanogenesis (Fig. 4) (42, 65). In this pathway, F420red generated by the oxidation of methyl groups to CO2 is oxidized by Frh to form H2. Diffusion of H2 to the external Vht hydrogenase active site allows electrons from F420red to enter the electron transport system by way of the hydrogen cycling mechanism discussed above (65). A mutant strain of M. barkeri that lacks the Frh hydrogenase is still able to utilize the methylotrophic pathway for methanogenesis; however, the growth rate and final yield are severely diminished, indicating that the Frh-Vht mediated hydrogen cycling mechanism is the preferred method of electron transport (42). Thus, Frh plays an important role in both the CO2 reduction and methylotrophic pathways.
The Methanophenazine-Reducing Hydrogenase
Only methanogens with cytochromes have an MP-reducing hydrogenase, which plays a critical role in all four methanogenic pathways. Vht (as the MP-reducing hydrogenase is designated in M. barkeri) catalyzes the exergonic reduction of MP with electrons obtained by the oxidation of H2 (ΔG°′ = −50 kJ per mole) (2). Energy is conserved by the production of 2 external H+ ions from H2 oxidation at the Vht active site on the outside of the cell, thereby contributing to the transmembrane proton gradient (60). The Vht-generated MPred is then used for reduction of the terminal electron acceptor common to all methanogenic pathways, CoM-CoB, as described for the H2-dependent electron transport system (48, 74). During methanogenesis via the CO2 reduction and methyl reduction pathways H2 is provided externally, and for the aceticlastic and methylotrophic pathways H2 is produced internally by Ech and Frh (37, 64, 65). The inability of an M. barkeri mutant strain lacking Vht to grow on any substrate supports the essentiality of Vht in Methanosarcina strains that rely on H2 as a substrate and for electron transport (65).
Vht was first characterized from cell extracts of Methanosarcina mazei and M. barkeri. Initial purification of Vht from membrane fractions indicated that it was a two-subunit, membrane-bound hydrogenase (134, 135). These subunits consisted of VhtA, the large subunit containing the [NiFe] active site, and VhtG, the small subunit containing three Fe-S clusters (Fig. 10). The third subunit, VhtC, was later identified by experiments searching for genes that encode Vht, as the vht operon contains genes for all three subunits (136, 137). VhtC is a membrane-spanning cytochrome b protein that contains two heme b prosthetic groups as the location for MP reduction. The VhtA and VhtG subunits were determined to be located on the outer face of the cell membrane based on the presence of a twin-arginine-translocation (Tat) signal peptide on the N terminus of VhtG and on the high level of homology to cytochrome b-containing hydrogenases from bacterial species that are located in the periplasm (3, 138, 139). Thus, the external active site of Vht allows for the extraction of electrons from H2 while simultaneously contributing to the proton gradient, which can be used to generate ATP.
FIG 10.
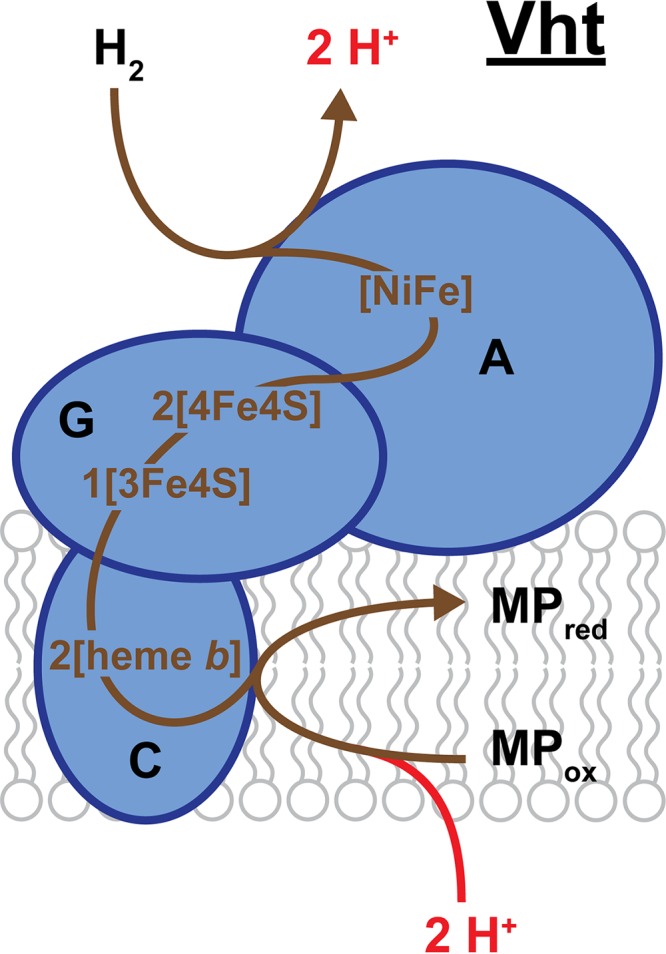
Vht of M. barkeri. The large hydrogenase subunit (VhtA) is located on the outside face of the cytoplasmic membrane and contains the bimetallic [NiFe] active site where H2 is oxidized. This reaction generates 2 external H+ ions that contribute to proton motive force (shown in red) and 2 electrons that are transported via Fe-S clusters ([4Fe4S] and [3Fe4S]) located in the small subunit (VhtG) to heme b groups in the membrane-bound cytochrome b subunit (VhtC). Reduction of the electron carrier MP by VhtC involves uptake of 2 H+ ions from the cytoplasm, which are released externally upon MP oxidation (not shown). Brown lines trace the putative pathway of electron transport.
CONCLUSION
Despite wide-ranging environmental and phylogenetic diversity, most methanogens lack cytochromes and are limited to a single process for methane production, the CO2 reduction pathway. However, overlapping metabolic pathways of cytochrome-containing methanogens, as exemplified by Methanosarcina species, allow this type of methanogen to use a comparatively large number of substrates for growth and methanogenesis. While the methanogenic pathways have been largely characterized, important aspects of energy conservation mechanisms in both types of methanogens, such as electron bifurcation and the involvement of hydrogenases in electron flow, are continually being discovered (2, 65, 83, 111). These concepts have a broad impact, as similar mechanisms have been found in multiple domains of life (65, 94). Additionally, investigation of newly identified methanogen classes, both within and outside of the Euryarchaeota phylum, may reveal novel methanogenic and energy conservation mechanisms. Thus, the unique biochemistry of methanogens continues to be a rich source for future studies uncovering fundamental properties of life.
