

Abstract
The last decade has brought unprecedented advances in our understanding of megakaryocyte (MK) biology and platelet production, processes that are strongly dependent on the cytoskeleton. Facilitated by technological innovations, such as new high-resolution imaging techniques (in vitro and in vivo) and lineage-specific gene knockout and reporter mouse strains, we are now able to visualize and characterize the molecular machinery required for MK development and proplatelet formation in live mice. Whole genome and RNA seq analysis of patients with rare platelet disorders, combined with targeted genetic interventions in mice, has led to the identification and characterization of numerous new genes important for MK development. Many of the genes important for proplatelet formation code for proteins that control cytoskeletal dynamics in cells, such as Rho GTPases and their downstream targets. In this review, we discuss how the final stages of MK development are controlled by the cellular cytoskeletons, and we compare changes in MK biology observed in patients and mice with mutations in cytoskeleton regulatory genes.
Introduction
Platelets are small anucleate cells that circulate in blood. They play a crucial role in securing vascular integrity upon disruption of the endothelial barrier[1,2]. A significant change in the platelet count can be a major risk factor for bleeding or thrombosis. In humans, platelets circulate in blood with an average lifespan of 7–10 days. Thus, the body must produce a great number of platelets daily to maintain a consistent circulating platelet count. Their precursor cell, the megakaryocyte (MK), is responsible for producing approximately 1011 platelets daily, with each individual MK being able to generate a thousand or more platelets[3]. In humans, MKs reside primarily in the bone marrow (BM). Pulmonary MKs can also be detected, but their relevance for thrombopoiesis remains controversial[4,5]. In mice, the spleen serves as an important hematopoietic organ in addition to the BM [6].
The classical MK development model starts with pluripotent hematopoietic stem cells (HSCs) undergoing commitment towards the lymphoid or the myeloid lineage[7]. The common myeloid progenitor (CMP) then transitions to a bipotent MK-erythrocyte progenitor (MEP), which gives rise to both erythroid and megakaryocytic progenitors (MKP). Interestingly, recent evidence suggests an alternative pathway to MKPs, where defined HSC subpopulations exist, including a MK-biased HSC that can differentiate into a MKP without transitioning through classical progenitor cell stages, such as the MEP [7–9]. Irrespective of how they developed, MKPs eventually lose the capacity to divide and begin the process of endoreplication, leading to polyploidization and cellular enlargement[10]. MKs that reach the mature stage are characterized by a highly polyploid nucleus, expanded size, and membrane structures called the demarcation membrane system (DMS) (Figure 1). The DMS provides the membranes required for the extension of proplatelet shafts, cellular protrusions that shed proplatelets from their tips. Proplatelet formation (PPF) requires dynamic remodeling and profound changes in the microtubule (MT) and actin cytoskeleton. For example, it was shown that the actin cytoskeleton is critical for DMS formation as it provides mechanical forces that drive invaginations at the plasma membrane [11]. The tubulin cytoskeleton guides proplatelet shaft initiation and elongation through continuous growth of MT plus-ends as well as sliding of adjacent MTs relative to one another[12]. Proplatelet branching, a process that determines how many platelets can be released per MK, is guided by non‐muscle myosin IIA and the actin cytoskeleton[13]. It is important to remember, however, that cellular cytoskeletons do not operate in isolation. Multidomain proteins, such as formins, can connect actin microfilaments and MTs and thus establish a link between different cytoskeleton-driven cellular responses[14]. Critical upstream regulators, such as Rho GTPases, control both actin and MT cytoskeletal dynamics by interacting with a variety of target proteins[15–18].
Figure 1. Schematic representation showing the final stages of MK development leading to platelet release into BM sinusoids.
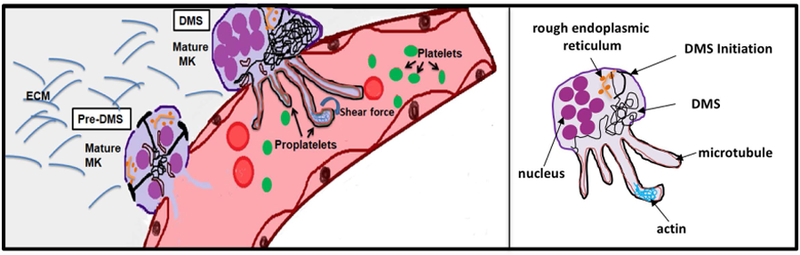
Terminal MKs enter the process of proplatelet formation by establishing an expansive and interconnected membranous network, called the Demarcation Membrane System (DMS). Signals initiated at the cell membrane lead to formation of focal tubular membrane invaginations and the generation of surface-connected intracellular membrane pools, called the pre-DMS, which are precisely located along the cleavage furrow of the endomitotic MK. Maturation of the pre-DMS into the DMS is dependent on the insertion of additional membranes. Once it is fully established, the DMS provides the membranes required for the extension of proplatelet shafts, cellular protrusions that shed proplatelets from their tips. Proplatelet formation (PPF) requires dynamic remodeling and profound changes in the microtubule (MT) and actin cytoskeleton. The release of proplatelet structures, which mature into platelets, into the blood stream is supported by shear force and turbulence. ECM: extracellular matrix.
Filamentous actin (F- actin) is a polymer of actin monomers (globular actin or G-actin), which align in a head-to-tail manner[19,20] (Figure 2A). Actin filaments are flexible fibers of ~7nm in diameter. Actin filaments have plus (also called barbed) and minus (pointed) ends that differ in their affinities for ATP-bound G-actin. There are six genes (3 α-, 1 β-, and 2 γ-actin genes) encoding for different actin isoforms. Platelets express β- and γ-actin at ~2 million copies per cell (0.55 mM, ~15% of total platelet protein). F-actin assembly and turnover is regulated by a large number of proteins, some of which will be discussed below. In humans, 9 genes each encode for a variety of α- and β-tubulin isoforms[21,22]. MKs and platelets express several of these of α- and β-tubulin isoforms, β1 tubulin being unique and critical for the tubulin cytoskeleton in these cells. The α- and β-tubulins share 40% amino-acid sequence identity, and both undergo a variety of posttranslational modifications. MTs are tubulin heterodimer polymers, which form rigid tubes of ~25nm in diameter (Figure 2B). MTs are polar structures with the plus-end being capable of rapid growth, while the minus end loses subunits if not stabilized[23,24]. Polymerization involves extension through the addition of free tubulin dimers to existing MTs or de novo formation of new MTs. Proplatelet extend outwardly at a steady rate of ~1 μm/min, and generally reach a length of ~0.5–1 mm. The MT coil at the periphery of resting platelets is critical for the maintenance of cellular size and shape.
Figure 2. DMS biogenesis and proplatelet elongation depend on the actin and microtubule cytoskeletons.
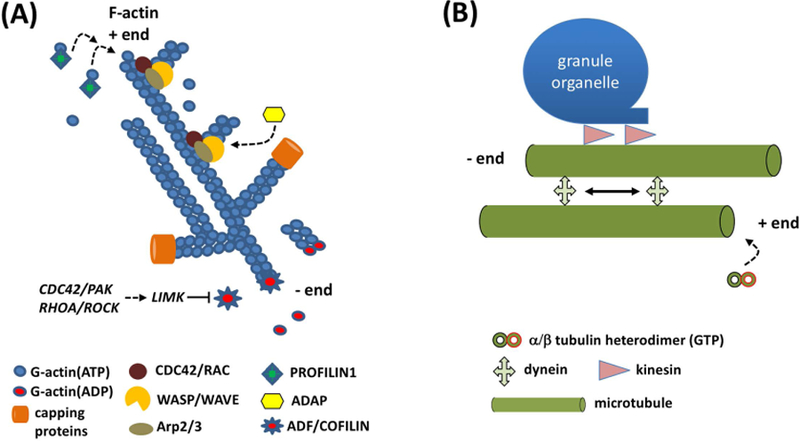
(A) The actin cytoskeleton is critical for DMS biogenesis. Filamentous (F-) actin is organized in a branched network. Plus (+) ends grow rapidly by addition of G-actin ATP monomers, facilitated by PROFILIN1. The ARP2/3 complex, controlled by the CDC42-WASP and/or the RAC/WAVE pathways, initiates branching of existing F-actin filaments. The adapter protein, ADAP, assists in this process. Binding of capping proteins to the +ends limits the growth of F-actin filaments. Actin depolymerizing factor (ADF)/COFILIN is a family of actin-binding proteins which disassembles actin filaments. Its activity is inhibited by LIM kinase (LIMK), which is controlled by the CDC42/PAK and RHOA/ROCK pathways.
