

Abstract
Misfolding, aggregation, and aberrant accumulation of proteins are central components in the progression of neurodegenerative disease. Cellular molecular chaperone systems modulate proteostasis, and, therefore, are primed to influence aberrant protein-induced neurotoxicity and disease progression. Molecular chaperones have a wide range of functions from facilitating proper nascent folding and refolding to degradation or sequestration of misfolded substrates. In disease states, molecular chaperones can display protective or aberrant effects, including the promotion and stabilization of toxic protein aggregates. This seems to be dependent on the aggregating protein and discrete chaperone interaction. Small heat shock proteins (sHsps) are a class of molecular chaperones that typically associate early with misfolded proteins. These interactions hold proteins in a reversible state that helps facilitate refolding or degradation by other chaperones and co-factors. These sHsp interactions require dynamic oligomerization state changes in response to diverse cellular triggers and, unlike later steps in the chaperone cascade of events, are ATP-independent. Here, we review evidence for modulation of neurodegenerative disease-relevant protein aggregation by sHsps. This includes data supporting direct physical interactions and potential roles of sHsps in the stewardship of pathological protein aggregates in brain. A greater understanding of the mechanisms of sHsp chaperone activity may help in the development of novel therapeutic strategies to modulate the aggregation of pathological, amyloidogenic proteins. sHsps-targeting strategies including modulators of expression or post-translational modification of endogenous sHsps, small molecules targeted to sHsp domains, and delivery of engineered molecular chaperones, are also discussed.
Keywords: neurodegeneration, proteostasis, molecular chaperone, sHsps, HspB, aging
Introduction
Maintenance of cellular protein homeostasis (proteostasis) is crucial for cell function and survival (Powers et al., 2009; Klaips et al., 2018; Yu et al., 2019). Neurons are particularly sensitive to dysregulated proteostasis as evidenced by the accumulation and aggregation of amyloidogenic proteins, which are a hallmark of neurodegenerative disease (Skibinski and Finkbeiner, 2013; Smith et al., 2015; Yerbury et al., 2016). Proteostasis is regulated by networks of interacting proteins that include translation machinery (Steffen and Dillin, 2016; Anisimova et al., 2018), secretory pathways (Plate and Wiseman, 2017; Remondelli and Renna, 2017; Wong et al., 2018), proteases (Wilson et al., 2015; Klein et al., 2018), the ubiquitin-proteasome system (UPS) (Bett, 2016), autophagic machinery (Carra et al., 2009; Menzies et al., 2015), and molecular chaperones (Balchin et al., 2016). In addition to their vital role in folding nascent proteins, molecular chaperones recognize and triage proteins if misfolding events occur. Chaperones can be classified as holdases (that bind and hold partially folded protein intermediates to prevent their aggregation), foldases (that assist the proper folding of proteins in an ATP-dependent manner), and unfoldases (that convert misfolded proteins into transiently unfolded intermediates to provide an opportunity for spontaneous proper refolding) (Hoffmann et al., 2004; Mattoo and Goloubinoff, 2014). ATP-dependent chaperones, like the 70 kDa heat shock protein (Hsp70) and the 90 kDa heat shock protein (Hsp90), facilitate refolding, degradation, or sequestration of these misfolded proteins. In mammalian systems, chaperones often work together in multisubunit heterocomplexes, comprised of an ATP-dependent chaperone as well as co-chaperone and accessory proteins. These discrete heterocomplexes allow for specific client selection, the dynamic modulation of these clients by regulating ATPase activity, or, if a protein is unable to refold, delivery to additional proteostasis machinery for degradation or sequestration (Sahasrabudhe et al., 2017; Chen et al., 2018; Freilich et al., 2018; Grousl et al., 2018). Here, we review evidence for modulation of neurodegenerative disease-relevant protein aggregation by a family of molecular chaperones, known as small heat shock proteins (sHsps).
Structural Plasticity of sHsps
sHsps are a special class of molecular chaperones that lack an ATPase domain. They are characterized as small proteins, between 12 and 43 kDa, containing a core α-crystallin domain (ACD) flanked by variable N-terminal and C-terminal domains (de Jong et al., 1998; Franck et al., 2004; Haslbeck et al., 2005; Mchaourab et al., 2009; Kriehuber et al., 2010; Haslbeck and Vierling, 2015; Treweek et al., 2015; Zhu and Reiser, 2018). There are 10 α-crystallin domain-containing mammalian sHsps, designated HspB1 (Hsp27), HspB2 (myotonic dystrophy kinase-binding protein, MKBP), HspB3 (Hsp17), HspB4 (αA-crystallin), HspB5 (αB-crystallin), HspB6 (Hsp20), HspB7 (cardiovascular Hsp, cvHsp), HspB8 (Hsp22), HspB9 (cancer/testis antigen 51, CT51) and HspB10 (outer dense fiber protein 1, ODFP1) (see Table 1 ) (Kappé et al., 2003; Kampinga et al., 2009). In general, sHsps interact with intermediately folded proteins through surface exposed hydrophobic residues in order to stabilize the protein and prevent further misfolding and/or aggregation (Sharma et al., 1997; van Montfort et al., 2001; Haslbeck et al., 2005; Jaya et al., 2009). Therefore, sHsps act early in the chaperone processing of misfolded proteins, often prior to refolding attempts by ATP-dependent chaperone complexes (Mchaourab et al., 2009; Bakthisaran et al., 2015; Fu, 2015; Haslbeck and Vierling, 2015). The dynamic nature of sHsp structure appears to be an important determinant for client protein binding and chaperone function (Leroux et al., 1997; Jaya et al., 2009; Mchaourab et al., 2009; Peschek et al., 2009; Benesch et al., 2010; Stengel et al., 2010). sHsp dynamics occur at five levels, including through 1) flexible domains flanking the ACD, 2) polydisperse self-multimerization, 3) multimerization with other sHSPs, 4) subunit exchange, and 5) regulation by the cellular environment including post-translational modifications (Fu, 2015), which are discussed in more detail below.
Table 1.
sHsp expression and relevance for neurodegenerative diseases.
↑, increased; ↓, reduced
The disordered nature of the N- and C-termini flanking the ACD allows for tertiary flexibility that likely supports dynamic interactions with client proteins as well as other sHsp subunits (Lindner et al., 2000; Stromer et al., 2004; Baldwin et al., 2011; Jehle et al., 2011; Sudnitsyna et al., 2012; Patel et al., 2014; Carver et al., 2017). Observations of intrinsic disorder propensity based on primary structure are presented in Figure 1 . A universal intrinsic disorder propensity within the C-terminal domains appears to be a common trait among human sHsp family members; while disorder propensity among N-terminal domains are more variable. HspB4 and HspB5 (the α-crystallins) distinguish themselves as having the most ordered N-terminal domains. The N-terminal domains of HspB1, HspB2, HspB3, HspB6, and HspB10 have discrete segments of disorder; while HspB7, HspB8, and HspB9 have a more widespread disorder propensity in the variable N-terminal domain ( Figure 1 ). It is notable that, for many of the sHsps, high disorder propensity also extends into the conserved ACDs, which are generally regarded to have a well-structured immunoglobulin-like fold of β-sheets (Bagnéris et al., 2009; Laganowsky et al., 2010; Carver et al., 2017), and may reflect differences in ACD flexibility among sHsps.
Figure 1.

Intrinsic disorder profiles generated for human HspB1 (UniProt ID: P04792), HspB2 (UniProt ID: Q16082), HspB3 (UniProt ID: Q12988), HspB4 (UniProt ID: P02489), HspB5 (UniProt ID: P02511), HspB6 (UniProt ID: O14558), HspB7 (UniProt ID: Q9UBY9), HspB8 (UniProt ID: Q9UJY1), HspB9 (UniProt ID: Q9BQS6), and HspB10 (UniProt ID: Q14990), visualized as the mean of 6 commonly used disorder predictors from the PONDR family, PONDR® VLXT, PONDR® VSL2, PONDR® VL3, PONDR® FIT, IUpred_short and IUpred_long. In these plots, disorder score exceeding the 0.5 threshold correspond to intrinsically disordered regions, whereas disorder score ranging from 0.2 to 0.5 show flexible regions. The light cyan shade around disorder predisposition curves represents error (SD) distribution. The grey shaded background corresponds to the ACD of each sHsp sequence to facilitate discrimination from the terminal domains.
sHsps utilize flexible hydrophobic surfaces to interact with exposed hydrophobic surfaces of client proteins, which are made available during misfolding or denaturation (Sharma et al., 1997; van Montfort et al., 2001; Haslbeck et al., 2005; Jaya et al., 2009). Both high affinity and dynamic low affinity interactions of sHsps with client proteins have been described (Bruinsma et al., 2011; McDonald et al., 2012; Mattoo and Goloubinoff, 2014; Ecroyd, 2015). Generally, high affinity interactions are associated with amorphous misfolding clients while sHsp effects on amyloid fibril formation are often due to weak or transient client interactions (Kulig and Ecroyd, 2012; Treweek et al., 2015). Expression of sHsps, with their own exposed hydrophobic surfaces, could potentially pose a cellular proteostasis problem of its own (Dobson, 1999; Seong and Matzinger, 2004; Díaz-Villanueva et al., 2015). One way for cells to address this problem is to restrict expression of sHsps until stress events (like heat shock or oxidative stress) necessitate chaperone activity. Another mechanism is to sequester exposed hydrophobic regions into large, dormant, multimeric structures (sHsp oligomers); keeping sHsp chaperones poised to handle early misfolding events prior to transcription and translation of other stress-inducible chaperones (Santhanagopalan et al., 2018). Indeed large, inactive, oligomeric sHsps disperse into smaller oligomers as they become active (Basha et al., 2012; Haslbeck et al., 2019). This transition can be precipitated by stress events, post-translational modifications, and by direct competition of client proteins with sHsp oligomeric interactions (Arrigo, 2017; Freilich et al., 2018).
Dynamic sHsp homodimers and polydisperse, homooligomeric structures regulate chaperone function as well as client protein specificity and binding (Leroux et al., 1997; Kim et al., 1998; van Montfort et al., 2001; Stromer et al., 2004; Horwitz, 2009; Jaya et al., 2009; Mchaourab et al., 2009; Peschek et al., 2009; Benesch et al., 2010; Jehle et al., 2010; Stengel et al., 2010; Baldwin et al., 2011; Patel et al., 2014). Many factors regulate the size of these structures. sHsp expression (or concentration) itself alters the size of oligomers in vitro (Shashidharamurthy et al., 2005). HspB1 and HspB5 each form higher order homooligomers of greater than 20 subunits in vitro, while other family members generally form smaller oligomers or dimers (Mymrikov et al., 2017). Moreover, sHsp homooligomers with a discrete number of subunits may actually be composed of a variety of dynamic structures, since polymorphic arrangement of the dimer and oligomer subunits have been reported (Feil et al., 2001; Hochberg et al., 2014; Fu, 2015).
Chaperone activity is also regulated by the formation of dynamic sHsp heterodimers and heterooligomers that undergo continuous subunit exchange (Bova et al., 2000; Mchaourab et al., 2009; Aquilina et al., 2013; Arrigo, 2013). Native α-crystallin in the eye lens is made up of polydisperse HspB4/HspB5 heterooligomeric complexes (Haley et al., 2000; Shi et al., 2006; Horwitz, 2009). The crystal structure of a tetrameric HspB2/HspB3 heterocomplex, which is found in muscle cells, was recently reported (den Engelsman et al., 2009; Clark et al., 2018). While a complete picture of the physiologically relevant heterooligomeric sHsp combinations has not been elucidated, in vitro evidence for a variety of heterooligomeric sHsp groupings is emerging, including: HspB1/HspB5 (Zantema et al., 1992; Aquilina et al., 2013), HspB1/HspB6 (Boros et al., 2004; Bukach et al., 2009; Heirbaut et al., 2017) and HspB2/HspB6 (Boros et al., 2004) heterooligomeric complexes as well as HspB8 interactions with HspB1, HspB2, HspB5, HspB6, and HspB7 (Sun et al., 2004; Fontaine et al., 2005). The relative expression levels of discrete sHsp family members will influence the complement of oligomers resulting from subunit exchange (Bova et al., 2000; Mchaourab et al., 2009; Aquilina et al., 2013; Arrigo, 2013), which may guide an interminable number of oligomeric combinations.
Changes in the cellular environment and post-translational modifications also regulate chaperone activity by affecting the structural and multimerization dynamics of sHsps (Eaton et al., 2002; Nagaraj et al., 2003; Mymrikov et al., 2011). Environmental influencers include alterations in temperature and pH (Lelj-Garolla and Mauk, 2006; Hayes et al., 2009; Clouser and Klevit, 2017). The most commonly studied post-translational modification of sHsps is phosphorylation within the N-terminal domain, which is generally considered to decrease oligomer size (Ito et al., 1997; van den IJssel et al., 1998; Lambert et al., 1999; Ito et al., 2001; Hayes et al., 2009; Thornell and Aquilina, 2015; Arrigo, 2017), but opposite effects have been reported (Shemetov et al., 2011). Phosphomimetics of HspB5 have been shown to form smaller but more polydisperse oligomers (Ecroyd et al., 2007; Peschek et al., 2013).
Dynamic sHsp oligomeric structures would be expected to reach equilibrium in a constant environment, but the current literature paints a picture of something more complicated. Since sHsp dynamics are sensitive to many factors, there may be no set equilibrium in the context of an ever-changing cellular environment (Bakthisaran et al., 2015; Haslbeck et al., 2015). This dynamic nature of sHsps can make pinning down a specific structure difficult; and any identified structure of a single oligomeric conformation will likely represent only a snapshot of the evolving assembly. Continuously morphing sHsp oligomers may facilitate the broad recognition of clients that this family of molecular chaperones regulates (Stengel et al., 2010; Fu, 2015).
sHsp Expression in the Brain
Of the 10 mammalian sHsp proteins, HspB1, HspB5, and HspB8 are expressed in the brain, as well as, HspB6, and HspB7, albeit at lower levels (Quraishe et al., 2008). HspB2 and HspB3 mRNA expression have also been detected at relatively low levels in adult brain (Kirbach and Golenhofen, 2011) ( Table 1 ). HspB4 expression appears to be restricted to the eye lens, while HspB9 expression is restricted to testes and heart, and HspB10 expression is restricted to testes, eye, and muscle; these sHsps show no or negligible gene and protein expression in the brain under normal conditions (Quraishe et al., 2008). In addition to the difference in tissue specificity, the regulation of expression is also varied among the HspB family members. Heat shock stress upregulates the expression of HspB1, HspB5, and HspB8 (Chowdary et al., 2004; Kirbach and Golenhofen, 2011). However, in vitro HspB8 responds to heat shock in only a subset of cell types (Gober et al., 2003; Chowdary et al., 2004), suggesting that stress-induced expression may not be a ubiquitous phenomenon across all cell types in vivo. Brain expression of sHSP family members may be induced by other stressors. For example, HspB1, HspB5, HspB6, and HspB8 were upregulated in response to oxidative stress and hyperosmotic stress in rat hippocampal neurons (Bartelt-Kirbach and Golenhofen, 2014) ( Table 1 ). Surprisingly, HspB4, which is not found in normal brain tissues, has been reported to have detectable mRNA and protein expression in cultured primary rat astrocytes, even in the absence of stress induction (Li et al., 2012). It is unclear whether this is reflective of HspB4 expression in a subpopulation of normal brain cells or if expression was induced by culture conditions; either way it is clear that sHsps not normally found in brain tissue have the potential to become expressed in response to stressors, which may include pathological situations (Zhu and Reiser, 2018). HspB1 and HspB5 are more predominantly expressed in glial cells under basal conditions, rather than in neurons (Uhlén et al., 2015; Golenhofen and Bartelt-Kirbach, 2016). Multiple reports demonstrate HspB1 and HspB5 upregulation in neurodegenerative disease (Renkawek et al., 1999; Dabir et al., 2004; Schwarz et al., 2010; Brownell et al., 2012; Liu et al., 2015; Leyns and Holtzman, 2017). For example, in the AD brain, along with other tauopathies including PSP, CBD, and FTDP-17, HspB1 and HspB5 are upregulated in reactive glial cells and can be found to colocalized with glial tau inclusions (Wilhelmus et al., 2006c; Björkdahl et al., 2008; Schwarz et al., 2010). HspB5 upregulation in the astrocytes of ALS patient spinal cord (Iwaki et al., 1992) and HspB1 upregulation in a transgenic mouse model of ALS (Vleminckx et al., 2002) have been reported.
