

Abstract
Objectives: Ninety percent of malignant ovarian cancers are epithelial and thought to arise from the ovarian surface epithelium (OSE). We hypothesized that biological characteristics of primary OSE cells would more closely resemble OSE in vivo if established as three‐dimensional (3D) cultures.
Materials and methods: OSE cells were cultured as multicellular spheroids (MCS) (i) in a rotary cell culture system (RCCS) and (ii) on polyHEMA‐coated plastics. The MCSs were examined by electron microscopy and compared to OSE from primary tissues and cells grown in 2D. Annexin V FACS analysis was used to evaluate apoptosis and expression of extracellular matrix (ECM) proteins was analysed by immunohistochemical staining.
Results: On polyHEMA‐coated plates, OSE spheroids had defined internal architecture. RCCS MCSs had disorganized structure and higher proportion of apoptotic cells than polyHEMA MCSs and the same cells grown in 2D culture. In 2D, widespread expression of AE1/AE3, laminin and vimentin were undetectable by immunohistochemistry, whereas strong expression of these proteins was observed in the same cells grown in 3D culture and in OSE on primary tissues.
Conclusions: Physiological and biological features of OSE cells grown in 3D culture more closely resemble characteristics of OSE cells in vivo than when grown by classical 2D approaches. It is likely that establishing in vitro 3D OSE models will lead to greater understanding of the mechanisms of neoplastic transformation in epithelial ovarian cancers.
Introduction
Human ovaries are covered with a monolayer of flat/cuboidal mesothelial‐type cells referred to as the ovarian surface epithelium (OSE). These cells are widely considered to be the origin of epithelial ovarian cancers, which represent about 90% of all malignant ovarian tumours (1, 2, 3). Understanding biological and molecular characteristics of OSE and the earliest stages of ovarian tumour development has been hampered in the past by lack of a suitable in vitro model of normal OSE (NOSE). This is partly because primary OSE cells have proved difficult to establish in culture and have a short lifespan in vitro. Since the first description of OSE cultures in 1984, optimization of collection techniques and culture media has increased in vitro lifespan of these cells (4, 5, 6). However, there remain limitations to culturing NOSE cells as standard two‐dimensional (2D) monolayers. For example, NOSE cells can lose some of their epithelial characteristics, even when cultured in enriched media (6). Primary NOSE cell cultures show considerable phenotypic plasticity and can exhibit both epithelial (for example, presence of desmosomes; collagen IV, laminin and cytokeratin production) and mesenchymal characteristics (collagen I, collagen III and vimentin production) (5, 7).
Three‐dimensional (3D) culture systems enable the propagation of cells in a microenvironment that resembles in vivo conditions more closely than traditional 2D cultures. In vivo, epithelial cells are surrounded by a complex extracellular matrix (ECM). They contact and communicate with a host of different cell types through receptors distributed throughout the entire cell surface. However, in 2D monocultures, clonally derived cells only communicate along a small proportion of their membrane. Cells that are traditionally difficult to culture in 2D, such as primary hepatocytes, can often be maintained in vitro for longer periods when grown in 3D cultures (8). There is now substantial evidence to suggest that 3D cultures more closely resemble the in vivo microenvironment than 2D cultures, and that culturing cells in 3D can cause phenotypic and molecular changes that reflect in vivo biology of the cells more closely than 2D (9, 10, 11).
Several different approaches can be used to establish 3D cell culture models. For example, cells can be grown in 3D using ECM protein gel scaffolds (such as collagen gels). Other techniques prevent cell adherence to tissue culture plastics and hence, encourage cells to adhere to each other; such systems include the rotary cell culture system (RCCS) or poly‐2‐hydroxyethyl methacrylate (polyHEMA)‐coated tissue culture plastics. However, few studies have directly compared different 3D culture techniques. In one study, Ghosh et al. cultured melanoma cells in collagen gels and found that gene expression profiles were similar to those of cells in 2D; yet when cells were grown on polyHEMA‐coated plates, they observed differential expression of > 150 genes (10) and many of the genes upregulated in polyHEMA 3D cultures were consistent with their upregulation in vivo (10). Hence, different 3D culture techniques are not equivalent in their effectiveness.
