

Summary
Progenitor heterogeneity and identities underlying tumor initiation and relapse in medulloblastomas remain elusive. Utilizing single-cell transcriptomic analysis, we demonstrated a developmental hierarchy of progenitor pools in Sonic Hedgehog (SHH)-medulloblastomas, and identified OLIG2-expressing glial progenitors as transit-amplifying cells at the tumorigenic onset. Although OLIG2+ cells become quiescent stem-like progenitors in full-blown tumors, they are highly enriched in therapy-resistant and recurrent medulloblastomas. Depletion of mitotic Olig2+ progenitors or Olig2-ablation impeded tumor initiation. Genomic profiling revealed that OLIG2 modulates chromatin landscapes and activates oncogenic networks including HIPPO-YAP/TAZ and AURORA-A/MYCN pathways. Co-targeting these oncogenic pathways induced tumor growth arrest. Together, our results indicate that glial lineage-associated OLIG2+ progenitors are tumor-initiating cells during medulloblastoma tumorigenesis and relapse, suggesting OLIG2-driven oncogenic networks as potential therapeutic targets.
Graphical Abstract
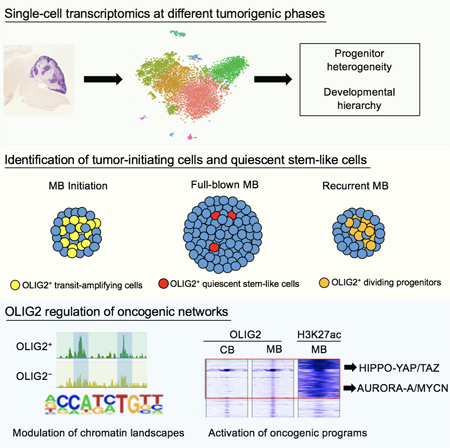
In Brief:
18–01020 Lu (Zhang et al.)
Zhang et al. show that OLIG2+ progenitor cells drive Sonic Hedgehog subtype medulloblastoma (SHH-MB) tumorigenesis in mice and are enriched in recurrent or resistant patient SHH-MB. OLIG2 activates HIPPO and AURORA-A/MYC signaling, and co-inhibition of these pathways arrests SHH-MB growth in mice.
INTRODUCTION
Medulloblastoma (MB) is the most common pediatric brain tumor in the posterior cranial fossa and accounts for approximately 25% of all pediatric brain tumors (Cavalli et al., 2017; Roussel and Hatten, 2011). Despite aggressive treatment, the prognosis for these patients is grim. Those who survive the primary tumor suffer severe side effects, and often have tumor relapse, which is generally fatal (Archer et al., 2017; Ramaswamy and Taylor, 2017).
Transcriptomic, genomic, epigenomic, and proteomic profiles indicate that human MBs are highly heterogeneous. These tumors are classified into four principal subgroups: Wingless (WNT), Sonic hedgehog (SHH), Group 3, and Group 4 (Cavalli et al., 2017; Taylor et al., 2012). Activation of the SHH signaling pathway due, for example, to loss of function of PTCH1 or gain-of-function of SMO, is detected in about 30% of human MB cases (Gilbertson and Ellison, 2008; Wang et al., 2018). GNAS, which encodes a G protein α subunit, is a tumor suppressor in a subgroup of aggressive SHH tumors (He et al., 2014). Loss-of-function mutations and low GNAS expression are associated with poor prognosis in SHH-MB patients (He et al., 2014; Huh et al., 2014).
MB tumorigenesis and recurrence are thought to be driven by a rare population of tumor initiating cells (Manoranjan et al., 2013; Wang et al., 2018); however, the identities of these cells are incompletely defined. SHH-MBs are traditionally considered neuronal progenitor derived (Roussel and Hatten, 2011; Wang et al., 2018); for instance, granule neuron progenitors (GNPs) can be transformed into cancerous cells in the cerebellum (Schuller et al., 2008; Yang et al., 2008). Although the MB tumors are biologically and anatomically distinct from other types of brain tumors, they often have characteristics of stem-like or glial progenitors (Goldberg-Stern et al., 1991; Mannoji et al., 1981). Refractory or relapsed disease likely occurs due to failure to eradicate the MB tumor stem/initiating cells (Manoranjan et al., 2013; Morrissy et al., 2016; Ramaswamy and Taylor, 2017). Recent genomic studies have yielded insight into the mutations and pathways that contribute to MB pathogenesis (Cavalli et al., 2017; Lin et al., 2016; Northcott et al., 2012; Robinson et al., 2012). Despite these advances, genetic and cellular drivers of many cases of MB are unknown, and intratumoral progenitor heterogeneity during tumorigenesis remains poorly defined at the single-cell level. It is also unclear how diverse progenitors propagate and transform into tumor cells. Here we sought to explore progenitor cell heterogeneity and identify tumor-initiating progenitors during MB tumorigenesis and recurrence.
RESULTS
Single-cell Transcriptomics Reveals a Neural Lineage Development Hierarchy in SHH-MB
Deletion of Ptch in neural progenitors leads to formation of SHH-MB in early postnatal stages in mice (Yang et al., 2008). We performed droplet-based single-cell RNA sequencing (scRNA-seq) on the 10X Genomics platform for the cells dissociated from cerebellar tumor-like tissues at post-natal day 10 (P10) of Ptchfl/fl;hGFAP-Cre (GFAP-Ptch) mice, where Ptch is deleted in hGFAP-Cre+ cerebellar progenitors (Zhuo et al., 2001). Tumorigenesis was confirmed by orthotopic transplantation assays (Figures S1A–S1D). Using principal component and t-SNE analyses, we identified nine transcriptionally distinct cell clusters (Figures 1A and 1B). Consistent with previous studies indicating that GNPs play a role in MB tumorigenesis (Schuller et al., 2008; Yang et al., 2008), GNP populations accounted for approximately 93% of the analyzed transcriptomes; we also identified GABAergic neurons (2.4%), oligodendrocytes (1.6%), glutamatergic neurons (1.3%), immune cells (0.9%), and astrocytes (0.8%) (Figures 1C and S1E).
Figure 1. Neural Lineage Developmental Hierarchy in Progressing MB.

(A) t-SNE analysis of cell clusters in GFAP-Ptch MB at P10.
(B) Heatmap of GFAP-Ptch MB cells. Columns, individual cells; rows, genes. OL, oligodendrocytes; IC, immune cells; AS, astrocytes.
(C) Relative proportions of distinct clusters in total cells.
(D) Dot plot displaying the expression level of selected marker genes in subpopulations. The size of the dot reflects the percentage of the cells that express the gene (pct.exp). Average expression levels (ave.exp.scale) of the genes are color-coded.
(E) t-SNE plots of expression of subpopulation markers.
(F) t-SNE plots of stem/progenitor-like markers.
(G) Pseudo-time ordering of distinct populations within neural cell lineages.
(H, I) The mean expression of representative genes of NSC-like, GNPs_cycling, GNPs_mature populations (H) and glia-related progenitors (I) for each tree node. Expression levels (expr) are color-coded.
See also Figures S1–S3.
Unbiased clustering with Seurat (Satija et al., 2015) subdivided the GNP populations into mitotic proliferating cells, which could be further differentiated by phases of the cell cycle, and mature post-mitotic populations (Figure 1D). The early GNP marker Atoh1 was predominantly expressed in proliferating GNPs, rather than in mature GNPs, whereas the committed neuronal lineage marker Pax6 was expressed by both proliferating and mature populations (Figures 1D and 1E). The late neuronal progenitor marker Dcx, which is not expressed in Sox2+ stem-like cells (Vanner et al., 2014), was mainly enriched in postmitotic GNP populations (Figures 1D and 1E). Consistent with previous data (Aruga et al., 1994; Yang et al., 2008), the GNP marker Zic1 was expressed in both proliferating and mature GNPs. The proliferating GNP populations were divided into three clusters depending on cell-cycle phase (Figure 1E). Postmitotic GNPs expressed high levels of mature neuronal markers such as Rbfox3, NeuroD1, and Tubb3 (Figure 1D, E), suggesting that these cells are maturing GNPs. These unbiased analyses revealed dynamic cell populations in SHH-MB that mirror neural lineage development in the cerebellum.
Lineage Trajectory Reveals OLIG2+ Progenitors in the Stem-like Population in MB
To determine the potential developmental trajectory of the neural lineage cells in tumor tissues, we performed unsupervised pseudo-time analysis using TSCAN (Ji and Ji, 2016), which links gene expression profiles with developmental stages. In the stem-like cells, we detected Prom1 (which encodes for CD133), Sox2, and Nes, which have been associated with stem cell compartments in MB (Li et al., 2013; Vanner et al., 2014). The Prom1+, Sox2+, or Nes+ population was present at a low frequency in the tumor tissues (Figure 1F).
TSCAN analysis revealed a developmental trajectory from stem-like cells expressing Sox2 and Nes to intermediate GNPs that express Ccnd2 and Mki67 (Ayrault et al., 2010), and into differentiated GNPs that express NeuroD1 and Cntn2 (Figures 1G and 1H). Strikingly, a set of genes (Olig2, Olig1, Hes1, Hey1, Bhlhe41, Etv5, Elk3, and Wdsub1) were enriched in the cluster of the stem-like population (Figure S1F). Unexpectedly, we found that the distribution pattern of the early neural/glial lineage marker Olig2 (Lu et al., 2000; Zhou et al., 2000) coincided with those of Sox2 and Nes (Figures 1F–1I). Similar cellular clusters were revealed by analysis of additional GFAP-Ptch tumor tissues (Figure S2). Levels of Olig1 and Olig2 were highest in stem-like progenitor populations and progressively decreased along the GNP lineage trajectory (Figure 1I), suggesting a potential role of these progenitors during initiation of MB tumorigenesis. To investigate the relevance of OLIG2+ stem-like progenitors in our mouse models to SHH-MB in patients, we performed unsupervised pseudo-time trajectory analysis of scRNA-seq data from SHH-MB patients (Vladoiu et al., 2019). Our analysis revealed the enrichment of Olig2+ progenitor cells within the neural stem cell-like population in human SHH-MBs (Figures S3A and S3B). We also analyzed scRNA-seq data with the ccRemover program, which removes cell-cycle variation (Barron and Li, 2016) and obtained results similar to Seurat analyses (Figures S3C–S3H), confirming that OLIG2+ progenitors are mainly restricted to stem-like cell populations (Figure S3I).
OLIG2+ Intermediate Progenitors are Present at Initiation of MB
We examined the OLIG2 expression pattern during early stages of MB tumorigenesis by immunostaining of control and tumor-forming cerebella at P5, early in MB tumorigenesis (Yang et al., 2008). In the normal cerebellum, OLIG2+ cells were mainly detected in oligodendrocyte lineage cells in the cerebellar white matter (Figure S4A–S4D); none were detected in the external granular layer (EGL) at P5 or P12. The majority of the OLIG2+ cells in the cerebellum were not proliferating as shown by BrdU incorporation, and the percentage of proliferating OLIG2+ cells decreased as mice aged (Figure S4E and S4F). In contrast, in tumor-forming cerebella of GFAP-Ptch animals at P5, we observed OLIG2+ cells in the EGL (Figures 2A and 2B) and in the cerebellar white matter (Figures S4G–S4I). A substantial proportion of the OLIG2+ cells in the EGL were proliferative (Figures 2B and S4J). In addition, a large population of OLIG2+ cells in the EGL at neonatal stages (~77%) expressed SOX2 but not ZIC1 (Figures 2C and 2D), in contrast to the absence of OLIG2+/SOX2+ cells in normal EGL (Figure S4K).
Figure 2. OLIG2+ Cells Propagate during Tumor Progression.

(A) Images of GFAP-Ptch MB at P5 (upper) and P12 (bottom) tumors stained with H&E and for OLIG2 and BrdU. Boxed areas are shown at high magnification in the right panels. Arrows, co-labeled cells.
(B) Percentage of OLIG2+ cells (left) and BrdU+/OLIG2+ cells (right) in GFAP-Ptch MB at indicated stages.
(C) Images of GFAP-Ptch MB at P5 (upper) and P12 (bottom) stained for OLIG2, SOX2, and ZIC1. Arrows, co-labeled cells.
(D) Percentage of OLIG2+ cells in indicated populations in GFAP-Ptch MB (left) or in GFAP-Gnas MB (right).
(E) Images of GFAP-Ptch MB at P5 stained for OLIG2, PDGFRα and CC1. Arrows, OLIG2+ cells.
(F) Images of GFAP-Gnas MB at P15 stained for OLIG2, ZIC1, and BrdU. Boxed area is shown at a high magnification in right panels.
(G) Images of GFAP-Gnas MB at P15 stained as indicated. Arrows, co-labeled cells.
(H) Percentage of OLIG2+ cells among indicated populations.
(I) Image of sphere formation by cells isolated from GFAP-Gnas;Olig2-GFP tumors.