HspB1 and HspB5 share many characteristics that differentiate them from other sHsps (see Figure 2 ). They both readily form higher order homooligomers of 20+ subunits, while other family members generally form smaller oligomers of less than 10 subunits or dimers (Mymrikov et al., 2017). Comparison of the number of human sHsp-interacting proteins also reveals that HspB1 has the largest interactome followed by HspB5 and HspB2 (BioGRID 3.5.170) ( Figure 2 ). HspB1 and HspB5 also exhibit chaperone activity across a wider spectrum of model aggregating proteins, compared to other sHsps (Mymrikov et al., 2017) ( Figure 2 ). HspB1 and HspB5 subunits comprise ubiquitous large, homooligomeric and, perhaps, heterooligomeric complexes (Kato et al., 1992; Mymrikov et al., 2012; Aquilina et al., 2013; Arrigo, 2013), while other family members appear to function as smaller (under 10 subunits) sHsp oligomers. Alternatively, sHsps that form smaller homooligomers may become incorporated into large sHsp heterooligomers through subunit exchange that correlates with their expression level and sHsp interactome; imbuing new interaction preferences for clients as well as additional proteostasis machinery. Known physical interactions between sHsp family members are summarized in Figure 3 (BioGRID 3.5.170).
Figure 2.
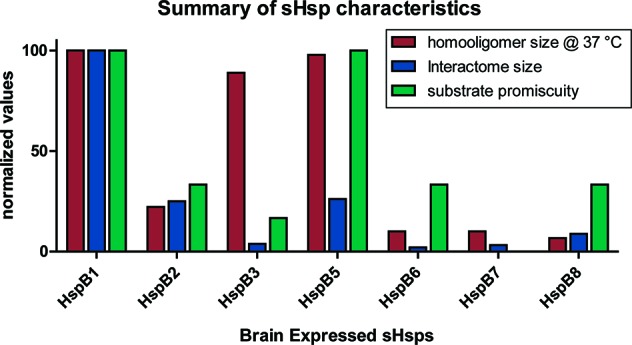
Comparison of specific characteristics of sHsps family members expressed in brain. The average homooligomer size at 37°C was adapted from the relative molecular weights of the peak elution profiles from size exclusion chromatography analysis found in Mymrikov et al., 2017. Chaperone promiscuity was defined as the proportion of six model substrates that demonstrated chaperone activity in a side by side comparison found in Mymrikov et al., 2017. Interactome size was determined by comparison of the number of protein interactors of human sHsps from the BioGRID database version 3.5.170. All values are normalized to HspB1.
Figure 3.
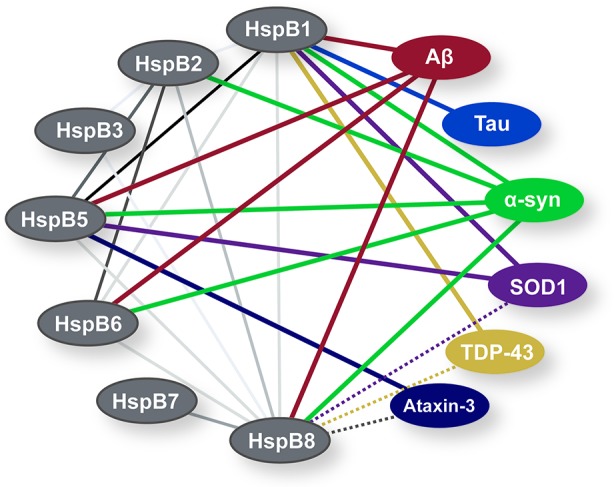
A map of interactions between sHsps expressed in brain and neurodegeneration-associated aggregation-prone proteins discussed in this review. Solid grey lines indicate interactions between sHsps, the darkness of the greyscale lines correlate to the level of evidence for an interaction in BioGRID database 3.5.170. Solid color lines identify physical interactions confirmed in vitro between sHsps and aggregation-prone proteins. Dotted color lines indicate presumed interactions based on increased autophagic clearance via HspB8/Bag3.
sHsps Influence Client Proteostatic Fate
sHsps interact with numerous client proteins, especially under stress conditions. Therefore, it can be difficult to determine whether cellular effects of sHsps result from direct interactions with a given client protein or indirectly through modulation of other proteins and/or signaling pathways. Indeed, cytoprotective roles for sHsps have been described, including inhibition of apoptotic signaling pathways, inhibition of neuroinflammation, and buffering against damage from oxidative stress (Mehlen et al., 1996; Préville et al., 1999; Oshita et al., 2010; McGreal et al., 2012; Bakthisaran et al., 2015). Therefore, sHsps will undoubtedly affect cellular proteostasis at multiple levels.
Direct sHsp interactions with misfolded proteins can prevent further irreversible misfolding events and facilitate client triage by other chaperones that more actively influence client fate through specific proteostasis pathways (Hartl et al., 2011; Vos et al., 2011; Ungelenk et al., 2016). Generally, HspB1, HspB4, and HspB5, which are all heat shock responsive, are commonly reported to play a role in substrate refolding (Ehrnsperger et al., 1997; Vos et al., 2010, Vos et al., 2016), while HspB6, HspB7, and HspB8 have been shown to preferentially promote the degradation of clients through autophagy (Fuchs et al., 2009; Vos et al., 2010; Vos et al., 2011; Carra et al., 2013; Vos et al., 2016). However, this simple categorization of HspBs does not hold true in many instances, including many examples discussed in this review. HspB8, the best characterized sHsp in regard to proteostasis mechanisms, forms a complex with the co-chaperone BCL2-Associated Athanogene 3 (Bag3), Hsp70, and C-terminus of Hsc70-interacting protein (CHIP), which appears to drive aggresome formation and promote autophagic degradation of client proteins (Hishiya et al., 2011; Seidel et al., 2012; Minoia et al., 2014; Rauch et al., 2017; Stürner and Behl, 2017). HspB8 overexpression has been reported to lead to enhanced proteasome expression and activity as well as influence its subcellular localization, at least in cardiac tissues (Hedhli et al., 2008). The authors also demonstrated that HspB8 was found in complex with proteasomes together with HspB1 and other chaperones. This may have a general, indirect effect on the degradation of aberrant proteins. HspB1, HspB5, and HspB6 have also been shown to interact with Bag3 (Rauch et al., 2017). In neurodegenerative disease, Bag3 and HspB8 are upregulated in astrocytes (Seidel et al., 2012), which may help clear protein aggregates. HspB6 has been reported to regulate autophagy through direct interaction and stabilization of Beclin 1, a key regulator of autophagosome formation (Liu et al., 2018). Hsp90 inhibition and proteasome inhibition both activate the heat shock response (Bush et al., 1997; Lee et al., 2013, 90), which upregulates HspB1, HspB4, and HspB5 (Bush et al., 1997; Awasthi and Wagner, 2005; Kumano et al., 2012). Proteasome inhibition has also been shown to upregulate HspB8 (Crippa et al., 2010a; Crippa et al., 2010b). Heat shock response has also been shown to promote HspB1 phosphorylation, regulate its oligomerization state (Bolhuis and Richter-Landsberg, 2010), and enhances UPS activity (Gan et al., 2010). Upregulated HspB1 in response to heat shock has been reported to promote proteasomal degradation of certain client proteins (Parcellier et al., 2003; Parcellier et al., 2006). Bag1 competition for Bag3 occupancy in Hsp70/CHIP complexes drives client degradation through the UPS (Lüders et al., 2000; Minoia et al., 2014; Cristofani et al., 2017; Klimek et al., 2017; Rauch et al., 2017; Stürner and Behl, 2017). Aging and oxidative stress correlates with a switch in expression from Bag1 to Bag3 that may shift the client proteins from UPS-mediated degradation to an autophagic pathway (Gamerdinger et al., 2009a; Gamerdinger et al., 2009b; Behl, 2011). Expression and activity of sHsps may influence shuttling misfolded proteins between proteasomal and autophagic degradation pathways, allowing for the alternative system to be utilized when the primary degradation pathway for a given client is impaired or insufficient.
sHsp Interactions With Amyloidogenic Proteins
Chaperone activity of sHsps was first demonstrated by suppression of stress-induced formation of amorphous aggregates (Horwitz, 1992). The first observation that sHsps also inhibit amyloid fibril formation under physiological conditions in vitro was reported using apolipoprotein C-II (Hatters et al., 2001). Since then, a role for sHsp modulation of amyloidogenic protein aggregation has been reported for a variety of client proteins relevant to neurodegeneration (Golenhofen and Bartelt-Kirbach, 2016; Kourtis and Tavernarakis, 2018). Here, we focus on selected amyloidogenic proteins in neurodegeneration with evidence for direct sHsp interactions and/or chaperone activity ( Figure 3 ). However, it should be noted that for cellular and in vivo applications, direct chaperone activity may be only part of the sHsp-associated mechanisms modifying aggregation or toxicity. Moreover, in disease, while many chaperones still support a healthy environment for the cell (Chen et al., 2018; Grousl et al., 2018), certain chaperone complexes appear to exacerbate the problem of protein misfolding by stabilizing or in some cases promoting the stabilization and accumulation of toxic, prefibrillar oligomers (Tortosa et al., 2009; Blair et al., 2013; Shelton et al., 2017).
Amyloid β
Amyloid β (Aβ) peptides are cleavage products of the amyloid precursor protein (APP) that are prone to aggregate into toxic oligomers, amorphous aggregates, and fibrils; extracellular deposition of Aβ-rich plaques is considered a hallmark of AD. Multiple sHsps have been reported to affect Aβ, including HspB1, HspB2, HspB5, HspB6, and HspB8. HspB1, HspB2, and HspB6 associate with senile plaques in AD brain tissue (Wilhelmus et al., 2006c). In vitro associations between Aβ and HspB1, HspB5, HspB6, and HspB8 have been described (Liang, 2000; Lee et al., 2006; Wilhelmus et al., 2006a; Wilhelmus et al., 2006b). While a connection between HspB1 and Aβ1-42 has been reported, little is known about the molecular details of this interaction. What is known is that, in cellular systems, extracellular Aβ1-42 induces the expression of HspB1; inversely, exogenous exposure of extracellular HspB1 dose-dependently ameliorated Aβ1-42 cellular toxicity (Ojha et al., 2011b). Recombinant protein studies have suggested that the role of HspB1 is to affect Aβ quaternary structure, sequestering toxic oligomers into less toxic, larger aggregates (Ojha et al., 2011b); while other studies have demonstrated a reduction in Aβ aggregation (Wilhelmus et al., 2006a; Yoshiike et al., 2008). A more detailed interaction between HspB5 and Aβ has been described. HspB5 interacts with the hydrophobic core of Aβ1-40 and competes for Aβ–Aβ interactions (Narayanan et al., 2006) HspB5 association with Aβ1-40 prevented amyloid fibril-associated cellular toxicity (Dehle et al., 2010). HspB5 also binds to Aβ1-42 with micromolar affinity and inhibits Aβ fibril elongation and the nucleation of Aβ seeds (Shammas et al., 2011). This work demonstrated that the N-terminal domain of HspB5 was not required for Aβ binding nor inhibition of Aβ fibrillation, whereas the β4–β8 groove of the ACD appears to be crucial for this interaction (Mainz et al., 2015). HspB6 interacts with Aβ and the physical interaction was mapped to residues adjacent to the KLVFF oligomerization domain within Aβ (Cameron et al., 2014). This interaction was strengthened by HspB6 N-terminal domain phosphorylation (Cameron et al., 2014; Cameron et al., 2017. Similar to HspB1, exogenous, extracellular HspB6 also reduced Aβ1-42 cellular toxicity. In addition, it was demonstrated that phosphorylation of endogenous HspB6 correlated with an attenuation of Aβ1-42 toxicity (Cameron et al., 2017). A direct interaction of HspB8 with Aβ1-40 and Aβ1-42 was demonstrated using surface plasmon resonance (SPR), with a significantly greater affinity to Aβ1-40 with the E22N Dutch mutation that correlates with cerebral amyloid angiopathy (Wilhelmus et al., 2006b). In vivo studies using a cross between HspB2/HspB5 knock-out and mutant APP mice revealed locomotor and sensory deficits, suggesting a negative synergy when these genotypes are combined. However, evaluation of changes to amyloid deposition was not possible, since the knock-out mice did not survive to an age wherein amyloid deposits can be detected (Morrison et al., 2004; Ojha et al., 2011a). More work is needed to fully understand the effects of discrete sHsps or sHsp combinations on Aβ deposition and toxicity.
Tau
The microtubule-associated protein tau (tau) is an intracellular intrinsically disordered protein that binds to and stabilizes axonal microtubules. Tau is the major constituent of neurofibrillary tangles (NFTs), protein aggregates that form in neurons and contribute to neurodegeneration seen in more than 20 tauopathic diseases, including AD (Götz et al., 2019). Only two sHsps have been characterized for their interactions with tau, HspB1 and HspB5. Interestingly, both HspB1 and HspB5 are upregulated and associated with pathological NFTs in tauopathies (Dabir et al., 2004; Björkdahl et al., 2008). HspB1 has been shown to bind directly to tau and reduce aggregation (Abisambra et al., 2010; Baughman et al., 2018; Freilich et al., 2018; Mok et al., 2018). HspB1 targets early tau aggregates and delays aggregation, but does not inhibit the fibril elongation process once started (Baughman et al., 2018). As recently revealed by nuclear magnetic resonance (NMR), HspB1 recognizes two aggregation-prone regions within the microtubule binding repeat regions of tau via both the N-terminal domain and the conserved β4–β8 cleft in the ACD (Baughman et al., 2018; Freilich et al., 2018). Interestingly, this is the same surface used for sHsp dimerization as well as interaction with certain other clients and co-chaperone proteins. The fact that HspB1 binds to several different proteins in the same region, and that this binding is relatively weak, suggests that competitive protein–protein interactions may regulate its chaperone activity toward tau (Freilich et al., 2018). This supports a hypothesis for large, inactive sHsp oligomers that convert to smaller, active oligomers through competitive tau interactions. Phosphorylation of the N-terminal domain, or the introduction of specific pseudophosphorylation mutations, reduces the size of HspB1 oligomeric structures and increases chaperone activity (Hayes et al., 2009; Jovcevski et al., 2015). HspB1 and a pseudophosphorylated mutant both bound tau and reduced or delayed tau aggregation in vitro, albeit at different levels (Abisambra et al., 2010; Baughman et al., 2018). Although there are no reports of a direct interaction of tau and HspB8, Bag3 overexpression in neurons reduces tau aggregation; which may implicate a role for HspB8 and autophagy (Lei et al., 2015). Additionally, tau predominantly aggregates in excitatory neurons, with lower Bag3 and HspB8 expression, relative to inhibitory neurons (Fu et al., 2019). HspB1 overexpression was evaluated in brain using a transgenic mouse model of tau aggregation via intracranial injection of an adeno-associated viral (AAV) vector (Abisambra et al., 2010). In this model, wild-type HspB1 reduced neuronal tau levels and prevented long-term potentiation (LTP) dysfunction, a cellular correlate of learning and memory. The pseudophosphorylated mutant had opposite effects; increasing neuronal tau levels and failing to prevent the LTP deficit (Abisambra et al., 2010). These results suggest that while reduction of sHsp oligomer size increases chaperone activity in vitro, sHsp oligomeric complex dynamics are crucial for proper chaperone activity in vivo. Further studies are needed to test other sHsps for their interactions with tau and for their effects in tauopathic mouse models.