(B) Proplatelet elongation is microtubule (MT)-driven. MTs are α/β tubulin heterodimer polymers, which form rigid tubes of ~25nm in diameter. MTs are polar structures that grow from their plus (+) end. Proplatelet shafts are elongated by two separate MT activities: the continuous growth of MT + ends and sliding of adjacent MTs relative to one another. Cytoplasmic dynein is a motor protein responsible for sliding of MTs relative to one another, the major mechanism underlying proplatelet growth. Kinesin is another motor protein required for cargo (organelles, granules) movement along the MTs.
The various members of the Rho family of small GTPases are critical upstream regulators of both the MT and actin cytoskeleton[18]. Rho GTPases cycle between two conformational forms, the GTP-bound active form and the GDP-bound inactive form[25,26]. Once activated, Rho GTPases bind to a variety of effectors, including protein kinases and proteins that bind and regulate the actin and tubulin cytoskeletons. The best-studied members of the mammalian Rho GTPase family are RHOA, RAC and CDC42, which transmit extracellular signals towards the formation of stress fibers, lamellipodia, and filopodia, respectively[25].
In this review, we will discuss the current state-of-the-art on how the cellular cytoskeletons control DMS biogenesis and PPF in MKs. A particular focus will be on the molecular mechanisms by which Rho GTPases and their downstream targets guide actin and MTs in these processes.
DMS biogenesis
Critical Role for GPIb-V-IX, Filamin A, and F-BAR proteins in DMS Initiation
During their maturation in the BM, MKs synthesize large amounts of proteins required for the generation of endoplasmic reticulum (ER), Golgi apparatus, vesicles, storage granules and the expansive and interconnected membranous network of the DMS[27]. Elegant studies by Eckly et al.[28] suggest a model where DMS biogenesis is initiated from the cell membrane, in areas rich in the GPIb-V-IX complex (Figures 1 & 3). Signals initiated in these GPIb-V-IX clusters lead to the generation of surface-connected intracellular membrane pools, called the pre-DMS, which are precisely located along the cleavage furrow of the endomitotic MK. Development of the pre-DMS starts by formation of focal tubular membrane invaginations, at the cell periphery, followed by the expansion of the tubular membranes by adding material delivered from the Golgi complex and by ER-mediated lipid transfer. The growth of the pre-DMS is then fueled by additional plasma membrane invaginations as well as insertion of vesicular membranes from intracellular stores such as the trans Golgi network[28]. The biogenesis of the DMS is strongly dependent on F-BAR domain proteins (FCH-BAR, Fes/CIP4 Homology-BAR)[29]. Members of the BAR (Bin-Amphiphysin-Rvs) domain superfamily bind to PI(4,5)P2 through polycationic regions and have diverse roles in cellular architecture and function through their ability to curve membranes[30]. The key role in DMS biogenesis for F-BAR proteins, such as CIP4 and PACSIN2, was shown in studies with knockout mice[31–33]. Cip4−/− mice display mild thrombocytopenia due to impaired PPF. Cip4−/− MKs exhibit several defects, including decreased cortical actin tension and plasma membrane stiffening, impaired endocytosis and marked alterations in DMS structure. CIP4 recruits and activates CDC42 for membrane tubulation and N-WASP to promote actin cytoskeletal reorganization[31]. PACSIN2 (syndapin 2, FAP52) is one of the most abundant F-BAR proteins in human and mouse MKs. It is composed of an N-terminal F-BAR domain and C-terminal Src homology 3 domains which interact with actin regulatory and endocytic proteins, required for normal MK development [32]. PACSIN2 co-localizes with Filamin-A and GPIb-V-IX in cluster-like focal membrane invaginations, important for the initiation of DMS biogenesis[34]. PACSIN2 associates preferentially with soluble Filamin-A to catalyze membrane projection and dissociates from Filamin-A once membrane projections elongate. The DMS in mature MKs isolated from FlnA−/− mice is less well defined than in control MKs [31]. The interaction of Filamin-A with GPIbα is also important for the regulation of platelet size, as well as for platelet adhesion and maintenance of membrane integrity under high shear[36]. Thus, the GPIb-V-IX receptor is not just a marker of DMS membranes, it also provides an important linkage to the sub-membranous actin cytoskeleton, mediated by its interaction with Filamin-A[36,37]. Consistently, impaired F-actin formation results in the disappearance of the DMS and disrupted association between GPIb-V-IX and the plasma membrane[11]. DMS assembly depends on actin cytoskeleton filaments to generate mechanical forces that drive invaginations at the plasma membrane. F-actin localizes around the DMS network, while Myosin-IIA delineates the DMS and links F-actin to the membrane to help actin turnover during PPF[11]. Moreover, F-actin, but not tubulin, drives the partitioning of the membrane and nuclei territories, required for PPF that follows.
Figure 3. Role of Rho GTPases and their downstream targets in DMS biogenesis and proplatelet formation.
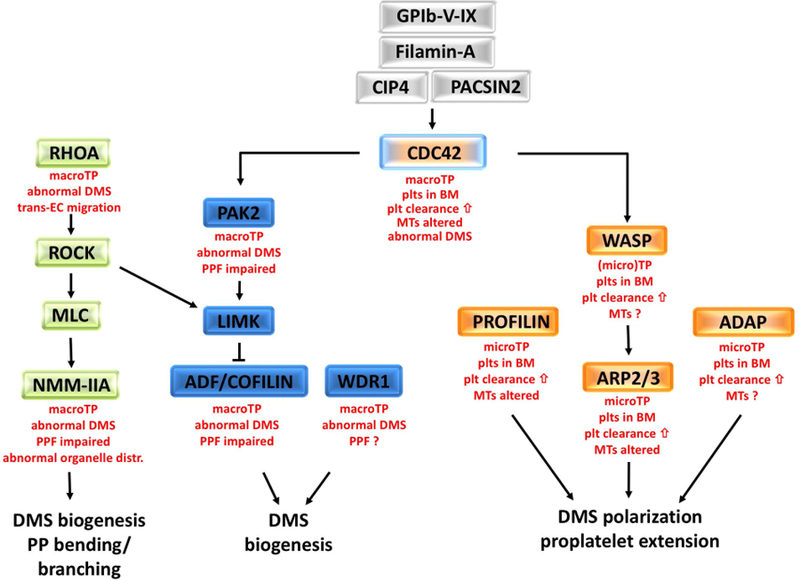
Small GTPases are important regulators of the MK cytoskeletons, DMS biogenesis, and proplatelet formation. Activation of CDC42 is dependent on the GPIb-V-IX complex, filamin A, and the F-BAR proteins, CIP4 and PACSIN2 (grey boxes). CDC42 affects the activity state of ADF/COFILIN via PAK2/LIMK signaling for proper DMS formation (blue boxes). WD40 repeat protein 1 (WDR1) forms a complex with COFILIN. Downstream of RHOA, ROCK controls F-actin levels via ADF/COFILIN and bending via MLC and non-muscle myosin (NMM-IIA) (green boxes). Macrothrombocytopenia (macroTP), an abnormal DMS, and impaired PPF are common to mutant mice with defects in RHOA/ROCK or CDC42/PAK2 signaling pathways. However, mice deficient in CDC42 show additional defects such as ectopic platelet release into the bone marrow niche and increased clearance of platelets from the circulation. Similar defects are observed in mice deficient in WASP, ARP2/3, PROFILIN1, and ADAP (orange boxes), suggesting that these proteins are part of the same signaling pathway. Micro-thrombocytopenia (microTP), an altered tubulin cytoskeleton in platelets, abnormal DMS polarization, and defective proplatelet extension are additional defects characteristic for these mice. The small GTPase RAC1 can partially compensate for deficiency in CDC42 (not shown). Listed in red font are the main MK and platelet phenotypes in mice with defects in the respective signaling molecules.
