

Abstract
In response to the growing threat posed by antibiotic-resistant bacterial strains, extensive research is currently focused on developing antimicrobial agents that target lipid II, a vital precursor in the biosynthesis of bacterial cell walls. The lantibiotic nisin and related peptides display unique and highly selective binding to lipid II. A key feature of the nisin–lipid II interaction is the formation of a cage-like complex between the pyrophosphate moiety of lipid II and the two thioether-bridged rings, rings A and B, at the N-terminus of nisin. To understand the important structural factors underlying this highly selective molecular recognition, we have used solid-phase peptide synthesis to prepare individual ring A and B structures from nisin, the related lantibiotic mutacin, and synthetic analogues. Through NMR studies of these rings, we have demonstrated that ring A is preorganized to adopt the correct conformation for binding lipid II in solution and that individual amino acid substitutions in ring A have little effect on the conformation. We have also analyzed the turn structures adopted by these thioether-bridged peptides and show that they do not adopt the tight α-turn or β-turn structures typically found in proteins.
Introduction
In recent years, the alarming rise in strains of bacteria that are resistant to antibiotics1 has prompted researchers to revisit natural products as lead structures for new antimicrobial therapies with novel modes of action.2 In particular, there has been a resurgence of interest in antimicrobial peptides (AMP),3,4 in part, due to their diversity and broad spectrum of activity.
One class of AMPs that has recently been extensively explored is the lantibiotics. This class of bacteriocins is produced by, and exerts their antibacterial effect on, Gram-positive bacteria.5 The need for potent new antibiotics has revived interest in this class of peptides, particularly as they have a well-characterized activity and low minimum inhibitory concentration (MIC) (μM or nM) against a number of clinically relevant species.6 These structurally complex peptides are distinguished by the presence of one or more thioether linkages between amino acid side chains. These thioether linkages are formed via Michael addition of cysteine to dehydroalanine (Dha) or dehydrobutyrine (Dhb), giving lanthionine (Lan) and β-methyl lanthionine (MeLan), respectively, during the post-translational modification of the ribosomally synthesized prepeptide precursor. Many lantibiotics bind to lipid II,7 which is the key precursor in the biosynthesis of the peptidoglycan cell wall for both Gram-positive and Gram-negative bacteria. AMPs that bind to lipid II and disrupt either the biosynthesis of peptidoglycan or the structure of the membrane8 will be key to developing the next generation of antimicrobial therapies with high selectivity for bacterial cells. As lipid II is both essential for bacterial growth and is unique to bacteria, it will be difficult for bacteria to evolve resistance against such AMPs.
The lantibiotic nisin (Figure 1) was first isolated in 19289 and is routinely used as a food preservative.10 Despite over four decades of use, there are very few examples of naturally occurring lantibiotic resistance, which may be attributed to its unique mode of action.11 Nisin binds selectively to lipid II and exerts its antibacterial action through two mechanisms: sequestration of lipid II, resulting in prevention of cell wall biosynthesis, and rapid and efficient formation of nisin–lipid II complexes, which lead to pores in the bacterial cell membrane.12 The pore complex is composed of eight nisin and four lipid II molecules.13 Pore formation begins with the binding of the N-terminal region of nisin (rings A and B: nisin(1–12)) to the pyrophosphate group of lipid II, forming a 1:1 complex. The NMR structure (PDB ID: 1WCO)14 of a 1:1 complex of nisin bound to a truncated analogue of lipid II in DMSO elucidated details of this interaction, showing that rings A and B form a cage structure with hydrogen bonds between the backbone amides and the pyrophosphate.
Figure 1.

Structures of nisin and lipid II.
Recently, Weingarth et al. reported the solid-state NMR of nisin in a 2:1 pore complex in DOPC liposomes15 and observed that the chemical shifts of nisin under these conditions differ drastically from the nisin–lipid II 1:1 DMSO structure.14 They proposed that the membrane environment considerably alters the nisin–lipid II complex structure and that the conformation adopted by nisin is very different in a pore complex with lipid II in membranes. Computational studies have proposed that the next stage of pore formation involves the binding of a second molecule of nisin to the pentapeptide of lipid II,16 causing the tail of the lipid to extend deeper into the cell membrane.17 The C-terminal region nisin(24–34) is thought to be partially embedded within the membrane17,18 with the C-terminus itself at the inner water–membrane interface.15 The hinge region nisin(21–23) plays a crucial role in the orientation and complexation of nisin and lipid II, which are still not well understood,13,16−18 although these residues appear to line the lumen of the pore.15 NMR studies of the interactions of lipid II with two unrelated lantibiotics, lacticin4b and mersacidin,19 also reveal cage structures formed by two lanthionine/methyllanthionine rings, which complex to the pyrophosphate group of lipid II.
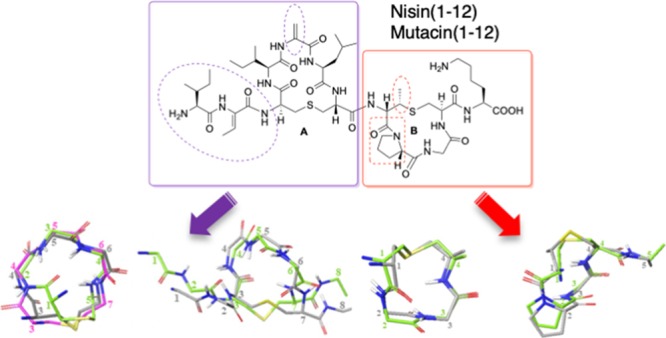
Several other naturally occurring lantibiotics20−25 bear the same N-terminal AB ring-bridging pattern as nisin (Table 1). It has been proposed that all of these form an A + B ring cage and bind the pyrophosphate group of lipid II in the same manner.7b Intriguingly, unlike the other nisin-like lantibiotics, ring B of mutacin I is also formed by a Lan residue,23c,23d and it bears a Leu rather than a Pro residue at position 9. Further differences are that, in mutacin I, the two dehydro residues are both Dha, unlike in other nisin-like peptides with two dehydro residues, which have one Dha and one Dhb. These small modifications to the A and B rings are interesting from both structural and synthetic perspectives.
Table 1. Comparison of the AB Rings of Lantibiotics from the Nisin Family.

Despite the recent resurgence of interest in lanthionine-containing peptides, the underlying conformational preferences of cyclic peptides incorporating lanthionine or methyllanthionine bridges are as yet not well understood. In addition, for those lantibiotics, which recognize and bind to the pyrophosphate group of lipid II, the effects of varying the amino acid sequences, and the presence or absence of dehydro residues, on the conformations of the individual rings A and B has also not been extensively studied. The synthetic challenges presented by the lantibiotics have meant that only a few groups have reported syntheses of ring A24,25 or ring B.25,26 Moreover, only two groups have previously studied the solution structures and conformations of these isolated rings, and analogues, by NMR.26b,27
In this paper, we have sought to further elucidate the key structural factors governing the binding of rings A and B of the nisin-type lantibiotics to the pyrophosphate group of lipid II. In particular, through a combination of synthesis of analogues and NMR studies of isolated rings, we wished to determine whether the individual rings are preorganized into the cage conformation or whether binding to lipid II induces a conformational change, as might be suggested by the presence of two ring B conformers in the isolated rings. We also wished to study further the effects of individual amino acid substitutions on ring conformation, in particular in ring A structures where a wider sequence variation is apparently tolerated, and to determine the extent to which the ring conformations depend on the unique lanthionine bridge versus the amino acid sequences within each ring.
Results and Discussion
Ring B: Synthesis of Wild-Type and Analogue Structures
The chemical synthesis of nisin and other lantibiotics poses a significant challenge due to their highly modified structure and oxidative instability. To date, only a limited number of effective approaches have been established.28 We have developed a powerful solid-phase peptide synthesis (SPPS) strategy for the synthesis of lanthionine-containing peptides.29 This is based on the incorporation of orthogonally protected lanthionine building blocks into linear peptides, followed by chemoselective deprotection, on-resin cyclization, and chain extension where required, leading to lanthionine-bridged peptides with complete control of stereochemistry at the α-positions (and β-positions) and complete regioselectivity of cyclization. This methodology is now widely used and has been exploited for the total synthesis of the lantibiotics lactocin S, lacticin 481, and lacticin 3147, as well as unnatural analogues of lacticin 3147 and epilancin 15X.30
To compare the effects on ring B conformation of the incorporation of MeLan versus Lan and also of the substitution of Pro for Leu, we synthesized three peptides: mutacin I ring B 1 (incorporating Lan8–11 and Leu9), nisin ring B 2 (incorporating MeLan8–11 and Pro9), and an analogue of nisin ring B 3 (substituting Lan8–11 but retaining Pro9) (Figure 2).
Figure 2.

Ring B structures synthesized.
For peptides 1 and 3, (Teoc, TMSE/Fmoc) Lan monomer 4 was required. This could be expediently prepared from Fmoc-Cys-OTce and Trt-β-iodoalanine-OTMSE via our previously published route;29b however, we have improved the procedure for the synthesis of the key intermediate Fmoc-Cys-OTce to allow this route to be carried out at the 0.5 g scale. To synthesize mutacin I ring B 1, a low-loading (0.18 mmol g–1) Fmoc-Thr(tBu)-Novasyn TGT resin 5 was used to avoid cross-linking between different peptides during intramolecular cyclization steps. (Teoc, TMSE/Fmoc) Lan 4 was then incorporated (Scheme 1) using a microwave coupling protocol to decrease the reaction time and ensure complete reaction.29c,31 The Gly and Leu residues were then coupled using standard Fmoc SPPS procedures, giving linear resin-bound peptide 6. The orthogonal silyl protecting groups were then removed with TBAF, and although it was expected that this would also remove the Fmoc protecting groups, the peptide was then treated with 40% piperidine to ensure complete Fmoc deprotection. The cyclization reaction to form 7 was effected by double coupling for 2 h with PyAOP, HOAt, and DIPEA, with 5 min of microwave irradiation. After completion of the synthesis, the peptide was cleaved from the resin and purified by reverse-phase HPLC to give mutacin I ring B peptide 1 in 34% yield. A similar protocol was used to prepare the Lan8,11 analogue of nisin ring B 3.
Scheme 1. SPPS of Mutacin Ring B 1.

Reagents and conditions: (i) piperidine/DMF; (ii) 4, HOAt, PyAOP, DIPEA, μwave, 60 °C, 5 min; (iii) piperidine/DMF; (iv) Fmoc-Gly-OH, HOAt, PyAOP, DIPEA, 2 h; (v) piperidine/DMF; (vi) Fmoc-Leu-OH, HOAt, PyAOP, DIPEA, 2 h; (vii) TBAF, DMF, 1 h; (viii) piperidine/DMF; (ix) HOAt, PyAOP, DIPEA, μwave, 60 °C, 5 min; (x) TFA/H2O/TIPS.
For peptide 2, (Alloc, allyl/Fmoc) MeLan monomer 8 was prepared. We used the procedure reported by Vederas et al.;30c however, in our hands, it proved expedient to use Fmoc-Cys-OTce in the key aziridine ring-opening reaction (Scheme S1).
This was attached to a low-loading (0.18 mmol g–1) Fmoc-Lys(Boc)-Novasyn TGT resin 9 using previously reported conditions,30a followed by chain elongation with Gly and Pro residues to give linear resin-bound peptide 10 (Scheme 2). Removal of the Alloc and allyl groups with Pd(PPh3)4, subsequent Fmoc deprotection, and cyclization with PyBOP, HOBt, and NMM gave 11. Cleavage and purification, as before, gave nisin ring B 2 in 37% yield.
Scheme 2. SPPS of Nisin Ring B 2.

Reagents and conditions: (i) piperidine/DMF; (ii) 8, HOBt, PyBOP, NMM, μwave, 60 °C, 5 min; (iii) piperidine/DMF; (iv) Fmoc-Gly-OH, HOAt, PyAOP, DIPEA, 2 h; (v) piperidine/DMF; (vi) Fmoc-Pro-OH, HOAt, PyAOP, DIPEA, 2 h; (vii) Pd(PPh3)4, PhSiH3, CH2Cl2:DMF (1:1), 2 h; (viii) piperidine/DMF; (ix) HOAt, PyAOP, DIPEA, μwave, 60 °C, 5 min, and then 1 h r.t.; (x) TFA/H2O/TIPS.
Ring A: Synthesis of Wild-Type and Analogue Structures
We next turned our attention to the synthesis of isolated ring A structures, mutacin ring A 12 and nisin ring A 13. Here, we envisaged that the main synthetic challenge would be the incorporation of Dha or Dhb residues at position 2 and Dha residues at position 5. As such, dehydro amino acids are a feature of the N-terminus of many, but not all, of the nisin-like lantibiotics; we wished to study what effect these residues had on the conformation of ring A. The structural and conformation properties of peptides containing dehydro amino acid residues have been extensively investigated.32 However, most previous studies focus on linear oligopeptides and/or peptides containing ΔPhe residues, and it is therefore more difficult to predict the effects of incorporating Dha or Dhb residues on the conformations of cyclic peptides. In peptides 12 and 13, we incorporated an Ala residue at position 8 as a non-oxidizable mimetic for the (methyl) lanthionine bridges of the B ring found in the native sequences. To determine whether the dehydro residues could be substituted with more stable33 saturated analogues, we sought to synthesize the mutacin ring A (Ser2, Ala5, and Ala8) analogue 14. Finally, to assess the effects of the N-terminal residues on the conformation of these cyclic peptides, we prepared the truncated analogue 15 (Figure 3).
Figure 3.

Ring A structures synthesized.
A variety of synthetic methods have been developed for the synthesis of dehydro amino acids and peptides containing dehydro residues.33a,34 Due to the reactive nature of the Dha residue, its direct incorporation into a growing peptide chain by SPPS is not feasible. Instead, it is usually necessary to incorporate a precursor residue into the peptide, which can be transformed at a later stage to reveal the Dha. For example, in their recent synthesis of dicarba analogues of nisin ring A, Slootweg et al. generated Dha residues by elimination from Ser under basic conditions using EDCI and CuCl.25b In the total synthesis of nisin, a 2,3-diaminopropionic acid residue was incorporated into the peptide at the desired position of dehydration, followed by methylation and Hofmann degradation to produce the Dha residue.24b Oxidative elimination of phenylselenocysteine (Sec(Ph)) residues has also been used to incorporate Dha into peptides,35a notably to produce a Dha-containing peptide used in a biomimetic synthesis of nisin ring B.35b A number of different methods have been reported for the generation of Dha from Cys residues; for example, Matteucci et al. reported the oxidation-elimination of S-methyl cysteine as part of their biomimetic synthesis of nisin ring B.26c However, this approach is incompatible with lantibiotic syntheses requiring orthogonally protected (methyl)lanthionines as the strongly oxidizing conditions needed to produce the intermediate sulfoxide would cause undesired oxidation of the thioether bridge. The introduction of Dha residues in peptides and proteins by the bis-S-alkylation and β-elimination of Cys residues has been widely explored by Davis et al.36a and has been used to introduce Dha residues in a recently reported synthesis of the lanthipeptide SapB.36b Morrison et al. have recently reported an improvement to this methodology using a new alkylation reagent, methyl 2,5-dibromopentanoate.37 This reagent enabled the simultaneous introduction of multiple Dha residues into peptides while avoiding the undesired cross-linking between Cys residues, which can occur when using other common bis-alkylation reagents such as dibromoadipamide.
By contrast, Dhb residues tend to be more resistant to unwanted Michael addition than Dha due to the presence of the β-methyl group. Therefore, this residue is normally incorporated into lantibiotics by prior synthesis of linear Dhb-containing pentapeptides30b or dipeptides.30c,30d The precursor peptides were prepared from analogues with (2S,3R)Thr incorporated and were stereoselectively dehydrated to afford Z-Dhb. However, both Shiba et al.24b and Liskamp et al.25b reported low yields when attempting to couple an Ile-Dhb dipeptide at the N-terminus of nisin or analogues and instead coupled a protected precursor Ile-Thr dipeptide, followed by stereoselective dehydration.
Fmoc-Ala-Novasyn TGT resin (loading, 0.17–0.21 mmol g–1) 16 was used for the synthesis of the simplified mutacin ring A analogue 14 (Scheme 3). After Fmoc deprotection, (Teoc, TMSE/Fmoc) Lan 4 was added using the same microwave coupling protocol. Further chain extension gave the linear resin-bound peptide intermediate 17. Silyl deprotection and cyclization were carried out as for the ring B analogues to give the cyclic resin-bound intermediate 18. Initially, the last two residues were coupled using PyAOP/HOAt, as before. However, after resin and protecting group cleavage and purification, although the desired peptide 14 could be detected by LCMS analysis of the crude sample, a truncated peptide 19, with no addition of the final two amino acids, was also observed. This suggested that, following the cyclization, the terminal amine may be less accessible, leading to inefficient coupling of the final two residues. HATU has been reported to give good results when coupling sterically hindered amino acids.38 However, in this case, only a small increase in the ratio of desired product 14 to truncated peptide 19 was observed with this reagent. Best results were achieved using amino acid fluorides, synthesized from the Fmoc amino acids with cyanuric fluoride39 and incorporated into the peptide by coupling with DIPEA in CH2Cl2. This further increased the ratio of 14 to 19. Although the reaction could not be pushed to completion, purification by HPLC gave the mutacin I ring A analogue 14 in 3% yield and truncated peptide 19 in 2% yield. A similar reaction sequence was used to afford the truncated mutacin ring A analogue 15 in 5% yield after purification.
