

Abstract
Background
In patients with chronic heart failure and chronic kidney disease, correction of anemia with erythropoietin‐stimulating agents targeting normal hemoglobin levels is associated with an increased risk of cardiovascular morbidity and mortality. Emerging data suggest a direct effect of erythropoietin on fibroblast growth factor 23 (FGF23), elevated levels of which have been associated with adverse outcomes. We investigate effects of erythropoietin‐stimulating agents in patients with both chronic heart failure and chronic kidney disease focusing on FGF23.
Methods and Results
In the EPOCARES (Erythropoietin in CardioRenal Syndrome) study, we randomized 56 anemic patients (median age 74 [interquartile range 69–80] years, 66% male) with both chronic heart failure and chronic kidney disease into 3 groups, of which 2 received epoetin beta 50 IU/kg per week for 50 weeks, and the third group served as control. Measurements were performed at baseline and after 2, 26, and 50 weeks. Data were analyzed using linear mixed‐model analysis. After 50 weeks of erythropoietin‐stimulating agent treatment, hematocrit and hemoglobin levels increased. Similarly, C‐terminal FGF23 levels, in contrast to intact FGF23 levels, rose significantly due to erythropoietin‐stimulating agents as compared with the controls. During median follow‐up for 5.7 (2.0–5.7) years, baseline C‐terminal FGF23 levels were independently associated with increased risk of mortality (hazard ratio 2.20; 95% CI, 1.35‐3.59; P=0.002).
Conclusions
Exogenous erythropoietin increases C‐terminal FGF23 levels markedly over a period of 50 weeks, elevated levels of which, even at baseline, are significantly associated with an increased risk of mortality. The current results, in a randomized trial setting, underline the strong relationship between erythropoietin and FGF23 physiology in patients with chronic heart failure and chronic kidney disease.
Clinical Trial Registration
URL: http://www.clinicaltrials.gov. Unique identifier: NCT00356733.
Keywords: chronic kidney disease, erythropoietin, fibroblast growth factor
Subject Categories: Heart Failure
Clinical Perspective
What Is New?
In the EPOCARES (Erythropoietin in CardioRenal Syndrome) study, which consists of 56 anemic patients with both chronic heart failure and chronic kidney disease who were randomized to 3 groups, of which 2 received epoetin beta for a period of 50 weeks and 1 was the control group, we show that administration of exogenous erythropoietin over time increases C‐terminal fibroblast growth factor 23, elevated baseline levels of which are associated with an increased risk of mortality in this population.
What Are the Clinical Implications?
Current results underline in a randomized trial setting the strong relationship between erythropoietin and fibroblast growth factor 23 physiology in patients with chronic heart failure and chronic kidney disease.
C‐terminal fibroblast growth factor 23 levels might be the potential link between the previously well‐established association of exogenous erythropoietin treatment with detrimental outcomes in this patient setting.
Introduction
Anemia is associated with diminished exercise capacity and quality of life in patients with chronic heart failure (CHF) and/or chronic kidney disease (CKD).1 Major contributing factors to development of anemia are impaired erythropoietin (EPO) production and response.2, 3, 4 Interestingly, large randomized trials in CHF and CKD striving for full correction of anemia with erythropoiesis‐stimulating agents (ESA) were associated with an increased risk of cardiovascular morbidity and mortality.5, 6, 7 To date, the mechanism linking ESA treatment and increased cardiovascular risk is unknown.
Recently, it has been established in animal models that exogenous EPO administration augments expression of fibroblast growth factor 23 (FGF23), an osteocyte‐derived phosphaturic hormone essential in bone and mineral metabolism.8, 9 Recent human and animal experimental studies describe an increase in C‐terminal FGF23 (cFGF23) levels following EPO treatment, while intact FGF23 (iFGF23) remained stable, which together is suggestive of upregulated production and concomitant cleavage of FGF23.10, 11, 12, 13 Preclinical studies demonstrated that FGF23 can induce left ventricular hypertrophy by binding to FGF23 receptor 4 in cardiac myocytes and promote endothelial dysfunction.14, 15 Elevated levels of cFGF23 have been shown to be associated with increased risk of cardiovascular mortality across different patient populations, including CKD patients and CHF patients, but also among healthy individuals.16, 17, 18
Furthermore, it is known that exogenous EPO treatment increases the need for iron by stimulating erythropoiesis. Iron stores frequently cannot be mobilized fast enough to meet the demand of increased erythropoiesis, resulting in functional iron deficiency.19 Recently, studies from our group and others have shown that iron deficiency results in increased production and concomitant upregulated cleavage of FGF23, resulting in elevated levels of cFGF23.20, 21, 22, 23
We analyzed the data of the EPOCARES (Erythropoietin in the CardioRenal Syndrome) study aiming to assess the effects of ESA therapy on red cell production, iron status, inflammation, and bone mineral homeostasis, including both iFGF23 and cFGF23.
Methods
Study Design and Patients
The data that support the findings of this study are available from the corresponding author on reasonable request. The EPOCARES study has been described in detail.24, 25 In brief, we conducted an open‐label, prospective, randomized trial to study effects of ESA in patients with CHF, CKD, and anemia. At enrollment, patients had to be at least 18 years of age and <85 years, have a renal function of 20 to 70 mL/min per 1.73 m2 calculated with the Cockroft‐Gault equation, and have hemoglobin levels between 10.2 and 12.7 g/dL for men and 12.0 g/dL for women. CHF was defined as New York Heart Association class II or higher, based on symptoms, signs, and objective evidence of an abnormality in cardiac structure or function according to the European Society of Cardiology guidelines. Key exclusion criteria constituted patients with an active systemic disease, malignancy, uncontrolled hypertension (ie, systolic blood pressure higher than 160 mm Hg or diastolic blood pressure higher than 100 mm Hg), uncontrolled diabetes mellitus (ie, a glycated hemoglobin A1c of more than 8.0%), EPO therapy in the previous 6 months, and anemia due to bleeding, hemolysis, or vitamin B12, folate, or iron deficiency. Follow‐up data about mortality have been retrieved at fixed time points from the patient medical records after the study was finished.