DMS Biogenesis Depends on the Actin Cytoskeleton and its Regulators
Since the integrity of the actin cytoskeleton is a prerequisite for DMS biogenesis, it is not surprising that mutations affecting proteins involved in actin regulation often give rise to thrombocytopenia (Figure 3). Deficiency in the small Rho GTPase, CDC42, severely impairs DMS biogenesis and GPIbα localization in MKs [11]. CDC42 regulates the phosphorylation of p21-activated kinases, PAK1 and PAK2. Mice deficient in PAK2 exhibit macrothrombocytopenia due to impaired platelet production. Pak2−/− MKs exhibit increased polyploidization and decreased PPF[38]. The downstream target of PAK2 is LIM domain kinase 1 (LIMK1) which regulates actin polymerization and inactivation of actin depolymerizing factor (ADF) and COFILIN. The ADF/COFILIN family spatially and temporally regulates F-actin turnover. When dephosphorylated, ADF/COFILIN lead to actin monomer formation by severing F-actin filaments (Figure 2A). LIMK1 can inhibit this activity by phosphorylating a regulatory serine residue[39]. MKs from mice deficient in ADF and COFILIN exhibit various abnormalities, including an abnormal DMS due to F-actin accumulation and altered granule distribution[40]. WD40 repeat protein 1 (WDR1) forms a complex with COFILIN. Mice hypomorphic for WDR1 exhibit profound megakaryocytosis and severe macrothrombocytopenia[41]. WDR1 mutant MKs show gross morphologic abnormalities, including altered DMS biogenesis and the presence of MK cytoplasmic fragments throughout the bone marrow and spleen (Figure 3).
Defects in DMS biogenesis are also observed in mice with defects in other regulators of the actin cytoskeleton. Tropomodulin-3 (TMOD3) is critical for actin polymerization and stability by capping F-actin pointed ends and by binding tropomyosins[42]. Deletion of Tmod3 in mice leads to embryonic lethality by E18.5, macrothrombocytopenia, hemorrhage and endothelial barrier dysfunction. Tmod3−/− MKs display an abnormal cytoplasmic organelle distribution and immature DMS leading to fewer and larger platelets[42]. Tropomyosin-4 (TPM4) plays an important role by binding actin filaments, which can generate proplatelet branches. TPM4 insufficiency causes macrothrombocytopenia in mice and humans in a gene dose-dependent manner[43]. Loss of TPM4 impairs binding of other proteins, such as Filamin-A and Actinin-1, to actin filaments. This results in an impairment of the complex actomyosin cytoskeletal network that drives proplatelet formation. Tropomyosins are implicated in the regulation of cell morphology, adhesion, migration, granule trafficking, cell division, and apoptosis [44].
An underdeveloped DMS with insufficient membrane for PPF is also observed in MKs with a disrupted spectrin cytoskeleton[45]. Both erythroid and nonerythroid spectrin isoforms accumulate in MKs within distinct locations, suggesting unique roles in platelet formation. The erythroid isoforms exhibit a more punctate localization in proplatelets, whereas the nonerythroid isoforms have a cytoskeletal localization that is enriched with F-actin at the cell periphery. However, the molecular details for how spectrin affects DMS biogenesis are not well understood.
In summary, the formation of the DMS is a critical step in MK development leading to proplatelet formation. DMS formation is initiated by engagement of the GPIb-V-IX complex and mediated by the actin cytoskeleton. Mutations in GPIb-V-IX and its downstream actin regulatory proteins lead to macrothombocytopenia in mice and humans. Once established, the DMS provides membranes for nascent platelets. The next section of this review will discuss how proplatelet formation is initiated and guided following DMS formation.
Proplatelet formation
Microtubules Drive Proplatelet Shaft Initiation and Elongation
Once the DMS is fully established, MKs need to form an extensive pseudopodial network in order to produce and release nascent platelets into bone marrow sinusoids. These pseudopodial extensions initiate with the formation of 100–500 µm long protrusions known as proplatelet shafts[46–49]. MTs, composed of αβ-tubulin heterodimers, play a key role in this process [50,51]. During the initial phase, the bulk of the proplatelet area is filled with MTs, collected and assembled in the cortex below the plasma membrane of the mature MK. Linear arrays of MTs then align and merge into thick linear bundles that fill the proplatelet shafts[52–54]. Next, these MTs form a loop at free proplatelet ends and re-enter the proplatelet shaft, giving rise to bulbous tips measuring 3–5 µm in diameter. Finally, platelets are released with a MT coil, which is formed at the tips of proplatelet shafts before the terminal release of platelets [53,55]. Proplatelet shafts are elongated by two separate MT activities: the continuous growth of MT plus-ends and sliding of adjacent MTs relative to one another (Figure 2B). The rate of MT growth in MKs depends on the maturation state of the MK. MT growth rates are biphasic in proplatelets: one population grows at rates of 5μm/min, while the other elongates much faster at rates of 14μm/min [13]. Time-lapse video fluorescence microscopy studies revealed that MTs polymerize in both directions in proplatelets, towards both the tips and the cell body [12]. Interestingly, the rate of proplatelet shaft elongation did not change in the presence of MT inhibitors, suggesting that this process requires MT sliding, but not polymerization of new MTs. Cytoplasmic dynein and kinesin are motor proteins that move on MTs and attach to cargo that is moved over MTs [13,56]. Localization of kinesin, dynein, and dynactin (a co-factor of dynein required to traverse the MT) was studied in murine MKs at different stages of differentiation [12,57]. Dynein and dynactin, but not kinesin, are localized to the proplatelet MT bundles. Cytoplasmic dynein is primarily responsible for sliding of MTs relative to one another in proplatelet shafts, while kinesin associates with granules and organelles to be transported into nascent proplatelets (Figure 2B). Individual MTs slide relative to one another at approximately 4 μm/min in either direction, a process that is inhibited in MKs lacking functional dynein. Recent work by Bender et al. further established that proplatelet shaft elongation is not a continuous process once initiated, but rather undergoes repetitive phases of extension, pause, and retraction back to the MK cell body[58]. Exposure to shear further increases the speed of MT sliding as shear force gets converted into biochemical signals that increase dynein function [59].
The predominant tubulin isoform in platelets and MKs is the β-tubulin, β1. In MKs, β1-tubulin is localized to the marginal bands of proplatelets. In platelets, it is wound in 8–12 coils at the periphery just below the plasma membrane[60,61]. β1 tubulin-deficient mice[61] and patients carrying a mutation in the TUBB1 gene[62] are macro-thrombocytopenic, in part due to impaired PPF. Tubulin-deficient platelets have uniform spherical shape caused by altered marginal bands that contain only two to three MT coils[63,64]. Ran binding protein 10 (RanBP10) is a β1 tubulin binding protein expressed in MKs and platelets[65]. It is found in the cytoplasm of mature MKs and platelets, concentrated on polymerized, noncentrosomal MTs. Ranbp10−/− mice exhibit slightly reduced PPF but normal peripheral platelet counts. Interestingly, Ranbp10−/− platelets show defects in MT filament numbers and localization[65]. Thus, the interaction of β1-tubulin with RanBP10 is critical for the structural organization of the platelet marginal band and for platelet discoid shape. While the unique role of β1-tubulin in MK and platelet biology is well established, little is known about critical α-tubulin isoforms in these cells. A recent study by Strassel et al. identified enrichment of α4A-tubulin, encoded by the TUBA4A gene, in MKs and platelets[66]. Both mice and a patient carrying missense mutations in TUBA4A displayed mild macrothrombocytopenia and platelet marginal band abnormalities due to MK maturation and PPF defect in mutant mice.
Microtubules provide tracks for transport of granules and organelles into nascent platelets
Platelets have important functions in various physiological processes, including hemostasis, angiogenesis, and immunity[67]. To fulfill these functions, they need to be equipped with organelles and granules, the latter serving as a storage compartment for important hemostatic, vasoactive and chemotactic mediators. MKs distribute a homogeneous amount of granules and organelles into the proplatelet bud before releasing platelets, facilitated by the tubulin cytoskeleton [56]. The MT motor protein, kinesin, colocalizes with cargo in proplatelet shafts and is likely the key driver of this process (Figure 2B), as kinesin-coated beads move bidirectionally on MTs of permeabilized [13][56]. MTs can also move bidirectionally in relation to other filaments, thereby contributing to cargo movement. Utilizing these mechanisms, organelles and granules are transferred to the proplatelet tip, where they continue to circle along the MT coil and finally detach within the nascent platelet. Myosin IIA and F-actin are not required for organelle movement along MT tracks, but they seem to play a role for their distribution within preplatelet fragments released into the sinusoids[68] (Figure 2).