Scheme 3. SPPS of Simplified Mutacin Ring A Analogue 14.

Reagents and conditions: (i) piperidine/DMF; (ii) 4, HOAt, PyAOP, DIPEA, μwave, 60 °C, 5 min; (iii) piperidine/DMF; (iv) Fmoc-Leu-OH, HOAt, PyAOP, DIPEA, 2 h; (v) piperidine/DMF; (vi) Fmoc-Ala-OH, HOAt, PyAOP, DIPEA, 2 h; (vii) piperidine/DMF; (viii) Fmoc-Leu-OH, HOAt, PyAOP, DIPEA, 2 h; (ix) TBAF, DMF, 1 h; (x) piperidine/DMF; (xi) HOAt, PyAOP, DIPEA, μwave, 60 °C, 5 min; (xii) Fmoc-Ser(OtBu)-F, DIPEA, 1 h; (xiii) piperidine/DMF; (xiv) Fmoc-Phe-F, DIPEA, 1 h; (xv) piperidine/DMF; (xvi) TFA, H2O, TIPS.
We then investigated different strategies to give wild-type ring A peptides containing Dha and Dhb residues. Initial attempts to convert the Ser residue of 15 to Dha by β-elimination using CuCl/EDCI25b were unsuccessful. As this method was developed for peptides with protected N- and C-termini, this may be due to oligomerization of the peptide. Incorporation of Sec(Ph) residues into model ring A sequences was then attempted. Fmoc-Sec(Ph)-OH 20 was prepared from Fmoc-Ser-OAllyl40a following the procedure reported by Levengood et al.40b Cyclic peptide 21 was then prepared using the same strategy (Scheme 4) via the linear resin-bound peptide 22 and the cyclic resin-bound peptide 23. Unfortunately, all attempts to carry out the oxidative elimination with NaIO4, as previously reported,40b resulted in oxidation of the lanthionine bridge, giving mixtures of 24 as well as the desired 25 (Figure S1, Supporting Information).
Scheme 4. Attempted Synthesis of Mutacin Ring A by Oxidative Elimination of Sec(Ph).

Reagents and conditions: (i) piperidine/DMF; (ii) HOAt, PyAOP, DIPEA, μwave, 60 °C, 5 min, 4; (iii) piperidine/DMF; (iv) Fmoc-Leu-OH, HOAt, PyAOP, DIPEA, 2 h; (v) piperidine/DMF; (vi) Fmoc-Sec(Ph)-OH 20, HOAt, PyAOP, DIPEA, 2 h; (vii) piperidine/DMF; (viii) Fmoc-Leu-OH, HOAt, PyAOP, DIPEA, 2 h; (ix) TBAF, DMF, 1 h; (x) piperidine/DMF; (xi) HOAt, PyAOP, DIPEA, μwave, 60 °C, 5 min; (xii) TFA, H2O, TIPS; (xiii) NaIO4, MeCN/H2O.
The alkylation-elimination methodology recently reported by Webb’s group37 proved to be more successful. For the synthesis of mutacin ring A 12 using this approach, the precursor peptide 26, containing Cys residues at positions 2 and 5, was first required. This was prepared in a similar manner to the simplified ring A analogues, again starting from Fmoc-Ala-Novasyn TGT resin 16 (Scheme 5). Synthesis of the linear resin-bound intermediate 27, followed by deprotection of the silyl and Fmoc groups and on-resin cyclization, gave the cyclic resin-bound intermediate 28. Gratifyingly, in this case, the N-terminal Cys and Phe residues were coupled using standard conditions without any of the problems encountered in the synthesis of 14, and the cyclic peptide 26 was obtained in 15% yield following purification by reverse-phase HPLC. It is possible that the bulky Cys(Trt) protecting group at residue 5 forces the cyclic resin-bound intermediate 28 into a different ring conformation in which the terminal amino group is more accessible. Subsequent treatment of 26 with TCEP, followed by the addition of a large excess of methyl 2,5-dibromopentanoate and K2CO3, gave mutacin ring A 12 in 11% yield (from the original resin loading).
Scheme 5. SPPS of Mutacin Ring A 12.

Reagents and conditions: (i) piperidine/DMF; (ii) 4, HOAt, PyAOP, DIPEA, μwave, 60 °C, 5 min; (iii) piperidine/DMF; (iv) Fmoc-Leu-OH, HOAt, PyAOP, DIPEA, 2 h; (v) piperidine/DMF; (vi) Fmoc-Cys(Trt)-OH, HOAt, PyAOP, DIPEA, 2 h; (vii) piperidine/DMF; (viii) Fmoc-Leu-OH, HOAt, PyAOP, DIPEA, 2 h; (ix) TBAF, DMF, 1 h; (x) piperidine/DMF; (xi) HOAt, PyAOP, DIPEA, μwave, 60 °C, 5 min; (xii) Fmoc-Cys(Trt)-OH, HOAt, PyAOP, DIPEA, 1 h; (xiii) piperidine/DMF; (xiv) Fmoc-Phe-OH, HOAt, PyAOP, DIPEA, 1 h; (xv) piperidine/DMF; (xvi) TFA, H2O, TIPS; (xvii) TCEP; (xviii) methyl 2,5-dibromopentanoate, K2CO3.
In view of this successful transformation, we hypothesized that this approach could be extended to generate Dhb in peptides. On the assumption that the bis-S-alkylation and β-elimination of Cys residues take place via an antiperiplanar transition state,41 we reasoned that incorporating (2R,3R)-β-MeCys in the peptide sequence should lead to the formation of the desired Z-dehydrobutyrine stereochemistry. We therefore adapted the previously reported route to protected MeCys derivatives via the ring opening with trityl thiol42a of a l-threonine-derived aziridine42b to give the appropriately protected (2R,3R)-Fmoc-β-MeCys(Trt)-OH 29. A similar synthetic route from Fmoc-Ala-Novasyn TGT resin 16 (Scheme 6), via the linear resin-bound intermediate 30, gave the cyclic resin-bound intermediate 31. Again, no problems were encountered with the coupling of Fmoc-β-MeCys(Trt)-OH 29 or with the subsequent Fmoc-Ile-OH residue, affording the precursor peptide 32 in 11% yield after cleavage and deprotection.
Scheme 6. SPPS of Nisin Ring A 13.

Reagents and conditions: (i) piperidine/DMF; (ii) 4, HOAt, PyAOP, DIPEA, μwave, 60 °C, 5 min; (iii) piperidine/DMF; (iv) Fmoc-Leu-OH, HOAt, PyAOP, DIPEA, 2 h; (v) piperidine/DMF; (vi) Fmoc-Cys(Trt)-OH, HOAt, PyAOP, DIPEA, 2 h; (vii) piperidine/DMF; (viii) Fmoc-Ile-OH, HOAt, PyAOP, DIPEA, 2 h; (ix) TBAF, DMF, 1 h; (x) piperidine/DMF; (xi) HOAt, PyAOP, DIPEA, μwave, 60 °C, 5 min; (xii) (2R,3R)-Fmoc-β-MeCys(Trt)-OH 29, HOAt, PyAOP, DIPEA, 1 h; (xiii) piperidine/DMF; (xiv) Fmoc-Ile-OH, HOAt, PyAOP, DIPEA, 1 h; (xv) piperidine/DMF; (xvi) TFA, H2O, TIPS; (xvii) TCEP; (xviii) methyl 2,5-dibromopentanoate, K2CO3.
The alkylation-elimination reaction was again carried out by treating 32 with TCEP, methyl 2,5-dibromopentanoate, and K2CO3. Gratifyingly, LCMS analysis showed complete conversion to nisin ring A 13 after 2 h at 37 °C, and the desired peptide was isolated in 1% yield (from the original resin loading) after purification by HPLC. Examination of the NOESY spectrum of 13 (Figure 4) revealed that the Z-Dhb residue was generated exclusively as only a cross-peak between Lan3 NH and Dhb βH was seen.
Figure 4.

Expected NOEs (red arrows) for possible geometries of Dhb in nisin ring A (13) and experimental NOE spectrum of nisin ring A. Positions of expected cross peaks with the Lan3 NH are shown in red boxes; only the Lan3 NH–Dhb βH cross-peak is seen.
NMR Studies of Ring A and B Cyclic Peptides
With all the desired peptides in hand, each was fully characterized by NMR. All peptide conformations were studied in DMSO-d6 as a mimic of the membrane environment in which the nisin–lipid II complex forms. Nisin–lipid II complexes are known to precipitate in aqueous solutions, while DMSO is often regarded as a reasonable membrane mimetic solvent, with a dielectric constant (ε = 47.2) in between that of water (ε = 80) and the interior of the membrane (ε = 2–4).14 Moreover, determining the conformations in DMSO enabled direct comparisons to be drawn with the published NMR structure of nisin in a 1:1 complex with lipid II, which was also carried out in DMSO.14 To enable structure calculation, each of the unusual amino acids first had to be parameterized for XPLOR-NIH. Solution state structures of the peptides were then calculated (see the Supporting Information for parameterization details and structure calculation protocol).43 Ensembles of the 15 lowest energy structures for each of the synthesized peptides are shown in Figures 5 and 6.
Figure 5.
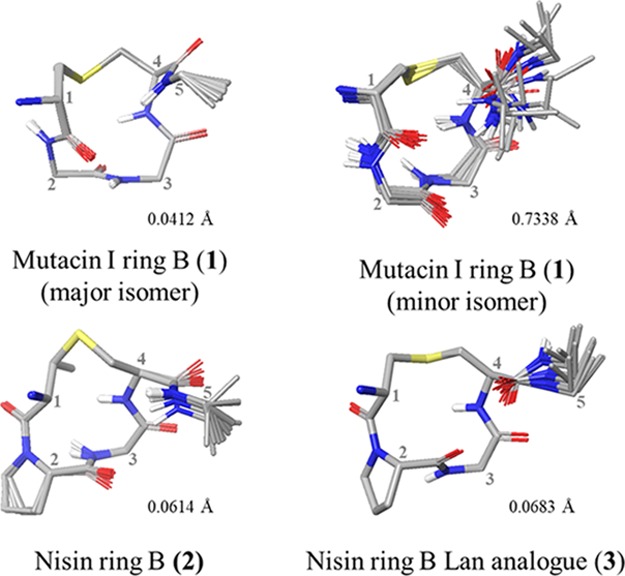
XPLOR ensembles of ring B peptides. Ensembles of the lowest-energy structures were produced in Maestro (version 11.4, Schrödinger, LLC) by alignment of αC and S atoms within the lantibiotic rings. Side chains (excluding Pro) and nonpolar hydrogens have been omitted for clarity. Residues are numbered from the full-length parent peptide.
Figure 6.
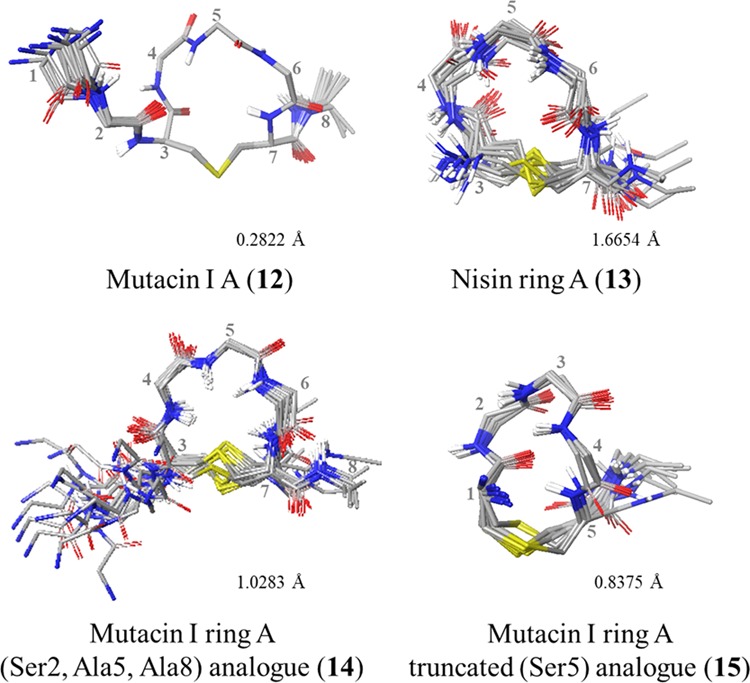
XPLOR ensembles of ring A peptides. Ensembles of the lowest-energy structures were produced in Maestro (version 11.4, Schrödinger, LLC) by alignment of αC and S atoms within the lantibiotic rings. Side chains (excluding Pro), nonpolar hydrogens, and residues 1 and 2 in 13 have been omitted for clarity. Residues are numbered from the full-length parent peptide.
The 1H NMR spectrum of mutacin I ring B 1 revealed the presence of two sets of resonances in a 3:1 ratio, which could be separately assigned. Running the NMR at elevated temperature confirmed that these corresponded to conformers rather than two different peptides or diastereomers as the peaks coalesced into one set of average resonances (Figure S2, Supporting Information).
On comparison of the calculated structures, the main difference between the two conformers seemed to be a peptide plane flip along the Leu–Gly amide bond (Figure 7A). Numerous examples of peptide plane flips have been reported in the literature.44 Toogood26b and Goodman27a have previously demonstrated that both the ring B of nisin and an analogue of ring B of epidermin with Lan substituted for MeLan exist as two slowly interconverting conformers at room temperature. These result from cis/trans isomerization at the Pro residue, with the trans isomer predominating. More recently, this plane flip has also been observed in the Pro–Gly bond of mutacin 1140.45 The existence of two conformers of mutacin ring B is surprising as it suggests that the MeLan and Pro residues found in the B rings of all other nisin-type lantibiotics are not the main determinants of peptide conformation.
Figure 7.
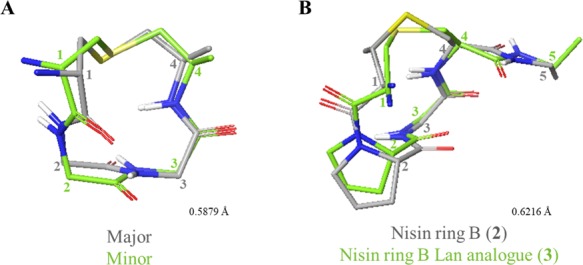
Comparisons of ring B peptides. (A) Comparison of major and minor conformations of 1. (B) Comparison of 2 and 3. Figures were produced in Maestro (version 11.4, Schrödinger, LLC) by alignment of αC and S atoms within the lantibiotic rings. Side chains (excluding Pro), nonpolar hydrogens, and residue 5 in 1 have been omitted for clarity. Representative structures (closest to the average) were used for comparison. Residues are numbered from the full-length parent peptide.
Unexpectedly, the spectra of the nisin ring B peptides 2 and 3 indicated the presence of only one conformer. Determination of the geometry of the (Me)Lan–Pro bonds using the Promega server indicated that the likelihood of this bond to exist in cis conformation was 99.4 and 91%, respectively.46 These predictions were supported by the short experimental distances observed between the Pro Hα and the Lan or MeLan Hα in 2 and 3, respectively (Figure 8). We hypothesized that this difference from the previously reported observations may result from the solid-phase synthetic strategy that we have adopted. It may suggest that the Pro residue must be cis in the linear resin-bound peptide intermediate in Schemes 1 and 2 for the cyclization step to take place. Other groups have reported that certain Pro-rich cyclic peptides demonstrate very different biological properties, dependent on whether they were prepared synthetically or isolated from natural sources, despite the structural and stereochemical integrity of the synthetic methodology being validated.47 For example, Albericio and co-workers showed that the macrolactamization methodology used for the synthesis of the phakellistatins resulted in these cyclic peptides being locked into an incorrect Pro conformer and thus into a biologically inactive structure.47c
Figure 8.

Experimental NOESY spectra of nisin ring B 2 and nisin ring B Lan analogue 3. Positions of cross-peaks characteristic of cis-Pro residues, between Pro Hα and either MeLan or Lan Hα, are shown in red boxes.
Again, comparison of the structures of 2 and 3 also revealed that replacement of Lan for MeLan does not significantly change the backbone conformation of the peptide (Figure 7B). In addition, Pattabiraman et al. have shown that a Lan analogue of lacticin 3147 A2 retains its synergistic activity with the A1 peptide.30a These observations suggest that MeLan could successfully be replaced by Lan in the synthesis of future analogues, providing faster access to peptides as only one orthogonally protected lanthionine would be necessary.
To examine the effect of the dehydro residues on solution conformation, the structures of the mutacin I ring A peptides 12, 14, and 15 were compared (Figure 9A). The most noticeable difference was that the absence of the two N-terminal residues caused truncated analogue 15 to have a less rounded, more elongated shape than the other mutacin peptides, indicating that these residues play an important role in restricting the accessible solution conformations. While in the previously reported nisin–lipid II complex (PDB ID: 1WCO),14 the N-terminal (Ile1, Dhb2) residues were not visible, it is known that these are a critical component of the biological activity of this peptide as modified or mutant nisin analogues with the N-terminus methylated or extended had severely reduced antimicrobial activity.48 Molecular dynamics simulations of the interaction between nisin and a phospholipid bilayer49 suggested that the N-terminal amine initially engages the negatively charged phospholipids, and recent ssNMR studies also propose that the Ile1 side chain plays an important role in the assembled nisin–lipid II pore.15
Figure 9.