α-Synuclein
α-Synuclein (α-syn) is an intrinsically disordered neuronal protein associated with the regulation of synaptic vesicles (Burré, 2015; Theillet et al., 2016). It is also the primary structural component in Lewy Bodies, large protein aggregates that develop in neurodegenerative synucleinopathies, such as Parkinson’s Disease (PD) or dementia with Lewy Bodies (DLB) (Spillantini et al., 1997; Martí et al., 2003). Point mutations (A30P, E46K, H50Q, and A53T) or whole gene triplication of the SNCA gene, encoding α-syn in humans, are associated with the familial forms of early onset PD (Flagmeier et al., 2016; Konno et al., 2016). sHsps interact with multiple species of α-syn, from monomeric partially folded intermediates to fully formed fibrils, modulating both aggregation and cellular toxicity (Cox et al., 2014; Cox et al., 2018). HspB5 was the first sHsp reported to bind α-syn with low affinity and has been known for some time to inhibit amyloid fibril formation (Rekas et al., 2004; Waudby et al., 2010). It has also been reported that α-Syn can interact with at least 5 different sHsps as determined by SPR, with varying affinities; in order of highest affinity for α-syn: HspB5 (Kd = ∼60 nM) > HspB1 and HspB6 (∼400 nM) > HspB2 and HspB8 (∼4 µM) (Bruinsma et al., 2011). Each of these five sHsps delayed or inhibited α-syn fibril formation in vitro. Remarkably, HspB8 had the most robust inhibition of α-syn fibrillization in vitro, but had one of the lowest affinities for α-syn. HspB1 and HspB5 reduced α-syn aggregation, which was attributed to weak transient interactions (Cox et al., 2016), similar to the relationship of HspB1 with tau. This suggests that transient, dynamic sHsp interactions may best ameliorate fibril formation. While HspB5 and HspB8 reduced fibril formation for wild-type α-syn as well as for three familial PD-linked mutants, other sHsps showed more varied effects on discrete α-syn mutants. Surprisingly, HspB2 and HspB6 actually increased the formation of α-syn A53T fibrils (Bruinsma et al., 2011). A recent study found that reduction of the disulfide-bond at the HspB1 β4–β8 dimer interface increased its chaperone activity toward multiple client proteins, including α-syn; perhaps implicating this interface as an important surface for interactions with α-syn (Alderson et al., 2019). This study also demonstrates that the ACD region appears rigid in the dimer, but becomes dynamic and partially unfolded as a monomer (Alderson et al., 2019); this is consistent with the predicted disorder propensity of the HspB1 ACD shown in Figure 1 . HspB5 expression rescued a rough eye phenotype in a transgenic α-syn fly model (Tue et al., 2012). However, HspB5 overexpression was shown to promote mutant α-syn accumulation in a mouse model of PD through suppression of astrocytic autophagy, while knock-down promoted α-syn clearance (Lu et al., 2019). Additional in vivo studies are needed to fully understand the relationship between sHsps and α-syn variants.
PolyQ Expanded Proteins
Genetic expansion of polyglutamine (polyQ) tracts in certain proteins promotes aggregation into fibrillar structures and the death of neurons in several diseases, including spinocerebellar ataxias (via ataxin proteins), spinal and bulbar muscular atrophy (via the androgen receptor protein), and Huntington’s disease (via the huntingtin protein) (Adegbuyiro et al., 2017). While sHsps have been implicated in an amelioration of aggregate formation in a number of cellular polyQ models, direct interaction between sHsps and polyQ domains has not been reported. This is not surprising as the polar polyQ tract would not directly provide hydrophobic surfaces for sHsp–client interactions. Effects of sHsps on polyQ toxicity may be attributed to direct interactions or secondary effects through an accentuation of autophagy via the interaction of HspB8 with the co-chaperone Bag3 (Carra et al., 2008; Carra et al., 2009; Rusmini et al., 2017) or a reduction in oxidative stress (Wyttenbach et al., 2002). HspB5 has been reported to bind to the Josephin domain flanking the polyQ expansion of the ataxin-3 protein, slowing polyQ-driven aggregation (Robertson et al., 2010). A protein engineering strategy similar to the action of HspB5, targeting polyQ-flanking regions with engineered single-chain antibody fragments, also successfully inhibited mutant huntingtin aggregation (Colby et al., 2004). Vos et al. performed an elegant comparison of all 10 sHsps on the aggregation of polyQ expanded huntingtin and ataxin-3, which demonstrated that HspB7 most potently reduced polyQ aggregation in a proteasome-independent manner (Vos et al., 2010). The well-studied and generally promiscuous sHsps, HspB1 and HspB5, were surprisingly not very effective at reducing polyQ aggregation (Vos et al., 2010). It is not yet known whether any of the polyQ huntingtin-active sHsps target a flanking region or act solely through secondary effects. In another study, HspB1 overexpression in a N-terminal huntingtin-fragment transgenic mouse model (R6/2) was unable to alter disease progression or huntingtin aggregation (Zourlidou et al., 2007). However, astrocyte-specific HspB5 overexpression in a full-length huntingtin transgenic model (BACHD) showed neuroprotective effects and slowed disease progression (Oliveira et al., 2016). Moreover, HspB7, ameliorated an eye degeneration phenotype in a fly model expressing polyQ expanded ataxin-3 (Vos et al., 2010) and has yet to be evaluated in mammalian polyQ disease models. More work is needed to understand the direct and indirect actions of sHsps on polyQ expanded aggregates in vivo.
SOD-1 and TDP-43
Superoxide dismutase-1 (SOD-1) is a free radical scavenging metalloenzyme that can contain aggregation-prone mutations linked to familial amyotrophic lateral sclerosis (ALS), a neurodegenerative motoneuron disease. HspB1, HspB5, and HspB8 can all affect the accumulation of this protein. HspB1 and HspB5 can directly interact with SOD-1 in vitro and suppress the aggregation of both wild-type and the familial ALS-linked G93A mutant SOD-1 (Yerbury et al., 2013). As with polyQ containing proteins, HspB8 has been shown to facilitate the autophagic removal of misfolded or aggregated SOD-1. In this context, HspB8 interacts with a multimeric complex that also includes Bag3, Hsp70, and CHIP (Crippa et al., 2010b; Crippa et al., 2016a; Crippa et al., 2016b), however there is not yet any direct evidence for direct HspB8 interactions or “holdase” chaperone activity for SOD-1. Similar observations of HspB8-linked enhancement of autophagic clearance have been made for another ALS-linked protein, transactive response DNA-binding protein 43 kDa (TDP-43) (Crippa et al., 2016a), without evidence for a direct interaction. However, there is evidence via high throughput co-fractionation data for an interaction of TDP-43 with HspB1 (Wan et al., 2015). Two other studies evaluated overexpression of HspB1 in a transgenic SOD-1G93A model of ALS; one showed no effect on disease progression (Krishnan et al., 2008), while the other showed a modest protection at early stages of disease, which were not sustained and resulted in no improvement at later stages (Sharp et al., 2008). Likewise, HspB5 knock-down in a transgenic SOD-1 mutant mouse model did not increase SOD-1 aggregation or disease progression (Sharp et al., 2008). However, differential expression of HspB5 ALS transgenic mouse models with varied background correlated with slower disease progression in these animals (Marino et al., 2015). The effects of other sHsp family members on ALS-related aggregates have not been reported.
sHsp Secretion and Protein Aggregation in the Brain
In addition to intracellular sHsp dynamics, some sHsps have been reported outside of the cell. HspB1, HspB5, and HspB6 have been found in the extracellular environment, in the absence of a traditional secretory signal sequence, and there is evidence for both exosomal and endolysosomal secretion mechanisms (Clayton et al., 2005; Sreekumar et al., 2010; Gangalum et al., 2011; Zhang et al., 2012; Bakthisaran et al., 2015; Batulan et al., 2016). Elevated HspB1 protein levels in serum have been detected in multiple sclerosis (Ce et al., 2011) and in both plasma and cerebrospinal fluid during ischemia (Hecker and McGarvey, 2011). The presence of HspB5 autoantibodies in the serum of AD and PD patients provides additional evidence for the presence of extracellular sHsps in neurodegenerative disease (Papuć et al., 2015; Papuć et al., 2016). sHsp secretion appears to be regulated by post-translational modifications. A phosphorylation deficient HspB1 mutant colocalized with the lysosome-associated membrane protein-1 (LAMP-1) and was preferentially secreted; while a pseudophosphorylated mutant demonstrated decreased secretion and decreased LAMP-1 colocalization (Lee et al., 2012). Evidence for functional roles of these secreted sHsps are emerging (Reddy et al., 2018). In a simple case of direct interaction, sHsps would need to be located outside of cells in order to associate with and reduce toxicity of extracellular misfolded proteins, like Aβ (Ojha et al., 2011b; Cameron et al., 2017); which has been described for HspB1, HspB5, and HspB8 in extracellular, classic, senile plaques in AD brain (Wilhelmus et al., 2006b; Wilhelmus et al., 2006c). However, extracellular sHsps have also been implicated in cell-to-cell communication, signaling, inflammation, and immunity (Binder et al., 2004; Schmitt et al., 2007; Rayner et al., 2008; Reddy et al., 2018), which suggests multiple indirect effects on neuronal health. For example, extracellular HspB5 functions as a stroke neuroprotectant by modulating the immune system (Arac et al., 2011).
Meanwhile, aggregation prone proteins that are traditionally considered to be intracellular, like tau, α-syn, TDP-43, and huntingtin, are also found to be secreted into the extracellular milieu, which may be a mechanism that enables pathological spreading from cell to cell (Hansen et al., 2011; Braak et al., 2013; Babcock and Ganetzky, 2015; Fontaine et al., 2016; Trajkovic et al., 2017; Pérez et al., 2018; Gibbons et al., 2019). There is growing evidence that this cell-to-cell transfer contributes to the anatomical pattern of pathology progression to distinct brain regions that typify specific neurodegenerative diseases (Braak and Braak, 1991; Dickson et al., 2010). Both sHsps and neurodegenerative amyloids are reported to be secreted from cells through similar mechanisms of exosomal release or endolysosomal secretion (Danzer et al., 2012; Batulan et al., 2016; Fontaine et al., 2016; Guix et al., 2018; Katsinelos et al., 2018; Lee et al., 2018; Pérez et al., 2018; Reddy et al., 2018; Sardar Sinha et al., 2018); other mechanisms, including direct transport and tunneling nanotubes, may also contribute (Abounit et al., 2016; Batulan et al., 2016). Physical interactions between sHsps and amyloidogenic proteins may also suggest that this class of molecular chaperones could play a regulatory role in their non-canonical secretion or perhaps buffer against the spread of toxic aggregates to additional cells.
sHsps and Liquid–Liquid Phase Separation
There is growing evidence that formation of membrane-less organelles can be influenced by some aggregation-prone proteins, including proteins linked to neurodegenerative disease (Patel et al., 2015; Ambadipudi et al., 2017). Membrane-less organelles are viscous, liquid, subcellular compartments containing proteins and often RNA that develop as localized pockets of condensed cellular material formed by a process called liquid–liquid phase-separation (LLPS) (Brangwynne, 2013; Boeynaems et al., 2018; Gomes and Shorter, 2019). Alterations in pH, ion concentrations, temperature, oxidative stress, molecular crowding, and post-translational modifications can trigger molecular supersaturation causing LLPS (Shelkovnikova et al., 2014; Bah and Forman-Kay, 2016; Chen and Liu, 2017; Wang et al., 2017; Boeynaems et al., 2018). Proteins with intrinsically disordered regions can also drive LLPS (Toll-Riera et al., 2012; Brangwynne, 2013; Zhu and Brangwynne, 2015), due to conformational flexibility that allows for a multitude of transient multivalent interactions (Toretsky and Wright, 2014; Boeynaems et al., 2018; Darling et al., 2018). AD, ALS, and frontotemporal dementia-related proteins, such as tau and TDP-43 can form LLPS droplets (Molliex et al., 2015; Patel et al., 2015; Conicella et al., 2016; Ambadipudi et al., 2017; Harrison and Shorter, 2017; Boeynaems et al., 2018; Wegmann et al., 2018; Ambadipudi et al., 2019; Jiang et al., 2019). There is evidence that aberrant membrane-less organelles can become more rigid over time, and this process can be accompanied (or preceded) by the misfolding and pathological aggregation of proteins residing in these organelles (Patel et al., 2015; Ambadipudi et al., 2017; Wegmann et al., 2018).
sHsps share a number of properties with proteins that undergo LLPS, including regions of high intrinsic disorder ( Figure 1 ), capability to transiently interact with many substrates, and self-oligomerization (Gomes and Shorter, 2019). The same aggregation-prone motifs in the microtuble-binding regions of tau that are implicated in driving LLPS are also targeted by HspB1 (Baughman et al., 2018; Freilich et al., 2018; Ambadipudi et al., 2019). HspB2 itself has been shown to undergo LLPS, through interactions of the disordered C-terminal domain, which results in the formation of liquid-like compartments in the nucleus that cause impaired nuclear homeostasis and transcription (Morelli et al., 2017). Remarkably, this droplet formation is abrogated by co-expression of HspB3 (Morelli et al., 2017), which forms heterooligomeric complexes with HspB2 (den Engelsman et al., 2009; Clark et al., 2018). Emerging evidence for sHsp residency in membrane-less organelles may suggest additional roles for sHsps in LLPS, including regulation of membrane-less organelle biogenesis and clearance. For example, HspB1, HspB5, and HspB7 have been shown to associate with nuclear speckles (van den IJssel et al., 1998; Bryantsev et al., 2007; Vos et al., 2009). Furthermore, HspB1 and HspB8 are recruited to cytoplasmic stress granules, which are membrane-less organelles (Ganassi et al., 2016). While HspB8 has a role in autophagy, the Hsp70-HspB8-Bag3 complex also plays a role in both the autophagy-independent disassembly of stress granules as well as the extraction of misfolded proteins, known as defective ribosomal products (DRiPs). Additionally, down-regulation of HspB8-Bag3 leads to conversion of stress granules to an aberrant state (Ganassi et al., 2016). HspB1 may also affect stress granule dynamics, but perhaps only after stress granules that have already progressed to an aberrant state (Ganassi et al., 2016).
Membrane-less organelles may provide a microenvironment for the seeding and/or propagation of neurodegeneration-related amyloids. The presence of sHsps in membrane-less organelles may be through a direct interaction with misfolded proteins or due to the intrinsic ability of discrete sHsps to phase separate. Either way, the commingling of neurodegenerative-associated proteins and sHsps in membrane-less organelles may represent a nexus for regulating aggregation. Therefore, modulation of chaperone protein expression or activity may be a potential therapeutic target to treat various neurodegenerative disease pathologies.
Therapeutic Opportunities
Understanding the interactions and chaperone functions of sHsps with amyloidogenic protein aggregates may better inform small molecule and engineered biologic therapeutic strategies for neurodegenerative proteinopathies.
sHsp Modulators
Potential therapeutic strategies that aim to modulate endogenous sHsp expression or phosphorylation generally suffer from a lack of specificity for the sHsp family, let alone for discrete sHsps. Heat stress-responsive sHsps can be activated by drugs that generate a challenge to proteostasis, which includes proteasome inhibitors (e.g. Bortezomib), Hsp90 inhibitors (e.g. 17-AAG), and oxidative stress inducers (e.g. terrecyclic acid) (Westerheide and Morimoto, 2005; Powers and Workman, 2007). However, these treatments also induce expression of other molecular chaperone families (e.g. Hsp70 and Hsp40) and are not specific for sHsp activation. Efforts to identify Hsp co-inducers, substances that potentiate stress responses without inducing a primary stress response on their own, may offer improved selectivity (Hargitai et al., 2003; Kasza et al., 2016). Arimoclomol is a co-inducer currently in clinical trials for ALS (NCT00706147, NCT03491462), which may be further enhanced by co-administration with a heat shock response inducer, like Celastrol (Deane and Brown, 2016). As phosphorylation of sHsps can modulate their oligomeric status and chaperone activity, identifying compounds that activate specific kinases or phosphatases that modulate sHsps phosphorylation may also be an attractive therapeutic strategy. In fact, inhibitors of phosphodiesterase 9 were found to induce phosphorylation of HspB6 and reduce Aβ cellular toxicity (Cameron et al., 2017); one of these compounds is currently being tested in an AD clinical trial (NCT00930059).
sHsp-Targeting Small Molecules
Small molecules that interact with sHsps may be a promising strategy for therapeutics, but the nature of this family of chaperones makes drugability difficult. There are no known small molecule ligands to use as a scaffold to start from. The dynamic and polydisperse nature of these proteins taunt the idea of engineering a high affinity binding drug; indeed, these promiscuous proteins likely have many client binding sites with a variety of conformations. The dimer interface of the ACD, regions within the N-terminal domain, and the IPV motif of the C-terminal domain are important for dimer, higher order oligomer, and client protein interactions. sHsp-client protein interactions compete for oligomeric sHsp–sHsp interactions and activate chaperone activity; these sites have also been proposed as potential drug discovery targets (Arrigo et al., 2007; Jaya et al., 2009; Peschek et al., 2013; Freilich et al., 2018). Oxysterols, like Lanesterol, are an example of potential sHsp-targeting small molecules reported to reverse HspB4 or HspB5 aberrant aggregation, which has been reported as a genetic cause of cataract formation in the eye lens. These compounds appear to reduce the formation of amyloid-like fibers through an interaction with the ACD-dimer interface (Makley et al., 2015; Zhao et al., 2015); however, the high concentrations required for this effect may suggest a lack of specificity for this interaction (Daszynski et al., 2019). sHsp peptide fragments or client binding domains may be promising scaffolds to initiate drug discovery efforts. Small molecules or peptides, with optimally tuned affinities triggering cycling between large oligomers and smaller species, may be more effective “activators” of sHsp chaperone activity than traditional static high affinity binders; as cycling itself has been determined to be crucial for proper chaperone activity in vivo with respect to tau aggregation (Abisambra et al., 2010).