Intervention
All eligible patients started with a standard run‐in treatment, at least 4 weeks before inclusion and randomization, consisting of oral iron supplementation and medical treatment according to CHF guidelines.26 If the subjects were still anemic after at least 4 weeks of oral iron supplementation, they were included and randomized into 3 different groups. Randomization was stratified for EPO resistance (defined as an observed or predicted log[serum EPO] ratio <0.6), and allocation was performed in blocks of 6 patients (block randomization) using a computerized table of random numbers. The first group received a fixed dose of 50 IU/kg per week of EPO (epoietin‐β, Neorecormon; Roche Pharmaceuticals, Mannheim, Germany) to increase the Hb level to a maximum of 13.7 g/dL for men and 13.4 g/dL for women (hemoglobin‐rise group). The second group also received 50 IU/kg per week EPO, but the hemoglobin levels in these patients were maintained at baseline level during 26 weeks by sequential blood withdrawal (hemoglobin‐stable group). The third, the control group only received standard care. Of the 62 patients included in the EPOCARES study, 5 withdrew their informed consent, and 1 was excluded because of presumed malignancy at the time of inclusion.
To maintain hemoglobin levels steady in the hemoglobin‐stable group, blood was drawn if hemoglobin levels exceeded 14.0 g/dL in men or 13.8 g/dL in women while the low dose of 50 IU/kg of EPO was maintained. Blood was drawn up to a maximum of 250 mL per session, to a maximum of 250 mL per 2 weeks. However, after 26 weeks the phlebotomies ceased in the hemoglobin‐stable group, and the hemoglobin was allowed to increase equal to that of the hemoglobin‐rise group according to a request of the institutional review board. In a prespecified subgroup, echocardiograms were performed according to study protocol, as described previously.24 The study protocol has been approved by the institutional review boards, and written informed consent was obtained from all subjects and adhered to the principles of the Declaration of Helsinki.
Laboratory Tests
All blood samples were drawn between 8 and 9 am. Serum ferritin, as a marker of iron stores, was determined using routine laboratory procedures. Iron status was further assessed by serum iron, transferrin, transferrin saturation, and serum hepcidin. Details of the methods used for biomarker analysis have been published.27 Intact FGF23 was measured using stored plasma samples by ELISA (Kainos Laboratories, Inc, Tokyo, Japan) and cFGF23 by ELISA (Immutopics/Quidel, Inc, San Clemente, CA). The cFGF23 immunometric assay uses 2 antibodies directed against different epitopes within the C‐terminal part of FGF23, which therefore detects both the intact hormone and the C‐terminal cleavage products. In contrast, the iFGF23 assay detects only the intact molecule.28 All variables were measured at baseline and after 2, 26, and 50 weeks.
Statistical Analyses
Intention‐to‐treat analyses included all randomized patients starting ESA treatment or standard of care in the control group. Data were analyzed using IBM SPSS software, version 23.0 (SPSS Inc, Chicago, IL). Normally distributed variables are presented as means±SD, whereas skewed distributed variables are shown as median with interquartile range. Categorical variables are shown as numbers with percentage. Baseline characteristics among the 3 groups were evaluated with a 1‐way ANOVA for normally distributed data, a Kruskal‐Wallis test for skewed distributed data, and a chi‐squared test for categorical variables. Cox proportional hazard regression analysis was performed to assess whether baseline cFGF23 levels were associated with risk of mortality over time. Adjustments were performed for age, sex, estimated glomerular filtration rate, presence of diabetes mellitus, hypertension, and smoking status as traditional mortality risk factors. In addition, we adjusted for iron status and red blood cell dynamics (ferritin, hemoglobin, and EPO), which could be considered confounders because of their relationship with both cFGF23 and mortality. Linear regression analysis based on intention‐to‐treat approach was performed to assess the association between baseline cFGF23 levels and measured ejection fraction by echocardiography at 50 weeks. Possible effect modification by group randomization on the association of cFGF23 with mortality and measured ejection fraction has been assessed. The difference in cFGF23 levels at baseline and 50 weeks among the 3 groups was assessed by means of contrast analysis in the linear mixed models. As sensitivity analysis, we performed a per‐protocol analysis of our primary association between ESA treatment and the effect on cFGF23 and iFGF23 by repeating the linear mixed‐model analysis.
To estimate the effect of EPO on the hemoglobin‐rise and hemoglobin‐stable groups compared with the control group, we performed a linear mixed‐effect model with “group,” “time” (as continuous variable), and “group×time” as fixed effects and patient identification number as random effect. In all analyses, skewed data were natural‐log transformed before analysis, and a 2‐sided P<0.05 was considered significant.
Results
Baseline Characteristics
Fifty‐six patients (median age 74 [interquartile 69–80] years, 66% males, mean estimated glomerular filtration rate of 36±15 mL/min per 1.73 m2) were included. Demographics and clinical characteristics of the 56 patients, subdivided by study group, are shown in Table 1. At baseline, no significant differences were observed for the main parameters. During the course of the study, 6 patients died (3 in the control group, 2 in the hemoglobin‐rise group, and 1 in the hemoglobin‐stable group); 3 of these patients died due to terminal heart failure, 1 due to abdominal sepsis, 1 due to an out‐of‐hospital cardiac arrest, and 1 due to ventricular fibrillation.
Table 1.