Rho GTPases and WASP/WAVE proteins are critical regulators of PPF
As outlined above, DMS generation and PPF require a profound reorganization of the MK actin and tubulin cytoskeleton, guided by changes in Rho GTPase activity (Figure 3). The RHO/ROCK pathway is a negative regulator of PPF, likely through phosphorylation and activity regulation of myosin light chain 2 (MLC2) [69]. The specific contribution of RAC1 and CDC42 during MK maturation and platelet production was studied in mice lacking these proteins in the MK lineage only (mKO). While deficiency in RAC1 did not alter the peripheral platelet count [70], moderate thrombocytopenia was observed in CDC42-mKO mice[71]. Deficiency in both RAC1 and CDC42 led to severe defects, including marked macrothrombocytopenia, increased platelet turnover, increased numbers of MKs in BM, altered DMS and tubulin structure and impaired PPF[72]. In control MKs, actin is homogenously distributed with organized tubulin bundles during pseudopodia formation. In contrast, in RAC1/CDC42-mKO MKs tubulin was organized in thick bundles, which only partially co-localized with actin. Mechanistic studies identified changes in COFILIN phosphorylation and IQGAP1 levels in MKs from these mice [73].
CDC42 and RAC1 activate downstream mediators of the Wiskott Aldrich Syndrome protein (WASP) family and the WASP family verprolin-homologous protein (WAVE) families, respectively. In the absence of CDC42 and phosphoinositide-4,5-bisphosphate (PIP2) binding, WASP is maintained in an autoinhibitory state, stabilized by WASP-interacting protein (WIP). Upon activation, WASP interacts with the ARP2/3 complex to mediate de novo actin nucleation and branched actin network formation [74]. A surprising observation in WASP-deficient mice was the premature release of platelet-like particles into the BM compartment[75]. However, a similar phenotype was recently reported for mice deficient in CDC42 [71], ARP2/3[76] or PROFILIN[77], highlighting the importance of this pathway for the directed release of proplatelets into BM sinusoids (Figure 3). In humans, Wiskott–Aldrich syndrome (WAS) is caused by mutations in the WAS gene and is characterized by immunodeficiency, eczema and microthrombocytopenia [78][79][80]. Patients carrying a mutation in the WIP gene or Wip−/− mice exhibit milder phenotypes when compared to WASP deficiency [81–83]. The WAVE family consists of three proteins, WAVE1, WAVE2, and WAVE3, all of which are regulators of the actin cytoskeleton[84]. WAVE1 and WAVE2 are expressed in MKs, with WAVE1 expression being dependent on the MK differentiation state [84]. In mice, genetic knockout of WAVE2, the predominant isoform in platelets and MKs, causes early embryonic lethality [85]. Wave2−/− embryonic stem cells differentiated into MKs exhibit defects in PPF and lamellipodia formation. Mice deficient in WAVE1 are viable and exhibit a normal peripheral platelet count. Wave1−/− platelets show a defect in spreading and integrin activation only when activated via the collagen receptor, GPVI [86].
The ARP2/3 complex consists of two actin-related proteins, ARP2 and ARP3, and five other subunits (ARPC1–5, actin-related protein complex components)[87]. Upon activation, the complex assists in the ATP-dependent nucleation of new actin branches from existing filaments[88][89][90]. ARP2/3 also plays a role in regulating MT dynamics, but the underlying mechanisms are not well understood. Havelkova et al. identified that the ARPC2 subunit can directly bind MTs in plants [91]. In mammalian cells, Arp2/3-branched actin affects the MT cytoskeleton by controlling HDAC6-mediated acetylation of tubulin[92]. However, while HDAC6 inhibition impairs PPF in human MKs[93], this effect was the result of altered acetylation of cortactin, not tubulin. Both cortactin and HDAC6 were dispensable for PPF in mice. The contribution of the ARP2/3 complex to MK biology was studied in Arpc2-mKO mice, which lack the entire ARP2/3 complex in MKs and platelets[88]. Mice deficient in ARP2/3 exhibited marked microthrombocytopenia, premature platelet release into the bone marrow compartment, impaired MK spreading, and marked alterations in the platelet ultrastructure, including a distortion of the peripheral tubulin ring. The altered MK and platelet phenotype described for ARP2/3 mutant mice is in agreement with findings in patients with mutations in the ARP2/3 complex [76].
PROFILIN1 (PFN1) is another protein that controls the actin nucleation and assembly process. PFN1 binds monomeric G-actin and accelerates the nucleotide-exchange from ADP to ATP, a key step in actin filament elongation (Figure 2A). In addition, PFN1 associates with MTs and regulates its (+)-end turnover [94][95]. Pfn1-mKO mice display microthrombocytopenia and premature platelet release into the BM [77]. Mutant MKs and platelets exhibit misarranged and hyperstable MTs, a defect that was also found in platelets from WAS patients [77]. PFN1 is functionally linked to MTs through formins (see below), suggesting that PFN1 might act as a direct activator of WASP or formin [96]. These results reveal an unexpected function of PFN1 as a regulator of MT organization.
Another clinical complication characterized by microthrombocytopenia is congenital autosomal-recessive small-platelet thrombocytopenia (CARST). CARST is caused by mutations in adhesion and degranulation-promoting adaptor protein (ADAP) [97]. In other cell types, ADAP indirectly connects with the WASP/ARP2/3 complex, and thus may be critical for F-actin branching. Mice deficient in ADAP show moderate microthrombocytopenia (Figure 3)[98]. Adap−/− MKs exhibit fragmentation and aberrant release of (pro)platelet-like particles into the BM compartment. In vitro, BM-derived MKs lacking ADAP showed reduced spreading on ECM proteins as well as reduced activation of β1 integrins, impaired podosome formation, and defective polarization of the DMS.
In summary, similar MK and platelet phenotypes were reported for mice deficient in CDC42, WASP, ARP2/3, PFN1, or ADAP, suggesting that these proteins are part of a critical pathway in these cells (Figure 3). The cellular defects include microthrombocytopenia, ectopic platelet release into the BM, impaired DMS polarization and podosome formation, altered MT structure, and premature platelet clearance from circulation. PPF, MK polyploidization, and MK localization within the BM were not significantly impaired. While there is a well-documented CDC42-WASp-ARP2/3 signaling axis, future studies will need to investigate how PFN1 and ADAP interact with these proteins.
Actin cytoskeleton and Non‐muscle myosin IIA are critical for proplatelet branching
The number of platelets released per MK depends upon the lengthening and branching of proplatelet ends. These ends are amplified, repeatedly bent and further bifurcate the proplatelet shaft to form new ends. The shaft bends from the site of the original pro-platelet and further branches to form a new daughter proplatelet shaft [99]. Actin-based forces are required for proplatelet bending as MKs treated with actin inhibitors like cytochalasin and latrunculin can extend long proplatelets but fail to branch and thus have fewer beads along the proplatelet shaft [100]. Proplatelet bending and branching is powered by a molecular motor, myosin (Figure 3). Human non‐muscle myosin IIA (NMMIIA), encoded by the MYH9 gene, a class II myosin acts as an ATPase motor that binds to actin filaments and generates force for contraction[68]. Inactivation of the mouse Myh9 gene (Myh9Δ) or its mutation in MYH9‐related diseases leads to macrothrombocytopenia[68,101,102]. Myh9Δ MKs exhibit various ultra-structural defects, including a decreased and disorganized DMS, suggesting that membranes accessible for the formation of the future platelets are a limiting factor [103,104]. RHOA and its effector ROCK control proplatelet bending and branching by phosphorylating Ser19 in myosin light chain 2 (MLC2) and inhibiting myosin phosphatase, upstream regulators of NMMIIA [105,106].
Formins are at the interface between the actin and the tubulin cytoskeleton
As outlined above, the tubulin cytoskeleton is critical for proplatelet extension and platelet release from MKs, whereas the actin cytoskeleton controls DMS generation and proplatelet branching. But both cytoskeletons do not operate in isolation. Formins are multidomain proteins, which can connect actin microfilaments and MTs to support cell adhesion, cell migration during development, and cellular responses to injury and infection[107]. The highest expressed formins in mammalian MKs and platelets are dishevelled-associated activator of morphogenesis 1 (DAAM1), protein diaphanous homolog 1 (DIAPH1, mDia1), FH1/FH2 domain-containing protein 1 (FHOD1), and inverted formin 2 (INF2)[108]. Formin activation requires the displacement of an autoinhibitory domain, a process that can occur in a Rho GTPase-dependent and -independent manner. Once activated, formins can contribute to actin nucleation and elongation, actin bundling, actin and MT alignment, MT stabilization, and MT bundling. The role of formins in MK function is still poorly defined. Knock-down of DIAPH1 in cultured MKs altered F-actin and MT dynamics, leading to increased PPF[109][107]. However, deletion of Diaph1 in mice did not affect the peripheral platelet count. Importantly, a gain-of-function mutation in DIAPH1 (R1213*) was recently identified in patients with macrothrombocytopenia and sensorineural hearing loss. Expression of constitutively active DIAPH1 led to abnormal cortical filamentous actin and reduced PPF in cultured MKs. Patient platelets also exhibited increased stability of MTs [110]. Further studies are required to establish how other formins contribute to MK development, and whether there is redundancy between individual formins that could explain why platelet counts are normal in Diaph1−/− mice.