Engineered Molecular Chaperones
Although we generally restricted this review to human sHsps that interact with amyloidogenic proteins involved in neurodegenerative disease, the diversity of sHsps from different organisms, from bacteria to humans, provides a rich set of ACD-containing proteins to explore for aggregation prevention activity (Abisambra et al., 2011). For example, a sHsp from a parasite was shown to be a potent inhibitor of Aβ fibrillation and reduced associated toxicity in a neuroblastoma cell model (Lee et al., 2006). Specific mutant or engineered sHsp variants, with altered oligomeric structure or client interactions, may prove to have increased chaperone activity towards amyloidogenic proteins (Abisambra et al., 2010). Additionally, effective holdase chaperones may be engineered as fusion proteins with domains from other chaperones (foldase or unfoldase) or co-chaperones. Small peptides derived from human HspB4 and HspB5 sequences, termed mini-chaperones, display chaperone-like activity (Raju et al., 2016). One of these constructs reduced cellular toxicity of Aβ (Raju et al., 2018). An important hurdle for eventual clinical translation of engineered molecular chaperones will be delivery to the site of aggregation. Preclinical evaluation of exogenous chaperone expression can be evaluated in mouse models through gene therapy delivery, as used to evaluate tau pathology by our group (Abisambra et al., 2010; Blair et al., 2013; Baker et al., 2017; Shelton et al., 2017); but clinical translation will rely on the continued development of clinical gene therapy or protein transduction domain strategies (Simonato et al., 2013; Naldini, 2015; Guidotti et al., 2017; Raju et al., 2018).
Author Contributions
JW, AD and VU wrote sections of the manuscript. JW, VU, and LB created the figures, JW and LB edited the manuscript. All authors contributed to the manuscript, read, and approved the submitted version.
Funding
This work was supported by the National Institute On Aging of the National Institutes of Health under Award Number RF1AG055088. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Abisambra J. F., Blair L. J., Hill S. E., Jones J. R., Kraft C., Rogers J., et al. (2010). Phosphorylation dynamics regulate Hsp27-mediated rescue of neuronal plasticity deficits in tau transgenic mice. J. Neurosci. 30, 15374–15382. 10.1523/JNEUROSCI.3155-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abisambra J. F., Jinwal U. K., Jones J. R., Blair L. J., Koren J., Dickey C. A. (2011). Exploiting the diversity of the heat-shock protein family for primary and secondary tauopathy therapeutics. Curr. Neuropharmacol. 9, 623–631. 10.2174/157015911798376226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abounit S., Wu J. W., Duff K., Victoria G. S., Zurzolo C. (2016). Tunneling nanotubes: a possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 10, 344–351. 10.1080/19336896.2016.1223003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adegbuyiro A., Sedighi F., Pilkington A. W., Groover S., Legleiter J. (2017). Proteins containing expanded polyglutamine tracts and neurodegenerative disease. Biochemistry 56, 1199–1217. 10.1021/acs.biochem.6b00936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderson T. R., Roche J., Gastall H. Y., Dias D. M., Pritišanac I., Ying J., et al. (2019). Local unfolding of the HSP27 monomer regulates chaperone activity. Nat. Commun. 10, 1068. 10.1038/s41467-019-08557-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambadipudi S., Biernat J., Riedel D., Mandelkow E., Zweckstetter M. (2017). Liquid–liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 8, 275. 10.1038/s41467-017-00480-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambadipudi S., Reddy J. G., Biernat J., Mandelkow E., Zweckstetter M. (2019). Residue-specific identification of liquid phase separation hot spots of the Alzheimer’s disease-related protein Tau. Chem. Sci. 10, 6503–6507 10.1039/C9SC00531E [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova A. S., Alexandrov A. I., Makarova N. E., Gladyshev V. N., Dmitriev S. E. (2018). Protein synthesis and quality control in aging. Aging (Albany NY) 10, 4269–4288. 10.18632/aging.101721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquilina J. A., Shrestha S., Morris A. M., Ecroyd H. (2013). Structural and functional aspects of hetero-oligomers formed by the small heat shock proteins αB-crystallin and HSP27. J. Biol. Chem. 288, 13602–13609. 10.1074/jbc.M112.443812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arac A., Brownell S. E., Rothbard J. B., Chen C., Ko R. M., Pereira M. P., et al. (2011). Systemic augmentation of alphaB-crystallin provides therapeutic benefit twelve hours post-stroke onset via immune modulation. Proc. Natl. Acad. Sci. U.S.A. 108, 13287–13292. 10.1073/pnas.1107368108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigo A.-P. (2013). Human small heat shock proteins: protein interactomes of homo- and hetero-oligomeric complexes: An update. FEBS Lett. 587, 1959–1969. 10.1016/j.febslet.2013.05.011 [DOI] [PubMed] [Google Scholar]
- Arrigo A.-P. (2017). Mammalian HspB1 (Hsp27) is a molecular sensor linked to the physiology and environment of the cell. Cell Stress Chaperones 22, 517–529. 10.1007/s12192-017-0765-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigo A.-P., Simon S., Gibert B., Kretz-Remy C., Nivon M., Czekalla A., et al. (2007). Hsp27 (HspB1) and αB-crystallin (HspB5) as therapeutic targets. FEBS Lett. 581, 3665–3674. 10.1016/j.febslet.2007.04.033 [DOI] [PubMed] [Google Scholar]
- Awasthi N., Wagner B. J. (2005). Upregulation of heat shock protein expression by proteasome inhibition: an antiapoptotic mechanism in the lens. Invest. Ophthalmol. Vis. Sci. 46, 2082–2091. 10.1167/iovs.05-0002 [DOI] [PubMed] [Google Scholar]
- Babcock D. T., Ganetzky B. (2015). Transcellular spreading of huntingtin aggregates in the Drosophila brain. Proc. Natl. Acad. Sci. U.S.A. 112, E5427–E5433. 10.1073/pnas.1516217112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnéris C., Bateman O. A., Naylor C. E., Cronin N., Boelens W. C., Keep N. H., et al. (2009). Crystal Structures of α-Crystallin Domain Dimers of αB-Crystallin and Hsp20. J. Mol. Biol. 392, 1242–1252. 10.1016/j.jmb.2009.07.069 [DOI] [PubMed] [Google Scholar]
- Bah A., Forman-Kay J. D. (2016). Modulation of intrinsically disordered protein function by post-translational modifications. J. Biol. Chem. 291, 6696–6705. 10.1074/jbc.R115.695056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J. D., Shelton L. B., Zheng D., Favretto F., Nordhues B. A., Darling A., et al. (2017). Human cyclophilin 40 unravels neurotoxic amyloids. PLoS Biol. 15, e2001336. 10.1371/journal.pbio.2001336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakthisaran R., Tangirala R., and Ch., M. (2015). Small heat shock proteins: role in cellular functions and pathology. Biochi. Biophys. Acta (BBA) Proteins Proteom. 1854, 291–319. 10.1016/j.bbapap.2014.12.019 [DOI] [PubMed] [Google Scholar]
- Balchin D., Hayer-Hartl M., Hartl F. U. (2016). In vivo aspects of protein folding and quality control. Science 353, aac4354–aac4354. 10.1126/science.aac4354 [DOI] [PubMed] [Google Scholar]
- Baldwin A. J., Lioe H., Hilton G. R., Baker L. A., Rubinstein J. L., Kay L. E., et al. (2011). The polydispersity of αB-crystallin is rationalized by an interconverting polyhedral architecture. Structure 19, 1855–1863. 10.1016/j.str.2011.09.015 [DOI] [PubMed] [Google Scholar]
- Bartelt-Kirbach B., Golenhofen N. (2014). Reaction of small heat-shock proteins to different kinds of cellular stress in cultured rat hippocampal neurons. Cell Stress Chaperones 19, 145–153. 10.1007/s12192-013-0452-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartelt-Kirbach B., Slowik A., Beyer C., Golenhofen N. (2017). Upregulation and phosphorylation of HspB1/Hsp25 and HspB5/αB-crystallin after transient middle cerebral artery occlusion in rats. Cell Stress Chaperones 22, 653–663. 10.1007/s12192-017-0794-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basha E., O’Neill H., Vierling E. (2012). Small heat shock proteins and α-crystallins: dynamic proteins with flexible functions. Trends Biochem. Sci. 37, 106–117. 10.1016/j.tibs.2011.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batulan Z., Pulakazhi Venu V. K., Li Y., Koumbadinga G., Alvarez-Olmedo D. G., Shi C., et al. (2016). Extracellular release and signaling by heat shock protein 27: role in modifying vascular inflammation. Front. Immunol. 7, 285. 10.3389/fimmu.2016.00285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman H. E. R., Clouser A. F., Klevit R. E., Nath A. (2018). HspB1 and Hsc70 chaperones engage distinct tau species and have different inhibitory effects on amyloid formation. J. Biol. Chem. 293, 2687–2700. 10.1074/jbc.M117.803411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl C. (2011). BAG3 and friends: Co-chaperones in selective autophagy during aging and disease. Autophagy 7, 795–798. 10.4161/auto.7.7.15844 [DOI] [PubMed] [Google Scholar]
- Benesch J. L. P., Aquilina J. A., Baldwin A. J., Rekas A., Stengel F., Lindner R. A., et al. (2010). The quaternary organization and dynamics of the molecular chaperone HSP26 are thermally regulated. Chem. Biol. 17, 1008–1017. 10.1016/j.chembiol.2010.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett J. S. (2016). Proteostasis regulation by the ubiquitin system. Essays Biochem. 60, 143–151. 10.1042/EBC20160001 [DOI] [PubMed] [Google Scholar]
- Binder R. J., Vatner R., Srivastava P. (2004). The heat-shock protein receptors: some answers and more questions. Tissue Antigens 64, 442–451. 10.1111/j.1399-0039.2004.00299.x [DOI] [PubMed] [Google Scholar]
- Björkdahl C., Sjögren M. J., Zhou X., Concha H., Avila J., Winblad B., et al. (2008). Small heat shock proteins Hsp27 or alphaB-crystallin and the protein components of neurofibrillary tangles: tau and neurofilaments. J. Neurosci. Res. 86, 1343–1352. 10.1002/jnr.21589 [DOI] [PubMed] [Google Scholar]
- Blair L. J., Nordhues B. A., Hill S. E., Scaglione K. M., O’Leary J. C., Fontaine S. N., et al. (2013). Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Invest. 123, 4158–4169. 10.1172/JCI69003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S., Alberti S., Fawzi N. L., Mittag T., Polymenidou M., Rousseau F., et al. (2018). Protein phase separation: a new phase in cell biology. Trends Cell Biol. 28, 420–435. 10.1016/j.tcb.2018.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolhuis S., Richter-Landsberg C. (2010). Effect of proteasome inhibition by MG-132 on HSP27 oligomerization, phosphorylation, and aggresome formation in the OLN-93 oligodendroglia cell line. J. Neurochem. 114, 960–971. 10.1111/j.1471-4159.2010.06600.x [DOI] [PubMed] [Google Scholar]
- Boros S., Kamps B., Wunderink L., Bruijn W., Jong W. W., et al. (2004). Transglutaminase catalyzes differential crosslinking of small heat shock proteins and amyloid-β. FEBS Lett. 576, 57–62. 10.1016/j.febslet.2004.08.062 [DOI] [PubMed] [Google Scholar]
- Bova M. P., Mchaourab H. S., Han Y., Fung B. K.-K. (2000). Subunit exchange of small heat shock proteins analysis of oligomer formation of αA-crystallin and Hsp27 by fluorescence resonance energy transfer and site-directed truncations. J. Biol. Chem. 275, 1035–1042. 10.1074/jbc.275.2.1035 [DOI] [PubMed] [Google Scholar]
- Braak H., Braak E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- Braak H., Brettschneider J., Ludolph A. C., Lee V. M., Trojanowski J. Q., Del Tredici K. (2013). Amyotrophic lateral sclerosis—a model of corticofugal axonal spread. Nat. Rev. Neurol. 9, 708–714. 10.1038/nrneurol.2013.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne C. P. (2013). Phase transitions and size scaling of membrane-less organelles. J. Cell Biol. 203, 875–881. 10.1083/jcb.201308087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehme M., Voisine C., Rolland T., Wachi S., Soper J. H., Zhu Y., et al. (2014). A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 9, 1135–1150. 10.1016/j.celrep.2014.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownell S. E., Becker R., Steinman L. (2012). The protective and therapeutic function of small heat shock proteins in neurological diseases. Front. Immunol. 3, 74. 10.3389/fimmu.2012.00074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruinsma I. B., Bruggink K. A., Kinast K., Versleijen A. A. M., Segers-Nolten I. M. J., Subramaniam V., et al. (2011). Inhibition of α-synuclein aggregation by small heat shock proteins. Proteins: Struct. Funct.Bioinform. 79, 2956–2967. 10.1002/prot.23152 [DOI] [PubMed] [Google Scholar]
- Bryantsev A. L., Chechenova M. B., Shelden E. A. (2007). Recruitment of phosphorylated small heat shock protein Hsp27 to nuclear speckles without stress. Exp. Cell Res. 313, 195–209. 10.1016/j.yexcr.2006.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukach O. V., Glukhova A. E., Seit-Nebi A. S., Gusev N. B. (2009). Heterooligomeric complexes formed by human small heat shock proteins HspB1 (Hsp27) and HspB6 (Hsp20). Biochem. Biophys. Acta 1794, 486–495. 10.1016/j.bbapap.2008.11.010 [DOI] [PubMed] [Google Scholar]
- Burré J. (2015). The Synaptic Function of α-Synuclein. J. Parkinsons Dis. 5, 699–713. 10.3233/JPD-150642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K. T., Goldberg A. L., Nigam S. K. (1997). Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J. Biol. Chem. 272, 9086–9092. 10.1074/jbc.272.14.9086 [DOI] [PubMed] [Google Scholar]
- Cameron R. T., Quinn S. D., Cairns L. S., MacLeod R., Samuel I. D. W., Smith B. O., et al. (2014). The phosphorylation of Hsp20 enhances its association with amyloid-β to increase protection against neuronal cell death. Mol. Cell. Neurosci. 61, 46–55. 10.1016/j.mcn.2014.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron R. T., Whiteley E., Day J. P., Parachikova A. I., Baillie G. S. (2017). Selective inhibition of phosphodiesterases 4, 5 and 9 induces HSP20 phosphorylation and attenuates amyloid beta 1–42-mediated cytotoxicity. FEBS Open Bio. 7, 64–73. 10.1002/2211-5463.12156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carra S., Brunsting J. F., Lambert H., Landry J., Kampinga H. H. (2009). HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2α phosphorylation. J. Biol. Chem. 284, 5523–5532. 10.1074/jbc.M807440200 [DOI] [PubMed] [Google Scholar]
- Carra S., Rusmini P., Crippa V., Giorgetti E., Boncoraglio A., Cristofani R., et al. (2013). Different anti-aggregation and pro-degradative functions of the members of the mammalian sHSP family in neurological disorders. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20110409. 10.1098/rstb.2011.0409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carra S., Seguin S. J., Landry J. (2008). HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy 4, 237–239. 10.4161/auto.5407 [DOI] [PubMed] [Google Scholar]