ACKNOWLEDGMENTS
We acknowledge the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy, for funding of this work through grant DE-FG02-02ER15296.
REFERENCES
- 1.Wolfe RS. 1991. My kind of biology. Annu Rev Microbiol 45:1–35. doi: 10.1146/annurev.mi.45.100191.000245. [DOI] [PubMed] [Google Scholar]
- 2.Thauer RK, Kaster A-K, Seedorf H, Buckel W, Hedderich R. 2008. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol 6:579–591. doi: 10.1038/nrmicro1931. [DOI] [PubMed] [Google Scholar]
- 3.Thauer RK, Kaster A-K, Goenrich M, Schick M, Hiromoto T, Shima S. 2010. Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H2 storage. Annu Rev Biochem 79:507–536. doi: 10.1146/annurev.biochem.030508.152103. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, Whitman WB. 2008. Metabolic, phylogenetic, and ecological diversity of the methanogenic Archaea. Ann N Y Acad Sci 1125:171–189. doi: 10.1196/annals.1419.019. [DOI] [PubMed] [Google Scholar]
- 5.Dean JF, Middelburg JJ, Röckmann T, Aerts R, Blauw LG, Egger M, Jetten MSM, de Jong AEE, Meisel OH, Rasigraf O, Slomp CP, in’t Zandt MH, Dolman AJ. 2018. Methane feedbacks to the global climate system in a warmer world. Rev Geophys 56:207–250. doi: 10.1002/2017RG000559. [DOI] [Google Scholar]
- 6.Nisbet EG, Dlugokencky EJ, Manning MR, Lowry D, Fisher RE, France JL, Michel SE, Miller JB, White JWC, Vaughn B, Bousquet P, Pyle JA, Warwick NJ, Cain M, Brownlow R, Zazzeri G, Lanoisellé M, Manning AC, Gloor E, Worthy DEJ, Brunke E-G, Labuschagne C, Wolff EW, Ganesan AL. 2016. Rising atmospheric methane: 2007–2014 growth and isotopic shift. Global Biogeochem Cycles 30:1356–1370. doi: 10.1002/2016GB005406. [DOI] [Google Scholar]
- 7.Enzmann F, Mayer F, Rother M, Holtmann D. 2018. Methanogens: biochemical background and biotechnological applications. AMB Express 8:1. doi: 10.1186/s13568-017-0531-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.US Department of Agriculture, US Environmental Protection Agency, US Department of Energy. 2014. Biogas opportunities roadmap. USDA, EPA, DOE, Washington, DC. [Google Scholar]
- 9.Sakai S, Imachi H, Hanada S, Ohashi A, Harada H, Kamagata Y. 2008. Methanocella paludicola gen. nov., sp. nov., a methane-producing archaeon, the first isolate of the lineage ‘Rice Cluster I’, and proposal of the new archaeal order Methanocellales ord. nov. Int J Syst Evol Microbiol 58:929–936. doi: 10.1099/ijs.0.65571-0. [DOI] [PubMed] [Google Scholar]
- 10.Iino T, Tamaki H, Tamazawa S, Ueno Y, Ohkuma M, Suzuki K, Igarashi Y, Haruta S. 2013. Candidatus Methanogranum caenicola: a novel methanogen from the anaerobic digested sludge, and proposal of Methanomassiliicoccaceae fam. nov. and Methanomassiliicoccales ord. nov., for a methanogenic lineage of the class Thermoplasmata. Microbes Environ 28:244–250. doi: 10.1264/jsme2.me12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sorokin DY, Merkel AY, Abbas B, Makarova KS, Rijpstra WIC, Koenen M, Sinninghe Damste JS, Galinski EA, Koonin EV, van Loosdrecht M. 2018. Methanonatronarchaeum thermophilum gen. nov., sp. nov. and ‘Candidatus Methanohalarchaeum thermophilum’, extremely halo(natrono)philic methyl-reducing methanogens from hypersaline lakes comprising a new euryarchaeal class Methanonatronarchaeia classis nov. Int J Syst Evol Microbiol 68:2199–2208. doi: 10.1099/ijsem.0.002810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans PN, Parks DH, Chadwick GL, Robbins SJ, Orphan VJ, Golding SD, Tyson GW. 2015. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 350:434–438. doi: 10.1126/science.aac7745. [DOI] [PubMed] [Google Scholar]
- 13.Nobu MK, Narihiro T, Kuroda K, Mei R, Liu WT. 2016. Chasing the elusive Euryarchaeota class WSA2: genomes reveal a uniquely fastidious methyl-reducing methanogen. ISME J 10:2478–2487. doi: 10.1038/ismej.2016.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vanwonterghem I, Evans PN, Parks DH, Jensen PD, Woodcroft BJ, Hugenholtz P, Tyson GW. 2016. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat Microbiol 1:16170. doi: 10.1038/nmicrobiol.2016.170. [DOI] [PubMed] [Google Scholar]
- 15.Borrel G, Adam PS, Gribaldo S. 2016. Methanogenesis and the Wood-Ljungdahl pathway: an ancient, versatile, and fragile association. Genome Biol Evol 8:1706–1711. doi: 10.1093/gbe/evw114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evans PN, Boyd JA, Leu AO, Woodcroft BJ, Parks DH, Hugenholtz P, Tyson GW. 2019. An evolving view of methane metabolism in the Archaea. Nat Rev Microbiol 17:219. doi: 10.1038/s41579-018-0136-7. [DOI] [PubMed] [Google Scholar]
- 17.Lyu Z, Liu Y. 2018. Diversity and taxonomy of methanogens. In Stams A, Sousa D (ed), Biogenesis of hydrocarbons. Handbook of hydrocarbon and lipid microbiology Springer, Cham, Switzerland. doi: 10.1007/978-3-319-53114-4_5-2. [DOI] [Google Scholar]
- 18.Miller TL, Wolin MJ. 1985. Methanosphaera stadtmaniae gen. nov., sp. nov.: a species that forms methane by reducing methanol with hydrogen. Arch Microbiol 141:116–122. doi: 10.1007/BF00423270. [DOI] [PubMed] [Google Scholar]