To date, there are no reports describing 3D culturing of NOSE cells. There are some reports of culturing ovarian cancer cells in 3D, in particular, tumour cell aggregates derived from ascites (11, 12, 13). The purpose of the current study was to evaluate biological effects of two different 3D culturing techniques on culture of primary normal ovarian surface epithelial cells and to establish a 3D model of NOSE cells that could be used to study the earliest stages of neoplastic transformation in epithelial ovarian cancer. In evaluating these 3D models, we compared morphological and biological characteristics of cells with the same cells grown in 2D cultures and of NOSE from primary tissue samples. We hypothesized that by allowing NOSE cells to form 3D structures, we would maintain a phenotype that more closely resembles NOSE cells in vivo than in 2D culture.
Materials and methods
Tissue samples
Primary NOSE cells were collected and established as previously described (6). Two primary cell isolates (NOSE4 and NOSE11) were established from cells obtained during total laparoscopic hysterectomy with bilateral salpingo‐oophorectomy procedures. NOSE4 cells were from a 62‐year‐old‐woman diagnosed with endometrioid endometrial cancer stage I and cervical clear cell carcinoma; NOSE11 cells were from a 49‐year‐old‐woman with ovarian stromal and endometrial hyperplasia. Another line, NOSE19L3 was derived from cells from an ovarian cytobrushing from a 39‐year old patient undergoing total abdominal hysterectomy for cervical cancer. In all cases, ovarian epithelia were verified as histologically normal with no evidence of hyperplasia nor neoplasia.
Cell culture and reagents
All NOSE cell cultures were maintained in medium (NOSE‐CM) comprising MCDB105:Medium 199 (1 : 1) supplemented with 15% foetal bovine serum, 10 ng/ml epidermal growth factor, 0.5 mg/ml hydrocortisone, 5 mg/ml insulin, and 34 mg protein/ml bovine pituitary extract, (all Sigma, St Louis, MO, USA). Primary NOSE cells were collected by brushing the surface of normal ovaries with a sterile cytobrush, which was then agitated in 5 ml of NOSE‐CM to release the cells. Suspensions were plated into 25‐cm2 tissue culture flasks and left for 7 days to allow cells to grow. Cell culture medium was changed every 2–3 days until cultures reached 80–90% confluence, at which point they were subcultured. Cells were grown as 3D multicellular spheroids (MSC) either in RCCS (Synthecon, Houston, TX, USA) or in plastic dishes coated with 2.5% solution of polyHEMA (Sigma), prepared in 95% ethanol, also from Sigma.
In vitro phenotypic analysis of cell cultures
Population doubling rates were measured for 1 × 105 cells, in triplicate. Cultures were passaged and population doublings (PD) calculated using the following formula:
| PD = log (total cell number at each passage/initial cell number)/log2. |
Assays for anchorage independent growth were performed by plating 2 × 104 cells in complete medium containing 0.3% Noble Agar (Sigma) over a base layer of complete medium containing 0.6% Noble Agar. Five replicates were plated for each primary NOSE cell line. As control, anchorage independent growth was evaluated simultaneously for the TOV112D endometrioid ovarian cancer cell line. After 4 weeks, cells were fixed with methanol and stained with 1% p‐iodonitrotetrazolium violet (Sigma). Colony formation was observed and number of colonies containing > 50 cells were counted. Colony forming efficiencies (CFE %) were calculated using the following formula:
| CFE = [(number of colonies counted)/(number of cells plated)] × 100%. |
Immunoflourescent cytochemistry/immunohistochemistry
All three primary cell isolates (NOSE4, NOSE11 and NOSE19L3) were confirmed as epithelial by immunofluorescent cytochemistry, performed using standard protocols. The following antibodies were used (all diluted 1 : 1000): AE1/AE3 (Dako Corporation, Carpinteria, CA, USA), cytokeratin 7 (CRUK, London, UK), BerEp4 (Dako), CA125 (Dako), E‐cadherin (Cell Signaling, Danvers, MA, USA), FSP (Sigma), and Factor VIII (Lab Vision, Fremont, CA, USA). All cell cultures tested were found to express epithelial markers (AE1/AE3, BerEP4, ck7), and did not express endothelial cell (Factor VIII) and fibro‐blast (FSP) markers. Cells were weakly positive for ovarian cancer‐associated marker CA125. This observation is consistent with findings of another study: 50% of NOSE cell cultures express CA125 in the first few passages (14). Staining for CA125 was also observed in primary ovaries from which NOSE4 and NOSE11 were derived. Immunohistochemistry for 2D and 3D cultures was performed using standard protocols at the CRUK Histology Service.