- Carver J. A., Grosas A. B., Ecroyd H., Quinlan R. A. (2017). The functional roles of the unstructured N- and C-terminal regions in αB-crystallin and other mammalian small heat-shock proteins. Cell Stress Chaperones 22, 627–638. 10.1007/s12192-017-0789-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ce P., Erkizan O., Gedizlioglu M. (2011). Elevated HSP27 levels during attacks in patients with multiple sclerosis. Acta Neurol. Scand. 124, 317–320. 10.1111/j.1600-0404.2010.01475.x [DOI] [PubMed] [Google Scholar]
- Chen J. Y., Parekh M., Seliman H., Bakshinskaya D., Dai W., Kwan K., et al. (2018). Heat shock promotes inclusion body formation of mutant huntingtin (mHtt) and alleviates mHtt-induced transcription factor dysfunction. J. Biol. Chem. 293, 15581–15593. 10.1074/jbc.RA118.002933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Liu B. (2017). Relationships between stress granules, oxidative stress, and neurodegenerative diseases. Oxid. Med. Cell. Longev. 2017, 189592 10.1155/2017/1809592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdary T. K., Raman B., Ramakrishna T., Rao C. M. (2004). Mammalian Hsp22 is a heat-inducible small heat-shock protein with chaperone-like activity. Biochem. J. 381, 379–387. 10.1042/BJ20031958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. R., Vree Egberts W., Kondrat F. D. L., Hilton G. R., Ray N. J., Cole A. R., et al. (2018). Terminal regions confer plasticity to the tetrameric assembly of human HspB2 and HspB3. J. Mol. Biol. 430, 3297–3310. 10.1016/j.jmb.2018.06.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton A., Turkes A., Navabi H., Mason M. D., Tabi Z. (2005). Induction of heat shock proteins in B-cell exosomes. J. Cell Sci. 118, 3631–3638. 10.1242/jcs.02494 [DOI] [PubMed] [Google Scholar]
- Clouser A. F., Klevit R. E. (2017). pH-dependent structural modulation is conserved in the human small heat shock protein HSBP1. Cell Stress Chaperones 22, 569–575. 10.1007/s12192-017-0783-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colby D. W., Garg P., Holden T., Chao G., Webster J. M., Messer A., et al. (2004). Development of a human light chain variable domain (V(L)) intracellular antibody specific for the amino terminus of huntingtin via yeast surface display. J. Mol. Biol. 342, 901–912. 10.1016/j.jmb.2004.07.054 [DOI] [PubMed] [Google Scholar]
- Conicella A. E., Zerze G. H., Mittal J., Fawzi N. L. (2016). ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity c-terminal domain. Structure 24, 1537–1549. 10.1016/j.str.2016.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D., Carver J. A., Ecroyd H. (2014). Preventing α-synuclein aggregation: the role of the small heat-shock molecular chaperone proteins. Biochim. Biophys. Acta (BBA) – Mol. Basis Dis. 1842, 1830–1843. 10.1016/j.bbadis.2014.06.024 [DOI] [PubMed] [Google Scholar]
- Cox D., Selig E., Griffin M. D. W., Carver J. A., Ecroyd H. (2016). Small heat-shock proteins prevent α-synuclein aggregation via transient interactions and their efficacy is affected by the rate of aggregation. J. Biol. Chem. 291, 22618–22629. 10.1074/jbc.M116.739250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D., Whiten D. R., Brown J. W. P., Horrocks M. H., San Gil R., Dobson C. M., et al. (2018). The small heat shock protein Hsp27 binds α-synuclein fibrils, preventing elongation and cytotoxicity. J. Biol. Chem. 293, 4486–4497. 10.1074/jbc.M117.813865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crippa V., Carra S., Rusmini P., Sau D., Bolzoni E., Bendotti C., et al. (2010. a). A role of small heat shock protein B8 (HspB8) in the autophagic removal of misfolded proteins responsible for neurodegenerative diseases. Autophagy 6, 958–960. 10.4161/auto.6.7.13042 [DOI] [PubMed] [Google Scholar]
- Crippa V., Cicardi M. E., Ramesh N., Seguin S. J., Ganassi M., Bigi I., et al. (2016. a). The chaperone HSPB8 reduces the accumulation of truncated TDP-43 species in cells and protects against TDP-43-mediated toxicity. Hum. Mol. Genet. 25, 3908–3924. 10.1093/hmg/ddw232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crippa V., D’Agostino V. G., Cristofani R., Rusmini P., Cicardi M. E., Messi E., et al. (2016. b). Transcriptional induction of the heat shock protein B8 mediates the clearance of misfolded proteins responsible for motor neuron diseases. Sci. Rep. 6, 22827. 10.1038/srep22827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crippa V., Sau D., Rusmini P., Boncoraglio A., Onesto E., Bolzoni E., et al. (2010. b). The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum. Mol. Genet. 19, 3440–3456. 10.1093/hmg/ddq257 [DOI] [PubMed] [Google Scholar]
- Cristofani R., Crippa V., Rusmini P., Cicardi M. E., Meroni M., Licata N. V., et al. (2017). Inhibition of retrograde transport modulates misfolded protein accumulation and clearance in motoneuron diseases. Autophagy 13, 1280–1303. 10.1080/15548627.2017.1308985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabir D. V., Trojanowski J. Q., Richter-Landsberg C., Lee V. M.-Y., Forman M. S. (2004). Expression of the small heat-shock protein αB-crystallin in tauopathies with glial pathology. Am. J. Pathol. 164, 155–166. 10.1016/S0002-9440(10)63106-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer K. M., Kranich L. R., Ruf W. P., Cagsal-Getkin O., Winslow A. R., Zhu L., et al. (2012). Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 7, 42. 10.1186/1750-1326-7-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling A. L., Liu Y., Oldfield C. J., Uversky V. N. (2018). Intrinsically disordered proteome of human membrane-less organelles. Proteomics 18, e1700193. 10.1002/pmic.201700193 [DOI] [PubMed] [Google Scholar]
- Daszynski D. M., Santhoshkumar P., Phadte A. S., Sharma K. K., Zhong H. A., Lou M. F., et al. (2019). Failure of oxysterols such as lanosterol to restore lens clarity from cataracts. Sci. Rep. 9, 1–14. 10.1038/s41598-019-44676-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong W. W., Caspers G.-J., Leunissen J. A. M. (1998). Genealogy of the α-crystallin—small heat-shock protein superfamily. Int. J. Biol. Macromol. 22, 151–162. 10.1016/S0141-8130(98)00013-0 [DOI] [PubMed] [Google Scholar]
- Deane C. A. S., Brown I. R. (2016). Induction of heat shock proteins in differentiated human neuronal cells following co-application of celastrol and arimoclomol. Cell Stress Chaperones 21, 837–848. 10.1007/s12192-016-0708-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehle F. C., Ecroyd H., Musgrave I. F., Carver J. A. (2010). αB-crystallin inhibits the cell toxicity associated with amyloid fibril formation by κ-casein and the amyloid-β peptide. Cell Stress Chaperones 15, 1013–1026. 10.1007/s12192-010-0212-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Engelsman J., Boros S., Dankers P. Y. W., Kamps B., Vree Egberts W. T., Böde C. S., et al. (2009). The Small Heat-Shock Proteins HSPB2 and HSPB3 Form Well-defined Heterooligomers in a Unique 3 to 1 Subunit Ratio. J. Mol. Biol. 393, 1022–1032. 10.1016/j.jmb.2009.08.052 [DOI] [PubMed] [Google Scholar]
- Díaz-Villanueva J. F., Díaz-Molina R., García-González V. (2015). Protein folding and mechanisms of proteostasis. Int. J. Mol. Sci. 16, 17193–17230. 10.3390/ijms160817193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson D. W., Uchikado H., Fujishiro H., Tsuboi Y. (2010). Evidence in favor of Braak staging of Parkinson’s disease. Mov. Disord. 25 Suppl 1, S78–S82. 10.1002/mds.22637 [DOI] [PubMed] [Google Scholar]
- Dobson C. M. (1999). Protein misfolding, evolution and disease. Trends Biochem. Sci. 24, 329–332. 10.1016/S0968-0004(99)01445-0 [DOI] [PubMed] [Google Scholar]
- Eaton P., Fuller W., Shattock M. J. (2002). S-thiolation of HSP27 regulates its multimeric aggregate size independently of phosphorylation. J. Biol. Chem. 277, 21189–21196. 10.1074/jbc.M200591200 [DOI] [PubMed] [Google Scholar]
- Ecroyd H. (2015). “Redefining the chaperone mechanism of sHsps: not just holdase chaperones,” in The big book on small heat shock proteins heat shock proteins. Eds. Tanguay R. M., Hightower L. E. (Cham: Springer International Publishing; ), 179–195. 10.1007/978-3-319-16077-1_7 [DOI] [Google Scholar]
- Ecroyd H., Meehan S., Horwitz J., Aquilina J. A., Benesch J. L., Robinson C. V., et al. (2007). Mimicking phosphorylation of alphaB-crystallin affects its chaperone activity. Biochem. J. 401, 129–141. 10.1042/BJ20060981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrnsperger M., Gräber S., Gaestel M., Buchner J. (1997). Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J. 16, 221–229. 10.1093/emboj/16.2.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil I. K., Malfois M., Hendle J., van der, Zandt H., Svergun D. I. (2001). A novel quaternary structure of the dimeric α-crystallin domain with chaperone-like activity. J. Biol. Chem. 276, 12024–12029. 10.1074/jbc.M010856200 [DOI] [PubMed] [Google Scholar]
- Flagmeier P., Meisl G., Vendruscolo M., Knowles T. P. J., Dobson C. M., Buell A. K., et al. (2016). Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proc. Natl. Acad. Sci. U.S.A. 113, 10328–10333. 10.1073/pnas.1604645113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine J.-M., Sun X., Benndorf R., Welsh M. J. (2005). Interactions of HSP22 (HSPB8) with HSP20, alphaB-crystallin, and HSPB3. Biochem. Biophys. Res. Commun. 337, 1006–1011. 10.1016/j.bbrc.2005.09.148 [DOI] [PubMed] [Google Scholar]
- Fontaine S. N., Zheng D., Sabbagh J. J., Martin M. D., Chaput D., Darling A., et al. (2016). DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 35, 1537–1549. 10.15252/embj.201593489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franck E., Madsen O., van Rheede T., Ricard G., Huynen M. A., de Jong W. W. (2004). Evolutionary diversity of vertebrate small heat shock proteins. J. Mol. Evol. 59, 792–805. 10.1007/s00239-004-0013-z [DOI] [PubMed] [Google Scholar]
- Freilich R., Betegon M., Tse E., Mok S.-A., Julien O., Agard D. A., et al. (2018). Competing protein–protein interactions regulate binding of Hsp27 to its client protein tau. Nat. Commun. 9, 4563. 10.1038/s41467-018-07012-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H., Possenti A., Freer R., Nakano Y., Villegas N. C. H., Tang M., et al. (2019). A tau homeostasis signature is linked with the cellular and regional vulnerability of excitatory neurons to tau pathology. Nat. Neurosci. 22, 47. 10.1038/s41593-018-0298-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X. (2015). “Insights into how small heat shock proteins bind a great diversity of substrate proteins: a super-transformer model,” in The big book on small heat shock proteins heat shock proteins. Eds. Tanguay R. M., Hightower L. E. (Cham: Springer International Publishing; ), 101–117. 10.1007/978-3-319-16077-1_4 [DOI] [Google Scholar]
- Fuchs M., Poirier D. J., Seguin S. J., Lambert H., Carra S., Charette S. J., et al. (2009). Identification of the key structural motifs involved in HspB8/HspB6-Bag3 interaction. Biochem. J. 425, 245–255. 10.1042/BJ20090907 [DOI] [PubMed] [Google Scholar]
- Gamerdinger M., Hajieva P., Behl C. (2009. a). A switch from BAG1 to BAG3 during ageing triggers the enhanced use of the autophagic-lysosomal system for the degradation of polyubiquitinated proteins. FASEB J. 23, 668, 1,–668. 1. [Google Scholar]
- Gamerdinger M., Hajieva P., Kaya A. M., Wolfrum U., Hartl F. U., Behl C. (2009. b). Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 28, 889–901. 10.1038/emboj.2009.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan N., Wu Y.-C., Brunet M., Garrido C., Chung F.-L., Dai C., et al. (2010). Sulforaphane activates heat shock response and enhances proteasome activity through up-regulation of Hsp27. J. Biol. Chem. 285, 35528–35536. 10.1074/jbc.M110.152686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganassi M., Mateju D., Bigi I., Mediani L., Poser I., Lee H. O., et al. (2016). A surveillance function of the HSPB8-BAG3-HSP70 chaperone complex ensures stress granule integrity and dynamism. Mol. Cell. 63, 796–810. 10.1016/j.molcel.2016.07.021 [DOI] [PubMed] [Google Scholar]
- Gangalum R. K., Atanasov I. C., Zhou Z. H., Bhat S. P. (2011). AlphaB-crystallin is found in detergent-resistant membrane microdomains and is secreted via exosomes from human retinal pigment epithelial cells. J. Biol. Chem. 286, 3261–3269. 10.1074/jbc.M110.160135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons G. S., Lee V. M. Y., Trojanowski J. Q. (2019). Mechanisms of cell-to-cell transmission of pathological tau: a review. JAMA Neurol. 76, 101–108. 10.1001/jamaneurol.2018.2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gober M. D., Smith C. C., Ueda K., Toretsky J. A., Aurelian L. (2003). Forced expression of the H11 heat shock protein can be regulated by DNA methylation and trigger apoptosis in human Cells. J. Biol. Chem. 278, 37600–37609. 10.1074/jbc.M303834200 [DOI] [PubMed] [Google Scholar]
- Golenhofen N., Bartelt-Kirbach B. (2016). The impact of small heat shock proteins (HspBs) in Alzheimer’s and other neurological diseases. Curr. Pharm. Des. 22, 4050–4062. 10.2174/1381612822666160519113339 [DOI] [PubMed] [Google Scholar]
- Gomes E., Shorter J. (2019). The molecular language of membraneless organelles. J. Biol. Chem. 294, 7115–7127. 10.1074/jbc.TM118.001192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter R. P., Stephenson J., Nutma E., Anink J., de Jonge J. C., Baron W., et al. (2018). Rapidly progressive amyotrophic lateral sclerosis is associated with microglial reactivity and small heat shock protein expression in reactive astrocytes. Neuropathol. Appl. Neurobiol. 45, 459–475 10.1111/nan.12525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz J., Halliday G., Nisbet R. M. (2019). Molecular pathogenesis of the tauopathies. Annu. Rev. Pathol.: Mech. Dis. 14, 239–261. 10.1146/annurev-pathmechdis-012418-012936 [DOI] [PubMed] [Google Scholar]
- Grousl T., Ungelenk S., Miller S., Ho C.-T., Khokhrina M., Mayer M. P., et al. (2018). A prion-like domain in Hsp42 drives chaperone-facilitated aggregation of misfolded proteins. J. Cell Biol. 217, 1269–1285. 10.1083/jcb.201708116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti G., Brambilla L., Rossi D. (2017). Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol. Sci. 38, 406–424. 10.1016/j.tips.2017.01.003 [DOI] [PubMed] [Google Scholar]
- Guix F. X., Corbett G. T., Cha D. J., Mustapic M., Liu W., Mengel D., et al. (2018). Detection of aggregation-competent tau in neuron-derived extracellular vesicles. Int. J. Mol. Sci. 19, 663. 10.3390/ijms19030663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley D. A., Bova M. P., Huang Q.-L., Mchaourab H. S., Stewart P. L. (2000). Small heat-shock protein structures reveal a continuum from symmetric to variable assemblies11Edited by W. Baumeister. J. Mol. Biol. 298, 261–272. 10.1006/jmbi.2000.3657 [DOI] [PubMed] [Google Scholar]