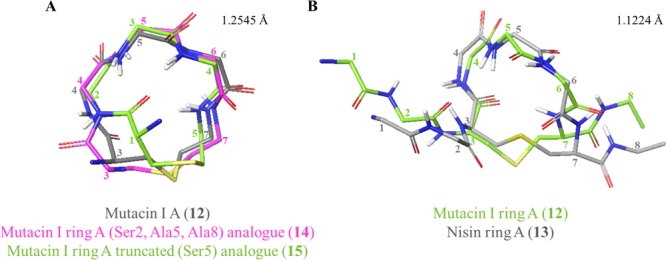
Comparisons of ring A peptides. (A) Comparison of WT mutacin I ring A (12) with analogues 14 and 15. All nonring residues have been omitted for clarity. (B) WT mutacin I ring A (12) with WT nisin ring A (13). Figures were produced in Maestro (version 11.4, Schrödinger, LLC) by alignment of αC and S atoms within the lantibiotic rings. Side chains and nonpolar hydrogens have been omitted for clarity. Representative structures (closest to the average) were used for comparison. Residues are numbered from the full-length parent peptide.
The (Ser2, Ala5, Ala8) analogue 14 and WT peptide 12 were fairly similar to each other, however, with most flexibility observed in the thioether bridge, as was also described by Lian et al. in nisin ring A.50 Comparison of all three mutacin peptides showed that the replacement of Dha5 in WT peptide 12 for either Ser or Ala did not significantly affect the overall conformation of the Leu4-Xaa5-Leu6 portion of ring A. In contrast, groups of Goodman and Shiba27 have also compared the NMR structure of wild-type ring A, with an analogue with Dha at position 5 and with the D-Ala5 and L-Ala5 analogues, and have determined that the conformational preferences of the saturated analogues differed from each other and from the wild-type sequence. However, there is evidence in the literature suggesting that substitution of either of the dehydro residues in WT nisin and mutacin I would be tolerated. For example, retention of bioactivity against Micrococcus luteus is observed for both nisin and mutacin 1140 with Dha5 replaced by Ala.;51a,51b Wiedemann et al. have shown that the replacement of Dhb2 in nisin with either Ser, Ala, or Val has little effect on MIC,12c and mutations at Dha5 in mutacin 1140 have been found to be moderately well accommodated with the Gly5 mutant, showing increased bioactivity.51c
Comparison of the two WT peptides, mutacin ring A 12 and nisin ring A 13, again showed most flexibility around the thioether bridge, although there was little difference between the two peptides in the rest of the ring (Figure 9B). This was perhaps to be expected as the only difference is at position 4 (Ile4 in nisin, Leu4 in mutacin). The lipid II binding amides within the ring (Ile4/Leu4 NH and Dha5 NH) adopt the same relative positions and orientations in both peptides.14
Finally, to determine whether the individual rings may be preorganized into the cage conformation in solution, each of the four synthesized WT rings was compared to the corresponding segment of the NMR structure of full-length nisin bound to lipid II in DMSO (PDB ID: 1WCO).14,52 Of the mutacin I ring B 1 conformers, the minor conformer was most similar to the published structure (Figure 10A,B). The largest difference between the two peptides was observed in the (Me)Lan-Leu/Pro section of the backbone and indicates that mutacin I may be more flexible in solution due to the absence of the Pro residue. Comparison of synthesized nisin ring B 2 to 1WCO again revealed that the two were most dissimilar along the MeLan-Pro backbone section, in this case, due to the cis-Pro in 2 (Figure 10C). This difference suggests that, when synthesized as described in this work, nisin ring B does not adopt the correct lipid II binding conformation in solution.
Figure 10.
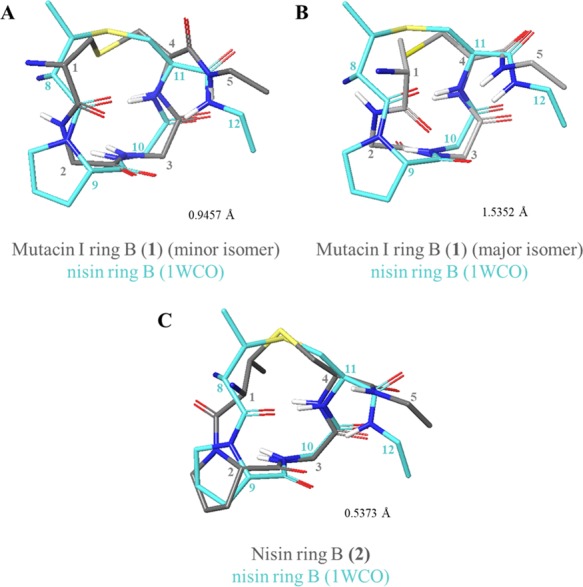
Comparisons of WT ring B peptides to 1WCO. (A) Comparison of 1WCO to 1 (minor conformer). (B) Comparison of 1WCO to 1 (major conformer). (C) Comparison of 1WCO to 2. Figures were produced in Maestro (version 11.4, Schrödinger, LLC) by alignment of αC and S atoms within the lantibiotic rings. Side chains (excluding Pro) and nonpolar hydrogens have been omitted for clarity. Representative structures (closest to the average), and a modified version of 1WCO,52 were used for comparison. Residues are numbered from the full-length parent peptide.
On the other hand, both of the WT ring A peptides 12 and 13 could be overlaid with 1WCO with low RMSD (Figure 11A,B) and the lipid II binding amides within the rings (Ile4/Leu4 NH and Dha5 NH) adopt the lipid II binding orientation observed in the 1WCO (Figure 11C,D). This implies that the WT A ring peptides may display some preorganization for lipid II binding in solution.
Figure 11.
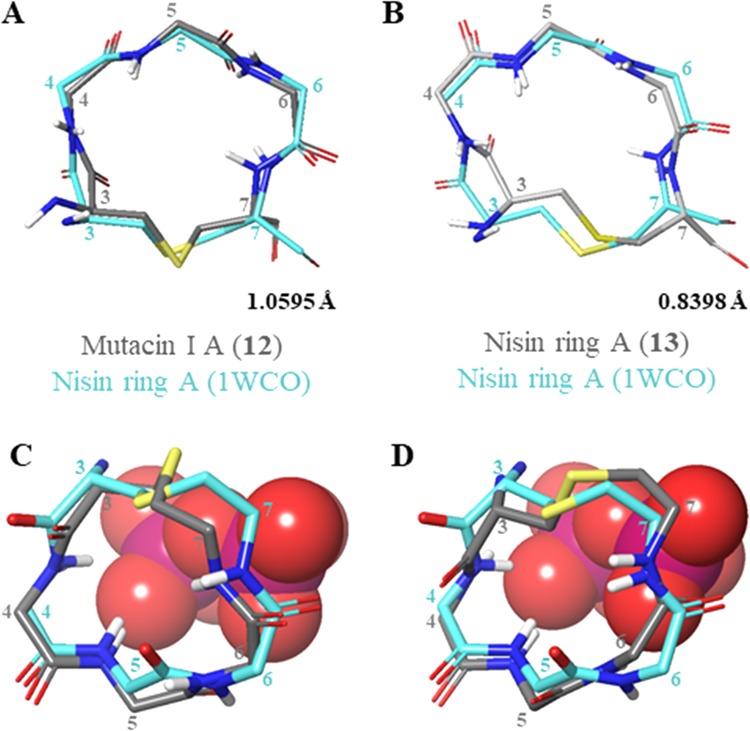
(A) Comparison of 1WCO to 12. (B) Comparison of 1WCO to 13. (C) Overlay of 12 with nisin ring A and the lipid II pyrophosphate from 1WCO. (D) Overlay of 13 with nisin ring A and the lipid II pyrophosphate from 1WCO. Pyrophosphate is shown in space-filling representation with phosphorus atoms in magenta and oxygen atoms in red. Figures were produced in Maestro (version 11.4, Schrödinger, LLC) by alignment of αC and S atoms within the lantibiotic rings. Side chains (excluding Pro), nonpolar hydrogens, and nonring residues have been omitted for clarity. Representative structures (closest to the average) were used for comparison. Residues are numbered from the full-length parent peptide.
Structure calculation also allowed an analysis of the turn structure of the peptides in DMSO solution. Dihedral angles and Cα(i)–Cα(i + 3) or Cα(i + 4) distances were measured in Maestro for each of the peptides and then compared to the values proposed for β- or α-turn structures reported by Chou.53 All three ring B peptides have a Cα(i)–Cα(i + 3) distance of less than 7 Å, which suggests the presence of a β-turn in ring B of both nisin and mutacin I. However, none of the measured dihedral angles for ring B peptides calculated in this work are within ±30° with those proposed for any typical β-turn structures (Table S19). This was unexpected as nisin ring B has previously been reported to display a type II β-turn in solution.27a,54 Similarly, a Cα(i)–Cα(i + 4) distance of less than 7 Å in all four of the ring A peptides suggests the presence of an α-turn, although none of the measured dihedral angles are within ±30° of those proposed for any α-turn structures reported by Chou (Table S20).53 This is in agreement with a previous report showing that no typical turns are present in nisin ring A.54b
Conclusions
While many advances in the understanding of the interaction between nisin and its bacterial target, lipid II, have been reported in recent years, a complete understanding of the unique molecular architecture of the nisin-lipid II pore will require the synthesis and structural evaluation of individual components and analogues of both the lantibiotic and its lipid partner. While significant progress has been made in the exploitation of the biosynthetic machinery for the in vitro preparation of libraries of lantipeptides, and of full-length lantibiotics incorporating unnatural amino acids,55 SPPS remains the strategy of choice to access individual lanthionine-bridged peptides, subunits of the naturally occurring lantibiotics, and lantipeptides containing significant numbers of nonproteinogenic amino acids as these are not readily accessible via a biotransformation approach.
As a first step toward elucidating the details of the nisin–lipid II interaction at the molecular level, in this paper, we have extended our previous SPPS methodology to prepare the wild-type sequences and synthetic analogues of individual rings A and B of the lantibiotics nisin and mutacin. In particular, we have shown that dehydro amino acid synthesis using methyl 2,5-dibromopentanoate has enabled the expedient synthesis of ring A analogues, overcoming some of the synthetic difficulties previously encountered in synthesizing this region of nisin. We have demonstrated for the first time that this approach can be used to exclusively introduce Z-Dhb residues into such peptides.
We have analyzed the conformational properties of these individual rings A and B by NMR. For the ring A peptides, the observed NMR structures give some insights into how the peptide sequence and conformation in DMSO of isolated single rings might influence the biological activity of this class of lantibiotics. We have confirmed the importance of the first two residues at the N-terminus, which appear to influence the conformation of ring A. The similarities between the wild-type mutacin ring A 12 and wild-type nisin ring A 13 structures and comparison with the previously published NMR structure of a 1:1 nisin–lipid II complex14 reinforce the accepted hypothesis that mutacin binds to lipid II in the same manner as nisin. We have also shown that the dehydro amino acids in the wild-type structures are not the main determinants of peptide conformation. Substitution of a Dha residue at position 5 and of Dha or Dhb at position 2 has little influence on the conformation of ring A, thus providing a rationale for why mutations at these positions are well tolerated. For the ring B peptides, it is clear from the structure of mutacin ring B 1 that neither the Pro9 residue nor the methyl group of MeLan8,12 is indispensible for the correct conformation to be adopted. This study has, however, revealed that the methodology adopted for the synthesis of this region of nisin may influence the conformational outcome.
These results must also be viewed in the light of a recently reported study of the nisin–lipid II pore by ssNMR.15 These indicate that the critical interactions between the ring A–ring B cage and the pyrophosphate moiety of lipid II in the 1:1 complex are altered in the 8:4 nisin–lipid II pore in lipid bilayer structures. The solution NMR studies that we report in this paper are complementary to this approach and represent a more accessible method to allow the structural comparison of the biologically active conformations, which, in turn, could be used to facilitate a screening approach.
However, the analysis of the conformational properties of individual rings cannot give a complete picture of the interactions of the intact peptide with lipid II and especially of the factors governing the molecular recognition of the pyrophosphate group by the ring A–ring B cage. We have therefore followed these studies by developing a synthetic route to the solid-phase synthesis of the entire bicyclic structure of analogues of nisin(1–12) and mutacin(1–12) and have carried out a detailed conformational study of wild-type nisin(1–12) and analogues.56 These studies, in turn, will enable a more detailed understanding of the molecular recognition of lipid II by the N-terminal portion of nisin and related lantibiotics.
A deeper understanding of how nisin interacts with its biological target will also enable the design of novel lipid II binding molecules, which could, in turn, be lead compounds for next-generation antibacterial agents. An important step toward this goal will be the design of simplified nisin- or mutacin-like structures that are synthetically accessible and are stable in vivo. In this paper, we have shown that antibacterial agents based on mutacin I (with only Lan bridges and only Dha residues) may be easier to produce using SPPS methods than many other lipid II binding lantibiotics. We have also confirmed that Dha5 can be replaced in ring A with saturated amino acids that will be more metabolically stable.
Finally, thioether-bridged peptides have general applicability in medicinal chemistry and chemical biology studies as nonreducible mimics of disulfide-bridged peptides. It is thus important that the structural consequences of incorporating a lanthionine bridge into cyclic peptides are well understood, particularly as our results imply that the presence of a thioether bridge between side chains is the key factor in determining the conformational properties of the resulting cyclic peptides. Our conformational analysis of the cyclic peptides presented in this paper shows that such Lan or MeLan bridges may allow access to turn structures and conformational space, which is not accessible using only the 20 proteinogenic amino acids.
Experimental Section
General Procedures
All chemicals were used as ordered unless otherwise stated, with reagents being purchased from Sigma-Aldrich Co. Ltd., Acros Organics, or Alfa Aesar. Boc-D-Ser(Bn)-OH was purchased from Santa Cruz Biotechnology. Pd(PPh3)4 was used as purchased from Sigma-Aldrich Co. Ltd. Caesium carbonate was dried in vacuo before use. pTsCl was recrystallized from warm hexane before use. Anhydrous THF, CH2Cl2, and DMF (dried over molecular sieves) were used as purchased from Acros Organics. Pet. ether refers to petroleum ether with 40–60 °C fractions. Ether refers to diethyl ether. All water used was distilled with an Elga Purelab Option R7 purifier. Solvent used for HPLC was all HPLC-grade and used directly from the bottle.
Reactions requiring anhydrous conditions were carried out under an argon atmosphere using oven-dried glassware. TLC analysis was on aluminum-backed Sigma-Aldrich TLC plates with an F254 fluorescent indicator, with UV visualization at 254 nm or visualization by staining with KMnO4, ninhydrin, or Ellman’s reagent (5,5′-dithiobis-(2-nitrobenzoic acid)). Flash column chromatography was carried out using Merck silica gel 60 (40–60 μm).
LCMS spectra were recorded on a Waters Acquity UPLC SQD using a linear gradient of 5–95% B over 5 min (A = water, B = acetonitrile, 0.1% formic acid) with a C8 column at a flow rate of 0.6 mL min–1. Analysis of the chromatograms was conducted using MassLynx software. HRMS spectra were recorded on a Waters LCT Premier XE instrument (TOF) with data analyzed using MassLynx software. Optical rotations were measured at 25 °C, unless otherwise stated, on a Perkin-Elmer Model 343 polarimeter. Specific rotations are given in 10–1 deg cm2 g–1. Melting points are noncorrected and were recorded using Stuart SMP11 Analogue melting point apparatus. Infrared spectra were recorded on a Perkin Elmer 100 FT-IR spectrometer.
1H, 13C, and all 2D NMR spectra were recorded on either a Bruker Avance 300, 500, or 600 spectrometer, with chemical shifts (δ) given in ppm relative to the solvent signal and coupling constants (J) given in Hz. Carbon signals were assigned from HSQC and HMBC cross-peaks. Where they could be distinguished, terminal protons of allyl groups (CH2CH=CH2) are labeled cis and trans with respect to the central CH (CH2CH=CH2). Where the resonances for symmetric carbon atoms in the Fmoc group could be distinguished, the shift of both is given. Abbreviations used in 1H NMR assignment are as follows: Ar = aromatic, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br s = broad singlet, br d = broad doublet, dd = doublet of doublets, dt = doublet of triplets, dq = doublet of quartets, ddt = doublet of doublet of triplets, td = triplet of doublets, tt = triplet of triplets, ttd = triplet of triplet of doublets, qd = quartet of doublets. Data processing was carried out using ACD/NMR Processor Academic Edition, version 12.01 (Advanced Chemistry Development Inc.).
Synthesis of Protected Amino Acids
(Teoc, TMSE/Fmoc) Lan 4
(Teoc, TMSE/Fmoc) Lan 4 was prepared following our previously published route,29b with the following modifications.