(J) Quantification of numbers of spheres.
(K) Images of tumors from NSG mice subcutaneously transplanted with OLIG2-GFP+ cells.
(L) Images of tumors from NSG mice stained with H&E stain and for SOX2, ZIC1, and Ki67.
(M) FACS sorting of GFP+ cells from GFAP-Ptch;Olig2-GFP tumors.
(N) Frequency of allograft formation at indicated doses.
(O) Representative tumor allograft from OLIG2+ cell transplants. Arrow, tumor tissue.
(P) Tumor allograft stained with H&E and for BrdU and ZIC1.
Data are means ± SEM from at least three independent experiments; **p<0.01, ***p<0.001; Student’s t test in H and J, one-way ANOVA with Tukey’s multiple-comparisons test in B and E. Scale bars in A (left, 500 µm, middle, right, 20 µm); in C, D, L, P, 20 µm; F (left, 300 µm; right, 100 µm); in G, 10 µm; in I, 100 µm; in O, 5 mm.
See also Figures S4 and S5.
At P12 and P25, the percentage of OLIG2+ cells or proliferative OLIG2+ cells was substantially lower than that at P5 in the tumor tissues of GFAP-Ptch animals (Figures 2A, 2B and S4G–S4I). These OLIG2+ progenitors were co-labeled with SOX2 but not ZIC1 (Figures 2C, 2D and S4K). Strikingly, OLIG2+ cells in tumor-like tissues expressed the oligodendrocyte progenitor (OPC) marker PDGFRα (Lu et al., 2000; Zhou et al., 2000) but not mature oligodendrocyte marker CC1 (Figure 2D and 2E). OLIG2 was observed in neural stem/progenitors in the subventricular zone and in the nuclear transitory zone (NTZ) of the developing cerebellum (Figure S4L–S4N) as previously reported (Ju et al., 2016; Seto et al., 2014), but not in the ATOH1-expressing GNP niche at the upper rhombic lip (Figure S4M).
Mice lacking Gnas (Gnasfl/fl;hGFAP-Cre or GFAP-Gnas mice) develop SHH-MB with 100% penetrance but exhibit later mortality (2–3 months) than GFAP-Ptch mice (4 weeks) (He et al., 2014; Yang et al., 2008). In GFAP-Gnas mice, OLIG2+ progenitors were detected in the EGL at P7 and reached the highest density at P15 before declining (Figure 2D and S4O). At P15, we detected OLIG2+ progenitors within the center region of expanded EGL devoid of ZIC1, and OLIG2+ cells coincided with BrdU+ proliferative populations (Figure 2F and 2G). In contrast, most ZIC1+ GNPs in the EGL were post-mitotic, akin to proliferative tumor-like cells, and were segregated from BrdU+ cells (Figures 2F and 2G). Similar to the GFAP-Ptch tumors, the majority of OLIG2+ cells in GFAP-Gnas tumors were co-labeled with BrdU and SOX2 but not neuronal markers ZIC1 or NEUROD1 (Figures 2G and 2H). These observations suggest that OLIG2+ cells are the major propagating cells at the tumor initiation stage in animal models of SHH-MB.
OLIG2+ Progenitors are MB Tumor-Initiating cells
To further investigate the self-renewal and tumorigenic capacity of OLIG2+ populations, we generated GFAP-Gnas mice carrying an Olig2-GFP reporter. We isolated GFP+ cells by FACS from MB tissues of these mice at P15 and performed neurosphere formation assays (Lu et al., 2016; Venugopal et al., 2012). GFP reporter expression coincided with OLIG2+ expression in tumor tissues (Figures S4P and S4Q). Compared to GFP– tumor cells, GFP+ tumor cells exhibited a much higher sphere-forming capacity (Figures 2I and 2J). Similarly, OLIG2-GFP+ cells exhibited a higher tumorigenicity than GFP– cells in an allograft model when dissociated cells from primary spheres were subcutaneously transplanted into NOD scid gamma (NSG) mice (Figure 2K). All OLIG2-GFP+ tumors were histologically similar to human MB (Louis et al., 2016) with the small, round, cellular morphology, and cells were highly proliferative as indicated by abundant Ki67 expression, and expressed ZIC1 and SOX2 (Figure 2L), characteristic features of MB.
To compare tumorigenic potentials of OLIG2+ and OLIG2– cells from GFAP-Ptch mice in orthotopic allografts, we sorted and transplanted GFP+ and GFP– cells into NSG mice at varying cell doses (Figures 2M and 2N). The OLIG2+ cells had much higher tumor-propagating potential, and tumors were highly proliferative and consisted of abundant ZIC1+ GNPs (Figures 2O and 2P). To evaluate the neoplastic nature of OLIG2+ cells, we performed an assay for transposase-accessible-chromatin with high-throughput sequencing (ATAC-seq) of GFP+ and GFP– cells from tumor tissues of GFAP-Ptch;Olig2-GFP mice. The transcriptomic profiling showed that OLIG2+ cells were enriched for stemness signatures and oncogenic pathways, such as SHH, HIPPO, NOTCH, and AURORA-A signaling (Figures S5A–S5E) compared with OLIG2– cells. Moreover, the chromatin accessibilities of the stemness genes and proto-oncogenes, but not the genes characteristic of mature GNPs, were higher in OLIG2+ than OLIG2– cells (Figures S5F–S5H). Collectively, these observations suggest that OLIG2+ cells are neoplastic rather than differentiated glial cells.
High OLIG2 Expression Predicts Poor Outcome in SHH-MB Patients
To investigate the correlation between OLIG2 levels and patient survival, we examined OLIG2 expression from a transcriptomic dataset of a human MB cohort (Cavalli et al., 2017). We found that high OLIG2 expression is significantly correlated with decreased overall survival for patients with SHH-MBs but not in WNT, Group 3, or Group 4 MBs (Figure 3A). OLIG2 was present at higher levels in treatment-resistant large-cell anaplastic (LCA) MB with poor prognosis (Kool et al., 2012), than in SHH-MB with classic morphology (Figure S5I).
Figure 3. High OLIG2 Signature Predicts Poor Outcome in SHH-MB Patients and Ablation of OLIG2+ Cells Reduces Tumor Growth in Mice.

(A) Kaplan-Meier survival curves of the patients in different MB subgroups based on OLIG2 expression levels.
(B) Cox’s multivariate survival analysis of OLIG2 expression, TP53 mutation status, anaplasia, and their interactions.
(C, D) Overall survival stratified by TP53 mutation (C) or large-cell anaplastic (LCA) (D) with OLIG2 high and low expression in SHH-MB. + indicates censored cases.
(E) Diagram showing GCV-mediated depletion of Olig2-TK+ cells.
(F) H&E-stained cerebellar sections of GFAP-Gnas and Olig2-TK tumors from mice treated with GCV from P5 to P40.
(G, H) Kaplan-Meier survival curves of mice treated with GCV from P5 to P40 (G) or from P15 to P35 (H).
(I) Images of GFAP-Gnas and Olig2-TK tumors at P35 stained for BrdU and DAPI.
(J) Percentage of BrdU+ cells in GFAP-Ptch and Olig2-TK tumors at P35 (n=5 animals/group).
(K) H&E-stained images of GFAP-Ptch and GFAP-Ptch;Olig2-TK tumors of mice treated with GCV from P5 to P10 and harvested at P18.
(L) Relative volumes of GFAP-Ptch and GFAP-Ptch;Olig2-TK tumors (n=5).
Data are means ± SEM; n.s., not significant, ***p<0.001; Student’s t test in J, L; log-rank test in A, C, D, G, H. Scale bars in F, L, 300 µm; in I, 20 µm.
See also Figures S5.
We also performed a Cox’s multivariate analysis of SHH-MBs with the log-rank test to assess different risk factors. Our analysis showed that of OLIG2 expression, TP53 mutation status, and LCA, only OLIG2 expression was significantly correlated with poor patient outcomes (p=0.042, log-rank test) (Figure 3B). The interactions of Olig2 expression with either TP53 mutation or both TP53 mutation and anaplasia were not correlated with overall survival (p=0.500 and p=0.159, respectively) (Figure 3B), suggesting that OLIG2 expression is an independent determinant for survival of SHH-MB patients. Further, high levels of Olig2 expression combined with TP53 mutation or LCA were associated with even poorer outcomes in SHH-MB patients (Figures 3C and 3D). Thus, Olig2 expression affects patient outcome independently and defines a subset of high-risk SHH-MBs.
Eradication of Mitotic OLIG2+ Progenitor Populations Inhibits MB Growth
To test whether OLIG2+ proliferative cells are necessary for MB growth, we utilized an in vivo cell suicide approach to deplete mitotic OLIG2+ progenitors. We generated GFAP-Gnas mice with the suicide gene Herpes simplex virus thymidine kinase (HSV-TK) knocked in at the Olig2 locus (Olig2-TK) (Figure 3E). HSV-TK kinase activity converts ganciclovir (GCV) into toxic triphosphates that inhibit DNA polymerase, eliminating actively dividing tumor cells while sparing normal post-mitotic OLIG2+ cells, such as mature oligodendrocytes (Lu et al., 2016). When GCV was administered to the GFAP-Gnas mice carrying Olig2-TK at P5 prior to the onset of tumor initiation, tumor initiation was blocked and lifespan significantly extended (Figures 3F and 3G). Administration of GCV at a later stage P15 extended lifespan but to a lesser extent than at P5 (Figure 3H). Ablation of OLIG2+ progenitors also reduced tumor cell proliferation (Figures 3I and 3J). Similarly, in the Olig2-TK bearing GFAP-Ptch mice, GCV administration inhibited tumor development (Figures 3K and 3L). These data suggest that OLIG2+ mitotic progenitors are critical for tumor initiation in animal models of SHH-MB.
OLIG2+ Cells are Quiescent in Late-stage MB Tumors
We next performed scRNA-seq of the tumor cells from GFAP-Ptch mice at the late stage of tumorigenesis P24. t-SNE analyses identified nine distinct cell clusters (Figure 4A and S6A and S6B). Of the total analyzed cells, 93.6% were GNPs (Figure 4B), comparable to the frequency in the tumor tissues at P10. The proportion of proliferating GNPs was approximately 54% in the late-stage MB, a decrease from about 75% in early-stage tumors. The percentage of cells expressing cell-cycle-associated genes was reduced in late-stage compared to early-stage MB (Figure 4C).
Figure 4. OLIG2+ Quiescent Stem Cells in Late-stage MB are Reactivated during Relapse.

(A) t-SNE plot of cell clusters from GFAP-Ptch MB at P24.
(B) Relative proportions of distinct clusters.
(C) Percentages of cells from GFAP-Ptch tumors expressing indicated genes at P10 and P24.
(D) Images of GFAP-Ptch MB tumors at P24 stained for NES, SOX2, and OLIG2. Arrows, co-labeled cells.
(E, F) Percentages of OLIG2+ cells in indicated GFAP-Ptch MB populations at P24 (E) and at P5 and P24 (F).
(G) Images of OLIG2 immunostained vehicle- and drug-treated Atoh1-Ptch tumors. Veh, Vehicle.
(H, I) Percentages of OLIG2+ (H) and BrdU+/OLIG2+ (I) cells in vehicle- and drug-treated Atoh1-Ptch tumors.
(J, K) MB patient tumors stained for OLIG2, Ki67, and SOX2 (J) and percentages of labeling cells among OLIG2+ cells (K). Arrowheads, OLIG2+ cells; arrows, co-labeled cells.
(L, M) Images of OLIG2 immunostaining (left) and the percentage of OLIG2+ cells (right) in the tumors of vehicle- and cyclophosphamide-treated TB13–5634 cells derived PDX mice (L) and palbociclib-treated Med-314FH cells derived PDX mice (M).
(N) OLIG2 mRNA expression in samples from primary and recurrent SHH-MBs from a publicly available patient cohort (n=5 matched pairs, permutation test).
(O) MRI images of paired primary (left) and recurrent (right) MB from the same patient. Arrows, tumors.
(P) Images of primary (upper) and recurrent (bottom) MB tumors stained for OLIG2.
(Q) The percentage of OLIG2+ cells in primary and recurrent MB tumors.
(R) MRI images of a MB patient with primary (left) and spinal cord metastasis (right) of MB tumors (arrows).
(S) Images of primary (left) and metastatic (right) MB tumors stained for OLIG2 and Ki67. Arrowheads, OLIG2+ cells; arrows, co-labeled cells.
(T) The percentage of OLIG2+ cells among tumor cells (left) or Ki67+ cells among OLIG2+ cells (right) in primary and metastatic patient MB sections.
(U) Box plot of OLIG2 mRNA expression in primary (n=134) and in metastatic (n=26) human SHH-MBs. Whiskers show the minimum and maximum, and boxes extend from the first to the third quartiles with midlines at the medians.