- 19.Fricke WF, Seedorf H, Henne A, Kruer M, Liesegang H, Hedderich R, Gottschalk G, Thauer RK. 2006. The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. J Bacteriol 188:642–658. doi: 10.1128/JB.188.2.642-658.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones WJ, Leigh JA, Mayer F, Woese CR, Wolfe RS. 1983. Methanococcus jannaschii sp. nov., an extremely thermophilic methanogen from a submarine hydrothermal vent. Arch Microbiol 136:254–261. doi: 10.1007/BF00425213. [DOI] [Google Scholar]
- 21.Miller JF, Shah NN, Nelson CM, Ludlow JM, Clark DS. 1988. Pressure and temperature effects on growth and methane production of the extreme thermophile Methanococcus jannaschii. Appl Environ Microbiol 54:3039–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imachi H, Sakai S, Nagai H, Yamaguchi T, Takai K. 2009. Methanofollis ethanolicus sp. nov., an ethanol-utilizing methanogen isolated from a lotus field. Int J Syst Evol Microbiol 59:800–805. doi: 10.1099/ijs.0.003731-0. [DOI] [PubMed] [Google Scholar]
- 23.Widdel F, Rouvière PE, Wolfe RS. 1988. Classification of secondary alcohol-utilizing methanogens including a new thermophilic isolate. Arch Microbiol 150:477–481. doi: 10.1007/BF00422290. [DOI] [Google Scholar]
- 24.Kurr M, Huber R, König H, Jannasch HW, Fricke H, Trincone A, Kristjansson JK, Stetter KO. 1991. Methanopyrus kandleri, gen. and sp. nov. represents a novel group of hyperthermophilic methanogens, growing at 110°C. Arch Microbiol 156:239–247. doi: 10.1007/BF00262992. [DOI] [Google Scholar]
- 25.Takai K, Nakamura K, Toki T, Tsunogai U, Miyazaki M, Miyazaki J, Hirayama H, Nakagawa S, Nunoura T, Horikoshi K. 2008. Cell proliferation at 122 degrees C and isotopically heavy CH4 production by a hyperthermophilic methanogen under high-pressure cultivation. Proc Natl Acad Sci U S A 105:10949–10954. doi: 10.1073/pnas.0712334105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dridi B, Fardeau ML, Ollivier B, Raoult D, Drancourt M. 2012. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int J Syst Evol Microbiol 62:1902–1907. doi: 10.1099/ijs.0.033712-0. [DOI] [PubMed] [Google Scholar]
- 27.Borrel G, Harris HMB, Tottey W, Mihajlovski A, Parisot N, Peyretaillade E, Peyret P, Gribaldo S, O’Toole PW, Brugère J-F. 2012. Genome sequence of “Candidatus Methanomethylophilus alvus” Mx1201, a methanogenic archaeon from the human gut belonging to a seventh order of methanogens. J Bacteriol 194:6944–6945. doi: 10.1128/JB.01867-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borrel G, Harris HM, Parisot N, Gaci N, Tottey W, Mihajlovski A, Deane J, Gribaldo S, Bardot O, Peyretaillade E, Peyret P, O’Toole PW, Brugere JF. 2013. Genome sequence of “Candidatus Methanomassiliicoccus intestinalis” Issoire-Mx1, a third Thermoplasmatales-related methanogenic archaeon from human feces. Genome Announc 1:e00453-13. doi: 10.1128/genomeA.00453-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kröninger L, Gottschling J, Deppenmeier U. 2017. Growth characteristics of Methanomassiliicoccus luminyensis and expression of methyltransferase encoding genes. Archaea 2017:2756573. doi: 10.1155/2017/2756573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakai S, Takaki Y, Shimamura S, Sekine M, Tajima T, Kosugi H, Ichikawa N, Tasumi E, Hiraki AT, Shimizu A, Kato Y, Nishiko R, Mori K, Fujita N, Imachi H, Takai K. 2011. Genome sequence of a mesophilic hydrogenotrophic methanogen Methanocella paludicola, the first cultivated representative of the order Methanocellales. PLoS One 6:e22898. doi: 10.1371/journal.pone.0022898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mayumi D, Mochimaru H, Tamaki H, Yamamoto K, Yoshioka H, Suzuki Y, Kamagata Y, Sakata S. 2016. Methane production from coal by a single methanogen. Science 354:222–225. doi: 10.1126/science.aaf8821. [DOI] [PubMed] [Google Scholar]
- 32.Berghuis BA, Yu FB, Schulz F, Blainey PC, Woyke T, Quake SR. 2019. Hydrogenotrophic methanogenesis in archaeal phylum Verstraetearchaeota reveals the shared ancestry of all methanogens. Proc Natl Acad Sci U S A 116:5037–5044. doi: 10.1073/pnas.1815631116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guss AM, Mukhopadhyay B, Zhang JK, Metcalf WW. 2005. Genetic analysis of mch mutants in two Methanosarcina species demonstrates multiple roles for the methanopterin-dependent C-1 oxidation/reduction pathway and differences in H2 metabolism between closely related species. Mol Microbiol 55:1671–1680. doi: 10.1111/j.1365-2958.2005.04514.x. [DOI] [PubMed] [Google Scholar]
- 34.de Poorter LM, Geerts WG, Theuvenet AP, Keltjens JT. 2003. Bioenergetics of the formyl-methanofuran dehydrogenase and heterodisulfide reductase reactions in Methanothermobacter thermautotrophicus. Eur J Biochem 270:66–75. doi: 10.1046/j.1432-1033.2003.03362.x. [DOI] [PubMed] [Google Scholar]
- 35.Bertram PA, Karrasch M, Schmitz RA, Bocher R, Albracht SP, Thauer RK. 1994. Formylmethanofuran dehydrogenases from methanogenic Archaea. Substrate specificity, EPR properties and reversible inactivation by cyanide of the molybdenum or tungsten iron-sulfur proteins. Eur J Biochem 220:477–484. doi: 10.1111/j.1432-1033.1994.tb18646.x. [DOI] [PubMed] [Google Scholar]
- 36.Bertram PA, Thauer RK. 1994. Thermodynamics of the formylmethanofuran dehydrogenase reaction in Methanobacterium thermoautotrophicum. Eur J Biochem 226:811–818. doi: 10.1111/j.1432-1033.1994.t01-1-00811.x. [DOI] [PubMed] [Google Scholar]