Apoptosis assays
MCSs from 3D cell clusters were washed in phosphate‐buffered saline and trypsinized at 37 °C to create single cell suspensions; 2D cultures were trypsinized and centrifuged, and cell pellets resuspended in FACS buffer, according to manufacturer's instructions (Roche, Basel, Switzerland). Samples were run on a Becton Dickinson FACS Scan (Franklin Lakes, NJ, USA) and annexin V‐positive cell population was measured.
Transmission electron microscopy/scanning electron microscopy
Cells were either grown on glass coverslips or, for 3D cultures, cell clusters were harvested and media aspirated. Cells were washed once with phosphate‐buffered saline from VWR (West Chester, PA, USA) then fixed with 2% paraformaldehyde, 1.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.3) for 1–2 h (all from Sigma). Samples were then washed in 0.1 M cacodylate buffer and post‐fixed with 1% osmium tetroxide (Sigma) in 0.1 M cacodylate buffer (pH 7.3) for 1 h at 4 °C, before washing twice in 0.1 M cacodylate buffer, then water for 5 min. Samples were stained with 0.5% uranyl acetate (Sigma) for 20 min then washed in water before dehydration with increasing concentrations of ethanol. Samples were then embedded in agar resin, sectioned and examined on a JEOL (JEOL Ltd, Tokyo, Japan) 1010 transmission electron microscope. For scanning electron microscopy, samples were fixed as for transmission electron microscopy, post‐fixed with 1% osmium tetroxide in 0.1 M cacodylate buffer (pH 7.3) for 45 min at 4 °C and dehydrated with increasing concentrations of ethanol. Samples were then critically point dried, mounted on carbon stubs and gold‐coated before viewing using a JEOL 7401 series FEGSEM.
Results
The aim of this study was to establish 3D in vitro models of NOSE cells and to compare morphological and biological characteristics with the same cells grown as 2D cultures, and with primary ovarian tissues. We first established and characterized three primary NOSE cell isolates (NOSE4, NOSE11, NOSE19L3) grown as 2D monolayers. Cell cultures in vitro had growth characteristics typical of NOSE cell cultures (Table 1) (6). NOSE11 cells had the shortest population doubling time and the longest lifespan of the three primary cell isolates. This may be because NOSE4 cells were derived from a postmenopausal woman and, therefore, are likely to have reduced lifespan in vitro. NOSE19L3 was a clone isolated from cell brushing thus, originates from a smaller initial population, hence the reduced lifespan observed in this culture.
Table 1.
Patient information and growth characteristics of normal primary ovarian epithelial cells (OSE)
| Primary cell line | Patient characteristics | Cells grown as 2D monolayers | 3D multicellular spheroids | |||
|---|---|---|---|---|---|---|
| Age | Menopausal status | PD time a (hours) | Maximum no. of PDs | CFE in soft agar b | Average MCS diameter (µm) (range) c | |
| NOSE4 | 62 | Post | 82.4 | 7.6 | 0 | 78.25 (32.70–152.00) n = 11 |
| NOSE11 | 48 | Peri | 57.2 | 17.8 | 0 | 91.053 (80.92–252.40) n = 11 |
| NOSE19L3 | 39 | Pre | 83.6 | 9.7 | 0 | 171.98 (63.90–125.60) n = 5 |
Population doubling (PD) times calculated from the exponential growth phase;
b colony forming efficiency (CFE);
cmulticellular spheroids (MCS) grown in 3D by polyHEMA coating of tissue culture plastics, size measured under the scanning electron microscope.
Establishing and characterizing 3D NOSE cell cultures
All three primary cell isolates formed MCSs when cultured for 14 days either on polyHEMA‐coated plates or in the Rotary Cell Culture System (Fig. 1). MCSs appeared solid and smooth by light microscopy. MCSs grown on polyHEMA‐coated plates (PH‐MCSs) were typically 70–170 µm in diameter (Table 1). NOSE19L3 cells formed significantly larger PH‐MCS than NOSE4 or NOSE11 cells (P < 0.0001 and P = 0.025, respectively; two‐tailed unpaired t‐test). MCSs formed in the RCCS (RCCS‐MCS) grew to 1–2 mm in diameter.
Figure 1.