Baseline Characteristics of 56 Patients With Chronic Heart Failure, Chronic Kidney Disease, and Anemia
| Hb‐Stable Group (n=18) | Hb‐Rise Group (n=19) | Control Group (n=19) | P Value | |
|---|---|---|---|---|
| Age, y | 78 (69–81) | 74 (70–80) | 72 (66–77) | 0.65 |
| Male sex, n (%) | 10 (56) | 13 (68) | 14 (74) | 0.49 |
| BMI, kg/m2 | 26.1±4.9 | 25.7±3.6 | 27.4±4.3 | 0.54 |
| eGFR, mL/min per 1.73 m2 | 36±14 | 35±12 | 34±16 | 0.94 |
| NT‐proBNP, pg/mL | 1767 (762–3127) | 1373 (524–2151) | 1680 (659–2610) | 0.78 |
| Etiology of heart failure | 0.43 | |||
| Ischemic, n (%) | 9 (50) | 13 (68) | 13 (68) | |
| Hypertensive, n (%) | 3 (17) | 3 (16) | 3 (16) | |
| Valvular, n (%) | 2 (11) | 1 (5) | 3 (16) | |
| Other, n (%) | 4 (22) | 2 (11) | 0 (0) | |
| Diabetes mellitus, n (%) | 5 (28) | 7 (37) | 7 (37) | 0.80 |
| Hypertension, n (%) | 14 (78) | 13 (68) | 16 (84) | 0.51 |
| Smoking status | 0.05 | |||
| Never smoker, n (%) | 10 (56) | 5 (26) | 3 (16) | |
| Former smoker, n (%) | 7 (39) | 13 (68) | 12 (63) | |
| Current smoker, n (%) | 1 (6) | 1 (5) | 4 (21) | |
| Hemoglobin, g/dL | 11.7±0.8 | 11.8±1.1 | 11.8±0.8 | 0.94 |
| Hematocrit, % | 36±3 | 35±4 | 35±3 | 0.89 |
| MCV, fL | 90±4 | 91±4 | 89±4 | 0.61 |
| Reticulocytes, % | 1.1±0.3 | 1.2±0.4 | 1.1±0.4 | 0.85 |
| RDW (%) | 14.5 (13.4–15.2) | 13.6 (13.2–14.3) | 14.2 (13.1–15.1) | 0.48 |
| EPO, IU/L | 13 (7–15) | 14 (10–19) | 15 (5–17) | 0.64 |
| Iron, μmol/L | 11.4±5.4 | 11.8±4.4 | 11.8±3.5 | 0.96 |
| Ferritin, μg/L | 127 (87–179) | 136 (71–307) | 128 (76–164) | 0.81 |
| TSAT, % | 22±13 | 23±9 | 22±7 | 0.99 |
| Hepcidin, ng/mL | 6.6 (2.8–8.7) | 6.6 (4.1–11.5) | 5.7 (3.3–7.9) | 0.28 |
| Calcium, mmol/L | 2.34±0.14 | 2.29±0.08 | 2.30±0.12 | 0.32 |
| Phosphate, mmol/L | 1.2±0.2 | 1.2±0.1 | 1.1±0.2 | 0.56 |
| PTH, pmol/L | 10.0 (6.0–11.2) | 11.9 (6.9–19.2) | 12.0 (6.6–20.1) | 0.34 |
| cFGF23, RU/mL | 162 (110–239) | 205 (69–442) | 315 (127–685) | 0.17 |
| iFGF23, pg/mL | 89 (53–114) | 118 (46–235) | 115 (77–248) | 0.11 |
| hs‐CRP, mg/dL | 2.8 (1.1–11.0) | 6.8 (1.7–11.4) | 4.3 (1.7–6.9) | 0.44 |
Mean±SD or median (interquartile range) are shown. Differences between groups were calculated with 1‐way ANOVA for normally distributed data, with Kruskal‐Wallis test for skewed distributed data, and chi‐squared test for categorical data. BMI indicates body mass index; cFGF23, C‐terminal fibroblast growth factor 23; eGFR, estimated glomerular filtration rate; EPO, erythropoietin; Hb, hemoglobin; hs‐CRP, high‐sensitivity CRP; iFGF23, intact fibroblast growth factor 23; MCV, mean corpuscular volume; NT‐proBNP, N‐terminal pro–brain natriuretic peptide; PTH, parathyroid hormone; RDW, red cell distribution width; TSAT, transferrin saturation.
Laboratory Results in Response to EPO Treatment
Table 2 summarizes laboratory values at the end of the 50‐week trial and shows treatment effects of EPO. After 50 weeks of treatment, hemoglobin levels in the EPO hemoglobin‐stable group increased from 11.7±0.84 to 13.1±0.8 g/dL, and in the EPO hemoglobin‐rise group it increased from 11.8±1.07 to 13.2±1.30 g/dL, whereas hemoglobin levels remained stable at 11.8±0.79 g/dL in the control group. Similarly, hematocrit increased due to EPO treatment. No significant differences were noticed in serum ferritin levels due to EPO treatment. In contrast, transferrin levels increased significantly in the EPO‐treated groups (Table 2). Surprisingly, transferrin saturation levels remained stable or even increased slightly due to EPO treatment, although not significantly. No significant differences due to EPO treatment were observed for renal function, electrolytes, or inflammatory parameters.
Table 2.