- 37.Meuer J, Kuettner HC, Zhang JK, Hedderich R, Metcalf WW. 2002. Genetic analysis of the archaeon Methanosarcina barkeri Fusaro reveals a central role for Ech hydrogenase and ferredoxin in methanogenesis and carbon fixation. Proc Natl Acad Sci U S A 99:5632–5637. doi: 10.1073/pnas.072615499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hedderich R. 2004. Energy-converting [NiFe] hydrogenases from archaea and extremophiles: ancestors of complex I. J Bioenerg Biomembr 36:65–75. doi: 10.1023/B:JOBB.0000019599.43969.33. [DOI] [PubMed] [Google Scholar]
- 39.Wagner T, Watanabe T, Shima S. 2018. Hydrogenotrophic methanogenesis. In Stams A, Sousa D (ed), Biogenesis of hydrocarbons. Handbook of hydrocarbon and lipid microbiology Springer, Cham, Switzerland. doi: 10.1007/978-3-319-53114-4_3-1. [DOI] [Google Scholar]
- 40.Kunow J, Shima S, Vorholt JA, Thauer RK. 1996. Primary structure and properties of the formyltransferase from the mesophilic Methanosarcina barkeri: comparison with the enzymes from thermophilic and hyperthermophilic methanogens. Arch Microbiol 165:97–105. doi: 10.1007/s002030050303. [DOI] [PubMed] [Google Scholar]
- 41.Te Brommelstroet BW, Hensgens CM, Geerts WJ, Keltjens JT, van der Drift C, Vogels GD. 1990. Purification and properties of 5,10-methenyltetrahydromethanopterin cyclohydrolase from Methanosarcina barkeri. J Bacteriol 172:564–571. doi: 10.1128/jb.172.2.564-571.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kulkarni G, Kridelbaugh DM, Guss AM, Metcalf WW. 2009. Hydrogen is a preferred intermediate in the energy-conserving electron transport chain of Methanosarcina barkeri. Proc Natl Acad Sci U S A 106:15915–15920. doi: 10.1073/pnas.0905914106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Te Brommelstroet BW, Hensgens CM, Keltjens JT, van der Drift C, Vogels GD. 1990. Purification and properties of 5,10-methylenetetrahydromethanopterin reductase, a coenzyme F420-dependent enzyme, from Methanobacterium thermoautotrophicum strain delta H. J Biol Chem 265:1852–1857. [PubMed] [Google Scholar]
- 44.te Brommelstroet BW, Hensgens CM, Keltjens JT, van der Drift C, Vogels GD. 1991. Purification and characterization of coenzyme F420-dependent 5,10-methylenetetrahydromethanopterin dehydrogenase from Methanobacterium thermoautotrophicum strain delta H. Biochim Biophys Acta 1073:77–84. doi: 10.1016/0304-4165(91)90185-j. [DOI] [PubMed] [Google Scholar]
- 45.Gottschalk G, Thauer RK. 2001. The Na+-translocating methyltransferase complex from methanogenic archaea. Biochim Biophys Acta 1505:28–36. doi: 10.1016/s0005-2728(00)00274-7. [DOI] [PubMed] [Google Scholar]
- 46.Thauer RK. 1998. Biochemistry of methanogenesis: a tribute to Marjory Stephenson. Microbiology 144:2377–2406. doi: 10.1099/00221287-144-9-2377. [DOI] [PubMed] [Google Scholar]
- 47.Wongnate T, Ragsdale SW. 2015. The reaction mechanism of methyl-coenzyme M reductase: how an enzyme enforces strict binding order. J Biol Chem 290:9322–9334. doi: 10.1074/jbc.M115.636761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deppenmeier U. 2004. The membrane-bound electron transport system of Methanosarcina species. J Bioenerg Biomembr 36:55–64. doi: 10.1023/B:JOBB.0000019598.64642.97. [DOI] [PubMed] [Google Scholar]
- 49.Ferry JG. 1997. Enzymology of the fermentation of acetate to methane by Methanosarcina thermophila. Biofactors 6:25–35. doi: 10.1002/biof.5520060104. [DOI] [PubMed] [Google Scholar]
- 50.Ferry JG. 1992. Methane from acetate. J Bacteriol 174:5489–5495. doi: 10.1128/jb.174.17.5489-5495.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hattori S. 2008. Syntrophic acetate-oxidizing microbes in methanogenic environments. Microbes Environ 23:118–127. doi: 10.1264/jsme2.23.118. [DOI] [PubMed] [Google Scholar]
- 52.Thauer RK, Jungermann K, Decker K. 1977. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol Rev 41:100–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zinder SH, Koch M. 1984. Non-aceticlastic methanogenesis from acetate: acetate oxidation by a thermophilic syntrophic coculture. Arch Microbiol 138:263–272. doi: 10.1007/BF00402133. [DOI] [Google Scholar]
- 54.Balk M, Weijma J, Stams AJ. 2002. Thermotoga lettingae sp. nov., a novel thermophilic, methanol-degrading bacterium isolated from a thermophilic anaerobic reactor. Int J Syst Evol Microbiol 52:1361–1368. doi: 10.1099/00207713-52-4-1361. [DOI] [PubMed] [Google Scholar]
- 55.Hattori S, Kamagata Y, Hanada S, Shoun H. 2000. Thermacetogenium phaeum gen. nov., sp. nov., a strictly anaerobic, thermophilic, syntrophic acetate-oxidizing bacterium. Int J Syst Evol Microbiol 50:1601–1609. doi: 10.1099/00207713-50-4-1601. [DOI] [PubMed] [Google Scholar]
- 56.Schnürer A, Schink B, Svensson BH. 1996. Clostridium ultunense sp. nov., a mesophilic bacterium oxidizing acetate in syntrophic association with a hydrogenotrophic methanogenic bacterium. Int J Syst Bacteriol 46:1145–1152. doi: 10.1099/00207713-46-4-1145. [DOI] [PubMed] [Google Scholar]
- 57.Garrity GM, Labeda DP, Oren A. 2011. Judicial Commission of the International Committee on Systematics of Prokaryotes XIIth International (IUMS) Congress of Bacteriology and Applied Microbiology. Int J Syst Evol Microbiol 61:2775–2780. doi: 10.1099/ijs.0.037366-0. [DOI] [Google Scholar]