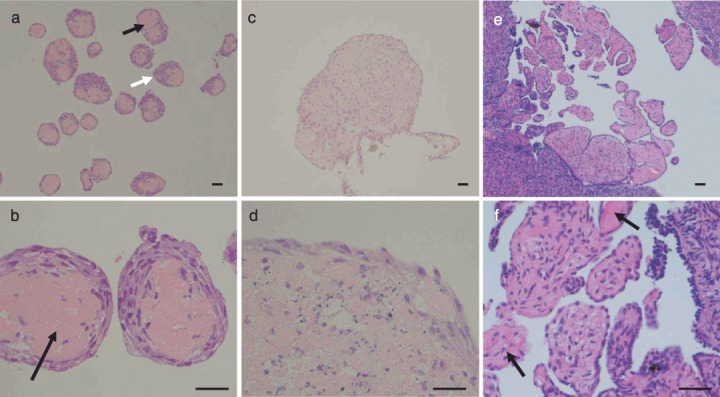
Haematoxylin and eosin‐stained sections of multicellular spheroids from three‐dimensional (3D) cultures. Normal ovarian surface epithelium (NOSE) cells form smaller 3D multicellular structures when cultured on polyHEMA‐coated plates (PH‐MCSs) (a,b) compared to culturing in the rotary cell culture system (RCCS‐MCSs) (c,d). The matrix cores of PH‐MCS are clearly visible (black arrows); cells form either a ring around the matrix or a ‘cap’ on one side (white arrow). PH‐MCSs show architectural resemblance to 3D structures observed on the surface of the ovary in vivo (e,f), where surface papillary projections consist of epithelial cells around stromal cores with matrix protein (black arrows). Scale bars represent 0.1 mm.
We examined the internal architecture of MCSs following paraffin wax embedding, sectioning, and staining with haematoxylin and eosin. For all three primary cell cultures, PH‐MCSs had defined internal architecture: a central core of matrix protein, surrounded by aligned elongated cells. The cells formed a ring around the matrix core, often with a ‘cap’ to one side, with resemblance to papillary structures sometimes observed on the surface of normal ovaries in vivo (Fig. 1). The matrix core often contained degenerate nuclear debris. When cells were grown in RCCS, resulting MCSs had a less organized arrangement of cells. Cells within RCCS spheroids tended to be more rounded with a 1–2 cell deep layer of cells around the edge of the clusters which had an elongated morphology (Fig. 1).
Ultrastructure of PH‐MCS
PH‐MCS were architecturally interesting, so we used transmission and scanning electron microscopy to study their ultrastructure and of the same cells grown in 2D. Desmosomes, which are characteristic of epithelial cells, were present in all 2D and 3D cultures (Fig. 2a). In PH‐MCSs, outer cells (up to 8 cell layers but sometimes only 1–2 cells thick) were elongated and aligned. Peripheral cells were also longer in shape compared to cells located towards the core of the spheroid, where cells were rounder and less densely packed (Fig. 2b). Transmission electron microscopy of PH‐MCSs revealed that cells towards the centre tended to have extensive and dilated rough endoplasmic reticulum, well‐developed Golgi apparatus and open nucleolus. Some cells appeared to have formed a discontinuous basement membrane and matrix‐like material in the extracellular space. This ECM material was abundant in the core of the MCSs.
Figure 2.

Transmission and scanning electron micrographs of normal ovarian surface epithelium (NOSE) cells in culture and from primary tissues. (a) A high‐power electron micrograph of two‐dimensional (2D) NOSE cultured cells. Bundles of intermediate filaments can be seen, with the formation of desmosomes between adjacent cells (white arrow). (b) A low‐power electron micrograph of multicellular structures cultured on polyHEMA‐coated plates (PH‐MCS) showing the flatter and closely opposed cells at the periphery forming concentric rings (such as cell B). Cells within the inner region of the PH‐MCS have a more rounded morphology and are less tightly packed (cell A). Note the cells towards the core of the MCS are full of electron dense vesicles (white arrow), which may illustrate a trend to a more secretary phenotype towards the centre of the multicellular clusters. Extracellular material (M) is abundant in the cores of PH‐MCS. (c) Section of quiescent ovarian epithelium. Note the cuboidal shape of cell, condensed chromatin within the nuclei (N), basement membrane (black arrow), desmosomes (white arrow) and age‐related storage: lipofuscin (L). In vivo NOSE cells are mostly quiescent (unless at the site of follicular rupture, following ovulation). (d) A low power SEM image of a PH‐MCS showing surface morphology. The flattened surface cells form a patchwork with no obvious orientation. The number of surface protrusions varied from a few to many between cells (black arrows).