Synthesis of (Fmoc-Cys-OTce)2
To a stirred solution of (Fmoc-Cys-OH)257 (8.98 g, 13.1 mmol) in benzene (250 mL) was added p-toluenesulfonic acid (5.99 g, 31.5 mmol) and 2,2,2-trichloroethanol (3.02 mL, 31.5 mmol). The solution was heated at reflux with a Dean and Stark trap for 48 h before cooling to 0 °C and filtering to remove excess acid. The solvent was then removed in vacuo, and the residue was washed with EtOAc (150 mL) and dried in vacuo to give (Fmoc-Cys-OTce)2 as a white solid (8.42 g, 8.89 mmol, 68%). Rf 0.35 (CH2Cl2); mp 189–201 °C; [α]D25 −8.57 (c 1.4 mg mL–1, CHCl3), [lit.29b [α]D −10.7 (c 1.4 mg mL–1, CHCl3)]; 1H NMR (600 MHz, CDCl3) δ 3.27 (d, J = 5.3 Hz, 4H), 4.22 (t, J = 6.3 Hz, 2H), 4.39–4.46 (m, 4H), 4.75–4.87 (m, 6H), 5.75 (d, J = 7.8 Hz, 2H), 7.31 (t, J = 7.5 Hz, 4H), 7.40 (t, J = 7.5 Hz, 4H), 7.59 (d, J = 7.2 Hz, 4H), 7.76 (d, J = 7.5 Hz, 4H); 13C{1H} (150 MHz, CDCl3) δ 40.8, 47.0, 53.3, 67.4, 74.8, 94.1, 120.0, 125.0, 127.1, 127.8, 141.3, 143.5, 155.6, 168.9; LCMS (ES+) m/z: calcd for C40H35N2O8Cl6S2 [M + H]+, 945.0; found, 945.4 [M(6 × 35Cl) + H]+, 947.4 [M(5 × 35Cl) + H]+, 969.3 [M(5 × 35Cl) + Na]+.
Synthesis of Fmoc-Cys-OTce
To a stirred solution of (Fmoc-Cys-OTce)2 (2.50 g, 2.64 mmol) in CH2Cl2 (50 mL) were added DTT (489 mg, 3.17 mmol) and Et3N (0.44 mL, 3.17 mmol). After 1 h, the reaction mixture was washed with saturated sodium bicarbonate (3 × 50 mL), brine (1 × 50 mL), and H2O (1 × 50 mL). The organic layer was then dried (MgSO4), and the solvent was removed in vacuo to give title compound as an off-white solid (2.42 g, 5.10 mmol, 96%). Rf 0.52 (CH2Cl2); mp 67–70 °C; [α]D25 −2.5 (c 17.5 mg mL–1, CHCl3), [lit.29b [α]D −2.4 (c 17.3 mg mL–1, CHCl3)]; 1H NMR (600 MHz, CDCl3) δ 1.47 (t, J = 9.0 Hz, 1H), 3.02–3.07 (m, 1H), 3.14–3.18 (m, 1H), 4.25 (t, J = 6.8 Hz, 1H), 4.46 (d, J = 6.8 Hz, 2H), 4.74 (d, J = 11.7 Hz, 1H), 4.83–4.86 (m, 1H), 4.94 (d, J = 12.0 Hz, 1H), 5.70 (d, J = 8.3 Hz, 1H), 7.34 (tdd, J = 7.4, 2.4, 0.9 Hz, 2H), 7.43 (t, J = 7.5 Hz, 2H), 7.62 (d, J = 7.5 Hz, 2H), 7.79 (d, J = 7.5 Hz, 2H); 13C{1H} (150 MHz, CDCl3) δ 26.9, 47.1, 55.1, 67.2, 74.6, 94.2, 120.0, 125.0, 127.1, 127.8, 141.3, 143.5, 143.8, 155.6, 168.7; LCMS (ES+) m/z: calcd for C20H19NO4Cl3S [M + H]+, 474.0; found, 474.3 [M(3 × 35Cl) + H]+, 476.2 [M(2 × 35Cl) + H]+, 496.2 [M(3 × 35Cl) + Na]+, 498.3 [M(2 × 35Cl) + Na]+, 296.1 [M + H-Fmoc]+.
(Alloc, Allyl/Fmoc) MeLan 8
(Alloc, allyl/Fmoc) MeLan 8 was prepared following the previously published route of Liu et al.,30c with the following modifications.
Synthesis of (DNs, Allyl/Fmoc, Tce) MeLan
A stirred solution of N-DNs/CO2allyl aziridine30c (100 mg, 0.25 mmol) in dry CH2Cl2 (1 mL) was cooled to −78 °C under Ar. Separate solutions of Fmoc-Cys-OTce (475 mg, 1.00 mmol) in dry CH2Cl2 (1 mL) and BF3·OEt2 (0.25 mL, 2.0 mmol) in dry CH2Cl2 (1 mL) were also prepared and cooled to −78 °C. The Fmoc-Cys-OTce solution was added dropwise to the aziridine, followed by the dropwise addition of the BF3·OEt2 solution. The reaction was then stirred at −78 °C for 15 min before being allowed to warm to r.t. and stirred for 48 h. The solvent was removed in vacuo before purification by flash column chromatography (pet. ether/EtOAc, 5:1), which yielded (DNs, allyl/Fmoc, Tce) MeLan as a yellow oil (30.0 mg, 0.035 mmol, 14%). Rf 0.55 (pet. ether/EtOAc, 7:3); [α]D25 +43.6 (c 5.0 mg mL–1, CHCl3); 1H NMR (600 MHz, CDCl3) δ 1.45 (d, J = 7.2 Hz, 3H), 2.94 (dd, J = 15.0, 7.2 Hz, 1H), 3.11 (dd, J = 14.0, 4.5 Hz, 1H), 3.52–3.57 (m, 1H), 4.25 (t, J = 6.9 Hz, 1H), 4.36 (dd, J = 9.6, 3.0 Hz, 1H), 4.41–4.50 (m, 4H), 4.71–4.77 (m, 2H), 4.86 (d, J = 12.0 Hz, 1H), 5.19 (dd, J = 10.2, 1.8 Hz, 1H), 5.21 (dd, J = 16.8, 1.2 Hz, 1H), 5.58 (d, J 7.8 Hz, 1H), 5.68–5.74 (m, 1H), 6.63 (d, J = 9.6 Hz, 1H), 7.33 (t, J = 7.5 Hz, 2H), 7.43 (t, J = 7.2 Hz, 2H), 7.61 (d, J = 6.0 Hz, 2H), 7.79 (d, J 7.2 Hz, 2H), 8.27 (d, J = 8.4 Hz, 1H), 8.49 (dd, J = 8.4, 2.4 Hz, 1H), 8.72 (d, J = 1.8 Hz, 1H); 13C{1H} (150 MHz, CDCl3) δ 19.7, 29.7, 43.5, 47.0, 53.7, 61.7, 66.9, 67.4, 74.7, 94.1, 120.1 (2 signals), 120.9, 125.0, 127.1 (2 signals), 127.8, 130.6, 131.9, 139.7, 141.3, 143.5, 147.7, 149.7, 155.7, 168.7, 168.9; HRMS (ES–) m/z: [M – H]− calcd for C33H30N4O12S2Cl3, 843.0367; found, 843.0362.
Synthesis of (Alloc, Allyl/Fmoc, Tce) MeLan
To a solution of (DNs, allyl/Fmoc, Tce) MeLan (22.5 mg, 0.027 mmol) in dry CH2Cl2 (300 μL) at 0 °C were added DIPEA (14 μL, 0.080 mmol) and thioglycolic acid (2.3 μL, 0.033 mmol), and the solution was stirred at r.t. for 2 h. After this time, the reaction was cooled to 0 °C, and DIPEA (14 μL, 0.080 mmol) and Alloc-Cl (4.5 μL, 0.042 mmol) were added. The solution was allowed to warm to r.t. and stirred for 15 h before removal of the solvent in vacuo. Purification by flash column chromatography (pet. ether/EtOAc, 4:1) yielded (Alloc, allyl/Fmoc, Tce) MeLan as a clear oil (11.0 mg, 0.016 mmol, 59%). Rf 0.71 (pet. ether/EtOAc, 7:3); [α]D25 −23.2 (c 5.0 mg mL–1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 1.36 (d, J = 7.0 Hz, 3H), 3.02 (dd, J = 13.5, 6.0 Hz, 1H), 3.14 (dd, J = 14.0, 4.5 Hz, 1H), 3.42–3.48 (m, 1H), 4.26 (t, J = 7.0 Hz, 1H), 4.45 (d, J = 7.0 Hz, 2H), 4.57–4.75 (m, 6H), 4.77 (d, J = 12.0 Hz, 1H), 4.87 (d, J = 12.0 Hz, 1H), 5.22–5.39 (m, 4H), 5.57 (d, J = 9.1 Hz, 1H), 5.69 (d, J = 9.0 Hz, 1H), 5.88–5.97 (m, 2H), 7.33 (td, J = 7.5, 0.9 Hz, 2H), 7.42 (t, J = 7.4 Hz, 2H), 7.62 (d, J = 6.1 Hz, 2H), 7.78 (d, J = 7.4 Hz, 2H); 13C{1H} (125 MHz, CDCl3) δ 19.4, 33.5, 44.0, 47.0, 53.7, 58.4, 66.1, 66.5, 67.4, 74.7, 94.2, 118.0, 119.5, 120.0, 125.0, 127.1, 127.8, 131.2, 132.4, 141.3, 143.6, 143.7, 155.7, 156.2, 168.9, 170.1; HRMS (ES+) m/z: [M + H]+ calcd for C31H34N2O8SCl3, 699.1102; found, 699.1100.
Synthesis of (Alloc, Allyl/Fmoc) MeLan 8
To a stirred solution of (Alloc, allyl/Fmoc, Tce) MeLan (48.0 mg, 0.069 mmol) in THF (9.6 mL) was slowly added zinc dust (44.8 mg, 0.69 mmol) followed by aqueous NH4OAc (1 M solution, 0.51 mL). The solution was stirred at r.t. for 5 h and filtered, and the solvent was removed in vacuo. The residue was redissolved in CH2Cl2 (25 mL), washed with brine (50 mL), and dried (MgSO4), and the solvent was removed in vacuo. Purification by flash column chromatography (CH2Cl2/MeOH, 9:1) yielded 8 as an off-white solid (30.9 mg, 0.054 mmol, 78%). Rf 0.59 (CH2Cl2/MeOH, 9:1); [α]D25 −1.38 (c 2.7 mg mL–1, CH2Cl2), [lit.30c [α]D −0.03 (c 4.0 mg mL–1, CH2Cl2)]; 1H NMR (600 MHz, CDCl3) δ 1.29 (d, J = 7.2 Hz, 3H), 2.88 (dd, J = 13.2, 7.2 Hz, 1H), 3.11 (dd, J = 15.0, 4.8 Hz, 1H), 3.36–3.38 (m, 1H), 4.19–4.30 (m, 3H), 4.42–4.44 (m, 2H), 4.54 (m, 2H), 4.63–4.65 (m, 2H), 5.17 (d, J = 10.2 Hz, 1H), 5.21 (d, J = 10.2 Hz, 1H), 5.31 (dd, J = 17.4, 1.2 Hz, 1H), 5.35 (dd, J = 17.4, 1.8 Hz, 1H), 5.89–5.97 (m, 2H), 7.31 (t, J = 7.2 Hz, 2H), 7.39 (t, J = 7.4 Hz, 2H), 7.68 (d, J = 7.2 Hz, 2H), 7.80 (d, J = 7.5 Hz, 2H); 13C{1H} (150 MHz, CDCl3) δ 19.9, 30.9, 44.0, 48.5, 57.3, 60.6, 67.0, 67.2, 68.1, 117.9, 119.3, 121.1, 126.4, 126.5, 128.4, 128.9, 133.3, 134.3, 142.7, 145.4, 145.5, 155.5, 155.9, 169.1, 171.9; LCMS (ES+) m/z: [M + H]+ calcd for C29H33N2O8S, 569.2; found, 569.2.
(2R,3R)-Fmoc-β-MeCys(Trt)-OH 29
(2R,3R)-Fmoc-β-MeCys(Trt)-OH 29 was prepared via the ring opening with trityl thiol of a l-threonine-derived aziridine,42a followed by deprotection, as follows.
Synthesis of (2R,3R)-Fmoc-β-MeCys(Trt)-OAllyl
To a stirred solution of (2S,3S)-allyl-3-methyl-1-(9-fluorenylmethoxycarbonyl)aziridine-2-carboxylate42a (1.29 g, 3.55 mmol) in dry CH2Cl2 (30 mL) under Ar at 0 °C was added triphenylmethanethiol (3.43 g, 12.4 mmol) followed by the dropwise addition of a solution of BF3·OEt2 (0.92 mL, 7.46 mmol) in dry CH2Cl2 (5.7 mL). The reaction was stirred for 3 h before quenching with saturated aqueous NaHCO3. The layers were separated, and the aqueous layer was further extracted with CH2Cl2 (2 × 20 mL). The organic layers were then combined and dried (MgSO4), and the solvent was removed in vacuo. Purification by flash column chromatography (pet. ether/EtOAc, 9:1 → 7:1) yielded the title compound as a white solid (730 mg, 1.14 mmol, 32%). Rf 0.29 (pet. ether/EtOAc, 5:1); mp 44–46 °C; [α]D20 +22.0 (c 12.0 mg mL–1, CHCl3); 1H NMR (600 MHz, CDCl3) δ 0.88 (d, J = 7.2 Hz, 3H), 2.77–2.81 (m, 1H), 4.26 (t, J = 6.9 Hz, 1H), 4.41 (d, J = 6.6 Hz, 2H), 4.42–4.48 (m, 2H), 4.66 (dd, J = 13.2, 6.0 Hz, 1H), 5.24 (dd, J = 10.2, 1.2 Hz, 1H), 5.31 (dd, J = 17.4, 1.2 Hz, 1H), 5.40 (d, J = 9.0 Hz, 1H), 5.80–5.86 (m, 1H), 7.22 (t, J = 7.2 Hz, 3H), 7.28–7.35 (m, 8H), 7.40–7.44 (m, 2H), 7.52 (d, J = 7.8 Hz, 6H), 7.63 (t, J = 7.2 Hz, 2H), 7.78 (t, J = 7.2 Hz, 2H); 13C{1H} (150 MHz, CDCl3) δ 20.1, 42.9, 47.1, 59.4, 66.2, 67.2, 67.7, 119.0, 120.0, 125.1, 126.7, 127.1, 127.7, 127.9, 129.6, 131.4, 141.3, 143.7, 144.6, 156.1, 170.1; νmax (cm–1) 3402, 3056, 2948, 1720; HRMS (ES+) m/z: [M + Na]+ calcd for C41H37NO4SNa, 662.2341; found, 662.2345.
Synthesis of (2R,3R)-Fmoc-β-MeCys(Trt)-OH
To a solution of (2R,3R)-Fmoc-β-Me-Cys(Trt)-OAllyl (730 mg, 1.14 mmol) in degassed CH2Cl2 (47 mL) under Ar were added PhSiH3 (0.282 mL, 2.28 mmol) and Pd(PPh3)4 (132 mg, 0.114 mmol). The solution was stirred in the dark for 2 h before removal of the solvent in vacuo. Purification by flash column chromatography (CH2Cl2/MeOH, 9:1) yielded 29 as a brown solid (162 mg, 0.270 mmol, 24%). Rf 0.61 (CH2Cl2/MeOH, 9:1); mp 113–116 °C; [α]D20 + 7.2 (c 13.2 mg mL–1, CHCl3); 1H NMR (600 MHz, CD3OD) δ 0.78 (d, J = 7.2 Hz, 3H), 2.66–2.70 (m, 1H), 4.22 (t, J = 6.9 Hz, 1H), 4.29–4.37 (m, 2H), 4.41 (dd, J = 10.8, 6.9 Hz, 1H), 7.19 (t, J = 7.2 Hz, 3H), 7.25–7.30 (m, 8H), 7.38 (q, J = 7.2 Hz, 2H), 7.49 (dd, J = 8.1, 0.9 Hz, 6H), 7.65 (dd, J = 10.8, 8.1 Hz, 2H), 7.78 (dd, J = 7.2, 2.7 Hz, 2H); 13C{1H} (150 MHz, CD3OD) δ 19.3, 43.8, 48.6, 61.1, 68.1, 69.0, 121.1, 126.5, 127.9, 128.3, 128.9, 129.0, 131.0, 142.7, 145.3, 146.4, 158.4, 173.7; νmax (cm–1) 3059, 2948, 1722; HRMS (ES–) m/z: [M – H]− calcd for C38H32NO4S, 598.2051; found, 598.2052.
General Procedures for Peptide Synthesis
Peptides were synthesized by hand using the Fmoc solid-phase synthesis strategy. All residues were added to the peptides manually. The resin was continually agitated throughout coupling, deprotection, and cleavage steps by shaking at 480 rpm on an IKA KS130 basic platform shaker. For reactions requiring heating as well as shaking, a Bioer Mixing Block MB-102 was used. Microwave reactions were conducted using a Personal Chemistry Smith Creator microwave-assisted organic synthesizer system in 5 mL reaction vials with maximum 300 W power. An Eppendorf centrifuge model 5810R was used for centrifugation of peptide products before freeze-drying by a SP Scientific VirTis BenchTop Pro. All steps not conducted in the microwave were performed while shaking at room temperature in a 5 mL PP reaction syringe with a frit. The resin was washed copiously with DMF following each coupling and deprotection. Washing the resin refers to the addition of solvent to the resin followed by immediate evacuation. Mini-cleave experiments were carried out at key steps to check the progress of the syntheses. One to 2 mg of beads was removed from the reaction vessel and cleaved using cleavage cocktail 1 or 2 (below), and the crude peptide was analyzed by LCMS. LCMS traces showing the presence of intermediates 21, 26, and 32 are presented in the Supporting Information.
