

Abstract
Background
α Carboxyl terminus 1 (αCT1) is a 25–amino acid therapeutic peptide incorporating the zonula occludens‐1 (ZO‐1)–binding domain of connexin 43 (Cx43) that is currently in phase 3 clinical testing on chronic wounds. In mice, we reported that αCT1 reduced arrhythmias after cardiac injury, accompanied by increases in protein kinase Cε phosphorylation of Cx43 at serine 368. Herein, we characterize detailed molecular mode of action of αCT1 in mitigating cardiac ischemia‐reperfusion injury.
Methods and Results
To study αCT1‐mediated increases in phosphorylation of Cx43 at serine 368, we undertook mass spectrometry of protein kinase Cε phosphorylation assay reactants. This indicated potential interaction between negatively charged residues in the αCT1 Asp‐Asp‐Leu‐Glu‐Iso sequence and lysines (Lys345, Lys346) in an α‐helical sequence (helix 2) within the Cx43‐CT. In silico modeling provided further support for this interaction, indicating that αCT1 may interact with both Cx43 and ZO‐1. Using surface plasmon resonance, thermal shift, and phosphorylation assays, we characterized a series of αCT1 variants, identifying peptides that interacted with either ZO‐1–postsynaptic density‐95/disks large/zonula occludens‐1 2 or Cx43‐CT, but with limited or no ability to bind both molecules. Only peptides competent to interact with Cx43‐CT, but not ZO‐1–postsynaptic density‐95/disks large/zonula occludens‐1 2 alone, prompted increased pS368 phosphorylation. Moreover, in an ex vivo mouse model of ischemia‐reperfusion injury, preischemic infusion only with those peptides competent to bind Cx43 preserved ventricular function after ischemia‐reperfusion. Interestingly, a short 9–amino acid variant of αCT1 (αCT11) demonstrated potent cardioprotective effects when infused either before or after ischemic injury.
Conclusions
Interaction of αCT1 with the Cx43, but not ZO‐1, is correlated with cardioprotection. Pharmacophores targeting Cx43‐CT could provide a translational approach to preserving heart function after ischemic injury.
Keywords: cardioprotection, connexin 43, ischemia‐reperfusion injury, S368 phosphorylation, zonula occludens‐1, α carboxyl terminus 1
Subject Categories: Basic Science Research, Translational Studies, Cell Biology/Structural Biology, Ion Channels/Membrane Transport
Clinical Perspective
What Is New?
α Carboxyl terminus 1 (αCT1) is a 25–amino acid peptide incorporating a sequence of amino acids from the gap junction protein connexin 43 that is currently in phase 3 clinical trials for chronic skin wounds.
In this study, we show that the molecular mode of action of αCT1 involves it binding to a distinct region of connexin 43 and prompting a specific phosphorylation of this molecule.
We further demonstrate that αCT1 provides cardioprotection from ischemia‐reperfusion injury via this mechanism and identify a short 9–amino acid variant of αCT1 (αCT11) that shows potent cardioprotective effects when infused after ischemic injury.
What Are the Clinical Implications?
In phase 2 testing, αCT1 displayed efficacy in promoting the healing of 2 types of chronic, slow‐healing skin wound: diabetic foot ulcers and venous leg ulcers. αCT1 is currently in phase 3 clinical trials on >500 patients as a treatment for diabetic foot ulcers (Multicenter Study of the Efficacy and Safety of Granexin Gel in the Treatment of Diabetic Foot Ulcer [GAIT 1]).
The insight into molecular mechanism(s) on αCT1 provided in this study informs the clinical safety profile of this therapeutic peptide.
The identification of a short variant of αCT1, called αCT11, as a novel therapeutic molecule could provide the basis of a cardioprotective therapy for patients who have experienced an acute myocardial infarction.
Introduction
Heart muscle cells are connected together by large numbers of gap junction (GJ) channels.1, 2 The main subunit protein of GJs in the mammalian ventricle muscle is connexin 43 (Cx43; encoded by GJA1), which is preferentially localized in intercalated disks (ie, zones of specialized electromechanical interaction between cardiomyocytes).3, 4 After myocardial infarction in patients with ischemic heart disease, Cx43 remodels from its normal distribution in muscle tissue bordering the necrotic injury, redistributing from intercalated disks at cardiomyocyte ends to lateral domains of sarcolemma.5 This process of lateralized remodeling of Cx43 within the cell membrane is a hallmark of ischemic heart disease in humans and is thought to contribute to the arrhythmia‐promoting characteristics of the infarct border zone.
Cx43 phosphorylated status has emerged as a factor of interest in pathogenic assignments of the protein in the wound healing response of cardiac muscle and other tissues, including skin.6 Pertinent to GJ remodeling in heart disease, the results of Ek‐Vitorin and coworkers indicated that Cx43 phosphorylated at a consensus PKC (protein kinase C) site, serine 368, was retained at intercalated disks during early ischemia and that this retention was probably associated with cardioprotection induced by ischemic preconditioning.7 This being said, the factors contributing to phosphorylation of Cx43 at serine 368 (Cx43 pS368) and how this phosphorylation relates to variation in the severity of ischemic injury remain to be fully elucidated.
Previously, we showed that a peptide mimetic of the Cx43 carboxyl terminus (CT), incorporating its postsynaptic density‐95/disks large/zonula occludens‐1 (ZO‐1) (PDZ)–binding domain reduced Cx43 GJ remodeling in injury border zone tissues after cryoinfarction of the left ventricle (LV) in mice.8 The decreases in Cx43 remodeling, prompted by treatment with this peptide (termed αCT1), were associated with a decreased propensity of the injured hearts to develop inducible arrhythmias8 and sustained improvements in ventricular contractile performance over an 8‐week study period.9 We further reported that the decreases in Cx43 lateralization observed in hearts treated with αCT1 were correlated with increased Cx43 pS368,8 in line with results from others suggesting that this posttranslational modification was correlated with reduced GJ remodeling and cardioprotection.7
Initially, we interpreted the induction of increased phosphorylation by αCT1 as a downstream consequence of the well‐characterized property of the peptide to disrupt interactions between Cx43 and its scaffolding protein, ZO‐1.10, 11 However, in simple biochemical assays involving a purified PKC enzyme, and a Cx43 CT substrate, we went on to show that αCT1 promoted S368 phosphorylation in vitro in a dose‐dependent manner, without recourse to interaction with ZO‐1.8 This result raised the prospect that αCT1 mode of action could have at least 2 independent aspects: one involving inhibition of interaction between Cx43 and ZO‐1 and the other a ZO‐1–independent mechanism, associated with PKC‐mediated changes in Cx43 phosphorylation status.
Characterization of αCT1 molecular mechanism is of translational significance as the peptide shows therapeutic potential and is presently undergoing clinical testing.12 In phase 2 clinical trials, αCT1 demonstrated efficacy in promoting the healing of 2 types of chronic, slow‐healing skin wounds.13, 14, 15 αCT1 is currently in phase 3 clinical trials on >500 patients, as a treatment for diabetic foot ulcers (Multicenter Study of the Efficacy and Safety of Granexin Gel in the Treatment of Diabetic Foot Ulcer [GAIT 1]).16 In the present study, we provide insights into the molecular mechanism of αCT1, showing that the protective effects of αCT1 in ischemic injury to the LV is not directly related to ZO‐1 binding, but is likely associated with interactions of the peptide with the Cx43 CT.
Materials and Methods
Animals
Male C57BL/6 mice, aged 3 months, were used. The experimental protocols were approved by Institutional Animal Care and Use Committee of Virginia Polytechnic Institute and State University and conform to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
The data that support the findings of this study are available from the corresponding author on reasonable request.
Reagents: Peptides, cDNA Expression Constructs, and Antibodies
Sequences and a brief description of each Cx43‐CT–based peptide used are shown in the Table. Peptides were synthesized and quality checked for fidelity and purity using high‐performance liquid chromatography and mass spectrometry (LifeTein, Hillsborough, NJ). Biotinylated peptides were used for surface plasmon resonance (SPR) experiments. Glutathione‐S‐transferase fusion protein constructs, composed of the Cx43‐CT (pGEX‐6‐P2 Cx43 CT amino acids 255–382), ZO‐1 PDZ1, PDZ2, and PDZ3 were isolated and purified from isopropy‐b‐d‐thiogalactoside–induced BL21 bacteria using standard procedures, described in our previous publications.8, 10, 11 The pGEX6p2‐Cx43 CT plasmid was obtained from Prof. Paul L. Sorgen (University of Nebraska Medical Center). Cx43 CT mutant (Cx43 CT‐KK/QQ; amino acids Lys345, Lys346 to Gln 345, Gln 346) was developed by site‐directed mutagenesis of the pGEX6p2‐Cx43 CT plasmid (Agilent Technologies, QuikChange II Site‐Directed Mutagenesis Kit). The mutation was verified by sequencing. For SPR experiments, the glutathione‐S‐transferase was removed using PreScission protease, yielding Cx43 CT protein (wild type or mutant).
Table 1.
Cx43 Mimetic and Variant Peptide Sequences Used
| Peptide | Antennapedia Cx43 CT | Modification | Interaction With | |
|---|---|---|---|---|
| Cx43 CT | ZO‐1 PDZ2 | |||
| αCT1 | RQPKIWFPNRRKPWKK RPRPDDLEI | Unmodified αCT1 | +++ | +++ |
| Variant M1: AALAI | RQPKIWFPNRRKPWKK RPRPAALAI | DD and E substituted with As | − | − |
| Variant M2: AALEI | RQPKIWFPNRRKPWKK RPRPAALEI | DD substituted with As | ± | + |
| Variant M3: DDLAI | RQPKIWFPNRRKPWKK RPRPDDLBI | E substituted with A | + | +++ |
| Variant M4: scrambled | RQPKIWFPNRRKPWKK LPAARIAPR | Scrambled control | − | − |
| αCT1‐I | RQPKIWFPNRRKPWKK RPRPDDLE | CT isoleucine deleted | +++ | − |
| αCT11 | RPRPDDLEI | No antennapedia sequence | +++ | +++ |
Boldfaced letters indicate alanine substitutions of negatively charged D and E amino acid residues occurring at the CT of wild‐type Cx43. The “Interaction With” column indicates peptide interaction characterization with Cx43 CT or the ZO‐1 PDZ2 domain, as determined by cross‐linking, molecular modeling, surface plasmon resonance, and thermal shift assays, provided in Figures 1, 2, 3 through 4 and Table S1 and Figures S3 and S5. CT indicates carboxyl terminus; Cx43, connexin 43; PDZ, postsynaptic density‐95/disks large/ZO‐1; ZO‐1, zonula occludens‐1.
+++ = high levels of interaction.
++ = Moderate levels of interaction.
+ = low levels of interaction.
± = possible interaction.
− = no evidence of interaction.
Antibodies used were as follow: phosphorylated Cx43 (Ser368) (Cell Signaling, Danvers, MA, 3511S); anti‐Cx43, produced in rabbit (Sigma, St Louis, MO, C6219); anti–glutathione‐S‐transferase, produced in goat (GE, Little Chalfont, UK, 27457701); and NeutrAvidin–horseradish peroxidase (Thermo, MA, 31030).
Western Blotting
Protein samples from all related experiments (PKC and 1‐ethyl‐3‐[‐3‐dimethylaminopropyl] carbodiimide hydrochloride [EDC] cross‐linking assays and Western blots on heart lysates) were processed in lithium dodecyl sulfate sample loading buffer (Bio‐Rad, CA, 1610737), heated at 95°C for 5 minutes. Samples from PKC and cross‐linking assays were loaded onto 18% Tris‐Glycine Stain‐Free gels (Bio‐Rad, CA, 5678073); samples from heart lysates were loaded onto 10% Tris‐Glycine Stain‐Free gel (Bio‐Rad, 5678033), resolved by SDS‐PAGE, and transferred to polyvinylidene difluoride membrane on a Turbo Transfer System (Bio‐Rad, 1704155). αCT1, eluted from cross‐linking reactions, was detected on blots against biotin with horseradish peroxidase–NeutrAvidin (ThermoFisher, MA, 31001). Signals were detected by horse radish peroxidase (HR)‐based chemiluminescence (ThermoFisher, MA, 34095), exposed to ECL Chemidoc (Bio‐Rad, 1708280), and digitized using Image Lab software (Bio‐Rad, 1709692). Detailed methods have also been previously described in our earlier articles.8, 10, 11
Surface Plasmon Resonance
Efficacy of the interaction of each αCT1 variant with Cx43 CT or Cx43 CT‐KK/QQ was tested using SPR, as described previously.17 In brief, SPR experiments were performed using a Biacore T200 (GE Healthcare). Equal amounts (response units [RUs]) of biotin‐αCT1 variants were immobilized on each flow cell of a streptavidin‐coated sensor chip (Biacore Inc) using immobilization buffer (in mmol/L: 10 HEPES, 1 EDTA, 100 NaCl, and 0.005% Tween‐20) at pH 7.4. Measurements with wild‐type Cx43 CT and mutant Cx43 CT‐KK/QQ analytes were done in running buffer (in mmol/L: 10 HEPES, 100 NaCl, pH 7.4) at a flow rate of 30 μL/min. Binding of analytes was verified at different concentrations, in random order (injection volume, 120 μL). Interacting proteins were then unbound by injection of 10 μL regeneration buffer (50 mmol/L NaOH and 1 mol/L NaCl) at a flow rate of 10 μL/min. Background levels were obtained from a reference cell containing a biotin‐control peptide in which the reversed sequence of the last 9 amino acids of Cx43 was fused to biotin‐antennapedia. The RU values obtained with biotin‐reverse control peptide were subtracted from the RU values obtained with the different biotin‐αCT1 variants (wild type or mutant) to generate the different response curves.
