

Abstract
Abstract. Adult bone tissue is continuously being remodelled and bone mass is maintained by a balance between osteoclastic bone resorption and osteoblastic bone formation. Alteration of osteoblastic cell proliferation may account in part for lack of balance between these two processes in bone loss of osteoporosis. There is calcium (Ca2+) control in numerous cellular functions; however, involvement of capacitative Ca2+ entry (CCE) in proliferation of bone cells is less well investigated. Objectives: The study described here was aimed to investigate roles of CCE in the proliferation of osteoblast‐like MG‐63 cells. Meterials and Methods: Pharmacological characterizations of CCE were undertaken in parallel, with evaluation of the expression of transient receptor potential canonical (TRPC) channels and of cell proliferation. Results: Intracellular Ca2+ store depletion by thapsigargin induced CCE in MG‐63 cells; this was characterized by a rapid transient increase of intracellular Ca2+ followed by significant CCE, induced by conditions that stimulated cell proliferation, namely serum and platelet‐derived growth factor. Inhibitors of store‐operated Ca2+ channels (2‐APB and SKF‐96365) prevented CCE, while voltage‐dependent Ca2+ channel blockers had no effect. Expression of various TRPC channels was shown in the cells, some having been shown to be responsible for CCE. Voltage‐dependent Ca2+ channel blockers had no effect on osteoblast proliferation while thapsigargin, 2‐APB and SKF‐96395, inhibited it. Cell cycle analysis showed that 2‐APB and SKF‐96395 lengthen the S and G2/M phases, which would account for the reduction in cell proliferation. Conclusions: Our results indicate that CCE, likely attributed to the activation of TRPCs, might be the main route for Ca2+ influx involved in osteoblast proliferation.
INTRODUCTION
Bone is a dynamic tissue continuously being remodeled. In the adult, bone mass homeostasis is maintained locally by a balance between osteoclastic bone resorption and osteoblastic bone formation. However, in many individuals, bone mass starts falling at midlife, leading to bone loss, osteoporosis and debilitating fractures. Both reduced bone formation by osteoblasts and increased bone resorption by osteoclasts have been associated with the development of osteoporosis. Osteoblasts ensure bone formation and mineralization through secretion of bone matrix components (type I collagen and non‐collagenous proteins) and they also provide factors essential for differentiation of osteoclasts, such as macrophage colony‐stimulating factor and receptor activator of nuclear factor kappa B ligand. By regulating osteoclast differentiation, osteoblasts play a central role not only in bone formation, but also in maintenance of bone remodeling's delicate balance (Mackie 2003). In this regard, adequate osteoblastic proliferation, differentiation, secretory functions and rate of any apoptosis are essential for both formation and resorption processes, and thereby bone remodelling equilibrium. Clinical and histomorphometric studies have demonstrated that ageing is associated with decrease in bone mass and that decreased bone formation is an important pathogenetic factor (Kragstrup et al. 1983; Parfitt 1991; Brockstedt et al. 1993). In rodent models of ageing research, studies have indicated a deficit of osteoprogenitor cells in the bone marrow, and a reduction in the number of osteoblasts (Liang et al. 1992; Roholl et al. 1994; Quarto et al. 1995; Bergman et al. 1996; Kotev‐Emeth et al. 2000; Chen 2004). Similar observations have been made in humans, showing defects in bone cell proliferation (Pfeilschifter et al. 1993; Kato et al. 1995; Neidlinger‐Wilke et al. 1995; Bergman et al. 1996; D’Ippolito et al. 1999; Martinez et al. 1999). Moreover, systemic ageing has been associated with decreased lifespan and accelerated senescence of human bone marrow osteoprogenitors and of osteoblasts in vitro (Kassem et al. 1997; Stenderup et al. 2003; Peterson et al. 2004). Therefore, during the ageing process, bone formation is likely to be affected by reduction of osteoblast proliferation, differentiation and lifespan (Chan & Duque 2002). Osteoporosis research aims to provide novel understanding of osteoblast proliferation that could provide new therapeutic approaches.
Calcium (Ca2+) influx is implicated in numerous cellular functions such as protein secretion, differentiation, apoptosis and more specifically in cell proliferation (Berridge et al. 2000b). It is generally accepted that capacitative Ca2+ entry (CCE) is triggered in order to ensure proper Ca2+ refilling of intracellular stores (Parekh & Putney 2005) and it is thought to be an essential component of long‐term responses of the cell, including proliferation. Activation of capacitative or store‐operated Ca2+ entry (CCE or SOCE) involves first rapid, transient inositol‐1,4,5‐trisphosphate (InsP3)‐mediated release of Ca2+ from stores, leading to a decrease in Ca2+ concentration in the lumen of the endoplasmic reticulum, followed by activation of plasma membrane Ca2+ channels to generate a sustained influx of Ca2+, CCE. In cells of the osteoblast lineage, Ca2+ channels have been shown to play fundamental roles in cellular responses to external stimuli including both mechanical forces and hormonal signals (Duncan et al. 1998; Iqbal & Zaidi 2005). Although functional and genetic evidence have argued for the expression of voltage‐dependent Ca2+ channels in osteoblasts, few studies have addressed the importance of CCE in osteoblastic functions. Wiemann et al. (1998) first reported CCE in primary cultures of osteoblasts. Induction of CCE by 1,25(OH)2D3 has been shown in rat osteoblast‐like cells (Baldi et al. 2002) and, recently, such CCE has been attributed to transient receptor potential canonical isoform 3 (TRPC3) channels (Baldi et al. 2003). However, the importance of CCE in cell proliferation and the expression of other TRPC channels has not been investigated in osteoblasts. TRPC channels were first identified and characterized in Drosophila (Montell & Rubin 1989) where it forms light‐sensitive and Ca2+‐permeable channels. Mammalian homologues have been identified in many non‐excitable cells. In general, TRPC channels are considered non‐selective Ca2+‐permeable cation channels, although the cation selectivity ratio varies significantly between different members of the family. TRPC channels have been proposed to encode components of store‐operated Ca2+ channels involved in CCE (Vazquez et al. 2004) and in cell proliferation (Golovina et al. 2001; Sweeney et al. 2002).
For this study, we sought to examine the relationship between conditions that stimulate cell proliferation and CCE in human osteoblast‐like MG‐63 cells. We conducted measurements of intracellular Ca2+ and performed pharmacological characterization of Ca2+ signals induced by conditions shown to stimulate cell proliferation, and investigated TRPC channel expression.
MATERIALS AND METHODS
Cell culture
MG‐63 and MC3T3‐E1 cell lines were obtained from the American Type Culture Collection (Rockville, MD, USA) and were grown in Dulbecco's modified Eagle's medium‐F12 without phenol (DMEM‐F12; Invitrogen, Burlington, Ontario, Canada) and α‐MEM (Invitrogen), respectively, supplemented with 10% foetal bovine serum (FBS; Cansera, Etobicoke, Ontario, Canada), 2 mm l‐glutamine, 0.1 mg/mL streptomycin, and 100 U/mL penicillin (ICN Biomedicals, Costa Mesa, CA, USA) at 37 °C in a 5% CO2 humidified atmosphere. Cells were passaged once a week with the aid of trypsin/EDTA solution (Sigma, Oakville, Ontario, Canada).
Measurements of intracellular calcium
MG‐63 cells were cultured in 4‐well Lab‐Tek (Nalge Nunc, Naperville, IL, USA) dishes for 5 days, in supplemented media. They were then transferred to HEPES‐buffered saline solution (mm) (121 NaCl, 5.4 KCl, 0.8 MgSO4, 25 HEPES, 1.8 CaCl2 and 6.0 NaHCO3 at pH 7.3) and loaded with 2 µm Fluo‐3 acetoxymethyl (Molecular Probes, Eugene, OR, USA) with an equivalent volume of 20% pluronic F127 (Molecular Probes) for 45 min at 37 °C in the dark. Thereafter, cells were washed with HEPES‐buffered saline solution and loaded dye was allowed to de‐esterify for 45 min at room temperature in the dark. Following transfer to a Ca2+‐free solution (HEPES‐buffered saline solution without CaCl2), additions were made in an open chamber configuration at room temperature. Cells were examined using a laser scanning confocal (Bio‐Rad Laboratories, Hercules, CA, USA) microscope (Nikon TE300, Tokyo, Japan) with an Apochromatic 40X N.A. 1.0 objective lens. Fluorescence was excited by an argon laser at 488 nm and emission was collected with a 515 filter. Release of Ca2+ from endoplasmic reticulum was induced by addition of 5 µm thapsigargin (Tg) (Sigma). Following addition of Ca2+ to the buffer (final concentration of 1.8 mm), a store‐operated influx could be obtained. Inhibition of store‐operated channels was accomplished by addition of 1‐{β‐[3‐(4‐methoxyphenyl)propoxy]‐4‐methoxyphenethy}‐1H‐imidazole hydrochloride (SKF‐96365) or 2‐aminoethoxydiphenylborate (2‐APB) before addition of Ca2+ to the media. Data were analysed with Laser Sharp 2.1T, Time Course 1.0 software (Hemel Hempstead, UK). The area under the curve was computed using GraphPad Prism version 4.00 for Macintosh (GraphPad Software, San Diego, CA, USA).