Data are means ± SEM from at least 3 independent experiments; *p<0.05; **p<0.01; ***p<0.001; Student’s t test in F, H, I, L, M, Q, T, U. Scale bars in D, J, L, M, S, 20 µm; in G, P, 100 µm.
See also Figures S6 and S7.
Progenitor cells expressing Olig1 or Olig2 were sparsely present in late-stage tumors. Olig1 and Olig2 had similar distribution patterns to Prom1+ and Sox2+ (Figure S6C). TSCAN analysis revealed a developmental trajectory from stem-like cells, to intermediate cycling GNPs, to differentiated GNPs (Figure S6D). Olig1 and Olig2 were expressed by stem-like progenitor populations but were progressively downregulated along the GNP lineage trajectory (Figures S6E and S6F). These data suggest that the OLIG2+ cells that co-express SOX2 are a rare stem-like population in full-blown MB tumors.
OLIG2+ Progenitors are Enriched in Therapy-Resistant and Relapsed Tumors
To characterize the rare OLIG2+ subpopulation in late-stage tumors, we investigated OLIG2 expression in different cell types in tumors of GFAP-Ptch mice at P24. Immunohistochemistry indicated that the majority of OLIG2+ cells were co-labeled with the stemness markers SOX2 or Nes (Figure 4D). OLIG2+ cells were excluded from differentiating GNP cells expressing NEUROD1 and NEUN, and did not express Ki67 or co-labeled with BrdU+ (Figures 4E, 4F and S6G). The OLIG2+ cells expressed OPC markers Sox10 and Ascl1 but not mature oligodendrocyte markers MAG and MOG (Figure S6H–S6J). These data suggest that in late-stage tumors, the OLIG2+ cells are non-cycling and have glial progenitor properties.
To investigate the responses of OLIG2+ populations to drug treatment, we treated tumor-bearing Atoh1-Ptch mice, in which Ptch1 is deleted in Atoh1+ cells, with chemotherapy drugs cisplatin and cyclophosphamide at P30 (Morfouace et al., 2014). These mice develop SHH-MB with 100% penetrance with a similar OLIG2 expression pattern but later mortality compared to GFAP-Ptch mice (Figures S6K and S6L). Compared to vehicle-treated mice, the percentage of OLIG2+ cells were significantly increased in the tumor tissues after drug treatment (Figure 4G and 4H), and a substantial proportion of OLIG2+ cells became proliferative (Figure 4I), indicating that treatment reactivated OLIG2+ cells.
To further examine OLIG2 expression in human primary SHH-MB tissues, we performed immunostaining for OLIG2, Ki67, SOX2, and oligodendrocyte lineage markers. Consistent with our single-cell data, in the primary advanced human SHH-MB, OLIG2+ cells were rare and co-labeled with the stemness marker SOX2 and oligodendrocyte progenitor markers SOX10 and ASCL1 but not with Ki67 or MAG or MOG (Figures 4J and 4K and S7A and S7B). This indicates that these OLIG2+ cells in fully developed human SHH-MB are quiescent, non-cycling cells with glial progenitor properties.
In patient-derived orthotopic xenografts (PDX), in which mice were orthotopically transplanted with human SHH-MB cells (TB13–5634) with MYCN amplification and mutant TP53, cyclophosphamide treatment led to a substantial increase in the number of OLIG2+ cells (Figure 4L). The robust enrichment of OLIG2+ cells in treatment-resistant and recurrent tumor tissues was also observed in another SHH-MB PDX model (Med-314FH) treated with the CDK4/6 inhibitor palbociclib (Brabetz et al., 2018) (Figure 4M). These results suggest that OLIG2+ progenitors re-emerged in therapy-resistant tumors.
The frequency of OLIG2+ cells varied among human MB subgroups and by tumor stage (Figures S7C and S7D). In matched-patient samples from primary and recurrent SHH-MB tumors (Cavalli et al., 2017), we found that OLIG2 mRNA expression levels were significantly higher in recurrent SHH-MB (Figure 4N). The frequencies of OLIG2+ cells were substantially increased in the recurrent relative to the primary tumors from human patients with SHH-MB (Figures 4O–4Q and S7E). In addition, we detected significantly increased OLIG2 expression and a larger fraction of OLIG2+ cells co-labeled with proliferation marker Ki67 in metastatic SHH-MB compared with primary SHH-MB (Figures 4R–4T). OLIG2 expression levels were significantly higher in metastatic tumors than primary tumors among SHH-MB cohorts (Figure 4U) (Cavalli et al., 2017). These data suggest that OLIG2+ progenitors are re-activated from a quiescent state during SHH-MB recurrence or metastasis.
Olig2 Deletion Inhibits MB Growth
To interrogate the function of OLIG2 in MB formation, we bred mice with a floxed Olig2 allele with GFAP-Ptch mice resulting in mice we refer to as Olig2cKO mice. The GFAP-Ptch mice had bulging cerebella with extensive tumor cell expansion in the EGL at P24. Olig2cKO mice had substantially smaller tumors (Figures 5A and 5B) and a significantly extended lifespan compared to the unablated GFAP-Ptch mice (Figure 5C). In contrast to robust OLIG2 expression in tumors of the GFAP-Ptch mice, in Olig2cKO mice OLIG2 was largely diminished during tumorigenesis (Figures 5D and 5E), although we detected OLIG2+ cells in the eventual tumors (Figures S7F and S7G). The stem cell marker SOX2 expression was modestly, but significantly, reduced in Olig2cKO tumors compared to the GFAP-Ptch tumors as was BrdU incorporation (Figures 5D–5G). In cells isolated from GFAP-Ptch and Olig2cKO tumors at P24, we found that BrdU+ GNPs were significantly reduced in the Olig2-deleted tumor cells (Figures 5H and 5I). In addition, inhibition of Olig2 expression using an siRNA in Daoy cells, a human SHH-MB cell line (Jacobsen et al., 1985; Sengupta et al., 2012), resulted in reduced expression of stemness-associated genes (Figure 5J). To assess the self-renewal capacity of Olig2-depleted tumor cells, we performed tumorsphere formation assays (Lu et al., 2016). Compared to GFAP-Ptch tumor cells, very few spheres were formed by Olig2cKO tumor cells during multiple passages in culture (Figures 5K and 5L).
Figure 5. Olig2 Deletion Inhibits Growth of Mouse MB.

(A) Photographs of GFAP-Ptch and Olig2cKO brains at P24. Arrows, cerebellum.
(B) H&E stain of GFAP-Ptch and Olig2cKO tumors at P24. Boxed area is shown at a high magnification.
(C) Kaplan-Meier survival of GFAP-Ptch (n=19) and Olig2cKO (n=16) mice.
(D,E) OLIG2 and SOX2 immunostain (D) and quantification (E) in GFAP-Ptch and Olig2cKO brains at P24.
(F,G) BrdU labeling (F) and the percentage of BrdU+ cells (G) of GFAP-Ptch and Olig2cKO tumors at P24.
(H,I) Immunostaining for BrdU and ZIC1 (H) and the percentage of BrdU+ cells among ZIC1+ GNPs (I) from GFAP-Ptch tumors and Olig2cKO tumors at P24.
(J) qRT-PCR analyses of indicated genes in scrambled control siRNA (Scr) or siOLIG2-treated Daoy cells.
(K, L) Neurosphere formation (K) and sphere numbers per 104 GNPs (L) from GFAP-Ptch and Olig2cKO tumor cells.
(M, N) GFAP-Ptch and Olig2cKO tumor appearance (M) and weight (N) from NSG mice.
(O, P) BrdU and ZIC1 immunostain (O) and the percentage of BrdU+/ZIC1+ cells (P) in tumor sections from NSG mice subcutaneously transplanted with GFAP-Ptch or Olig2cKO tumor cells
Data are means ± SEM from at least 3 independent experiments. *p<0.05, **p<0.01, ***p<0.001; log-rank test in C, Student’s t test in panels E, G, I, J, L, N, P. Scale bars in A, 5 mm; in B, 300 µm; in D, F, H, O, 20 µm; in K, 100 µm.
We next evaluated the tumorigenicity of cells from Olig2cKO tumors in an allograft model. Dissociated primary tumor cells from GFAP-Ptch and Olig2cKO tumors were subcutaneously transplanted into NSG mice. The tumors formed by Olig2cKO tumor cells had significantly lower size and weight than those originating from GFAP-Ptch mice (Figures 5M and N). Consistent with reduced tumor growth, the BrdU+ proliferating tumor cells were also substantially reduced in Olig2cKO tumors (Figures 5O and 5P). Thus, OLIG2 is an important regulator for MB cell growth and tumor progression.
OLIG2 Modulates Chromatin Landscape to Drive Oncogenic Programs
To gain insight into the potential mechanisms through which OLIG2 induces cellular reprogramming and tumor growth, we performed transcriptome profiling of GFAP-Ptch tumors and Olig2cKO tumors and identified sets of differentially regulated genes (Figures 6A and 6B). By interrogating gene expression signatures from the Molecular Signatures Database (Subramanian et al., 2005), we determined that the SHH pathway, the E2F1-mediated signaling pathway, and cell cycle-associated pathways were substantially downregulated (Figures 6C and 6D), whereas the pathways associated with normal neuronal differentiation were upregulated in Olig2cKO tumors (Figures 6C and 6E).
Figure 6. OLIG2 Controls a Network that Regulates Tumor Cell Proliferation and Stemness.

(A) Heatmap of differentially expressed genes in Olig2cKO vs. GFAP-Ptch tumors.
(B) Volcano plot of significantly altered genes (p<0.05).
(C) GSEA analysis of top differentially regulated genes.
(D) GSEA plots of downregulated SHH and E2F1 pathway gene sets in Olig2cKO vs. GFAP-Ptch tumors.
(E) GSEA plots of neuronal signaling and axon guidance gene sets in Olig2cKO vs. GFAP-Ptch tumors.
(F) Heatmaps of OLIG2 and H3K27ac ChIP-seq signals in normal cerebellum (CB) and MB tumors. Box: MB-specific target sites.
(G) H3K27Ac ChIP-seq enrichment profiles around OLIG2-bound regions in M tumors.
(H) The most significantly enriched motifs in OLIG2-bound regions in MB tumors.
(I) ToppGene analysis of OLIG2 and H3K27ac co-targeted pathways in MB tumors. hES, human embryonic stem cells.
(J) OLIG2 and H3K27ac occupancy on genes associated with stemness.
(K) OLIG2 and H3K27ac occupancy on genes associated with cell proliferation and on proto-oncogenes.
See also Figure S8.
To identify the target genes directly regulated by OLIG2, we performed genome-wide ChIP-seq in OLIG2-expressing GFAP-Ptch tumors and normal developing cerebella. Strikingly, OLIG2 targeted a specific set of promoter and enhancer elements that are highly enriched with the activating histone mark H3K27ac (Creyghton et al., 2010) in tumor tissues but not normal cerebellum (Figures 6F and 6G). Thus, OLIG2 appears to alter the chromatin landscape during tumorigenesis. Motif enrichment analysis revealed overrepresentation in the consensus binding sites of Tead, SOX2, Sox9, and MYCN within OLIG2 binding peaks (Figure 6H). OLIG2 may coordinate with these transcription factors to control tumorigenesis.
Gene enrichment analysis using the ToppGene suite (Chen et al., 2009) showed that OLIG2-bound genes with strong H3K27ac enrichment in tumor tissues were overrepresented in tumorigenesis-associated pathways (e.g., HIPPO, Myc, MAPK signaling pathways) and stemness-associated pathways (Figure 6I). Among the genes bound by OLIG2 were those pertinent to stemness (Sox2, Nes, Ntsr2, Pou3f2, Slc1a3, Egfr) and pro-oncogenic activity (Pdgfrα, Nek2, Gli2, Otx2, Cdk6, Myc) (Figures 6J, 6K and S8A and S8B). Consistently, the chromatin accessibility in the loci of stemness genes and proto-oncogenes was higher in OLIG2+ cells than in OLIG2– cells isolated from tumor tissues (Figures S5G and S5H). Together, these data indicate that OLIG2 targets de novo enhancers to regulate the chromatin landscape, and activates stemness and cell growth pathways to promote oncogenic transformation during MB tumorigenesis.
Activation of HIPPO-YAP/TAZ Signaling by OLIG2 Promotes MB Growth
OLIG2 is a nuclear transcriptional factor difficult to target with a small-molecule drug. OLIG2 downstream effectors might be druggable targets, however. We found that Yap, Taz, Tead1–3, and Ctgf, which promote cell growth and tumorigenesis in various tissues (Harvey et al., 2013; Zanconato et al., 2016), were marked by H3K27ac and were directly targeted by OLIG2 (Figure 7A). ATAC-seq analysis indicated a higher chromatin accessibility within these gene loci in OLIG2+ cells than in OLIG2– cells from tumor tissues (Figure 7B). Moreover, TSCAN analysis showed that expression of Olig2 and HIPPO components was enriched in populations associated with stemness (Figure 7C). These data suggest that OLIG2 may control MB growth, at least in part, through the HIPPO-YAP/TAZ signaling pathway. In support of this, HIPPO components were downregulated in Olig2cKO tumors compared to GFAP-Ptch tumors (Figure S8C).