PKCε Cx43 CT S368 Phosphorylation Assay
PKC assay conditions were used to evaluate the PKCε phosphorylation of Cx43‐CT substrate at Ser368, as we have described previously, with modifications.8 PKCε, 400 ng/mL (Life, Carlsbad, CA, 37717L), was prediluted in enzyme dilution buffer (10 mmol/L HEPES, pH 7.4, 0.01% 3‐cholamidopropyl dimethylammonio 1‐propanesulfonate (CHAPS), and 5 mmol/L dithiothreitol) and assayed in 20 mmol/L HEPES, pH 7.4, 10 mmol/L MgCl2, 0.1 mmol/L EGTA, 1× lipid mix (200 μg/mL phosphatidylserine [Avanti Polar Lipids, 840032C]), 20 μg/mL diacylglycerol (Avanti Polar Lipids), 1 mmol/L HEPES, pH 7.4, 0.03% CHAPS, 500 μmol/L ATP (Sigma, A6419), and 14 μg/mL Cx43‐CT substrate. Kinase assay buffer was supplemented with peptides to produce final concentrations of the reaction constituents, as indicated in figure legends. The mixture was incubated at 37°C for 12 minutes and quenched by addition of Lithium Dodecyl Sulfate (LDS) sample loading buffer (Bio Rad, 1610791). “XT sample buffer” is what was shown on the product label, whereas the component is similar as regular LDS buffer, containing ≈5% to 10% lithium dodecyl sulphate. The reaction was Western blotted for pS368 Cx43 using the phosphorylated Cx43 (Ser368) antibody from Cell Signaling. Proteins were eluted off by stripping buffer (Millipore, 2504) and reprobed for total Cx43 using the Sigma anti‐rabbit antibody. Percentage phosphorylation (% P) was quantified using equation 1 and normalized with control group (PKC+, no peptide added).
EDC Cross‐Linking Assay
To characterize the interaction between the Cx43 CT substrate and peptides, the in vitro kinase assay was performed as above with modification and the constituents were then subjected to a cross‐linking reaction. The assay buffer used was 20 mmol/L 3‐(N‐morpholino) propanesulfonic acid, pH 7.2. The Cx43 CT substrate concentration was 30 μg/mL, and peptide concentrations varied, as indicated in figure legends. All other reagents present in the kinase reaction were maintained as described above. The reaction was allowed to proceed at 37°C for 15 minutes. Afterwards, the carbodiimide cross‐linker, EDC (Thermo, 22980), was added to each solution for a final concentration of 20 mmol/L. The solution was cross‐linked for 1 hour at room temperature. The reaction was stopped by the addition of 4× LDS loading buffer, boiled for 5 minutes, and subsequently separated by PAGE. The resulting gel was stained in Coomassie brilliant blue (Sigma, B0770) for 2 hours and destained in a solution of 4% methanol and 7% acetic acid overnight. Gel bands were subsequently excised for mass spectrometric analysis. For the direct interaction between protein and peptide, PBS, pH 7.5, was used as the coupling buffer. The protein (50 μg/mL) and peptide (25 μmol/L) were allowed to react at room temperature for 1 hour before EDC was added to the reaction mixture. The reaction was Western blotted for Cx43, glutathione‐S‐transferase, or NeutrAvidin.
Tandem Mass Spectrometry
Gel bands corresponding to cross‐linked Cx43 and αCT1 were excised, cut into 1‐mm square pieces, and destained with 3 consecutive washes with a 50:50 mixture of 50 mmol/L ammonium bicarbonate and acetonitrile for 10 minutes. Dithiothreitol, 50 μL of 10 mmol/L, was then added to the gel pieces; and the gel pieces were incubated at 56°C for 1 hour. Iodoacetamide, 50 μL of 55 mmol/L, was then added to the sample to alkylate cysteines. The sample was incubated at 25°C in the dark for 45 minutes. The gel was then dehydrated with 3 consecutive washes with a 50:50 mixture of 50 mmol/L ammonium bicarbonate and acetonitrile for 10 minutes and completely dehydrated with 100% acetonitrile and dried in a SpeedVac. Gel pieces were rehydrated in 10 to 15 μL of solution containing 20 ng/μL trypsin (Promega, Madison, WI) in 50 mmol/L ammonium bicarbonate for 15 minutes. Ammonium bicarbonate buffer, 30 μL of 50 mmol/L, was added to each sample; and the samples were incubated at 37°C for 18 hours. Peptides were extracted using 20% Acetonitrile (ACN)/0.1% Trifluoroacetic acid (TFA) once, 60% ACN/0.1% TFA twice, and 80% (ACN)/0.1% (TFA) once. The extracted samples were pooled and dried in a SpeedVac and reconstituted in 0.1% formic acid for subsequent liquid chromatography–tandem mass spectrometry (MS/MS) analysis.
For liquid chromatography–MS/MS analysis, tryptic peptides were directly separated on a 1‐dimensional fused silica capillary column (150 mm×100 μm) packed with Phenomenex Jupiter resin (3‐μm mean particle size, 300‐Å pore size). One‐dimensional liquid chromatography was performed using the following gradient at a flow rate of 0.5 μL/min: 0 to 10 minutes, 2% ACN (0.1% formic acid); 10 to 50 minutes, 2% to 35% ACN (0.1% formic acid); 50 to 60 minutes, 35% to 0% ACN (0.1% formic acid) balanced with 0.1% formic acid. The eluate was directly infused into an LTQ Velos mass spectrometer (ThermoFisher, San Jose, CA) equipped with a nanoelectrospray source. The instruments were operated in a data‐dependent mode, with the top 5 most abundant ions in each MS scan selected for fragmentation in the LTQ instrument. Dynamic exclusion (exclude after 2 spectra, release after 30 seconds, and exclusion list size of 150) was enabled.18
Molecular Modeling
Structural information for the Cx43 CT domain, truncated at G251, was obtained from the Worldwide Protein Data Bank (DOI: 10.2210/pdb1r5s/pdb). The protonated structure of the αCT1 peptide was obtained by truncating the 9 carboxyl terminal amino acids of the Cx43 CT. To model the interaction of the Cx43 CT with αCT1, the publicly available protein‐protein docking sever, ZDOCK (http://zdock.umassmed.edu/help.html), and the Maestro suite of Schrodinger were used to dock αCT1 with the Cx43 CT in silico in low‐energy conformations. ZDOCK is a fast fourier transform–based protein docking program. Both αCT1 and the Cx43 CT were submitted to the ZDOCK server for possible binding modes in the translational and rotational space. Each pose was evaluated using an energy‐based scoring function.19 In Schrodinger, conformational generation was undertaken, followed by a round of standard precision peptide docking. Further refinements using an MM‐GBSA minimization were used to calculate the enthalpic contribution of binding. Peptide ligands were docked in a 40‐Å grid generated using PHASE focused around the Cx43 CT helix 2 (H2) domain, allowing the minimization of protein residues within 5 Å of predicted binding poses. Root Mean Square Deviation (RMSD) values (representative of changes in average atomic distances) were obtained for αCT1 and variant peptides using the input geometry of the initial Protein Data Bank (PDB) protein structure.
Protein Thermal Shift Assay
Thermal stability of recombinant glutathione‐S‐transferase–PDZ2 or Cx43 CT in the presence or absence of peptides was determined in a 96‐well format. Each assay well was composed of 500 μg/mL protein and 25 to 100 μmol/L of each peptide in PBS buffer, pH 7.4. All assays were performed independently 6 times. Samples were generally prepared in 96‐well plates at final volumes of 20 μL. The fluorescent dye SYPRO Orange (5000× concentrate in dimethyl sulfoxide; ThermoFisher, S6650) was added to a final concentration of 8×. Reactions were run on QuantStudio 6 Flex Real‐Time PCR system (Applied Biosystems, part of Life Technologies Corporation, CA), according to the manufacturer's recommendations, using a melt protocol in 0.05‐°C/s increments from 25°C to 95°C. The Reporter Dye was “ROX,” and quencher dye and passive reference were selected as “none” for the melt curve, according to manufacturer's instructions. The data were analyzed using Protein Thermal Shift Software, v1.3 package (Applied Biosystems).
Ischemia‐Reperfusion Injury Model and LV Contractility
Male, 3‐month‐old, body weight 25±5 g, C57BL/6 mice were used for this study and obtained from Charles River. Animals were randomly assigned to experimental groups, and LV function was measured and myocardial ischemia‐reperfusion (I/R) injury was induced, as previously described.20 Briefly, 15 minutes after the injection of heparin at a dose of 200 U/10 g body weight, the mouse was anesthetized by inhalation of isoflurane vapor and subjected to cervical dislocation on the cessation of respiration. Thoracotomy was immediately performed, and the heart was excised. The heart was arrested in ice‐cold Krebs‐Henseleit buffer (in mmol/L: 25 NaHCO3, 0.5 EDTA, 5.3 KCl, 1.2 MgSO4, 0.5 pyruvate, 118 NaCl, 10 glucose, 2.5 CaCl2). The aorta was isolated and cannulated in a Langendorff perfusion system. The heart was then perfused at a constant pressure of 75 mm Hg with Krebs‐Henseleit buffer, which was continually bubbled with 5% CO2/95% O2 at 37°C. Effluent from the Thebesian veins was drained by a thin polyethylene tube (PE‐10) pierced through the apex of the LV. A water‐filled balloon made of polyvinylchloride film was inserted in the LV and connected to a blood pressure transducer (Harvard Apparatus, MA, 733866). After a 30‐minute stabilization period, a balloon volume (BV), generating an LV end‐diastolic pressure of 0 mm Hg, was determined for the heart. The BV was then increased stepwise up through 1‐, 2‐, 5‐, 8‐, 10‐, 12‐, 15‐, 18‐, 20‐, 25‐, and 30‐μL increments of 1 to 5 μL, and contractile performance was recorded for 10 seconds at each step. The indexes of cardiac function were amplified by a Transducer Amplifier Module (Harvard Apparatus, 730065). Data were recorded and analyzed using PowerLab 4/35 (ADInstruments, CO, PL3504) and LabChart V7 (ADInstruments). The BV was then adjusted to set end‐diastolic pressure at ≈8 to 10 mm Hg and held constant during the ensuing steps of the protocol. Baseline function (determined by end‐diastolic pressure at ≈8–10 mm Hg) was recorded for 5 minutes. The perfused beating heart was then treated with freshly prepared peptide stocks (0.2 mmol/L), which were infused using syringe pump (Kent Sci, CT) into the perfusion buffer in a mixing chamber above the heart at 5% of coronary flow rate, to deliver final concentrations of 10 to 50 μmol/L or equivalent vehicle for 20 minutes. At the end of the peptide infusion period, hearts were subjected to global, no‐flow normothermic ischemia by turning off the perfusion flow for 20 minutes, followed by a reperfusion phase for 40 minutes. BV was retaken through the stepwise sequence of 1‐ to 5‐μL increments between 1 and 30 μL, with contractile performance again being recorded for 10 seconds at each step. Cardiac LV function was recorded throughout the procedure. In the case of postischemic treatment with αCT11 peptide, peptide infusion was begun at the initiation of the reperfusion phase and continued for 20 minutes, and then contractile function by BV increments was taken as per the other hearts. A set of hearts was freeze clamped immediately after peptide infusion for Western blotting. The protocol is illustrated in Figure S1.
Laser Scanning Confocal Microscopy and Fluorescence Quantification of Peptide‐perfused Hearts
LV samples were Langendorff perfused with vehicle control, αCT1 solution, and αCT11 solution, as described above and as summarized in Figure S1. Immunofluorescent labeling and detection and quantification of biotinylated peptide were performed, as previously described,8, 10, 11, 21 on 10‐μm cryosections of tissue. Samples were colabeled with a rabbit antibody against either Cx43 (Sigma, C6219, 1:250) or 4′,6‐diamidino‐2‐phenylindole and streptavidin conjugated to AlexaFluor 647 (1:4000; ThermoFisher Scientific). Cx43 primary antibodies were detected by goat anti‐rabbit AlexaFluor 488 (1:4000; ThermoFisher Scientific) secondary antibodies. Confocal imaging was performed using a TCS SP8 confocal microscope. Quantification of fluorescence intensity levels relative to background was performed using National Institutes of Health ImageJ software.
Statistical Analysis
Data were expressed as a mean±SE, unless otherwise noted. Differences among treatments were compared by 1‐way, 2‐way, or repeated‐measures ANOVA, followed by post hoc tests using Bonferonni's correction for multiple comparisons, as appropriate. Probability values of P<0.05 were considered significantly different. No strong evidence of divergence (P>0.05) from normality was found. Data analysis was performed using GraphPad7 (GraphPad Software, LaJolla, CA).
Results
αCT1 Shows Evidence of Interaction With the Cx43 CT
The 25‐mer Cx43 mimetic peptide αCT1 incorporates a 16–amino acid N‐terminal antennapedia sequence, followed by the CT‐most 9 amino acids of Cx43: Arg‐Pro‐Arg‐Pro‐Asp‐Asp‐Leu‐Glu‐Iso or RPRPDDLEI (Figure 1A and Table). The last 4 amino acids of this sequence (DLEI) comprise a class II PDZ‐binding motif, which has been shown to mediate a specific interaction with the second of the 3 PDZ (PDZ2) domains of ZO‐1.10, 11, 22, 23, 24, 25, 26, 27 We have previously reported on binding of αCT1 with ZO‐1, and the selectivity of this interaction for the ZO‐1 PDZ2 domain over that of ZO‐1 PDZ1 and PDZ3.11 This selectivity of αCT1 for ZO‐1 PDZ2 is illustrated in Figure 1B. Consistent with reports by others,25 deletion of the CT isoleucine of the DLEI binding motif (eg, as in the deletion of the CT isoleucine in αCT1‐I peptide; Table) abrogates interaction with ZO‐1 PDZ2 (Figure 1B). We also have previously shown that αCT1 upregulates a PKCε‐mediated phosphorylation of Cx43 at serine 368 (S368).8 This increase in pS368 by αCT1 was observed both in vivo in a LV injury model and in a biochemical assay of PKCε activity in vitro.8
Figure 1.