Concluding paragraph
In this review, we provide a broad overview of how the MK cytoskeletons control DMS biogenesis and PPF, with a special emphasis on tubulin and actin. Extensive studies in thrombocytopenic patients, transgenic mice and other model systems identified signaling molecules, which control the tubulin and actin cytoskeleton. Dysregulation of their function leads to thrombocytopenia and often a change in platelet size and/or morphology. Signaling pathways emerge, based on the observed phenotypes, which provide novel mechanistic insight into the underlying regulatory networks. Many important questions arise from this new knowledge. What explains the smaller platelet size in patients and mice with defects in proteins like WASP, ARP2/3, or PROFILIN? How are the actin and tubulin cytoskeletons linked in MKs? How do formins control cytoskeletal reorganization in MKs? How are MK Rho GTPases regulated in time and space? Do other small GTPases affect MK development, and if yes, is there cross-talk with Rho GTPases. Answering these and other questions related to the MK cytoskeletons will be important to obtain a more complete understanding of MKs, and it will guide efforts to (1) correct the platelet count, size and function in thrombocytopenic patients, and (2) optimize platelet production in culture by manipulating MKs.
Acknowledgement
We thank Ellen C O’Shaughnessy for helpful discussions. This work was supported by NIH grants R01 HL133668 and R35 HL144976 (to W.B.).
Footnotes
Conflicts of interest
The authors have no conflicts of interest to declare.
References-
- 1.Tomaiuolo M, Brass LF, Stalker TJ. Regulation of Platelet Activation and Coagulation and Its Role in Vascular Injury and Arterial Thrombosis. Interventional Cardiology Clinics 2017. [DOI] [PMC free article] [PubMed]
- 2.Boulaftali Y, Hess PR, Getz TM, Cholka A, Stolla M, Mackman N, Owens AP, Ware J, Kahn ML, Bergmeier W. Platelet ITAM signaling is critical for vascular integrity in infammation. J Clin Invest 2013; 123: 908–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grozovsky R, Giannini S, Falet H, Hoffmeister KM. Regulating billions of blood platelets: Glycans and beyond. Blood 2015. [DOI] [PMC free article] [PubMed]
- 4.Kaufman RM, Airo R, Pollack S, Crosby WH. Circulating megakaryocytes and platelet release in the lung. Blood 1965; 26(6): 720–31. [PubMed] [Google Scholar]
- 5.Lefrançais E, Ortiz-Muñoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, Thornton EE, Headley MB, David T, Coughlin SR, Krummel MF, Leavitt AD, Passegué E, Looney MR. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017; 544(7648): 105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slayton WB, Georgelas A, Pierce LJ, Elenitoba-Johnson KS, Perry SS, Marx M, Spangrude GJ. The spleen is a major site of megakaryopoiesis following transplantation of murine hematopoietic stem cells. Blood 2002; 100(12): 3975–82. [DOI] [PubMed] [Google Scholar]
- 7.Woolthuis CM, Park CY. Hematopoietic stem/progenitor cell commitment to the megakaryocyte lineage. Blood 2016. [DOI] [PMC free article] [PubMed]
- 8.Miyawaki K, Iwasaki H, Jiromaru T, Kusumoto H, Yurino A, Sugio T, Uehara Y, Odawara J, Daitoku S, Kunisaki Y, Mori Y, Arinobu Y, Tsuzuki H, Kikushige Y, Iino T, Kato K, Takenaka K, Miyamoto T, Maeda T, Akashi K. Identification of unipotent megakaryocyte progenitors in human hematopoiesis. Blood 2017; 129(25): 3332–43. [DOI] [PubMed] [Google Scholar]
- 9.Wilson NK, Kent DG, Buettner F, Shehata M, Macaulay IC, Calero-Nieto FJ, Sánchez Castillo M, Oedekoven CA, Diamanti E, Schulte R, Ponting CP, Voet T, Caldas C, Stingl J, Green AR, Theis FJ, Göttgens B. Combined Single-Cell Functional and Gene Expression Analysis Resolves Heterogeneity within Stem Cell Populations. Cell Stem Cell 2015; 16(6): 712–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazzi S, Lordier L, Debili N, Raslova H, Vainchenker W. Megakaryocyte and polyploidization. Experimental Hematology 2018. [DOI] [PubMed]
- 11.Antkowiak A, Viaud J, Severin S, Zanoun M, Ceccato L, Chicanne G, Strassel C, Eckly A, Leon C, Gachet C, Payrastre B, Gaits-Iacovoni F. Cdc42-dependent F-actin dynamics drive structuration of the demarcation membrane system in megakaryocytes. J Thromb Haemost 2016; 14(6): 1268–84. [DOI] [PubMed] [Google Scholar]
- 12.Patel SR, Richardson JL, Schulze H, Kahle E, Galjart N, Drabek K, Shivdasani R a, Hartwig JH, Italiano JE, Dc W. Differential roles of microtubule assembly and sliding in proplatelet formation by megakaryocytes Differential roles of microtubule assembly and sliding in proplatelet formation by megakaryocytes 2012; 106: 4076–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Italiano JE, Patel-Hett S, Hartwig JH, Review I Mechanics of proplatelet elaboration. J Thromb Haemost 2007; 5: 18–23. [DOI] [PubMed] [Google Scholar]
- 14.Zuidscherwoude M, Green HLH, Thomas SG. Formin proteins in megakaryocytes and platelets: regulation of actin and microtubule dynamics. Platelets 2019; 30: 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geddis AE. The regulation of proplatelet production. Haematologica 2009; 94: 756–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wojnacki J, Quassollo G, Marzolo M-P, Cáceres A. Rho GTPases at the crossroad of signaling networks in mammals: impact of Rho-GTPases on microtubule organization and dynamics. Small GTPases 2014; 5: e28430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pleines I, Cherpokova D, Bender M, Pleines I, Cherpokova D, Bender M. Rho GTPases and their downstream effectors in megakaryocyte biology Rho GTPases and their downstream effectors in megakaryocyte biology. Platelets Taylor & Francis; 2018; 00: 1–8. [Google Scholar]
- 18.Tapon N, Hall A. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr Opin Cell Biol 1997; 9: 86–92. [DOI] [PubMed] [Google Scholar]
- 19.Rottner K, Faix J, Bogdan S, Linder S, Kerkhoff E. Actin assembly mechanisms at a glance. J Cell Sci 2017; 130: 3427–35. [DOI] [PubMed] [Google Scholar]
- 20.Svitkina T The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb Perspect Biol NIH Public Access; 2018; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brouhard GJ, Rice LM. The contribution of αβ-tubulin curvature to microtubule dynamics. J Cell Biol 2014; 207: 323–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wade RH. Microtubules: an overview. Methods Mol Med 2007; 137: 1–16. [DOI] [PubMed] [Google Scholar]
- 23.Horio T, Murata T. The role of dynamic instability in microtubule organization. Front Plant Sci 2014; 5: 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akhmanova A, Steinmetz MO. Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol 2015; 16: 711–26. [DOI] [PubMed] [Google Scholar]
- 25.Hall A Rho GTPases and the control of cell behaviour. Biochem Soc Trans 2005; 33: 891–5. [DOI] [PubMed] [Google Scholar]
- 26.Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators. Nature Reviews Molecular Cell Biology 2016. [DOI] [PubMed]
- 27.Nakao K, Angrist AA. Membrane surface specialization of blood platelet and megakaryocyte. Nature 1968. [DOI] [PubMed]