Figure 7. OLIG2-mediated Regulation of HIPPO Signaling is Critical for MB Growth.

(A) OLIG2 and H3K27ac occupancy on HIPPO effector genes and target loci.
(B) ATAC-seq profiles on HIPPO effector genes and target loci.
(C) Pseudo-time analysis of HIPPO effectors and targets in neural stem cell-like populations.
(D) YAP and DAPI stain of normal cerebellum and GFAP-Ptch MB at P18.
(E) Immunoblot for YAP in extracts of normal cerebellum and GFAP-Ptch MB. GAPDH, a loading control.
(F,G) YAP, OLIG2, SOX2, BrdU labeling (F) and the percentage of labeled cells among YAP+ cells (G) in GFAP-Ptch MB at P18 mice (n=4).
(H) Images of H&E-stained cerebellar sections of control (Lats1/2fl/fl) and Atoh1-Lats1/2 mice at P18. Arrows, tumor lesions.
(I) Kaplan-Meier survival curves of control (n=20) and Atoh1-Lats1/2 mice (n=22) mice.
(J) GSEA plots of genes differentially regulated in Atoh1-Lats1/2 tumors vs. control normal cerebellum.
(K) PCA analysis of correlation among control normal cerebella, Atoh1-Lats1/2 and GFAP-Ptch tumors.
(L–N) Images of ZIC1, Ki67, YAP, BrdU, OLIG2, SOX2 labeling (L, M) and percentage of YAP+ cells among labeled cells (N) in Atoh1-Lats1/2 tumors at P18 (n=4).
Data are means ± SEM; ***p<0.001; log-rank test in I. Scale bars in D, 50 µm; in F, L, M, 20 µm; in H, 300 µm.
See also Figure S8.
In GFAP-Ptch MB tumors, immunostaining and western blotting showed higher frequencies of YAP+ cells and a higher level of YAP expression compared to normal cerebellum (Figures 7D and 7E). In addition, the majority of YAP+ cells were co-labeled with OLIG2 and SOX2 (Figures 7F and 7G), suggesting that YAP+ cells possess stemness characteristics. We hypothesized that the elevation of the HIPPO effectors is sufficient to induce MB. To test it, we ablated Lats1/2 to induce HIPPO-YAP/TAZ signaling. LATS1 and LATS2 are negative regulators of HIPPO signaling effectors (Hong and Guan, 2012; Varelas, 2014). Non-synonymous mutations in Lats1 and Lats2 have been reported in human recurrent SHH-MB (Morrissy et al., 2016; Parsons et al., 2011). Mice with floxed Lats1 and Lats2 were bred with the Atoh1-Cre (He et al., 2014) to result in mice without LATS1/2 expression in GNPs. Strikingly, all resulting Atoh1-Lats1/2 succumbed to MB tumors by 4 weeks of age (Figure 7H and 7I).
Consistent with the inhibitory function of LATS1/2 on the HIPPO pathway, we found activated HIPPO signaling and SHH pathway in Lats1/2-deficient tumors by RNA-seq analysis (Figure 7J). Furthermore, principal component analysis showed similar gene expression signatures in Lats1/2-deficient mice and GFAP-Ptch mice (Figure 7K), suggesting the formation of SHH-subtype MB in Atoh1-Lats1/2 mice. The tumor tissues from the Atoh1-Lats1/2 mice had high density of ZIC1+ GNPs and an OLIG2+/SOX2+ cell population, and neoplastic cells were highly proliferative (Figure 7L). Ablation of Lats1/2 also led to hyperactivity of HIPPO effector YAP, as indicated by immunostaining in tumor tissues (Figure 7M). Within the MB tissue, YAP-expressing cells co-labeled with BrdU and stemness markers OLIG2 and SOX2 (Figures 7M and 7N), suggesting that YAP+ progenitors are proliferative stem-like progenitor cells.
Co-inhibition of AURORA-A/MYCN and HIPPO Signaling Impedes MB Progression
The proto-oncogene MYCN is mis-expressed in various human tumors (Brockmann et al., 2013; Morrissy et al., 2016; Otto et al., 2009), and AURORA-A/MYCN pathway gene amplification and activation have been reported in MB (Hill et al., 2015; Swartling et al., 2012). Enhancers of AURORA-A/MYCN pathway genes were directly targeted by OLIG2 and were marked with H3K27ac (Figure 8A). These regulatory elements were bound by OLIG2 in MB tumor cells but not in cells from gliomas (Figure S8D and S8E). The levels of these genes were higher in GFAP-Ptch MB tissue than normal cerebellum (Figure 8B and S8F). Olig2 deletion significantly reduced the expression of these genes in Olig2cKO tumors (Figures 8C and 8D). Furthermore, siRNA-mediated inhibition of OLIG2 in Daoy cells downregulated the expression of AURORA-A/MYCN pathway components (Figure S8G). To determine the effect of inhibition of AURORA-A/MYCN-mediated signaling on MB development, we treated Daoy cells with the CD532, which disrupts the conformation of AURORA-A resulting in proteasomal degradation of MYCN (Gustafson et al., 2014). CD532 treatment significantly inhibited MCYN expression (Figures S8H and S8I) and reduced proliferation (Figures 8E and S8J). These data suggest that OLIG2 controls MB growth at least in part through regulating the AURORA-A/MYCN pathway.
Figure 8. Co-inhibition of HIPPO and AURORA-A/MYCN Signaling Impedes MB Progression.

(A) OLIG2 and H3K27ac occupancy on AURORA-A/MYCN pathway gene loci in GFAP-Ptch tumors.
(B) qRT-PCR analyses of AURORA-A/MYCN pathway genes in GFAP-Ptch tumors.
(C) Heatmap of expression of AURORA-A/MYCN pathway genes in Olig2cKO vs. GFAP-Ptch tumors.
(D) qRT-PCR analyses of AURORA-A/MYCN pathway genes in Olig2cKO vs. GFAP-Ptch tumors.
(E) BrdU labeling of Daoy cells treated with indicated inhibitors. Veh, vehicle; VP, verteporfin.
(F,G) Images (F) and percentage of BrdU+ cells among ZIC1+ cells (G) in GFAP-Ptch primary tumor cells treated with indicated inhibitors.
(H, I) BrdU labeling (H) and percentage of BrdU+ cells (I) of tumor sections from Atoh1-Ptch mice treated with indicated inhibitors.
(J,K) Cleaved Caspase 3 (cl-Casp3) labeling (J) and percentage of cl-Casp3+ cells (K) in tumor sections from Atoh1-Ptch mice treated with indicated inhibitors.
(L,M) Senescence-associated beta-galactosidase staining (SA-β-Gal) (L) and percentage of SA-β-Gal+ cells (M) in tumor sections from Atoh1-Ptch mice treated with indicated inhibitors.
(N) Kaplan-Meier survival curves of Atoh1-Ptch mice (n≥8 per group) treated with indicated inhibitors.
Data are means ± SEM; *p<0.05, **p<0.01, ***p<0.001; log-rank test in N, Student’s t test in B, D; one-way ANOVA with Tukey’s multiple-comparisons test in G, I, K, M. Scale bars in E, F, H, J, L, 20 µm.
See also Figure S8.
We then examined whether co-targeting of HIPPO and AURORA-A/MYCN signaling would reduce tumor cell proliferation more than either individual regimen. Combined treatment of Daoy cells with CD532 and YAP inhibitor verteporfin (VP) (Wu et al., 2018) more effectively inhibited cell proliferation than did treatment with either compound alone (Figures 8E and S8J). Primary tumor cells were also sensitive to inhibition of HIPPO and AURORA-A/MYCN signaling: Combined treatment of GFAP-Ptch tumor cells with verteporfin and CD532 additively decreased proliferation of ZIC1+ tumor cells relative to individual treatments (Figures 8F and 8G).
To assess therapeutic potential in vivo, we treated Atoh1-Ptch mice daily with verteporfin (100 mg/kg) (Wu et al., 2018), twice per week with CD532 (25 mg/kg) (Gustafson et al., 2014), or both. Treatment with both decreased tumor cell proliferation to a greater extent than either single agent (Figures 8H and 8I) and increased apoptosis-associated cleaved Caspase 3 levels as well as senescence-associated beta-galactosidase expression (Figures 8J–8M), indicative of growth arrest (Burton and Faragher, 2015; Shay and Roninson, 2004). In addition, concurrent inhibition of both HIPPO and AURORA-A/MYCN signaling resulted in a higher survival rate than single-agent treatments (Figure 8N).
DISCUSSION
Tumorigenesis is thought to result from dysregulated reprogramming of normal developmental processes following pathological insults (Hanahan and Weinberg, 2011; Wang et al., 2018). Our single-cell transcriptomics of murine models of SHH-MB identified a stem-like progenitor pool in tumors and distinct cerebellar GNP populations, suggesting that the developmental hierarchy during tumorigenesis mirrors the developmental neurogenesis program in the cerebellum. Strikingly, we found that OLIG2+ progenitors are a prominent actively cycling population during the initial phase of tumorigenesis in SHH-MB animal models. SHH-MBs are generally considered to arise from neuronal progenitors such as GNPs (Schuller et al., 2008; Yang et al., 2008); however, the OLIG2+ progenitors within MB tumors exhibited OPC-associated gene expression characteristics and did not express markers for neuronal progenitors or differentiated oligodendrocytes. These observations raise the unanticipated possibility that OLIG2+ glial progenitors or OPC-like progenitors are tumor-initiating cells in MB. These OLIG2+ progenitors may eventually transition into GNPs, which form the bulk of the MB tumor, consistent with the transition of OLIG2+ progenitors to GNPs during cerebellar development (Schuller et al., 2008). Therefore, OLIG2+ OPC-like progenitors may function as transit-amplifying cells prior to the establishment of GNP identity early in MB tumorigenesis.
Olig2 deletion resulted in a substantial reduction of MB tumor-cell proliferation and tumor growth, however, Olig2cKO mice eventually developed MB. This may be due to the observed incomplete deletion given the mosaicism in the hGFAP-Cre line or tumor cells may develop a mechanism to compensate for the loss of OLIG2.
Although we found that OLIG2 was co-expressed with SOX2 during tumorigenesis, SOX2 is not essential for tumor growth in the animal model of SHH-MB (Ahlfeld et al., 2013; Vanner et al., 2014). Notably, our ChIP-seq data indicate that OLIG2 directly binds to the enhancers of Sox2 and that Olig2 deletion downregulates Sox2 expression, suggesting that OLIG2 may be an upstream regulator of SOX2 or cooperate with SOX2 to promote tumorigenesis (Suva et al., 2014).
The nature of the stem-cell niche may vary among MB subgroups. In contrast to previous indication of OLIG2 expression in GNPs of the upper rhombic lip (Schuller et al., 2008), we and others observed OLIG2 in neural stem/progenitors in the subventricular zone and in the NTZ of the developing cerebellum (Ju et al., 2016; Seto et al., 2014). Since NTZ progenitors are thought to be a cell of origin for Group 4 MBs (Lin et al., 2016), it will be of interest to determine whether OLIG2 is important in initiation and progression of other MB subtypes. It is noteworthy that OLIG2+ OPC-like progenitors have been linked to gliomagenesis and the cells of origin in glioblastomas (Ligon et al., 2007; Liu et al., 2011; Lu et al., 2016). That OLIG2+ progenitors are a potential tumor-initiating cell population in both glioma and MB suggests that OPC-related OLIG2+ progenitors may be transformed by oncogenic insults in genetically and anatomically distinct types of brain tumors.
Integrative analyses of expression profiles and genome occupancy revealed that OLIG2 targets a specific set of genes enriched with H3K27ac in MB tumors but not normal cerebellum or glioma cells, suggesting that OLIG2 potentially modulates the chromatin landscape to activate a unique oncogenic program during MB tumorigenesis. The specific OLIG2-regulated oncogenic pathways may serve as potential therapeutic targets for MB treatment. Although OLIG2 marks anatomically distinct progenitors between gliomas and MB (Ligon et al., 2007; Lu et al., 2016), eradication of mitotic Olig2+ progenitors or ablation of Olig2 inhibits tumor initiation of both glioma and MB, suggesting a conserved role of OLIG2 in tumorigenesis in these genetically distinct brain tumors (Lu et al., 2019).
Analysis of our single-cell dataset further revealed that HIPPO signaling components YAP and TAZ are expressed in the stem-like population. YAP was previously shown to be amplified and up-regulated in SHH-MB tumorigenesis (Fernandez et al., 2009), however, the capacity of YAP elevation for inducing tumorigenesis remains elusive. We found that elevation of YAP/TAZ expression by inhibiting LATS1/2 promotes SHH-MB tumorigenesis. We found that concurrent inhibition of HIPPO and AURORA-A/MYCN signaling improved survival more than either single-agent regimen. Given that multiple oncogenic pathways are upregulated in MB tumors, identification of additional druggable targets and evaluation of other combination therapies is warranted.