- 58.Schlegel K, Müller V. 2013. Evolution of Na+ and H+ bioenergetics in methanogenic archaea. Biochem Soc Trans 41:421–426. doi: 10.1042/BST20120294. [DOI] [PubMed] [Google Scholar]
- 59.Deppenmeier U. 2002. The unique biochemistry of methanogenesis. Prog Nucleic Acids Res Mol Biol 71:223–283. doi: 10.1016/S0079-6603(02)71045-3. [DOI] [PubMed] [Google Scholar]
- 60.Welte C, Deppenmeier U. 2014. Bioenergetics and anaerobic respiratory chains of aceticlastic methanogens. Biochim Biophys Acta 1837:1130–1147. doi: 10.1016/j.bbabio.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 61.Grahame DA. 1991. Catalysis of acetyl-CoA cleavage and tetrahydrosarcinapterin methylation by a carbon monoxide dehydrogenase-corrinoid enzyme complex. J Biol Chem 266:22227–22233. [PubMed] [Google Scholar]
- 62.Gencic S, Duin EC, Grahame DA. 2010. Tight coupling of partial reactions in the acetyl-CoA decarbonylase/synthase (ACDS) multienzyme complex from Methanosarcina thermophila: acetyl C-C bond fragmentation at the A cluster promoted by protein conformational changes. J Biol Chem 285:15450–15463. doi: 10.1074/jbc.M109.080994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Welte C, Krätzer C, Deppenmeier U. 2010. Involvement of Ech hydrogenase in energy conservation of Methanosarcina mazei. FEBS J 277:3396–3403. doi: 10.1111/j.1742-4658.2010.07744.x. [DOI] [PubMed] [Google Scholar]
- 64.Meuer J, Bartoschek S, Koch J, Künkel A, Hedderich R. 1999. Purification and catalytic properties of Ech hydrogenase from Methanosarcina barkeri. Eur J Biochem 265:325–335. doi: 10.1046/j.1432-1327.1999.00738.x. [DOI] [PubMed] [Google Scholar]
- 65.Kulkarni G, Mand TD, Metcalf WW. 2018. Energy conservation via hydrogen cycling in the methanogenic archaeon Methanosarcina barkeri. mBio 9:e01256-18. doi: 10.1128/mBio.01256-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang M, Tomb JF, Ferry JG. 2011. Electron transport in acetate-grown Methanosarcina acetivorans. BMC Microbiol 11:165. doi: 10.1186/1471-2180-11-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Q, Li L, Rejtar T, Lessner DJ, Karger BL, Ferry JG. 2006. Electron transport in the pathway of acetate conversion to methane in the marine archaeon Methanosarcina acetivorans. J Bacteriol 188:702–710. doi: 10.1128/JB.188.2.702-710.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schlegel K, Welte C, Deppenmeier U, Müller V. 2012. Electron transport during aceticlastic methanogenesis by Methanosarcina acetivorans involves a sodium-translocating Rnf complex. FEBS J 279:4444–4452. doi: 10.1111/febs.12031. [DOI] [PubMed] [Google Scholar]
- 69.Sowers KR, Nelson MJ, Ferry JG. 1984. Growth of acetotrophic, methane-producing bacteria in a pH auxostat. Curr Microbiol 11:227–229. doi: 10.1007/BF01567165. [DOI] [Google Scholar]
- 70.Deppenmeier U, Müller V. 2007. Life close to the thermodynamic limit: How methanogenic Archaea conserve energy. Results Probl Cell Differ 45:123–152. [DOI] [PubMed] [Google Scholar]
- 71.Bose A, Pritchett MA, Metcalf WW. 2008. Genetic analysis of the methanol- and methylamine-specific methyltransferase 2 genes of Methanosarcina acetivorans C2A. J Bacteriol 190:4017–4026. doi: 10.1128/JB.00117-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Keltjens JT, Vogels GD. 1993. Conversion of methanol and methylamines to methane and carbon dioxide, p 253–303. In Ferry JG. (ed), Methanogenesis: ecology, physiology, biochemistry & genetics. Springer US, Boston, MA. doi: 10.1007/978-1-4615-2391-8_6. [DOI] [Google Scholar]
- 73.Guss AM, Kulkarni G, Metcalf WW. 2009. Differences in hydrogenase gene expression between Methanosarcina acetivorans and Methanosarcina barkeri. J Bacteriol 191:2826–2833. doi: 10.1128/JB.00563-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ide T, Bäumer S, Deppenmeier U. 1999. Energy conservation by the H2:heterodisulfide oxidoreductase from Methanosarcina mazei Gö1: identification of two proton-translocating segments. J Bacteriol 181:4076–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bäumer S, Ide T, Jacobi C, Johann A, Gottschalk G, Deppenmeier U. 2000. The F420H2 dehydrogenase from Methanosarcina mazei is a redox-driven proton pump closely related to NADH dehydrogenases. J Biol Chem 275:17968–17973. doi: 10.1074/jbc.M000650200. [DOI] [PubMed] [Google Scholar]
- 76.Blaut M, Gottschalk G. 1984. Coupling of ATP synthesis and methane formation from methanol and molecular hydrogen in Methanosarcina barkeri. Eur J Biochem 141:217–222. doi: 10.1111/j.1432-1033.1984.tb08178.x. [DOI] [PubMed] [Google Scholar]
- 77.Hoedt EC, Cuív PÓ, Evans PN, Smith WJ, McSweeney CS, Denman SE, Morrison M. 2016. Differences down-under: alcohol-fueled methanogenesis by archaea present in Australian macropodids. ISME J 10:2376–2388. doi: 10.1038/ismej.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Perski H-J, Moll J, Thauer RK. 1981. Sodium dependence of growth and methane formation in Methanobacterium thermoautotrophicum. Arch Microbiol 130:319–321. doi: 10.1007/BF00425947. [DOI] [Google Scholar]