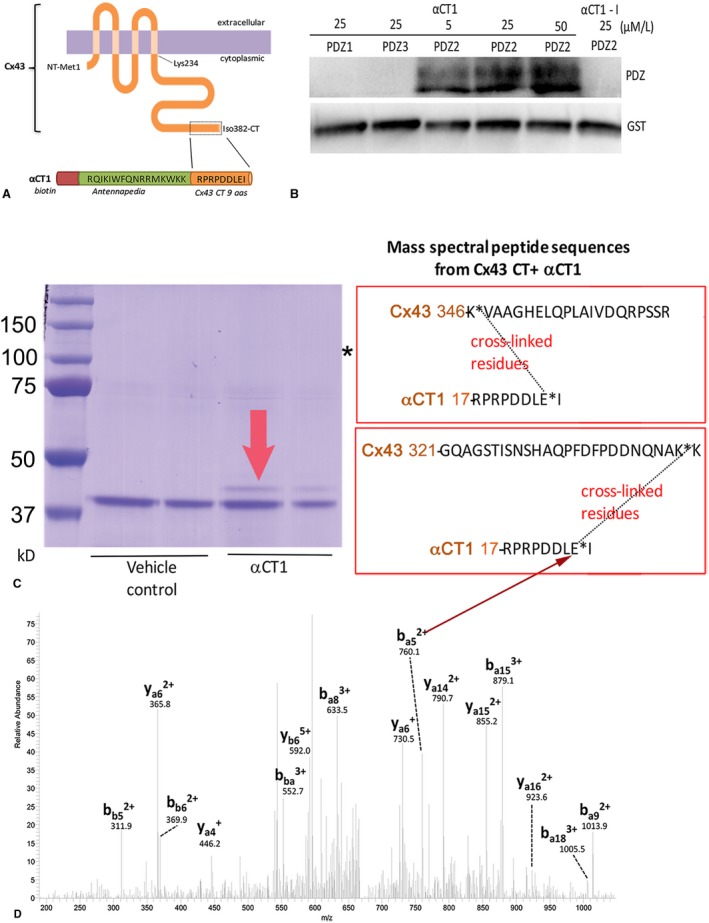
α Carboxyl terminus 1 (αCT1) interacts with zonula occludens‐1 (ZO‐1) postsynaptic density‐95/disks large/ZO‐1 (PDZ) 2 domain and the connexin 43 (Cx43) CT. A, Schematics of full‐length Cx43 and αCT1 peptide. B, αCT1 interaction with ZO‐1 PDZ domains, as indicated by 1‐ethyl‐3‐(3‐dimethylaminopropyl) carbodiimide hydrochloride (EDC) zero‐length cross‐linking to glutathione S‐transferase (GST) fusion PDZ1, PDZ2, and PDZ3 polypeptides and NeutrAvidin labeling of biotin‐tagged peptide at concentrations of 5, 25, and 50 μmol/L. The deletion of the CT isoleucine in αCT1‐I renders this peptide incompetent to interact with the ZO‐1 PDZ2 domain. C, Coomassie blue gel of EDC cross‐linked products of kinase reaction mixtures containing GST‐Cx43 CT and GST–protein kinase C (PKC)ε, with (αCT1) and without (vehicle) αCT1. The fainter band above GST‐Cx43 bands (indicated by red arrow) in the αCT1 lanes was cut from gels and analyzed by tandem mass spectrometry (MS/MS). The boxes to right of gel show Cx43 CT peptides identified by MS/MS as being cross‐linked to αCT1. Asterisk indicates a molecular mass consistent with GST‐PKCε. D, Tandem mass spectrum of a quintuply charged cross‐linked peptide (m/z, 674.1) between Cx43 345 to 366 (a‐chain) and αCT1 peptide through Cx43 K346 and E8 in αCT1 (b‐chain). Only the b‐ and y‐sequence specific ions are labeled. Arrow indicates ion (ba5 2+) consistent with cross‐linkage between Cx43 CT lysine K346 and the glutamic acid (E) residue of αCT1 at position ‐1.
To determine molecular factors contributing to αCT1 upregulation of S368 phosphorylation, the zero‐length cross‐linker, EDC, was introduced into the in vitro PKCε phosphorylation assay. Zero‐length cross‐linking covalently bonds directly interacting proteins, enabling identification of closely associating polypeptides in the reaction mixture. The components of the reaction were then separated by SDS‐PAGE, and MS/MS was performed (Figure 1C and 1D). Although no evidence for interaction between glutathione‐S‐transferase–PKCε (molecular weight=110 kDa) and biotin‐αCT1 was observed, on the basis of streptavidin detection of cross‐linked biotinylated peptide on blots of the reaction mixture (Figure S2), MS/MS revealed complexes between the Cx43 CT and αCT1 on bands cut out from gels corresponding to the molecular mass of the cross‐linked complex (Figure 1C). Moreover, it was determined that a negatively charged glutamic acid (E381) within the PDZ‐binding domain of αCT1 and a pair of aspartic acids (D378 and D379) were involved in bonding with a pair of positively charged lysines (K) at positions K345 and K346 of the Cx43 CT (Figure 1C and 1D). The site of this interaction was further confirmed by streptavidin labeling of cross‐linked products from kinase reaction mixtures containing Cx43 CT and αCT1 (Figure S3, left blots) or a scrambled peptide (M4) unable to bind Cx43 CT (Figure S3, middle blots), as well as in reaction mixtures containing a mutated Cx43 CT (glutathione‐S‐transferase–Cx43 CT KK/QQ) (Figure S3, right blots), in which the pair of positively charged lysine residues at K345 and K346 was substituted with neutral glutamines (Q). Although no evidence of cross‐linking between the scrambled peptide and Cx43 CT, or between αCT1 and the Cx43 CT KK/QQ mutant substrate, was found, αCT1 was covalently linked by EDC to Cx43 CT in a concentration‐dependent manner.
αCT1 Potentially Interacts With the Cx43 CT H2 Domain
Structural studies by Sorgen and coworkers have shown that K345 and K346 fall within a short α‐helical sequence along the Cx43 CT called H2.28, 29 Figure 2A provides a schematic of the secondary structure of the Cx43 CT showing the location of H2 (from Sosinsky et al27), together with a second nearby stretch of the α‐helical sequence (helix 1 [H1]). To model potential associations between αCT1 and Cx43 CT in silico, we submitted the interacting complex to the ZDOCK protein modeling server,19 initially fixing the interaction between the glutamic acid (E) at position ‐1 of αCT1 (ie, E381 in full‐length Cx43) and the K346 residue of Cx43, as indicated by the MS/MS data (ie, Figure 1C). The interaction pose shown in Figure 2B represents that based on the lowest energy minimization score from >1800 possible variants of the complex. Using Schrodinger, we next generated a molecular model of the interaction within a centralized grid of 40 Å in the vicinity of the lysine residues but allowed the peptide to dock anywhere within this volume postconformational search. This encompassed both the H1 and H2 α‐helical domains, with the structure used to generate the docking grid being the most ordered of those reported from nuclear magnetic resonance studies (eg, Figure S4A).28 The Schrodinger model indicated that αCT1 was optimally configured in an antiparallel orientation, with its available side chains arrayed along the H2 sequence (Figure 2C and 2D). The in silico model also predicted that a salt bridge would form between the αCT1 glutamic acid (E) residue and Cx43 K346, consistent with MS/MS observations. The modeled interaction further suggested the occurrence of hydrogen bonding between the side chains of 4 amino acids arrayed along αCT1 (RPRPDDLEI; amino acids involved in hydrogen bonds are bolded) and 4 amino acids between Q340 and E360 along the H2 sequence (Figure 2D). We concluded that molecular modeling indicated a potential interaction between αCT1 and the H2 region of the Cx43 CT. This being said, it is recognized that this does not preclude other regions of the Cx43 that may have affinity for αCT1; and these in silico data also do not necessarily mean that this potential is realized in vivo.
Figure 2.
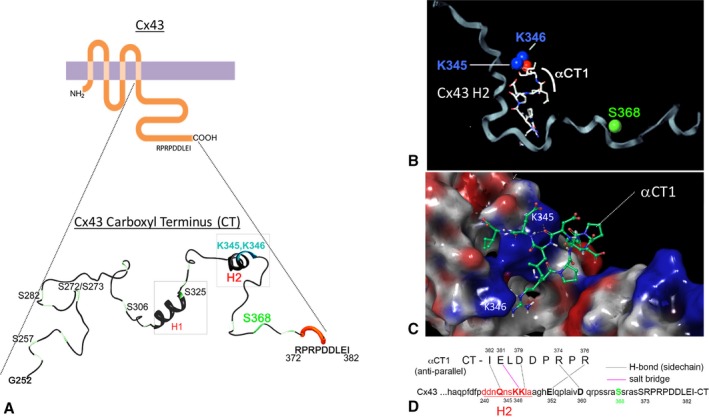
Molecular modeling of the α carboxyl terminus 1 (αCT1) and connexin 43 (Cx43) CT complex. A, Schematics of Cx43 and the secondary structure of Cx43 CT from amino acid residues glycine 252 (G252) through to isoleucine 382 (I382). The depiction of secondary structure has been modified from a diagram originally provided by Sosinsky and coworkers.27 B and C, ZDOCK (B) and Schrodinger (C) molecular modeling analysis of the structure of a proposed αCT1‐Cx43 CT complex. The protonated structure of αCT1 peptide and Cx43 CT (PDB: 1r5s), constrained by a salt‐bridge interaction between lysine 346 (K346) in the Cx43 CT and the glutamic acid (E) at position ‐1 of αCT1. The αCT1‐Cx43 interaction shown represents that based on the lowest energy minimization score determined in the model. D, From Schrodinger modeling: a 2‐dimensional map of αCT1‐Cx43 CT in antiparallel orientation showing location of amino acids predicted to bond to each other and the type of bond that is predicted to occur. In the case of Schrodinger simulations, docking was not constrained by specifying a salt bridge between Cx43 CT K346 and glutamic acid of αCT1.
Substitution of Negatively Charged Amino Acids in αCT1 Results in Loss of Cx43 CT Binding
To further probe the αCT1/Cx43 CT complex, 3 variant peptides based on αCT1 were prepared that were rationally designed to have abrogated Cx43 CT binding potential. To accomplish this goal, negatively charged E and D amino acids in the RPRPDDLEI sequence of αCT1 (ie, those indicated by MS/MS to be likely involved in Cx43 CT interaction) were substituted by neutral alanines. These αCT1 variant peptides had the sequences RPRPAALAI, RPRPAALEI, and RPRPDDLAI and are referred to as M1, M2, and M3, respectively. As a first step in the characterization of these peptides, we calculated the predicted binding enthalpy of each peptide with the Cx43 CT (MM‐GBSA ΔG [Molecular Mechanics Generalized‐Born surface area ΔG]) based on their lowest energy poses from a standard precision peptide docking in the PHASE module of Schrodinger. Modeling indicated that binding enthalpies were consistent with decreased potential for interaction with Cx43 CT in silico for all αCT1 variants (Table S1). RMSD atomic position values from overlaying the input protein structure (PDB: 1r5s) with MM‐GBSA–minimized docking complexes were within 1 Å (Table S1).
Next, SPR was used to analyze interactions of biotinylated versions of αCT1 and the αCT1 variant peptides, immobilized to streptavidin‐coated sensor chips, with the Cx43 CT and Cx43 CT‐KK/QQ proteins as analytes (Figure 3A through 3F, Figure S5A and S5B). The curves shown are obtained by subtracting control peptide RU signals from those of αCT1, and negative values indicate that the protein displays elevated binding to the control compared with the target peptide, consistent with nonspecific binding. The concentration of the analyte was varied between 0.5 and 15 μmol/L. A concentration‐dependent increase in RUs was observed for Cx43 CT binding to biotin‐αCT1 (Figure 3A). M1 showed loss of Cx43 CT binding competence, consistent with having all negatively charged amino acids substituted with alanine (Figure 3C). Substitution of D378A/D379A (M2) or E381A (M3) residues by alanines also abrogated peptide interaction with Cx43 CT (Figure 3E and 3F, Figure S5A and S5B). In complementary observations, SPR confirmed that the Cx43 CT KK/QQ mutant polypeptide showed minimal interaction with αCT1, M1, M2, or M3 (Figure 3B, 3D, and 3F), consistent with the pair of lysines at K345 and K346 in H2 being involved in interactions between Cx43 and αCT1.
Figure 3.