Current therapies for MB do not effectively eliminate rare quiescent cancer stem cells, which may lead to drug resistance and tumor recurrence (Ramaswamy et al., 2016; Smoll, 2012; Taylor et al., 2012). OLIG2+ progenitors are rapidly dividing transit-amplifying cells at the onset of SHH-MB tumorigenesis but are rare in fully developed tumors in murine models, suggesting that they are quiescent, slow-cycling stem-like cells in advanced tumors. Strikingly, we found that OLIG2+ cells are enriched in a set of recurrent tumors of human SHH-MBs, suggesting that OLIG2 expression can be re-activated during tumor relapse. That OLIG2+ progenitors may become a dominant clone during recurrence is in agreement with notion that primary, recurrent, and metastatic MB tumors have distinct genetic makeups likely due to clonal selection (Morrissy et al., 2016; Ramaswamy et al., 2013; Wu et al., 2012).
Unlike other stemness-associated factors, such as SOX2, which are broadly expressed in many tissues, OLIG2 expression is restricted to the central nervous system (Lu et al., 2000; Zhou et al., 2000). That OLIG2+ glial progenitor cells are critical tumor-initiating cells might have important implications for design of therapies to target cell lineage vulnerability during MB tumorigenesis and recurrence. The functional role of OLIG2 as an activator of oncogenic signaling as well as a vehicle for tumor recurrence suggests that targeting OLIG2 and downstream pathways might minimize the onset of therapeutic resistance and improve the outcomes of patients with SHH-MB.
STAR★Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Qing Richard Lu (Richard.Lu@cchmc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Mice homozygous for floxed alleles of Ptchfl/fl (Yang et al., 2008), Gnasfl/fl (He et al., 2014) and Lats1/2fl/fl (Heallen et al., 2011) were crossed with mice carrying hGFAP-Cre+/− (Yang et al., 2008) or Atoh1-Cre+/− (Yang et al., 2008) to generate corresponding control and mutant offsprings. Olig2fl/fl (Yue et al., 2006) mice were crossed with Ptchfl/+;hGFAP-Cre+/− to generate Olig2cKO (Ptchfl/fl;Olig2fl/fl;hGFAP-Cre+/− ) mice. The above control mice developed and behaved the same as wild-type mice. We bred Olig2-TK (Lu et al., 2016) mice with Gnasfl/fl mice to produce GFAP-Gnas;Olig2-TK mice. Olig2-GFP (MMRRC, Jackson Laboratory) mice were also bred with Gnasfl/fl mice to monitor Olig2-expressing cells. We used both male and female mice for the study. The mouse strains used in this study were generated and maintained on a mixed C57BL/6; 129Sv; CD-1 background and were housed in a vivarium with a 12-hr light–12-hr dark cycle. Immunodeficient NSG mice (Charles River) were used for allograft transplantation. All studies complied with all relevant animal use guidelines and ethical regulations. All animal use and study protocols were approved by the Institutional Animal Care and Use Committee at the Cincinnati Children′s Hospital Medical Center, Ohio, USA.
Human Tumor Samples
All human patient samples were obtained with consent under approval and oversight by the Institutional Review Board (IRB) committees of Cincinnati Children’s Hospital Medical Center, the Hospital for Sick Children at the University of Toronto, St. Jude Children’s Research Hospital, and Seattle Children’s Hospital. The SHH subtypes of human MB samples were defined by immunohistochemistry GAB1 and YAP.
Human MB Single Cell RNA-seq Datasets and Gene Expression Datasets
We analyzed the single cell RNA-seq expression profiles of human MB from the European Genome-phenome Archive (EGA; https://www.ebi.ac.uk/ega/studies/) repositories and the accession number is EGAS00001003170 (Vladoiu et al., 2019). We performed unsupervised trajectory analysis by R package TSCAN (https://zhiji.shinyapps.io/TSCAN/). Human MB patient survival and gene expression data were analyzed from R2 Genomics Analysis and Visualization Platform (https://hgserver1.amc.nl/cgi-bin/r2/main.cgi?&species=hs).
Mouse and Human Tumor Cell Culture
For primary tumor cells, microdissected tumor tissues were digested with Trypsin/DNase (1 mg/ml; Worthington), triturated to obtain a single-cell suspension, and then centrifuged through a 35–65% Percoll gradient (Sigma) as described previously (Yang et al., 2008). Cells from the 35–65% interface were suspended in the GNP culture medium (Neurobasal (Gibco) with 2 mM L-glutamine, 0.45% D-glucose, B27 supplement (without vitamin A, Invitrogen, Carlsbad, CA), 16 µg/ml N-acetyl-l-cysteine and penicillin/streptomycin). We pre-plated GNPs onto poly-d-lysine (100 µg/ml)-coated plates for 1 h at 37 °C twice, and then tran sferred them to Poly-DL-ornithine (Sigma-Aldrich, P8638) coated plates for culture. We treated GNPs with verteporfin (MedChemExpress, HY-B0146) or CD532 (Millipore, 532650) at concentrations indicated in the text for 24 h. For in vitro proliferation assays, we labeled GNPs with BrdU (50 µg/ml) for 48 h followed by immunostaining.
Daoy cells which were derived from biopsy material taken from a tumor in the posterior fossa of a 4 year old boy, were cultured with complete standard growth medium (high glucose Dulbecco’s Modified Eagle’s Medium, 1% Pen/Strep, 1% L-Glutamine, 10% Fetal Bovine Serum) and incubated at 37°C in a suitable incubator with a 5% CO2 in air atmosphere. The cell line was purchased from ATCC (https://www.atcc.org) and was tested and authenticated. We treated Daoy cells with verteporfin or CD532 at concentrations indicated in the text for 24 h. For in vitro proliferation assays, we labeled Daoy cells with BrdU (50 µg/ml) for 3 h followed by immunostaining.
For the sphere formation assay, the microdissected tumor tissues were dissociated into single cell suspensions and cultured in the serum-free growth medium (1 × 104 cells/ml) containing B27 supplement, 2-mM glutamine, 1% penicillin/streptomycin, human-EGF (20 ng/ml, Peprotech) and human-bFGF (10 ng/mL, Peprotech).
METHOD DETAILS
Tissue Processing, Antibodies and Immunostaining
Mouse brains at defined ages were dissected and fixed overnight in 4% (w/v) paraformaldehyde and processed for cryosectioning or paraffin embedding and sectioning. The procedure for immunostaining was described previously (Lu et al., 2000). Briefly, for tissue immunostaining, cryosections or pre-deparaffinized tissue sections were incubated overnight in primary antibodies diluted in block solution (PBS with 5% v/v normal goat serum (Sigma-Aldrich) and 0.3% v/v Triton X-100). After washing with PBS for five times, sections were then either incubated overnight with the biotinylated goat anti-mouse IgG antibody (Vector Laboratories, BA-9200), followed by using the ABC avidin/biotin method to visualize staining signals under light microscopy with the peroxidase/diaminobenzidine (DAB) method, or incubated with corresponding fluorophore-conjugated secondary antibodies (Jackson ImmunoResearch) under fluorescent microscopy.
For cell immunostaining, cultured cells were fixed with 4% PFA for 10 min and washed five times with PBS, then placed in blocking solution for 30 min. We incubated primary antibodies in blocking solution with proper dilutions and stained cells for 1 h at room temperature. For BrdU staining, cells or tissue sections were denatured with 0.1N HCl for 1 h in 37 °C water bath. After denaturation, sections were neutralized with 0.1 M Borax, pH 8.5 (Sigma) for 10 min. Sections were washed with 0.3% Triton X-100/1×PBS (wash buff er) 3 times and blocked with 5% normal donkey serum (Sigma-Aldrich) in wash buffer for 1 h at room temperature. Mouse-anti BrdU (BD Bioscience, 1:500) antibody was used to label BrdU overnight at 4 °C. DAPI counterstain was included in the final washes before the samples were mounted in Fluoromount G (SouthernBiotech) for microscopy.
Primary antibodies used in this study were as follows: Nestin (Abcam, ab22035), OLIG2 (Millipore, ab9610; MABN50), SOX2 (Santa Cruz Biotechnology, sc-17320), NEUROD1 (Abcam, ab16508), NEUN (Millipore, MAB377), Ki67 (Thermo Sci, clone SP6), BrdU (BD Bioscience 347580; Abcam, ab6326), ZIC1 (Rockland, 200–401-159), YAP (Cell Signaling, 4912; Abcam, Cat# ab52771), GAB-1 (Santa Cruz Biotechnology, sc-133191); MYCN (Abcam, ab24193), Cleaved Caspase 3 (Cell Signaling, 9661), PDGFRα (BD Bioscience 558774), CC1 (Oncogene Research, OP80), MAG (Millipore, MAB1567), MOG (DSHB, 8–18C5), Sox10 (Abcam, ab180862), Ascl1(BD Bioscience, 556604) and ATOH1 (Abcam, ab85513).
Single-cell Isolation and Library Preparation for scRNA-seq
To minimize the intra-tumor variability, rather than performing single-cell RNA-seq for each tumor/mouse, we pooled the tumor tissues from multiple GFAP-Ptch mice (at least 3 animals) used for single-cell RNA-seq at the indicated stages. The tissues were digested by TrypLE added with collagenase type I, and single cell suspension was treated by Red Blood Cell Lysis Buffer (Sigma, 11814389001), the sequencing followed 10X Genomics Chromium and Drop-seq protocols as described previously (Weng et al., 2019).
For 10X Genomics single cell gene expression, we followed Chromium™ Single cell 3’ User Guide. A Single Cell Gel Bead kit (120217), Single cell chip kit (120219) and Single cell library kit (120218) were used along with a 10× GemCode Single Cell Instrument, per the manufacturer’s specifications and manuals (document CG00011; revision B). Base on the distribution of cells ordered by percentage of mitochondrial genes and detected gene numbers, we excluded those cells with either more than 5000 detected genes or less than 500, and an average mitochondrial expression level more than 0.5. For Drop-seq (highly parallel droplet-based single-cell transcriptomics), we followed Macosko procedure (Macosko et al., 2015), briefly, monodisperse droplets ∼1 nl in size were generated using the microfluidic device, in which barcoded microparticles, suspended in lysis buffer. As soon as droplet generation was complete, droplets were broken with perfluorooctanol in 30 ml of 6× SSC. Th e addition of a large aqueous volume to the droplets reduces hybridization events after droplet breakage. The beads were then washed and resuspended in a reverse transcriptase mix, followed by a treatment with exonuclease I to remove unextended primers, and PCR amplified. The PCR reactions were purified and pooled, and the amplified cDNA quantified on a BioAnalyzer High Sensitivity Chip (Agilent). cDNA size selection was performed to reduce reads that end up just giving PolyA tail sequences. The cDNA was then fragmented and amplified for sequencing with the Nextera XT DNA sample prep kit (Illumina) using custom primers that enabled the specific amplification of only the 3’ ends. The libraries were purified, quantified, and then sequenced on the Illumina Hiseq 2500. Base on the distribution of cells ordered by percentage of mitochondrial genes and detected gene numbers, we excluded those cells with either more than 3000 detected genes or less than 500, and an average mitochondrial expression level more than 0.5.
Single Cell RNA-seq Data Analysis
For 10X Genomics Chromium scRNA-seq, we performed unsupervised clustering by R package Seurat (Satija et al., 2015) for the scRNA-seq data. The highly variable genes were identified from these cells using Seurat with the default setting for mouse datasets, which were used for principle component analysis (PCA). scRNA-seq data that met quality control criteria were used for transcriptomic analysis as previously described (Weng et al., 2019). The statistically significant PCs were used for two-dimension t-distributed stochastic neighbor embedding (t-SNE). We used the neural cell type markers (Zhang et al., 2014) for the identification of non-GNP cell types in t-SNE clusters. To analyze the trajectory development of tumor cells, an unsupervised pseudo developmental timeline of single cells was calculated with the package TSCAN (Ji and Ji, 2016), using the most variable genes as time ordering genes. Based on the established differentiation direction of granule cells, the direction of pseudotime axis was determined.
Transient Transfections
For siRNA knockdown in Daoy cells, we used Lipofectamine RNAiMAX (Life Technologies) per the manufacturer’s instructions; cells were harvested after 72 hr and then subjected to qRT-PCR. siOlig2: SASI_Hs01_00092187 and SASI_Hs02_00340317; scrambled control siRNA: MISSION siRNA Universal Negative Control #1 (SIC001).