When examined by scanning electron microscopy (SEM), cells on the outer surface of PH‐MCS had a flattened morphology and very few surface features. Although microvilli were absent, there were often many surface projections connecting adjacent cells amongst PH‐MCS. Two‐dimensional monolayers also had very few surface features; cells were unremarkable, with the exception of long surface projections extending between cells, similar to those seen on PH‐MCSs. Absence of microvilli may have been due to loss of polarization as a result of culturing the cells on glass. It may be possible to overcome this by growing NOSE cells with oestrogen, ovarian stromal fibroblasts or on collagen or fibronectin‐coated plates to attempt to maintain polarization and microvilli formation (15, 16). Dead cells were observed both on the surface of the PH‐MCS and in 2D cultures. Early‐stage cell death was identifiable by holes in the membrane and later‐stage degraded cells were observed budding off the PH‐MCSs and 2D cultures.
Expression of ECM proteins in 2D and 3D cultures
Previous studies have shown that culturing cells in 3D may alter cell–cell interactions, including expression of ECM proteins, receptors and corresponding degradative enzymes. NOSE cells reportedly produce a variety of ECM molecules in vitro (17). Therefore, we used immunohistochemistry to determine whether any of these proteins were differentially expressed between 2D and 3D cultures (Fig. 3). Fibronectin was expressed in 2D and 3D cultures in all three cell cultures. For 2 out of the 3 cell cultures, weak focal vimentin staining was observed in 2D cultures around some mitoses. However, strong positive vimentin characterized all 3D cultures and OSE in vivo. Laminin and AE1/AE3 were not expressed in 2D cultures, but strong expression of these markers was observed PH‐MCSs, RCCS‐MCSs and in epithelium of normal ovarian tissue. Fibrous fibronectin, laminin and vimentin filaments were observed between cells, suggesting that these proteins play a role in maintaining the structure of MCSs. For the markers tested, staining intensity did not vary according to spheroid size.
Figure 3.

Expression analysis of candidate markers by immunohistochemistry of normal ovarian surface epithelium (NOSE) cells from representative two‐ (2D) and three‐dimensional (3D) cultures and in primary NOSE from tissue sections of normal ovaries. A summary of the results of these analyses are given in Table 2. Here, staining patterns suggest that primary NOSE from normal ovarian tissues express a range of extracellular matrix proteins (e.g. vimentin, fibronectin, laminin). Staining profiles of 3D cultures more closely resemble that of primary OSE than 2D cultures. Scale bars represent 0.1 mm.
Table 2.
Differential expression of pan‐cytokeratin (AE1/AE3), laminin, vimentin and collagen IV, detected by immunohistochemistry in two‐ (2D) and three‐dimensional (3D) cultures and in primary normal ovarian tissues. Graded shading denotes extent of staining. White denotes negative staining; crosshatched grey denotes weak or focal staining; light grey represents that 20–50% cells stain positive; and dark grey shading indicates over 50% cells stain positive.

Apoptosis in 2D and 3D cultures
We characterized the proportion of apoptotic cells in the cultures using annexin V and propidium iodide staining, followed by FACS analysis (Fig. 4). Propidium iodide staining identifies the necrotic component of cell cultures; annexin V recognizes externalized phosphatidylserine, which is a measure of early stages of apoptosis. In RCCS‐MCSs, the proportion of cells positive for annexin V was at least 2.5‐fold that of 2D cultures and PH‐MCSs. For two cell cultures, we did not observe any significant difference in proportion of apoptotic cells in PH‐MCSs compared to 2D cultures. For one primary cell isolate (NOSE19L3), there was significantly more apoptosis in PH‐MCS (P = 0.0470 using two‐tailed paired t‐test) compared to 2D cultures. This may be due to the shorter population doubling time for this culture or due to loss of apoptotic cells from 2D cultures during washing and harvesting. We found significant differences in proportions of annexin V‐positive cells in RCCS‐MCSs compared to 2D cultures and PH‐MCS for all three cell cultures (NOSE4, P = 0.0176; NOSE11, P = 0.0164; and NOSE19L3, P = 0.0152, using two‐tailed paired t‐tests). For one culture (NOSE11), we observed by propidium iodide staining, a significantly larger necrotic component in RCCS‐MCSs compared to 2D and PH‐MCSs (P = 0.0242, two‐tailed paired t‐test). Thus, for this culture there was a statistically significant increase in the proportion of cells at all stages of apoptosis in RCCS cultures compared to cultures grown in 2D or on polyHEMA‐coated plates. Indeed, cells at all stages of apoptosis were distributed throughout the RCCS‐MCSs (Fig. 4).