All peptides were purified by preparative reverse-phase HPLC on a Dionex 580 HPLC System with a PDA-100 photodiode array detector, P580 Pump, and a model ASI-100 automated sample injector. A Phenomenex Onyx C18 100 × 10 mm column, a Dr. Maisch GmbH Reprosil Gold 200 C8 5 μm 150 × 10 mm column, or an Agilent Zorbax 300SB-C18 5 μm 250 × 9.4 mm column was used (as stated), with detection at 214 and 254 nm. Water (0.1% TFA) and acetonitrile (0.1% TFA) were used as solvents. Chromatograms were analyzed using Chromeleon software version 2.0. Analytical HPLC of peptides was performed on the above-described machine or an Agilent Technologies 1260 Infinity system using either a Fluka Analytical Discovery BIO C18-10 25 × 4.6 mm column, an ACE5 C18-300 150 × 4.6 mm column, or a Dr. Maisch GmbH Reprosil Gold 200 C8 5μm 250 × 4.6 mm column (as stated), with detection at 214 and 254 nm. A linear solvent gradient of 2–98% MeCN (0.1% TFA) in H2O (0.1% TFA) over 15 min was used at a flow rate of 1 mL min–1.
General Methods for Peptide Synthesis
The coupling, deprotection, and cyclization protocols used for all peptide syntheses are described in this section. The exact masses and volumes of amino acid, coupling reagent, and base used per coupling step are indicated in the individual procedures for each peptide synthesized.
Swelling the Resin
DMF (2 mL) was added to the resin in a syringe and shaken for 30 min. The DMF was then evacuated, and the resin was washed with DMF (2 × 2 mL).
Fmoc Deprotection
A solution of piperidine in DMF (40% v/v, 1.5 mL) was added to the resin and left to shake for 3 min. After this time, the syringe was evacuated. Another portion of piperidine in DMF (20% v/v, 1.5 mL) was then added to the resin and left to stir for 10 min. This was evacuated, and the resin was washed with DMF (6 × 2 mL).
Coupling Steps for Lanthionine-Containing Peptides
(Teoc, TMSE/Fmoc) Lan 4 (3 equiv), HOAt (5 equiv), and PyAOP (5 equiv) were dissolved in DMF (2 mL) in a glass vial, and DIPEA (10 equiv) was added. This solution was left to preactivate for 2 min and then, along with the resin, was transferred to a microwave vial and coupled in the microwave at 60 °C for 5 min followed by a further 1 h stirring at r.t. The resin and coupling solution were then transferred back to the reaction syringe, the coupling solution was removed, and the resin was washed thoroughly with DMF (4 × 2 mL).
Coupling Steps for Methyllanthionine-Containing Peptides
The same procedure was followed exactly for methyllanthionine-containing peptides, except that (Alloc, allyl/Fmoc) MeLan 8 (2.5 equiv), HOBt (5 equiv), PyBOP (5 equiv), and NMM (10 equiv) were used in place of (Teoc, TMSE/Fmoc) Lan 4, HOAt, PyAOP, and DIPEA.
Double Coupling of Normal Fmoc-Protected Amino Acids
The desired Fmoc-protected amino acid (5 equiv), HOAt (5 equiv), and PyAOP (5 equiv) were dissolved in DMF (1.5 mL for 50 mg scale reactions and 2 mL for 100 and 150 mg scale reactions), and DIPEA (10 equiv) was added. This solution was left to preactivate for 2 min and then added to the syringe containing the resin. The suspension was stirred at r.t. for 2 h before removal of the coupling solution. A fresh sample of the same preactivated coupling solution was then added to the resin and left to stir for 2 h before evacuation. The resin was then washed with DMF (4 × 2 mL).
Ring Closing Steps
The silyl or allyl protecting groups were first removed. To remove silyl groups, a solution of TBAF (1 M in THF, 1 mL) in DMF (1 mL) was added to the resin and left to stir at r.t. under Ar for 1 h. After this time, the TBAF solution was removed, and the resin was washed with DMF (6 × 2 mL). To remove allyl groups, a solution of Pd(PPh3)4 (2 equiv) and PhSiH3 (10 equiv) in CH2Cl2/DMF (1:1, 2 mL) was added to the resin and stirred in the dark for 2 h. The deprotection solution was then removed, and the resin was washed with CH2Cl2 (5 × 2 mL), sodium diethyldithiocarbamate (0.5% w/v in DMF, 5 × 3 mL), and DMF (5 × 2 mL). The terminal Fmoc group was then removed as described above.
Lanthionine-Containing Peptides
A solution of HOAt (5 equiv), PyAOP (5 equiv), and DIPEA (10 equiv) in DMF (1.5 mL for 50 mg scale reactions and 2 mL for 100 and 150 mg scale reactions) was preactivated and then, along with the resin, was transferred to a microwave vial and coupled in the microwave at 60 °C for 5 min followed by a further 1 h stirring at r.t. The resin and coupling solution were then transferred back to the reaction syringe, and the coupling solution was evacuated before adding a fresh solution of activated coupling reagents to the resin and leaving to stir for 2 h. The coupling solution was then removed, and the resin was washed with DMF (4 × 2 mL).
Methyllanthionine-Containing Peptides
A solution of HOBt (5 equiv), PyAOP (5 equiv), and NMM (10 equiv) in DMF (2 mL) was preactivated and then, along with the resin, was transferred to a microwave vial and coupled in the microwave at 60 °C for 5 min followed by a further 1 h stirring at r.t. The resin and coupling solution were then transferred back to the reaction syringe, and the coupling solution was evacuated before adding a fresh sample of the same preactivated coupling solution to the resin and leaving to stir for 2 h. The solution was then evacuated, and the same 2 h r.t. coupling was repeated one further time before evacuating the solution and washing the resin with DMF (4 × 2 mL).
Cleavage
Cleavage cocktail 1 (for cysteine-containing peptides) is composed of TFA (940 μL), EDT (25 μL), water (25 μL), and TIPS (10 μL), and cleavage cocktail 2 (for peptides containing no cysteine) is composed of TFA (965 μL), water (25 μL), and TIPS (10 μL).
The resin was washed with CH2Cl2 (3 × 2 mL), MeOH (2 × 2 mL), and ether (2 × 2 mL) and then dried in vacuo for 30 min before cleavage. The cleavage cocktail (1 mL) was premixed in a glass vial before addition to the resin. This was left to shake for 40 min before evacuating directly into a 15 mL Falcon tube containing cold ether (7 mL). A further portion of cleavage cocktail (1 mL) was then also added to the resin and left to shake for 30 min before adding to the Falcon tube. The volume was then made up to 14 mL with more cold ether. This was centrifuged at 4000 rpm at 5 °C for 15 min, after which time a precipitate formed. The ether was poured off, and fresh ether was added to resuspend the pellet before a further round of centrifugation (4000 rpm, 5 °C, 10 min). The resuspension and centrifugation process was repeated once more. The resultant pellet was dissolved in water (3 mL) and lyophilized to yield the crude peptide.
Elimination of Cysteine and β-Methylcysteine in Solution To Form Dha and Dhb
TCEP (0.44 equiv) was added to a solution of the purified cysteine-containing peptide in water (2.3 mg mL–1), and this was shaken on a shaker plate at 480 rpm at r.t. for 45 min. A solution of methyl 2,5-dibromopentanoate (60 equiv) in DMSO (2.3 mg mL–1) was then added to the peptide, followed by K2CO3 (150 equiv), and this was incubated at 37 °C while shaking at 500 rpm for 2 h. Following the reaction, the solution was filtered with a syringe filter, diluted with water, and purified directly by HPLC.
Peptide Synthesis
Mutacin I Ring B WT 1
Fmoc-Thr(tBu)-NovaSyn TGT resin (150 mg, 27.0 μmol) was swollen in DMF, and the Fmoc group was removed. (Teoc, TMSE/Fmoc) Lan 4 (55 mg, 81 μmol) was coupled using the microwave procedure, and the Fmoc group was then removed. The peptide chain was elongated using the double-coupling procedure described: Fmoc-Gly-OH (40 mg, 135 μmol) was added first, and the Fmoc group was then removed, followed by the addition of Fmoc-Leu-OH (48 mg, 135 μmol). The cyclization reaction was then carried out as described above. The silyl protecting groups were removed, followed by removal of the final Fmoc group. The cyclization reaction was then carried out as described for lanthionine-containing peptides. Following completion of the synthesis, the peptide was cleaved from the resin using the procedure described above, with cleavage cocktail 2. The crude peptide was purified by preparative reverse-phase HPLC using a semi-prep Phenomenex Onyx C18 100 × 10 mm column. A linear solvent gradient of 5–30% MeCN (0.1% TFA) in H2O (0.1% TFA) over 40 min at a flow rate of 1 mL min–1 was used. The fractions containing the target peptide were collected (retention time, 11.13 min) and lyophilized to give the pure sample as a fluffy white powder (4.2 mg, 7.31 μmol, 34%). HRMS (ES+) m/z: [M + H]+ calcd for C18H32N5O7S, 462.2022; found, 462.2020. 1H and 13C NMR data and assignments are tabulated in Tables S2–S5 (Supporting Information).
Nisin Ring B Lan Analogue 3
Fmoc-Lys(Boc)-NovaSyn TGT resin (100 mg, 18.0 μmol) was swollen in DMF, and the Fmoc group was removed. (Teoc, TMSE/Fmoc) Lan 4 (37 mg, 54 μmol) was coupled using the microwave procedure, and the Fmoc group was then removed. The peptide chain was elongated using the double-coupling procedure described: Fmoc-Gly-OH (27 mg, 90 μmol) was added first, and the Fmoc group was removed, followed by the addition of Fmoc-Pro-OH (30 mg, 90 μmol). The cyclization reaction was then carried out as described above. The silyl protecting groups were removed, followed by removal of the final Fmoc group. The cyclization reaction was then carried out as described for lanthionine-containing peptides. Following completion of the synthesis, the peptide was cleaved from the resin using the procedure described above, with cleavage cocktail 2. The crude peptide was purified by preparative reverse-phase HPLC using a semi-prep Agilent Zorbax 300SB-C18 5 μm 250 × 9.4 mm column. A linear solvent gradient of 5–40% MeCN (0.1% TFA) in H2O (0.1% TFA) over 25 min at a flow rate of 2 mL min–1 was used. The fractions containing the target peptide were collected (retention time, 9 min) and lyophilized to give the pure sample as a fluffy white powder (600 μg, 0.859 μmol, 7%). HRMS (ES+) m/z: [M + H]+ calcd for C19H33N6O6S, 473.2182; found, 473.2173. 1H and 13C NMR data and assignments are tabulated in Tables S6 and S7 (Supporting Information).
Nisin Ring B WT 2
Fmoc-Lys(Boc)-NovaSyn TGT resin (50 mg, 9.0 μmol) was swollen in DMF, and the Fmoc group was removed. (Alloc, allyl/Fmoc) MeLan 8 (15 mg, 23 μmol) was coupled using the microwave procedure, and the Fmoc group was then removed. The peptide chain was elongated using the double-coupling procedure described: Fmoc-Gly-OH (14 mg, 45 μmol) was added first, and the Fmoc group was removed, followed by the addition of Fmoc-Pro-OH (15 mg, 45 μmol). The cyclization reaction was then carried out as described above. The allyl protecting groups were first removed, followed by removal of the final Fmoc group. The cyclization reaction was then carried out as described for methyllanthionine-containing peptides. Following completion of the synthesis, the peptide was cleaved from the resin using the procedure described above with cleavage cocktail 2. The crude peptide was purified by preparative reverse-phase HPLC using a semi-prep Agilent Zorbax 300SB-C18 5 μm 250 × 9.4 mm column. A linear solvent gradient of 5–40% MeCN (0.1% TFA) in H2O (0.1% TFA) over 30 min at a flow rate of 2 mL min–1 was used. The fractions containing the target peptide were collected (retention time, 10 min) and lyophilized to give the pure sample as a fluffy white powder (1.6 mg, 2.25 μmol, 37%). HRMS (ES+) m/z: [M + H]+ calcd for C20H35N6O6S, 487.2339; found, 487.2339. 1H and 13C NMR data and assignments are tabulated in Tables S8 and S9 (Supporting Information).
Mutacin I Ring A Ser/Ala Analogue 14
Fmoc-Ala-NovaSyn TGT resin (50 mg, 8.5 μmol) was swollen in DMF, and the Fmoc group was removed. (Teoc, TMSE/Fmoc) Lan 4 (17 mg, 26 μmol) was coupled using the microwave procedure, and the Fmoc group was then removed. The peptide chain was elongated using the double-coupling procedure described: Fmoc-Leu-OH (15 mg, 43 μmol) was added first, and the Fmoc group was removed, followed by the addition of Fmoc-Ala-OH (14 mg, 43 μmol), removal of the Fmoc group, and coupling of a second Fmoc-Leu-OH (15 mg, 43 μmol). The cyclization reaction was then carried out as described above. The silyl protecting groups were removed, followed by removal of the final Fmoc group. The cyclization reaction was then carried out as described for lanthionine-containing peptides. For coupling of the final two amino acids of this peptide, best results were achieved by double-coupling with amino acid fluorides. To a solution of Fmoc-Ser(OtBu)-F39a,39b (10 mg, 25.5 μmol, 3 equiv) in dry CH2Cl2 (2 mL), DIPEA (4 μL, 25.5 μmol, 3 equiv) was added. This solution was then added to the resin and stirred at r.t. for 1 h. The solution was then evacuated, and the same coupling reaction was repeated. This solution was then evacuated, and the resin was washed with CH2Cl2 (2 × 2 mL) and DMF (4 × 2 mL). The Fmoc group was then removed. The same double-coupling procedure was employed to add the phenylalanine using solutions of Fmoc-Phe-F39a−39c (10 mg, 25.5 μmol, 3 equiv) and DIPEA (4 μL, 25.5 μmol, 3 equiv) in dry CH2Cl2 (2 mL). The final Fmoc group was then removed. Following completion of the synthesis, the peptide was cleaved from the resin using cleavage cocktail 2. The crude peptide was purified by preparative reverse-phase HPLC using a semi-prep Phenomenex Onyx C18 100 × 10 mm column. A linear solvent gradient of 5–40% MeCN (0.1% TFA) in H2O (0.1% TFA) over 40 min at a flow rate of 2 mL min–1 was used. The fractions containing the target peptide were collected (retention time, 22 min) and lyophilized to give the pure sample as a fluffy white powder (200 μg, 0.221 μmol, 3%). HRMS (ES+) m/z: [M + H]+ calcd for C36H57N8O10S, 793.3918; found, 793.3928. 1H and 13C NMR data and assignments are tabulated in Tables S10 and S11 (Supporting Information).
Mutacin I Ring A Ser Analogue 15
Fmoc-Ala-NovaSyn TGT resin (150 mg, 25.5 μmol) was swollen in DMF, and the Fmoc group was removed. (Teoc, TMSE/Fmoc) Lan 4 (52 mg, 77 μmol) was coupled using the microwave procedure, and the Fmoc group was then removed. The peptide chain was elongated using the double-coupling procedure described; Fmoc-Leu-OH (45 mg, 128 μmol) was added first, and the Fmoc group was removed, followed by the addition of Fmoc-Ser(tBu)-OH (49 mg, 128 μmol), removal of the Fmoc group, and coupling of a second Fmoc-Leu-OH (45 mg, 128 μmol). The cyclization reaction was then carried out as described above. The silyl protecting groups were first removed, followed by removal of the final Fmoc group. The cyclization reaction was then carried out as described for lanthionine-containing peptides. Following completion of the synthesis, the peptide was cleaved from the resin using cleavage cocktail 2. The crude peptide was purified by preparative reverse-phase HPLC using a semi-prep Phenomenex Onyx C18 100 × 10 mm column. A linear solvent gradient of 5–50% MeCN (0.1% TFA) in H2O (0.1% TFA) over 20 min at a flow rate of 2 mL min–1 was used. The fractions containing the target peptide were collected (retention time, 9 min) and lyophilized to give the pure sample as a fluffy white powder (800 μg, 1.16 μmol, 5%). HRMS (ES+) m/z: [M + H]+ calcd for C24H43N6O8S, 575.2863; found, 575.2859. 1H and 13C NMR data and assignments are tabulated in Tables S12 and S13 (Supporting Information).
