

Abstract
Despite advances in chemotherapies that improve cancer survival, most patients who relapse succumb to the disease due to the presence of cancer stem cells (CSCs), which are highly chemoresistant. The pluripotency factor PRDM14 plays a key role in initiating many types of cancer. Normally, PRDM14 uses epigenetic mechanisms to establish and maintain the pluripotency of embryonic cells, and its role in cancer is similar. This important link between cancer and induced pluripotency is a key revelation for how CSCs may form: pluripotency genes like Prdm14 can expand stem-like cells as they promote ongoing DNA damage. PRDM14 and its protein-binding partners, the ETO/CBFA2T family, are ideal candidates for eliminating CSCs from relevant cancers, preventing relapse and improving long-term survival.
Keywords: Pluripotency, Cancer Stem Cell, Epigenetics, DNA damage, PRDM14, ETO/CBFA2
Cancer heterogeneity
The genetic and epigenetic heterogeneity of cancer challenges treatment efforts. Chemotherapy that eliminates the rapidly growing cells that make up the bulk of the tumour often results in recurrence due to the presence of a heterogeneous population of “cancer stem cells” (CSCs) (Glossary Box). CSCs are strictly defined as those cells that can repopulate the original tumour in serial transplantation assays[1]. Later, the term tumour initiating cells (TICs) came into use because many of the cells that could reconstitute the tumour in xenograft models were not strictly tested for all stem cell properties. True CSCs share many features with normal tissue-resident stem cells, including self-renewal and asymmetric cell divisions, which give rise to the more differentiated cells that compose the bulk of the tumour[2]. Stem cells often occupy a “niche” or an environment where they remain undifferentiated or quiescent until they are activated. As a cell becomes a CSC, it may undergo a mesenchymal to epithelial morphological transition, consistent with its change of state[3]. The resistance of CSCs to traditional chemotherapies is associated with this dormant state of residence in a niche. Normal stem cells seed differentiated tissue-specific derivatives when in their resident environment by receiving signals at the appropriate times and places; however, CSCs or TICs will give rise to diseased tissue that is not under appropriate regulatory control.
Glossary Box:
Bilateria: A major group of animals that have two symmetric left and right sides, and are derived from three germ layers, the endoderm, mesoderm and ectoderm.
Cancer stem cell: A tumour-repopulating cell that has stem cell characteristics, strictly defined as those cells that can repopulate the original tumour in serial transplantation assays.
Epiblast: The pluripotent layer of the embryo that gives rise to all cells of the embryo proper, and contributes to some extraembryonic tissues.
Homologous recombination: A type of recombination that uses a template to incorporate precise nucleotides by exchange into DNA after double strand breaks occur during replication. HR may use a sister chromatid as a template to ensure precise incorporation of nucleotides, so it is considered to be the most error-free of DNA repair mechanisms.
Immunophenotype: The cell surface properties of blood cells that are identified through flow cytometry or mass spectrometry, which have been correlated with their functional or developmental behaviour.
Imprinting: An epigenetic phenomenon in diploid mammals that allows genes to be expressed from only one copy when inherited from either the maternal or paternal chromosome. Marks that confer such an expression pattern must be erased during potency reprogramming, and then reset, requiring that the genome “remember” its origin through multiple generations.
Mutation signature: Cancer genome sequencing has revealed patterns of mutations that can predict the cause and outcome of a given tumour; for example, mutations in DNA mismatch repair proteins can cause cancer with a signature of hypermutation, which has a very high mutation load.
Non-homologous end-joining: A type of DNA repair that occurs after the formation of a double stranded break that allows a free end of DNA to combine with other ends, which is a form of imprecise editing that often creates deletions, insertions or duplications.
Oncogene: A gene that when mis- or over-expressed can give rise to a cancer.
Pioneer transcription factors: Those factors that can open chromatin from a condensed or inaccessible state, regulating DNA methylation and chromatin marks to recruit other transcription factors. POU5f1, SOX2, PRDM14 and NANOG are pioneers.
Pluripotent: A cell that has the potential to become many differentiated derivatives, and thus, can give rise to most cells of the embryo.
Primed epigenetic state: A characteristic of promoters and enhancers that contain chromatin marks that are poised or ready for rapid transcriptional activation. Such regions contain both open and repressive histone marks together.
Subclones: Cells present in a tumour that differ in genetic or physical properties from each other. Based on the presence of mutations, some subclones may grow faster, or evade the immune system more efficiently than others.
Tetrapods: The class of vertebrate animal species that have four limbs.
Totipotent: A cell that is able to give rise to any cell. Only one cell in the two cell embryo is truly totipotent.
Tumour initiating cell: A cell that may be rare or common in a tumour and can reconstitute the tumour characteristics in xenograft models; such cells have not been strictly tested for all CSC characteristics.
Tumour suppressor gene: A gene that when eliminated can give rise to a cancer.
Xenograft: A non-species graft of human tumour cells that is placed into an immunodeficient host, allowing for propagation of the tumour.
Three models have been proposed to explain the origin of CSCs: 1) resident stem cells receive aberrant signals that allow them to lose context with their surrounding niche to result in uncontrolled growth; 2) mis-regulation of oncogenes or tumour suppressor genes may hijack a stem cell pathway in partially differentiated progenitor cells to promote a program of self-renewal and potency; 3) mis-regulation of oncogenes or tumour suppressors drives a de-differentiation program in fully differentiated cells that allow them to acquire a self-renewing capacity (Figure 1). Some studies suggest that CSCs are transient, and others that they are rare resident cells that are primed to seed recurrences or metastases at a time subsequent to the initial event[4]. A variety of combinations of all the above models may be present in different tumours.