Effect of Erythropoietin Treatment in Hemoglobin‐Stable and Hemoglobin‐Rise Patients Compared With Control Patients
| Values After 50 Weeks of Treatment | Treatment Effect | ||||
|---|---|---|---|---|---|
| EPO‐Hb‐Stable (n=18) | EPO‐Hb‐Rise (n=19) | Control (n=19) | EPO‐Hb‐Stable vs Control | EPO‐Hb‐Rise vs Control | |
| Red blood cell and iron status | |||||
| Hemoglobin, g/dL | 13.1±0.8 | 13.2±1.3 | 11.8±1.2 | 1.0 (0.17 to 1.83)* | 1.2 (0.61 to 1.79)*** |
| Hematocrit, % | 40.4±2.2 | 39.8±3.8 | 36.0±3.6 | 4.0 (1.0 to 6.6)** | 4.0 (2.0 to 6.0)*** |
| MCV, fL | 92.2±5.1 | 89.4±4.3 | 90.4±3.2 | 2.0 (−0.01 to 4.01) | −0.3 (−1.8 to 1.2) |
| Reticulocytes, % | 1.2±0.3 | 1.2±0.4 | 1.0±0.4 | 0.002 (−0.01 to 0.01) | 0.003 (−0.005 to 0.01) |
| RDW (%) | 14.5 (13.6 to 15.5) | 13.9 (13.5 to 14.4) | 13.8 (13.2 to 14.6) | 0.8 (−1.0 to 2.5) | 0.7 (−0.8 to 2.1) |
| EPO,† IU/L | 32 (25 to 46) | 35 (26 to 50) | 10 (7 to 13) | 6.0 (−16.2 to 28.2) | 10.0 (−5.7 to 25.7) |
| Iron, μmol/L | 12.8±4.5 | 10.9±2.5 | 11.4±2.7 | −7.0 (−19.4 to 5.5) | −6.0 (−14.8 to 2.8) |
| Ferritin,† μg/L | 84 (47 to 102) | 99 (68 to 139) | 139 (61 to 232) | 0.61 (0.23 to 1.62) | 0.47 (0.11 to 2.05) |
| Transferrin, g/L | 2.4±0.4 | 2.2±0.2 | 2.2±0.3 | 0.8 (0.06 to 1.44)* | 0.5 (0.01 to 0.99)* |
| TSAT, % | 24±10 | 21±6 | 22±6 | 2.5 (−21.1 to 26.1) | 0 (−17 to 17) |
| Hepcidin,† ng/mL | 2.8 (1.3 to 5.0) | 6.0 (2.9 to 7.9) | 6.2 (5.1 to 9.2) | 0.29 (0.02 to 4.58) | 0.45 (0.07 to 3.36) |
| Renal function and heart failure | |||||
| Urea,† mmol/L | 11.9 (8.3 to 17.8) | 13.5 (11.3 to 23.1) | 14.1 (9.1 to 23.8) | 0.70 (0.53 to 0.93)* | 0.82 (0.67 to 1.00)* |
| Creatinine, μmol/L | 152 (118 to 231) | 189 (126 to 279) | 176 (143 to 334) | 0.93 (0.82 to 1.05) | 0.96 (0.88 to 1.04) |
| eGFR,‡ mL/min per 1.73 m2 | 36±14 | 32±14 | 33±17 | 2.5 (−2.1 to 7.1) | 1.95 (−1.4 to 5.3) |
| NT‐proBNP,† pg/mL | 1756 (888 to 2713) | 1017 (666 to 1925) | 1355 (373 to 2220) | 0.74 (0.05 to 10.5) | 0.78 (0.11 to 5.54) |
| Bone and mineral metabolism | |||||
| Calcium, mmol/L | 2.36±0.13 | 2.34±0.09 | 2.29±0.08 | −0.03 (−0.10 to 0.04) | 0.01 (−0.04 to 0.06) |
| Phosphate, mmol/L | 1.1±0.2 | 1.2±0.2 | 1.2±0.2 | −0.20 (−0.34 to −0.06)** | −0.1 (−0.2 to −0.002) |
| PTH,† pmol/L | 7.9 (5.6 to 13.9) | 11.4 (7.8 to 20.2) | 11.4 (9.1 to 14.3) | 1.16 (0.79 to 1.72) | 1.12 (0.64 to 1.95) |
| cFGF23,† RU/mL | 306 (231 to 443) | 322 (187 to 685) | 178 (132 to 424) | 1.72 (1.02 to 2.90)* | 1.49 (1.01 to 2.21)* |
| iFGF23,† pg/mL | 129 (60 to 200) | 206 (73 to 572) | 120 (113 to 288) | 1.28 (0.85 to 1.95) | 1.22 (0.91 to 1.64) |
| Electrolytes | |||||
| Sodium, mmol/L | 142±2 | 139±4 | 140±3 | −2.5 (−15.0 to 10.0) | −2.5 (−11.3 to 6.3) |
| Potassium, mmol/L | 4.4±0.3 | 4.5±0.4 | 4.4±0.4 | 0.33 (−0.09 to 0.75) | 0.03 (−0.26 to 0.32) |
| Inflammation | |||||
| hs‐CRP,† mg/dL | 3.0 (2.0 to 7.5) | 3.0 (1.3 to 7.0) | 5.5 (2.0 to 10.8) | 1.65 (0.31 to 8.90) | 1.28 (0.48 to 3.42) |
| IL‐6,† pg/mL | 3.14 (2.67 to 6.62) | 3.69 (1.65 to 6.81) | 3.13 (2.76 to 3.76) | 1.42 (0.82 to 2.47) | 1.22 (0.83 to 1.81) |
Mean±SD or median (interquartile range) are shown. Samples were collected at weeks 0, 2, 26, and 50. cFGF23 indicates C‐terminal fibroblast growth factor 23; eGFR, estimated glomerular filtration rate; EPO, erythropoietin; Hb, hemoglobin; hs‐CRP, high‐sensitive C‐reactive protein; iFGF23, intact fibroblast growth factor 23; IL‐6, interleukin‐6; MCV, mean corpuscular volume; NT‐proBNP, N‐terminal‐pro–brain natriuretic peptide; PTH, parathyroid hormone; RDW, red cell distribution width; TSAT, transferrin saturation.
P‐values: ***<0.001, **<0.01, *<0.05.
†Due to skewed distribution, the treatment effect is seen as a relative increase on a natural logarithm scale.
‡Calculated with Modification of Diet in Renal Disease equation.
Intact and C‐Terminal FGF23 in Response to EPO Treatment
After 50 weeks of EPO treatment, cFGF23 levels increased significantly in the EPO hemoglobin‐stable group from 162 (110–239) to 306 (231–443) RU/mL and in the EPO hemoglobin‐rise group from 205 (69–442) to 322 (187–685) RU/mL, whereas the levels decreased in the control group from 315 (127–685) to 178 (132–424) RU/mL (Figure, upper panel). Intact FGF23 levels in both the EPO hemoglobin‐stable and EPO hemoglobin‐rise groups were not different between baseline and after 50 weeks of treatment (89 [53–114] to 129 [60–200] pg/mL and 118 [46–235] to 206 [73–572] pg/mL, respectively), and the level remained stable in the control group (Figure, lower panel). Phosphate levels decreased in both EPO‐treated groups, significantly in the EPO hemoglobin‐stable group. Calcium and PTH levels did not significantly change after EPO treatment.
Figure 1.