RNA Isolation and Real-time RT-PCR analysis
RNAs were isolated with the RNeasy Plus Mini kit (Qiagen) from cultured cells or snap-frozen tumors. cDNA was synthesized from 1 mg RNA using iScript Reverse Transcription Supermix (BioRad) according to the manufacturer’s instructions. qRT-PCR was performed using the StepOnePlus Real-Time PCR System (Applied Biosystems). qRT-PCR was performed using quantitative SYBR green PCR mix (BioRad).
Western Blotting
Tumor tissues were lysed in modified RIPA buffer (50 mM Na-Tris, pH 7.4, 150 mM NaCl, 1% (v/v) NP-40, 0.25% sodium deoxycholate, 1 mM dithiothreitol, 10 mM NaF, 1 mM active sodium vanadate, 1 mM PMSF and 1× a cocktail of cOmplete p rotease inhibitors (Roche Applied Science) and centrifuged at 13,000 r.p.m. for 15 min at 4 °C. After the determination of protein concentration (Bio-Rad), the lysates were separated by 4–12% SDS-PAGE. We used antibodies against YAP (Cell Signaling Technology, 4912), MYCN (Abcam, ab24193) and GAPDH (Millipore, MAB374). Bands were visualized with secondary antibodies conjugated to horseradish peroxidase (Bio-Rad) and ECL western blotting detection reagents (Pierce) per the manufacturer’s instructions.
ChIP-Seq, ATAC-seq and Data Analysis
ChIP assays were performed as described previously with minor modifications (Wu et al., 2018). Dissociated tissues were fixed for 15 min at room temperature with 1% formaldehyde-containing medium. Nuclei were isolated and sonicated in sonication buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 0.5 mM EGTA and protease inhibitor cocktail). Sonicated chromatin (~300 µg) was used for immunoprecipitation by incubation with appropriate antibodies (4 mg) overnight at 4 °C. Pre-rinsed magnetic protein A/G beads (50 ml) were added to each ChIP reaction and reactions were incubated for 1 hr at 4 °C. The beads were then incubated in 200 ml elution buffer at 65 °C for 20 min to elute immunoprecipitated materials. The ChIP-seq libraries were prepared using NEBNext ChIP-seq Library Prep Master Mix Set for Illumina (NEB catalogue number E6240L) and then run on the Illumina sequencer HiSeq 2500. We used antibodies against OLIG2 (Millipore, ab9610) and H3K27ac (Rabbit, Abcam, 4729) for ChIP. The crosslinked and sonicated chromatins without immunoprecipitation were used as input controls. For ChIP-seq in tumor tissues, we performed three biological repeats for OLIG2 and two biological repeats for H3K27ac. For ChIP-seq in the normal cerebellum (from three animals), we have performed it once for OLIG2 and H3K27ac. To minimize the intra-tumor variability, we pooled the tumor tissues from multiple GFAP-Ptch mice (at least three animals) using for ChIP-seq. The signals from independent experiments are consistent with each other.
ATAC-seq was performed as previously described (Buenrostro et al., 2015). Briefly, dissociated tumor cells (50,000 cells) were spun down at 500 x g for 5 min at 4 °C, and lysed in cold Lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630). After spinning down at 500 x g for 10 min at 4 °C, nuclei were resuspended in transposition mix containing TD (2x reaction buffer), TDE1 (Nextera Tn5 Transposase) at 37 °C for 30 min. Immediately following transposition, DNA were purified using a Qiagen MinElute PCR Purification Kit. Transposed DNA fragments were subsequently amplified and the amplified library was purified using Qiagen MinElute PCR Purification Kit. Libraries were generated using the Ad1_noMX and Ad2.1–2.2 barcoded primers (Buenrostro et al., 2015) and were amplified for 11 total cycles. Libraries were purified with AMPure beads (Agencourt) to remove contaminating primer dimers. All libraries were sequenced on the Illumina HiSeq 2500 with 75 bp single-end reads.
All sequencing data were mapped to mouse genome assembly mm10, and ChIP-seq peak calling was performed as previously described using model-based analysis of ChIP-seq (MACS, version 1.4.2; http://liulab.dfci.harvard.edu/MACS) with default parameters to get primary binding regions. To ensure that our data were of high quality and reproducibility, we called peaks with enrichment ≥10-fold over control (p ≤ 10−9) and compared the peak sets using the ENCODE overlap rules. The identified primary regions were further filtered using the following criteria, to define a more stringent protein-DNA interactome: (1) the p value cutoff was set to ≤10−9; and (2) we required an enrichment of six-fold and peak height >5. The genome-wide distribution of protein-binding regions was determined by HOMER (http://homer.salk.edu/homer/index.html) in reference to UCSC mm10. For all ChIP-seq data sets, WIG files were generated with MACS, which were subsequently visualized using Mochiview v1.46. Occupancy was analyzed with Pearson’s correlation and ToppCluster (https://toppcluster.cchmc.org/). ChIP-seq heat maps were ordered by strength of binding. The heat maps were drawn using the Heatmap tools provided by Cistrome (http://cistrome.org/ap).
RNA Sequencing and Data Analysis
RNA from the normal cerebella, tumor tissues, OLIG2-GFP+ cells and OLIG2-GFP- cells were extracted using TRIzol (Life Technologies), followed by purification using a RNeasy Mini Kit (Qiagen). RNA-seq was performed using two individual animals for GFAP-Ptch tumors and Olig2cKO tumors, three individual animals for normal cerebella and Atoh1-Lats1/2 tumors, and three individual animals for OLIG2-GFP+ and OLIG2-GFP- cells. RNA-seq libraries were prepared using the Illumina TruSeq RNA Library Prep Kit v2 and sequenced by a HiSeq 2500 sequencer. RNA-seq reads were aligned to mm10 using TopHat with default settings (http://tophat.cbcb.umd.edu/). We used Cuff-diff to (1) estimate fragments per kilobase of transcript per million mapped reads (FPKM) values for known transcripts and to (2) analyze differentially expressed transcripts. In all differential expression tests, a difference was considered significant if the q value was less than 0.05 (Cuff-diff default). A heat map of gene expression was generated using the R language (version 3.2.1) and was generated on the basis of log2 (FPKM). Gene ontology analysis of gene expression changes was performed using Gene Set Enrichment Analysis (GSEA; http://www.broadinstitute.org/gsea/index.jsp).
Allograft Transplantations
Immunodeficient NSG mice (Charles River) were used for allograft transplantation. Isolated tumor cells were plated in GNP culture medium and harvested with 0.25% trypsin and 0.02% EDTA for 2 min, washed twice with Hank’s balanced salt solution (HBSS), and resuspended in Ca2+ and Mg2+-free HBSS. Cell viability was determined by trypan blue exclusion. Only single-cell suspensions with more than 90% viability were used for in vivo allograft studies. 1 × 106 tumor cells diluted in Matrigel (Corning) and GNP culture medium: 1:2 ratio were injected subcutaneously into the flanks. For intracranial transplantation, OLIG2-GFP+ cells or OLIG2-GFP- cells were injected into the cerebella of NSG mice using a stereotaxic frame with a mouse adaptor. 6–9 week old NSG mice were anaesthetized using gaseous isoflurane and immobilized in a stereotaxic head frame. An incision was made in the midline of the scalp over the cerebellum, and a small hole was made in the skull using a beveled 18G needle 1mm lateral and 2mm posterior to bregma. Freshly sorted cells were injected 2mm deep to the surface of the skull over the course of 2 min, and the needle was left in place for another 2 min to avoid reflux. After removing the mouse from the frame, 1–2 drops of 0.25% (2.5 mg/ml) bupivicaine were applied along the incision for postoperative analgesia and the incision was sutured using catgut sutures.
In Vivo Drug Administration
Atoh1-Ptch mutant mice at P15 were randomized to receive vehicle, verteporfin (100 mg/kg) or CD532 (25 mg/kg) administered via intraperitoneal injection. Verteporfin was dissolved in DMSO (100 mg/ml), aliquoted, and stored at −80 °C. Worki ng solution was prepared at 10 mg/ml in PBS freshly before use. Mice were administered at a dose of 100 mg/kg every other day. CD532 was formulated in 5% DMSO and 95% polyethylene glycol 300 (PEG 300) and administered at a dose of 25 mg/kg twice per week. For cisplatin and cyclophosphamide treatment, Atoh1-Ptch mutant mice at P30 were randomized to receive vehicle, cisplatin (5 mg/kg) or cyclophosphamide (130 mg/kg). Cisplatin (Cayman Chemical) was diluted in saline at 1 mg/mL and injected i.v. for day 1 and cyclophosphamide (Cayman Chemical) was dissolved in saline at 16.25 mg/mL and injected i.p. for day 2–6. Brain tissues were harvested around 2 weeks after treatment and subjected to immunostaining. For GCV treatment, animals were administrated with GCV (50 mg/kg) by i.p. injection twice daily at the indicated period. Brain tissues were harvested at indicated stages and subjected to immunostaining.
Senescence-associated β-galactosidase Staining
For senescence-associated β-galactosidase (SA-β-Gal) staining, we used Senescence β-Gal Staining Kit (Cell Signaling Technology, 9860) following the manufacturer’s instructions.
QUANTIFICATION AND STATISTICAL ANALYSIS
All analyses were done using GraphPad Prism 6.00 (San Diego, CA; http://www.graphpad.com/). Data are shown in dot plots or bar graphs as mean ± SEM. p < 0.05 is deemed statistically significant. Data distribution was assumed to be normal, but this was not formally tested. Statistical analysis was performed using two-tailed unpaired Student’s t-tests between two samples and one-way ANOVA with Tukey’s post hoc analysis for multiple comparisons, log-rank test for survivals or as indicated. Permutation test was carried out based on the paired t-test statistic and 100,000 permutations. Quantifications were performed from at least three experimental groups in a blinded fashion. The n value was defined as the number of experiments that were repeated independently with similar results. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those generally employed in the field. No randomization was used to collect all the data, but data were quantified with blinding. No animals or data points were excluded from analyses.