Figure 1. The origin of CSCs: three models.
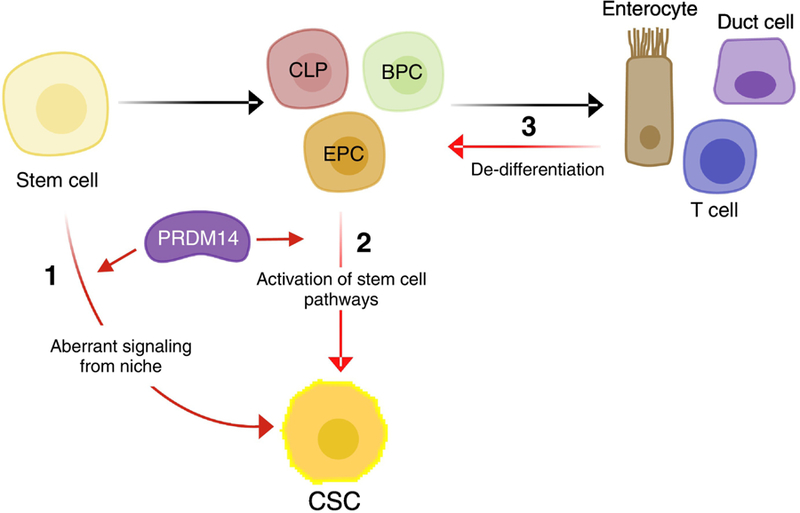
The normal developmental hierarchy (black arrows) from a stem cell to a mature cell can be corrupted by mutations that lead to the establishment of cancer stem cells (CSCs; red arrows). CSCs may arise from: 1) self-renewing tissue stem cells that improperly respond to or receive abnormal niche signals, 2) progenitor cells that receive abnormal signals to reactivate stem cell programs and self-renewal or 3) differentiated cells that de-differentiate based on abnormal signals to obtain self-renewing ability. Experimental evidence suggests that aberrant expression of PRDM14 is capable of promoting models 1 and 2, but not 3. CLP, common lymphoid progenitor; BPC, breast ductal progenitor cell; EPC, enterocyte progenitor cell.
Many of the molecular pathways that confer pluripotency to embryonic cells are present in CSCs (Box 1). Both pluripotent stem cells and CSCs express pioneer transcription factors such as Pou5f1 (Oct3/4), Sox2, and Nanog, as they have DNA and chromatin marks consistent with a primed epigenetic state[5]. Consistently, mutations that cause dysregulation of canonical stem cell pathways including NOTCH and WNT can lead to neoplastic transformation[6]. The first solid evidence of a CSC came from human Acute Myeloid Leukemias (AML) that contained a rare population of stem-like cells that could re-constitute the AML when transplanted into immuno-deficient mice[1]. Methods to study CSCs include isolating the rare cells in a population using sorting by cell surface markers. The identification and isolation of CSCs or TICs from a large variety of tumours has led to disappointing results, since TICs do not always have the cell surface properties as true stem cells for a given tissue[6]. Thus, the promise of cancer eradication based on the identification and targeting of CSCs has fallen short of early expectations[7].
Box 1: Potency and the primed epigenetic state:
One of the first decisions made in the early mammalian embryo is whether a cell will be a part of the embryo proper or contribute to extraembryonic tissues. The mouse blastocyst contains a compacted inner cell mass (ICM), which will populate the epiblast and differentiate into the embryo proper, and an outer extraembryonic layer, the trophectoderm (TE). The pluripotent ICM cells can be cultured to derive embryonic stem cells (ESCs), which can give rise to any cell in the organism. All pluripotent cells express genes that represent a signature of stem cell characteristics, and a primed epigenetic state, which is associated with global demethylation, “poised” chromatin marks, intact genomic imprints and two active X chromosomes[66]. DNA methylation is regulated by de novo DNA methyltransferases Dnmt3a and Dnmt3b and maintenance methyltransferase Dnmt1. DNA methyl groups are removed by passive depletion during replication or by an active process of base excision repair (BER). Histone tails may be ‘written’ by a number of highly regulated enzymes that confer chromatin marks that are associated with a closed, open, or poised transcriptional state. Pluripotent cells contain many bi-valent promoters, which contain the active histone H3 mark tri-methylation of lysine 4 (H3K4me3), along with the repressive mark, H3K27me3; thus, such promoters are “primed” to repress or activate gene expression[67].
ESCs cultured in leukemia inhibitory factor (LIF) are heterogeneous, with some bearing transcriptional signatures, epigenetic states, and marker expression similar to naïve ICM cells and others resembling primed post-implantation epiblast-like cells (EpiLCs)[68]. Notably, Prdm14 expression is enriched in the ICM-like fraction[69]. An ESC culture condition termed “2i” combines a MAPK/ERK inhibitor and a GSK3 inhibitor to maintain ESCs in a homogenous ICM-like “ground” state. Cells cultured in 2i with LIF have uniform expression of Prdm14[70].