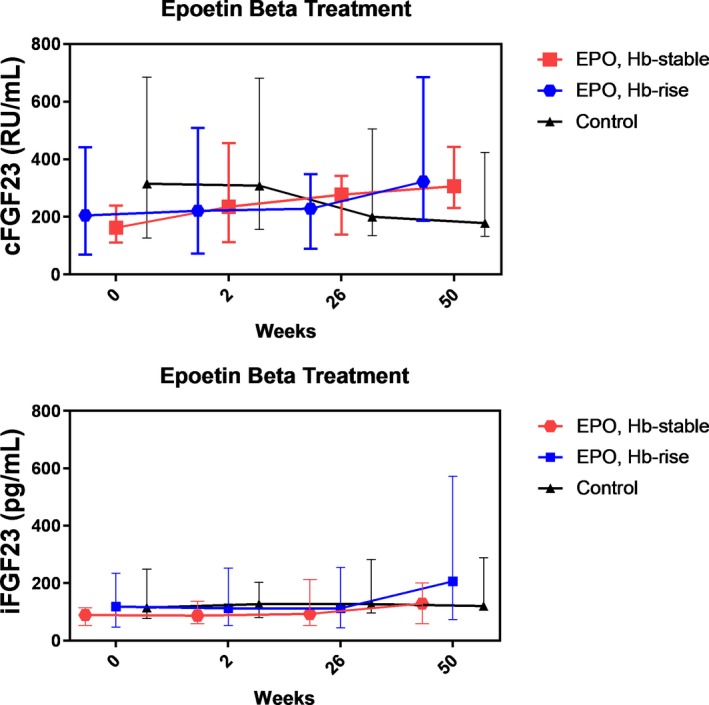
Effect of erythropoietin on C‐terminal fibroblast growth factor 23 and intact fibroblast growth factor 23. Median levels with interquartile range of both cFGF23 and iFGF23 levels are shown over time. cFGF23 indicates C‐terminal fibroblast growth factor 23; EPO, erythropoietin; Hb, hemoglobin; iFGF23, intact fibroblast growth factor 23.
Association of Baseline cFGF23 and iFGF23 With Prospective Outcomes
During a median follow‐up of 5.7 (2.0–5.7) years, 27 (48%) patients died, which is in line with survival rates previously reported in this patient setting.29 Baseline natural log‐transformed (ln) cFGF23 was univariately positively associated with increased mortality risk (hazard ratio [HR] 1.85; 95% CI, 1.27–2.70; P=0.001). No effect modification by group randomization was noted (P=0.27). After adjustment for age, sex, and estimated glomerular filtration rate, the association between baseline ln cFGF23 and mortality remained materially unchanged (HR 2.02; 95% CI, 1.35–3.00; P=0.001). Further adjustment for the presence of diabetes mellitus, hypertension, and smoking did also not materially alter the positive association between ln cFGF23 and mortality (HR 2.44; 95% CI, 1.54–3.87; P<0.001). Finally, the positive association between ln cFGF23 and mortality remained independent of additional adjustment for ln ferritin, hemoglobin, and ln EPO levels (HR 2.20; 95% CI, 1.35–3.59; P=0.002). In contrast, ln iFGF23 levels were univariately not associated with increased risk of mortality (HR 1.17; 95% CI, 0.69–2.00; P=0.57).
In linear regression analyses, baseline ln cFGF23 levels were inversely associated with biplane left ventricular ejection measurement by echocardiography after 50 weeks (β=−0.50, P<0.001) as assessed in a subset of 28 patients. No effect modification by group randomization was noted (P=0.96). After adjustment for age, sex, and estimated glomerular filtration rate, baseline cFGF23 levels remained inversely associated with ejection fraction (β=−0.49, P=0.01). As for mortality, ln iFGF23 levels were univariately not associated with ejection fraction (β=0.03, P=0.88).
Sensitivity Analyses
As a sensitivity analysis, we performed a per‐protocol analysis (49 patients by excluding the 6 patients who were lost to follow‐up during the study) instead of intention‐to‐treat analysis and reassessed the mixed models analysis on the association of EPO treatment with cFGF23 and iFGF23. Again, after 50 weeks of EPO treatment cFGF23 levels significantly increased from 188 (100–358) RU/mL to 311 (210–541) RU/mL as a response to EPO treatment (P<0.05), whereas iFGF23 levels increased nonsignificantly from 98 (47–165) pg/mL to 149 (67–394) pg/mL (P=0.14). The rise in cFGF23 between 50 weeks and baseline assessed by contrast analysis was significant in the EPO hemoglobin‐rise group (P=0.03) and a statistical trend in the EPO hemoglobin‐stable group (P=0.07), whereas the decline of cFGF23 levels in the control group was not significant (P=0.53).
Discussion
In this study we have shown that exogenous EPO is associated with increased cFGF23 levels out of proportion to iFGF23 levels, implicating an upregulated production and concomitant increased cleavage of FGF23. As expected, the effect of EPO treatment resulted in an increment in hematocrit and hemoglobin level and a tendency to decrease in ferritin level.30 No important differences were observed in parameters representing inflammation, kidney function, and electrolytes. The current study underlines the essential role of EPO in FGF23 physiology and provides a speculative mechanism, linking the use of exogenous EPO with a higher risk of cardiovascular events because increased cFGF23 levels were associated with increased mortality risk in the current study, reiterating the association of elevated cFGF23 levels with many other reported adverse outcomes.
To date, the underlying mechanism of the association between use of exogenous EPO and detrimental outcomes is unknown. In 2007 Fishbane and Besarab suggested a set of hypotheses that could explain the link between exogenous EPO and adverse outcomes, all of which currently still appear to be valid.31 The hypotheses are that the detrimental outcomes are the result of either the achieved hemoglobin level itself or the (high‐dose) ESA therapy in EPO‐resistant patients. In the current study we add to these proposed mechanisms that ESA therapy increases levels of cFGF23, which is known to be strongly associated with increased cardiovascular disease events, kidney disease progression, and death among individuals with CKD.17, 32, 33 The current study is the first to extend these findings to a human setting with combined CKD and CHF. Also in the current patient setting, baseline cFGF23 levels were associated with an increased risk of adverse outcomes and reduced left ventricular ejection fraction, emphasizing the effect of EPO treatment in further increasing cFGF23 levels.