Supplementary Material
Table S1. Primer sequences for q-PCR and genotyping. Related to STAR Methods.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Nestin | Abcam | Cat# ab22035; RRID: AB_446723 |
| Mouse Monoclonal anti-OLIG2 | Millipore | Cat# MABN50; RRID:AB_10807410 |
| Rabbit Polyclonal anti-OLIG2 | Millipore | Cat# AB9610; RRID:AB_10141047 |
| Goat polyclonal anti-SOX2 | Santa Cruz Biotechnology | Cat# sc-17320; RRID:AB_2286684 |
| Rabbit polyclonal anti-NEUROD1 | Abcam | Cat# ab16508; RRID:AB_470254 |
| Mouse Monoclonal anti-NEUN | Millipore | Cat# MAB377; RRID:AB_2298772 |
| Rabbit Monoclonal anti-Ki67 | Thermo Fisher Scientific | Cat# MA5–14520; RRID:AB_1097948 |
| Rat Monoclonal anti-BrdU | Abcam | Cat# ab6326; RRID: AB_305426 |
| Rabbit Polyclonal anti-ZIC1 | Rockland Immunochemicals Inc. | Cat# 200–401-159; RRID:AB_2219354 |
| Rabbit Polyclonal anti-YAP | Cell Signaling | Cat# 4912; RRID: AB_2218911 |
| Mouse monoclonal anti-GAB1 | Santa Cruz Biotechnology | Cat# sc-133191; RRID:AB_2107855 |
| Rabbit polyclonal anti-MYCN | Abcam | Cat# ab24193; RRID:AB_776816 |
| Rabbit Polyclonal anti-Cleaved Caspase 3 | Cell Signaling | Cat# 9661; RRID:AB_2341188 |
| Rat Monoclonal anti-PDGFRα | BD Bioscience | Cat# 558774; RRID:AB_397117 |
| Mouse monoclonal anti-APC (CC1) | Oncogene Research | Cat# OP80; RRID:AB_2057371 |
| Mouse monoclonal anti-MAG | Millipore | Cat# MAB1567; RRID:AB_2137847 |
| Mouse monoclonal anti-MOG | Millipore | Cat# MAB5680; RRID:AB_1587278 |
| Rabbit monoclonal anti-SOX10 | Abcam | Cat# ab180862; RRID: AB_2721184 |
| Mouse monoclonal anti-ASCL1 | BD Bioscience | Cat# 556604; RRID:AB_396479 |
| Rabbit polyclonal anti-ATOH1 | Abcam | Cat# ab85513; RRID:AB_1924798 |
| Mouse Monoclonal anti-GAPDH | Millipore | Cat# MAB374; RRID: AB_2107445 |
| Rabbit Polyclonal anti-H3K27ac | Active motif | Cat# 39135, RRID:AB_2614979 |
| Alexa Fluor® 488 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch Inc. | Cat# 711–545-152 |
| Cy™3 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch Inc. | Cat# 711–165-152 |
| Cy™5 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch Inc. | Cat# 711–175-152 |
| Alexa Fluor® 488 AffiniPure Donkey Anti-Rat IgG (H+L) | Jackson ImmunoResearch Inc. | Cat# 712–545-153 |
| Cy™5 AffiniPure Donkey Anti-Rat IgG (H+L) | Jackson ImmunoResearch Inc. | Cat# 712–175-150 |
| Alexa Fluor® 488 AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson ImmunoResearch Inc. | Cat# 705–545-147 |
| Cy™3 AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson ImmunoResearch Inc. | Cat# 705–165-147 |
| Cy™5 AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson ImmunoResearch Inc. | Cat# 705–175-147 |
| Cy™3 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson ImmunoResearch Inc. | Cat# 715–165-150 |
| Alexa Fluor® 488 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson ImmunoResearch Inc. | Cat# 715–545-150 |
| Cy™5 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson ImmunoResearch Inc. | Cat# 715–175-151 |
| Biotinylated Goat Anti-Mouse IgG Antibody | Vector Laboratories | Cat# BA-9200 |
| Biological Samples | ||
| Mouse cerebellum | This study | N/A |
| Mouse MB tumors | This study | N/A |
| Human MB tumors | Cincinnati Children’s Hospital | N/A |
| Human MB tumors | The Hospital for Sick Children | N/A |
| PDX tumors derived from human TB13–5634 cell line | St. Jude Children’s Research Hospital | N/A |
| PDX tumors derived from human Med-314FH cell line | Fred Hutchinson Cancer Research Center | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 5-bromo-2’-deoxyuridine (BrdU) | Sigma-Aldrich | Cat# B5002 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| Verteporfin | Selleck Chemicals | Cat# S1786 |
| Ganciclovir | APP Pharmaceuticals, LLC | Cat# 315110 |
| Cisplatin | Cayman Chemical | Cat# 13119 |
| Cyclophosphamide (hydrate) | Cayman Chemical | Cat# 13849 |
| Palbociclib | LC Labs | Cat# P-7788 |
| Bupivicaine | Hospira | Cat# 00409361301 |
| CD532 | Millipore | Cat# 532605 |
| Red Blood Cell Lysis Buffer | Sigma-Aldrich | Cat# 11814389001 |
| iTaq™ Universal SYBR® Green Supermix | Bio-rad Inc. | Cat# 1725120 |
| RNAiMAX | ThermoFisher Scientific | Cat# 13778030 |
| TRIzol reagent | ThermoFisher Scientific | Cat# 15596018 |
| iScript™ Reverse Transcription Supermix | Bio-rad Inc. | Cat# 170–8841 |
| EGF | Peprotech | Cat# 100–15 |
| bFGF | PeproTech | Cat# 100–18B |
| TrypLE | GIBCO | Cat# A12177 |
| Collagenase type I | ThermoFisher Scientific | Cat# 17018029 |
| Neurobasal | ThermoFisher Scientific | Cat# 21103049 |
| B27 | Invitrogen | Cat# 12587010 |
| poly-D-lysine | ThermoFisher Scientific | Cat# A3890401 |
| poly-DL-ornithine | Sigma-Aldrich | Cat# P8638 |
| Critical Commercial Assays | ||
| Chromium Single Cell 3’ Library & Gel Bead Kit v2, 16 rxns PN | 10x Genomics | Cat# 120237 |
| Senescence β-Gal Staining Kit | Cell Signaling Technology | Cat# 9860 |
| Chromium Single Cell A Chip Kit, 48 rxns PN | 10x Genomics | Cat# 120236 |
| Chromium i7 Multiplex Kit, 96 rxns PN | 10x Genomics | Cat# 120262 |
| NEBNext ChIP-seq Library Prep Master Mix Set for Illumina | New England Biolabs | Cat# E6240L |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat# FC-131–1024 |
| RNeasy Plus Mini Kit | Qiagen | Cat# 74134 |
| TDE1 | Illumina | Cat# FC-121–1030 |
| Agencourt AMPure XP | Beckman coulter | Cat# A64880 |
| DAB Peroxidase (HRP) Substrate Kit | Vector Laboratories | Cat# SK-4100 |
| TruSeq RNA Library Prep Kit v2 | Illumina | Cat# RS-122–2001 |
| MinElute PCR Purification Kit | Qiagen | Cat# 28004 |
| Deposited Data | ||
| Raw and processed data | This paper | GSE120974 |
| Expression array data of human MB | (Cavalli et al., 2017) | GEO: GSE85218 |
| Single-cell RNA-seq data of human MB | (Vladoiu et al., 2019) | EGAS00001003170 |
| OLIG2 ChIP-seq in Glioma | (Lu et al., 2016) | GEO: GSE80089 |
| Experimental Models: Cell Lines | ||
| Human: Daoy | ATCC | Cat# HTB-186 |
| Mouse: GNPs | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Ptchlox/lox | (Yang et al., 2008) | N/A |
| Mouse: Gnaslox/lox | (He et al., 2014) | N/A |
| Mouse: Lats1lox/lox | (Wu et al., 2018) | N/A |
| Mouse: Lats2lox/lox | (Wu et al., 2018) | N/A |
| Mouse: Olig2lox/lox | (Lu et al., 2016) | N/A |
| Mouse: hGFAP-Cre | (Yang et al., 2008) | N/A |
| Mouse: Atoh1-Cre | (Yang et al., 2008) | N/A |
| Mouse: Olig2-GFP (B6;FVB-Tg(Olig2-EGFP/Rpl10a)JD97Htz/J) | Jackson Laboratory | Stock No: 030265; RRID:IMSR_JAX:030265 |
| Mouse: Olig2-TK | (Lu et al., 2016) | N/A |
| Mouse: NOD SCID Mouse | Charles River | Stock No:394; RRID:IMSR_CRL:394 |
| Oligonucleotides | ||
| OLIG2 siRNA-1: GGACAAGAAGCAAAUGACA[dT][dT] | Sigma-Aldrich | SASI_Hs01_00092187 |
| OLIG2 siRNA-2: GAUGUUCUCUCCGGGACCU[dT][dT] | Sigma-Aldrich | SASI_Hs02_00340317 |
| MISSION siRNA Universal Negative Control #1 | Sigma-Aldrich | Cat#: SIC001 |
| Ad1_noMX: AATGATACGGCGACCACCGAGATCTACACTCGTCGGCAGCGTCAGATGTG | (Buenrostro et al., 2015) | N/A |
| Ad2.1: CAAGCAGAAGACGGCATACGAGATTCGCCTTAGTCTCGTGGGCTCGGAGATGT | (Buenrostro et al., 2015) | N/A |
| Ad2.2: CAAGCAGAAGACGGCATACGAGATCTAGTACGGTCTCGTGGGCTCGGAGATGT | (Buenrostro et al., 2015) | N/A |
| See Table S1 for the primer sequences for q-PCR and genotyping | N/A | N/A |
| Software and Algorithms | ||
| R language | R Core Team (2016) The R Project for Statistical Computing | http://www.r-project.org |
| Seurat | (Macosko et al., 2015) | http://satijalab.org/seurat/ |
| Mochiview v1.46 | University of California, San Francisco | http://www.johnsonlab.ucsf.edu/mochiview |
| Cistrome | Harvard University | http://cistrome.org/ |
| Altanalyze | Cincinnati Children’s Hospital Medical Center | http://www.altanalyze.org/ |
| TopHat | Computational Biology at the Johns Hopkins University | http://tophat.cbcb.umd.edu |
| GraphPad Prism 6.00 | GraphPad | www.graphpad.com |
| Toppcluster | Cincinnati Children’s Hospital Medical Center | https://toppcluster.cchmc.org/ |
| HOMER | Integrative Genomics and Bioinformatics core at the Salk Institute | http://homer.ucsd.edu/homer/ |
| TSCAN | Johns Hopkins University | https://zhiji.shinyapps.io/TSCAN/ |
| MACS version 1.4.2 | Harvard University | http://liulab.dfci.harvard.edu/MACS |
| Gene Set Enrichment Analysis (GSEA) | Broad Institute | http://software.broadinstitute.org/gsea/index.jsp |
| R2 | The Academic Medical Center (AMC) in Amsterdam | http://r2.amc.nl |
| ccRemover | Barron and Li, 2016 | https://cran.r-project.org/web/packages/ccRemover/index.html |
| Bowtie2 | Johns Hopkins University | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
Significance.
By single-cell transcriptomics of Sonic Hedgehog (SHH)-medulloblastoma, we found unexpectedly that OLIG2+ glial lineage progenitors are the rapidly propagating cells during the initial phase of tumorigenesis, although in full-blown tumors these cells are quiescent and have stem-like properties. OLIG2+ progenitors are enriched in therapy-resistant and recurrent tumors and may serve as a niche for tumor re-initiation. Eradication of proliferative OLIG2+ progenitors abrogated medulloblastoma initiation and growth. OLIG2-driven reprogramming activates HIPPO and AURORA-A/MYCN pathways to promote tumorigenesis, and blocking these pathways reduced tumor growth. Demonstrating clinical relevance, high levels of OLIG2 expression are predictive of poor outcome in human patients with SHH-medulloblastomas. Thus, our studies provide insights into tumor stem/initiating cell niche and signaling network therapeutic vulnerabilities of medulloblastoma.
Highlights:
Single-cell-omics reveal OLIG2+ glial progenitors as tumor-initiating cells in MB
OLIG2+ cells are quiescent stem-like in full-blown MB but re-emerge during relapse
Ablation of mitotic OLIG2+ cells or deletion of OLIG2 impedes MB tumor initiation
OLIG2 activates HIPPO-YAP and AURORA-A/MYCN oncogenic networks to promote MB growth
ACKNOWLEDGMENTS
The authors thank Randy Johnson for Lats1/2 floxed mice, Robert Wechsler-Reya for Ptch1 floxed mice, Lee Weinstein for Gnas floxed mice, Bernd Fritzsch for Atoh1-Cre line, Ed Hurlock for critical reading, and Lily Qian, Sean Ogurek and Yunhan Yang for technical support. This study was funded by grants from the NIH (R01 NS078092 and R01 NS075243) to Q.R.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
Accession Codes
All high-throughput data mentioned in the paper are publicly available from GEO under accession GSE120974.