Fully differentiated somatic cells can become ESC-like when exposed to four factors - Pou5f1 (Oct3/4), Sox2, c-Myc and Klf4[71]. These Yamanaka factors reprogram the pluripotency network to create induced Pluripotent Stem Cells (iPSCs), which are important in regenerative medicine because of their capacity to be engineered to replace diseased cells, while not requiring embryonic tissue. Notably, PRDM14 alone can reprogram stem cells derived from the epiblast of the mouse embryo. PRDM14’s role in demethylation and potentiation of the pluripotency program underscores its importance in stem cell reprogramming and maintenance of the naïve pluripotent state.
Advances in sequencing have uncovered a complex cancer genetic landscape that suggests that the timing and number of mutations in any cancer likely has a critical effect on disease progression. Much of our knowledge of the origins and evolution of tumours came from studies of gastrointestinal tumours, which arise in a step-wise fashion[8]. Colon epithelial cells first acquire “gatekeeper” mutations (e.g. in APC) that confer a proliferative advantage. The slow-growing adenoma then acquires secondary mutations in genes such as KRAS and, subsequently, TP53, allowing for enhanced clonal expansion and genomic instability. Subsequently, the tumour obtains a host of additional mutations that result in uncontrolled growth, evasion of the immune system, and failure of apoptosis after DNA damage, along with passenger mutations, which do not bestow a competitive advantage, but may create a mutation signature for that tumour type[9].
Proof of a multistep model has come from lineage-tracing of an early leukemia progenitor cell. To this end, deep-sequencing delineated an originating or driver mutation in AMLs with both DNMT3A and NPM1c mutations. Here, DNMT3A mutations were found in non-malignant T-cells, suggesting that a common progenitor cell harboured the pioneering DNMT3A mutation. High-resolution sorting of stem and progenitor cell populations led to the identification of an ancestral pre-leukemic CSC with hematopoietic stem cell (HSC)-like properties that lacked the NPM1c mutation, suggesting that the DNMT3A mutation was the cancer driver. DNMT3A-mutant CSCs harboured a competitive growth advantage over immunophenotype matched HSCs and persisted in remission samples, suggesting that they were resistant to chemotherapy. Importantly, these results highlight that a mutation in a progenitor ancestral pre-leukemic HSC can initiate disease[10]. Although sequencing has provided a wealth of information about the genetic origin of tumours and their evolution, a correlation of the genetic events with disease progression in real time can only be studied in model organisms.
PRDM14 is implicated in cancer initiation
PRDM14, a member of the PR domain (PRDM) superfamily (Box 2), is a candidate oncogene with functional properties that can establish CSCs. PRDM14’s normal role is to reset and maintain pluripotency in embryonic cells. Prdm14 expression has not been detected in adult tissues; however, genomic amplification, methylation and mis-expression implicate PRDM14 in an increasing number of human tumours (Table 1). This wide involvement suggests that PRDM14 may function as a tumour-initiating oncogene in many different cell types.
Box 2: PRDM proteins.
The PRDM protein family consists of epigenetic modifiers and adapters that play critical roles in cellular differentiation and disease. Each family member has an N-terminal domain first described in positive regulatory domain I-binding factor 1 (PRDI-BF1) and retinoblastoma protein-interacting zinc finger protein 1 (RIZ1), named the “PR” domain. The PRDM family consists of 17 members in humans, all with N-terminal PR domains[72]. Three to seventeen Cys2-His2 (C2H2) zinc finger repeats, which likely mediate DNA-protein, RNA-protein, or protein-protein interactions, follow the PR domain in all but one family member (PRDM11). The PR domain shares 20–30% amino acid sequence identity with the Suppressor of variegation 3–9, Enhancer of zeste, and Trithorax (SET) domain, which is the catalytic domain of histone lysine methyltransferases[73]. Notable SET-domain proteins include KMT2A/MLL, which is the target of recurrent chromosomal translocations in approximately 10% of human leukemias[74] and EZH2 or PRC2, which are also recurrent translocation partners and frequently overexpressed in leukemia[75]. Some PRDM family members have histone methyltransferase activity (e.g., PRDM2, PRDM6 PRDM8, PRDM9, and PRDM16), whereas others serve as scaffolds to recruit epigenetic modifiers to specific loci (e.g., PRDM3)[68]. PRDM14 contains six zinc finger motifs, which bind the same consensus DNA sequence in both mouse and human[23, 24], yet has no methyltransferase activity[39, 76]. Instead, PRDM14 regulates gene expression through protein interaction partners.
PRDM proteins function in cellular differentiation and are frequently deregulated in hematological malignancies and solid cancers where they function as both tumor suppressors and oncogenes. PRDM1, or B-lymphocyte-induced maturation protein 1 (BLIMP1), plays critical roles at multiple stages of hematopoiesis as a transcriptional repressor of anti-terminal differentiation genes[77]. PRDM1 is a tumour suppressor in diffuse large B cell lymphoma (DLBCL), but may also act as an oncogene in plasmacytoma and multiple myeloma[78]. PRDM2 (RIZ) has PR-containing and PR-lacking isoforms, with the former serving as a tumor suppressor in both DLBCL and chronic myeloid leukemia[79]. PRDM3, also called MDS1 and EVI1 complex locus (MECOM) may be a tumour suppressor or an oncogene in myeloid disease, depending on the presence of the PR domain[80]. PRDM3 is a regulator of LT-HSC identity, as its targeted disruption drives stem cells from quiescence into active cycling, abrogating long-term repopulation capability[81]. PRDM16 (MDS1/EVI1-like gene1 MEL1) plays roles in many stem cells[82] and is a master regulator of brown fat determination[83]. Therefore, the activity of PRDM proteins is highly context-dependent, and differs based on functional isoforms.