- Hansen C., Angot E., Bergström A.-L., Steiner J. A., Pieri L., Paul G., et al. (2011). α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 121, 715–725. 10.1172/JCI43366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargitai J., Lewis H., Boros I., Rácz T., Fiser A., Kurucz I., et al. (2003). Bimoclomol, a heat shock protein co-inducer, acts by the prolonged activation of heat shock factor-1. Biochem. Biophys. Res. Commun. 307, 689–695. 10.1016/S0006-291X(03)01254-3 [DOI] [PubMed] [Google Scholar]
- Harrison A. F., Shorter J. (2017). RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 474, 1417–1438. 10.1042/BCJ20160499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl F. U., Bracher A., Hayer-Hartl M. (2011). Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332. 10.1038/nature10317 [DOI] [PubMed] [Google Scholar]
- Haslbeck M., Franzmann T., Weinfurtner D., Buchner J. (2005). Some like it hot: the structure and function of small heat-shock proteins. Nat. Struct. Mol. Biol. 12, 842–846. 10.1038/nsmb993 [DOI] [PubMed] [Google Scholar]
- Haslbeck M., Vierling E. (2015). A first line of stress defense: small heat shock proteins and their function in protein homeostasis. J. Mol. Biol. 427, 1537–1548. 10.1016/j.jmb.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslbeck M., Weinkauf S., Buchner J. (2015). “Regulation of the chaperone function of small Hsps,” in The big book on small heat shock proteins heat shock proteins. Eds. Tanguay R. M., Hightower L. E. (Cham: Springer International Publishing; ), 155–178. 10.1007/978-3-319-16077-1_6 [DOI] [Google Scholar]
- Haslbeck M., Weinkauf S., Buchner J. (2019). Small heat shock proteins: Simplicity meets complexity. J. Biol. Chem. 294, 2121–2132. 10.1074/jbc.REV118.002809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatters D. M., Lindner R. A., Carver J. A., Howlett G. J. (2001). The molecular chaperone, alpha-crystallin, inhibits amyloid formation by apolipoprotein C-II. J. Biol. Chem. 276, 33755–33761. 10.1074/jbc.M105285200 [DOI] [PubMed] [Google Scholar]
- Hayes D., Napoli V., Mazurkie A., Stafford W. F., Graceffa P. (2009). Phosphorylation dependence of hsp27 multimeric size and molecular chaperone function. J. Biol. Chem. 284, 18801–18807. 10.1074/jbc.M109.011353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker J. G., McGarvey M. (2011). Heat shock proteins as biomarkers for the rapid detection of brain and spinal cord ischemia: a review and comparison to other methods of detection in thoracic aneurysm repair. Cell Stress Chaperones 16, 119–131. 10.1007/s12192-010-0224-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedhli N., Wang L., Wang Q., Rashed E., Tian Y., Sui X., et al. (2008). Proteasome activation during cardiac hypertrophy by the chaperone H11 Kinase/Hsp22. Cardiovasc. Res. 77, 497–505. 10.1093/cvr/cvm054 [DOI] [PubMed] [Google Scholar]
- Heirbaut M., Lermyte F., Martin E. M., Beelen S., Sobott F., Strelkov S. V., et al. (2017). Specific sequences in the N-terminal domain of human small heat-shock protein HSPB6 dictate preferential hetero-oligomerization with the orthologue HSPB1. J. Biol. Chem. 292, 9944–9957. 10.1074/jbc.M116.773515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishiya A., Salman M. N., Carra S., Kampinga H. H., Takayama S. (2011). BAG3 directly interacts with mutated alphab-crystallin to suppress its aggregation and toxicity. PLOS One 6, e16828. 10.1371/journal.pone.0016828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg G. K. A., Ecroyd H., Liu C., Cox D., Cascio D., Sawaya M. R., et al. (2014). The structured core domain of αB-crystallin can prevent amyloid fibrillation and associated toxicity. Proc. Natl. Acad. Sci. U. S. A. 111, E1562–E1570. 10.1073/pnas.1322673111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann J. H., Linke K., Graf P. C., Lilie H., Jakob U. (2004). Identification of a redox-regulated chaperone network. EMBO J. 23, 160–168. 10.1038/sj.emboj.7600016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz J. (1992). Alpha-crystallin can function as a molecular chaperone. Proc. Natl. Acad. Sci. U.S.A. 89, 10449–10453. 10.1073/pnas.89.21.10449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz J. (2009). Alpha crystallin: The quest for a homogeneous quaternary structure. Exp. Eye Res. 88, 190–194. 10.1016/j.exer.2008.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H., Kamei K., Iwamoto I., Inaguma Y., Nohara D., Kato K. (2001). Phosphorylation-induced change of the oligomerization state of αB-crystallin. J. Biol. Chem. 276, 5346–5352. 10.1074/jbc.M009004200 [DOI] [PubMed] [Google Scholar]
- Ito H., Okamoto K., Nakayama H., Isobe T., Kato K. (1997). Phosphorylation of alphaB-crystallin in response to various types of stress. J. Biol. Chem. 272, 29934–29941. 10.1074/jbc.272.47.29934 [DOI] [PubMed] [Google Scholar]
- Iwaki T., Wisniewski T., Iwaki A., Corbin E., Tomokane N., Tateishi J., et al. (1992). Accumulation of alpha B-crystallin in central nervous system glia and neurons in pathologic conditions. Am. J. Pathol. 140, 345–356. [PMC free article] [PubMed] [Google Scholar]
- Jaya N., Garcia V., Vierling E. (2009). Substrate binding site flexibility of the small heat shock protein molecular chaperones. Proc. Natl. Acad. Sci. U.S.A. 106, 15604–15609. 10.1073/pnas.0902177106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jehle S., Rajagopal P., Bardiaux B., Markovic S., Kühne R., Stout J. R., et al. (2010). Solid-state NMR and SAXS studies provide a structural basis for the activation of αB-crystallin oligomers. Nat. Struct. Mol. Biol. 17, 1037–1042. 10.1038/nsmb.1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jehle S., Vollmar B. S., Bardiaux B., Dove K. K., Rajagopal P., Gonen T., et al. (2011). N-terminal domain of αB-crystallin provides a conformational switch for multimerization and structural heterogeneity. Proc. Natl. Acad. Sci. U. S. A. 108, 6409–6414. 10.1073/pnas.1014656108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L., Ash P. E. A., Maziuk B. F., Ballance H. I., Boudeau S., Abdullatif A. A., et al. (2019). TIA1 regulates the generation and response to toxic tau oligomers. Acta Neuropathol. 137, 259–277. 10.1007/s00401-018-1937-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovcevski B., Kelly M. A., Rote A. P., Berg T., Gastall H. Y., Benesch J. L. P., et al. (2015). Phosphomimics destabilize Hsp27 oligomeric assemblies and enhance chaperone activity. Chem. Biol. 22, 186–195. 10.1016/j.chembiol.2015.01.001 [DOI] [PubMed] [Google Scholar]
- Kampinga H. H., Hageman J., Vos M. J., Kubota H., Tanguay R. M., Bruford E. A., et al. (2009). Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 14, 105–111. 10.1007/s12192-008-0068-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappé G., Franck E., Verschuure P., Boelens W. C., Leunissen J. A. M., de Jong W. W. (2003). The human genome encodes 10 alpha-crystallin-related small heat shock proteins: HspB1-10. Cell Stress Chaperones 8, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasza Á., Hunya Á., Frank Z., Fülöp F., Török Z., Balogh G., et al. (2016). Dihydropyridine derivatives modulate heat shock responses and have a neuroprotective effect in a transgenic mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 53, 557–571. 10.3233/JAD-150860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K., Shinohara H., Goto S., Inaguma Y., Morishita R., Asano T. (1992). Copurification of small heat shock protein with alpha B crystallin from human skeletal muscle. J. Biol. Chem. 267, 7718–7725. [PubMed] [Google Scholar]
- Katsinelos T., Zeitler M., Dimou E., Karakatsani A., Müller H.-M., Nachman E., et al. (2018). Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep. 23, 2039–2055. 10.1016/j.celrep.2018.04.056 [DOI] [PubMed] [Google Scholar]
- Kim K. K., Kim R., Kim S. H. (1998). Crystal structure of a small heat-shock protein. Nature 394, 595–599. 10.1038/29106 [DOI] [PubMed] [Google Scholar]
- Kirbach B. B., Golenhofen N. (2011). Differential expression and induction of small heat shock proteins in rat brain and cultured hippocampal neurons. J. Neurosci. Res. 89, 162–175. 10.1002/jnr.22536 [DOI] [PubMed] [Google Scholar]
- Klaips C. L., Jayaraj G. G., Hartl F. U. (2018). Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 217, 51–63. 10.1083/jcb.201709072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein T., Eckhard U., Dufour A., Solis N., Overall C. M. (2018). Proteolytic cleavage-mechanisms, function, and “omic” approaches for a near-ubiquitous posttranslational modification. Chem. Rev. 118, 1137–1168. 10.1021/acs.chemrev.7b00120 [DOI] [PubMed] [Google Scholar]
- Klimek C., Kathage B., Wördehoff J., Höhfeld J. (2017). BAG3-mediated proteostasis at a glance. J. Cell Sci. 130, 2781–2788. 10.1242/jcs.203679 [DOI] [PubMed] [Google Scholar]
- Konno T., Ross O. A., Puschmann A., Dickson D. W., Wszolek Z. K. (2016). Autosomal dominant Parkinson’s disease caused by SNCA duplications. Parkinsonism Relat. Disord. 22 Suppl 1, S1–S6. 10.1016/j.parkreldis.2015.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtis N., Tavernarakis N. (2018). Small heat shock proteins and neurodegeneration: recent developments. Biomol. Concepts 9, 94–102. 10.1515/bmc-2018-0009 [DOI] [PubMed] [Google Scholar]
- Kriehuber T., Rattei T., Weinmaier T., Bepperling A., Haslbeck M., Buchner J. (2010). Independent evolution of the core domain and its flanking sequences in small heat shock proteins. FASEB J. 24, 3633–3642. 10.1096/fj.10-156992 [DOI] [PubMed] [Google Scholar]
- Krishnan J., Vannuvel K., Andries M., Waelkens E., Robberecht W., Van Den Bosch L. (2008). Over-expression of Hsp27 does not influence disease in the mutant SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Neurochem. 106, 2170–2183. 10.1111/j.1471-4159.2008.05545.x [DOI] [PubMed] [Google Scholar]
- Kulig M., Ecroyd H. (2012). The small heat-shock protein αB-crystallin uses different mechanisms of chaperone action to prevent the amorphous versus fibrillar aggregation of α-lactalbumin. Biochem. J. 448, 343–352. 10.1042/BJ20121187 [DOI] [PubMed] [Google Scholar]
- Kumano M., Furukawa J., Shiota M., Zardan A., Zhang F., Beraldi E., et al. (2012). Cotargeting stress-activated Hsp27 and autophagy as a combinatorial strategy to amplify endoplasmic reticular stress in prostate cancer. Mol. Cancer Ther. 11, 1661–1671 10.1158/1535-7163.MCT-12-0072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganowsky A., Benesch J. L. P., Landau M., Ding L., Sawaya M. R., Cascio D., et al. (2010). Crystal structures of truncated alphaA and alphaB crystallins reveal structural mechanisms of polydispersity important for eye lens function. Protein Sci 19, 1031–1043. 10.1002/pro.380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert H., Charette S. J., Bernier A. F., Guimond A., Landry J. (1999). HSP27 multimerization mediated by phosphorylation-sensitive intermolecular interactions at the amino terminus. J. Biol. Chem. 274, 9378–9385. 10.1074/jbc.274.14.9378 [DOI] [PubMed] [Google Scholar]
- Lee J., Xu Y., Zhang T., Cui L., Saidi L., Ye Y. (2018). Secretion of misfolded cytosolic proteins from mammalian cells is independent of chaperone-mediated autophagy. J. Biol. Chem. 293, 14359–14370. 10.1074/jbc.RA118.003660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-H., Gao J., Kosinski P. A., Elliman S. J., Hughes T. E., Gromada J., et al. (2013). Heat shock protein 90 (HSP90) inhibitors activate the heat shock factor 1 (HSF1) stress response pathway and improve glucose regulation in diabetic mice. Biochem. Biophys. Res. Commun. 430, 1109–1113. 10.1016/j.bbrc.2012.12.029 [DOI] [PubMed] [Google Scholar]
- Lee S., Carson K., Rice-Ficht A., Good T. (2006). Small heat shock proteins differentially affect Aβ aggregation and toxicity. Biochem. Biophys. Res. Commun. 347, 527–533. 10.1016/j.bbrc.2006.06.128 [DOI] [PubMed] [Google Scholar]
- Lee Y.-J., Lee H.-J., Choi S.-H., Jin Y. B., An H. J., Kang J.-H., et al. (2012). Soluble HSPB1 regulates VEGF-mediated angiogenesis through their direct interaction. Angiogenesis 15, 229–242. 10.1007/s10456-012-9255-3 [DOI] [PubMed] [Google Scholar]
- Lei Z., Brizzee C., Johnson G. V. W. (2015). BAG3 facilitates the clearance of endogenous tau in primary neurons. Neurobiol. Aging 36, 241–248. 10.1016/j.neurobiolaging.2014.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelj-Garolla B., Mauk A. G. (2006). Self-association and chaperone activity of Hsp27 are thermally activated. J. Biol. Chem. 281, 8169–8174. 10.1074/jbc.M512553200 [DOI] [PubMed] [Google Scholar]
- Leroux M. R., Melki R., Gordon B., Batelier G., Candido E. P. M. (1997). Structure-function studies on small heat shock protein oligomeric assembly and interaction with unfolded polypeptides. J. Biol. Chem. 272, 24646–24656. 10.1074/jbc.272.39.24646 [DOI] [PubMed] [Google Scholar]
- Leyns C. E. G., Holtzman D. M. (2017). Glial contributions to neurodegeneration in tauopathies. Mol. Neurodegener. 12, 50. 10.1186/s13024-017-0192-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R., Zhu Z., Reiser G. (2012). Specific phosphorylation of αA-crystallin is required for the αA-crystallin-induced protection of astrocytes against staurosporine and C2-ceramide toxicity. Neurochem. Int. 60, 652–658. 10.1016/j.neuint.2012.02.031 [DOI] [PubMed] [Google Scholar]
- Liang J. J.-N. (2000). Interaction between β-amyloid and lens αB-crystallin. FEBS Lett. 484, 98–101. 10.1016/S0014-5793(00)02136-0 [DOI] [PubMed] [Google Scholar]
- Lindner R. A., Carver J. A., Ehrnsperger M., Buchner J., Esposito G., Behlke J., et al. (2000). Mouse Hsp25, a small heat shock protein. Eur. J. Biochem. 267, 1923–1932. 10.1046/j.1432-1327.2000.01188.x [DOI] [PubMed] [Google Scholar]
- Liu G.-S., Zhu H., Cai W.-F., Wang X., Jiang M., Essandoh K., et al. (2018). Regulation of BECN1-mediated autophagy by HSPB6: Insights from a human HSPB6S10F mutant. Autophagy 14, 80–97. 10.1080/15548627.2017.1392420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zhou Q., Tang M., Fu N., Shao W., Zhang S., et al. (2015). Upregulation of alphaB-crystallin expression in the substantia nigra of patients with Parkinson’s disease. Neurobiol. Aging 36, 1686–1691. 10.1016/j.neurobiolaging.2015.01.015 [DOI] [PubMed] [Google Scholar]
- Lu S., Guo Y., Liang P., Zhang S., Yin S., Yin Y., et al. (2019). Suppression of astrocytic autophagy by αB-crystallin contributes to α-synuclein inclusion formation. Transl. Neurodegeneration 8, 3. 10.1186/s40035-018-0143-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüders J., Demand J., Höhfeld J. (2000). The ubiquitin-related BAG-1 provides a link between the molecular chaperones Hsc70/Hsp70 and the proteasome. J. Biol. Chem. 275, 4613–4617. 10.1074/jbc.275.7.4613 [DOI] [PubMed] [Google Scholar]
- Mainz A., Peschek J., Stavropoulou M., Back K. C., Bardiaux B., Asami S., et al. (2015). The chaperone αB-crystallin uses different interfaces to capture an amorphous and an amyloid client. Nat. Struct. Mol. Biol. 22, 898–905. 10.1038/nsmb.3108 [DOI] [PubMed] [Google Scholar]