Our study is in line with recent experimental studies describing the positive association between EPO and cFGF23. Clinkenbeard et al have shown in experimental models that recombinant EPO acutely increases circulating FGF23 levels in mice with a normal kidney function and in mice with diminished kidney function.8 The authors described that EPO stimulated FGF23 production in hematopoietic progenitor cells and in cortical bone. Furthermore, exogenous EPO was shown to increase FGF23 levels in humans with normal kidney function. Recently, Rabadi et al showed that acute blood loss with a subsequent increase in EPO levels increases cFGF23 levels. In addition, exogenous EPO administration led to an increase in cFGF23 levels similar to the effect of acute blood loss.10 In keeping with this finding Flamme et al identified that administration of exogenous EPO in experimental rat models induces a steep increase in cFGF23 levels within 1 hour following intravenous administration. FGF23 mRNA expression was strongly induced in bone and bone marrow after recombinant EPO treatment and was even independent of 2‐week pretreatment with EPO or saline.11 Furthermore, Toro et al reported that exogenous EPO increased bone marrow FGF23 mRNA in vivo and in vitro via EPO receptor activity in erythroid progenitor cells; they further extended this result with the notion that blockade of the EPO receptor prevented induction of FGF23 and suppressed circulating FGF23 levels.12 Intriguingly, Agoro et al recently described a converse direct relationship between cFGF23 and EPO in CKD mice in which inhibition of FGF23 signaling decreased erythroid cell apoptosis and induced renal and bone marrow EPO expression by creating a hypoxic environment that activated EPO‐induced erythropoiesis.34 Furthermore, FGF23 inhibition ameliorated iron deficiency by reducing inflammation, and hence decreasing serum hepcidin, leading to restoration of iron status parameters. The present findings together with the reported studies point at pivotal direct relationships among EPO, iron deficiency, and FGF23.
In our study, EPO increased cFGF23 out of proportion to iFGF23. These elevated cFGF23 levels represent mainly C‐terminal fragments because the cFGF23 immunometric assay measures both the intact molecule as the C‐terminal fragments, whereas the iFGF23 assay detects only the intact molecule. The C‐terminal fragments are allegedly assumed to be inactive. Contrary to this prevailing view are observations made by Goetz et al that showed that C‐terminal FGF23 fragments may function as FGF23 antagonists by competing with iFGF23 for binding to the FGF23 receptor.35 Furthermore, it has been shown in vitro by Courbebaisse et al that C‐terminal FGF23 in itself can increase the cell surface area of adult rat ventricular cardiomyocytes by binding to the FGF23 receptor.36 Future studies will need to further unravel the biologic activity of the C‐terminal fragments. Finally, because the net result of EPO administration resulted in a decrease in phosphate levels after 50 weeks, this suggests that EPO administration led to an increased production of FGF23 with somewhat increased iFGF23 levels (that are physiologically active) along with out‐of‐proportion increases in cFGF23 levels, which implies an increased cleavage of the intact molecule.
Our study has both strengths and limitations. The major strength of the study is that it comprises a randomized trial setting in which we could visualize by means of multiple consecutive blood samples the effect of EPO treatment for 50 weeks. For about half of the treatment duration, the groups were treated distinctly, and we performed all analyses stratified for the 3 groups to prevent an unrecognized effect due to difference in hemoglobin handling that could have been introduced by pooling the 2 EPO treatment groups. Furthermore, because iron status decreased in the EPO treatment arms of the randomized controlled trial, it might be that the increment in cFGF23 levels is at least partly due to induced iron deficiency. As a limitation, the association between FGF23 and mortality in the current study can be considered a post hoc analysis. Furthermore, the current study comprises a relatively small sample size, albeit the largest number of patients with both CHF and CKD in which this association has been investigated to date. Due to the relatively small sample size, we cannot exclude that more modest effects of EPO on iFGF23 would have been identified with a greater number of subjects. The small sample size and missing values in follow‐up did not let us perform a useful ΔcFGF23 analysis to assess whether ΔFGF23 was a stronger predictor of mortality than baseline FGF23 levels alone, as shown by Isakova et al.37 Finally, we cannot exclude the possibility that renal phosphate handling might have influenced the currently identified results of FGF23 induction and cleavage, although phosphate levels were similar at baseline between the arms of the trial.
In conclusion, we have demonstrated that administration of exogenous EPO over a time course of 50 weeks is associated with increased cFGF23 levels out of proportion to iFGF23 levels. Baseline cFGF23 levels were strongly associated with an increased risk of mortality. The currently identified association between exogenous EPO and cFGF23 levels could be the potential link between exogenous EPO and detrimental outcomes in this patient setting. Further research is needed to establish whether adverse outcomes associated with EPO treatment are truly attributable to a direct effect of exogenous EPO on cFGF23 levels.
Sources of Funding
This work was supported by the Dutch Heart Foundation, The Hague, the Netherlands (grant number 2005B192) and by an unrestricted grant from Roche, the Netherlands.
Disclosures
None.
(J Am Heart Assoc. 2019;8:e011130 DOI: 10.1161/JAHA.118.011130.)