Figure 4.

Variation in levels of apoptosis in two‐ (2D) and three‐dimensional (3D) cultures. (a) Levels of apoptosis (as measured by annexin V expression) are significantly higher in 3D multicellular spheroids (MCS) grown in the rotary cell culture system (RCCS‐MCS) compared to 2D cultures and MCS grown on polyHEMA‐coated plates (PH‐MCS). Error bars = standard error of the mean (SEM). (b) Apoptotic cells (arrow) are visible throughout the RCCS‐MCS, and are not localized to a necrotic ‘core’ of MCSs. Scale bar represents 0.1 mm.
Discussion
To the best of our knowledge, this paper describes for the first time the development and characterization of a 3D model of normal ovarian surface epithelial cells. The OSE is typically described as a monolayer of cells, but it nonetheless has a 3D architecture that cannot be replicated by culturing primary NOSE cells in 2D on adherent plastic surfaces. In this study, we show that NOSE cells form 3D structures (MCSs) when cells are prevented from adhering to tissue culture surfaces either by chemical treatment of tissue culture vessels or by maintaining cells in constant rotation. In the RCCS, NOSE cell clusters were larger and had a chaotic internal structure compared to cells grown on polyHEMA‐coated plastics. NOSE cells grown in polyHEMA‐coated vessels formed concentric layers around a core of matrix protein. These data suggest that in a static microenvironment, NOSE cells spontaneously form organized 3D multicellular structures.
There are other reports describing 3D cell aggregates of other cell types in which two distinct regions of cells are observed: an inner area of cells that are smaller and less proliferative than surrounding cells on the periphery of MCS (10, 18). In one study, Freyer and Sutherland found that cells on the surface of MCSs formed from mouse mammary tumour cells were similar in size to an exponentially growing 2D cell culture, but with 60% less cells in S phase (19). Our observations of the structure of RCCS‐MCSs (by light microscopy) and PH‐MCSs (by light and transmission electron microscopy) are consistent with these findings: instead of a continual reduction in cell size from the outer to inner core of spheroids, we found clear distinction in morphology between cells on the outer layers compared to cells within the spheroids. Cells within MCS also appeared to be rounder in shape compared to elongated cells at the periphery. Other studies have found differences in cell size and morphology at different regions of MCSs (10, 19).
Measurements of apoptosis between 2D and 3D cultures suggest a greater proportion of cells grown in the RCCS are apoptotic when compared to 2D and polyHEMA 3D cultures. This was confirmed by the observation of many apoptotic nuclei distributed throughout the RCCS‐MCS. An explanation for this is unclear, but it could be that the continual motion of spinning the cells in the RCCS causes mechanical damage which in turn induces apoptosis. Thus, increased levels of apoptosis observed in RCCS‐MCSs may be an experimental artefact. Apoptosis is generally low in the ovarian epithelium in vivo since these cells are usually quiescent (except immediately after rupture of a mature follicle). Unlike some cancer cell spheroids, extensive apoptosis and necrosis is not localized to the cores of the clusters. Conditions at the centre of the RCCS‐MCS are unlikely to be hypoxic in comparison to the in vivo microenvironment – the oxygen concentration in the pelvis is 5.5% (measured at the cervix) and cells were cultured in 20% oxygen (20), further suggesting that increased rates of apoptosis observed in RCCS‐MCS were a consequence of mechanical damage. Indeed, we have observed that prolonged culture in the RCCS can result in spheroids that consist only of cells that have degenerated (unpublished data).
Transmission electron microscopy of PH‐MCSs revealed that cells contained swollen endoplasmic reticulum and Golgi apparatus and had an open nucleolus. Cells within PH‐MCS were actively producing and appeared to be secreting a basement‐membrane‐like matrix, which was abundant in the core of the MCSs. ECM molecules play a vital role in tissue architecture and are vital for the formation of hepatoma spheroids (21). Both RCCS‐MCSs and PH‐MCSs produced an abundance of ECM proteins laminin, fibronectin and vimentin, but did not express collagen IV. Two‐dimensional cultures showed widespread expression of fibronectin and collagen IV but not laminin, and vimentin was only expressed focally at mitoses in 2 out of the 3 primary cell cultures. These patterns of protein expression reflect the phenotypic plasticity that is characteristic of NOSE cells: in both 2D and 3D, cells show both epithelial (collagen IV, laminin, cytokeratin) and mesenchymal (vimentin) features.