Cell proliferation
Cells were seeded at 2000 cells/cm2 and were cultured for 3 days in supplemented DMEM‐F12. To evaluate serum‐regulated proliferation, cells in 24‐well plates (Sarstedt, Montréal, Québec, Canada) were incubated with or without 10% FBS until day 7 and cell counts were performed on trypsinized cells at different intervals of time. Otherwise, cell proliferation was determined in 96‐well plates (Sarstedt) using the microtiter tetrazolium (MTT) assay after 24 or 48 h of treatment. Briefly, 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrasodium bromide (MTT, Sigma) was added to the media at a final concentration of 0.5 mg/mL. Four hours later, formazan crystals generated by cellular reduction of the MTT reagent were dissolved in dimethyl sulfoxide (Sigma) for 30 min at 37 °C and absorbance was determined at 575 nm. Results are expressed as the percentage of MTT values of treated versus control conditions for which initial MTT [absorbance values of the initial day of treatment (MTTi)] were subtracted according to the equation:
| Cell proliferation (%) = [(OD575nm treated – MTTi)/(OD575nm control – MTTi)] × 100. |
Cell cycle analysis
MG‐63 cells were cultured in 100 mm dishes (Sarstedt) and were incubated with Ca2+ channel blockers or appropriate vehicle substance. They were fixed overnight in 70% ethanol and incubated at 37 °C for 30 min in phosphate‐buffered saline (PBS; 2.7 mm KCl, 1.4 mm KH2PO4, 0.5 mm MgCl2, 140 mm NaCl et 10 mm Na2HPO4 at pH 7.4) containing 1.12% citrate with 125 U RNase (GE Healthcare, Baie d’Urfé, Québec, Canada). Cells were then incubated for 1 h with 12.5 µg propidium iodide (PI) (Sigma) and data were acquired by a FACScan flow cytometer (Becton Dickinson, Mountain View, CA, USA) using CellQuest software and analysed with the ModFit LT version 3.0 software (Verity Software House Inc., Topsham, ME, USA).
Determination of expression of TRPC and voltage‐dependent Ca2+ channel by reverse transcription‐polymerase chain reaction
Total RNA was extracted using Trizol (Invitrogen) according to the manufacturer's instructions. Complementary DNA was synthesized using Omniscript reverse transcription (RT) kit (Qiagen, Mississauga, Ontario, Canada) with random hexamer primers. Oligonucleotide primers for polymerase chain reaction (PCR) were designed from coding regions of human TRPC channels and voltage‐dependent Ca2+ channels (VDCC) and special care was paid that complementary sequences were in different exons (1, 2). PCRs were performed with a MultiGene thermal cycler (Labnet, IR System, Edison, NJ, USA) using the Taq PCR core kit (Qiagen). The mixture was initially denatured at 94 °C (3 min) followed by 40 cycles consisting in denaturation at 94 °C (1 min), annealing at 58 °C (30 s) and extension at 72 °C (1 min). PCR products were subjected to electrophoresis on 2% agarose gel and amplicons were visualized by ethidium bromide staining. Cleavage of amplicons by restriction enzymes was used to confirm the specificity of the PCR amplifications.
Table 1.
Primer sequences for the detection of transient receptor potential canonical (TRPC) channel by RT‐PCR
| Gene | Accession number | Primer orientation | Primer sequence | Product size (bp) | Location in sequence |
|---|---|---|---|---|---|
| hTRPC1 | NM_003304 | Sens | 5′‐GTTTCATGATTTTGCTGATCGGA‐3′ | 472 | 1472–1493 |
| Antisens | 5′‐AGCACAATCACAACCACGACATT‐3′ | 1924–1944 | |||
| hTRPC3 | NM_003305 | Sens | 5′‐TCCAAGACGCTGAACGTCAAC‐3′ | 404 | 608–628 |
| Antisens | 5′‐CACTTGCAGAAATAGTCGTGCG‐3′ | 990–1011 | |||
| hTRPC4 | NM_016179 | Sens | 5′‐AGAAGTCGTCGGAGCTGTTGAG‐3′ | 1211 | 549–570 |
| Antisens | 5′‐CCAGAGATATTTGCAGAGGTCCC‐3′ | 1737–1759 | |||
| hTRPC6 | NM_004621 | Sens | 5′‐AGAAGGGGAGAAGGTTAGCTAATCG‐3′ | 692 | 648–671 |
| Antisens | 5′‐CAGTTCATTGCTAAGTTCTAAAGCCG‐3′ | 1324–1339 |
Table 2.
Primer sequences for the detection of voltage‐dependent Ca2+ channel by RT‐PCR
| Gene | Accession number | Primer orientation | Primer sequence | Product size (bp) | Location in sequence |
|---|---|---|---|---|---|
| α1D | NM_000720 | Sens | 5′‐TGCAAGATGACGAGCCTGAG‐3′ | 245 | 5163–5182 |
| Antisens | 5′‐ATGGTTATGATGGTTATGACAC‐3′ | 5386–5407 | |||
| α1C | NM_000719 | Sens | 5′‐CTACCCCAGCACGGTCAGCAC‐3′ | 437 | 5614–5634 |
| Antisens | 5′‐CCTGATGATGAACCAGATGCAAG‐3′ | 6028–6050 | |||
| α1B | NM_000718 | Sens | 5′‐CTCATCCTCCTCCTCGGAGAAG‐3′ | 607 | 6496–6517 |
| Antisens | 5′‐GTCCCAGGGTGCAGTGGTAAC‐3′ | 7083–7103 | |||
| α1A | NM_000068 | Sens | 5′‐CTGATGCTGAATCTCTTTGTCGC‐3′ | 512 | 5683–5705 |
| Antisens | 5′‐CAGTTCATTGCTAAGTTCTAAAGCCG‐3′ | 6174–6194 | |||
| α1E | NM_000721 | Sens | 5′‐ACCTTGTTTGGCAACTACACGC‐3′ | 792 | 2224–2245 |
| Antisens | 5′‐TTCATCCAGACTGCGTTCCTG‐3′ | 2995–3015 |
Western blot analysis
To prepare crude membranes, cells were retrieved in ice‐cold hypotonic lysis buffer (10 mm NaCl, 10 mm Tris pH 7.4, 1 mm EDTA and 3 mm MgCl2) containing protease inhibitors (1 mm Na2VO3, 0.1 mm phenylmethylsulphonyl fluoride, 5 µg/mL leupeptin, 0.5 mm benzamidine, 5 µg/mL aprotinin and 1 mm dithiothreitol), homogenized with a Teflon‐glass homogenizer and were centrifuged at 10 000 g. Supernatants were then centrifuged at 100 000 g for 1 h and pellets were re‐suspended in buffer containing 1% Triton, 150 mm NaCl, 50 mm Tris pH 7.4, 1 mm EDTA, 0.5% Nonidet P‐40 and 60 mm octylglucoside with protease inhibitors. Protein concentration was determined by a MicroBCA protein assay (Pierce, Rockford, IL, USA). Proteins were subjected to sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (7.5% polyacrylamide gel) and were transferred to polyvinylidene difluoride membranes (NEN Life Science Products Inc., Boston, MA, USA). After overnight incubation at 4 °C with 2% blocking agent (GE Healthcare) in Tris‐buffered saline and 0.1% Tween‐20 (TBS‐T), polyvinylidene difluoride membranes were incubated with affinity‐purified polyclonal antibodies specific for various TRPC channels (Chemicon, Temecula, CA, USA; Ab‐5446 for TRPC1, Ab‐5576 for TRPC3, Ab‐5812 for TRPC4 and Ab‐5574 for TRPC6) at room temperature. Membranes were then washed with TBS‐T, were incubated with antirabbit horseradish peroxidase‐conjugated immunoglobulin G and immunoreactive proteins were revealed by enhanced luminol‐linked chemiluminescence detection system (ECL Advance, GE Healthcare). Human brain cell lysate (Calbiochem, La Jolla, CA, USA) was used as positive control for specificity of the antibodies. Also, immunodetection with TRPC antibodies pre‐incubated with the antigen was used to ascertain the specificity of the detected TRPC bands.
Statistical analysis
Statistical comparisons were performed using anova or Student's t‐test (using GraphPad Prism version 3.02). Differences were considered to be significant when P < 0.05.