Attempted Synthesis of Mutacin I Ring A 25 via Sec(Ph) Elimination
Fmoc-Ala-NovaSyn TGT resin (50 mg, 8.5 μmol) was swollen in DMF, and the Fmoc group was removed. (Teoc, TMSE/Fmoc) Lan 4 (17 mg, 26 μmol) was coupled using the microwave procedure, and the Fmoc group was then removed. The peptide chain was then elongated. Fmoc-Leu-OH (15 mg, 43 μmol) was added first using the double-coupling procedure described above. The Fmoc group was removed, followed by the addition of Fmoc-Sec(Ph)-OH (16 mg, 34 μmol, 4 equiv). This residue was coupled at r.t. for 2 h using HOBt (4 equiv) and DIC (4 equiv) in DMF (1.5 mL). After evacuation of the coupling solution, the resin was washed with DMF (2 × 4 mL). The Fmoc group was then removed, and a second Fmoc-Leu-OH (15 mg, 43 μmol) was added using the double-coupling procedure. The cyclization reaction was then carried out as described above. The silyl protecting groups were removed, followed by removal of the final Fmoc group. The cyclization reaction was then carried out as described for lanthionine-containing peptides, except that COMU (5 equiv) was used in place of PyAOP and HOAt. Following completion of the synthesis, the peptide was cleaved from the resin using cleavage cocktail 2. The crude peptide was purified by preparative reverse-phase HPLC using a semi-prep Phenomenex Onyx C18 100 × 10 mm column. A linear solvent gradient of 20–60% MeCN (0.1% TFA) in H2O (0.1% TFA) over 30 min at a flow rate of 2 mL min–1 was used. The fractions containing the target peptide were collected (retention time, 13 min) and lyophilized, yielding a fluffy white solid (200 μg, 0.3 μmol).
To form the dehydroalanine residue, a solution of the peptide in water (50 μL) and MeCN (30 μL) was first cooled to 0 °C in an ice bath before the addition of NaIO4 (40 μL of a 1.25 mg mL–1 stock solution in water). This was stirred for 1 h before filtering the reaction mixture with a syringe filter and purifying it directly by preparative reverse-phase HPLC. A semi-prep Phenomenex Onyx C18 100 × 10 mm column was used with a linear solvent gradient of 2–98% MeCN (0.1% TFA) in H2O (0.1% TFA) over 20 min at a flow rate of 2 mL min–1. The fractions containing the peptide were collected and lyophilized to give the pure sample as a white powder (100 μg, 0.149 μmol, 2%). HRMS (ES+) m/z: [M + H]+ calcd for C24H41N6O7S, 557.2758; found, 557.2760.
Mutacin I Ring A WT 12
Fmoc-Ala-NovaSyn TGT resin (100 mg, 17 μmol) was swollen in DMF, and the Fmoc group was removed. (Teoc, TMSE/Fmoc) Lan 4 (17 mg, 26 μmol) was coupled using the microwave procedure, and the Fmoc group was then removed. The peptide chain was then elongated. Fmoc-Leu-OH (30 mg, 85 μmol) was added first, and the Fmoc group was removed, followed by the addition of Fmoc-Cys(Trt)-OH (50 mg, 85 μmol), removal of the Fmoc group, and coupling of a second Fmoc-Leu-OH (30 mg, 85 μmol). The cyclization reaction was then carried out. The silyl protecting groups were removed, followed by removal of the final Fmoc group. The cyclization reaction was then carried out as described for lanthionine-containing peptides. The peptide chain was then further elongated using the double-coupling procedure. First, Fmoc-Cys(Trt)-OH (50 mg, 85 μmol) was added, and the Fmoc group was removed, followed by the addition of Fmoc-Phe-OH (33 mg, 85 μmol) and removal of the final Fmoc group. Following completion of the synthesis, the peptide was cleaved from the resin using cleavage cocktail 1. The crude peptide was purified by preparative reverse-phase HPLC using a semi-prep Phenomenex Onyx C18 100 × 10 mm column. A linear solvent gradient of 20–80% MeCN (0.1% TFA) in H2O (0.1% TFA) over 30 min at a flow rate of 2 mL min–1 was used. The fractions containing the target peptide were collected (retention time, 11 min) and lyophilized to give a fluffy white powder (2 mg, 2.4 μmol).
The dehydroalanine residues were formed by elimination of cysteine according to General Procedures above. The peptide was then purified by preparative reverse-phase HPLC using a semi-prep Phenomenex Onyx C18 100 × 10 mm column. A linear solvent gradient of 20–55% MeCN (0.1% TFA) in H2O (0.1% TFA) over 30 min at a flow rate of 2 mL min–1 was used. The fractions containing the peptide were collected (retention time, 13 min) and lyophilized to give the pure sample as a fluffy white powder (1.5 mg, 1.69 μmol, 11%). HRMS (ES+) m/z: [M + H]+ calcd for C36H53N8O9S, 773.3656; found, 773.3660. 1H and 13C NMR data and assignments are tabulated in Tables S14 and S15 (Supporting Information).
Nisin Ring A WT 13
Fmoc-Ala-NovaSyn TGT resin (100 mg, 21 μmol) was swollen in DMF, and the Fmoc group was removed. (Teoc, TMSE/Fmoc) Lan 4 (43 mg, 63 μmol) was coupled using the microwave procedure, and the Fmoc group was then removed. The peptide chain was then elongated. Fmoc-Leu-OH (37 mg, 105 μmol) was added first, and the Fmoc group was removed, followed by the addition of Fmoc-Cys(Trt)-OH (62 mg, 105 μmol), removal of the Fmoc group, and coupling of Fmoc-Ile-OH (37 mg, 105 μmol). The cyclization reaction was then carried out. The silyl protecting groups were removed, followed by removal of the final Fmoc group. The cyclization reaction was then carried out as described for lanthionine-containing peptides. The peptide chain was then further elongated using the double-coupling procedure described in Coupling Steps for Lanthionine-Containing Peptides. First, Fmoc-β-Me-Cys(Trt)-OH (2 equiv (12 mg, 42 μmol) per coupling) was added, and the Fmoc group was removed, followed by the addition of Fmoc-Ile-OH (37 mg, 105 μmol) and removal of the final Fmoc group. Following completion of the synthesis, the peptide was cleaved from the resin using cleavage cocktail 1. The crude peptide was purified by preparative reverse-phase HPLC using a semi-prep Phenomenex Onyx C18 100 × 10 mm column. A linear solvent gradient of 20–80% MeCN (0.1% TFA) in H2O (0.1% TFA) over 30 min at a flow rate of 2 mL min–1 was used. The fractions containing the target peptide were collected (retention time, 11 min) and lyophilized to give a fluffy white powder (2 mg, 2.4 μmol).
The dehydro residues were formed according to General Procedures above. The peptide was then purified by preparative reverse-phase HPLC using a semi-prep Agilent Zorbax 300SB-C18 5 μm 250 × 9.4 mm column. A linear solvent gradient of 20–70% MeCN (0.1% TFA) in H2O (0.1% TFA) over 28 min at a flow rate of 2 mL min–1 was used. The fractions containing the peptide were collected (retention time, 22 min) and lyophilized to give the pure sample as a fluffy white powder (200 μg, 0.231 μmol, 1%). HRMS (ES+) m/z: [M + H]+ calcd for C34H57N8O9S, 753.3969; found, 753.3948. 1H and 13C NMR data and assignments are tabulated in Tables S16 and S17 (Supporting Information).
Peptide Structure Calculation Protocol
For restraint generation, distance restraints for structure calculation were determined from 2D 1H–1H NOESY spectra (mixing time, 0.6 s) using the “Make Distance Restraints” tool in CCPN Analysis.58 Only inter-residue restraints were used for structure calculation. Backbone ϕ and ψ angle restraints were either calculated from 3JHN–Hα coupling constants measured directly from 1D 1H spectra using constants for the Karplus equation reported by Vögeli et al.59 or predicted using the TALOS-N server from Bax’s group.60,61 Geometry of Xaa–Pro peptide bonds was determined by examination of the Pro β and γ 13C shifts and prediction using the Promega server from Bax and Shen.46
Structure Calculation
The additional topology and parameters required for the unusual amino acids (lanthionine, methyllanthionine, dehydroalanine, and dehydrobutyrine) were based on the parameterization used by Turpin et al.49 and were added to the XPLOR .top and .par files. Patch residues were created for the Lan bridge from two cysteines and for the MeLan bridge from an α-aminobutyric acid (Abu) and a cysteine. New topology for the Abu residue itself was based on the topology of Thr, with the OH group replaced with an additional βH.19 New topologies then needed to be built for the dehydro residues to accurately describe the effect of the α,β-unsaturation. The parameterization is described in full in the Supporting Information. All of the bond lengths, angles, impropers, and dihedrals for these residues were based on the CHARMM force field parameters for Dha and Dhb developed by Turpin et al.49 The angle and distance restraints were then used in structure calculation in XPLOR-NIH version 2.45.43,62 The calculation procedure was as follows: a .psf file was created from the peptide sequence, followed by calculation of an initial extended structure. A family of 100–250 structures was then generated with simulated annealing using NOE and dihedral restraints (heating to 1000 K over 6000 time steps of 0.005 or 0.003 ps, followed by cooling over 3000 time steps). This family of structures was then refined using a similar simulated annealing protocol (cooling was conducted over 2000 time steps to a final temperature of 100 K) over two to five rounds of refinement, followed by selection of the 15 lowest-energy structures for analysis. Figures were generated using PyMOL (The PyMOL Molecular Graphics System, version 1.8, Schrödinger, LLC). Ensembles of the lowest-energy structures were validated using the Protein Structure Validation Software Suite (PSVS) version 1.5 (http://psvs-1_5-dev.nesg.org/).63
Acknowledgments
We thank the EPSRC for a Ph.D. studentship (EP/L504889/1) and for the award of an EPSRC Doctoral Prize Fellowship (EP/N509577/1) (to R.D.) and the Department of Chemistry, UCL, for the award of a Ph.D. studentship (to S.A.M.). The Wellcome Trust (101569/z/13/z) and the BBSRC (BB/R000255/1) are acknowledged for supporting the ISMB NMR facility at University College London. We would also like to thank Shang-Te Danny Hsu (Institute of Biological Chemistry, Academia Sinica, Taiwan) and Alexandre Bonvin (Utrecht University, The Netherlands) for helpful discussions and for providing the revised structure of the 1:1 nisin–lipid II complex.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.9b01253.
Figures S1 and S2; Scheme S1; 1H NMR spectra for 29 and key synthetic intermediates; spectra parameters for 2D spectra; NMR, mass spectra, and HPLC data for all peptides after purification; XPLOR-NIH parameterization data; structure statistics; and dihedral angle data (PDF)
Coordinates for peptide 14 (CIF)
Coordinates for peptide 12 (CIF)
Coordinates for peptide 15 (CIF)
Coordinates for peptide 13 (CIF)
Coordinates for peptide 1 (minor conformer) (CIF)
Coordinates for peptide 1 (major conformer) (CIF)
Coordinates for peptide 3 (CIF)
Coordinates for peptide 2 (CIF)
Accession Codes
The coordinates for the structures of the cyclic peptides have been deposited in the Protein Data Bank with accession codes 6QTF (1, major conformer) and 6QYR (1, minor conformer), 6QYS (2), 6QM1 (3), 6QYU (12), 6QYW (13), 6QYV (14), and 6QYT (15).
The authors declare no competing financial interest.
Supplementary Material
References
- a Smith R.; Coast J. The true cost of antimicrobial resistance. Br. Med. J. 2013, 346, f1493. 10.1136/bmj.f1493. [DOI] [PubMed] [Google Scholar]; b World Health Organisation . Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: 2014. [Google Scholar]; c O’Neill J.Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; HM Government: London, UK, 2016. [Google Scholar]
- a Lewis K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]; b Harvey A. L.; Edrada-Ebel R.; Quinn R. J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. 10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]; c Mousa W. K.; Athar B.; Merwin N. J.; Magarvey N. A. Antibiotics and specialized metabolites from the human microbiota. Nat. Prod. Rep. 2017, 34, 1302–1331. 10.1039/C7NP00021A. [DOI] [PubMed] [Google Scholar]; d Adnani N.; Rajski S. R.; Bugni T. S. Symbiosis-inspired approaches to antibiotic discovery. Nat. Prod. Rep. 2017, 34, 784–814. 10.1039/C7NP00009J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Henninot A.; Collins J. C.; Nuss J. M. The Current State of Peptide Drug Discovery: Back to the Future?. J. Med. Chem. 2018, 61, 1382–1414. 10.1021/acs.jmedchem.7b00318. [DOI] [PubMed] [Google Scholar]; b Zipperer A.; Konnerth M. C.; Laux C.; Berscheid A.; Janek D.; Weidenmaier C.; Burian M.; Schilling N. A.; Slavetinsky C.; Marchal M.; Willmann M.; Kalbacher H.; Schittek B.; Brötz-Oesterheld H.; Grond S.; Peschel A.; Krismer B. Human commensals producing a novel antibiotic impair pathogen colonization. Nature 2016, 535, 511–516. 10.1038/nature18634. [DOI] [PubMed] [Google Scholar]; c Chu J.; Vila-Farres X.; Inoyama D.; Ternei M.; Cohen L. J.; Gordon E. A.; Reddy B. V. B.; Charlop-Powers Z.; Zebroski H. A.; Gallardo-Macias R.; Jaskowski M.; Satish S.; Park S.; Perlin D. S.; Freundlich J. S.; Brady S. F. Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat. Chem. Biol. 2016, 12, 1004–1006. 10.1038/nchembio.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ling L. L.; Schneider T.; Peoples A. J.; Spoering A. L.; Engels I.; Conlon B. P.; Mueller A.; Schäberle T. F.; Hughes D. E.; Epstein S.; Jones M.; Lazarides L.; Steadman V. A.; Cohen D. R.; Felix C. R.; Fetterman K. A.; Millett W. P.; Nitti A. G.; Zullo A. M.; Chen C.; Lewis K. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bakhtiary A.; Cochrane S. A.; Mercier P.; McKay R. T.; Miskolzie M.; Sit C. S.; Vederas J. C. Insights into the Mechanism of Action of the Two-Peptide Lantibiotic Lacticin 3147. J. Am. Chem. Soc. 2017, 139, 17803–17810. 10.1021/jacs.7b04728. [DOI] [PubMed] [Google Scholar]
- Chatterjee C.; Paul M.; Xie L.; van der Donk W. A. Biosynthesis and mode of action of lantibiotics. Chem. Rev. 2005, 105, 633–684. 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]
- a Knerr P. J.; van der Donk W. A. Discovery, Biosynthesis, and Engineering of Lantipeptides. Annu. Rev. Biochem. 2012, 81, 479–505. 10.1146/annurev-biochem-060110-113521. [DOI] [PubMed] [Google Scholar]; b Smith L.; Hillman J. D. Therapeutic potential of type A (I) lantibiotics, a group of cationic peptide antibiotics. Curr. Opin. Microbiol. 2008, 11, 401–408. 10.1016/j.mib.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Breukink E.; de Kruijff B. Lipid II as a target for antibiotics. Nat. Rev. Drug. Discov. 2006, 5, 321–323. 10.1038/nrd2004. [DOI] [PubMed] [Google Scholar]; b Hasper H. E.; Kramer N. E.; Smith J. L.; Hillman J. D.; Zachariah C.; Kuipers O. P.; de Kruijff B.; Breukink E. An alternative bactericidal mechanism of action for lantibiotic peptides that target lipid II. Science 2006, 313, 1636–1637. 10.1126/science.1129818. [DOI] [PubMed] [Google Scholar]; c Münch D.; Sahl H.-G. Structural variations of the cell wall precursor lipid II in Gram-positive bacteria - Impact on binding and efficacy of antimicrobial peptides. Biochim. Biophys. Acta 2015, 1848, 3062–3071. 10.1016/j.bbamem.2015.04.014. [DOI] [PubMed] [Google Scholar]; d Oppedijk S. F.; Martin N. I.; Breukink E. Hit ’em where it hurts: The growing and structurally diverse family of peptides that target lipid-II. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 947–957. 10.1016/j.bbamem.2015.10.024. [DOI] [PubMed] [Google Scholar]