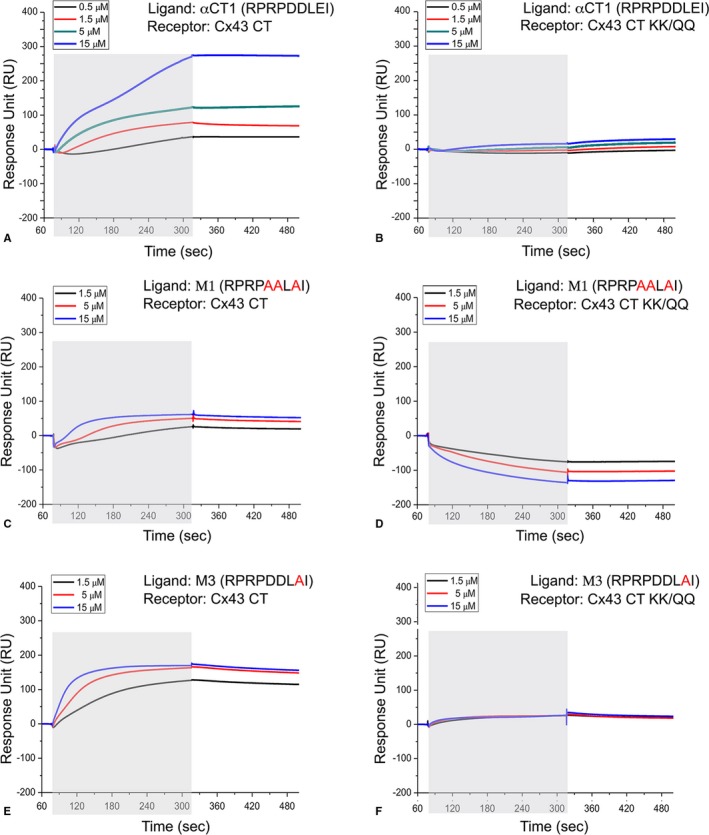
α Carboxyl terminus 1 (αCT1) variants with alanine substitutions of negatively charged amino acids show abrogated ability to bind connexin 43 (Cx43) CT. A through F, Surface plasmon resonance was used to analyze interactions of biotin‐αCT1 and biotin‐αCT1 variant peptides, immobilized to streptavidin‐coated chips, with the Cx43 CT (Cx43‐CT: amino acids 255–382) and Cx43 CT‐KK/QQ as analytes, respectively. The mean of 3 runs is plotted for each analyte concentration. The exposure of the sensor chip to the specific analyte is indicated by the gray area. Sensorgrams obtained for the following: Cx43 CT and biotin‐αCT1 (A), Cx43 CT‐KK/QQ and biotin‐αCT1 (B), and Cx43 CT and biotin‐M1 (C). D, Cx43 CT‐KK/QQ and biotin‐M1. E, Cx43 CT and biotin‐M3. F, Cx43 CT‐KK/QQ and biotin‐M3.
Inspection of SPR curves in Figure 3 provided further insight into the nature of the interaction. Cx43 CT/αCT1 dissociation rates appeared to be low (Figure 3A), suggesting a strong interaction once the complex is formed. The binding affinity of Cx43 CT/αCT1 interaction also appeared to be low. Consistent with this, no steady‐state binding was observed during the time course of the association phase and the binding of Cx43 CT to αCT1 was not saturated by increasing the concentration of Cx43 CT from 5 to 15 μmol/L (Figure 3A). This indicated that equilibrium dissociation constant (KD) is likely to be at least ≥5 μmol/L. Moreover, the binding of Cx43 CT to αCT1 at 15 μmol/L displayed a biphasic response. This suggested that at higher concentrations, Cx43 CT may not only bind to αCT1, but also interact with other Cx43 CT polypeptides already captured by peptide immobilized on the chip. One explanation of this could be the tendency of Cx43 CT to dimerize.29 Together with the previous complexities revealed by SPR, this biphasic profile hampered calculation of KD values. Finally, the binding of Cx43 CT to M3, despite displaying a lower ultimate RU than αCT1, had a higher initial on rate (Figure 3E), suggesting that the interaction surface is not only complex, but binding kinetics to the remaining negatively charged residues in M3 were fast.
To further investigate the potential for interaction between αCT1 and Cx43 CT, we synthesized a 43–amino acid peptide mimetic encompassing amino acids Y313 through A348 of the Cx43 CT, containing the H1 and H2 α‐helical regions (Figure S4A through S4D). In SPR assays, Cx43 Y313‐A348 showed levels of interaction with αCT1 comparable to the full Cx43 CT sequence (≈150 amino acids; Figure S4D). Nuclear magnetic resonance solutions have indicated that ordered arrangements of the H1 and H2 α helices may include the formation of a loop‐like domain near the middle of the CT sequence28 (eg, Figure S4A). Cx43 Y313‐A348 was designed to have cysteines at its N‐terminal and CT, which were used to disulfide cross‐link the peptide into a cyclized conformation (Figure S4B). SPR indicated that disulfide linkage of Cx43 Y313‐A348 resulted in a complete loss of αCT1 binding (Figure S4D), suggesting that interaction required a degree of conformational flexibility.
Substitution of Negatively Charged Amino Acids in αCT1 Fully and Partially Abrogates Interaction With Cx43 CT and ZO‐1 PDZ2, Respectively
To further characterize the Cx43‐binding characteristics of αCT1 and the αCT1 variants, thermal shift assays30 of peptide‐protein interactions were performed (Figure 4). This assay takes advantage of a change in fluorescence arising when SYPRO Orange binds to hydrophobic residues that become exposed when a protein undergoes thermal unfolding prompted by temperature change in a real‐time quantitative polymerase chain reaction machine. Binding of a ligand to a protein, such as when αCT1 interacts with the ZO‐1 PDZ2 domain, alters the thermal stability of the polypeptide, which is reflected by a shift between the thermal denaturation curves of the unliganded protein and the liganded protein. Thus, this assay provides data on the potential effect of ligand interaction on protein structural stability, with significantly increased or decreased thermal stability (as opposed to no change) being diagnostic of interaction. In line with the known stabilizing effect of the last 10 amino acids of Cx43 CT on ZO‐1 PDZ2,31 αCT1 concentrations of 25, 50, and 100 μmol/L increased the melt temperature of PDZ2 in a dose‐dependent manner (Figure 4A). Thermal shift assays indicated that the peptides from the Table fell into 2 classes with respect to Cx43 CT interaction: those that provided evidence of interaction with Cx43 CT and those that were Cx43 CT interaction incompetent (Figure 4B). Consistent with the SPR results, M1, M2, and M3 showed no propensity to alter Cx43 CT thermal stability, demonstrating no significant variance from Cx43 CT alone or Cx43 CT in the presence of the scrambled control peptide M4. By contrast, αCT1, αCT1‐I, and a short variant of αCT1 comprising the Cx43 CT 9‐mer sequence RPRPDDLEI (αCT11) all caused highly significant decreases in melt temperature, consistent with interaction causing reductions in the thermal stability of the Cx43 CT.
Figure 4.
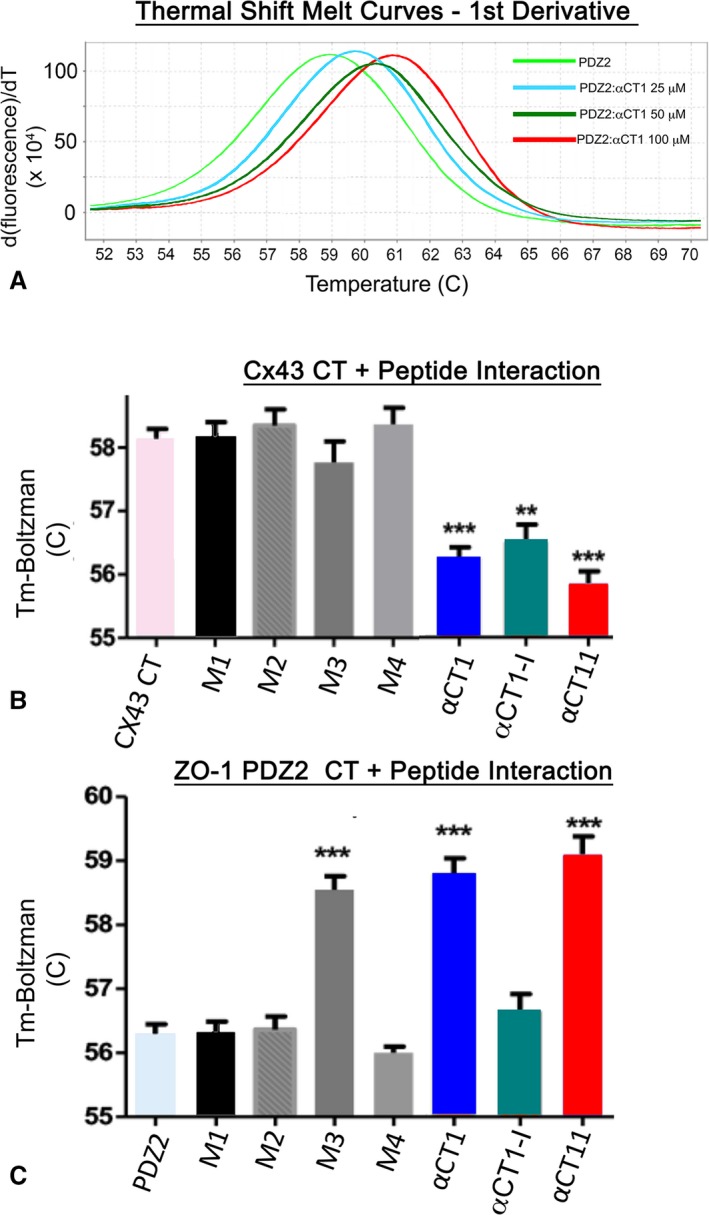
α Carboxyl terminus 1 (αCT1) interaction stabilizes postsynaptic density‐95/disks large/zonula occludens‐1 (ZO‐1) (PDZ) 2 and destabilizes connexin 43 (Cx43) CT structure. A, Representative first derivatives of melt curves (bottom) for ZO‐1 PDZ2 at 500 μg/mL alone or in combination with αCT1 at concentrations of 25, 50, and 100 μmol/L. B, Temperature maxima (Tm) from Boltzman curves from left to right of Cx43 CT (Cx43‐CT: amino acids 255–382) alone, Cx43 CT in combination with αCT1, and the αCT1 variants, including M1, M2, M3, M4 scrambled, deletion of the CT isoleucine in αCT‐I, and αCT11. αCT1, αCT1‐I, and αCT11 show similar abilities to destabilize (ie, significantly decrease the Tm of) Cx43 CT. **P<0.01, ***P<0.002 (N=6). C, Tm from Boltzman curves from left to right of PDZ2 alone and PDZ2 in combination with αCT1 and αCT1 variants, including M1, M2, M3, M4 scrambled, αCT‐I, and αCT11. M3, αCT1, and αCT11 show similar abilities to stabilize (ie, significantly increase the Tm of) PDZ2. Changes in Tm assayed from SYPRO orange fluorescence variation do not reflect changes in the direction of heat flow due to the reaction per se, but how interaction of a given peptide ligand alters PDZ2 or Cx43 CT stability in response to imposed temperature changes (ie, by the real‐time quantitative polymerase chain reaction machine). ***P<0.01 (N=6).
We also examined the effects of the αCT1 variants on thermal stability of ZO‐1 PDZ2. Unlike in presence of the parent peptide αCT1 (Figure 4A and 4C), M1 and M2 did not alter the melt temperature of PDZ2 (Figure 4C), neither of which differed significantly from PDZ2 alone or PDZ2 in the presence of either scrambled peptide (M4) or αCT1‐I (the 2 PDZ2‐interaction incompetent peptides) (Figure 1). The results were consistent with M1 or M2 having no, or limited, propensity to interact with the ZO‐1 domain. However, M3, the most conservative substitution variant, showed evidence of significant interaction with the ZO‐1–binding domain, with its effects on the thermal stability of PDZ2 being similar to those of αCT1 and αCT11 (Figure 4C). Thus, although M3 had no or limited competence to interact with Cx43 CT in this assay, the peptide did show evidence of ZO‐1 PDZ2‐binding activity at levels similar to unmodified αCT1.
Substitution of Negatively Charged Amino Acids in αCT1 Abrogates S368 Phosphorylation
Next, we examined the variant peptides in the PKCε kinase assay. Unlike αCT1, M1, M2, or M3 did not increase Cx43 S368 phosphorylation above levels detected in the absence of peptide (PKCε+plus lanes of Figure 5A) or in the presence of scrambled control peptide (M4; Figure 5A, Figure S6A). Quantification of blots indicated that the ability of unmodified αCT1 to induce S368 phosphorylation was ~ 3‐fold greater than that of either the PKCε+plus control reaction (P<0.001) or reactions including M1 or M4 peptides (Figure 5B). It was further determined that a 9–amino acid peptide comprising only RPRPDDLEI (ie, αCT11=αCT1 with its 16–amino acid N‐terminal antennapedia sequence truncated) significantly increased pS368 levels over control (versus PKCε+plus control; P<0.001) (Figure 5B). αCT1‐I, the ZO‐1–binding deficient peptide with the CT isoleucine truncated, also prompted a significant increase in PKCε‐mediated phosphorylation of Cx43 CT (P<0.05 versus PKCε+plus control; Figure 5B, Figure S6A). Neither αCT1 nor M1 prompted an increase in phosphorylation of the Cx43 CT KK/QQ mutant substrate in the PKCε assay, consistent with interaction of αCT1 with these positively charged residues being required for upregulated pS368 (Figure S6B). These results from the kinase assay indicated that only those αCT1‐based peptides competent to interact Cx43 CT (ie, αCT1, αCT11, and αCT1‐I), but not those unable to (ie, M1, M2, M3, and M4), increased pS368 above control levels. Also, given that M3 is unable to prompt Cx43 pS368 increase, but does retain PDZ2 interaction ability, the assay results suggested that ZO‐1–binding activity is dispensable for this phosphorylation.
Figure 5.