RESULTS
Stimulation of CCE by thapsigargin‐induced store depletion
In order to assess the presence of CCE in human osteoblast‐like MG‐63 cells, we performed fluorescence measurements of intracellular Ca2+ from MG‐63 cells loaded with Fluo‐3 acetoxymethyl. The fluorescence of 30–40 cells per field was analysed. The individual fluorescence intensities of treated cells were compared to the fluorescence intensity before the stimulation and were used to obtain a mean fluorescence ratio for each experiment. Thapsigargin is an inhibitor of endoplasmic reticulum Ca2+‐ATPase and in the absence of external Ca2+, 5 µm Tg induced a transient increase in intracellular Ca2+ concentration ([Ca2+]i) (Fig. 1), which corresponds to the classical depletion of intracellular Ca2+ stores. When extracellular Ca2+ was reintroduced, a rapid rise in Ca2+ was evoked, which is typical of CCE. In contrast, the cells showed no response to addition of 1.8 mm Ca2+ in the buffer without prior stimulation with Tg (Fig. 1, inset), suggesting no significant Ca2+ influx following the addition of Ca2+ at basal conditions. The majority of cells (77–98%) responded to the addition of Tg and Ca2+. In order to confirm activation of CCE, we used two well‐known inhibitors of store‐operated Ca2+ entry, namely 2‐APB and SKF‐96365. Following Ca2+ store depletion with 5 µm Tg in the absence of extracellular Ca2+, addition of 1.8 mm Ca2+ and 100 µm 2‐APB to the incubation media strongly reduced Ca2+ entry – by 87.3 ± 9.6% (Fig. 1) (Dunnett's t‐test; P < 0.01). Furthermore, similar reduction of CCE was observed in the presence of 30 µm SKF‐96365 (86.8 ± 5.4%) (Dunnett's t‐test; P < 0.01). No significant difference was demonstrated for the percentage of cells that responded to Tg in the presence of channel blockers. In contrast, verapamil, a VDCC antagonist, had no effect on Tg‐induced CCE (data not shown). Altogether, these results reveal the presence of CCE in MG‐63 cells and the efficiency of 2‐APB and SKF‐96365 to inhibit Ca2+ entry.
Figure 1.
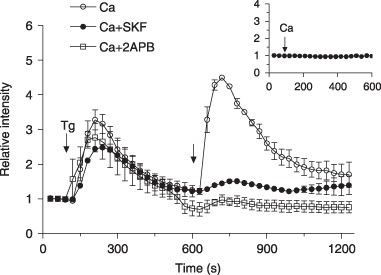
Induction of capacitative calcium (Ca2+) entry in MG‐63 cells. Fluo‐3‐loaded cells were treated with 5 µm thapsigargin (Tg) in Ca2+‐free HEPES‐buffered saline solution. After agonist‐mediated intracellular Ca2+ release, Ca2+ (final concentration of 1.8 mm, right arrow) were introduced into the buffer alone (○) or in the presence of (□) 2‐APB (100 µm) or ( ) SKF‐96365 (30 µm). Each response shown is from at least three experiments with cumulative analysis of between 30 and 40 cells per field. Inset: addition of calcium to Ca2+‐free HEPES‐buffered saline solution without prior incubation with Tg.
) SKF‐96365 (30 µm). Each response shown is from at least three experiments with cumulative analysis of between 30 and 40 cells per field. Inset: addition of calcium to Ca2+‐free HEPES‐buffered saline solution without prior incubation with Tg.
Cell proliferation and CCE induced by serum and platelet‐derived growth factor‐BB
In order to evaluate the roles of CCE in MG‐63 cells, we first determined conditions that stimulate cell proliferation. We performed cell counts over a 7‐day culture period with or without FBS (Fig. 2a). A typical exponential growth curve was observed for cells cultured in media supplemented with 10% FBS, resulting in a doubling time of approximately 28 h. Cell proliferation was almost completely prevented in serum‐free medium for the last 4 days of culture, with a cell doubling time of approximately 60 h. As shown in Fig. 2b, cell proliferation was also stimulated by platelet‐derived growth factor‐BB (PDGF‐BB) with an EC50 of 9 ng/mL and 85% stimulation was reached at 25 ng/mL. The subsequent experiments were therefore conducted after 5 days of culture in media supplemented with 10% FBS or 25 ng/mL PDGF‐BB.
Figure 2.
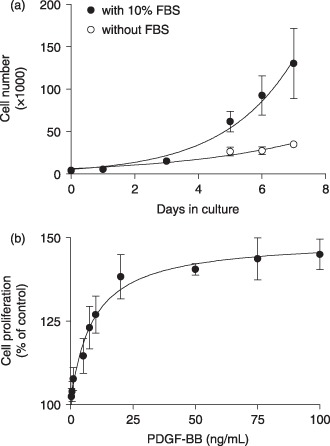
Cell proliferation induced by foetal bovine serum (FBS) and platelet‐derived growth factor‐BB (PDGF‐BB). Cells were seeded with DMEM‐F12 containing 10% FBS at 2000 cells/cm2 in 24‐well plates (for cell counts) or 96‐well plates (for MTT assay). (a) At day 3 of culture, cells were incubated in supplemented DMEM‐F12 with or without 10% FBS until day 7 and media were refreshed at day 5. Counts were performed in triplicate on trypsinized cells from 4 to 6 separate cultures and data are mean ± SEM (two‐way anova; P < 0.004). (b) After 4 days of culture, cells were serum‐starved for 24 h and were incubated for 48 h in DMEM‐F12 without FBS and with increasing concentrations of PDGF‐BB. Cell proliferation was determined by MTT assay and data are expressed as the mean ± SEM of percentage of control conditions without growth factors from four individual experiments (anova; P < 0.0001, Dunnett's t‐test; P < 0.01 from 7.5 ng/mL).
We investigated cellular Ca2+ signals induced by factors that stimulate MG‐63 cell proliferation. As shown in Fig. 3, PDGF‐BB induced a rapid transient increase of [Ca2+]i followed by a sustained influx following Ca2+ addition in the incubation medium. The increase in [Ca2+]i following addition of extracellular Ca2+ was prevented by 2‐APB and SKF‐96365 (Fig. 3) (Dunnett's t‐test; P < 0.01) indicating that stimulation of the cells by PDGF‐BB is associated with the induction of CCE. A similar response was obtained when cells were stimulated with 10% FBS and serum‐induced CCE was also inhibited by store‐operated Ca2+ channel (SOCC) blockers (data not shown). Verapamil had no effect on PDGF‐induced CCE (Fig. 3, inset). Furthermore, the rapid Ca2+ spike induced by PDGF‐BB was abolished when thapsigargin‐sensitive stores were depleted before addition of growth factors (data not shown) indicating the solicitation of identical intracellular Ca2+ stores.
Figure 3.
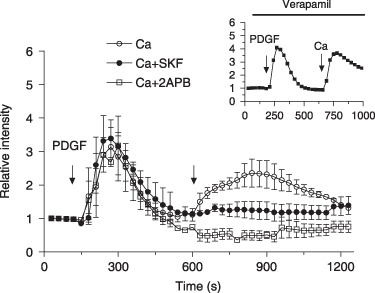
Capacitative calcium (Ca2+) entry induced by platelet‐derived growth factor‐BB (PDGF‐BB). Fluo‐3‐loaded cells were treated with 25 ng/mL PDGF‐BB in HEPES‐buffered saline solution and intracellular Ca2+ measurements were performed without (○) or with (□) 100 µm 2‐APB and () 30 µm SKF‐96365. Each response shown is representative of four experiments cumulating analysis of between 30 and 40 cells per field. Inset: representative data of intracellular Ca2+ measurement from cells stimulated with 25 ng/mL PDGF‐BB in the presence of 100 µm verapamil.
Expression of VDCCs and TRPC channels
Because TRPC channels have been associated with CCE (Birnbaumer et al. 1996; Berridge et al. 2000a), we sought to determine expression of such channels in MG‐63 cells. Messenger RNA for TRPC1, TRPC3, as well as TRPC4 and TRPC6 were revealed by RT‐PCR (Fig. 4a). Interestingly, amplification with primers for TRPC4 showed three major bands that likely correspond to different splicing variants previously reported as TRPC4 and α, β, ɛ (1200 bp, NM_016179, AF421358, AF421359, AF421360), TRPC4η (900 bp, AF421361), TRPC4ζ (700 bp, AF421362). Genetic expression of TRPC5 and TRPC7 were not revealed here by RT‐PCR using total or messenger RNA.
Figure 4.
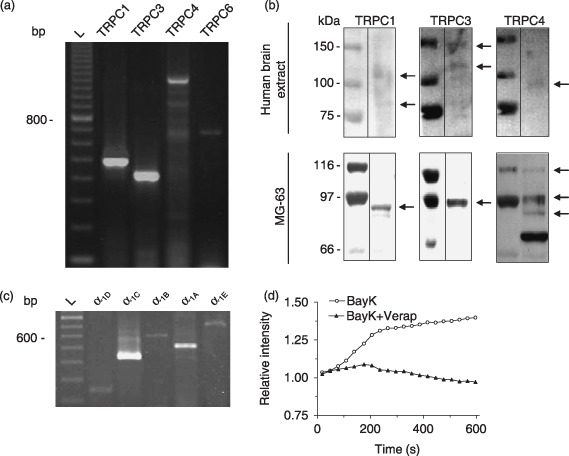
Expression of transient receptor potential canonical (TRPC) and voltage‐dependent Ca2+ channel (VDCC) in MG‐63 cells. (a) Total RNA from MG‐63 cells was used for RT‐PCR with specific primers for TRPC. Amplicons were revealed by 2% agarose gel electrophoresis and are representative results from at least three experiments. (L: 100 bp marker). (b) Protein extracts from human brain cell lysate or MG‐63 cells were subjected to SDS‐PAGE on 7.5% acrylamide gel. Proteins were electrotransfered on polyvinylidene difluoride membranes and immunodetection was performed as described in the materials and methods section. (c) Total RNA from MG‐63 cells was used for RT‐PCR with specific primers for L‐type (neuroendocrine α1D and cardiac α1C isoforms), N‐type (α1B), P/Q‐type (α1A) and R‐type (α1E) VDCC. Amplicons were revealed by 2% agarose gel electrophoresis and are representative results from at least three experiments. (L: 100 bp marker). (d) Fluo‐3‐loaded cells were treated with 50 µm BayK 8644 without or with 100 µm verapamil in HEPES‐buffered saline solution and intracellular Ca2+ measurements were performed.