Table 1.
PRDM14 is implicated in many human neoplasms.
| Cancer | Implications | Reference |
|---|---|---|
| Breast | ● Intragenic methylation correlates with increased expression ● Genomic amplification correlates with increased expression, high mitotic index and high histological grade ● mRNA and protein overexpression (34–75%) ● Expression linked to resistance to chemotherapy ● Higher PRDM14 copy numbers in metastases, highest in brain metastases ● High PRDM14 expression correlates with poor prognosis |
[11, 13, 84, 85] |
| Bladder | ● Gene methylation associated with high-grade tumours ● mRNA overexpression |
[13, 86] |
| Blood | ● mRNA overexpression in high hyperdiploid precursor B-ALL (75%*) and T-ALL (58%*) paediatric cases | [17] |
| Cervical | ● Gene methylation associated with high-grade carcinomas ● Protein overexpression (18.4%*) |
[13, 87] |
| Colorectal | ● Gene methylation associated with tumours | [88] |
| Gastric | ● mRNA overexpression (42%*) | [11] |
| Germ Cell Tumour (GCT) | ● Intracranial GCT: copy number gains (50%*) correlated with overexpression ● Mixed GCTs (embryonal carcinoma): protein expression (100%*) ● Testicular GCT: associated with susceptibility ● Testicular seminoma: protein expression (100%*) |
[89–91] |
| Head & neck | ● Genomic amplification (16%*) | [92] |
| Non-small cell carcinoma (NSCLC | ● Genomic amplification (41.5%) ● Protein overexpression (25.6%) ● High expression correlates with poor disease-free and overall survival ● PRDM14 inhibition decreases metastasis ● Gene methylation associated with tumours; diagnoses early NSCLC |
[13, 93–95] |
| Ovarian | ● mRNA and protein overexpression in ovarian cancer cell lines (27%*) and primary tumours (37.3%) | [11, 13] |
| Pancreatic | ● mRNA and protein overexpression (29.3%)Protein overexpression in premalignant precursor lesions (pancreatic intraepithelial neoplasia subtype) ● Chronic pancreatitis, a risk factor for pancreatic cancer |
[13–15] |
| Prostate | ● Protein overexpression (15.4%) | [13] |
| Renal | ● mRNA and protein overexpression (38.8%) | [13] |
Percentages indicate the proportion of tumours tested that had overexpression or amplification of PRDM14
The 8q13.3 region containing PRDM14 is amplified by copy number variation (CNV) in many human breast cancers, resulting in its mis-expression at early stages that suggests a role in tumour initiation[11, 12]. Consistently, silencing PRDM14 in breast cancer cells inhibits their stem-cell-like properties, ability to cause tumours and prevents metastasis in an immuno-deficient mouse model[13]. Expression of PRDM14 is also associated with progression to pancreatic cancer[14], and its inhibition suppresses pancreatic cancer cell metastasis in an immuno-deficient mouse model[15]. Prdm14 was also identified as the driver mutation in retrovirally-induced mouse lymphoid leukemias. Its mis-expression causes aggressive lymphoblastic leukemias after expanding pre-leukemic progenitor cells in mouse bone marrow, suggesting that it is capable of tumour initiation[16]. Notably, fulfilling the definition of a CSC, PRDM14-expressing cells can reconstitute leukemias in serial transplantation assays[17].
PRDM14 reprograms cells to the pluripotent state
During embryonic development, Prdm14 is the first zygotic gene to be expressed asymmetrically in one cell of the first cellular division. The first lineage choice in the developing embryo at the 4-cell stage is also influenced by PRDM14: PRDM14-expressing cells will adopt the pluripotent embryonic inner cell mass (ICM) fate rather than the more differentiated extraembryonic trophectoderm fate. The expression of PRDM14 is lost by the 16-cell stage, yet expression recurs in the pluripotent ICM of the blastocyst (Box 1)[18]. PRDM14 maintains potency in both mouse and human ESCs through epigenetic changes. In mouse ESCs, PRDM14 directly represses the de novo DNA methyltransferases Dnmt3a and Dnmt3b to influence passive demethylation[19]. PRDM14 also promotes active demethylation at pluripotency and germline-specific genes through an active base excision repair (BER) mechanism of ten-eleven translocation (TET)-modified nucleotides[20, 21]. PRDM14 plays similar roles in human ESCs, which more closely resemble primed mouse Epiblast Stem Cells (EpiSCs) than mESCs[22]. In human cells, PRDM14 enhances the reprogramming efficiency of fibroblasts to induced Pluripotent Stem Cells (iPSCs) and can replace KLF4, but not POU5F1, SOX2, or c-MYC as reprogramming factors (Box 1)[23]. The consensus DNA-binding sequence for PRDM14 is nearly identical for mESCs and hESCs, regulating Pou5f1 through a distal enhancer in mESCs, and a proximal enhancer in hESCs[23, 24]. After activating expression of POU5F1, PRDM14 recruits the POU5F1 protein to the promoters of many genes to activate the pluripotency network through promoter demethylation and recruitment of other transcription factors[25].