In general, cells demonstrate considerable plasticity and have an ability to respond dramatically to their environment. A transcriptome that promotes growth within an organism is unlikely to provide the cell with an optimal phenotype for growth in 2D. Since the 1970s, 3D culture techniques have been considered to be an invaluable tool for studying cancer cell growth. Some studies have suggested that multicellular tumour spheroids grown either in spinner flasks or on polyHEMA‐coated plates are representative of the early stages of tumour growth prior to vascular involvement (10, 22). Furthermore, many studies point to the downstream pathways of cancer cell interactions with the ECM as important therapeutic targets (23). Thus, 3D models, such as those presented here, are likely to represent useful tools to examine the roles of different genes involved in the earliest stages of tumorigenesis. More complex, heterotypic cultures containing ovarian stromal fibroblasts and endothelial cells will assist in developing our understanding of the role of interactions between different cell types during epithelial tumourigenesis. There is a need for more biologically relevant in vitro models of epithelial ovarian cancer since in vivo modelling of this disease has had limited success (24, 25). Despite the discovery of ovary‐specific Mullerian‐inhibiting substance receptor type II, an in vivo model that accurately reflects epithelial ovarian cancer: a disease that is late‐onset and histologically heterogeneous, has yet to be established. By mimicking the processes of early ovarian tumour development in an microenvironment that closely resembles this process in vivo, it may be possible to identify new proteins associated with tumour progression that represent biomarkers for early detection of disease and/or novel therapeutic targets.
Acknowledgements
This work is supported by an Medical Research Council studentship (Kate Lawrenson), the Eve Appeal Gynaecology Cancer Research Fund and the Rosetrees Trust (via The Eve Appeal). We also thank Ken Choi for his help with fluorescence activated cell sorting analysis.
References
- 1. Scully RE (1995) Pathology of ovarian cancer precursors. J. Cell. Biochem. 23, 208–218. [DOI] [PubMed] [Google Scholar]
- 2. Auersperg N, Wong AS, Choi KC, Kang SK, Leung PC (2001) Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr. Rev. 22, 255–288. [DOI] [PubMed] [Google Scholar]
- 3. Okamura H, Katabuchi H (2001) Detailed morphology of human ovarian surface epithelium focusing on its metaplastic and neoplastic capability. Ital. J. Anat. Embryol. 106(2 Suppl. 2), 263–276. [PubMed] [Google Scholar]
- 4. Auersperg N, Siemens CH, Myrdal SE (1984) Human ovarian surface epithelium in primary culture. In Vitro 20, 743–755. [DOI] [PubMed] [Google Scholar]
- 5. Auersperg N, Maines‐Bandiera SL, Dyck HG, Kruk PA (1994) Characterization of cultured human ovarian surface epithelial cells: phenotypic plasticity and premalignant changes. Lab. Invest. 71, 510–518. [PubMed] [Google Scholar]
- 6. Li NF, Wilbanks G, Balkwill F, Jacobs IJ, Dafou D, Gayther SA (2004) A modified medium that significantly improves the growth of human normal ovarian surface epithelial (OSE) cells in vitro . Lab. Invest. 84, 923–931. [DOI] [PubMed] [Google Scholar]
- 7. Dyck HG, Hamilton TC, Godwin AK, Lynch HT, Maines‐Bandiera S, Auersperg N (1996) Autonomy of the epithelial phenotype in human ovarian surface epithelium: changes with neoplastic progression and with a family history of ovarian cancer. Int. J. Cancer 69, 429–436. [DOI] [PubMed] [Google Scholar]
- 8. Khaoustov VI, Darlington GJ, Soriano HE, Krishnan B, Risin D, Pellis NR et al (1999) Induction of three‐dimensional assembly of human liver cells by simulated microgravity. In. Vitro. Cell Dev. Biol. Anim. 35, 501–509. [DOI] [PubMed] [Google Scholar]