- Ng V.; Chan W. C. New Found Hope for Antibiotic Discovery: Lipid II Inhibitors. Chem. – Eur. J. 2016, 22, 12606–12616. 10.1002/chem.201601315. [DOI] [PubMed] [Google Scholar]
- a Rogers L. A. The inhibiting effect of Streptococcus lactis on Lactobacillus bulgaricus. J. Bacteriol. 1928, 16, 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gross E.; Morell J. L. Structure of nisin. J. Am. Chem. Soc. 1971, 93, 4634–4635. 10.1021/ja00747a073. [DOI] [PubMed] [Google Scholar]; c Morell J. L.; Gross E. Configuration of the β-carbon atoms of the β-methyllanthionine residues in nisin. J. Am. Chem. Soc. 1973, 95, 6480–6481. 10.1021/ja00800a069. [DOI] [PubMed] [Google Scholar]
- a De Arauz L. J.; Jozala A. F.; Mazzola P. G.; Penna T. C. V. Nisin biotechnological production and application: a review. Trends Food Sci. Technol. 2009, 20, 146–154. 10.1016/j.tifs.2009.01.056. [DOI] [Google Scholar]; b Cleveland J.; Montville T. J.; Nes I. F.; Chikindas M. L. Bacteriocins: safe, natural antimicrobials for food preservation. Int. J. Food Microbiol. 2001, 71, 1–20. 10.1016/S0168-1605(01)00560-8. [DOI] [PubMed] [Google Scholar]
- a Zhou H.; Fang J.; Tian Y.; Lu X. Y. Mechanisms of nisin resistance in Gram-positive bacteria. Ann. Microbiol. 2014, 64, 413–420. 10.1007/s13213-013-0679-9. [DOI] [Google Scholar]; b Draper L. A.; Cotter P. D.; Hill C.; Ross R. P. Lantibiotic Resistance. Microbiol. Mol. Biol. Rev. 2015, 79, 171–191. 10.1128/MMBR.00051-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Brötz H.; Josten M.; Wiedemann I.; Schneider U.; Götz F.; Bierbaum G.; Sahl H.-G. Role of lipid-bound peptidoglycan precursors in the formation of pores by nisin, epidermin and other lantibiotics. Mol. Microbiol. 1998, 30, 317–327. 10.1046/j.1365-2958.1998.01065.x. [DOI] [PubMed] [Google Scholar]; b Breukink E.; Wiedemann I.; van Kraaij C.; Kuipers O. P.; Sahl H.-G.; de Kruijff B. Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic. Science 1999, 286, 2361–2364. 10.1126/science.286.5448.2361. [DOI] [PubMed] [Google Scholar]; c Wiedemann I.; Breukink E.; van Kraaij C.; Kuipers O. P.; Bierbaum G.; de Kruijff B.; Sahl H.-G. Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J. Biol. Chem. 2001, 276, 1772–1779. 10.1074/jbc.M006770200. [DOI] [PubMed] [Google Scholar]
- a Breukink E.; van Heusden H. E.; Vollmerhaus P. J.; Swiezewska E.; Brunner L.; Walker S.; Heck A. J. R.; de Kruijff B. Lipid II is an intrinsic component of the pore induced by nisin in bacterial membranes. J. Biol. Chem. 2003, 278, 19898–19903. 10.1074/jbc.M301463200. [DOI] [PubMed] [Google Scholar]; b Hasper H. E.; de Kruijff B.; Breukink E. Assembly and stability of nisin-Lipid II pores. Biochemistry 2004, 43, 11567–11575. 10.1021/bi049476b. [DOI] [PubMed] [Google Scholar]
- Hsu S.-T. D.; Breukink E.; Tischenko E.; Lutters M. A. G.; de Kruijff B.; Kaptein R.; Bonvin A. M. J. J.; van Nuland N. A. J. The nisin-lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat. Struct. Mol. Biol. 2004, 11, 963–967. 10.1038/nsmb830. [DOI] [PubMed] [Google Scholar]
- Medeiros-Silva J.; Jekhmane S.; Paioni A. L.; Gawarecka K.; Baldus M.; Swiezewska E.; Breukink E.; Weingarth M. High-resolution NMR studies of antibiotics in cellular membranes. Nat. Commun. 2018, 9, 3963. 10.1038/s41467-018-06314-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland S.; Turpin E. R.; Bonev B. B.; Hirst J. D. Docking and molecular dynamics simulations of the ternary complex nisin2:lipid II. Sci. Rep. 2016, 6, 21185. 10.1038/srep21185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch D. C.; Schmidt T. H.; Sahl H.-G.; Kubitscheck U.; Kandt C. Structural dynamics of the cell wall precursor lipid II in the presence and absence of the lantibiotic nisin. Biochim. Biophys. Acta 2014, 1838, 3061–3068. 10.1016/j.bbamem.2014.07.024. [DOI] [PubMed] [Google Scholar]
- van Heusden H. E.; de Kruijff B.; Breukink E. Lipid II induces a transmembrane orientation of the pore-forming peptide lantibiotic nisin. Biochemistry 2002, 41, 12171–12178. 10.1021/bi026090x. [DOI] [PubMed] [Google Scholar]
- Hsu S. T. D.; Breukink E.; Bierbaum G.; Sahl H. G.; de Kruijff B.; Kaptein R.; van Nuland N. A. J.; Bonvin A. M. J. J. NMR study of mersacidin and lipid II interaction in dodecylphosphocholine micelles - Conformational changes are a key to antimicrobial activity. J. Biol. Chem. 2003, 278, 13110–13117. 10.1074/jbc.M211144200. [DOI] [PubMed] [Google Scholar]
- a Mulders J. W. M.; Boerrigter I. J.; Rollema H. S.; Siezen R. J.; Vos W. M. Identification and characterization of the lantibiotic nisin Z, a natural nisin variant. Eur. J. Biochem. 1991, 201, 581–584. 10.1111/j.1432-1033.1991.tb16317.x. [DOI] [PubMed] [Google Scholar]; b Zendo T.; Fukao M.; Ueda K.; Higuchi T.; Nakayama J.; Sonomoto K. Identification of the lantibiotic Nisin Q, a new natural nisin variant produced by Lactococcus lactis61-14 isolated from a river in Japan. Biosci., Biotechnol., Biochem. 2003, 67, 1616–1619. 10.1271/bbb.67.1616. [DOI] [PubMed] [Google Scholar]; c Wirawan R. E.; Klesse N. A.; Jack R. W.; Tagg J. R. Molecular and genetic characterization of a novel nisin variant produced by Streptococcus uberis. Appl. Environ. Microbiol. 2006, 72, 1148–1156. 10.1128/AEM.72.2.1148-1156.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Rink R.; Wierenga J.; Kuipers A.; Kluskens L. D.; Driessen A. J. M.; Kuipers O. P.; Moll G. N. Dissection and Modulation of the Four Distinct Activities of Nisin by Mutagenesis of Rings A and B and by C-Terminal Truncation. Appl. Environ. Microbiol. 2007, 73, 5809–5816. 10.1128/AEM.01104-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; e de Kwaadsteniet M.; ten Doeschate K.; Dicks L. M. T. Characterization of the Structural Gene Encoding Nisin F, a New Lantibiotic Produced by a Lactococcus lactis subsp. lactis Isolate from Freshwater Catfish (Clarias gariepinus). Appl. Environ. Microbiol. 2008, 74, 547–549. 10.1128/AEM.01862-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Zhang Q.; Yu Y.; Vélasquez J. E.; van der Donk W. A. Evolution of lanthipeptide synthetases. Proc. Natl. Acad. Sci. 2012, 109, 18361–18366. 10.1073/pnas.1210393109. [DOI] [PMC free article] [PubMed] [Google Scholar]; g O’Connor P. M.; O’Shea E. F.; Guinane C. M.; O’Sullivan O.; Cotter P. D.; Ross R. P.; Hill C. Nisin H Is a New Nisin Variant Produced by the Gut-Derived Strain Streptococcus hyointestinalis DPC6484. Appl. Environ. Microbiol. 2015, 81, 3953–3960. 10.1128/AEM.00212-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Parisot J.; Carey S.; Breukink E.; Chan W. C.; Narbad A.; Bonev B. Molecular mechanism of target recognition by subtilin, a class I lanthionine antibiotic. Antimicrob. Agents Chemother. 2008, 52, 612–618. 10.1128/AAC.00836-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Stein T.; Borchert S.; Conrad B.; Feesche J.; Hofemeister B.; Hofemeister J.; Entian K. D. Two Different Lantibiotic-Like Peptides Originate from the Ericin Gene Cluster of Bacillus subtilis A1/3. J. Bacteriol. 2002, 184, 1703–1711. 10.1128/JB.184.6.1703-1711.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Schnell N.; Entian K.-D.; Schneider U.; Götz F.; Zähner H.; Kellner R.; Jung G. Prepeptide sequence of epidermin, a ribosomally synthesized antibiotic with four sulphide-rings. Nature 1988, 333, 276–278. 10.1038/333276a0. [DOI] [PubMed] [Google Scholar]; b Bonelli R. R.; Schneider T.; Sahl H.-G.; Wiedemann I. Insights into In Vivo Activities of Lantibiotics from Gallidermin and Epidermin Mode-of-Action Studies. Antimicrob. Agents Chemother. 2006, 50, 1449–1457. 10.1128/AAC.50.4.1449-1457.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Daly K. M.; Upton M.; Sandiford S. K.; Draper L. A.; Wescombe P. A.; Jack R. W.; O’Connor P. M.; Rossney A.; Götz F.; Hill C.; Cotter P. D.; Ross R. P.; Tagg J. R. Production of the Bsa Lantibiotic by Community-Acquired Staphylococcus aureus Strains. J. Bacteriol. 2010, 192, 1131–1142. 10.1128/JB.01375-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bouhss A.; Al-Dabbagh B.; Vincent M.; Odaert B.; Aumont-Nicaise M.; Bressolier P.; Desmadril M.; Mengin-Lecreulx D.; Urdaci M. C.; Gallay J. Specific Interactions of Clausin, a New Lantibiotic, with Lipid Precursors of the Bacterial Cell Wall. Biophys. J. 2009, 97, 1390–1397. 10.1016/j.bpj.2009.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Paiva A. D.; Irving N.; Breukink E.; Mantovani H. C. Interaction with Lipid II Induces Conformational Changes in Bovicin HC5 Structure. Antimicrob. Agents Chemother. 2012, 56, 4586–4593. 10.1128/AAC.00295-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mota-Meira M.; Lacroix C.; LaPointe G.; Lavoie M. C. Purification and structure of mutacin B-Ny266: A new lantibiotic produced by Streptococcus mutans. FEBS Lett. 1997, 410, 275–279. 10.1016/S0014-5793(97)00425-0. [DOI] [PubMed] [Google Scholar]; b Hillman J. D.; Novák J.; Sagura E.; Gutierrez J. A.; Brooks T. A.; Crowley P. J.; Hess M.; Azizi A.; Leung K.-P.; Cvitkovitch D.; Bleiweis A. S. Genetic and biochemical analysis of mutacin 1140, a lantibiotic from Streptococcus mutans. Infect. Immun. 1998, 66, 2743–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Qi F.; Chen P.; Caufield P. W. Purification and biochemical characterization of mutacin I from the group I strain of Streptococcus mutans, CH43, and genetic analysis of mutacin I biosynthesis genes. Appl. Environ. Microbiol. 2000, 66, 3221–3229. 10.1128/AEM.66.8.3221-3229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Qi F.; Chen P.; Caufield P. W. The group I strain of Streptococcus mutans, UA140, produces both the lantibiotic mutacin I and a nonlantibiotic bacteriocin, mutacin IV. Appl. Environ. Microbiol. 2001, 67, 15–21. 10.1128/AEM.67.1.15-21.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Previous syntheses of ring A of; a nisin:Wakamiya T.; Shimbo K.; Sano A.; Fukase K.; Shiba T. Synthetic Study on Peptide Antibiotic Nisin. I. The Synthesis of Ring A. Bull. Chem. Soc. Jpn. 1983, 56, 2044–2049. 10.1246/bcsj.56.2044. [DOI] [Google Scholar]; b nisin:Fukase K.; Kitazawa M.; Sano A.; Shimbo K.; Horimoto S.; Fujita H.; Kubo A.; Wakamiya T.; Shiba T. Synthetic Study on Peptide Antibiotic Nisin.. V. Total Synthesis of Nisin. Bull. Chem. Soc. Jpn. 1992, 65, 2227–2240. 10.1246/bcsj.65.2227. [DOI] [Google Scholar]; c analogues:Manzor K.; ó Proinsias K.; Kelleher F. Solid-phase peptide synthesis of analogues of the N-terminus A-ring fragment of the lantibiotic nisin: Replacements for the dehydroalanine (Dha) residue at position 5 and the first incorporation of a thioamide residue. Tetrahedron Lett. 2017, 58, 2959–2963. 10.1016/j.tetlet.2017.06.052. [DOI] [Google Scholar]
- Previous syntheses of both rings A and B and analogues of nisin and subtilisin:; a Burrage S.; Raynham T.; Williams G.; Essex J. W.; Allen C.; Cardno M.; Swali V.; Bradley M. Biomimetic synthesis of lantibiotics. Chem.–Eur. J. 2000, 6, 1455–1466. . [DOI] [PubMed] [Google Scholar]; b Slootweg J. C.; van Herwerden E. F.; van Doremalen M. F. M.; Breukink E.; Liskamp R. M. J.; Rijkers D. T. S. Synthesis of nisin AB dicarba analogs using ring-closing metathesis: influence of sp3 versus sp2 hybridization of the α-carbon atom of residues dehydrobutyrine-2 and dehydroalanine-5 on the lipid II binding affinity. Org. Biomol. Chem. 2015, 13, 5997–6009. 10.1039/C5OB00336A. [DOI] [PubMed] [Google Scholar]
- Previous syntheses of ring B of; a nisin:Fukase K.; Wakamiya T.; Shiba T. Synthetic Study on Peptide Antibiotic Nisin. II. The Synthesis of Ring B. Bull. Chem. Soc. Jpn. 1986, 59, 2505–2508. 10.1246/bcsj.59.2505. [DOI] [Google Scholar]; b epidermin:Toogood P. L. Model studies of lantibiotic biogenesis. Tetrahedron Lett. 1993, 34, 7833–7836. 10.1016/S0040-4039(00)61488-0. [DOI] [Google Scholar]; c nisin:Matteucci M.; Bhalay G.; Bradley M. Cystine mimetics - solid phase lanthionine synthesis. Tetrahedron Lett. 2004, 45, 1399–1401. 10.1016/j.tetlet.2003.12.074. [DOI] [Google Scholar]; d subtilin:Zhou H.; van der Donk W. A. Biomimetic stereoselective formation of methyllanthionine. Org. Lett. 2002, 4, 1335–1338. 10.1021/ol025629g. [DOI] [PubMed] [Google Scholar]; e nisin:Goto Y.; Iwasaki K.; Torikai K.; Murakami H.; Suga H. Ribosomal synthesis of dehydrobutyrine- and methyllanthionine-containing peptides. Chem. Commun. 2009, 3419–3421. 10.1039/b904314d. [DOI] [PubMed] [Google Scholar]
- Structural studies of isolated rings:; a Palmer D. E.; Mierke D. F.; Pattaroni C.; Goodman M.; Wakamiya T.; Fukase K.; Kitazawa M.; Fujita H.; Shiba T. Interactive NMR and Computer Simulation Studies of Lanthionine-Ring Structures. Biopolymers 1989, 28, 397–408. 10.1002/bip.360280137. [DOI] [PubMed] [Google Scholar]; b Palmer D. E.; Pattaroni C.; Nunami K.; Chadha R. K.; Goodman M.; Wakamiya T.; Fukase K.; Horimoto S.; Kitazawa M.; Fujita H.; Kubo A.; Shiba T. Effects of Dehydroalanine on Peptide Conformations. J. Am. Chem. Soc. 1992, 114, 5634–5642. 10.1021/ja00040a024. [DOI] [Google Scholar]
- a Tabor A. B. The challenge of the lantibiotics: synthetic approaches to thioether-bridged peptides. Org. Biomol. Chem. 2011, 9, 7606–7628. 10.1039/c1ob05946g. [DOI] [PubMed] [Google Scholar]; b Tabor A. B. Recent advances in synthetic analogues of lantibiotics: What can we learn from these?. Bioorg. Chem. 2014, 55, 39–50. 10.1016/j.bioorg.2014.04.004. [DOI] [PubMed] [Google Scholar]; c Ongey E. L.; Neubauer P. Lanthipeptides: chemical synthesis versus in vivo biosynthesis as tools for pharmaceutical production. Microb. Cell Fact. 2016, 15, 97. 10.1186/s12934-016-0502-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Denoël T.; Lemaire C.; Luxen A. Progress in Lanthionine and Protected Lanthionine Synthesis. Chem. – Eur. J. 2018, 24, 15421–15441. 10.1002/chem.201801115. [DOI] [PubMed] [Google Scholar]
- a Bregant S.; Tabor A. B. Orthogonally Protected Lanthionines: Synthesis and Use for the Solid-Phase Synthesis of an Analogue of Nisin Ring C. J. Org. Chem. 2005, 70, 2430–2438. 10.1021/jo048222t. [DOI] [PubMed] [Google Scholar]; b Mothia B.; Appleyard A. N.; Wadman S.; Tabor A. B. Synthesis of Peptides Containing Overlapping Lanthionine Bridges on the Solid Phase: An Analogue of Rings D and E of the Lantibiotic Nisin. Org. Lett. 2011, 13, 4216–4219. 10.1021/ol201548m. [DOI] [PubMed] [Google Scholar]; c Wright Z. V. F.; McCarthy S.; Dickman R.; Reyes F. E.; Sanchez-Martinez S.; Cryar A.; Kilford I.; Hall A.; Takle A. K.; Topf M.; Gonen T.; Thalassinos K.; Tabor A. B. The Role of Disulfide Bond Replacements in Analogues of the Tarantula Toxin ProTx-II and Their Effects on Inhibition of the Voltage-Gated Sodium Ion Channel Nav1.7. J. Am. Chem. Soc. 2017, 139, 13063–13075. 10.1021/jacs.7b06506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pattabiraman V. R.; McKinnie S. M. K.; Vederas J. C. Solid-Supported Synthesis and Biological Evaluation of the Lantibiotic Peptide Bis(desmethyl) Lacticin 3147 A2. Angew. Chem. Int. Ed. 2008, 47, 9472–9475. 10.1002/anie.200802919. [DOI] [PubMed] [Google Scholar]; b Ross A. C.; Liu H.; Pattabiraman V. R.; Vederas J. C. Synthesis of the Lantibiotic Lactocin S Using Peptide Cyclizations on Solid Phase. J. Am. Chem. Soc. 2010, 132, 462–463. 10.1021/ja9095945. [DOI] [PubMed] [Google Scholar]; c Liu W.; Chan A. S. H.; Liu H.; Cochrane S. A.; Vederas J. C. Solid Supported Chemical Syntheses of Both Components of the Lantibiotic Lacticin 3147. J. Am. Chem. Soc. 2011, 133, 14216–14219. 10.1021/ja206017p. [DOI] [PubMed] [Google Scholar]; d Knerr P. J.; van der Donk W. A. Chemical Synthesis and Biological Activity of Analogues of the Lantibiotic Epilancin 15X. J. Am. Chem. Soc. 2012, 134, 7648–7651. 10.1021/ja302435y. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Knerr P. J.; van der Donk W. A. Chemical Synthesis of the Lantibiotic Lacticin 481 Reveals the Importance of Lanthionine Stereochemistry. J. Am. Chem. Soc. 2013, 135, 7094–7097. 10.1021/ja4014024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Thakkar A.; Trinh T. B.; Pei D. Global Analysis of Peptide Cyclization Efficiency. ACS Comb. Sci. 2013, 15, 120–129. 10.1021/co300136j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pedersen S. L.; Tofteng A. P.; Malik L.; Jensen K. J. Microwave heating in solid-phase peptide synthesis. Chem. Soc. Rev. 2012, 41, 1826–1844. 10.1039/C1CS15214A. [DOI] [PubMed] [Google Scholar]