- Makley L. N., McMenimen K. A., DeVree B. T., Goldman J. W., McGlasson B. N., Rajagopal P., et al. (2015). Pharmacological chaperone for α-crystallin partially restores transparency in cataract models. Science 350, 674–677. 10.1126/science.aac9145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino M., Papa S., Crippa V., Nardo G., Peviani M., Cheroni C., et al. (2015). Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol. Aging 36, 492–504. 10.1016/j.neurobiolaging.2014.06.026 [DOI] [PubMed] [Google Scholar]
- Martí M. J., Tolosa E., Campdelacreu J. (2003). Clinical overview of the synucleinopathies. Mov. Disord. 18 Suppl 6, S21–S27. 10.1002/mds.10559 [DOI] [PubMed] [Google Scholar]
- Mattoo R. U. H., Goloubinoff P. (2014). Molecular chaperones are nanomachines that catalytically unfold misfolded and alternatively folded proteins. Cell Mol. Life Sci. 71, 3311–3325. 10.1007/s00018-014-1627-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald E. T., Bortolus M., Koteiche H. A., Mchaourab H. S. (2012). Sequence, structure, and dynamic determinants of Hsp27 (HspB1) equilibrium dissociation are encoded by the N-terminal domain. Biochemistry 51, 1257–1268. 10.1021/bi2017624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGreal R. S., Kantorow W. L., Chauss D. C., Wei J., Brennan L. A., Kantorow M. (2012). αB-crystallin/sHSP protects cytochrome c and mitochondrial function against oxidative stress in lens and retinal cells. Biochem. Biophys. Acta 1820, 921–930. 10.1016/j.bbagen.2012.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mchaourab H. S., Godar J. A., Stewart P. L. (2009). Structure and mechanism of protein stability sensors: the chaperone activity of small heat-shock proteins. Biochemistry 48, 3828–3837. 10.1021/bi900212j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlen P., Schulze-Osthoff K., Arrigo A.-P. (1996). Small stress proteins as novel regulators of apoptosis heat shock protein 27 blocks fas/apo-1- and staurosporine-induced cell death. J. Biol. Chem. 271, 16510–16514. 10.1074/jbc.271.28.16510 [DOI] [PubMed] [Google Scholar]
- Menzies F. M., Fleming A., Rubinsztein D. C. (2015). Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 16, 345–357. 10.1038/nrn3961 [DOI] [PubMed] [Google Scholar]
- Minoia M., Boncoraglio A., Vinet J., Morelli F. F., Brunsting J. F., Poletti A., et al. (2014). BAG3 induces the sequestration of proteasomal clients into cytoplasmic puncta: implications for a proteasome-to-autophagy switch. Autophagy 10, 1603–1621. 10.4161/auto.29409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok S.-A., Condello C., Freilich R., Gillies A., Arhar T., Oroz J., et al. (2018). Mapping interactions with the chaperone network reveals factors that protect against tau aggregation. Nat. Struct. Mol. Biol. 25, 384–393. 10.1038/s41594-018-0057-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A., Temirov J., Lee J., Coughlin M., Kanagaraj A. P., Kim H. J., et al. (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133. 10.1016/j.cell.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli F. F., Verbeek D. S., Bertacchini J., Vinet J., Mediani L., Marmiroli S., et al. (2017). Aberrant compartment formation by HSPB2 mislocalizes lamin A and compromises nuclear integrity and function. Cell Rep. 20, 2100–2115. 10.1016/j.celrep.2017.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison L. E., Whittaker R. J., Klepper R. E., Wawrousek E. F., Glembotski C. C. (2004). Roles for alphaB-crystallin and HSPB2 in protecting the myocardium from ischemia-reperfusion-induced damage in a KO mouse model. Am. J. Physiol. Heart Circ. Physiol. 286, H847–H855. 10.1152/ajpheart.00715.2003 [DOI] [PubMed] [Google Scholar]
- Mymrikov E. V., Daake M., Richter B., Haslbeck M., Buchner J. (2017). The chaperone activity and substrate spectrum of human small heat shock proteins. J. Biol. Chem. 292, 672–684. 10.1074/jbc.M116.760413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mymrikov E. V., Seit-Nebi A. S., Gusev N. B. (2011). Large potentials of small heat shock proteins. Physiol. Rev. 91, 1123–1159. 10.1152/physrev.00023.2010 [DOI] [PubMed] [Google Scholar]
- Mymrikov E. V., Seit-Nebi A. S., Gusev N. B. (2012). Heterooligomeric complexes of human small heat shock proteins. Cell Stress Chaperones 17, 157–169. 10.1007/s12192-011-0296-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj R. H., Oya-Ito T., Padayatti P. S., Kumar R., Mehta S., West K., et al. (2003). Enhancement of chaperone function of alpha-crystallin by methylglyoxal modification. Biochemistry 42, 10746–10755. 10.1021/bi034541n [DOI] [PubMed] [Google Scholar]
- Naldini L. (2015). Gene therapy returns to centre stage. Nature 526, 351–360. 10.1038/nature15818 [DOI] [PubMed] [Google Scholar]
- Narayanan S., Kamps B., Boelens W. C., Reif B. (2006). αB-crystallin competes with Alzheimer’s disease β-amyloid peptide for peptide–peptide interactions and induces oxidation of Abeta-Met35. FEBS Lett. 580, 5941–5946. 10.1016/j.febslet.2006.09.063 [DOI] [PubMed] [Google Scholar]
- Ojha J., Karmegam R. V., Masilamoni J. G., Terry A. V., Cashikar A. G. (2011. a). Behavioral defects in chaperone-deficient alzheimer’s disease model mice. PLoS One 6, e16550. 10.1371/journal.pone.0016550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojha J., Masilamoni G., Dunlap D., Udoff R. A., Cashikar A. G. (2011. b). Sequestration of toxic oligomers by HspB1 as a cytoprotective mechanism. Mol. Cell. Biol. 31, 3146–3157. 10.1128/MCB.01187-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira A. O., Osmand A., Outeiro T. F., Muchowski P. J., Finkbeiner S. (2016). αB-Crystallin overexpression in astrocytes modulates the phenotype of the BACHD mouse model of Huntington’s disease. Hum. Mol. Genet. 25, 1677–1689. 10.1093/hmg/ddw028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshita S. E., Chen F., Kwan T., Yehiely F., Cryns V. L. (2010). The small heat shock protein HspB2 is a novel anti-apoptotic protein that inhibits apical caspase activation in the extrinsic apoptotic pathway. Breast Cancer Res. Treat. 124, 307–315. 10.1007/s10549-010-0735-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papuć E., Krupski W., Kurys-Denis E., Rejdak K. (2016). Antibodies against small heat-shock proteins in Alzheimer’s disease as a part of natural human immune repertoire or activation of humoral response? J. Neura. Transm. 123, 455–461. 10.1007/s00702-015-1477-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papuć E., Kurys-Denis E., Krupski W., Rejdak K. (2015). Humoral response against small heat shock proteins in Parkinson’s disease. PLOS One 10, e0115480. 10.1371/journal.pone.0115480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parcellier A., Brunet M., Schmitt E., Col E., Didelot C., Hammann A., et al. (2006). HSP27 favors ubiquitination and proteasomal degradation of p27Kip1 and helps S-phase re-entry in stressed cells. FASEB J. 20, 1179–1181. 10.1096/fj.05-4184fje [DOI] [PubMed] [Google Scholar]
- Parcellier A., Schmitt E., Gurbuxani S., Seigneurin-Berny D., Pance A., Chantôme A., et al. (2003). HSP27 is a ubiquitin-binding protein involved in I-κBα proteasomal degradation. Mol. Cell. Biol. 23, 5790–5802. 10.1128/MCB.23.16.5790-5802.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A., Lee H. O., Jawerth L., Maharana S., Jahnel M., Hein M. Y., et al. (2015). A Liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077. 10.1016/j.cell.2015.07.047 [DOI] [PubMed] [Google Scholar]
- Patel S., Vierling E., Tama F. (2014). Replica exchange molecular dynamics simulations provide insight into substrate recognition by small heat shock proteins. Biophys. J. 106, 2644–2655. 10.1016/j.bpj.2014.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez M., Medina M., Hernández F., Avila J. (2018). Secretion of full-length Tau or Tau fragments in cell culture models. Propagation of Tau in vivo and in vitro. Biomol. Concepts 9, 1–11. 10.1515/bmc-2018-0001 [DOI] [PubMed] [Google Scholar]
- Peschek J., Braun N., Franzmann T. M., Georgalis Y., Haslbeck M., Weinkauf S., et al. (2009). The eye lens chaperone α-crystallin forms defined globular assemblies. Proc. Natl. Acad. Sci. U. S. A. 106, 13272–13277. 10.1073/pnas.0902651106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschek J., Braun N., Rohrberg J., Back K. C., Kriehuber T., Kastenmüller A., et al. (2013). Regulated structural transitions unleash the chaperone activity of αB-crystallin. Proc. Natl. Acad. Sci. U.S.A. 110, E3780–E3789. 10.1073/pnas.1308898110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plate L., Wiseman R. L. (2017). Regulating secretory proteostasis through the unfolded protein response: from function to therapy. Trends Cell Biol. 27, 722–737. 10.1016/j.tcb.2017.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers E. T., Morimoto R. I., Dillin A., Kelly J. W., Balch W. E. (2009). Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 78, 959–991. 10.1146/annurev.biochem.052308.114844 [DOI] [PubMed] [Google Scholar]
- Powers M. V., Workman P. (2007). Inhibitors of the heat shock response: biology and pharmacology. FEBS Lett. 581, 3758–3769. 10.1016/j.febslet.2007.05.040 [DOI] [PubMed] [Google Scholar]
- Préville X., Salvemini F., Giraud S., Chaufour S., Paul C., Stepien G., et al. (1999). Mammalian small stress proteins protect against oxidative stress through their ability to increase glucose-6-phosphate dehydrogenase activity and by maintaining optimal cellular detoxifying machinery. Exp. Cell Res. 247, 61–78. 10.1006/excr.1998.4347 [DOI] [PubMed] [Google Scholar]
- Quraishe S., Asuni A., Boelens W. C., O’Connor V., Wyttenbach A. (2008). Expression of the small heat shock protein family in the mouse CNS: differential anatomical and biochemical compartmentalization. Neuroscience 153, 483–491. 10.1016/j.neuroscience.2008.01.058 [DOI] [PubMed] [Google Scholar]
- Raju M., Santhoshkumar P., Krishna K. S. (2016). Alpha-crystallin-derived peptides as therapeutic chaperones. Biochim. Biophys. Acta 1860, 246–251. 10.1016/j.bbagen.2015.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju M., Santhoshkumar P., Sharma K. K. (2018). Cell-penetrating chaperone peptide prevents protein aggregation and protects against cell apoptosis. Adv. Biosyst. 2, 1700095. 10.1002/adbi.201700095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch J. N., Tse E., Freilich R., Mok S.-A., Makley L. N., Southworth D. R., et al. (2017). BAG3 is a modular, scaffolding protein that physically links heat shock protein 70 (Hsp70) to the small heat shock proteins. J. Mol. Biol. 429, 128–141. 10.1016/j.jmb.2016.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner K., Chen Y.-X., McNulty M., Simard T., Zhao X., Wells D. J., et al. (2008). Extracellular release of the atheroprotective heat shock protein 27 is mediated by estrogen and competitively inhibits acLDL binding to scavenger receptor-A. Circ. Res. 103, 133–141. 10.1161/CIRCRESAHA.108.172155 [DOI] [PubMed] [Google Scholar]
- Reddy V. S., Madala S. K., Trinath J., Reddy G. B. (2018). Extracellular small heat shock proteins: exosomal biogenesis and function. Cell Stress Chaperones 23, 441–454. 10.1007/s12192-017-0856-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekas A., Adda C. G., Andrew Aquilina J., Barnham K. J., Sunde M., Galatis D., et al. (2004). Interaction of the molecular chaperone alphaB-crystallin with alpha-synuclein: effects on amyloid fibril formation and chaperone activity. J. Mol. Biol. 340, 1167–1183. 10.1016/j.jmb.2004.05.054 [DOI] [PubMed] [Google Scholar]
- Remondelli P., Renna M. (2017). The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Front. Mol. Neurosci. 10, 187. 10.3389/fnmol.2017.00187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renkawek K., Stege G. J., Bosman G. J. (1999). Dementia, gliosis and expression of the small heat shock proteins hsp27 and αB-crystallin in Parkinson’s disease. NeuroReport 10, 2273. 10.1097/00001756-199908020-00009 [DOI] [PubMed] [Google Scholar]
- Robertson A. L., Headey S. J., Saunders H. M., Ecroyd H., Scanlon M. J., Carver J. A., et al. (2010). Small heat-shock proteins interact with a flanking domain to suppress polyglutamine aggregation. PNAS 107, 10424–10429. 10.1073/pnas.0914773107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusmini P., Cristofani R., Galbiati M., Cicardi M. E., Meroni M., Ferrari V., et al. (2017). The role of the heat shock protein B8 (HSPB8) in motoneuron diseases. Front. Mol. Neurosci. 10, 176. 10.3389/fnmol.2017.00176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahasrabudhe P., Rohrberg J., Biebl M. M., Rutz D. A., Buchner J. (2017). The plasticity of the Hsp90 co-chaperone system. Mol. Cell 67, 947–961.e5. 10.1016/j.molcel.2017.08.004 [DOI] [PubMed] [Google Scholar]
- Santhanagopalan I., Degiacomi M. T., Shepherd D. A., Hochberg G. K. A., Benesch J. L. P., Vierling E. (2018). It takes a dimer to tango: Oligomeric small heat shock proteins dissociate to capture substrate. J. Biol. Chem. 293, 19511–19521. 10.1074/jbc.RA118.005421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardar Sinha M., Ansell-Schultz A., Civitelli L., Hildesjö C., Larsson M., Lannfelt L., et al. (2018). Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 136, 41–56. 10.1007/s00401-018-1868-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt E., Gehrmann M., Brunet M., Multhoff G., Garrido C. (2007). Intracellular and extracellular functions of heat shock proteins: repercussions in cancer therapy. J. Leukoc. Biol. 81, 15–27. 10.1189/jlb.0306167 [DOI] [PubMed] [Google Scholar]
- Schwarz L., Vollmer G., Richter-Landsberg C. (2010). The small heat shock protein HSP25/27 (HspB1) is abundant in cultured astrocytes and associated with astrocytic pathology in progressive supranuclear palsy and corticobasal degeneration. Int. J. Cell Biol. 2010, 717520. 10.1155/2010/717520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel K., Vinet J., Dunnen W. F. A., Brunt E. R., Meister M., Boncoraglio A., et al. (2012). The HSPB8-BAG3 chaperone complex is upregulated in astrocytes in the human brain affected by protein aggregation diseases. Neuropathol. Appl. Neurobiol. 38, 39–53. 10.1111/j.1365-2990.2011.01198.x [DOI] [PubMed] [Google Scholar]
- Seong S.-Y., Matzinger P. (2004). Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat.Rev. Immunol. 4, 469. 10.1038/nri1372 [DOI] [PubMed] [Google Scholar]
- Shammas S. L., Waudby C. A., Wang S., Buell A. K., Knowles T. P. J., Ecroyd H., et al. (2011). Binding of the molecular chaperone αB-Crystallin to Aβ amyloid fibrils inhibits fibril elongation. Biophys. J. 101, 1681–1689. 10.1016/j.bpj.2011.07.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K. K., Kaur H., Kester K. (1997). Functional elements in molecular chaperone alpha-crystallin: identification of binding sites in alpha B-crystallin. Biochem. Biophys. Res. Commun. 239, 217–222. 10.1006/bbrc.1997.7460 [DOI] [PubMed] [Google Scholar]