References
- 1. Kalra PR, Bolger AP, Francis DP, Genth‐Zotz S, Sharma R, Ponikowski PP, Poole‐Wilson PA, Coats AJ, Anker SD. Effect of anemia on exercise tolerance in chronic heart failure in men. Am J Cardiol. 2003;91:888–891. [DOI] [PubMed] [Google Scholar]
- 2. van der Putten K, Braam B, Jie KE, Gaillard CA. Mechanisms of disease: erythropoietin resistance in patients with both heart and kidney failure. Nat Clin Pract Nephrol. 2008;4:47–57. [DOI] [PubMed] [Google Scholar]
- 3. van der Meer P, Lok DJ, Januzzi JL, de la Porte PW, Lipsic E, van Wijngaarden J, Voors AA, van Gilst WH, van Veldhuisen DJ. Adequacy of endogenous erythropoietin levels and mortality in anaemic heart failure patients. Eur Heart J. 2008;29:1510–1515. [DOI] [PubMed] [Google Scholar]
- 4. Westenbrink BD, Voors AA, de Boer RA, Schuringa JJ, Klinkenberg T, van der Harst P, Vellenga E, van Veldhuisen DJ, van Gilst WH. Bone marrow dysfunction in chronic heart failure patients. Eur J Heart Fail. 2010;12:676–684. [DOI] [PubMed] [Google Scholar]
- 5. Drueke TB, Locatelli F, Clyne N, Eckardt KU, Macdougall IC, Tsakiris D, Burger HU, Scherhag A; CREATE Investigators . Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med. 2006;355:2071–2084. [DOI] [PubMed] [Google Scholar]
- 6. Singh AK, Szczech L, Tang KL, Barnhart H, Sapp S, Wolfson M, Reddan D; CHOIR Investigators . Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355:2085–2098. [DOI] [PubMed] [Google Scholar]
- 7. Swedberg K, Young JB, Anand IS, Cheng S, Desai AS, Diaz R, Maggioni AP, McMurray JJ, O'Connor C, Pfeffer MA, Solomon SD, Sun Y, Tendera M, van Veldhuisen DJ; RED‐HF Committees, RED‐HF Investigators . Treatment of anemia with darbepoetin alfa in systolic heart failure. N Engl J Med. 2013;368:1210–1219. [DOI] [PubMed] [Google Scholar]
- 8. Clinkenbeard EL, Hanudel MR, Stayrook KR, Appaiah HN, Farrow EG, Cass TA, Summers LJ, Ip CS, Hum JM, Thomas JC, Ivan M, Richine BM, Chan RJ, Clemens TL, Schipani E, Sabbagh Y, Xu L, Srour EF, Alvarez MB, Kacena MA, Salusky IB, Ganz T, Nemeth E, White KE. Erythropoietin stimulates murine and human fibroblast growth factor‐23, revealing novel roles for bone and bone marrow. Haematologica. 2017;102:e427–e430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hasegawa H, Nagano N, Urakawa I, Yamazaki Y, Iijima K, Fujita T, Yamashita T, Fukumoto S, Shimada T. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early‐stage chronic kidney disease. Kidney Int. 2010;78:975–980. [DOI] [PubMed] [Google Scholar]
- 10. Rabadi S, Udo I, Leaf DE, Waikar S, Christov M. Acute blood loss stimulates fibroblast growth factor 23 production. Am J Physiol Renal Physiol. 2018;314:F132–F139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Flamme I, Ellinghaus P, Urrego D, Kruger T. FGF23 expression in rodents is directly induced via erythropoietin after inhibition of hypoxia inducible factor proline hydroxylase. PLoS One. 2017;12:e0186979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Toro L, Barrientos V, Leon P, Rojas M, Gonzalez M, Gonzalez‐Ibanez A, Illanes S, Sugikawa K, Abarzua N, Bascunan C, Arcos K, Fuentealba C, Tong AM, Elorza AA, Pinto ME, Alzamora R, Romero C, Michea L. Erythropoietin induces bone marrow and plasma fibroblast growth factor 23 during acute kidney injury. Kidney Int. 2018;93:1131–1141. [DOI] [PubMed] [Google Scholar]
- 13. Hanudel MR, Eisenga MF, Rappaport M, Chua K, Qiao B, Jung G, Gabayan V, Gales B, Ramos G, de Jong MA, van Zanden JJ, de Borst MH, Bakker SJL, Nemeth E, Salusky IB, Gaillard CAJM, Ganz T. Effects of erythropoietin on fibroblast growth factor 23 in mice and humans. Nephrol Dial Transplant. 2018; Jul 10. Available at: https://academic.oup.com/ndt/advance-article/doi/10.1093/ndt/gfy189/5051711. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutierrez OM, Aguillon‐Prada R, Lincoln J, Hare JM, Mundel P, Morales A, Scialla J, Fischer M, Soliman EZ, Chen J, Go AS, Rosas SE, Nessel L, Townsend RR, Feldman HI, St John Sutton M, Ojo A, Gadegbeku C, Di Marco GS, Reuter S, Kentrup D, Tiemann K, Brand M, Hill JA, Moe OW, Kuro‐O M, Kusek JW, Keane MG, Wolf M. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Silswal N, Touchberry CD, Daniel DR, McCarthy DL, Zhang S, Andresen J, Stubbs JR, Wacker MJ. FGF23 directly impairs endothelium‐dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. Am J Physiol Endocrinol Metab. 2014;307:E426–E436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Souma N, Isakova T, Lipiszko D, Sacco RL, Elkind MS, DeRosa JT, Silverberg SJ, Mendez AJ, Dong C, Wright CB, Wolf M. Fibroblast growth factor 23 and cause‐specific mortality in the general population: the Northern Manhattan Study. J Clin Endocrinol Metab. 2016;101:3779–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, Wahl P, Gutierrez OM, Steigerwalt S, He J, Schwartz S, Lo J, Ojo A, Sondheimer J, Hsu CY, Lash J, Leonard M, Kusek JW, Feldman HI, Wolf M; Chronic Renal Insufficiency Cohort (CRIC) Study Group . Fibroblast growth factor 23 and risks of mortality and end‐stage renal disease in patients with chronic kidney disease. JAMA. 2011;305:2432–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Poelzl G, Trenkler C, Kliebhan J, Wuertinger P, Seger C, Kaser S, Mayer G, Pirklbauer M, Ulmer H, Griesmacher A. FGF23 is associated with disease severity and prognosis in chronic heart failure. Eur J Clin Invest. 2014;44:1150–1158. [DOI] [PubMed] [Google Scholar]