- a Jain R.; Chauhan V. S. Conformational characteristics of peptides containing α,β-dehydroamino acid residues. Biopolymers 1996, 40, 105–119. . [DOI] [PubMed] [Google Scholar]; b Mathur P.; Ramakumar S.; Chauhan V. S. Peptide design using α,β-dehydro amino acids: From β-turns to helical hairpins. Biopolymers 2004, 76, 150–161. 10.1002/bip.10571. [DOI] [PubMed] [Google Scholar]; c Siodłak D. α,β-Dehydroamino acids in naturally occurring peptides. Amino Acids 2015, 47, 1–17. 10.1007/s00726-014-1846-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Humphrey J. M.; Chamberlin A. R. Chemical synthesis of natural product peptides: Coupling methods for the incorporation of noncoded amino acids into peptides. Chem. Rev. 1997, 97, 2243–2266. 10.1021/cr950005s. [DOI] [PubMed] [Google Scholar]; b Chan W. C.; Bycroft B. W.; Lian L. Y.; Roberts G. C. K. Isolation and characterisation of two degradation products derived from the peptide antibiotic nisin. FEBS Lett. 1989, 252, 29–36. 10.1016/0014-5793(89)80884-1. [DOI] [Google Scholar]
- a Schmidt U.; Lieberknecht A.; Wild J. Didehydroamino Acids (DDAA) and Didehydropeptides (DDP). Synthesis 1988, 159–172. 10.1055/s-1988-27503. [DOI] [Google Scholar]; b Bonauer C.; Walenzyk T.; König B. α,β-Dehydroamino acids. Synthesis 2006, 1–20. 10.1055/s-2005-921759. [DOI] [Google Scholar]; c Jiang J.; Ma Z.; Castle S. L. Bulky α,β-dehydroamino acids: their occurrence in nature, synthesis, and applications. Tetrahedron 2015, 71, 5431–5451. 10.1016/j.tet.2015.06.001. [DOI] [Google Scholar]
- a Hashimoto K.; Sakai M.; Okuno T.; Shirahama H. β-Phenylselenoalanine as a dehydroalanine precursor-efficient synthesis of alternariolide (AM-toxin I). Chem. Commun. 1996, 1139–1140. 10.1039/CC9960001139. [DOI] [Google Scholar]; b Okeley N. M.; Zhu Y.; van der Donk W. A. Facile chemoselective synthesis of dehydroalanine-containing peptides. Org. Lett. 2000, 2, 3603–3606. 10.1021/ol006485d. [DOI] [PubMed] [Google Scholar]
- a Chalker J. M.; Gunnoo S. B.; Boutureira O.; Gerstberger S. C.; Fernández-González M.; Bernardes G. J. L.; Griffin L.; Hailu H.; Schofield C. J.; Davis B. G. Methods for converting cysteine to dehydroalanine on peptides and proteins. Chem. Sci. 2011, 2, 1666–1676. 10.1039/c1sc00185j. [DOI] [Google Scholar]; b Chen H.; Zhang Y.; Li Q.-Q.; Zhao Y.-F.; Chen Y.-X.; Li Y.-M. De NovoDesign To Synthesize Lanthipeptides Involving Cascade Cysteine Reactions: SapB Synthesis as an Example. J. Org. Chem. 2018, 83, 7528–7533. 10.1021/acs.joc.8b00259. [DOI] [PubMed] [Google Scholar]
- Morrison P. M.; Foley P. J.; Warriner S. L.; Webb M. E. Chemical generation and modification of peptides containing multiple dehydroalanines. Chem. Commun. 2015, 51, 13470–13473. 10.1039/C5CC05469A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Carpino L. A. 1-Hydroxy-7-azabenzotriazole. An efficient peptide coupling additive. J. Am. Chem. Soc. 1993, 115, 4397–4398. 10.1021/ja00063a082. [DOI] [Google Scholar]; b Albericio F.; Bofill J. M.; El-Faham A.; Kates S. A. Use of onium salt-based coupling reagents in peptide Synthesis1. J. Org. Chem. 1998, 63, 9678–9683. 10.1021/jo980807y. [DOI] [Google Scholar]; c Montalbetti C. A. G. N.; Falque V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. 10.1016/j.tet.2005.08.031. [DOI] [Google Scholar]
- a Carpino L. A.; Sadat-Aalaee D.; Chao H. G.; DeSelms R. H. [(9-Fluorenylmethyl)oxy]carbonyl (FMOC) Amino Acid Fluorides. Convienient New Peptide Coupling Reagents Applicable to the FMOC/tert-Butyl Strategy for Solution and Solid-Phase Syntheses. J. Am. Chem. Soc. 1990, 112, 9651–9652. 10.1021/ja00182a041. [DOI] [Google Scholar]; b van Lierop B. J.; Jackson W. R.; Robinson A. J. 5,5-Dimethylproline dipeptides: an acid-stable class of pseudoproline. Tetrahedron 2010, 66, 5357–5366. 10.1016/j.tet.2010.05.068. [DOI] [Google Scholar]; c Raz R.; Rademann J. Fmoc-Based Synthesis of Peptide Thioesters for Native Chemical Ligation Employing a tert-Butyl Thiol Linker. Org. Lett. 2011, 13, 1606–1609. 10.1021/ol1029723. [DOI] [PubMed] [Google Scholar]
- a Brimble M. A.; Kowalczyk R.; Harris P. W. R.; Dunbar P. R.; Muir V. J. Synthesis of fluorescein-labelled O-mannosylated peptides as components for synthetic vaccines: comparison of two synthetic strategies. Org. Biomol. Chem. 2008, 6, 112–121. 10.1039/B712926B. [DOI] [PubMed] [Google Scholar]; b Levengood M. R.; van der Donk W. A. Dehydroalanine-containing peptides: preparation from phenylselenocysteine and utility in convergent ligation strategies. Nat. Protoc. 2006, 1, 3001–3010. 10.1038/nprot.2006.470. [DOI] [PubMed] [Google Scholar]
- Somekh L.; Shanzer A. Stereospecific synthesis of α,β-dehydroamino acids from β-hydroxy α-amino acid derivatives. J. Org. Chem. 1983, 48, 907–908. 10.1021/jo00154a040. [DOI] [Google Scholar]
- a Maynard S. J.; Almeida A. M.; Yoshimi Y.; Gellman S. H. New Charge-Bearing Amino Acid Residues That Promote β-Sheet Secondary Structure. J. Am. Chem. Soc. 2014, 136, 16683–16688. 10.1021/ja510265e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu H.; Pattabiraman V. R.; Vederas J. C. Stereoselective syntheses of 4-oxa diaminopimelic acid and its protected derivatives via aziridine ring opening. Org. Lett. 2007, 9, 4211–4214. 10.1021/ol701742x. [DOI] [PubMed] [Google Scholar]
- Schwieters C. D.; Kuszewski J. J.; Tjandra N.; Clore G. M. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003, 160, 65–73. 10.1016/S1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- a Hayward S. Peptide-plane flipping in proteins. Protein Sci. 2001, 10, 2219–2227. 10.1110/ps.23101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Touw W. G.; Joosten R. P.; Vriend G. Detection of trans-cis flips and peptide-plane flips in protein structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 1604–1614. 10.1107/S1399004715008263. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Milner-White J. E.; Watson J. D.; Qi G.; Hayward S. Amyloid formation may involve α- to β sheet interconversion via peptide plane flipping. Structure 2006, 14, 1369–1376. 10.1016/j.str.2006.06.016. [DOI] [PubMed] [Google Scholar]; d Moynié L.; Giraud M. F.; Breton A.; Boissier F.; Daignan-Fornier B.; Dautant A. Functional significance of four successive glycine residues in the pyrophosphate binding loop of fungal 6-oxopurine phosphoribosyltransferases. Protein Sci. 2012, 21, 1185–1196. 10.1002/pro.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L.; Zachariah C.; Thirumoorthy R.; Rocca J.; Novák J.; Hillman J. D.; Edison A. S. Structure and dynamics of the lantibiotic mutacin 1140. Biochemistry 2003, 42, 10372–10384. 10.1021/bi034490u. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Bax A. Prediction of Xaa-Pro peptide bond conformation from sequence and chemical shifts. J. Biomol. NMR 2010, 46, 199–204. 10.1007/s10858-009-9395-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pettit G. R.; Taylor S. R. Synthesis of the marine sponge cycloheptapeptide stylopeptide 11. J. Org. Chem. 1996, 61, 2322–2325. 10.1021/jo951986b. [DOI] [Google Scholar]; b Mechnich O.; Messier G.; Kessler H.; Bernd M.; Kutscher B. Cyclic heptapeptides axinastatin 2, 3, and 4: Conformational analysis and evaluation of the biological potential. Helv. Chim. Acta 1997, 80, 1338–1354. 10.1002/hlca.19970800503. [DOI] [Google Scholar]; c Pelay-Gimeno M.; Meli A.; Tulla-Puche J.; Albericio F. Rescuing Biological Activity from Synthetic Phakellistatin 19. J. Med. Chem. 2013, 56, 9780–9788. 10.1021/jm401520x. [DOI] [PubMed] [Google Scholar]; d Shaheen F.; Jabeen A.; Ashraf S.; Nadeem-ul-Haque M.; Shah Z. A.; Ziaee M. A.; Dastagir N.; Ganesan A. Total synthesis, structural, and biological evaluation of stylissatin A and related analogs. J. Pept. Sci. 2016, 22, 607–617. 10.1002/psc.2909. [DOI] [PubMed] [Google Scholar]; e Meli A.; Tedesco C.; Della Sala G.; Schettini R.; Albericio F.; De Riccardis F.; Izzo I. Phakellistatins: An Underwater Unsolved Puzzle. Marine Drugs 2017, 15, 78. 10.3390/md15030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kuipers O. P.; Rollema H. S.; de Vos W. M.; Siezen R. J. Biosynthesis and secretion of a precursor of nisin Z by Lactococcus lactis, directed by the leader peptide of the homologous lantibiotic subtilin from Bacillus subtilis. FEBS Lett. 1993, 330, 23–27. 10.1016/0014-5793(93)80911-D. [DOI] [PubMed] [Google Scholar]; b Bonev B. B.; Breukink E.; Swiezewska E.; de Kruijff B.; Watts A. Targeting extracellular pyrophosphates underpins the high selectivity of nisin. FASEB J. 2004, 18, 1862–1869. 10.1096/fj.04-2358com. [DOI] [PubMed] [Google Scholar]
- Turpin E. R.; Mulholland S.; Teale A. M.; Bonev B. B.; Hirst J. D. New CHARMM force field parameters for dehydrated amino acid residues, the key to lantibiotic molecular dynamics simulations. RSC Adv. 2014, 4, 48621–48631. 10.1039/C4RA09897H. [DOI] [Google Scholar]
- Lian L. Y.; Chan W. C.; Morley S. D.; Roberts G. C.; Bycroft B. W.; Jackson D. Solution structures of nisin A and its two major degradation products determined by n.m.r. Biochem. J. 1992, 283, 413–420. 10.1042/bj2830413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chan W. C.; Dodd H. M.; Horn N.; Maclean K.; Lian L. Y.; Bycroft B. W.; Gasson M. J.; Roberts G. C. Structure-activity relationships in the peptide antibiotic nisin: Role of dehydroalanine 5. Appl. Environ. Microbiol. 1996, 62, 2966–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chen S.; Wilson-Stanford S.; Cromwell W.; Hillman J. D.; Guerrero A.; Allen C. A.; Sorg J. A.; Smith L. Site-Directed Mutations in the Lanthipeptide Mutacin 1140. Appl. Environ. Microbiol. 2013, 79, 4015–4023. 10.1128/AEM.00704-13. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Geng M.; Smith L. Modifying the Lantibiotic Mutacin 1140 for Increased Yield, Activity, and Stability. Appl. Environ. Microbiol. 2018, 84, e00830 10.1128/AEM.00830-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In the published 1WCO structure, the stereochemistry of the MeLan diastereoisomer in nisin ring B is incorrectly shown as (2S,3R,6R), rather than the native (2S,3S,6R) stereochemistry determined by Gross and Morell.9c We are very grateful to Shang-Te Danny Hsu and Alexandre Bonvin for providing us with the recalculated structures with the correct chirality, which we have used in the comparisons in Figure 8. It is notable that the conformation of ring B is very similar in the revised structure, giving further weight to the hypothesis that the Me group of MeLan has little effect on the conformation of ring B structures.
- Chou K. C. Prediction of tight turns and their types in proteins. Anal. Biochem. 2000, 286, 1–16. 10.1006/abio.2000.4757. [DOI] [PubMed] [Google Scholar]
- a Ven F. J. M.; Hooven H. W.; Konings R. N. H.; Hilbers C. W. NMR studies of lantibiotics: The structure of nisin in aqueous solution. Eur. J. Biochem. 1991, 202, 1181–1188. 10.1111/j.1432-1033.1991.tb16488.x. [DOI] [PubMed] [Google Scholar]; b Hooven H. W.; Doeland C. C. M.; Kamp M.; Konings R. N. H.; Hilbers C. W.; Ven F. J. M. Three-dimensional structure of the lantibiotic nisin in the presence of membrane-mimetic micelles of dodecylphosphocholine and of sodium dodecylsulphate. Eur. J. Biochem. 1996, 235, 382–393. 10.1111/j.1432-1033.1996.00382.x. [DOI] [PubMed] [Google Scholar]
- a Levengood M. R.; Knerr P. J.; Oman T. J.; van der Donk W. A. In Vitro Mutasynthesis of Lantibiotic Analogues Containing Nonproteinogenic Amino Acids. J. Am. Chem. Soc. 2009, 131, 12024–12025. 10.1021/ja903239s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zambaldo C.; Luo X.; Mehta A. P.; Schultz P. G. Recombinant Macrocyclic Lanthipeptides Incorporating Non-Canonical Amino Acids. J. Am. Chem. Soc. 2017, 139, 11646–11649. 10.1021/jacs.7b04159. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kakkar N.; Perez J. G.; Liu W. R.; Jewett M. C.; van der Donk W. A. Incorporation of Nonproteinogenic Amino Acids in Class I and II Lantibiotics. ACS Chem. Biol. 2018, 13, 951–957. 10.1021/acschembio.7b01024. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Yang X.; Lennard K. R.; He C.; Walker M. C.; Ball A. T.; Doigneaux C.; Tavassoli A.; van der Donk W. A. A lanthipeptide library used to identify a protein-protein interaction inhibitor. Nat. Chem. Biol. 2018, 14, 375–380. 10.1038/s41589-018-0008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman R.; Danelius E.; Mitchell S. A.; Hansen D. F.; Erdélyi M.; Tabor A. B.. A chemical biology approach to understanding molecular recognition of lipid II by nisin(1-12): Synthesis and NMR ensemble analysis of nisin(1-12) and analogues. Chem. – Eur. J. 2019, in press. DOI 10.1002/chem.201902814. [DOI] [PMC free article] [PubMed]
- Boger D. L.; Ichikawa S.; Tse W. C.; Hedrick M. P.; Jin Q. Total Syntheses of Thiocoraline and BE-22179 and Assessment of Their DNA Binding and Biological Properties. J. Am. Chem. Soc. 2001, 123, 561–568. 10.1021/ja003602r. [DOI] [PubMed] [Google Scholar]
- Vranken W. F.; Boucher W.; Stevens T. J.; Fogh R. H.; Pajon A.; Llinas M.; Ulrich E. L.; Markley J. L.; Ionides J.; Laue E. D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins: Struct., Funct., Bioinf. 2005, 59, 687–696. 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- Vögeli B.; Ying J.; Grishaev A.; Bax A. Limits on variations in protein backbone dynamics from precise measurements of scalar couplings. J. Am. Chem. Soc. 2007, 129, 9377–9385. 10.1021/ja070324o. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Bax A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013, 56, 227–241. 10.1007/s10858-013-9741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Delaglio F.; Cornilescu G.; Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 2009, 44, 213–223. 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters C. D.; Kuszewski J. J.; Clore G. M. Using Xplor-NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 48, 47–62. 10.1016/j.pnmrs.2005.10.001. [DOI] [Google Scholar]
- Bhattacharya A.; Tejero R.; Montelione G. T. Evaluating protein structures determined by structural genomics consortia. Proteins: Struct., Funct., Bioinf. 2007, 66, 778–795. 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.