Germ cells are the fundamental units of reproduction, heredity, and propagation of most multicellular species. Their earliest derivatives in mammals, the primordial germ cells (PGCs), undergo unique reprogramming steps controlled by PRDM1 and PRDM14 to regain potency during derivation from somatic-primed cells of the epiblast at embryonic day (E) 7.5 in the mouse. PRDM1 is required to repress somatic differentiation[26], while PRDM14 is required to reset the epigenetic state[27]. Mice lacking Prdm14 are sterile, because they lose PGCs by E12.5 due to a failure of germ cells to migrate and colonize the gonads. Mutant PGCs in Prdm14−/− mice do not upregulate potency genes including Sox2, and histone methyl marks do not change from a repressive to a primed transcriptional state, causing epigenetic reprogramming to fail[27]. The transcription factor Tfap2c (AP2γ) is first activated by PRDM1, then maintained by PRDM14 to play an additional role in PGC specification[28]. In vitro, PGC-like-cells, capable of completely reconstituting gametogenesis, can be derived from embryonic stem cells (ESCs) by first inducing cells to transit through an epiblast-like cell (EpiLC) state[29]. PRDM14, PRDM1, and AP2γ, and to a lesser extent PRDM14 alone, can robustly induce the PGC state in EpiLCs[30], reinforcing the importance of the transcriptional circuitry for pluripotency that is regulated by PRDM1 and PRDM14. Thus, PRDM14 initiates and then maintains a transcriptional network for the pluripotent state (Key Figure 2).
Key Figure 2. Prdm14 promotes pluripotency programs during normal development and cancer initiation.
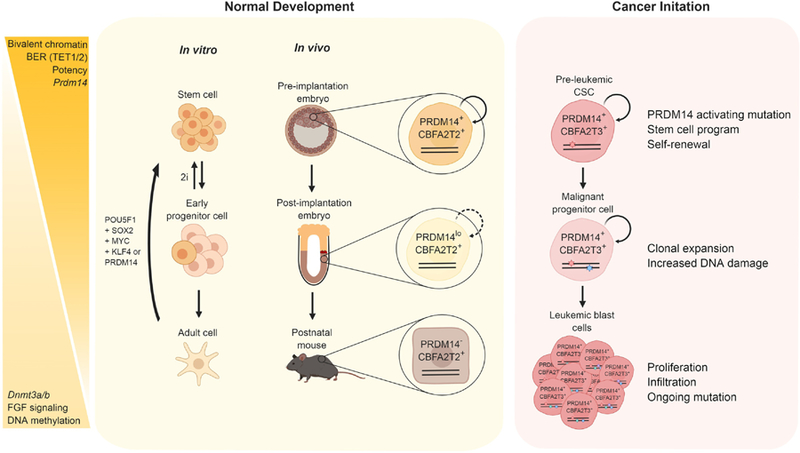
Prdm14 plays a critical role in early mammalian development (yellow box) by regulating pluripotency and establishing PGCs through epigenetic changes that include regulation of DNA demethylation, activation of potency genes., inhibition of differentiation genes (such as the Fibroblast growth factor (FGF) family) and establishing bivalent chromatin marks at promoters, Such “primed” chromatin allows for global remodeling during the establishment of pluripotency, or differentiation from a stem cell. Forced expression of Prdm14 in vitro along with three of the four Yamanaka factors (Box 1) can reprogram adult cells back into pluripotent stem cells. In vivo, PRDM14 interacts with CBFA2T2 in pluripotent cells including ESCs and PGCs to stabilize a protein complex on chromatin, recruit additional complex members and regulate target gene expression. Upon mis-expression in adult progenitor cells, (red box), Prdm14 can hijack ETO/CBFA2T family member CBFA2T3 present in hematopoietic stem cells to establish CSCs that initiate leukemia. Cancer initiation involves multiple downstream steps including continual clonal expansion and DNA damage leading to uncontrolled tumour growth. Stars represent individual mutations. +, gene and protein expression present; -, gene and protein expression absent; lo, gene and protein have low expression; BER, base excision repair; Dmnt3a/3b: DNA methyltransferases. (Made in BioRender.com)
PRDM14 exists in different protein complexes that repress or activate transcription in different cell types and stages of differentiation. For example, in ESCs, PRDM14 may interact with 1) the Polycomb Repressor Complex 2 (PRC2), which confers a repressive H3K27me3 mark[31], 2) Coactivator Associated Arginine Methyltransferase 1 (CARM1), a histone arginine methyltransferase important for cell fate specification in the early embryo[18], or 3) TET1 or 2, which oxidize 5-methylcytosine to 5-hydroymethylcytosine as a first step in BER demethylation[32]. However, in ESCs and germ cell derivatives, PRDM14’s primary interaction partner is the ETO-family member Core-binding factor, runt domain, alpha subunit 2 translocated to 2 (CBFA2T2) (Key Figure 2)[33, 34]. In this context, the CBFA2T2-PRDM14 protein interaction stabilizes the complex on chromatin and regulates gene expression through the recruitment of additional proteins. Mice carrying loss of function alleles of Cbfa2t2 lack PGCs and are sterile, similar to Prdm14 null mutants. CBFA2T2 and PRDM14 also regulate many of the same genes in a germ cell line and in ESCs, including POU5F1. In general, the PRDM14/CBFA2T2 complex represses genes involved in differentiation, as it activates potency genes. In the germ cell line, knocking down CBFA2T2 prevented PRDM14 from binding its target genes, suggesting that CBFA2T2 is an active partner in transcriptional repression and activation of target genes by PRDM14[34]. CBFA2T2 and PRDM14’s target genes overlap many of the same transcription factors, including those involved in lineage commitment (e.g. Fgfr1, Fgfr2), as well as chromatin regulators that are also bound by the potency factors POU5F1, SOX2 and NANOG. Although CBFA2T2 does not bind DNA directly, it is absolutely required for PRDM14’s function by serving as a scaffold to secure the transcription factor complex on DNA targets bound by PRDM14. The complex represses the euchromatic histone methyltransferase EHMT1, which regulates the balance between di- and tri-methylation of histone 3 lysine 9. Importantly, EHMT1 catalyzes the H3K9me2 methyl mark that is required for PGC establishment and embryonic development, and regulates the amount of H3K9me3 that is deposited by other histone methyltransferases, perhaps by blocking their binding[35]. Together, the data suggest that global chromatin changes controlled by PRDM14/CBFA2T2 regulate the balance between lineage specification and self-renewal in pluripotent cells.