- 28.Eckly A, Heijnen H, Pertuy F, Geerts W, Proamer F, Rinckel JY, Léon C, Lanza F, Gachet C. Biogenesis of the demarcation membrane system (DMS) in megakaryocytes. Blood 2014; 123(6): 921–30. [DOI] [PubMed] [Google Scholar]
- 29.Heath RJW, Insall RH. F-BAR domains: multifunctional regulators of membrane curvature. J Cell Sci 2008; 121(12): 1951–4. [DOI] [PubMed] [Google Scholar]
- 30.Frost A, Unger VM, De Camilli P. The BAR Domain Superfamily: Membrane-Molding Macromolecules. Cell 2009. [DOI] [PMC free article] [PubMed]
- 31.Chen Y, Aardema J, Corey SJ. Biochemical and functional significance of F-BAR domain proteins interaction with WASP/N-WASP. Seminars in Cell and Developmental Biology 2013. [DOI] [PubMed]
- 32.Chitu V, Stanley ER. PACSIN2: A BAR-rier forming the megakaryocyte DMS. Blood 2015; 126(1): 5–6. [DOI] [PubMed] [Google Scholar]
- 33.Chen Y, Aardema J, Kale S, Whichard ZL, Awomolo A, Blanchard E, Chang B, Myers DR, Ju L, Tran R, Reece D, Christensen H, Boukour S, Debili N, Strom TS, Rawlings D, Vazquez FX, Voth GA, Zhu C, Kahr WHA, et al. Loss of the F-BAR protein CIP4 reduces platelet production by impairing membrane-cytoskeleton remodeling. Blood 2013; 122(10): 1695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Begonja AJ, Pluthero FG, Suphamungmee W, Giannini S, Christensen H, Leung R, Lo RW, Nakamura F, Lehman W, Plomann M, Hoffmeister KM, Kahr WHA, Hartwig JH, Falet H. FlnA binding to PACSIN2 F-BAR domain regulates membrane tubulation in megakaryocytes and platelets. Blood 2015;. [DOI] [PMC free article] [PubMed]
- 35.Begonja AJ, Hoffmeister KM, Hartwig JH, Falet H. FlnA-null megakaryocytes prematurely release large and fragile platelets that circulate poorly. Blood 2011; 118(8): 2285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanaji T, Ware J, Okamura T, Newman PJ. GPIbα regulates platelet size by controlling the subcellular localization of filamin. Blood 2012; 119(12): 2906–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strassel C, Eckly A, Léon C, Petitjean C, Freund M, Cazenave JP, Gachet C, Lanza F. Intrinsic impaired proplatelet formation and microtubule coil assembly of megakaryocytes in a mouse model of Bernard-Soulier syndrome. Haematologica 2009;. [DOI] [PMC free article] [PubMed]
- 38.Kosoff RE, Aslan JE, Kostyak JC, Dulaimi E, Chow HY, Prudnikova TY, Radu M, Kunapuli SP, McCarty OJT, Chernoff J. Pak2 restrains endomitosis during megakaryopoiesis and alters cytoskeleton organization. Blood 2015;. [DOI] [PMC free article] [PubMed]
- 39.Goyal P, Pandey D, Brünnert D, Hammer E, Zygmunt M, Siess W. Cofilin Oligomer Formation Occurs In Vivo and Is Regulated by Cofilin Phosphorylation. PLoS One 2013; 8(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bender M, Eckly A, Hartwig JH, Elvers M, Pleines I, Gupta S, Krohne G, Jeanclos E, Gohla A, Gurniak C, Gachet C, Witke W, Nieswandt B. ADF/n-cofilin-dependent actin turnover determines platelet formation and sizing. Blood 2010;. [DOI] [PubMed]
- 41.Kile BT, Panopoulos AD, Stirzaker RA, Hacking DF, Tahtamouni LH, Willson TA, Mielke LA, Henley KJ, Zhang JG, Wicks IP, Stevenson WS, Nurden P, Watowich SS, Justice MJ. Mutations in the cofilin partner Aip1/Wdr1 cause autoinflammatory disease and macrothrombocytopenia. Blood 2007; 110(7): 2371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sui Z, Nowak RB, Sanada C, Halene S, Krause DS, Fowler VM. Regulation of actin polymerization by tropomodulin-3 controls megakaryocyte actin organization and platelet biogenesis. Blood 2015;. [DOI] [PMC free article] [PubMed]
- 43.Pleines I, Woods J, Chappaz S, Kew V, Foad N, Ballester-Beltrán J, Aurbach K, Lincetto C, Lane RM, Schevzov G, Alexander WS, Hilton DJ, Astle WJ, Downes K, Nurden P, Westbury SK, Mumford AD, Obaji SG, Collins PW, BioResource N, et al. Mutations in tropomyosin 4 underlie a rare form of human macrothrombocytopenia. J Clin Invest 2017; 127(3): 814–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gunning PW, Hardeman EC, Lappalainen P, Mulvihill DP. Tropomyosin - master regulator of actin filament function in the cytoskeleton. J Cell Sci 2015; 128(16): 2965–74. [DOI] [PubMed] [Google Scholar]
- 45.Patel-Hett S, Wang H, Begonja AJ, Thon JN, Alden EC, Wandersee NJ, An X, Mohandas N, Hartwig JH, Italiano JE. The spectrin-based membrane skeleton stabilizes mouse megakaryocyte membrane systems and is essential for proplatelet and platelet formation. Blood 2011; 118(6): 1641–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi ES, Nichol JL, Hokom MM, Hornkohl AC, Hunt P. Platelets generated in vitro from proplatelet-displaying human megakaryocytes are functional. Blood 1995; 85: 402–13. [PubMed] [Google Scholar]
- 47.Hartwig J, Italiano J. The birth of the platelet. J Thromb Haemost 2003; 1: 1580–6. [DOI] [PubMed] [Google Scholar]
- 48.Italiano JE, Shivdasani RA. Megakaryocytes and beyond: the birth of platelets. J Thromb Haemost 2003; 1: 1174–82. [DOI] [PubMed] [Google Scholar]
- 49.Kosaki G In vivo platelet production from mature megakaryocytes: does platelet release occur via proplatelets? Int J Hematol 2005; 81: 208–19. [DOI] [PubMed] [Google Scholar]
- 50.Handagama PJ, Feldman BF, Jain NC, Farver TB, Kono CS. In vitro platelet release by rat megakaryocytes: effect of metabolic inhibitors and cytoskeletal disrupting agents. Am J Vet Res 1987; 48: 1142–6. [PubMed] [Google Scholar]
- 51.Stenberg PE, McDonald TP, Jackson CW. Disruption of microtubules in vivo by vincristine induces large membrane complexes and other cytoplasmic abnormalities in megakaryocytes and platelets of normal rats like those in human and Wistar Furth rat hereditary macrothrombocytopenias. J Cell Physiol 1995; 162: 86–102. [DOI] [PubMed] [Google Scholar]
- 52.Patel SR, Richardson JL, Schulze H, Kahle E, Galjart N, Drabek K, Shivdasani RA, Hartwig JH, Italiano JE. Differential roles of microtubule assembly and sliding in proplatelet formation by megakaryocytes. Blood 2005;. [DOI] [PMC free article] [PubMed]
- 53.Thon JN, Montalvo A, Patel-Hett S, Devine MT, Richardson JL, Ehrlicher A, Larson MK, Hoffmeister K, Hartwig JH, Italiano JE. Cytoskeletal mechanics of proplatelet maturation and platelet release. J Cell Biol 2010; 191: 861–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thon JN, MacLeod H, Begonja AJ, Zhu J, Lee KC, Mogilner A, Hartwig JH, Italiano JE. Microtubule and cortical forces determine platelet size during vascular platelet production. Nat Commun Nature Publishing Group; 2012; 3: 852–9. [DOI] [PubMed] [Google Scholar]
- 55.Italiano JE, Lecine P, Shivdasani RA, Hartwig JH. Blood platelets are assembled principally at the ends of proplatelet processes produced by differentiated megakaryocytes. J Cell Biol 1999; 147: 1299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Richardson JL, Shivdasani RA, Boers C, Hartwig JH, Italiano JE. Mechanisms of organelle transport and capture along proplatelets during platelet production. Blood 2005; 106: 4066–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karki S, Holzbaur EL. Cytoplasmic dynein and dynactin in cell division and intracellular transport. Curr Opin Cell Biol Elsevier Current Trends; 1999; 11: 45–53. [DOI] [PubMed] [Google Scholar]