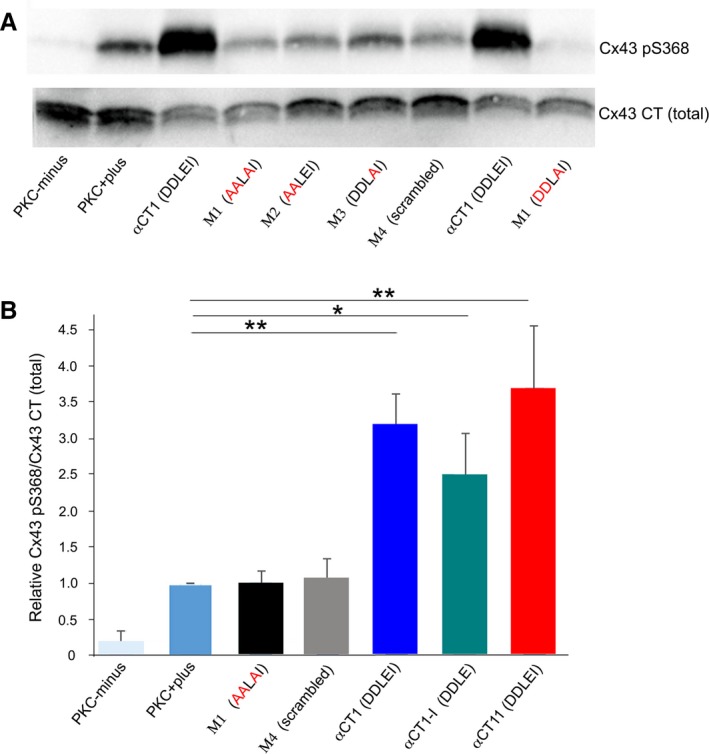
Connexin 43 (Cx43) mimetic peptides that retain Cx43‐binding capability are able to induce phosphorylation of Cx43 carboxyl terminus (CT) at serine 368 (S368). A, Blots of Cx43 phosphorylated at S368 (pS368) (top) and total Cx43 (bottom) in kinase reactions mixtures, including no‐kinase controls with substrate (Cx43‐CT: amino acids 255–382), but no protein kinase C (PKC)ε (PKC‐minus); Cx43‐CT substrate with PKCε (PKC‐plus); and mixtures containing PKCε, Cx43 CT, biotin‐tagged αCT1, biotin‐tagged αCT1 mutant peptides with alanine substitutions (M1, M2, M3), and biotin‐tagged M4 scrambled. B, Chart showing that the ability of unmodified αCT1 and the Cx43 CT interaction‐competent peptides biotin–deletion of the CT isoleucine in αCT1‐I or biotin‐αCT11 to induce S368 phosphorylation was 3‐ to 5‐fold greater than that of non‐Cx43 CT interacting peptides. *P<0.05, **P<0.01 (N=5 [αCT1 and M4], N=3 [other peptides]).
Only Peptides Interacting With Cx43 CT Protect Hearts From Ischemic Injury
The biochemical characterizations indicated that αCT1 is capable of 2 distinct protein‐protein interactions: one with ZO‐1 PDZ2 and the other with the Cx43 CT. This raised the question as to whether the previously characterized effects of αCT1 in cardiac injury models,8, 9 or its wound healing effects at large,13, 14, 15, 21 could be accounted for by one or another of these protein‐protein interactions. The series of αCT1‐based variant peptides generated for the present study provided an opportunity to address this question. Although αCT1‐I is not competent to interact with ZO‐1 PDZ2, this variant does bind the Cx43 CT and upregulates pS368. Conversely, although M3 showed diminished ability to either bind Cx43 CT or increase pS368, this peptide retained affinity for the ZO‐1 PDZ2 domain. Finally, M1 showed no evidence of interaction with either PDZ2 or Cx43 CT and demonstrated no ability to increase pS368 in the in vitro assay. We, thus, used the variant peptides, together with unmodified αCT1 in mouse hearts subjected to an I/R protocol, to systematically assess which aspect of mode of action (ie, peptide interaction with ZO‐1 versus Cx43) accounted for modulation of the I/R injury response by the Cx43 mimetic peptides.
The protocol and experimental design for the cardiac I/R injury model is illustrated in Figure S1. In summary, the protocol involved a 20‐minute period of no flow ischemia, followed by 40 minutes of reperfusion. For treatment, peptides were infused into hearts over a 20‐minute period just before the ischemic episode. Representative pressure traces from a vehicle control and αCT1‐treated hearts are shown in Figure 6A and 6B, from which it can be qualitatively appreciated that preischemic infusion of αCT1 results in preservation of LV contractile function on reperfusion relative to vehicle control.
Figure 6.
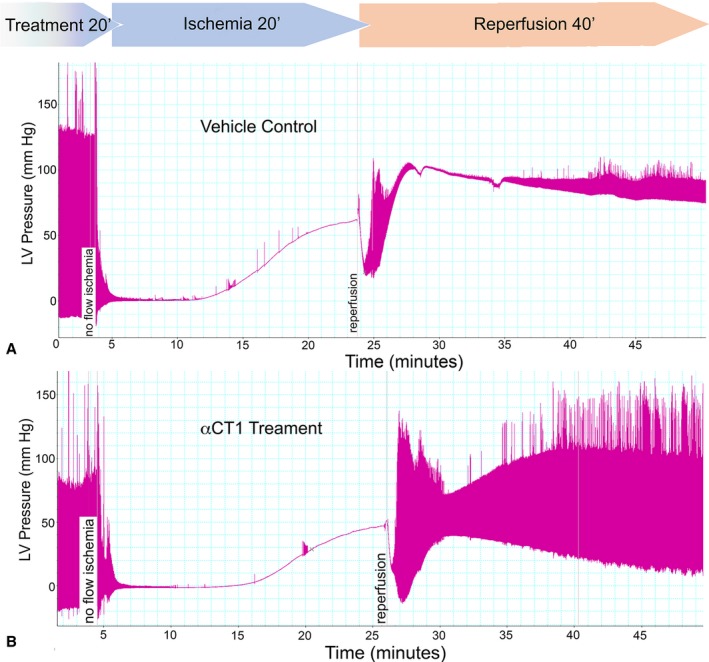
Preischemic treatment with peptides competent to interact with connexin 43 carboxyl terminus (CT) protect hearts from ischemia‐reperfusion (I/R) injury. Langendorff I/R protocols were performed on adult mouse hearts instrumented to monitor left ventricular (LV) function (protocol in Figure S1). Representative pressure traces from hearts from vehicle control (A) and 10 μmol/L αCT1 infused hearts (B). The αCT1 treatment results in notable recovery of LV function during reperfusion.
The effects of the αCT1 and the αCT1 variants on LV systolic and diastolic contractile function were correlated with the Cx43 CT‐binding potential of peptides (Figures 6 and 7A through 7D and Figure S7A through S7C). Although peptides with no or diminished Cx43 CT‐interacting capabilities showed no ability to improve recovery of either systolic (Figure 7A and 7B, Figure S7A) or diastolic (Figure 7C, Figure S7B and S7C) LV contractile performance during reperfusion, hearts pretreated with the Cx43 CT‐interacting peptides αCT1, αCT11, and αCT1‐I demonstrated significant functional recovery after I/R injury, compared with vehicle control mice (Figure 7A through 7D, Figure S7A through S7C). Furthermore, as αCT1‐I is able to interact with Cx43 CT, but not PDZ2, the results suggested that ZO‐1 binding was dispensable for induction of functional cardioprotection. More important, all Cx43 CT‐binding peptides resulted in highly significant 3‐ to 5‐fold improvements in functional recovery of LV contractile function after ischemic injury relative to vehicle control and the non‐Cx43 CT‐interacting peptides (Figure 7D). In line with the observations of the in vitro kinase assays (Figure 5, Figure S5), LV samples taken for Western blotting after preischemic treatment of Langendorff‐perfused mouse hearts with αCT1, αCT11, and αCT1‐I showed significant increases in phosphorylation at the Cx43 S368 relative to vehicle control perfused hearts (Figure 7E). By contrast, hearts exposed to peptides with abrogated ability to interact with Cx43 CT (ie, M1 and M3) showed no propensity to upregulate S368 phosphorylation (Figure 7E).
Figure 7.
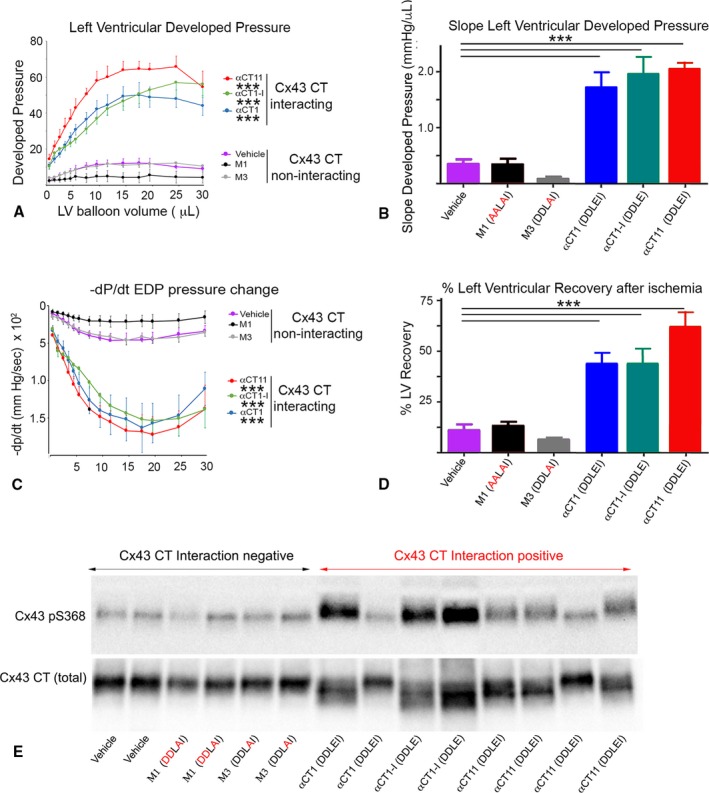
Preischemic treatment with peptides interacting with connexin 43 (Cx43) carboxyl terminus (CT) protects hearts from ischemia‐reperfusion (I/R) injury in association with increased phosphorylation at Cx43 serine 368 (pS368) in left ventricular (LV) myocardium. Langendorff I/R injury protocols were performed on adult mouse hearts instrumented to monitor LV contractility (protocol in Figure S1). Plots of LV systolic developed pressure against balloon volume (A); maximal systolic elastance (ie, the slope from A (B); rate of relaxation (‐dP/dt) against balloon volume (C); and percentage of LV contractile function recovery postischemia relative to baseline level (D). Data shown are mean±SE. Number of hearts/group: vehicle=8; αCT1=7; deletion of the CT isoleucine αCT1‐I=6; αCT11=4; M1=5; and M3=5 hearts. E, Blots of Cx43 pS368 (top) and total Cx43 (bottom) of LV samples infused with peptide for 20 minutes, according to the protocol in Figure S1. For hearts used in Western blots, the protocol did not proceed to the ischemia and reperfusion phases, being terminated after the peptide infusion step. Only those peptides competent to interact with Cx43 CT increase pS368 levels relative to total Cx43 above vehicle control. *P<0.05, ***P<0.001.
Postischemic Treatment With the 9‐Mer Peptide αCT11 Preserves LV Function
αCT1 is in phase 3 clinical testing in humans for pathologic skin wounds.16 The results of Figure 7 indicated that pretreatment with Cx43 CT‐binding peptides provided protection from injury in the ex vivo model studied. However, to be clinically useful to patients, such as those experiencing a myocardial infarction, a drug would typically need to be given after an ischemic insult to the heart (ie, after a myocardial infarction has been diagnosed). We, thus, treated hearts during the reperfusion phase after ischemic injury with αCT1, but determined that this did not result in significant recovery of LV function (data not shown). As αCT1 showed no evidence of postinfarction efficacy, we decided to explore an alternative approach. It was notable that the most striking recovery of postischemic LV function resulted from preischemic treatment with the 9‐mer Cx43 CT‐binding peptide αCT11 (Table and Figure 7A through 7D). This is illustrated in Figure 7A, where the curve for LV developed pressure for αCT11 conspicuously overarches that of the other 2 Cx43‐interacting peptides, αCT1 and αCT1‐I. This can also be observed in Figure 7D, where the percentage of LV function recovery associated with αCT11 preinfusion significantly exceeds that of αCT1 or αCT1‐I (P<0.05). On the basis of these results suggestive of increased potency, we sought to determine whether αCT11 had a postischemic cardioprotective effect.
αCT11 demonstrated an ability to significantly improve recovery of both systolic (Figure 8A and 8B, Figure S7D) and diastolic (Figure 8C and 8D, Figure S7E) LV contractile performance when infused in hearts during reperfusion after ischemic injury. The level of cardioprotection achieved by this postischemic treatment was not as high as when αCT11 was provided before insult, but it was similar to that resulting from preischemic treatment with αCT1 (Figure 8A through 8D, Figure S7D and S7E). Given that αCT11 is missing a cell penetration sequence, we were curious to determine whether the 9‐mer peptide (molecular weight=1110 Da) was being taken up into cardiomyocytes. We, thus, examined uptake of αCT11 in ventricular muscle in mouse hearts that had been perfused with a biotinlylated αCT11 under the protocol summarized in Figure S1. Cardiomyocytes showed uptake of the 9‐mer αCT11 sequence, as detected by Fluor‐conjugated streptavidin, and as compared with vehicle control perfused hearts (Figure 8E, Figure S7F). The cytoplasmic staining of αCT11 in ventricular myocytes, revealed by Laser Scanning Confocal Microscope (LSCM) optical sectioning, was diffuse; and we were unable to resolve increased localization of signals in association with Cx43 puncta. Nonetheless, relative to vehicle control, quantified levels of uptake of αCT11 in cells were comparable to those of αCT1, as indicated by measurement of relative fluorescence intensity levels in ventricular myocardial tissues (Figure S7F).
Figure 8.