- 79.Perski HJ, Schönheit P, Thauer RK. 1982. Sodium dependence of methane formation in methanogenic bacteria. FEBS Lett 143:323–326. doi: 10.1016/0014-5793(82)80126-9. [DOI] [Google Scholar]
- 80.Becher B, Müller V, Gottschalk G. 1992. N5-methyl-tetrahydromethanopterin:coenzyme M methyltransferase of Methanosarcina strain Gö1 is an Na+-translocating membrane protein. J Bacteriol 174:7656–7660. doi: 10.1128/jb.174.23.7656-7660.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Upadhyay V, Ceh K, Tumulka F, Abele R, Hoffmann J, Langer J, Shima S, Ermler U. 2016. Molecular characterization of methanogenic N5-methyl-tetrahydromethanopterin: coenzyme M methyltransferase. Biochim Biophys Acta 1858:2140–2144. doi: 10.1016/j.bbamem.2016.06.011. [DOI] [PubMed] [Google Scholar]
- 82.Müller V, Blaut M, Gottschalk G. 1988. The transmembrane electrochemical gradient of Na+ as driving force for methanol oxidation in Methanosarcina barkeri. Eur J Biochem 172:601–606. doi: 10.1111/j.1432-1033.1988.tb13931.x. [DOI] [PubMed] [Google Scholar]
- 83.Buckel W, Thauer RK. 2013. Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochim Biophys Acta 1827:94–113. doi: 10.1016/j.bbabio.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 84.Kaster AK, Moll J, Parey K, Thauer RK. 2011. Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proc Natl Acad Sci U S A 108:2981–2986. doi: 10.1073/pnas.1016761108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stojanowic A, Mander GJ, Duin EC, Hedderich R. 2003. Physiological role of the F420-non-reducing hydrogenase (Mvh) from Methanothermobacter marburgensis. Arch Microbiol 180:194–203. doi: 10.1007/s00203-003-0577-9. [DOI] [PubMed] [Google Scholar]
- 86.Hedderich R, Thauer RK. 1988. Methanobacterium thermoautotrophicum contains a soluble enzyme system that specifically catalyzes the reduction of the heterodisulfide of coenzyme M and 7-mercaptoheptanoylthreonine phosphate with H2. FEBS Lett 234:223–227. doi: 10.1016/0014-5793(88)81339-5. [DOI] [Google Scholar]
- 87.Buan NR, Metcalf WW. 2010. Methanogenesis by Methanosarcina acetivorans involves two structurally and functionally distinct classes of heterodisulfide reductase. Mol Microbiol 75:843–853. doi: 10.1111/j.1365-2958.2009.06990.x. [DOI] [PubMed] [Google Scholar]
- 88.Costa KC, Wong PM, Wang T, Lie TJ, Dodsworth JA, Swanson I, Burn JA, Hackett M, Leigh JA. 2010. Protein complexing in a methanogen suggests electron bifurcation and electron delivery from formate to heterodisulfide reductase. Proc Natl Acad Sci U S A 107:11050–11055. doi: 10.1073/pnas.1003653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Milton RD, Ruth JC, Deutzmann J, Spormann AM. 2018. Methanococcus maripaludis employs three functional heterodisulfide reductase complexes for flavin-based electron bifurcation using hydrogen and formate. Biochemistry 57:4848. doi: 10.1021/acs.biochem.8b00662. [DOI] [PubMed] [Google Scholar]
- 90.Rouvière PE, Wolfe RS. 1988. Novel biochemistry of methanogenesis. J Biol Chem 263:7913–7916. [PubMed] [Google Scholar]
- 91.Lie TJ, Costa KC, Lupa B, Korpole S, Whitman WB, Leigh JA. 2012. Essential anaplerotic role for the energy-converting hydrogenase Eha in hydrogenotrophic methanogenesis. Proc Natl Acad Sci U S A 109:15473–15478. doi: 10.1073/pnas.1208779109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Thauer RK. 2012. The Wolfe cycle comes full circle. Proc Natl Acad Sci U S A 109:15084–15085. doi: 10.1073/pnas.1213193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yan Z, Wang M, Ferry JG. 2017. A ferredoxin- and F420H2-dependent, electron-bifurcating, heterodisulfide reductase with homologs in the domains Bacteria and Archaea. mBio 8:e02285-16. doi: 10.1128/mBio.02285-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Buckel W, Thauer RK. 2018. Flavin-based electron bifurcation, ferredoxin, flavodoxin, and anaerobic respiration with protons (Ech) or NAD+ (Rnf) as electron acceptors: a historical review. Front Microbiol 9:401. doi: 10.3389/fmicb.2018.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Müller V, Lemker T, Lingl A, Weidner C, Coskun Ü, Grüber G. 2005. Bioenergetics of archaea: ATP synthesis under harsh environmental conditions. J Mol Microbiol Biotechnol 10:167–180. doi: 10.1159/000091563. [DOI] [PubMed] [Google Scholar]
- 96.Schlegel K, Leone V, Faraldo-Gómez JD, Müller V. 2012. Promiscuous archaeal ATP synthase concurrently coupled to Na+ and H+ translocation. Proc Natl Acad Sci U S A 109:947–952. doi: 10.1073/pnas.1115796109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jasso-Chavez R, Apolinario EE, Sowers KR, Ferry JG. 2013. MrpA functions in energy conversion during acetate-dependent growth of Methanosarcina acetivorans. J Bacteriol 195:3987–3994. doi: 10.1128/JB.00581-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tietze M, Beuchle A, Lamla I, Orth N, Dehler M, Greiner G, Beifuss U. 2003. Redox potentials of methanophenazine and CoB-S-S-CoM, factors involved in electron transport in Methanogenic archaea. Chembiochem 4:333–335. doi: 10.1002/cbic.200390053. [DOI] [PubMed] [Google Scholar]