TRPC1, TRPC3 and TRPC4 proteins were detected in crude membrane extracts from MG‐63 cells (Fig. 4b, bottom). TRPC1 immunodetection revealed the presence of an 85–90 kDa band. An 85–90 kDa band was also revealed in the human brain cell extract that was consistent with protein weight reported in literature. TRPC3 immunodetection showed a single‐specific band at 95 kDa that was consistent with the predicted weight between 85 and 115 kDa for non‐glycosylated TRPC3 protein and protein glucosylated to various degrees (Fig. 4b). Furthermore, TRPC4 immunodetection revealed the presence of three different bands that could represent protein isoforms corresponding to splice variants that we previously revealed by RT‐PCR. Unfortunately, TRPC6 protein could not be detected in MG‐63 cells and may indicate that the protein level was beneath our limit of detection.
Because VDCCs have been shown to be involved in osteoblastic functions (Guggino et al. 1989; Moreau et al. 1996, 1997; Bergh et al. 2004), we evaluated the genetic expression of VDCC in MG‐63 cells. As shown in Fig. 4c, presence of messenger RNAs for L‐type (neuroendocrine α1C, 437 bp; cardiac α1D, 245 bp), for N‐type (α1B, 607 bp), for P/Q‐type (α1A, 512 bp) and for R‐type (α1E) VDCC was revealed by RT‐PCR. No expression for the L‐type skeletal isoform (α1S) was revealed. Expression of T‐type VDCC was not investigated. Moreover, addition of the agonist of L‐type VDCC, BayK 8644, led to an increase in [Ca2+]i that was prevented by the L‐type VDCC antagonist verapamil (Fig. 4d) confirming the expression and activity of VDCC.
Implication of Ca2+ influx in MG‐63 cell proliferation
We first determined the effect of Tg‐mediated intracellular Ca2+ store depletion on proliferation of MG‐63 cells. As shown in Fig. 5, low concentrations of Tg significantly reduced this cell proliferation of after 48 h of incubation (anova; P < 0.0001; Dunnett's t‐test; P < 0.001 from 0.005 µm) and further impaired cell survival from 0.1 µm (Tukey's test; P < 0.05, compared to 100.3 ± 6.6 µm for initial MTT) and higher concentrations. To characterize Ca2+ influx involved in proliferation, it was evaluated in the presence of different Ca2+ channel blockers. Cells were incubated for 24 or 48 h in the presence of 10% FBS with increasing concentrations of VDCC blockers (verapamil and nifedipine). These blockers exerted no significant effect on serum‐mediated cell proliferation even at the highest concentrations used (Fig. 6a,b). We then evaluated effects of well‐known SOCC inhibitors, 2‐APB and SKF‐96365. As shown in Fig. 6c,d, both inhibitors decreased MG‐63 proliferation in a dose‐ and time‐dependent manner. When the cells were cultured for 48 h in the presence of 100 µm 2‐APB, a significant decrease in cell proliferation was observed, reaching 55% (Fig. 6c). In the presence of 20 µm SKF‐96365, strong inhibition (90%) of cell proliferation was also observed at 48 h (Fig. 6d). Moreover, proliferation of non‐transformed MC3T3‐E1 murine osteoblastic cells was also prevented by 100 µm 2‐APB and 10 µm SKF‐96365 (relative proliferation to control: 1.75 ± 0.04 versus 1.36 ± 0.27 and 1.04 ± 0.15 with 2‐APB and SKF‐96365, respectively), whereas 67 µm verapamil had no effect (1.75 ± 0.04 versus 1.88 ± 0.11 with verapamil). Furthermore, VDCC blockers were without effect on cell proliferation induced by PDGF‐BB (Fig. 7), while both 2‐APB and SKF‐96365 inhibited PDGF‐induced MG‐63 cell proliferation.
Figure 5.
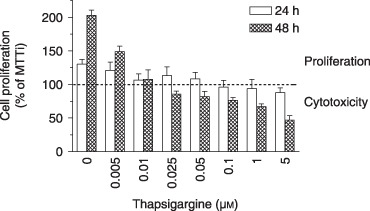
Effect of thapsigargin on the proliferation and survival of MG‐63 cells. Cells were incubated with increasing concentrations of thapsigargin (Tg) for 24 and 48 h in DMEM‐F12 supplemented with 10% foetal bovine serum. Cell proliferation was determined by MTT assay and data are the mean ± SEM of percentage of cell proliferation from three independent experiments.
Figure 6.
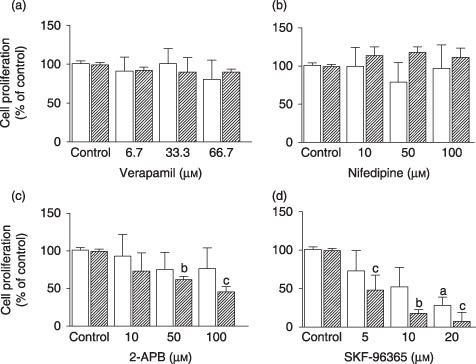
Effect of calcium channel blockers on cell proliferation induced by serum. Osteoblasts were cultured for 24 h (□) or 48 h ( ) in DMEM‐F12 supplemented with 10% FBS and increasing concentrations of (a) verapamil, (b) nifedipine (c) 2‐APB (d) SKF‐96365 or vehicle alone (control). Cell proliferation was determined by MTT assay and data are the mean ± SEM of the percentage of cell proliferation from three to six independent experiments (anova; P < 0.001, Dunnett's t‐test; a
P < 0.05; b
P < 0.01; c
P < 0.001 compared to control for the same incubation period).
) in DMEM‐F12 supplemented with 10% FBS and increasing concentrations of (a) verapamil, (b) nifedipine (c) 2‐APB (d) SKF‐96365 or vehicle alone (control). Cell proliferation was determined by MTT assay and data are the mean ± SEM of the percentage of cell proliferation from three to six independent experiments (anova; P < 0.001, Dunnett's t‐test; a
P < 0.05; b
P < 0.01; c
P < 0.001 compared to control for the same incubation period).
Figure 7.
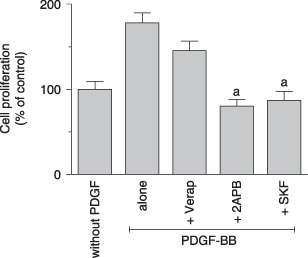
Effect of calcium channel blockers on MG‐63 proliferation induced by platelet‐derived growth factor‐BB (PDGF‐BB). Cells were incubated for 48 h in supplemented DMEM‐F12 containing 25 ng/mL PDGF‐BB in the absence or presence of 100 µm verapamil, 100 µm 2‐APB or 10 µm SKF‐96365. Cell proliferation was measured by MTT assay and data are expressed as mean ± SEM of percentage of the control without PDGF. Student's t‐test; a P < 0.001 compared to PDGF alone.
Although significant cytotoxicity with SOCC blockers was not seen under our experimental conditions, we evaluated the effect of SKF‐96365 under culture conditions without serum or PDGF‐BB, where cells have a reduced rate of proliferation, in order to establish the specific effect of the SKF‐96365 on this parameter. At a concentration of 10 µm, SKF‐96365 caused significant inhibition of cell population growth by 80% in the presence of 10% FBS (1.76 ± 0.11 versus 1.15 ± 0.05 with SKF‐96365), while it had no significant effect under low cell proliferation rates in serum‐free conditions (1.21 ± 0.05 versus 1.07 ± 0.10 with SKF‐96365). Moreover, 50 µm 2‐APB had no significant effect in the absence of serum (1.08 ± 0.04 versus 1.02 ± 0.05 with 2‐APB). Because reduction of osteoblast proliferation may promote differentiation, we determined alkaline phosphatase acitivity, a well‐known osteoblastic marker. The alkaline phosphatase activity of MG‐63 cells was not significantly different in the presence of 10 µm SKF‐96365 (63.0 ± 13.1 versus control 75.1 ± 3.0 nmol PNP/mg protein/1 h) indicating that inhibitors of CCE did not promote osteoblastic differentiation nor did they impair cell viability.
Some studies have reported potential side effects of SKF‐96365, not attributable to its inhibition of Ca2+ influx; it has been reported to modulate prostaglandin E2 (PGE2) pathway acting as an inhibitor of cyclooxygenase (Leis et al. 1995). Therefore, we investigated whether inhibition of cell proliferation by SKF‐96365 resulted from reduction of PGE2 production, using the cyclooxygenase inhibitor indomethacin. Treatment of MG‐63 cells with indomethacin had no significant effect on cell proliferation and survival (OD575nm of 0.61 ± 0.18 in the presence of 30 µm indomethacin versus control 0.63 ± 0.16) indicating that the antiproliferative effect of SKF‐96365 was not related to the reduction of PGE2 synthesis. Compounds related to SKF‐96365 such as imidazole antimycotics, econazole and miconazole, have been used as potent inhibitors of cytochrome P450 and CCE, leading to the proposed involvement of cytochrome P450 in CCE (Alvarez et al. 1992). In order to determine the involvement of cytochrome P450 inhibition in the reduction of cell proliferation by SKF‐96365, we evaluated cell proliferation in the presence of non‐imidazole inhibitor of cytochrome P450, SKF‐525 A. MG‐63 cell proliferation was not affected by SKF‐525 A (OD575nm of 1.37 ± 0.32 in the presence of 50 µm SKF‐525 A versus control 1.15 ± 0.18) indicating that inhibition of cytochrome P450 was not responsible for the observed prevention of cell proliferation by SKF‐96365.