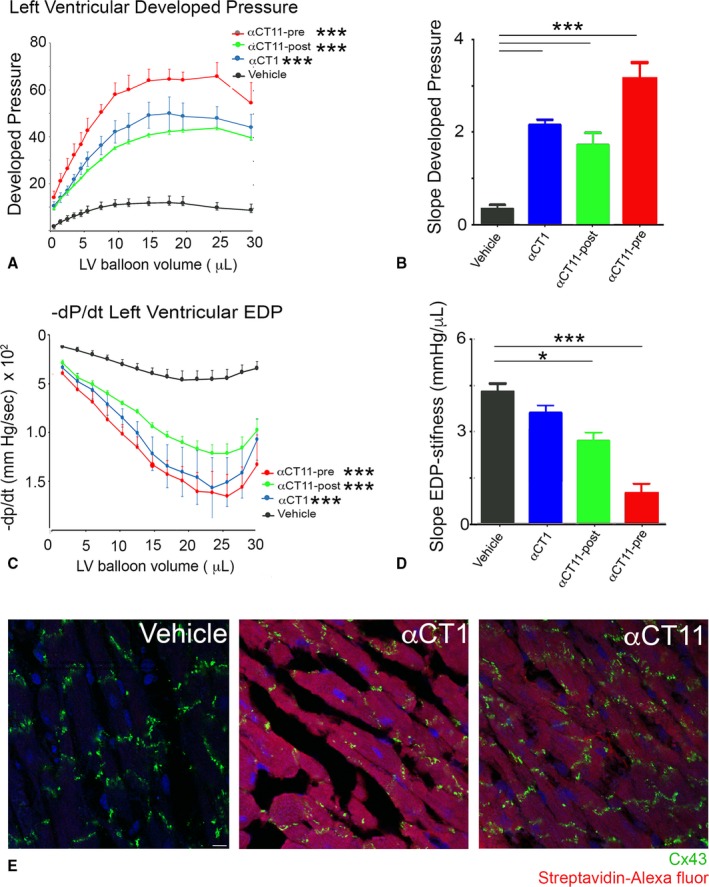
Preischemic and postischemic treatment with α carboxyl terminus 11 (αCT11) protects hearts from ischemia‐reperfusion (I/R) injury. Langendorff I/R protocols were performed on adult mouse hearts instrumented to monitor left ventricular (LV) contractility. Protocol used was as in Figure S1, except that a 20‐minute peptide infusion was begun after ischemic injury at the initiation of reperfusion (ie, a postischemic treatment as opposed to preischemic treatment). A, Plots of LV developed systolic pressure against balloon volume. B, Maximal systolic elastance (ie, the slope from A). C, Maximal rate of diastolic relaxation (‐dP/dt) against balloon volume. D, LV stiffness: The reciprocal of the slope of LV end‐diastolic pressure (EDP) against balloon volume (see Figure S8B). Number of hearts/group: vehicle postischemia=4; αCT11 postischemia=4; αCT11 preischemia=4; and αCT1 preischemia=7 hearts. E, Laser scanning confocal microscopic fields of ventricular sections from vehicle control, αCT1, and αCT11 treated hearts stained for connexin 43 (Cx43; green), nuclei (4′,6‐diamidino‐2‐phenylindole; blue), and Alexa647‐conjugated streptavidin (red). *P<0.05, ***P<0.001.
Discussion
This study demonstrates that peptide mimetic sequences incorporating the CT‐most 9 amino acids of Cx43 (amino acids R374 to I382) complex with the Cx43 CT in an interaction potentially involving sequences located between amino acids Y313 and A348 of Cx43. This interaction causes changes in the thermal stability of Cx43 CT, potentially indicative of alterations in polypeptide structural stability, and is associated with increases in a PKC‐mediated phosphorylation in a serine residue at position 368 of Cx43 (pS368). Moreover, evidence is provided that the cardioprotective properties of Cx43 CT mimetic peptides, such as αCT1, may be explained to a significant degree by their propensity to interact with the Cx43 CT. This conclusion is supported by our results indicating that Cx43 CT‐binding competent peptides αCT1, αCT1‐I, and αCT11 preserve LV function after ischemic injury, whereas Cx43 interaction deficient variants of αCT1, M1 and M3, do not. Interestingly, αCT1 interaction may involve a spatially distinct region of the Cx43 CT, including its H2 sequence, and as such points to the possibility that interactions between or within Cx43 molecules may be involved in regulating phosphorylation.
Our observations on the relationship between PKCε‐mediated phosphorylation of S368 and cardioprotection are consistent with long‐standing literature.6, 7, 8, 32, 33, 34, 35, 36, 37, 38, 39 Phosphorylation of Cx43 at S368 is correlated with reduced activity of Cx43‐formed hemichannels.6, 40, 41, 42 Proinflammatory and injury spread signals, resulting from unregulated opening of hemichannels in the myocyte sarcolemma, are thought to be determinants of the severity of I/R damage to the heart.43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54 Cx43 activity and S368 phosphorylation events associated with mitochondrial membranes have also been linked to I/R injury severity.44, 55, 56 Interestingly, it has been reported that Cx43 CT sequences incorporating the Cx43 H2‐binding sequence of interest herein result from alternative translation of the GJA1 gene.57 These include a 20‐kDa GJ isoform, which has been found to be enriched at the interface between mitochondria and microtubules.58 Similar to the results achieved with synthetic Cx43 CT mimetic sequences herein, exogenous provision of the 20‐kDa GJ isoform reduces infarct size in mouse hearts subjected to I/R injury.59 Ongoing experiments would usefully address the extent to which treatment regimens, based on αCT peptides and 20‐kDa GJ isoform, share aspects of molecular mechanisms.
The pH‐dependent gating of Cx43‐formed channels has been proposed to involve the Cx43 CT in a “ball‐and chain” mechanism.60, 61 The demonstration that the CT‐most 10 amino acids of Cx43 (S373‐I382; aka, CT10) interact with a region of the cytoplasmic loop domain of Cx43, referred to as L2, resulting in channel closure under acidic conditions, provides evidence supporting this hypothesis.62 We demonstrate in the current article that a near‐identical sequence to CT10, contained in αCT1 (ie, R374‐I382), may also interact with the H2 region of the Cx43 CT, potentially doing so via the same negatively charged amino acids required for L2 interaction.17 In addition to the shared potential affinity of the CT‐most 9 amino acids of Cx43 for L2 and H2, comparison of L2 and H2 indicate other notable parallels. The L2 and H2 sequences of Cx43 have related secondary structures, both being marked by short stretches of α‐helix. Furthermore, L2 and H2 incorporate a pair of lysine (KK) residues. As we demonstrate herein, these lysines appear to be required for αCT1 interaction, as substitution of K345 and K346 with neutral glutamines, as in the Cx43 CT KK/QQ construct, results in a loss of αCT1 binding to Cx43 CT, as well as a loss of the ability of the peptide to prompt Cx43 pS368.
Taken together, the evidence suggests that the 9–amino acid CT sequence of Cx43, mimicked by αCT1, is a multivalent ligand that participates in several protein‐protein interactions. In addition to affinity for other Cx43 regions (eg, L2), this short segment of Cx43 includes the PDZ‐binding ligand necessary for linkage to ZO‐1,10, 11, 22, 23, 24, 25, 26 as well as amino acids required for interaction with 14‐3‐3θ.63 Immediately proximal are consensus recognition sites for protein kinase B (S373),64 PKCε (S368),6, 7, 8, 65, 66 and T‐cell protein tyrosine phosphatase.67 The potential for complex patterns of protein‐protein interaction occurring within such short stretch of the Cx43 primary sequence raises intriguing possibilities for future work. Of relevance are questions as to the timing of association of partnering proteins during the Cx43 life cycle and the nature of the secondary and tertiary structures involved. With respect to the latter, it would be interesting to explore whether Cx43 S368 accessibility to PKCε could be governed by intramolecular interactions within Cx43 or involves intermolecular interactions between Cx43 molecules. For example, dimers of Cx43 molecules in overlapping interactions between the CT‐most amino acids of one Cx43 molecule and the H2 domain of an adjacent Cx43 within a connexon are suggested as one hypothesis as to how accessibility to S368 might be regulated. This being said, an important caveat bears repeating. Although our data suggest that the CT‐most 9 amino acids of Cx43 have the potential to interact with the Cx43 H2 region, the evidence we provide does not preclude other regions of the Cx43 also having affinity for the peptide. Further work is required to determine whether this interaction is realized in vivo.
The results of this study indicate that the Cx43 CT‐binding activity of αCT1, and not ZO‐1 PDZ2 interaction, explains the cardioprotective effects of αCT1, at least in the model used. Although Cx43–ZO‐1 interaction does not appear to have been a direct factor in our ex vivo model, potential roles for ZO‐1 in regulating Cx43 phosphorylation status and hemichannel availability in vivo, including during ischemic injury, should not be discounted. ZO‐1 is located at the edge of Cx43 GJs in a specialized zone of cell membrane known as the perinexus.10 In earlier studies, we have shown that high densities of hemichannels are found in this perijunctional region10, 68, 69, 70 and that PDZ‐based interactions between ZO‐1 and Cx43 govern the rate at which undocked connexons dock with connexons from apposed cells to form gap junctional channels, thereby regulating GJ size, as well as hemichannel availability within the cell membrane.10, 11 Recent work by 2 other groups has provided data supporting this hypothesis and has also shown that phosphorylation at Cx43 S368 and S373 is central to how ZO‐1 controls the accrual of perinexal hemichannels to the GJ.64, 71, 72 The potential for regulatory interplay between PKCε and ZO‐1 at the Cx43‐CT is further suggested by earlier studies indicating that the presence of ZO‐1 PDZ2 domain in the test tube–based PKC assay efficiently acted as a competitive inhibitor of αCT1 enhancement of S368 phosphorylation.8
A key question raised by our study is whether the αCT1 Cx43‐targeting mechanism, determined as necessary for preservation of LV function, also explains the primary mode of action of this therapeutic peptide in other tissues. In skin wounding experiments in mice and pigs, αCT1 has been shown to reduce inflammation, increase wound healing rates, and decrease granulation tissue formation.21 In related observations in phase 2 clinical testing of humans, αCT1 treatment increased the healing rate of slow‐healing skin wounds, including diabetic foot ulcers and venous leg ulcers.13, 14 Given the current results in heart, it will be of interest to determine whether the mode of action of αCT1 in wounded skin also involves Cx43 CT interaction and/or increased pS368. As the GAIT1 phase 3 clinical trial on αCT1 moves forward on >500 patients with diabetic foot ulcers,16 such insight on molecular mode of action will be useful in understanding the basis of any clinical efficacy identified in humans, as well as a step in building a safety profile for this therapeutic peptide.
Of further translational note are our findings on the cardioprotective effect of postischemic treatment by the short αCT1 variant αCT11, a result that may have clinical implications. This effect is mediated within a 20‐minute perfusion of αCT11 (at 10 μmol/L) into isolated heart preparations and is correlated with diffuse concentrations of αCT11 within the cytoplasm of ventricular myocytes in confocal optical sections. At present, we cannot not discriminate whether αCT11 is colocalized with Cx43 or accumulates in specific cell compartments, such as the nucleus. Techniques, such as polymerization ligation assays localizing codistribution of αCT11 with specific proteins (eg, Cx43), could provide such insights in future work. Interestingly, αCT11 does not have a cell penetration sequence, but it nonetheless is efficiently internalized by LV cardiomyocytes after intravascular perfusion in the ex vivo model used herein. The mechanism of this cellular uptake is presently under study by our group, but it may be explained by the small size (molecular weight=1110 Da) and linear, random, coiled‐coil 3‐dimensional structure of αCT11 (Figure 2A). Neijssen and coworkers reported that linear peptides with molecular masses <1800 Da readily diffuse through Cx43‐formed channels.73 Given αCT11 is a linear peptide with a molecular mass well <1800 Da, and that hemichannel opening is induced by ischemic insult,43 the interesting prospect is raised that αCT11 reaches its cytoplasmic target (ie, the CT domain of Cx43) via a short (<20‐nm) transit through an open Cx43 hemichannel pore. Future work would usefully test this hypothesis, as well as undertake testing of αCT11 in preclinical models of cardiac I/R injury in vivo as a prelude to phase 1 testing of this novel therapeutic in human patients with acute myocardial infarction.
Author Contributions
Drs Jiang, Palatinus, He, Bultynck, Schey, Poelzing, Hoagland, McGowan, and Gourdie were responsible for experimental design and data interpretation. Drs Jiang, Palatinus, He, Iyyathurai, Jiang, and Wang conducted experiments. Dr Gourdie was largely responsible for manuscript preparation, with editing assistance from Drs Jiang, He, Bultynck, Hoagland, Zhang, Schey, Poelzing, and McGowan.
Sources of Funding
This work was supported by National Institutes of Health (NIH) R01 grant HL56728 to Dr Gourdie, NIH R01 grant HL141855 to Dr Poelzing, and NIH R01 grant HL141855 to Drs Gourdie and Poelzing.
Disclosures
Dr Gourdie is a nonremunerated member of the Scientific Advisory Board of FirstString Research, which licensed α carboxyl terminus 1. Dr Gourdie and L.J. Jourdan have modest (<3% of total stock) ownership interests in the company. The remaining authors have no disclosures to report.
Supporting information
Table S1. ΔG of Binding After a Molecular Mechanics Energy Combined With Generalized Born and Surface Area Continuum Solvation (MM‐GBSA) Minimization of the Protein:Peptide Complex Using a 5 Å Distance Threshold Between Docked Peptide and Protein
Figure S1. The Ischemia Reperfusion (I/R) injury model/protocol used in this study involved a 20‐minute period of no flow ischemia period followed by 40 minutes of reperfusion, left ventricular (LV) contractile function was monitored throughout the whole process.
Figure S2. Blots of products of kinase reaction mixtures containing glutathione S‐transferase (GST)‐Connexin 43 (Cx43) carboxyl terminus (CT), GST‐Protein Kinase C (PKC)ε and biotinylated αCT1 (20 μmol/L).