PRDM14 requires protein binding partners for cancer initiation
It follows that PRDM14 mis-expression could lead to cancer development by promoting epigenetic reprogramming, self-renewal and pluripotency in somatic cells. Mouse models have been essential to understand the role of PRDM14 in cancer initiation and progression. Prdm14 was first identified as the oncogene overexpressed in mouse strains bearing retroviral insertions at ecotropic viral integration site 32 (Evi32)[16]. When Prdm14 was mis-expressed in mouse bone marrow (BM) using transduction of HSCs, the recipients succumbed to lymphoid leukemias after cells bearing a common lymphoid progenitor (CLP) immunophenotype expanded in the BM prior to leukemia onset[17]. Microarray expression analysis of the CLP-like cells identified overexpression of genes involved in pluripotency or stem cell function (Pou5f1, c-Kit, Cbfa2t3), tumor initiation (Myb, mTor, Tcf3), and genes within the pluripotency-associated imprinted Dlk1-Dio3 locus (Meg3, Dlk1)[17]. The mammalian Dlk1-Dio3 locus encodes three protein-coding genes (Dlk1, Rtl1 and Dio3) if inherited from the paternal genome, or a large cluster of imprinted microRNAs, small nucleolar RNAs, and long non-coding RNAs (Meg3, RIAN) if inherited from the maternal genome[36]. A failure of imprinting at this locus is associated with many cancers and pathological processes.
PRDM14 is an unusual oncogene in that it plays a role in tumour initiation in many different cell types. Genetic tools in the mouse allow for inducing Prdm14 expression in any cell for which there is a Cre driver (Figure 3)[37] for real time modelling of tumour initiation. The ROSA26 locus was exploited to spatially and temporally mis-express Prdm14 in HSCs and mature T-cells[38]. Mice expressing Prdm14 in HSCs succumbed to a completely penetrant T-cell acute lymphoblastic leukemia (T-ALL) with a highly infiltrative CD8+ immature single positive T-cell immunophenotype. Subsequent work showed that activating mutations in Notch1 occurred in all T-ALLs[39]. Strikingly, the T-ALLs developed very rapidly, within 42 – 64 days after Prdm14’s expression, faster than any other NOTCH-driven model[39]. NOTCH1 is involved in normal stem cell self-renewal and causes neoplastic proliferation when dysregulated; it is implicated in over 50% of all human T-ALL cases. In contrast, when Prdm14 was expressed in mature T-cells, mice remained healthy without any signs of leukemia[38], suggesting that Prdm14 requires an additional factor present in progenitor cells to act as an oncogene.
Figure 3. Mouse models for Prdm14-driven cancer initiation.
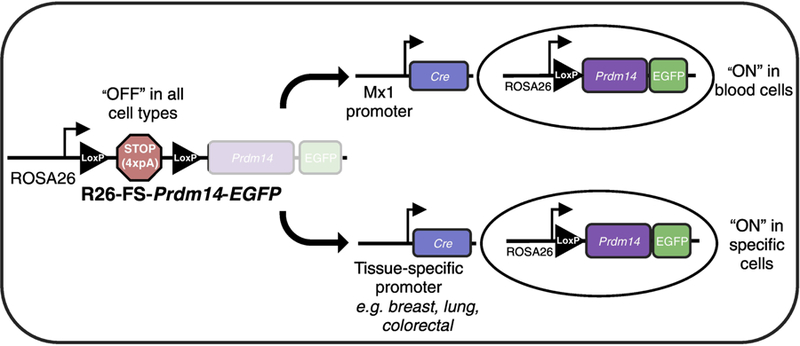
To allow for spatiotemporal control of Prdm14 expression, a mouse line was engineered to carry a transgene inserted downstream of the ubiquitously and constitutively active ROSA26 (R26) promoter. A loxP-STOP-loxP “floxed-STOP” (FS) cassette consisting of a 4X-repeated polyA sequence (pA) lies upstream of the mouse Prdm14 coding sequence, an internal ribosomal entry site and enhanced green fluorescent protein (EGFP). In this configuration, transcription in R26-FS-Prdm14-EGFP mice does not proceed past the STOP cassette and Prdm14 is not expressed. To model an initiating event where Prdm14 becomes aberrantly expressed, R26-FS-Prdm14-EGFP mice are crossed with mice engineered to express the Cre recombinase enzyme under the control of tissue specific promoters. Within a specific cell type, Cre catalyzes site-specific recombination between the loxP sites to excise the STOP cassette and allow for transcription of Prdm14 and EGFP. To study Prdm14-induced leukemogenesis, transgenic mice carrying the Mx1-Cre transgene were mated to mice carrying R26-FS-Prdm14-EGFP. The Mx1 promoter is activated using polyinosinic-polycytidylic acid, leading to the expression of Cre, which deletes the FS cassette, and allows for Prdm14 expression in HSCs. This system can be expanded into other cell types to model many different Prdm14-initiated cancers.