- 58.Bender M, Thon JN, Ehrlicher AJ, Wu S, Mazutis L, Deschmann E, Sola-Visner M, Italiano JE, Hartwig JH. Microtubule sliding drives proplatelet elongation and is dependent on cytoplasmic dynein. Blood 2015; 125: 860–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mazharian A, Senis YA. Proplatelets slip slidin’ away. Blood 2015; 125: 747–8. [DOI] [PubMed] [Google Scholar]
- 60.Lecine P, Italiano JE, Kim SW, Villeval JL, Shivdasani RA. Hematopoietic-specific beta 1 tubulin participates in a pathway of platelet biogenesis dependent on the transcription factor NF-E2. Blood 2000; 96: 1366–73. [PubMed] [Google Scholar]
- 61.Schwer HD, Lecine P, Tiwari S, Italiano JE, Hartwig JH, Shivdasani RA. A lineage-restricted and divergent beta-tubulin isoform is essential for the biogenesis, structure and function of blood platelets. Curr Biol 2001; 11: 579–86. [DOI] [PubMed] [Google Scholar]
- 62.Kunishima S, Kobayashi R, Itoh TJ, Hamaguchi M, Saito H. Mutation of the β1-tubulin gene associated with congenital macrothrombocytopenia affecting microtubule assembly. Blood 2009;. [DOI] [PubMed]
- 63.Kunishima S, Nishimura S, Suzuki H, Imaizumi M, Saito H. TUBB1 mutation disrupting microtubule assembly impairs proplatelet formation and results in congenital macrothrombocytopenia. Eur J Haematol 2014; 92: 276–82. [DOI] [PubMed] [Google Scholar]
- 64.Italiano JE, Bergmeier W, Tiwari S, Falet H, Hartwig JH, Hoffmeister KM, André P, Wagner DD, Shivdasani RA. Mechanisms and implications of platelet discoid shape. Blood 2003; 101: 4789–96. [DOI] [PubMed] [Google Scholar]
- 65.Kunert S, Meyer I, Fleischhauer S, Wannack M, Fiedler J, Shivdasani RA, Schulze H. The microtubule modulator RanBP10 plays a critical role in regulation of platelet discoid shape and degranulation. Blood 2009; 114: 5532–40. [DOI] [PubMed] [Google Scholar]
- 66.Strassel C, Magiera MM, Dupuis A, Batzenschlager M, Hovasse A, Pleines I, Guéguen P, Eckly A, Moog S, Mallo L, Kimmerlin Q, Chappaz S, Strub J-M, Kathiresan N, de la Salle H, Van Dorsselaer A, Ferec C, Py J-Y, Gachet C, Schaeffer-Reiss C, et al. An essential role for α4A-tubulin in platelet biogenesis. Life Sci Alliance 2019;. [DOI] [PMC free article] [PubMed]
- 67.Ho-Tin-Noé B, Boulaftali Y, Camerer E. Platelets and vascular integrity: How platelets prevent bleeding in inflammation. Blood 2018. [DOI] [PubMed]
- 68.Pertuy F, Eckly A, Weber J, Proamer F, Rinckel JY, Lanza F, Gachet C, León C. Myosin IIA is critical for organelle distribution and F-actin organization in megakaryocytes and platelets. Blood 2014;. [DOI] [PubMed]
- 69.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 1996; 273: 245–8. [DOI] [PubMed] [Google Scholar]
- 70.McCarty OJT, Larson MK, Auger JM, Kalia N, Atkinson BT, Pearce AC, Ruf S, Henderson RB, Tybulewicz VLJ, Machesky LM, Watson SP. Rac1 is essential for platelet lamellipodia formation and aggregate stability under flow. J Biol Chem 2005; 280: 39474–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pleines I, Eckly A, Elvers M, Hagedorn I, Eliautou S, Bender M, Wu X, Lanza F, Gachet C, Brakebusch C, Nieswandt B. Multiple alterations of platelet functions dominated by increased secretion in mice lacking Cdc42 in platelets. Blood 2010; 115: 3364–73. [DOI] [PubMed] [Google Scholar]
- 72.Pleines I, Dütting S, Cherpokova D, Eckly A, Meyer I, Morowski M, Krohne G, Schulze H, Gachet C, Debili N, Brakebusch C, Nieswandt B. Defective tubulin organization and proplatelet formation in murine megakaryocytes lacking Rac1 and Cdc42. Blood 2013; 122: 3178–87. [DOI] [PubMed] [Google Scholar]
- 73.Le Clainche C, Schlaepfer D, Ferrari A, Klingauf M, Grohmanova K, Veligodskiy A, Didry D, Le D, Egile C, Carlier M-F, Kroschewski R. IQGAP1 Stimulates Actin Assembly through the N-Wasp-Arp2/3 Pathway. J Biol Chem 2007; 282: 426–35. [DOI] [PubMed] [Google Scholar]
- 74.Thomas SG, Poulter NS, Bem D, Finney B, Machesky LM, Watson SP. The actin binding proteins cortactin and HS1 are dispensable for platelet actin nodule and megakaryocyte podosome formation. Platelets Taylor & Francis; 2017; 28: 372–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Prislovsky A, Marathe B, Hosni A, Bolen AL, Nimmerjahn F, Jackson CW, Weiman D, Strom TS. Rapid platelet turnover in WASP(−) mice correlates with increased ex vivo phagocytosis of opsonized WASP(−) platelets. Exp Hematol 2008; 36: 609–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kahr WHA, Pluthero FG, Elkadri A, Warner N, Drobac M, Chen CH, Lo RW, Li L, Li R, Li Q, Thoeni C, Pan J, Leung G, Lara-Corrales I, Murchie R, Cutz E, Laxer RM, Upton J, Roifman CM, Yeung RSM, et al. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nat Commun 2017; 8: 14816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bender M, Stritt S, Nurden P, Van Eeuwijk JMM, Zieger B, Kentouche K, Schulze H, Morbach H, Stegner D, Heinze K, Dütting S, Gupta S, Witke W, Falet H, Fischer A, Hartwig JH, Nieswandt B. Megakaryocyte-specific Profilin1-deficiency alters microtubule stability and causes a Wiskott-Aldrich syndrome-like platelet defect. Nat Commun 2014; 5. [DOI] [PubMed] [Google Scholar]
- 78.Reddy SS, Binnal A. Wiscott Aldrich syndrome with oral involvement: A case report. J Dent Child 2011; 78: 49–52. [PubMed] [Google Scholar]
- 79.Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich syndrome: a comprehensive review. Ann N Y Acad Sci 2013; 1285: 26–43. [DOI] [PubMed] [Google Scholar]
- 80.Kamuran K, Çetin M, Geylan H, Karaman S, Demir N, Yurekturk E, Yavuz İ, Yavuz G, Tuncer O. Wiskott–Aldrich syndrome: Two case reports with a novel mutation. Pediatr Hematol Oncol 2017; 34: 286–91. [DOI] [PubMed] [Google Scholar]
- 81.Lanzi G, Moratto D, Vairo D, Masneri S, Delmonte O, Paganini T, Parolini S, Tabellini G, Mazza C, Savoldi G, Montin D, Martino S, Tovo P, Pessach IM, Massaad MJ, Ramesh N, Porta F, Plebani A, Notarangelo LD, Geha RS, et al. A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP. J Exp Med 2012; 209: 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Curcio C, Pannellini T, Lanzardo S, Forni G, Musiani P, Antón IM. WIP null mice display a progressive immunological disorder that resembles Wiskott-Aldrich syndrome. J Pathol 2007; 211: 67–75. [DOI] [PubMed] [Google Scholar]
- 83.Sabri S, Foudi A, Boukour S, Franc B, Charrier S, Jandrot-Perrus M, Farndale RW, Jalil A, Blundell MP, Cramer EM, Louache F, Debili N, Thrasher AJ, Vainchenker W. Deficiency in the Wiskott-Aldrich protein induces premature proplatelet formation and platelet production in the bone marrow compartment. Blood 2006; 108: 134–40. [DOI] [PubMed] [Google Scholar]
- 84.Kashiwagi H, Shiraga M, Kato H, Honda S, Sako M, Kurata Y, Kanakura Y, Tomiyama Y. Expression and subcellular localization of WAVE isoforms in the megakaryocyte/platelet lineage. J Thromb Haemost 2005;. [DOI] [PubMed]