Figure S3. Blots of cross‐linked products of kinase reaction mixtures containing glutathione S‐transferase (GST)‐Connexin 43 (Cx43) carboxyl terminus (CT), GST‐Cx43 CT QQ/KK in which the lysine (K) residues were mutated to neutral glutamines (Q), Protein Kinase C (PKC)ε and αCT1 (at 5, 10 and 25 μmol/L) and a scrambled αCT1 (M4 scr) variant at the same concentrations.
Figure S4. A, Cartoon depiction of the two alpha helical regions of the Connexin 43 (Cx43) carboxyl terminus (CT), H1 and H2 from Sorgen et al.28. B, Schematic representation of the Cx43 Y313‐A348 peptide synthesized for a binding surface surrogate with linkable cysteine (Cys) on the amino terminus and CT. C, Single letter amino acid sequence of Cx43 Y313‐A348 peptide with predicted helix secondary structure underlined. D, Surface Plasmon Resonance (SPR) analysis of substrate captured αCT1 (700–1000 RUs) binding recombinant Cx43 CT (100 μmol/L, yellow), unlinked Cx43 Y313‐A348 peptide (25 μmol/L, Blue), and disulfide linked Cx43 Y313‐A348 (25 μmol/L, green). SPR indicates that non‐disulfide linked Cx43 Y313‐A348 peptide shows levels of interaction with αCT1 comparable to the full Cx43 CT polypeptide sequence ≈150 amino acids). Disulfide cross‐linking Cx43 Y313‐A348 into a looped conformation results in a loss of αCT1 binding, suggesting that αCT1 interaction with this peptide requires a degree conformational flexibility.
Figure S5. The αCT1 variant peptide M2 AALEI shows limited ability to bind Connexin 43 (Cx43) carboxyl terminus (CT). Surface Plasmon Resonance (SPR) was used to analyze interactions of biotin‐M2 AALEI with the Cx43 CT (A) and Cx43 CT‐KK/QQ (B) as respective analytes. The mean of three runs is plotted for each analyte concentration. The exposure of the sensor chip to the specific analyte is indicated by the gray area.
Figure S6. A, Blots of Connexin 43 (Cx43) phosphorylated at S368 (pS368‐top) and total Cx43 (bottom) in kinase reactions mixtures including no‐kinase controls with Cx43 carboxyl terminus (CT) substrate, but no Protein Kinase C (PKC)ε (PKC‐minus); Cx43‐CT substrate with PKC‐ε (PKC‐plus); and mixtures containing PKC‐ε, Cx43 CT, and biotin‐αCT1, biotin‐αCT1‐ I or biotin‐αCT11 (RPRPDDLEI with no antennapedia sequence at peptide amino terminus) and biotin‐M4 scrambled peptide. B) Blots of Cx43‐pS368 (top) and total Cx43 (bottom) in kinase reactions mixtures including no‐kinase controls with Cx43 CT and Cx43 CT KK/QQ substrates, but no PKC‐ε (PKC‐minus); Cx43‐CT and Cx43 CT KK/QQ substrates with PKC‐ε (PKC‐plus); and in mixtures containing CT substrates, PKC‐ε, and biotin‐αCT1(20 μmol/L).
Figure S7. A through C, Langendorff Ischemia‐Reperfusion (I/R) injury protocols were performed on adult mouse hearts pre‐treated with peptides and instrumented to monitor left ventricular (LV) contractility as shown in Figure 7 Plots of (A) Maximal systolic elastance (Emax)—ie, the slope from plots shown in Figure 7A in the manuscript; (B) LV end diastolic pressure (EDP) against balloon volume; (C) Maximal systolic elastance (Emax), the slopes from Figure S7B. D and E, Langendorff I/R injury protocols were performed on adult mouse hearts pre‐and post‐treated with peptides and instrumented to monitor LV contractility as shown in Figure 8. Plots of (D) Maximal systolic elastance (Emax)—ie, the slope from plots shown in Figure 7A in the manuscript; and (E) LV end diastolic pressure (EDP) against balloon volume; Data shown in Figure S7A through S7E are mean±SE *P<0.05, ***P<0.001, N hearts/group: N hearts/group: Vehicle pre ischemia=8; Vehicle post ischemia=4; αCT1 pre ischemia=7; αCT1‐I pre ischemia=6; αCT11 pre and post ischemia=4; M1 pre ischemia=5; and M3 pre ischemia=5 hearts. F, Average intensities of biotinylated peptide (indicated by streptavidin Alexa647 fluorescence intensity in ventricular myocytes level relative to background) in frozen sections from ventricles in the Vehicle control, αCT1, and αCT11 treated groups. *P<0.05; not significant (ns) N=5 hearts/group.
Acknowledgments
We thank Greg Hoeker, PhD, and Randy Strauss, BS, for reading and providing suggestions on the manuscript. Jacob Bond, BS, is thanked for his technical assistance in surface plasmon resonance imaging studies. We thank Linda Collins for her editorial contribution to this article.
(J Am Heart Assoc. 2019;8:e012385 DOI: 10.1161/JAHA.119.012385.)
Contributor Information
Zhiwei Zhang, Email: drzhangzw@sohu.com.
Robert G. Gourdie, Email: gourdier@vtc.vt.edu.
References
- 1. Laird DW, Lampe PD, Johnson RG. Cellular small talk. Sci Am. 2015;312:70–77. [DOI] [PubMed] [Google Scholar]
- 2. Desplantez T, Dupont E, Severs NJ, Weingart R. Gap junction channels and cardiac impulse propagation. J Membr Biol. 2007;218:13–28. [DOI] [PubMed] [Google Scholar]
- 3. Jansen JA, van Veen TA, de Bakker JM, van Rijen HV. Cardiac connexins and impulse propagation. J Mol Cell Cardiol. 2010;48:76–82. [DOI] [PubMed] [Google Scholar]
- 4. Martin PE, Evans WH. Incorporation of connexins into plasma membranes and gap junctions. Cardiovasc Res. 2004;62:378–387. [DOI] [PubMed] [Google Scholar]
- 5. Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008;80:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Solan JL, Lampe PD. Spatio‐temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim Biophys Acta. 2018;1860:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ek‐Vitorin JF, King TJ, Heyman NS, Lampe PD, Burt JM. Selectivity of connexin 43 channels is regulated through protein kinase C‐dependent phosphorylation. Circ Res. 2006;98:1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. O'Quinn MP, Palatinus JA, Harris BS, Hewett KW, Gourdie RG. A peptide mimetic of the Connexin43 carboxyl terminus reduces gap junction remodeling and induced arrhythmia following ventricular injury. Circ Res. 2011;108:704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ongstad EL, O'Quinn MP, Ghatnekar GS, Yost MJ, Gourdie RG. A Connexin43 mimetic peptide promotes regenerative healing and improves mechanical properties in skin and heart. Adv Wound Care (New Rochelle). 2013;2:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rhett JM, Jourdan J, Gourdie RG. Connexin43 connexon to gap junction transition is regulated by zonula occludens‐1. Mol Biol Cell. 2011;22:1516–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hunter AW, Barker RJ, Zhu C, Gourdie RG. Zonula occludens‐1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell. 2005;16:5686–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Laird DW, Lampe PD. Therapeutic strategies targeting connexins. Nat Rev Drug Discov. 2018;17:905–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grek CL, Prasad GM, Viswanathan V, Armstrong DG, Gourdie RG, Ghatnekar GS. Topical administration of a connexin43‐based peptide augments healing of chronic neuropathic diabetic foot ulcers: a multicenter, randomized trial. Wound Repair Regen. 2015;23:203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grek CL, Montgomery J, Sharma M, Ravi A, Rajkumar JS, Moyer KE, Gourdie RG, Ghatnekar GS. A multicenter randomized controlled trial evaluating a Cx43‐mimetic peptide in cutaneous scarring. J Invest Dermatol. 2017;137:620–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ghatnekar GS, Grek CL, Armstrong DG, Desai SC, Gourdie RG. The effect of a connexin43‐based peptide on the healing of chronic venous leg ulcers: a multicenter, randomized trial. J Invest Dermatol. 2015;135:289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. ClinicalTrials.gov . A study of granexin (αCT1) gel in the treatment of diabetic foot ulcer. https://clinicaltrials.gov/ct2/show/NCT02667327. GAIT 1 Phase 3 Clinical Trial. Accessed July 11, 2019.
- 17. D'Hondt C, Iyyathurai J, Wang N, Gourdie RG, Himpens B, Leybaert L, Bultynck G. Negatively charged residues (Asp378 and Asp379) in the last ten amino acids of the C‐terminal tail of Cx43 hemichannels are essential for loop/tail interactions. Biochem Biophys Res Commun. 2013;432:707–712. [DOI] [PubMed] [Google Scholar]
- 18. Wu SY, Zou P, Fuller AW, Mishra S, Wang Z, Schey KL, McHaourab HS. Expression of cataract‐linked gamma‐crystallin variants in zebrafish reveals a proteostasis network that senses protein stability. J Biol Chem. 2016;291:25387–25397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein‐protein complexes and symmetric multimers. Bioinformatics. 2014;30:1771–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. He H, Javadpour MM, Latif F, Tardiff JC, Ingwall JS. R‐92L and R‐92W mutations in cardiac troponin T lead to distinct energetic phenotypes in intact mouse hearts. Biophys J. 2007;93:1834–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ghatnekar GS, O'Quinn MP, Jourdan LJ, Gurjarpadhye AA, Draughn RL, Gourdie RG. Connexin43 carboxyl‐terminal peptides reduce scar progenitor and promote regenerative healing following skin wounding. Regen Med. 2009;4:205–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens‐1 protein. Curr Biol. 1998;8:931–934. [DOI] [PubMed] [Google Scholar]
- 23. Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Hori M, Tada M. Direct association of the gap junction protein connexin‐43 with ZO‐1 in cardiac myocytes. J Biol Chem. 1998;273:12725–12731. [DOI] [PubMed] [Google Scholar]
- 24. Ambrosi C, Ren C, Spagnol G, Cavin G, Cone A, Grintsevich EE, Sosinsky GE, Sorgen PL. Connexin43 forms supramolecular complexes through non‐overlapping binding sites for drebrin, tubulin, and ZO‐1. PLoS One. 2016;11:e0157073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jin C, Martyn KD, Kurata WE, Warn‐Cramer BJ, Lau AF. Connexin43 PDZ2 binding domain mutants create functional gap junctions and exhibit altered phosphorylation. Cell Commun Adhes. 2004;11:67–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bouvier D, Spagnol G, Chenavas S, Kieken F, Vitrac H, Brownell S, Kellezi A, Forge V, Sorgen PL. Characterization of the structure and intermolecular interactions between the connexin40 and connexin43 carboxyl‐terminal and cytoplasmic loop domains. J Biol Chem. 2009;284:34257–34271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sosinsky GE, Solan JL, Gaietta GM, Ngan L, Lee GJ, Mackey MR, Lampe PD. The C‐terminus of connexin43 adopts different conformations in the Golgi and gap junction as detected with structure‐specific antibodies. Biochem J. 2007;408:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sorgen PL, Duffy HS, Sahoo P, Coombs W, Delmar M, Spray DC. Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c‐Src and zonula occludens‐1. J Biol Chem. 2004;279:54695–54701. [DOI] [PubMed] [Google Scholar]
- 29. Sorgen PL, Duffy HS, Spray DC, Delmar M. pH‐dependent dimerization of the carboxyl terminal domain of Cx43. Biophys J. 2004;87:574–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Semisotnov GV, Rodionova NA, Razgulyaev OI, Uversky VN, Gripas AF, Gilmanshin RI. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 1991;31:119–128. [DOI] [PubMed] [Google Scholar]
- 31. Chen J, Pan L, Wei Z, Zhao Y, Zhang M. Domain‐swapped dimerization of ZO‐1 PDZ2 generates specific and regulatory connexin43‐binding sites. EMBO J. 2008;27:2113–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hawat G, Baroudi G. Differential modulation of unapposed connexin 43 hemichannel electrical conductance by protein kinase C isoforms. Pflugers Arch. 2008;456:519–527. [DOI] [PubMed] [Google Scholar]
- 33. Hatanaka T, Shimizu R, Hildebrand D. Expression of a Stokesia laevis epoxygenase gene. Phytochemistry. 2004;65:2189–2196. [DOI] [PubMed] [Google Scholar]
- 34. Hund TJ, Lerner DL, Yamada KA, Schuessler RB, Saffitz JE. Protein kinase Cepsilon mediates salutary effects on electrical coupling induced by ischemic preconditioning. Heart Rhythm. 2007;4:1183–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miura T, Miki T, Yano T. Role of the gap junction in ischemic preconditioning in the heart. Am J Physiol Heart Circ Physiol. 2010;298:H1115–H1125. [DOI] [PubMed] [Google Scholar]
- 36. Naitoh K, Yano T, Miura T, Itoh T, Miki T, Tanno M, Sato T, Hotta H, Terashima Y, Shimamoto K. Roles of Cx43‐associated protein kinases in suppression of gap junction‐mediated chemical coupling by ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2009;296:H396–H403. [DOI] [PubMed] [Google Scholar]
- 37. Jeyaraman MM, Srisakuldee W, Nickel BE, Kardami E. Connexin43 phosphorylation and cytoprotection in the heart. Biochim Biophys Acta. 2012;1818:2009–2013. [DOI] [PubMed] [Google Scholar]
- 38. Jozwiak J, Dhein S. Local effects and mechanisms of antiarrhythmic peptide AAP10 in acute regional myocardial ischemia: electrophysiological and molecular findings. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:459–470. [DOI] [PubMed] [Google Scholar]