REFERENCES:
- Ahlfeld J., Favaro R., Pagella P., Kretzschmar HA., Nicolis S., and Schuller U. (2013). SOX2 requirement in sonic hedgehog-associated medulloblastoma. Cancer research 73, 3796–3807. [DOI] [PubMed] [Google Scholar]
- Archer TC., Mahoney EL., and Pomeroy SL. (2017). Medulloblastoma: Molecular Classification-Based Personal Therapeutics. Neurotherapeutics 14, 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aruga J., Yokota N., Hashimoto M., Furuichi T., Fukuda M., and Mikoshiba K. (1994). A Novel Zinc-Finger Protein, Zic, Is Involved in Neurogenesis, Especially in the Cell Lineage of Cerebellar Granule Cells. J Neurochem 63, 1880–1890. [DOI] [PubMed] [Google Scholar]
- Ayrault O., Zhao H., Zindy F., Qu C., Sherr CJ., and Roussel MF. (2010). Atoh1 inhibits neuronal differentiation and collaborates with Gli1 to generate medulloblastoma-initiating cells. Cancer research 70, 5618–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron M., and Li J. (2016). Identifying and removing the cell-cycle effect from single-cell RNA-Sequencing data. Sci Rep 6, 33892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabetz S., Leary SES., Grobner SN., Nakamoto MW., Seker-Cin H., Girard EJ., Cole B., Strand AD., Bloom KL., Hovestadt V., et al. (2018). A biobank of patient-derived pediatric brain tumor models. Nat Med 24, 1752–1761. [DOI] [PubMed] [Google Scholar]
- Brockmann M., Poon E., Berry T., Carstensen A., Deubzer HE., Rycak L., Jamin Y., Thway K., Robinson SP., Roels F., et al. (2013). Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell 24, 75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD., Wu B., Chang HY., and Greenleaf WJ. (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton DG., and Faragher RG. (2015). Cellular senescence: from growth arrest to immunogenic conversion. Age (Dordr) 37, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli FMG., Remke M., Rampasek L., Peacock J., Shih DJH., Luu B., Garzia L., Torchia J., Nor C., Morrissy AS., et al. (2017). Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 31, 737–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Bardes EE., Aronow BJ., and Jegga AG. (2009). ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic acids research 37, W305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP., Cheng AW., Welstead GG., Kooistra T., Carey BW., Steine EJ., Hanna J., Lodato MA., Frampton GM., Sharp PA., et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez A., Northcott PA., Dalton J., Fraga C., Ellison D., Angers S., Taylor MD., and Kenney AM. (2009). YAP1 is amplified and up-regulated in hedgehog-associated medulloblastomas and mediates Sonic hedgehog-driven neural precursor proliferation. Gene Dev 23, 2729–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson RJ., and Ellison DW. (2008). The origins of medulloblastoma subtypes. Annu Rev Pathol 3, 341–365. [DOI] [PubMed] [Google Scholar]
- Goldberg-Stern H., Gadoth N., Stern S., Cohen IJ., Zaizov R., and Sandbank U. (1991). The prognostic significance of glial fibrillary acidic protein staining in medulloblastoma. Cancer 68, 568–573. [DOI] [PubMed] [Google Scholar]
- Gustafson WC., Meyerowitz JG., Nekritz EA., Chen J., Benes C., Charron E., Simonds EF., Seeger R., Matthay KK., Hertz NT., et al. (2014). Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell 26, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., and Weinberg RA. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Harvey KF., Zhang X., and Thomas DM. (2013). The HIPPO pathway and human cancer. Nature reviews Cancer 13, 246–257. [DOI] [PubMed] [Google Scholar]
- He X., Zhang L., Chen Y., Remke M., Shih D., Lu F., Wang H., Deng Y., Yu Y., Xia Y., et al. (2014). The G protein alpha subunit Galphas is a tumor suppressor in Sonic hedgehog-driven medulloblastoma. Nat Med 20, 1035–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heallen T., Zhang M., Wang J., Bonilla-Claudio M., Klysik E., Johnson RL., and Martin JF. (2011). HIPPO pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332, 458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RM., Kuijper S., Lindsey JC., Petrie K., Schwalbe EC., Barker K., Boult JK., Williamson D., Ahmad Z., Hallsworth A., et al. (2015). Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell 27, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong W., and Guan KL. (2012). The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian HIPPO pathway. Seminars in cell & developmental biology 23, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JY., Kwon MJ., Seo KY., Kim MK., Chae KY., Kim SH., Ki CS., Yoon MS., and Kim DH. (2014). Novel nonsense GNAS mutation in a 14-month-old boy with plate-like osteoma cutis and medulloblastoma. J Dermatol 41, 319–321. [DOI] [PubMed] [Google Scholar]
- Jacobsen PF., Jenkyn DJ., and Papadimitriou JM. (1985). Establishment of a human medulloblastoma cell line and its heterotransplantation into nude mice. J Neuropathol Exp Neurol 44, 472–485. [DOI] [PubMed] [Google Scholar]
- Ji Z., and Ji H. (2016). TSCAN: Pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic acids research 44, e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju J., Liu Q., Zhang Y., Liu Y., Jiang M., Zhang L., He X., Peng C., Zheng T., Lu QR., et al. (2016). OLIG2 regulates Purkinje cell generation in the early developing mouse cerebellum. Sci Rep 6, 30711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool M., Korshunov A., Remke M., Jones DT., Schlanstein M., Northcott PA., Cho YJ., Koster J., Schouten-van Meeteren A., van Vuurden D., et al. (2012). Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta neuropathologica 123, 473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Du F., Yuelling LW., Lin T., Muradimova RE., Tricarico R., Wang J., Enikolopov G., Bellacosa A., Wechsler-Reya RJ., et al. (2013). A population of Nestin-expressing progenitors in the cerebellum exhibits increased tumorigenicity. Nat Neurosci 16, 1737–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon KL., Huillard E., Mehta S., Kesari S., Liu H., Alberta JA., Bachoo RM., Kane M., Louis DN., Depinho RA., et al. (2007). Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 53, 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY., Erkek S., Tong Y., Yin L., Federation AJ., Zapatka M., Haldipur P., Kawauchi D., Risch T., Warnatz HJ., et al. (2016). Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 530, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Sage JC., Miller MR., Verhaak RG., Hippenmeyer S., Vogel H., Foreman O., Bronson RT., Nishiyama A., Luo L., et al. (2011). Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN., Perry A., Reifenberger G., von Deimling A., Figarella-Branger D., Cavenee WK., Ohgaki H., Wiestler OD., Kleihues P., and Ellison DW. (2016). The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta neuropathologica 131, 803–820. [DOI] [PubMed] [Google Scholar]
- Lu F., Chen Y., Zhao C., Wang H., He D., Xu L., Wang J., He X., Deng Y., Lu EE., et al. (2016). Olig2-Dependent Reciprocal Shift in PDGF and EGF Receptor Signaling Regulates Tumor Phenotype and Mitotic Growth in Malignant Glioma. Cancer Cell 29, 669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR., Qian L., and Zhou X. (2019). Developmental origins and oncogenic pathways in malignant brain tumors. Wiley Interdiscip Rev Dev Biol 8, e342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR., Yuk DI., Alberta JA., Zhu ZM., Pawlitzky I., Chan J., McMahon AP., Stiles CD., and Rowitch DH. (2000). Sonic hedgehog-regulateld oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron 25, 317–329. [DOI] [PubMed] [Google Scholar]
- Macosko EZ., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas AR., Kamitaki N., Martersteck EM., et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannoji H., Takeshita I., Fukui M., Ohta M., and Kitamura K. (1981). Glial fibrillary acidic protein in medulloblastoma. Acta neuropathologica 55, 63–69. [DOI] [PubMed] [Google Scholar]
- Manoranjan B., Venugopal C., McFarlane N., Doble BW., Dunn SE., Scheinemann K., and Singh SK. (2013). Medulloblastoma stem cells: modeling tumor heterogeneity. Cancer Lett 338, 23–31. [DOI] [PubMed] [Google Scholar]
- Morfouace M., Shelat A., Jacus M., Freeman BB., Turner D., Robinson S., Zindy F., Wang YD., Finkelstein D., Ayrault O., et al. (2014). Pemetrexed and Gemcitabine as Combination Therapy for the Treatment of Group3 Medulloblastoma. Cancer Cell 25, 516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissy AS., Garzia L., Shih DJ., Zuyderduyn S., Huang X., Skowron P., Remke M., Cavalli FM., Ramaswamy V., Lindsay PE., et al. (2016). Divergent clonal selection dominates medulloblastoma at recurrence. Nature 529, 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA., Shih DJ., Peacock J., Garzia L., Morrissy AS., Zichner T., Stutz AM., Korshunov A., Reimand J., Schumacher SE., et al. (2012). Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 488, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T., Horn S., Brockmann M., Eilers U., Schuttrumpf L., Popov N., Kenney AM., Schulte JH., Beijersbergen R., Christiansen H., et al. (2009). Stabilization of N-Myc is a critical function of Aurora-A in human neuroblastoma. Cancer Cell 15, 67–78. [DOI] [PubMed] [Google Scholar]
- Parsons DW., Li M., Zhang X., Jones S., Leary RJ., Lin JC., Boca SM., Carter H., Samayoa J., Bettegowda C., et al. (2011). The genetic landscape of the childhood cancer medulloblastoma. Science 331, 435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy V., Remke M., Adamski J., Bartels U., Tabori U., Wang X., Huang A., Hawkins C., Mabbott D., Laperriere N., et al. (2016). Medulloblastoma subgroup-specific outcomes in irradiated children: who are the true high-risk patients? Neuro Oncol 18, 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy V., Remke M., Bouffet E., Faria CC., Perreault S., Cho YJ., Shih DJ., Luu B., Dubuc AM., Northcott PA., et al. (2013). Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol 14, 1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy V., and Taylor MD. (2017). Medulloblastoma: From Myth to Molecular. J Clin Oncol 35, 2355–2363. [DOI] [PubMed] [Google Scholar]
- Robinson G., Parker M., Kranenburg TA., Lu C., Chen X., Ding L., Phoenix TN., Hedlund E., Wei L., Zhu X., et al. (2012). Novel mutations target distinct subgroups of medulloblastoma. Nature 488, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel MF., and Hatten ME. (2011). Cerebellum development and medulloblastoma. Curr Top Dev Biol 94, 235–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R., Farrell JA., Gennert D., Schier AF., and Regev A. (2015). Spatial reconstruction of single-cell gene expression data. Nature biotechnology 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller U., Heine VM., Mao J., Kho AT., Dillon AK., Han YG., Huillard E., Sun T., Ligon AH., Qian Y., et al. (2008). Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 14, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta R., Dubuc A., Ward S., Yang L., Northcott P., Woerner BM., Kroll K., Luo J., Taylor MD., Wechsler-Reya RJ., et al. (2012). CXCR4 activation defines a new subgroup of Sonic hedgehog-driven medulloblastoma. Cancer research 72, 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto Y., Nakatani T., Masuyama N., Taya S., Kumai M., Minaki Y., Hamaguchi A., Inoue YU., Inoue T., Miyashita S., et al. (2014). Temporal identity transition from Purkinje cell progenitors to GABAergic interneuron progenitors in the cerebellum. Nat Commun 5, 3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay JW., and Roninson IB. (2004). Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 23, 2919–2933. [DOI] [PubMed] [Google Scholar]
- Smoll NR. (2012). Relative survival of childhood and adult medulloblastomas and primitive neuroectodermal tumors (PNETs). Cancer 118, 1313–1322. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha VK., Mukherjee S., Ebert BL., Gillette MA., Paulovich A., Pomeroy SL., Golub TR., Lander ES., et al. (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. P Natl Acad Sci USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suva ML., Rheinbay E., Gillespie SM., Patel AP., Wakimoto H., Rabkin SD., Riggi N., Chi AS., Cahill DP., Nahed BV., et al. (2014). Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 157, 580–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartling FJ., Savov V., Persson AI., Chen J., Hackett CS., Northcott PA., Grimmer MR., Lau J., Chesler L., Perry A., et al. (2012). Distinct Neural Stem Cell Populations Give Rise to Disparate Brain Tumors in Response to N-MYC. Cancer Cell 21, 601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MD., Northcott PA., Korshunov A., Remke M., Cho YJ., Clifford SC., Eberhart CG., Parsons DW., Rutkowski S., Gajjar A., et al. (2012). Molecular subgroups of medulloblastoma: the current consensus. Acta neuropathologica 123, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanner RJ., Remke M., Gallo M., Selvadurai HJ., Coutinho F., Lee L., Kushida M., Head R., Morrissy S., Zhu X., et al. (2014). Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 26, 33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varelas X. (2014). The HIPPO pathway effectors TAZ and YAP in development, homeostasis and disease. Development 141, 1614–1626. [DOI] [PubMed] [Google Scholar]
- Venugopal C., McFarlane NM., Nolte S., Manoranjan B., and Singh SK. (2012). Processing of primary brain tumor tissue for stem cell assays and flow sorting. J Vis Exp. [DOI] [PMC free article] [PubMed]
- Vladoiu MC., El-Hamamy I., Donovan LK., Farooq H., Holgado BL., Sundaravadanam Y., Ramaswamy V., Hendrikse LD., Kumar S., Mack SC., et al. (2019). Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature, 10.1038/s41586-41019-41158-41587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Garancher A., Ramaswamy V., and Wechsler-Reya RJ. (2018). Medulloblastoma: From Molecular Subgroups to Molecular Targeted Therapies. Annu Rev Neurosci 41, 207–232. [DOI] [PubMed] [Google Scholar]
- Weng Q., Wang J., Wang J., He D., Cheng Z., Zhang F., Verma R., Xu L., Dong X., Liao Y., et al. (2019). Single-Cell Transcriptomics Uncovers Glial Progenitor Diversity and Cell Fate Determinants during Development and Gliomagenesis. Cell Stem Cell 24, 707–723 e708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LMN., Deng Y., Wang J., Zhao C., Wang J., Rao R., Xu L., Zhou W., Choi K., Rizvi TA., et al. (2018). Programming of Schwann Cells by Lats1/2-TAZ/YAP Signaling Drives Malignant Peripheral Nerve Sheath Tumorigenesis. Cancer Cell 33, 292–308 e297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Northcott PA., Dubuc A., Dupuy AJ., Shih DJ., Witt H., Croul S., Bouffet E., Fults DW., Eberhart CG., et al. (2012). Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 482, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZJ., Ellis T., Markant SL., Read TA., Kessler JD., Bourboulas M., Schuller U., Machold R., Fishell G., Rowitch DH., et al. (2008). Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell 14, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue T., Xian K., Hurlock E., Xin M., Kernie SG., Parada LF., and Lu QR. (2006). A critical role for dorsal progenitors in cortical myelination. J Neurosci 26, 1275–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F., Cordenonsi M., and Piccolo S. (2016). YAP/TAZ at the Roots of Cancer. Cancer Cell 29, 783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Chen KN., Sloan SA., Bennett ML., Scholze AR., O’Keeffe S., Phatnani HP., Guarnieri P., Caneda C., Ruderisch N., et al. (2014). An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. Journal of Neuroscience 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q., Wang S., and Anderson DJ. (2000). Identification of a novel family of oligodendrocyte lineage-specific basic helix-loop-helix transcription factors. Neuron 25, 331–343. [DOI] [PubMed] [Google Scholar]
- Zhuo L., Theis M., Alvarez-Maya I., Brenner M., Willecke K., and Messing A. (2001). hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis 31, 85–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sequences for q-PCR and genotyping. Related to STAR Methods.