- 85.Eto K, Nishikii H, Ogaeri T, Suetsugu S, Kamiya A, Kobayashi T, Yamazaki D, Oda A, Takenawa T, Nakauchi H. The WAVE2/Abi1 complex differentially regulates megakaryocyte development and spreading: implications for platelet biogenesis and spreading machinery. Blood 2007; 110: 3637–47. [DOI] [PubMed] [Google Scholar]
- 86.Calaminus SDJ, McCarty OJT, Auger JM, Pearce AC, Insall RH, Watson SP, Machesky LM. A major role for Scar/WAVE-1 downstream of GPVI in platelets. J Thromb Haemost 2007; 5: 535–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pollard TD, Beltzner CC. Structure and function of the Arp2/3 complex. Curr Opin Struct Biol 2002; 12: 768–74. [DOI] [PubMed] [Google Scholar]
- 88.Paul DS, Casari C, Wu C, Piatt R, Pasala S, Campbell RA, Poe KO, Ghalloussi D, Lee RH, Rotty JD, Cooley BC, Machlus KR, Italiano JE, Weyrich AS, Bear JE, Bergmeier W. Deletion of the Arp2/3 complex in megakaryocytes leads to microthrombocytopenia in mice. Blood Adv 2017; 1: 1398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li Z, Kim ES, Bearer EL. Arp2/3 complex is required for actin polymerization during platelet shape change. Blood 2002; 99: 4466–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Poulter NS, Pollitt AY, Davies A, Malinova D, Nash GB, Hannon MJ, Pikramenou Z, Rappoport JZ, Hartwig JH, Owen DM, Thrasher AJ, Watson SP, Thomas SG. Platelet actin nodules are podosome-like structures dependent on Wiskott-Aldrich syndrome protein and ARP2/3 complex. Nat Commun 2015; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Havelková L, Nanda G, Martinek J, Bellinvia E, Sikorová L, Šlajcherová K, Seifertová D, Fischer L, Fišerová J, Petrášek J, Schwarzerová K. Arp2/3 complex subunit ARPC2 binds to microtubules. Plant Sci 2015; 241: 96–108. [DOI] [PubMed] [Google Scholar]
- 92.Shi P, Wang Y, Huang Y, Zhang C, Li Y, Liu Y, Li T, Wang W, Liang X, Wu C. Arp2/3-branched actin regulates microtubule acetylation level and affects mitochondrial distribution. J Cell Sci 2019;. [DOI] [PubMed]
- 93.Messaoudi K, Ali A, Ishaq R, Palazzo A, Sliwa D, Bluteau O, Souquère S, Muller D, Diop KM, Rameau P, Lapierre V, Marolleau JP, Matthias P, Godin I, Pierron G, Thomas SG, Watson SP, Droin N, Vainchenker W, Plo I, et al. Critical role of the HDAC6-cortactin axis in human megakaryocyte maturation leading to a proplatelet-formation defect. Nat Commun 2017;. [DOI] [PMC free article] [PubMed]
- 94.Nejedla M, Sadi S, Sulimenko V, de Almeida FN, Blom H, Draber P, Aspenström P, Karlsson R. Profilin connects actin assembly with microtubule dynamics. Blanchoin L, editor. Mol Biol Cell 2016; 27: 2381–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rotty JD, Wu C, Haynes EM, Suarez C, Winkelman JD, Johnson HE, Haugh JM, Kovar DR, Bear JE. Profilin-1 Serves as a Gatekeeper for Actin Assembly by Arp2/3-Dependent and -Independent Pathways. Dev Cell 2015; 32: 54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kubota H, Miyazaki M, Ogawa T, Shimozawa T, Kinosita K, Ishiwata S. Biphasic Effect of Profilin Impacts the Formin mDia1 Force-Sensing Mechanism in Actin Polymerization. Biophys J 2017; 113: 461–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Levin C, Koren A, Pretorius E, Rosenberg N, Shenkman B, Hauschner H, Zalman L, Khayat M, Salama I, Elpeleg O, Shalev S. Deleterious mutation in the FYB gene is associated with congenital autosomal recessive small-platelet thrombocytopenia. J Thromb Haemost 2015; 13: 1285–92. [DOI] [PubMed] [Google Scholar]
- 98.Spindler M, van Eeuwijk JMM, Schurr Y, Nurden P, Nieswandt B, Stegner D, Reinhold A, Bender M. ADAP deficiency impairs megakaryocyte polarization with ectopic proplatelet release and causes microthrombocytopenia. Blood 2018; : blood-2018–01-829259. [DOI] [PubMed]
- 99.Machlus KR, Thon JN, Italiano JE. Interpreting the developmental dance of the megakaryocyte: a review of the cellular and molecular processes mediating platelet formation. Br J Haematol 2014; 165: 227–36. [DOI] [PubMed] [Google Scholar]
- 100.Rojnuckarin P, Kaushansky K, Dc W. Actin reorganization and proplatelet formation in murine megakaryocytes : the role of protein kinase C α Actin reorganization and proplatelet formation in murine megakaryocytes : the role of protein kinase C ␣ 2013; 97: 154–61. [DOI] [PubMed] [Google Scholar]
- 101.Pecci A, Malara A, Badalucco S, Bozzi V, Torti M, Balduini CL, Balduini A. Megakaryocytes of patients with MYH9-related thrombocytopenia present an altered proplatelet formation. Thromb Haemost 2009; 102: 90–6. [DOI] [PubMed] [Google Scholar]
- 102.Eckly A, Strassel C, Freund M, Cazenave JP, Lanza F, Gachet C, Léon C. Abnormal megakaryocyte morphology and proplatelet formation in mice with megakaryocyte-restricted MYH9 inactivation. Blood 2009; 113: 3182–9. [DOI] [PubMed] [Google Scholar]
- 103.Chen Y, Boukour S, Milloud R, Favier R, Saposnik B, Schlegel N, Nurden A, Raslova H, Vainchenker W, Balland M, Nurden P, Debili N. The abnormal proplatelet formation in MYH9-related macrothrombocytopenia results from an increased actomyosin contractility and is rescued by myosin IIA inhibition. J Thromb Haemost 2013; 11: 2163–75. [DOI] [PubMed] [Google Scholar]
- 104.Chen Z, Shivdasani RA. Regulation of platelet biogenesis: insights from the May-Hegglin anomaly and other MYH9-related disorders. J Thromb Haemost 2009; 7 Suppl 1: 272–6. [DOI] [PubMed] [Google Scholar]
- 105.Chang Y, Aurade F, Larbret F, Zhang Y, Le Couedic JP, Momeux L, Larghero J, Bertoglio J, Louache F, Cramer E, Vainchenker W, Debili N. Proplatelet formation is regulated by the Rho / ROCK pathway. Blood 2007; 109: 4229–36. [DOI] [PubMed] [Google Scholar]
- 106.Suzuki A, Shin J-W, Wang Y, Min SH, Poncz M, Choi JK, Discher DE, Carpenter CL, Lian L, Zhao L, Wang Y, Abrams CS. RhoA is essential for maintaining normal megakaryocyte ploidy and platelet generation. Freson K, editor. PLoS One 2013; 8: e69315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chesarone MA, DuPage AG, Goode BL. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol 2010; 11: 62–74. [DOI] [PubMed] [Google Scholar]
- 108.Zuidscherwoude M, Green HLH, Thomas SG, Zuidscherwoude M, Green HLH, Thomas SG, Zuidscherwoude M, Thomas SG. Formin proteins in megakaryocytes and platelets : regulation of actin and microtubule dynamics Formin proteins in megakaryocytes and platelets : regulation of actin and microtubule dynamics. Platelets Taylor & Francis; 2018; 00: 1–8. [Google Scholar]
- 109.Pan J, Lordier L, Meyran D, Rameau P, Lecluse Y, Kitchen-Goosen S, Badirou I, Mokrani H, Narumiya S, Alberts AS, Vainchenker W, Chang Y. The formin DIAPH1 (mDia1) regulates megakaryocyte proplatelet formation by remodeling the actin and microtubule cytoskeletons. Blood 2014;. [DOI] [PubMed]
- 110.Stritt S, Nurden P, Turro E, Greene D, Jansen SB, Westbury SK, Petersen R, Astle WJ, Marlin S, Bariana TK, Kostadima M, Lentaigne C, Maiwald S, Papadia S, Kelly AM, Stephens JC, Penkett CJ, Ashford S, Tuna S, Austin S, et al. A gain-of-function variant in DIAPH1 causes dominant macrothrombocytopenia and hearing loss. Blood 2016; 127: 2903–14. [DOI] [PubMed] [Google Scholar]