- 39. Morel S, Christoffersen C, Axelsen LN, Montecucco F, Rochemont V, Frias MA, Mach F, James RW, Naus CC, Chanson M, Lampe PD, Nielsen MS, Nielsen LB, Kwak BR. Sphingosine‐1‐phosphate reduces ischaemia‐reperfusion injury by phosphorylating the gap junction protein Connexin43. Cardiovasc Res. 2016;109:385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol. 2000;149:1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bao X, Altenberg GA, Reuss L. Mechanism of regulation of the gap junction protein connexin 43 by protein kinase C‐mediated phosphorylation. Am J Physiol Cell Physiol. 2004;286:C647–C654. [DOI] [PubMed] [Google Scholar]
- 42. Fiori MC, Figueroa V, Zoghbi ME, Saez JC, Reuss L, Altenberg GA. Permeation of calcium through purified connexin 26 hemichannels. J Biol Chem. 2012;287:40826–40834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shintani‐Ishida K, Uemura K, Yoshida K. Hemichannels in cardiomyocytes open transiently during ischemia and contribute to reperfusion injury following brief ischemia. Am J Physiol Heart Circ Physiol. 2007;293:H1714–H1720. [DOI] [PubMed] [Google Scholar]
- 44. Boengler K, Stahlhofen S, van de Sand A, Gres P, Ruiz‐Meana M, Garcia‐Dorado D, Heusch G, Schulz R. Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res Cardiol. 2009;104:141–147. [DOI] [PubMed] [Google Scholar]
- 45. Morel S, Kwak BR. Roles of connexins in atherosclerosis and ischemia‐reperfusion injury. Curr Pharm Biotechnol. 2012;13:17–26. [DOI] [PubMed] [Google Scholar]
- 46. Gill R, Kuriakose R, Gertz ZM, Salloum FN, Xi L, Kukreja RC. Remote ischemic preconditioning for myocardial protection: update on mechanisms and clinical relevance. Mol Cell Biochem. 2015;402:41–49. [DOI] [PubMed] [Google Scholar]
- 47. Retamal MA, Schalper KA, Shoji KF, Orellana JA, Bennett MV, Saez JC. Possible involvement of different connexin43 domains in plasma membrane permeabilization induced by ischemia‐reperfusion. J Membr Biol. 2007;218:49–63. [DOI] [PubMed] [Google Scholar]
- 48. Gourdie RG, Dimmeler S, Kohl P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat Rev Drug Discov. 2016;15:620–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Saez JC, Schalper KA, Retamal MA, Orellana JA, Shoji KF, Bennett MV. Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp Cell Res. 2010;316:2377–2389. [DOI] [PubMed] [Google Scholar]
- 50. Wang N, De Bock M, Decrock E, Bol M, Gadicherla A, Vinken M, Rogiers V, Bukauskas FF, Bultynck G, Leybaert L. Paracrine signaling through plasma membrane hemichannels. Biochim Biophys Acta. 2013;1828:35–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li F, Sugishita K, Su Z, Ueda I, Barry WH. Activation of connexin‐43 hemichannels can elevate [Ca(2+)]i and [Na(+)]i in rabbit ventricular myocytes during metabolic inhibition. J Mol Cell Cardiol. 2001;33:2145–2155. [DOI] [PubMed] [Google Scholar]
- 52. Kondo RP, Wang SY, John SA, Weiss JN, Goldhaber JI. Metabolic inhibition activates a non‐selective current through connexin hemichannels in isolated ventricular myocytes. J Mol Cell Cardiol. 2000;32:1859–1872. [DOI] [PubMed] [Google Scholar]
- 53. Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;277:10482–10488. [DOI] [PubMed] [Google Scholar]
- 54. Clarke TC, Williams OJ, Martin PE, Evans WH. ATP release by cardiac myocytes in a simulated ischaemia model: inhibition by a connexin mimetic and enhancement by an antiarrhythmic peptide. Eur J Pharmacol. 2009;605:9–14. [DOI] [PubMed] [Google Scholar]
- 55. Boengler K, Dodoni G, Rodriguez‐Sinovas A, Cabestrero A, Ruiz‐Meana M, Gres P, Konietzka I, Lopez‐Iglesias C, Garcia‐Dorado D, Di Lisa F, Heusch G, Schulz R. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc Res. 2005;67:234–244. [DOI] [PubMed] [Google Scholar]
- 56. Rodriguez‐Sinovas A, Boengler K, Cabestrero A, Gres P, Morente M, Ruiz‐Meana M, Konietzka I, Miro E, Totzeck A, Heusch G, Schulz R, Garcia‐Dorado D. Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90‐dependent TOM pathway and its importance for cardioprotection. Circ Res. 2006;99:93–101. [DOI] [PubMed] [Google Scholar]
- 57. Smyth JW, Shaw RM. Autoregulation of connexin43 gap junction formation by internally translated isoforms. Cell Rep. 2013;5:611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fu Y, Zhang SS, Xiao S, Basheer WA, Baum R, Epifantseva I, Hong T, Shaw RM. Cx43 isoform GJA1‐20k promotes microtubule dependent mitochondrial transport. Front Physiol. 2017;8:905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Basheer WA, Xiao S, Epifantseva I, Fu Y, Kleber AG, Hong T, Shaw RM. GJA1‐20k arranges actin to guide Cx43 delivery to cardiac intercalated discs. Circ Res. 2017;121:1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Morley GE, Taffet SM, Delmar M. Intramolecular interactions mediate pH regulation of connexin43 channels. Biophys J. 1996;70:1294–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Moreno AP, Chanson M, Elenes S, Anumonwo J, Scerri I, Gu H, Taffet SM, Delmar M. Role of the carboxyl terminal of connexin43 in transjunctional fast voltage gating. Circ Res. 2002;90:450–457. [DOI] [PubMed] [Google Scholar]
- 62. Ponsaerts R, De Vuyst E, Retamal M, D'Hondt C, Vermeire D, Wang N, De Smedt H, Zimmermann P, Himpens B, Vereecke J, Leybaert L, Bultynck G. Intramolecular loop/tail interactions are essential for connexin 43‐hemichannel activity. FASEB J. 2010;24:4378–4395. [DOI] [PubMed] [Google Scholar]
- 63. Smyth JW, Zhang SS, Sanchez JM, Lamouille S, Vogan JM, Hesketh GG, Hong T, Tomaselli GF, Shaw RM. A 14‐3‐3 mode‐1 binding motif initiates gap junction internalization during acute cardiac ischemia. Traffic. 2014;15:684–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dunn CA, Lampe PD. Injury‐triggered Akt phosphorylation of Cx43: a ZO‐1‐driven molecular switch that regulates gap junction size. J Cell Sci. 2014;127:455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Doble BW, Ping P, Kardami E. The epsilon subtype of protein kinase C is required for cardiomyocyte connexin‐43 phosphorylation. Circ Res. 2000;86:293–301. [DOI] [PubMed] [Google Scholar]
- 66. Srisakuldee W, Jeyaraman MM, Nickel BE, Tanguy S, Jiang ZS, Kardami E. Phosphorylation of connexin‐43 at serine 262 promotes a cardiac injury‐resistant state. Cardiovasc Res. 2009;83:672–681. [DOI] [PubMed] [Google Scholar]
- 67. Li H, Spagnol G, Naslavsky N, Caplan S, Sorgen PL. TC‐PTP directly interacts with connexin43 to regulate gap junction intercellular communication. J Cell Sci. 2014;127:3269–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Barker RJ, Price RL, Gourdie RG. Increased association of ZO‐1 with connexin43 during remodeling of cardiac gap junctions. Circ Res. 2002;90:317–324. [DOI] [PubMed] [Google Scholar]
- 69. Rhett JM, Ongstad EL, Jourdan J, Gourdie RG. Cx43 associates with Na(v)1.5 in the cardiomyocyte perinexus. J Membr Biol. 2012;245:411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Veeraraghavan R, Lin J, Hoeker GS, Keener JP, Gourdie RG, Poelzing S. Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: an experimental and modeling study. Pflugers Arch. 2015;467:2093–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Baker SM, Kim N, Gumpert AM, Segretain D, Falk MM. Acute internalization of gap junctions in vascular endothelial cells in response to inflammatory mediator‐induced G‐protein coupled receptor activation. FEBS Lett. 2008;582:4039–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Thevenin AF, Margraf RA, Fisher CG, Kells‐Andrews RM, Falk MM. Phosphorylation regulates connexin43/ZO‐1 binding and release, an important step in gap junction turnover. Mol Biol Cell. 2017;28:3595–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Neijssen J, Herberts C, Drijfhout JW, Reits E, Janssen L, Neefjes J. Cross‐presentation by intercellular peptide transfer through gap junctions. Nature. 2005;434:83–88. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. ΔG of Binding After a Molecular Mechanics Energy Combined With Generalized Born and Surface Area Continuum Solvation (MM‐GBSA) Minimization of the Protein:Peptide Complex Using a 5 Å Distance Threshold Between Docked Peptide and Protein
Figure S1. The Ischemia Reperfusion (I/R) injury model/protocol used in this study involved a 20‐minute period of no flow ischemia period followed by 40 minutes of reperfusion, left ventricular (LV) contractile function was monitored throughout the whole process.
Figure S2. Blots of products of kinase reaction mixtures containing glutathione S‐transferase (GST)‐Connexin 43 (Cx43) carboxyl terminus (CT), GST‐Protein Kinase C (PKC)ε and biotinylated αCT1 (20 μmol/L).
Figure S3. Blots of cross‐linked products of kinase reaction mixtures containing glutathione S‐transferase (GST)‐Connexin 43 (Cx43) carboxyl terminus (CT), GST‐Cx43 CT QQ/KK in which the lysine (K) residues were mutated to neutral glutamines (Q), Protein Kinase C (PKC)ε and αCT1 (at 5, 10 and 25 μmol/L) and a scrambled αCT1 (M4 scr) variant at the same concentrations.
Figure S4. A, Cartoon depiction of the two alpha helical regions of the Connexin 43 (Cx43) carboxyl terminus (CT), H1 and H2 from Sorgen et al.28. B, Schematic representation of the Cx43 Y313‐A348 peptide synthesized for a binding surface surrogate with linkable cysteine (Cys) on the amino terminus and CT. C, Single letter amino acid sequence of Cx43 Y313‐A348 peptide with predicted helix secondary structure underlined. D, Surface Plasmon Resonance (SPR) analysis of substrate captured αCT1 (700–1000 RUs) binding recombinant Cx43 CT (100 μmol/L, yellow), unlinked Cx43 Y313‐A348 peptide (25 μmol/L, Blue), and disulfide linked Cx43 Y313‐A348 (25 μmol/L, green). SPR indicates that non‐disulfide linked Cx43 Y313‐A348 peptide shows levels of interaction with αCT1 comparable to the full Cx43 CT polypeptide sequence ≈150 amino acids). Disulfide cross‐linking Cx43 Y313‐A348 into a looped conformation results in a loss of αCT1 binding, suggesting that αCT1 interaction with this peptide requires a degree conformational flexibility.
Figure S5. The αCT1 variant peptide M2 AALEI shows limited ability to bind Connexin 43 (Cx43) carboxyl terminus (CT). Surface Plasmon Resonance (SPR) was used to analyze interactions of biotin‐M2 AALEI with the Cx43 CT (A) and Cx43 CT‐KK/QQ (B) as respective analytes. The mean of three runs is plotted for each analyte concentration. The exposure of the sensor chip to the specific analyte is indicated by the gray area.
Figure S6. A, Blots of Connexin 43 (Cx43) phosphorylated at S368 (pS368‐top) and total Cx43 (bottom) in kinase reactions mixtures including no‐kinase controls with Cx43 carboxyl terminus (CT) substrate, but no Protein Kinase C (PKC)ε (PKC‐minus); Cx43‐CT substrate with PKC‐ε (PKC‐plus); and mixtures containing PKC‐ε, Cx43 CT, and biotin‐αCT1, biotin‐αCT1‐ I or biotin‐αCT11 (RPRPDDLEI with no antennapedia sequence at peptide amino terminus) and biotin‐M4 scrambled peptide. B) Blots of Cx43‐pS368 (top) and total Cx43 (bottom) in kinase reactions mixtures including no‐kinase controls with Cx43 CT and Cx43 CT KK/QQ substrates, but no PKC‐ε (PKC‐minus); Cx43‐CT and Cx43 CT KK/QQ substrates with PKC‐ε (PKC‐plus); and in mixtures containing CT substrates, PKC‐ε, and biotin‐αCT1(20 μmol/L).
Figure S7. A through C, Langendorff Ischemia‐Reperfusion (I/R) injury protocols were performed on adult mouse hearts pre‐treated with peptides and instrumented to monitor left ventricular (LV) contractility as shown in Figure 7 Plots of (A) Maximal systolic elastance (Emax)—ie, the slope from plots shown in Figure 7A in the manuscript; (B) LV end diastolic pressure (EDP) against balloon volume; (C) Maximal systolic elastance (Emax), the slopes from Figure S7B. D and E, Langendorff I/R injury protocols were performed on adult mouse hearts pre‐and post‐treated with peptides and instrumented to monitor LV contractility as shown in Figure 8. Plots of (D) Maximal systolic elastance (Emax)—ie, the slope from plots shown in Figure 7A in the manuscript; and (E) LV end diastolic pressure (EDP) against balloon volume; Data shown in Figure S7A through S7E are mean±SE *P<0.05, ***P<0.001, N hearts/group: N hearts/group: Vehicle pre ischemia=8; Vehicle post ischemia=4; αCT1 pre ischemia=7; αCT1‐I pre ischemia=6; αCT11 pre and post ischemia=4; M1 pre ischemia=5; and M3 pre ischemia=5 hearts. F, Average intensities of biotinylated peptide (indicated by streptavidin Alexa647 fluorescence intensity in ventricular myocytes level relative to background) in frozen sections from ventricles in the Vehicle control, αCT1, and αCT11 treated groups. *P<0.05; not significant (ns) N=5 hearts/group.