- 9. Knuechel R, Keng P, Hofstaedter F, Langmuir V, Sutherland RM, Penney DP (1990) Differentiation patterns in two‐ and three‐dimensional culture systems of human squamous carcinoma cell lines. Am. J. Pathol. 137, 725–736. [PMC free article] [PubMed] [Google Scholar]
- 10. Ghosh S, Spagnoli GC, Martin I, Ploegert S, Demougin P, Heberer M et al (2005) Three‐dimensional culture of melanoma cells profoundly affects gene expression profile: a high density oligonucleotide array study. J. Cell. Physiol. 204, 522–531. [DOI] [PubMed] [Google Scholar]
- 11. Zietarska M, Maugard CM, Filali‐Mouhim A, Alam‐Fahmy M, Tonin PN, Provencher DM et al (2007) Molecular description of a 3D in vitro model for the study of epithelial ovarian cancer (EOC). Mol. Carcinog. 46, 872–885. [DOI] [PubMed] [Google Scholar]
- 12. Burleson KM, Casey RC, Skubitz KM, Pambuccian SE, Oegema TR Jr, Skubitz AP (2004) Ovarian carcinoma ascites spheroids adhere to extracellular matrix components and mesothelial cell monolayers. Gynecol. Oncol. 93, 170–181. [DOI] [PubMed] [Google Scholar]
- 13. Burleson KM, Boente MP, Pambuccian SE, Skubitz AP (2006) Disaggregation and invasion of ovarian carcinoma ascites spheroids. J. Transl. Med. 4, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Auersperg N, Maines‐Bandiera SL, Dyck HG (1997) Ovarian carcinogenesis and the biology of ovarian surface epithelium. J. Cell. Physiol. 173, 261–265. [DOI] [PubMed] [Google Scholar]
- 15. Saridogan E, Djahanbakhch O, Kervancioglu ME, Kahyaoglu F, Shrimanker K, Grudzinskas JG (1997) Placental protein 14 production by human Fallopian tube epithelial cells in vitro . Hum. Reprod. 12, 1500–1507. [DOI] [PubMed] [Google Scholar]
- 16. Bai W, Oliveros‐Saunders B, Wang Q, Acevedo‐Duncan ME, Nicosia SV (2000) Estrogen stimulation of ovarian surface epithelial cell proliferation. In. Vitro. Cell Dev. Biol. Anim. 36, 657–666. [DOI] [PubMed] [Google Scholar]
- 17. Kruk PA, Uitto VJ, Firth JD, Dedhar S, Auersperg N (1994) Reciprocal interactions between human ovarian surface epithelial cells and adjacent extracellular matrix. Exp. Cell. Res. 215, 97–108. [DOI] [PubMed] [Google Scholar]
- 18. Sutherland RM, Sordat B, Bamat J, Gabbert H, Bourrat B, Mueller‐Klieser W (1986) Oxygenation and differentiation in multicellular spheroids of human colon carcinoma. Cancer Res. 46, 5320–5329. [PubMed] [Google Scholar]
- 19. Freyer JP, Sutherland RM (1980) Selective dissociation and characterization of cells from different regions of multicell tumor spheroids. Cancer Res. 40, 3956–3965. [PubMed] [Google Scholar]
- 20. Juul N, Jensen H, Hvid M, Christiansen G, Birkelund S (2007) Characterization of in vitro chlamydial cultures in low‐oxygen atmospheres. J. Bacteriol. 189, 6723–6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin RZ, Chou LF, Chien CC, Chang HY (2006) Dynamic analysis of hepatoma spheroid formation: roles of E‐cadherin and β1‐integrin. Cell Tissue Res. 324, 411–422. [DOI] [PubMed] [Google Scholar]
- 22. Sutherland RM, Durand RE (1976) Radiation response of multicell spheroids – an in vitro tumour model. Curr. Top. Radiat. Res. Q 11, 87–139. [PubMed] [Google Scholar]
- 23. Ahmed AA, Mills AD, Ibrahim AE, Temple J, Blenkiron C, Vias M et al (2007) The extracellular matrix protein TGFBI induces microtubule stabilization and sensitizes ovarian cancers to paclitaxel. Cancer Cell 12, 514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Connolly DC, Bao R, Nikitin AY, Stephens KC, Poole TW, Hua X et al (2003) Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res. 63, 1389–1397. [PubMed] [Google Scholar]
- 25. Garson K, Shaw TJ, Clark KV, Yao DS, Vanderhyden BC (2005) Models of ovarian cancer – are we there yet? Mol. Cell. Endocrinol. 239, 15–26. [DOI] [PubMed] [Google Scholar]