- Sharp P. S., Akbar M. T., Bouri S., Senda A., Joshi K., Chen H.-J., et al. (2008). Protective effects of heat shock protein 27 in a model of ALS occur in the early stages of disease progression. Neurobiol. Dis. 30, 42–55. 10.1016/j.nbd.2007.12.002 [DOI] [PubMed] [Google Scholar]
- Shashidharamurthy R., Koteiche H. A., Dong J., Mchaourab H. S. (2005). Mechanism of chaperone function in small heat shock proteins dissociation of the hsp27 oligomer is required for recognition and binding of destabilized t4 lysozyme. J. Biol. Chem. 280, 5281–5289. 10.1074/jbc.M407236200 [DOI] [PubMed] [Google Scholar]
- Shelkovnikova T. A., Robinson H. K., Southcombe J. A., Ninkina N., Buchman V. L. (2014). Multistep process of FUS aggregation in the cell cytoplasm involves RNA-dependent and RNA-independent mechanisms. Hum. Mol. Genet. 23, 5211–5226. 10.1093/hmg/ddu243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton L. B., Baker J. D., Zheng D., Sullivan L. E., Solanki P. K., Webster J. M., et al. (2017). Hsp90 activator Aha1 drives production of pathological tau aggregates. Proc. Natl. Acad. Sci. U.S.A. 114, 9707–9712. 10.1073/pnas.1707039114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemetov A. A., Seit-Nebi A. S., Gusev N. B. (2011). Phosphorylation of human small heat shock protein HspB8 (Hsp22) by ERK1 protein kinase. Mol. Cell. Biochem. 355, 47–55. 10.1007/s11010-011-0837-y [DOI] [PubMed] [Google Scholar]
- Shi J., Koteiche H. A., Mchaourab H. S., Stewart P. L. (2006). Cryoelectron microscopy and EPR analysis of engineered symmetric and polydisperse Hsp16.5 assemblies reveals determinants of polydispersity and substrate binding. J. Biol. Chem. 281, 40420–40428. 10.1074/jbc.M608322200 [DOI] [PubMed] [Google Scholar]
- Shimura H., Miura-Shimura Y., Kosik K. S. (2004). Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival. J. Biol. Chem. 279, 17957–17962. 10.1074/jbc.M400351200 [DOI] [PubMed] [Google Scholar]
- Simonato M., Bennett J., Boulis N. M., Castro M. G., Fink D. J., Goins W. F., et al. (2013). Progress in gene therapy for neurological disorders. Nat. Rev. Neurol. 9, 277–291. 10.1038/nrneurol.2013.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski G., Finkbeiner S. (2013). Longitudinal measures of proteostasis in live neurons: features that determine fate in models of neurodegenerative disease. FEBS Lett. 587, 1139–1146. 10.1016/j.febslet.2013.02.043 [DOI] [PubMed] [Google Scholar]
- Smith H. L., Li W., Cheetham M. E. (2015). Molecular chaperones and neuronal proteostasis. Semin Cell Dev. Biol. 40, 142–152. 10.1016/j.semcdb.2015.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini M. G., Schmidt M. L., Lee V. M.-Y., Trojanowski J. Q., Jakes R., Goedert M. (1997). α-synuclein in Lewy bodies. Nature 388, 839. 10.1038/42166 [DOI] [PubMed] [Google Scholar]
- Sreekumar P. G., Kannan R., Kitamura M., Spee C., Barron E., Ryan S. J., et al. (2010). αB crystallin is apically secreted within exosomes by polarized human retinal pigment epithelium and provides neuroprotection to adjacent cells. PLoS One 5, e12578. 10.1371/journal.pone.0012578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen K. K., Dillin A. (2016). A ribosomal perspective on proteostasis and aging. Cell Metabol. 23, 1004–1012. 10.1016/j.cmet.2016.05.013 [DOI] [PubMed] [Google Scholar]
- Stengel F., Baldwin A. J., Painter A. J., Jaya N., Basha E., Kay L. E., et al. (2010). Quaternary dynamics and plasticity underlie small heat shock protein chaperone function. PNAS 107, 2007–2012. 10.1073/pnas.0910126107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromer T., Fischer E., Richter K., Haslbeck M., Buchner J. (2004). Analysis of the regulation of the molecular chaperone Hsp26 by temperature-induced dissociation the N-terminal domain is important for oligomer assembly and the binding of unfolding proteins. J. Biol. Chem. 279, 11222–11228. 10.1074/jbc.M310149200 [DOI] [PubMed] [Google Scholar]
- Stürner E., Behl C. (2017). The role of the multifunctional BAG3 protein in cellular protein quality control and in disease. Front. Mol. Neurosci. 10, 177. 10.3389/fnmol.2017.00177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudnitsyna M. V., Mymrikov E. V., Gusev N.B. and Seit-Nebi A.S. (2012). The role of intrinsically disordered regions in the structure and functioning of small heat shock proteins. Curr. Protein Pept. Sci. 13, 76–85 10.2174/138920312799277875 [DOI] [PubMed] [Google Scholar]
- Sun X., Fontaine J.-M., Rest J. S., Shelden E. A., Welsh M. J., Benndorf R. (2004). Interaction of human HSP22 (HSPB8) with other small heat shock proteins. J. Biol. Chem. 279, 2394–2402. 10.1074/jbc.M311324200 [DOI] [PubMed] [Google Scholar]
- Theillet F.-X., Binolfi A., Bekei B., Martorana A., Rose H. M., Stuiver M., et al. (2016). Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 530, 45–50. 10.1038/nature16531 [DOI] [PubMed] [Google Scholar]
- Thornell E., Aquilina A. (2015). Regulation of αA- and αB-crystallins via phosphorylation in cellular homeostasis. Cell. Mol. Life Sci. 72, 4127–4137. 10.1007/s00018-015-1996-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll-Riera M., Radó-Trilla N., Martys F., Albà M. M. (2012). Role of low-complexity sequences in the formation of novel protein coding sequences. Mol. Biol. Evol. 29, 883–886. 10.1093/molbev/msr263 [DOI] [PubMed] [Google Scholar]
- Toretsky J. A., Wright P. E. (2014). Assemblages: functional units formed by cellular phase separation. J. Cell Biol. 206, 579–588. 10.1083/jcb.201404124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortosa E., Santa-Maria I., Moreno F., Lim F., Perez M., Avila J. (2009). Binding of Hsp90 to tau promotes a conformational change and aggregation of tau protein. J. Alzheimers Dis. 17, 319–325. 10.3233/JAD-2009-1049 [DOI] [PubMed] [Google Scholar]
- Trajkovic K., Jeong H., Krainc D. (2017). Mutant huntingtin is secreted via a late endosomal/lysosomal unconventional secretory pathway. J. Neurosci. 37, 9000–9012. 10.1523/JNEUROSCI.0118-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treweek T. M., Meehan S., Ecroyd H., Carver J. A. (2015). Small heat-shock proteins: important players in regulating cellular proteostasis. Cell. Mol. Life Sci. 72, 429–451. 10.1007/s00018-014-1754-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tue N. T., Shimaji K., Tanaka N., Yamaguchi M. (2012). Effect of αB-crystallin on protein aggregation in drosophila. BioMed. Res. Int. 2012, 252049 10.1155/2012/252049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- Ungelenk S., Moayed F., Ho C.-T., Grousl T., Scharf A., Mashaghi A., et al. (2016). Small heat shock proteins sequester misfolding proteins in near-native conformation for cellular protection and efficient refolding. Nat. Commun. 7,. 10.1038/ncomms13673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. (2017). Protein intrinsic disorder-based liquid-liquid phase transitions in biological systems: complex coacervates and membrane-less organelles. Adv. Colloid Interface Sci. 239, 97–114. 10.1016/j.cis.2016.05.012 [DOI] [PubMed] [Google Scholar]
- van den IJssel P. R., Overkamp P., Bloemendal H., de Jong W. W. (1998). Phosphorylation of alphaB-crystallin and HSP27 is induced by similar stressors in HeLa cells. Biochem. Biophys. Res. Commun. 247, 518–523. 10.1006/bbrc.1998.8699 [DOI] [PubMed] [Google Scholar]
- van Montfort R. L., Basha E., Friedrich K. L., Slingsby C., Vierling E. (2001). Crystal structure and assembly of a eukaryotic small heat shock protein. Nat. Struct. Biol. 8, 1025–1030. 10.1038/nsb722 [DOI] [PubMed] [Google Scholar]
- Vleminckx V., Van Damme P., Goffin K., Delye H., Van Den Bosch L., Robberecht W. (2002). Upregulation of HSP27 in a transgenic model of ALS. J. Neuropathol. Exp. Neurol. 61, 968–974. 10.1093/jnen/61.11.968 [DOI] [PubMed] [Google Scholar]
- Vos M. J., Carra S., Kanon B., Bosveld F., Klauke K., Sibon O. C. M., et al. (2016). Specific protein homeostatic functions of small heat-shock proteins increase lifespan. Aging Cell 15, 217–226. 10.1111/acel.12422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M. J., Kanon B., Kampinga H. H. (2009). HSPB7 is a SC35 speckle resident small heat shock protein. Biochim. Biophys. Acta (BBA). Mol. Cell Res. 1793, 1343–1353. 10.1016/j.bbamcr.2009.05.005 [DOI] [PubMed] [Google Scholar]
- Vos M. J., Zijlstra M. P., Carra S., Sibon O. C. M., Kampinga H. H. (2011). Small heat shock proteins, protein degradation and protein aggregation diseases. Autophagy 7, 101–103. 10.4161/auto.7.1.13935 [DOI] [PubMed] [Google Scholar]
- Vos M. J., Zijlstra M. P., Kanon B., van Waarde-Verhagen M. A. W. H., Brunt E. R. P., Oosterveld-Hut H. M. J., et al. (2010). HSPB7 is the most potent polyQ aggregation suppressor within the HSPB family of molecular chaperones. Hum. Mol. Genet. 19, 4677–4693. 10.1093/hmg/ddq398 [DOI] [PubMed] [Google Scholar]
- Wan C., Borgeson B., Phanse S., Tu F., Drew K., Clark G., et al. (2015). Panorama of ancient metazoan macromolecular complexes. Nature 525, 339–344. 10.1038/nature14877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Lomakin A., Kanai S., Alex R., Benedek G. B. (2017). Liquid–liquid phase separation in oligomeric peptide solutions. Langmuir 33, 7715–7721. 10.1021/acs.langmuir.7b01693 [DOI] [PubMed] [Google Scholar]
- Waudby C. A., Knowles T. P. J., Devlin G. L., Skepper J. N., Ecroyd H., Carver J. A., et al. (2010). The interaction of alphaB-crystallin with mature alpha-synuclein amyloid fibrils inhibits their elongation. Biophys. J. 98, 843–851. 10.1016/j.bpj.2009.10.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann S., Eftekharzadeh B., Tepper K., Zoltowska K. M., Bennett R. E., Dujardin S., et al. (2018). Tau protein liquid–liquid phase separation can initiate tau aggregation. EMBO J. 37, e98049. 10.15252/embj.201798049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide S. D., Morimoto R. I. (2005). Heat shock response modulators as therapeutic tools for diseases of protein conformation. J. Biol. Chem. 280, 33097–33100. 10.1074/jbc.R500010200 [DOI] [PubMed] [Google Scholar]
- Wilhelmus M. M. M., Boelens W. C., Otte-Höller I., Kamps B., de Waal R. M. W., Verbeek M. M. (2006. a). Small heat shock proteins inhibit amyloid-β protein aggregation and cerebrovascular amyloid-β protein toxicity. Brain Res. 1089, 67–78. 10.1016/j.brainres.2006.03.058 [DOI] [PubMed] [Google Scholar]
- Wilhelmus M. M. M., Boelens W. C., Otte-Höller I., Kamps B., Kusters B., Maat-Schieman M. L. C., et al. (2006. b). Small heat shock protein HspB8: its distribution in Alzheimer’s disease brains and its inhibition of amyloid-β protein aggregation and cerebrovascular amyloid-β toxicity. Acta Neuropathol. 111, 139–149. 10.1007/s00401-005-0030-z [DOI] [PubMed] [Google Scholar]
- Wilhelmus M. M. M., Otte-Höller I., Wesseling P., Waal R. M. W. D., Boelens W. C., Verbeek M. M. (2006. c). Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer’s disease brains. Neuropathol. Appl. Neurobiol. 32, 119–130. 10.1111/j.1365-2990.2006.00689.x [DOI] [PubMed] [Google Scholar]
- Wilson C. H., Shalini S., Filipovska A., Richman T. R., Davies S., Martin S. D., et al. (2015). Age-related proteostasis and metabolic alterations in Caspase-2-deficient mice. Cell Death Dis. 6, e1597. 10.1038/cddis.2014.567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M. Y., DiChiara A. S., Suen P. H., Chen K., Doan N.-D., Shoulders M. D. (2018). Adapting secretory proteostasis and function through the unfolded protein response. Curr. Top. Microbiol. Immunol. 414, 1–25. 10.1007/82_2017_56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyttenbach A., Sauvageot O., Carmichael J., Diaz-Latoud C., Arrigo A.-P., Rubinsztein D. C. (2002). Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum. Mol. Genet. 11, 1137–1151. 10.1093/hmg/11.9.1137 [DOI] [PubMed] [Google Scholar]
- Yerbury J. J., Gower D., Vanags L., Roberts K., Lee J. A., Ecroyd H. (2013). The small heat shock proteins αB-crystallin and Hsp27 suppress SOD1 aggregation in vitro. Cell Stress Chaperones 18, 251–257. 10.1007/s12192-012-0371-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerbury J. J., Ooi L., Dillin A., Saunders D. N., Hatters D. M., Beart P. M., et al. (2016). Walking the tightrope: proteostasis and neurodegenerative disease. J. Neurochem. 137, 489–505. 10.1111/jnc.13575 [DOI] [PubMed] [Google Scholar]
- Yoshiike Y., Minai R., Matsuo Y., Chen Y.-R., Kimura T., Takashima A. (2008). Amyloid oligomer conformation in a group of natively folded proteins. PLOS One 3, e3235. 10.1371/journal.pone.0003235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A., Fox S. G., Cavallini A., Kerridge C., O’Neill M. J., Wolak J., et al. (2019). Tau protein aggregates inhibit the protein-folding and vesicular trafficking arms of the cellular proteostasis network. J. Biol. Chem. 294, 7917–7930 10.1074/jbc.RA119.007527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L., Petyuk V. A., Gaiteri C., Mostafavi S., Young-Pearse T., Shah R. C., et al. (2018). Targeted brain proteomics uncover multiple pathways to Alzheimer’s dementia. Ann. Neurol. 84, 78–88. 10.1002/ana.25266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zantema A., Vries M. V.-D., Maasdam D., Bol S., van der Eb A. (1992). Heat shock protein 27 and alpha B-crystallin can form a complex, which dissociates by heat shock. J. Biol. Chem. 267, 12936–12941. [PubMed] [Google Scholar]
- Zhang X., Wang X., Zhu H., Kranias E. G., Tang Y., Peng T., et al. (2012). Hsp20 functions as a novel cardiokine in promoting angiogenesis via activation of VEGFR2. PLoS One 7, e32765. 10.1371/journal.pone.0032765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., James M., Middleton F. A., Davis R. L. (2005). Transcriptional analysis of multiple brain regions in Parkinson’s disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am. J. Med. Genet. Part B: Neuropsychiatric Genet. 137B, 5–16. 10.1002/ajmg.b.30195 [DOI] [PubMed] [Google Scholar]
- Zhao L., Chen X.-J., Zhu J., Xi Y.-B., Yang X., Hu L.-D., et al. (2015). Lanosterol reverses protein aggregation in cataracts. Nature 523, 607–611. 10.1038/nature14650 [DOI] [PubMed] [Google Scholar]
- Zhu L., Brangwynne C. P. (2015). Nuclear bodies: the emerging biophysics of nucleoplasmic phases. Curr. Opin. Cell Biol. 34, 23–30. 10.1016/j.ceb.2015.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., Reiser G. (2018). The small heat shock proteins, especially HspB4 and HspB5 are promising protectants in neurodegenerative diseases. Neurochem. Int. 115, 69–79. 10.1016/j.neuint.2018.02.006 [DOI] [PubMed] [Google Scholar]
- Zourlidou A., Gidalevitz T., Kristiansen M., Landles C., Woodman B., Wells D. J., et al. (2007). Hsp27 overexpression in the R6/2 mouse model of Huntington’s disease: chronic neurodegeneration does not induce Hsp27 activation. Hum. Mol. Genet. 16, 1078–1090. 10.1093/hmg/ddm057 [DOI] [PubMed] [Google Scholar]