Despite evidence that protein-binding partners are critical for PRDM14’s function, PRDM14’s functional partners in a cancer model have only recently been described [40]. Interestingly, PRDM14’s primary interacting partner in the T-ALL mouse model is CBFA2T3, a related family member to PRDM14’s partner in PGCs, CBFA2T2. CBFA2T3 is a master hematopoietic regulator that is crucial for HSC quiescence and hematopoietic lineage decisions. Homozygous Cbfa2t3 null mice are viable and fertile, developing only a mild anemia due to myeloid lineage perturbations[41]. Homozygous Cbfa2t3 null mice also have defects in stem cell self-renewal with fewer quiescent HSCs[42, 43], and their BM fails to repopulate the T-lineage after transplantation, because they have a reduced number of lymphoid progenitors[44]. CBFA2T3 controls the expression of a hematopoietic transcription factor complex that regulates long term (LT)-HSC quiescence, which includes the leukemia-initiating transcription factors T-cell acute lymphocytic leukemia 1 (TAL1) and LIM domain only 2 (LMO2)[45, 46]. Progenitor cells that resemble LT-HSCs and CLPs expand in the BM after expression of Prdm14, which is consistent with overexpression, rather than deletion, of Cbfa2t3. Consistently, Prdm14-expressing pre-leukemia BM cells overexpress Cbfa2t3 compared to immunophenotype-matched cells[17].
The eight-twenty-one (ETO) family of chromatin-associated proteins includes CBFA2T3 (MTG16, ETO-2), CBFA2T2 (MTGR1), and myeloid translocation gene 8 (MTG8, ETO). Each of the three ETO/CBFA2T family members participates in oncogenic translocations with RUNX1 in AML[47, 48]. ETO/CBFA2T proteins contain four highly conserved Nervy Homology Region (NHR) domains, whose name is derived from the Drosophila orthologue nervy, a gene involved in axonal guidance[49]. In mammals, the different family members can form hetero-dimeric and tetrameric complexes with each other through the NHR2 domains. These large complexes serve as a bridge to chromatin by recruiting DNA-binding E-proteins through the NHR1 domain and chromatin modifiers such as NCOR1/2 and HDACs through the NHR3 and 4 domains[46]. The interaction between PRDM14 and ETO/CBFA2T is conserved throughout vertebrate evolution, but was adapted from motor neurons in a common ancestor of the bilateria group of animals into the pluripotent cells of tetrapods[50]. This degree of evolutionary conservation suggests that the different family members may interact with a common set of transcription factors and co-repressors through common protein domains. In mice and humans, it is likely that specificity arises because each family member is expressed in different cell types[51]: CBFA2T2 is expressed at higher levels in stem and germ cells, whereas CBFA2T3 is expressed in at high levels in hematopoietic stem and progenitor cells. In mouse HSCs, Cbfa2t3 is expressed at twice the level of Cbfa2t2[52, 53]. Thus, when PRDM14 is mis-expressed in HSCs, it preferentially associates with the predominant ETO/CBFA2T family member, hijacking its normal functions to start a program of self-renewal, and skewing lineage decisions based on its expression in the wrong cellular context (Key Figure 2).
PRDM14 thus provides a model for the origin and behaviour of CSCs. When a pluripotency factor such as PRDM14 is mis-expressed outside its normal environment in the embryo, a related family binding partner present in a progenitor cell can functionally substitute to activate the stem cell program, reprogramming the cell to a state of self-renewal without quiescence, and expanding the number of progenitor cells that are poised to initiate cancer. Interestingly, CBFA2T3 is expressed in breast ductal epithelial cells[54], while CBFA2T2 is expressed in gastrointestinal stem and progenitor cells[55]. Thus, the presence of the ETO proteins in other stem and progenitor cell types suggests that they could act as PRDM14’s partner in the initiation of other cancers.
Model of stem cell expansion and increased DNA damage
Cancer does not develop solely due to the reactivation of pluripotency in adult cells. Mutations that promote genomic instability often occur early in the multi-step process of tumorigenesis. Although expression of Prdm14 to expand progenitor cells that have tumour-reconstituting potential is an initiating event, it is likely that additional mutations lead to malignant disease (Key Figure 2). Stem cells have several unique qualities that ensure genomic integrity, because they must give rise to every differentiated cell, while avoiding devastating consequences such as cancer[56]. First, they have a very efficient DNA damage response (DDR) system that becomes less efficient upon cell differentiation. For example, stem cells repair double stranded breaks (DSBs) in DNA through homologous recombination, a relatively error-proof method of ensuring DNA integrity during replication, whereas differentiated cells are more likely to employ other methods that are more error-prone, including non-homologous end joining. Second, stem cells employ anaerobic metabolism, consistent with their presence in a niche, but which also makes them less susceptible to oxidative stress that can induce DSBs. Not surprisingly then, activation of pluripotency in somatic cells can lead to aneuploidy and copy number alterations during iPS cell generation[57]. Moreover, ESCs tend to become aneuploid in culture.