Effect of capacitative Ca2+ channel blockers on the cell cycle
Because we observed that 2‐APB and SKF‐96365 inhibit osteoblastic cell proliferation, we investigated the effect of such blockers on the progression of cell cycle. Our results show that 2‐APB and SKF‐96365 had similar effects. Treatments for 4 h with either 50 µm 2‐APB or 5 µm SKF‐96365 reduced cell numbers in G0/G1 phase, and increased proportion of cells in S and G2/M phases (Fig. 8). After 24 h, treatment with both antagonists also resulted in augmentation of the proportion of cells in S and G2/M phases. This effect was also perceptible using confocal microscopy following PI staining where we observed many cells with double DNA content (data not shown). Taken together, these results suggest that SOCC blockers lengthen S and G2/M phases rather than arresting the cell cycle.
Figure 8.
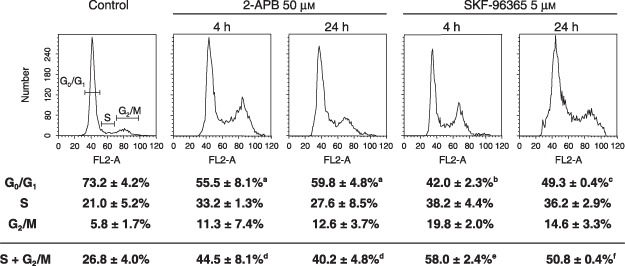
Effect of 2‐APB and SKF‐96365 on the cell cycle. Cells were incubated with 50 µm 2‐APB, 5 µm SKF‐96365 or vehicle alone for 4 h or 24 h in supplemented DMEM‐F12 media containing 10% FBS (control of 24 h). Cells were re‐suspended and stained with PI as described in the materials and methods section. Cells were then analysed using a FACScan flow cytometer. Profiles of DNA content are representative results from three independent experiments. Student's t‐test; a P < 0.05, b P < 0.03 and c P < 0.006 when compared to G0/G1 from the control; d P < 0.03, e P < 0.003 and f P < 0.006 when compared to S and G2/M from the control.
DISCUSSION
In the current study, we show that depletion of internal Ca2+ stores and inhibition of CCE prevented MG‐63 cell proliferation while inhibition of VDCC was without effect. Moreover, for the first time we have demonstrated expression of TRPC1, 3, 4 and 6 in the human osteoblast‐like MG‐63 cells. Our results support an important role of capacitative Ca2+ influx, more likely attributable to TRPC channels, in the proliferation of osteoblastic cells.
Treatment of MG‐63 cells with Tg in Ca2+‐free buffer led to depletion of internal Ca2+ stores with concomitant stimulation of CCE observed by addition of extracellular Ca2+. Such CCE has been reported in primary cultures of rat osteoblast cells (Wiemann et al. 1998) and in rat osteoblast‐like ROS 17/2.8 cells (Baldi et al. 2002). Capacitative Ca2+ influx was not prevented by VDCC inhibitors, suggesting that the latter channels were not solicited. CCE was prevented by SKF‐96365 and 2‐APB, which argues for the involvement of SOCC. 2‐APB has been shown to inhibit sarco/endoplasmic reticulum Ca2+‐ATPase and InsP3 receptors (Bilmen & Michelangeli 2002; Bilmen et al. 2002). Inhibition of sarco/endoplasmic reticulum Ca2+‐ATPase by 2‐APB would lead to a Ca2+ release from intracellular stores, an effect that was not seen in MG‐63 cells with the concentrations used. Furthermore, inhibition of InsP3 receptors by 2‐APB would prevent thapsigargin‐mediated Ca2+ release from intracellular stores, an effect that was not seen under our experimental conditions. Our results demonstrate that MG‐63 cells exhibit CCE induced by internal Ca2+ store depletion and that this Ca2+ influx is completely inhibited by SOCC blockers such as 2‐APB and SKF‐96365. A variety of mitogenic agents stimulate cell population growth by increasing [Ca2+]i. Our results indicate that growth factors from serum and PDGF‐BB induced an initial Ca+ release from Tg‐sensitive internal stores in MG‐63 cells that is followed by CCE. Previous addition of Tg in Ca2+‐free buffer prevents initial intracellular Ca2+ increase induced by serum and PDGF‐BB, indicating solicitation of similar internal stores. Sandy et al. (1998) reported that serum and PDGF induced IP3 synthesis in MG‐63 cells. Inositol production, following growth factor activation of tyrosine kinase receptors in osteoblasts, has been related to activation of phospholipase C gamma (Bornfeldt et al. 1995). Such IP3 production with subsequent depletion of IP3‐sensitive internal Ca2+ stores is a prerequisite for activation of CCE (Zitt et al. 2002).
Because Ca2+ signals are involved in numerous cell functions, we investigated the effect of different Ca2+ channel inhibitors on MG‐63 cell proliferation. The cells showed a doubling time of approximately 28 h in the presence of 10% FBS that is consistent with observations of Mohiti et al. (1997). In the absence of serum, they pursue their basal level of proliferation but with a population doubling time of 60 h. In addition, PDGF‐BB also stimulated cell proliferation. Under serum‐ or PDGF‐mediated proliferation conditions, VDCC blockers had no significant effect on proliferation levels, indicating that Ca2+ influx through L‐type VDCC is not involved or has only a minor contribution to serum‐ or PDGF‐induced cell proliferation. Similar results were obtained with non‐transformed MC3T3 murine osteoblastic cells. In contrast, Kim et al. (1991) reported that verapamil inhibits MC3T3 DNA synthesis and decreased cell proliferation. However, other studies reported that VDCC blockers were without effect on MC3T3 cell proliferation (Sugimoto et al. 1994). Interestingly, verapamil was shown to inhibit cell proliferation of electrically stimulated MC3T3 cells but not cell proliferation under control conditions (Lorich et al. 1998). The discrepancy concerning the effect of Verapamil may correspond to stimulation conditions and factors involved. Activation of L‐type VDCC has been reported for osteoblastic responses to external stimuli and mechanical stress. Accordingly, expression of VDCC, both L‐type and non‐L‐type isoforms (P/Q, N, R, T) has been demonstrated in osteoblasts (Barry 2000), and we confirmed expression and activity of VDCC in MG‐63 cells. Parathyroid hormone and PGE2 triggered an initial L‐type‐mediated Ca2+ influx in the MG‐63 cells that subsequently resulted in activation of potassium channels (Moreau et al. 1996). Calcitonin gene‐related peptide stimulates a cyclic adenosine monophosphate‐dependent Ca2+ influx that is inhibited by L‐type VDCC blocker verapamil (Burns et al. 2004). Fluorescence monitoring of intracellular Ca2+ concentrations indicated that L‐type VDCC are important in early mechanical stress transduction in primary osteoblasts from rat femora (Walker et al. 2000) and in murine osteoblasts MC3T3‐E1 (Genetos et al. 2005). Moreover, a range of studies has indicated that Ca2+ influx via L‐type VDCC is involved in synthesis and secretion of various bone proteins such as osteocalcin, osteopontin and osteoprotegerin (Guggino et al. 1989; Moreau et al. 1997; You et al. 2001; Bergh et al. 2004). It should be noted that verapamil and nifedipine strongly inhibit L‐type channel at low concentrations while at high concentrations, these two blockers also inhibit non‐L‐type currents (Diochot et al. 1995). Because our results have indicated that high concentrations of VDCC blockers had no effect on MG‐63 cell proliferation, it is more likely that VDCC, both L‐type and non‐L‐type, play a minimal role in osteoblast proliferation. In that respect, voltage‐dependent Ca2+ currents identified in osteoblasts are more likely to be implicated in synthesis and secretion of bone specific proteins with functional contributions in bone remodeling.
On the other hand, MG‐63 cell population growth was reduced by inhibition of CCE with 2‐APB and SKF‐96365. Concentrations of SOCC blockers exerting a significant effect on MG‐63 cell proliferation were consistent with concentrations shown to inhibit serum‐ and PDGF‐induced CCE under our experimental conditions. Furthermore, previous studies have reported significant inhibition of cell proliferation by 2‐APB and SKF‐96365 at concentrations of 30–100 µm and of 1–20 µm, respectively, depending on cell type (Magnier‐Gaubil et al. 1996; Sugioka et al. 1999; Enfissi et al. 2004). It should be noted that 2‐APB and SKF‐96365 had no significant effect on cells in the absence of serum or PDGF, suggesting that capacitative current is mainly triggered by growth factors and that SKF‐96365 had no side effect and did not induce apoptosis in growth arrested cells.