- 99.Deppenmeier U, Blaut M, Mahlmann A, Gottschalk G. 1990. Reduced coenzyme F420: heterodisulfide oxidoreductase, a proton-translocating redox system in methanogenic bacteria. Proc Natl Acad Sci U S A 87:9449–9453. doi: 10.1073/pnas.87.23.9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Welte C, Deppenmeier U. 2011. Re-evaluation of the function of the F420 dehydrogenase in electron transport of Methanosarcina mazei. FEBS J 278:1277–1287. doi: 10.1111/j.1742-4658.2011.08048.x. [DOI] [PubMed] [Google Scholar]
- 101.Bäumer S, Murakami E, Brodersen J, Gottschalk G, Ragsdale SW, Deppenmeier U. 1998. The F420H2:heterodisulfide oxidoreductase system from Methanosarcina species. 2-Hydroxyphenazine mediates electron transfer from F420H2 dehydrogenase to heterodisulfide reductase. FEBS Lett 428:295–298. doi: 10.1016/s0014-5793(98)00555-9. [DOI] [PubMed] [Google Scholar]
- 102.Beifuss U, Tietze M, Bäumer S, Deppenmeier U. 2000. Methanophenazine: structure, total synthesis, and function of a new cofactor from methanogenic Archaea. Angew Chem Int Ed Engl 39:2470–2472. doi:. [DOI] [PubMed] [Google Scholar]
- 103.Greening C, Ahmed FH, Mohamed AE, Lee BM, Pandey G, Warden AC, Scott C, Oakeshott JG, Taylor MC, Jackson CJ. 2016. Physiology, biochemistry, and applications of F420- and Fo-dependent redox reactions. Microbiol Mol Biol Rev 80:451–493. doi: 10.1128/MMBR.00070-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Simianu M, Murakami E, Brewer JM, Ragsdale SW. 1998. Purification and properties of the heme- and iron-sulfur-containing heterodisulfide reductase from Methanosarcina thermophila. Biochemistry 37:10027–10039. doi: 10.1021/bi9726483. [DOI] [PubMed] [Google Scholar]
- 105.Künkel A, Vaupel M, Heim S, Thauer RK, Hedderich R. 1997. Heterodisulfide reductase from methanol-grown cells of Methanosarcina barkeri is not a flavoenzyme. Eur J Biochem 244:226–234. doi: 10.1111/j.1432-1033.1997.00226.x. [DOI] [PubMed] [Google Scholar]
- 106.Hedderich R, Hamann N, Bennati M. 2005. Heterodisulfide reductase from methanogenic archaea: a new catalytic role for an iron-sulfur cluster. Biol Chem 386:961–970. doi: 10.1515/BC.2005.112. [DOI] [PubMed] [Google Scholar]
- 107.Suharti S, Wang M, de Vries S, Ferry JG. 2014. Characterization of the RnfB and RnfG subunits of the Rnf complex from the archaeon Methanosarcina acetivorans. PLoS One 9:e97966. doi: 10.1371/journal.pone.0097966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Benedict MN, Gonnerman MC, Metcalf WW, Price ND. 2012. Genome-scale metabolic reconstruction and hypothesis testing in the methanogenic archaeon Methanosarcina acetivorans C2A. J Bacteriol 194:855–865. doi: 10.1128/JB.06040-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Biegel E, Schmidt S, González JM, Müller V. 2011. Biochemistry, evolution and physiological function of the Rnf complex, a novel ion-motive electron transport complex in prokaryotes. Cell Mol Life Sci 68:613–634. doi: 10.1007/s00018-010-0555-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Deppenmeier U, Lienard T, Gottschalk G. 1999. Novel reactions involved in energy conservation by methanogenic archaea. FEBS Lett 457:291–297. doi: 10.1016/s0014-5793(99)01026-1. [DOI] [PubMed] [Google Scholar]
- 111.Mand TD, Kulkarni G, Metcalf WW. 2018. Genetic, biochemical, and molecular characterization of Methanosarcina barkeri mutants lacking three distinct classes of hydrogenase. J Bacteriol 200:e00342-18. doi: 10.1128/JB.00342-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Peters JW, Schut GJ, Boyd ES, Mulder DW, Shepard EM, Broderick JB, King PW, Adams M. 2015. [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim Biophys Acta 1853:1350–1369. doi: 10.1016/j.bbamcr.2014.11.021. [DOI] [PubMed] [Google Scholar]
- 113.Vignais PM, Billoud B. 2007. Occurrence, classification, and biological function of hydrogenases: an overview. Chem Rev 107:4206–4272. doi: 10.1021/cr050196r. [DOI] [PubMed] [Google Scholar]
- 114.Zirngibl C, Hedderich R, Thauer RK. 1990. N5,N10-Methylenetetrahydromethanopterin dehydrogenase from Methanobacterium thermoautotrophicum has hydrogenase activity. FEBS Lett 261:112–116. doi: 10.1016/0014-5793(90)80649-4. [DOI] [Google Scholar]
- 115.Shima S, Pilak O, Vogt S, Schick M, Stagni MS, Meyer-Klaucke W, Warkentin E, Thauer RK, Ermler U. 2008. The crystal structure of [Fe]-hydrogenase reveals the geometry of the active site. Science 321:572–575. doi: 10.1126/science.1158978. [DOI] [PubMed] [Google Scholar]
- 116.Afting C, Hochheimer A, Thauer RK. 1998. Function of H2-forming methylenetetrahydromethanopterin dehydrogenase from Methanobacterium thermoautotrophicum in coenzyme F420 reduction with H2. Arch Microbiol 169:206–210. doi: 10.1007/s002030050562. [DOI] [PubMed] [Google Scholar]
- 117.Afting C, Kremmer E, Brucker C, Hochheimer A, Thauer RK. 2000. Regulation of the synthesis of H2-forming methylenetetrahydromethanopterin dehydrogenase (Hmd) and of HmdII and HmdIII in Methanothermobacter marburgensis. Arch Microbiol 174:225–232. doi: 10.1007/s002030000197. [DOI] [PubMed] [Google Scholar]
- 118.Vignais PM, Billoud B, Meyer J. 2001. Classification and phylogeny of hydrogenases. FEMS Microbiol Rev 25:455–501. doi: 10.1111/j.1574-6976.2001.tb00587.x. [DOI] [PubMed] [Google Scholar]
- 119.Hedderich R, Forzi L. 2005. Energy-converting [NiFe] hydrogenases: more than just H2 activation. J Mol Microbiol Biotechnol 10:92–104. doi: 10.1159/000091557. [DOI] [PubMed] [Google Scholar]