- 19. Camaschella C. Iron deficiency: new insights into diagnosis and treatment. Hematology Am Soc Hematol Educ Program. 2015;2015:8–13. [DOI] [PubMed] [Google Scholar]
- 20. Wolf M, Koch TA, Bregman DB. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J Bone Miner Res. 2013;28:1793–1803. [DOI] [PubMed] [Google Scholar]
- 21. Hanudel MR, Chua K, Rappaport M, Gabayan V, Valore E, Goltzman D, Ganz T, Nemeth E, Salusky IB. Effects of dietary iron intake and chronic kidney disease on fibroblast growth factor 23 metabolism in wild type and hepcidin knockout mice. Am J Physiol Renal Physiol. 2016;311:F1369–F1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Farrow EG, Yu X, Summers LJ, Davis SI, Fleet JC, Allen MR, Robling AG, Stayrook KR, Jideonwo V, Magers MJ, Garringer HJ, Vidal R, Chan RJ, Goodwin CB, Hui SL, Peacock M, White KE. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor‐23 (Fgf23) knock‐in mice. Proc Natl Acad Sci USA. 2011;108:E1146–E1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eisenga MF, van Londen M, Leaf DE, Nolte IM, Navis G, Bakker SJL, de Borst MH, Gaillard CAJM. C‐terminal fibroblast growth factor 23, iron deficiency, and mortality in renal transplant recipients. J Am Soc Nephrol. 2017;28:3639–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van der Putten K, Jie KE, Emans ME, Verhaar MC, Joles JA, Cramer MJ, Velthuis BK, Meiss L, Kraaijenhagen RJ, Doevendans PA, Braam B, Gaillard CA. Erythropoietin treatment in patients with combined heart and renal failure: objectives and design of the EPOCARES study. J Nephrol. 2010;23:363–368. [PubMed] [Google Scholar]
- 25. van der Putten K, Jie KE, van den Broek D, Kraaijenhagen RJ, Laarakkers C, Swinkels DW, Braam B, Gaillard CA. Hepcidin‐25 is a marker of the response rather than resistance to exogenous erythropoietin in chronic kidney disease/chronic heart failure patients. Eur J Heart Fail. 2010;12:943–950. [DOI] [PubMed] [Google Scholar]
- 26. Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW, Yancy CW, Antman EM, Smith SC Jr, Adams CD, Anderson JL, Faxon DP, Fuster V, Halperin JL, Hiratzka LF, Jacobs AK, Nishimura R, Ornato JP, Page RL, Riegel B; American College of Cardiology, American Heart Association Task Force on Practice Guidelines, American College of Chest Physicians, International Society for Heart and Lung Transplantation, Heart Rhythm Society . ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation. 2005;112:e154–e235. [DOI] [PubMed] [Google Scholar]
- 27. Emans ME, van der Putten K, van Rooijen KL, Kraaijenhagen RJ, Swinkels D, van Solinge WW, Cramer MJ, Doevendans PA, Braam B, Gaillard CA. Determinants of red cell distribution width (RDW) in cardiorenal patients: RDW is not related to erythropoietin resistance. J Card Fail. 2011;17:626–633. [DOI] [PubMed] [Google Scholar]
- 28. Wolf M, White KE. Coupling fibroblast growth factor 23 production and cleavage: iron deficiency, rickets, and kidney disease. Curr Opin Nephrol Hypertens. 2014;23:411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Edner M, Benson L, Dahlstrom U, Lund LH. Association between renin‐angiotensin system antagonist use and mortality in heart failure with severe renal insufficiency: a prospective propensity score‐matched cohort study. Eur Heart J. 2015;36:2318–2326. [DOI] [PubMed] [Google Scholar]
- 30. Van Wyck DB, Stivelman JC, Ruiz J, Kirlin LF, Katz MA, Ogden DA. Iron status in patients receiving erythropoietin for dialysis‐associated anemia. Kidney Int. 1989;35:712–716. [DOI] [PubMed] [Google Scholar]
- 31. Fishbane S, Besarab A. Mechanism of increased mortality risk with erythropoietin treatment to higher hemoglobin targets. Clin J Am Soc Nephrol. 2007;2:1274–1282. [DOI] [PubMed] [Google Scholar]
- 32. Gutierrez OM, Mannstadt M, Isakova T, Rauh‐Hain JA, Tamez H, Shah A, Smith K, Lee H, Thadhani R, Juppner H, Wolf M. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359:584–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seiler S, Reichart B, Roth D, Seibert E, Fliser D, Heine GH. FGF‐23 and future cardiovascular events in patients with chronic kidney disease before initiation of dialysis treatment. Nephrol Dial Transplant. 2010;25:3983–3989. [DOI] [PubMed] [Google Scholar]
- 34. Agoro R, Montagna A, Goetz R, Aligbe O, Singh G, Coe LM, Mohammadi M, Rivella S, Sitara D. Inhibition of fibroblast growth factor 23 (FGF23) signaling rescues renal anemia. FASEB J. 2018;32:3752–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, Shi M, Eliseenkova AV, Razzaque MS, Moe OW, Kuro‐o M, Mohammadi M. Isolated C‐terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23‐FGFR‐Klotho complex formation. Proc Natl Acad Sci USA. 2010;107:407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Courbebaisse M, Mehel H, Petit‐Hoang C, Ribeil JA, Sabbah L, Tuloup‐Minguez V, Bergerat D, Arlet JB, Stanislas A, Souberbielle JC, Le Clesiau H, Fischmeister R, Friedlander G, Prie D. Carboxy‐terminal fragment of fibroblast growth factor 23 induces heart hypertrophy in sickle cell disease. Haematologica. 2017;102:e33–e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Isakova T, Cai X, Lee J, Xie D, Wang X, Mehta R, Allen NB, Scialla JJ, Pencina MJ, Anderson AH, Talierco J, Chen J, Fischer MJ, Steigerwalt SP, Leonard MB, Hsu CY, de Boer IH, Kusek JW, Feldman HI, Wolf M; Chronic Renal Insufficiency Cohort (CRIC) Study Investigators . Longitudinal FGF23 trajectories and mortality in patients with CKD. J Am Soc Nephrol. 2018;29:579–590. [DOI] [PMC free article] [PubMed] [Google Scholar]