Unfortunately, blockers tend to be selective for a group rather than specific to one type of channel and side effects are often reported. Leis et al. (1995) reported that SKF‐96365 impairs the PGE2 pathway. The latter has been reported to induce osteoblast proliferation (Conconi et al. 2002; Tsai et al. 2004; Ghayor et al. 2005). Treatments of MG‐63 cells with concentrations up to 30 µm indomethacin, a cyclooxygenase inhibitor, showed no significant effect on cell proliferation indicating that the PGE2 pathway was not implicated in proliferation of MG‐63 cells under our conditions. Moreover, imidazole antimycotics such as econazole and miconazole are potent inhibitors of cytochrome P450 and have been associated with inhibition of CCE (Alvarez et al. 1992). Our results indicate that non‐imidazole cytochrome P450 inhibitor SKF‐525 A did not affect MG‐63 cell proliferation, demonstrating that potential inhibition of cytochrome P450 by SKF‐96365 did not account for prevention of cell proliferation. Therefore, the inhibition of proliferation by the SKF‐96365 in our experiments is more likely due to inhibition of SOCC‐dependent Ca2+ entry. Interestingly, 2‐APB was less efficient than SKF‐96365 to inhibit proliferation of MG‐63 cells. It should be noted that 2‐APB has also been shown to activate TRPV5 and TRPV6, two highly Ca2+‐selective channels (Hu et al. 2004). Expression of TRPV channels has not yet been investigated in osteoblasts.
Maintenance of a sufficient reserve of Ca2+ in the endoplasmic reticulum is critical for normal cell function and these reserves are needed at least for progression of cells through the cell cycle (Takuwa et al. 1995). Our data showed that treatment with 50 µm 2‐APB or 5 µm SKF‐96365 resulted in reduction of cell number in G0/G1 phase, and an increase in the proportion of cells in S and G2/M phases (Fig. 8). Although Mitsui‐Saito et al. (2000) showed, in vitro, polymerization inhibition of microtubule protein isolated from bovine brain by SKF‐96365 that could result in mitotic spindle dysfunction and G2/M arrest, concentrations used in our cell cycle assay are lower than those used in their study. Moreover, we report that 2‐APB also affects cell cycle progression of MG‐63 cells although no alteration in microtubule dynamics has been reported for 2‐APB. Calmodulin, an intracellular Ca2+ binding protein, may regulate cell cycle phase transitions, affects the activation state of cyclin‐dependent kinase (cdk) complexes and cytokinesis (Kahl & Means 2003). Further studies on the effect of SOCC blockers on cdk/cyclin functions, and on the duration of cell cycle phases are essential to reveal the SOCC blocker mechanism related to cell cycle lengthening.
Our results indicate for the first time, presence of TRPC1, TRPC3, TRPC4 and TRPC6 mRNA in MG‐63 cells, although neither TRPC5 nor TRPC7 were detected by RT‐PCR on isolated mRNA. On the basis of sequence homology and functional similarities, members of the mammalian TRPC family can be divided into four subfamilies: TRPC1, TRPC2, TRPC3/6/7 and TRPC4/5, and in humans, TRPC2 is a pseudogene (Wes et al. 1995). It should be noted that expression of at least one member of each subfamily was revealed in MG‐63 cells. Both TRPC1 and TRPC4 have been shown to form SOCC (Liu et al. 2003; Wang et al. 2004). TRPC3 at endogenous low‐level expression behave as store‐operated cation channels (Trebak et al. 2003). In contrast, overexpression of TRPC3 led to currents that correspond to the receptor‐operated cation channel pattern. Substantial evidence has indicated that TRPC6 is more likely to be a receptor‐operated cation channel activated by the diacylglycerol, and insensitive towards store depletion (Boulay et al. 1997; Hofmann et al. 1999; Trebak et al. 2003). Nevertheless, complexity in functional characterization of TRPCs arises from the fact that TRPCs can exist as homo‐ and hetero‐tetramers, both in vivo and when heterologously expressed (Birnbaumer et al. 1996; Clapham et al. 2001). Therefore, we suggest that TRPC channels expressed in MG‐63 cells might be responsible of CCE and be further involved in osteoblastic cell proliferation. This would be consistent with other studies in which a similar implication of TRPCs in cell proliferation has been observed (Golovina et al. 2001; Sweeney et al. 2002). Further studies are warranted in order to establish exactly which TRPC is involved in the CCE of osteoblasts. Interestingly, Maroto et al. (2005) have reported that TRPC1 forms a stretch‐activated channel. Such stretch‐activated ion channels have been associated with mechanotransduction and mechanical loading has been shown to be essential for skeletal homeostasis and bone formation (Duncan & Turner 1995; Harada & Rodan 2003). In this respect, osteoblasts are directly activated by loading, and influx of extracellular Ca2+ has been shown to play a significant role in the early response of osteoblasts, such as increased proliferation, differentiation and matrix synthesis, to mechanical stimuli (Walker et al. 2000; Ziros et al. 2002; Danciu et al. 2003).
One hypothesis of ageing proposes that age‐dependent, subtle dysregulation of Ca2+ homeostasis, resulting in small changes of intracellular Ca2+‐free concentration sustained over extended period of time, would account for age‐related changes in cellular function (Khachaturian 1989). Accordingly, Huang et al. (2000) have reported that defects in the IP3‐mediated Ca2+ release pathway contribute to deficient Ca2+ signalling in cells undergoing replicative senescence, with endogenous Ca2+ store depletion. With regard to bone metabolism, Zahanich et al. (2005) have shown that in the majority of human cells of the osteoblastic lineage, Ca2+ entry through the plasma membrane is mediated by channels other than VDCCs. Our results are in agreement with the importance of other channels, likely to be TRPCs, in osteoblasts, especially in cell proliferation. Because some studies have demonstrated that ageing is associated with a defect in proliferation of cells of the osteoblastic lineage, further studies on the alteration of CCE and an ageing‐associated defect in osteoblast proliferation seem promising.
In this study, we have demonstrated for the first time, the presence of CCE in human osteoblast‐like MG‐63 cells, which is inhibited by SOCC antagonists. We also demonstrated the expression of TRPC1, 3, 4 and 6 in these cells, some of which have been implicated in CCE in other cell types. Inhibition of CCE here lengthened the cell cycle time and reduced cell proliferation rate. Taken together, our results suggest that stimulation of osteoblast proliferation is associated with induction of CCE more likely being attributable to TRPC channels. The results shed light on a possible new signalling pathway controlling transition from stationery to proliferative population growth in osteoblasts involving members of the TRPC channel family. Therefore, these findings could be of great importance in revealing the mechanism associated with osteoblastic proliferation in order to understand its importance in pathophysiology, such as in osteoporosis, and could lead to new insights in the development of treatment.
ACKNOWLEDGEMENTS
We wish to thank Denis Flipo for technical assistance in cytometry analysis. This work was supported by the Natural Sciences and Engineering Research Council of Canada and the Fonds Québécois de la Recherche sur la Nature et les Technologies.