- 120.Künkel A, Vorholt JA, Thauer RK, Hedderich R. 1998. An Escherichia coli hydrogenase-3-type hydrogenase in methanogenic archaea. Eur J Biochem 252:467–476. doi: 10.1046/j.1432-1327.1998.2520467.x. [DOI] [PubMed] [Google Scholar]
- 121.Bott M, Thauer RK. 1987. Proton-motive-force-driven formation of CO from CO2 and H2 in methanogenic bacteria. Eur J Biochem 168:407–412. doi: 10.1111/j.1432-1033.1987.tb13434.x. [DOI] [PubMed] [Google Scholar]
- 122.Bott M, Eikmanns B, Thauer RK. 1986. Coupling of carbon monoxide oxidation to CO2 and H2 with the phosphorylation of ADP in acetate-grown Methanosarcina barkeri. Eur J Biochem 159:393–398. doi: 10.1111/j.1432-1033.1986.tb09881.x. [DOI] [PubMed] [Google Scholar]
- 123.Bott M, Thauer RK. 1989. Proton translocation coupled to the oxidation of carbon monoxide to CO2 and H2 in Methanosarcina barkeri. Eur J Biochem 179:469–472. doi: 10.1111/j.1432-1033.1989.tb14576.x. [DOI] [PubMed] [Google Scholar]
- 124.Forzi L, Koch J, Guss AM, Radosevich CG, Metcalf WW, Hedderich R. 2005. Assignment of the [4Fe-4S] clusters of Ech hydrogenase from Methanosarcina barkeri to individual subunits via the characterization of site-directed mutants. FEBS J 272:4741–4753. doi: 10.1111/j.1742-4658.2005.04889.x. [DOI] [PubMed] [Google Scholar]
- 125.Jacobson FS, Daniels L, Fox JA, Walsh CT, Orme-Johnson WH. 1982. Purification and properties of an 8-hydroxy-5-deazaflavin-reducing hydrogenase from Methanobacterium thermoautotrophicum. J Biol Chem 257:3385–3388. [PubMed] [Google Scholar]
- 126.Fox JA, Livingston DJ, Orme-Johnson WH, Walsh CT. 1987. 8-Hydroxy-5-deazaflavin-reducing hydrogenase from Methanobacterium thermoautotrophicum: 1. Purification and characterization. Biochemistry 26:4219–4227. doi: 10.1021/bi00388a007. [DOI] [PubMed] [Google Scholar]
- 127.Muth E, Morschel E, Klein A. 1987. Purification and characterization of an 8-hydroxy-5-deazaflavin-reducing hydrogenase from the archaebacterium Methanococcus voltae. Eur J Biochem 169:571–577. doi: 10.1111/j.1432-1033.1987.tb13647.x. [DOI] [PubMed] [Google Scholar]
- 128.Baron SF, Ferry JG. 1989. Purification and properties of the membrane-associated coenzyme F420-reducing hydrogenase from Methanobacterium formicicum. J Bacteriol 171:3846–3853. doi: 10.1128/jb.171.7.3846-3853.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fiebig K, Friedrich B. 1989. Purification of the F420-reducing hydrogenase from Methanosarcina barkeri (strain Fusaro). Eur J Biochem 184:79–88. doi: 10.1111/j.1432-1033.1989.tb14992.x. [DOI] [PubMed] [Google Scholar]
- 130.de Poorter LM, Geerts WJ, Keltjens JT. 2005. Hydrogen concentrations in methane-forming cells probed by the ratios of reduced and oxidized coenzyme F420. Microbiology 151:1697–1705. doi: 10.1099/mic.0.27679-0. [DOI] [PubMed] [Google Scholar]
- 131.Alex LA, Reeve JN, Orme-Johnson WH, Walsh CT. 1990. Cloning, sequence determination, and expression of the genes encoding the subunits of the nickel-containing 8-hydroxy-5-deazaflavin reducing hydrogenase from Methanobacterium thermoautotrophicum delta H. Biochemistry 29:7237–7244. doi: 10.1021/bi00483a011. [DOI] [PubMed] [Google Scholar]
- 132.Lünsdorf H, Niedrig M, Fiebig K. 1991. Immunocytochemical localization of the coenzyme F420-reducing hydrogenase in Methanosarcina barkeri Fusaro. J Bacteriol 173:978–984. doi: 10.1128/jb.173.3.978-984.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mills DJ, Vitt S, Strauss M, Shima S, Vonck J. 2013. De novo modeling of the F420-reducing [NiFe]-hydrogenase from a methanogenic archaeon by cryo-electron microscopy. Elife 2:e00218. doi: 10.7554/eLife.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kemner JM, Zeikus JG. 1994. Purification and characterization of membrane-bound hydrogenase from Methanosarcina barkeri MS. Arch Microbiol 161:47–54. doi: 10.1007/BF00248892. [DOI] [Google Scholar]
- 135.Deppenmeier U, Blaut M, Schmidt B, Gottschalk G. 1992. Purification and properties of a F420-nonreactive, membrane-bound hydrogenase from Methanosarcina strain Gö1. Arch Microbiol 157:505–511. [DOI] [PubMed] [Google Scholar]
- 136.Deppenmeier U. 1995. Different structure and expression of the operons encoding the membrane-bound hydrogenases from Methanosarcina mazei Gö1. Arch Microbiol 164:370–376. doi: 10.1007/BF02529985. [DOI] [PubMed] [Google Scholar]
- 137.Deppenmeier U, Blaut M, Lentes S, Herzberg C, Gottschalk G. 1995. Analysis of the vhoGAC and vhtGAC operons from Methanosarcina mazei strain Gö1, both encoding a membrane-bound hydrogenase and a cytochrome b. Eur J Biochem 227:261–269. doi: 10.1111/j.1432-1033.1995.tb20383.x. [DOI] [PubMed] [Google Scholar]
- 138.Wu LF, Chanal A, Rodrigue A. 2000. Membrane targeting and translocation of bacterial hydrogenases. Arch Microbiol 173:319–324. doi: 10.1007/s002030000144. [DOI] [PubMed] [Google Scholar]
- 139.Eismann K, Mlejnek K, Zipprich D, Hoppert M, Gerberding H, Mayer F. 1995. Antigenic determinants of the membrane-bound hydrogenase in Alcaligenes eutrophus are exposed toward the periplasm. J Bacteriol 177:6309–6312. doi: 10.1128/jb.177.21.6309-6312.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]