REFERENCES
- Alvarez J, Montero M, Garcia‐Sancho J (1992) Cytochrome P450 may regulate plasma membrane Ca2+ permeability according to the filling state of the intracellular Ca2+ stores. FASEB J. 6, 786–792. [DOI] [PubMed] [Google Scholar]
- Baldi C, Vazquez G, Boland R (2002) Characterization of a 1,25(OH)2‐vitamin D3‐responsive capacitative Ca2+ entry pathway in rat osteoblast‐like cells. J. Cell. Biochem. 86, 678–687. [DOI] [PubMed] [Google Scholar]
- Baldi C, Vazquez G, Calvo JC, Boland R (2003) TRPC3‐like protein is involved in the capacitative cation entry induced by 1alpha,25‐dihydroxy‐vitamin D3 in ROS 17/2.8 osteoblastic cells. J. Cell. Biochem. 90, 197–205. [DOI] [PubMed] [Google Scholar]
- Barry EL (2000) Expression of mRNAs for the alpha 1 subunit of voltage‐gated calcium channels in human osteoblast‐like cell lines and in normal human osteoblasts. Calcif. Tissue Int. 66, 145–150. [DOI] [PubMed] [Google Scholar]
- Bergh JJ, Xu Y, Farach‐Carson MC (2004) Osteoprotegerin expression and secretion are regulated by calcium influx through the 1‐type voltage‐sensitive calcium channel. Endocrinology 145, 426–436. [DOI] [PubMed] [Google Scholar]
- Bergman RJ, Gazit D, Kahn AJ, Gruber H, McDougall S, Hahn TJ (1996) Age‐related changes in osteogenic stem cells in mice. J. Bone Miner. Res. 11, 568–577. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD (2000a) Signal transduction. The calcium entry pas de deux. Science 287, 1604–1605. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD (2000b) The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21. [DOI] [PubMed] [Google Scholar]
- Bilmen JG, Michelangeli F (2002) Inhibition of the type 1 inositol 1,4,5‐trisphosphate receptor by 2‐aminoethoxydiphenylborate. Cell. Signal. 14, 955–960. [DOI] [PubMed] [Google Scholar]
- Bilmen JG, Wootton LL, Godfrey RE, Smart OS, Michelangeli F (2002) Inhibition of SERCA Ca2+ pumps by 2‐aminoethoxydiphenyl borate (2‐APB). 2‐APB reduces both Ca2+ binding and phosphoryl transfer from ATP, by interfering with the pathway leading to the Ca2+‐binding sites. Eur. J. Biochem. 269, 3678–3687. [DOI] [PubMed] [Google Scholar]
- Birnbaumer L, Zhu X, Jiang M, Boulay G, Peyton M, Vannier B, Brown D, Platano D, Sadeghi H, Stefani E, Birnbaumer M (1996) On the molecular basis and regulation of cellular capacitative calcium entry: roles for Trp proteins. Proc. Natl. Acad. Sci. USA 93, 15195–15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornfeldt KE, Raines EW, Graves LM, Skinner MP, Krebs EG, Ross R (1995) Platelet‐derived growth factor. Distinct signal transduction pathways associated with migration versus proliferation. Ann. N. Y. Acad. Sci. 766, 416–430. [DOI] [PubMed] [Google Scholar]
- Boulay G, Zhu X, Peyton M, Jiang M, Hurst R, Stefani E, Birnbaumer L (1997) Cloning and expression of a novel mammalian homolog of Drosophila transient receptor potential (Trp) involved in calcium entry secondary to activation of receptors coupled by the Gq class of G protein. J. Biol. Chem. 272, 29672–29680. [DOI] [PubMed] [Google Scholar]
- Brockstedt H, Kassem M, Eriksen EF, Mosekilde L, Melsen F (1993) Age‐ and sex‐related changes in iliac cortical bone mass and remodeling. Bone 14, 681–691. [DOI] [PubMed] [Google Scholar]
- Burns DM, Stehno‐Bittel L, Kawase T (2004) Calcitonin gene‐related peptide elevates calcium and polarizes membrane potential in MG‐63 cells by both cAMP‐independent and cAMP‐dependent mechanisms. Am. J. Physiol. Cell Physiol. 287, C457–C467. [DOI] [PubMed] [Google Scholar]
- Chan GK, Duque G (2002) Age‐related bone loss: old bone, new facts. Gerontology 48, 62–71. [DOI] [PubMed] [Google Scholar]
- Chen TL (2004) Inhibition of growth and differentiation of osteoprogenitors in mouse bone marrow stromal cell cultures by increased donor age and glucocorticoid treatment. Bone 35, 83–95. [DOI] [PubMed] [Google Scholar]
- Clapham DE, Runnels LW, Strubing C (2001) The TRP ion channel family. Nat. Rev. Neurosci. 2, 387–396. [DOI] [PubMed] [Google Scholar]
- Conconi MT, Tommasini M, Baiguera S, De Coppi P, Parnigotto PP, Nussdorfer GG (2002) Effects of prostaglandins E1 and E2 on the growth and differentiation of osteoblast‐like cells cultured in vitro . Int. J. Mol. Med. 10, 451–456. [PubMed] [Google Scholar]
- D’Ippolito G, Schiller PC, Ricordi C, Roos BA, Howard GA (1999) Age‐related osteogenic potential of mesenchymal stromal stem cells from human vertebral bone marrow. J. Bone Miner. Res. 14, 1115–1122. [DOI] [PubMed] [Google Scholar]
- Danciu TE, Adam RM, Naruse K, Freeman MR, Hauschka PV (2003) Calcium regulates the PI3K‐Akt pathway in stretched osteoblasts. FEBS Lett. 536, 193–197. [DOI] [PubMed] [Google Scholar]
- Diochot S, Richard S, Baldy‐Moulinier M, Nargeot J, Valmier J (1995) Dihydropyridines, phenylalkylamines and benzothiazepines block N‐, P/Q‐ and R‐type calcium currents. Pflugers Arch. 431, 10–19. [DOI] [PubMed] [Google Scholar]
- Duncan RL, Turner CH (1995) Mechanotransduction and the functional response of bone to mechanical strain. Calcif. Tissue Int. 57, 344–358. [DOI] [PubMed] [Google Scholar]
- Duncan RL, Akanbi KA, Farach‐Carson MC (1998) Calcium signals and calcium channels in osteoblastic cells. Semin. Nephrol. 18, 178–190. [PubMed] [Google Scholar]
- Enfissi A, Prigent S, Colosetti P, Capiod T (2004) The blocking of capacitative calcium entry by 2‐aminoethyl diphenylborate (2‐APB) and carboxyamidotriazole (CAI) inhibits proliferation in Hep G2 and Huh‐7 human hepatoma cells. Cell Calcium 36, 459–467. [DOI] [PubMed] [Google Scholar]
- Genetos DC, Geist DJ, Liu D, Donahue HJ, Duncan RL (2005) Fluid shear‐induced ATP secretion mediates prostaglandin release in MC3T3–E1 osteoblasts. J. Bone Miner. Res. 20, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghayor C, Rey A, Caverzasio J (2005) Prostaglandin‐dependent activation of ERK mediates cell proliferation induced by transforming growth factor beta in mouse osteoblastic cells. Bone 36, 93–100. [DOI] [PubMed] [Google Scholar]
- Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, Rubin LJ, Yuan JX (2001) Upregulated TRP and enhanced capacitative Ca2+ entry in human pulmonary artery myocytes during proliferation. Am. J. Physiol. Heart Circ. Physiol. 280, H746–H755. [DOI] [PubMed] [Google Scholar]
- Guggino SE, Lajeunesse D, Wagner JA, Snyder SH (1989) Bone remodeling signaled by a dihydropyridine‐ and phenylalkylamine‐ sensitive calcium channel. Proc. Natl. Acad. Sci. USA 86, 2957–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada S, Rodan GA (2003) Control of osteoblast function and regulation of bone mass. Nature 423, 349–355. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G (1999) Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397, 259–263. [DOI] [PubMed] [Google Scholar]
- Hu HZ, Gu Q, Wang C, Colton CK, Tang J, Kinoshita‐Kawada M, Lee LY, Wood JD, Zhu MX (2004) 2‐aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J. Biol. Chem. 279, 35741–35748. [DOI] [PubMed] [Google Scholar]
- Huang MS, Adebanjo OA, Awumey E, Biswas G, Koval A, Sodam BR, Sun L, Moonga BS, Epstein J, Goldstein S, Lai FA, Lipschitz D, Zaidi M (2000) IP(3), IP(3) receptor, and cellular senescence. Am. J. Physiol. Renal. Physiol. 278, F576–F584. [DOI] [PubMed] [Google Scholar]
- Iqbal J, Zaidi M (2005) Molecular regulation of mechanotransduction. Biochem. Biophys. Res. Commun. 328, 751–755. [DOI] [PubMed] [Google Scholar]
- Kahl CR, Means AR (2003) Regulation of cell cycle progression by calcium/calmodulin‐dependent pathways. Endocr. Rev. 24, 719–736. [DOI] [PubMed] [Google Scholar]
- Kassem M, Ankersen L, Eriksen EF, Clark BF, Rattan SI (1997) Demonstration of cellular aging and senescence in serially passaged long‐term cultures of human trabecular osteoblasts. Osteoporos. Int. 7, 514–524. [DOI] [PubMed] [Google Scholar]
- Kato H, Matsuo R, Komiyama O, Tanaka T, Inazu M, Kitagawa H, Yoneda T (1995) Decreased mitogenic and osteogenic responsiveness of calvarial osteoblasts isolated from aged rats to basic fibroblast growth factor. Gerontology 41 (Suppl. 1), 20–27. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS (1989) The role of calcium regulation in brain aging: reexamination of a hypothesis. Aging (Milano.) 1, 17–34. [DOI] [PubMed] [Google Scholar]
- Kim YS, Yang IM, Kim SW, Kim JW, Kim KW, Choi YK (1991) Responses of osteoblastic cell line MC3T3–E1 cell to the calcium channel blocker diltiazem and verapamil. Contrib. Nephrol. 91, 43–49. [DOI] [PubMed] [Google Scholar]
- Kotev‐Emeth S, Savion N, Pri‐chen S, Pitaru S (2000) Effect of maturation on the osteogenic response of cultured stromal bone marrow cells to basic fibroblast growth factor. Bone 27, 777–783. [DOI] [PubMed] [Google Scholar]
- Kragstrup J, Melsen F, Mosekilde L (1983) Thickness of bone formed at remodeling sites in normal human iliac trabecular bone: variations with age and sex. Metab. Bone Dis. Relat. Res. 5, 17–21. [DOI] [PubMed] [Google Scholar]
- Leis HJ, Zach D, Huber E, Ziermann L, Gleispach H, Windischhofer W (1995) On the inhibition of prostanoid formation by SK & F 96365, a blocker of receptor‐operated calcium entry. Br. J. Pharmacol. 114, 598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CT, Barnes J, Seedor JG, Quartuccio HA, Bolander M, Jeffrey JJ, Rodan GA (1992) Impaired bone activity in aged rats: alterations at the cellular and molecular levels. Bone 13, 435–441. [DOI] [PubMed] [Google Scholar]
- Liu X, Singh BB, Ambudkar IS (2003) TRPC1 is required for functional store‐operated Ca2+ channels. Role of acidic amino acid residues in the S5–S6 region. J. Biol. Chem. 278, 11337–11343. [DOI] [PubMed] [Google Scholar]
- Lorich DG, Brighton CT, Gupta R, Corsetti JR, Levine SE, Gelb ID, Seldes R, Pollack SR (1998) Biochemical pathway mediating the response of bone cells to capacitive coupling. Clin. Orthop. Relat. Res. 246–256. [PubMed]
- Mackie EJ (2003) Osteoblasts: novel roles in orchestration of skeletal architecture. Int. J. Biochem. Cell Biol. 35, 1301–1305. [DOI] [PubMed] [Google Scholar]
- Magnier‐Gaubil C, Herbert JM, Quarck R, Papp B, Corvazier E, Wuytack F, Levy‐Toledano S, Enouf J (1996) Smooth muscle cell cycle and proliferation. Relationship between calcium influx and sarco‐endoplasmic reticulum Ca2+‐ATPase regulation. J. Biol. Chem. 271, 27788–27794. [DOI] [PubMed] [Google Scholar]
- Maroto R, Raso A, Wood TG, Kurosky A, Martinac B, Hamill OP (2005) TRPC1 forms the stretch‐activated cation channel in vertebrate cells. Nat. Cell Biol. 7, 179–185. [DOI] [PubMed] [Google Scholar]
- Martinez ME, Del Campo MT, Medina S, Sanchez M, Sanchez‐Cabezudo MJ, Esbrit P, Martinez P, Moreno I, Rodrigo A, Garces MV, Munuera L (1999) Influence of skeletal site of origin and donor age on osteoblastic cell growth and differentiation. Calcif. Tissue Int. 64, 280–286. [DOI] [PubMed] [Google Scholar]
- Mitsui‐Saito M, Nakahata N, Ohizumi Y (2000) Inhibition of microtubule polymerization by SK & F 96365, a blocker of receptor‐linked Ca2+ entry. Jpn. J. Pharmacol. 82, 269–271. [DOI] [PubMed] [Google Scholar]
- Mohiti J, Caswell AM, Walker JH (1997) The nuclear location of annexin V in the human osteosarcoma cell line MG‐63 depends on serum factors and tyrosine kinase signaling pathways. Exp. Cell Res. 234, 98–104. [DOI] [PubMed] [Google Scholar]
- Montell C, Rubin GM (1989) Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron 2, 1313–1323. [DOI] [PubMed] [Google Scholar]
- Moreau R, Aubin R, Lapointe JY, Lajeunesse D (1997) Pharmacological and biochemical evidence for the regulation of osteocalcin secretion by potassium channels in human osteoblast‐like MG‐63 cells. J. Bone Miner. Res. 12, 1984–1992. [DOI] [PubMed] [Google Scholar]
- Moreau R, Hurst AM, Lapointe JY, Lajeunesse D (1996) Activation of maxi‐K channels by parathyroid hormone and prostaglandin E2 in human osteoblast bone cells. J. Membr. Biol. 150, 175–184. [DOI] [PubMed] [Google Scholar]
- Neidlinger‐Wilke C, Stalla I, Claes L, Brand R, Hoellen I, Rubenacker S, Arand M, Kinzl L (1995) Human osteoblasts from younger normal and osteoporotic donors show differences in proliferation and TGF beta‐release in response to cyclic strain. J. Biomech. 28, 1411–1418. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Putney JW Jr (2005) Store‐operated calcium channels. Physiol. Rev. 85, 757–810. [DOI] [PubMed] [Google Scholar]
- Parfitt AM (1991) Bone forming cells in clinical conditions In: Hall BK, ed. Bone, the Osteoblast and Osteocyte. London: Telford Press, pp. 351–426. [Google Scholar]
- Peterson WJ, Tachiki KH, Yamaguchi DT (2004) Serial passage of MC3T3–E1 cells down‐regulates proliferation during osteogenesis in vitro . Cell Prolif. 37, 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeilschifter J, Diel I, Pilz U, Brunotte K, Naumann A, Ziegler R (1993) Mitogenic responsiveness of human bone cells in vitro to hormones and growth factors decreases with age. J. Bone Miner. Res. 8, 707–717. [DOI] [PubMed] [Google Scholar]
- Quarto R, Thomas D, Liang CT (1995) Bone progenitor cell deficits and the age‐associated decline in bone repair capacity. Calcif. Tissue Int. 56, 123–129. [DOI] [PubMed] [Google Scholar]
- Roholl PJ, Blauw E, Zurcher C, Dormans JA, Theuns HM (1994) Evidence for a diminished maturation of preosteoblasts into osteoblasts during aging in rats: an ultrastructural analysis. J. Bone Miner. Res. 9, 355–366. [DOI] [PubMed] [Google Scholar]
- Sandy J, Davies M, Prime S, Farndale R (1998) Signal pathways that transduce growth factor‐stimulated mitogenesis in bone cells. Bone 23, 17–26. [DOI] [PubMed] [Google Scholar]
- Stenderup K, Justesen J, Clausen C, Kassem M (2003) Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone 33, 919–926. [DOI] [PubMed] [Google Scholar]
- Sugimoto T, Kanatani M, Kano J, Kobayashi T, Yamaguchi T, Fukase M, Chihara K (1994) IGF‐I mediates the stimulatory effect of high calcium concentration on osteoblastic cell proliferation. Am. J. Physiol. 266, E709–E716. [DOI] [PubMed] [Google Scholar]
- Sugioka M, Zhou WL, Hofmann HD, Yamashita M (1999) Ca2+ mobilization and capacitative Ca2+ entry regulate DNA synthesis in cultured chick retinal neuroepithelial cells. Int. J. Dev. Neurosci. 17, 163–172. [DOI] [PubMed] [Google Scholar]
- Sweeney M, Yu Y, Platoshyn O, Zhang S, McDaniel SS, Yuan JX (2002) Inhibition of endogenous TRP1 decreases capacitative Ca2+ entry and attenuates pulmonary artery smooth muscle cell proliferation. Am. J. Physiol. Lung Cell. Mol. Physiol. 283, L144–L155. [DOI] [PubMed] [Google Scholar]
- Takuwa N, Zhou W, Takuwa Y (1995) Calcium, calmodulin and cell cycle progression. Cell. Signal. 7, 93–104. [DOI] [PubMed] [Google Scholar]
- Trebak M, Vazquez G, Bird GS, Putney JW (2003) The TRPC3/6/7 subfamily of cation channels. Cell Calcium 33, 451–461. [DOI] [PubMed] [Google Scholar]
- Tsai KS, Yang RS, Liu SH (2004) Benzo[a]pyrene regulates osteoblast proliferation through an estrogen receptor‐related cyclooxygenase‐2 pathway. Chem. Res. Toxicol. 17, 679–684. [DOI] [PubMed] [Google Scholar]
- Vazquez G, Wedel BJ, Aziz O, Trebak M, Putney JW Jr (2004) The mammalian TRPC cation channels. Biochim. Biophys. Acta 1742, 21–36. [DOI] [PubMed] [Google Scholar]
- Walker LM, Publicover SJ, Preston MR, Said Ahmed MA, El Haj AJ (2000) Calcium‐channel activation and matrix protein upregulation in bone cells in response to mechanical strain. J. Cell. Biochem. 79, 648–661. [DOI] [PubMed] [Google Scholar]
- Wang X, Pluznick JL, Wei P, Padanilam BJ, Sansom SC (2004) TRPC4 forms store‐operated Ca2+ channels in mouse mesangial cells. Am. J. Physiol. Cell Physiol. 287, C357–C364. [DOI] [PubMed] [Google Scholar]
- Wes PD, Chevesich J, Jeromin A, Rosenberg C, Stetten G, Montell C (1995) TRPC1, a human homolog of a Drosophila store‐operated channel. Proc. Natl. Acad. Sci. USA 92, 9652–9656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemann M, Busselberg D, Schirrmacher K, Bingmann D (1998) A calcium release activated calcium influx in primary cultures of rat osteoblast‐like cells. Calcif. Tissue Int. 63, 154–159. [DOI] [PubMed] [Google Scholar]
- You J, Reilly GC, Zhen X, Yellowley CE, Chen Q, Donahue HJ, Jacobs CR (2001) Osteopontin gene regulation by oscillatory fluid flow via intracellular calcium mobilization and activation of mitogen‐activated protein kinase in MC3T3–E1 osteoblasts. J. Biol. Chem. 276, 13365–13371. [DOI] [PubMed] [Google Scholar]
- Zahanich I, Graf EM, Heubach JF, Hempel U, Boxberger S, Ravens U (2005) Molecular and functional expression of voltage‐operated calcium channels during osteogenic differentiation of human mesenchymal stem cells. J. Bone Miner. Res. 20, 1637–1646. [DOI] [PubMed] [Google Scholar]
- Ziros PG, Gil AP, Georgakopoulos T, Habeos I, Kletsas D, Basdra EK, Papavassiliou AG (2002) The bone‐specific transcriptional regulator Cbfa1 is a target of mechanical signals in osteoblastic cells. J. Biol. Chem. 277, 23934–23941. [DOI] [PubMed] [Google Scholar]
- Zitt C, Halaszovich CR, Luckhoff A (2002) The TRP family of cation channels: probing and advancing the concepts on receptor‐activated calcium entry. Prog. Neurobiol. 66, 243–264. [DOI] [PubMed] [Google Scholar]