Notably, PRDM14-expressing tumours have hallmarks of genomic instability, namely a high degree of aneuploidy along with one of the highest copy number variation (CNV) profiles recorded in mouse tumours, with recurring deletions and duplications consistent with a failure of DSB repair that recapitulate those found in human T-ALLs[58]. Consistently, PRDM14-induced pre-leukemic cells show decreased expression of genes involved in chromosomal stability and DNA repair[17]. Such a profile of increased DNA damage with decreased DDR factors is also found in RUNX1-ETO driven leukemias[59]. Moreover, RUNX1-ETO driven tumours have altered DNA methylation profiles, presumably in cooperation with an altered TET2. Changes in methylation and bi-valent promoter occupation were also associated with PRDM14 expression in breast cancer cells[13]. It is possible that the ETO fusion proteins elevate expression levels of a pluripotency factor such as PRDM14, triggering genome instability; alternatively, it is possible that PRDM14-driven tumours have genome instability due to mis-appropriation of the ETO/CBFA2T family members. It is difficult to separate the CBFA2T3 and PRDM14 functions in mouse leukemias, since mice lacking CBFA2T3 do not have expanded hematopoietic progenitor cells and do not develop leukemia when PRDM14 is mis-expressed[40]. Certainly, the association of CSCs with genetic events that take place in progenitor cells rather than more differentiated cells may be explained by the hijacking of resident proteins. Therefore these findings are consistent with the establishment of CSCs by the previously proposed models 1 or 2, but not by full de-differentiation as proposed in model 3 (Figure 1).
In either case, outside the normal environment of a stem cell that must maintain genomic integrity, the expression of a potency factor such as PRDM14 likely leads to accelerated genetic damage without repair. As PRDM14 uses CBFA2T3 to expand progenitor cells in the pre-leukemia stage, expression of PRDM14 in the absence of factors needed for regulation of damage and apoptosis could enhance cancer progression. Ongoing genomic instability in these CSCs or TICs could subsequently allow for outgrowth of unique subclones, which acquire additional driver mutations that endow them with a selective growth advantage and subsequently give rise to heterogeneous tumour cell populations. The cause of the damage is not clear at this time; it is possible that unchecked BER within progenitor cells leads to double stranded breaks (DSBs) that are not repaired. Alternatively, Prdm14 or ETO/CBFA2T family member overexpression may lead to mis-appropriation of DNA methylation or chromatin marks, which could enhance inter- and intra-chromosomal recombination in somatic cells, resulting in unchecked recombination and perturbation of cell cycle checkpoints. Another PRDM family member, PRDM9, confers H3K4me3 marks to recruit the recombination protein Spo11 at synaptic junctions during meiosis[60]; therefore, PRDM14 may also hijack such a function through association with different binding partners when mis-expressed in cancer. Alternatively, its overexpression may lead to inappropriate H3K4me3 marks at PRDM14 target genes due to direct down regulation of EHMT1. Notably, Prdm14-driven pre-leukemia cells also show increased expression of Spo11, whose mis-expression could lead to mitotic recombination events that may result in changes in copy number, loss of heterozygosity, and chromosome mis-segregation[17]. In lymphoid cells, the presence of the recombination activating genes 1 or 2 (RAG1/2), which are required for antibody receptor rearrangements, also creates an environment of genomic instability[61]. The RAG1/2 endonuclease complex is essential for the development of PRDM14-induced T-ALLs by mediating recombination between cryptic recombination signal sequences (cRSSs) at NOTCH1 to drive tumour growth. The cRSSs show elevated levels of H3K4me3 in PRDM14-expressing pre-leukemia cells, a mark that is critical for recruiting RAG enzymes to target regions[39]. In the future, it will be interesting to determine if other tumour types caused by PRDM14 mis-expression have a similar degree of DNA damage.
Concluding remarks and Future Perspectives
Together, the data support the idea that Prdm14-expression promotes CSC self-renewal, expanding a progenitor cell population that is susceptible to genomic rearrangements that “enable” cancer development, a process that can occur in many different cell types. This hypothesis would explain cancer heterogeneity and evolution. For example, in lymphoid disease, a subclone from expanded progenitors may outcompete the original CSCs to carry different translocations and genetic alterations from the original tumor after cancer relapse post chemotherapy[62–64]. Similarly, in breast or colorectal cancer, relapsed and/or metastatic cancers may carry distinct genetic signatures from the primary tumour at diagnosis. Altogether, PRDM14’s mis-expression is consistent with the behaviour of a CSC: a rare population of cells may self-renew as they also accumulate genetic damage, leading to tumour heterogeneity and resistance to chemotherapy.
The resistance of CSCs to conventional cancer treatments is responsible for indolent and relapsed disease. Thus, identifying factors that drive CSC growth and evolution, as well as developing therapeutics for the targeted eradication of CSCs is critical for ensuring a sustained decline in cancer mortality. Eliminating PRDM14 in tumours may be a strategy to eradicate CSCs[65]. In mice, eliminating CBFA2T3 prevented the expansion of CSCs and the development of T-ALL when PRDM14 was mis-expressed. Notably, CBFA2T3 is also a therapeutic target in cancers, and may serve as an alternate for CSC therapy. The PRDM14-CBFA2T3 model, however, may not be specific to these two proteins. Other proteins that confer stem-cell like properties may also find a partner in progenitor cells that can lead to the establishment of CSCs. The understanding of precise genetic events prior to the development of an extant cancer may lead to logical therapies to completely eradicate CSCs and impede cancer recurrence and metastasis.
Outstanding questions:
How can PRDM14’s involvement in human cancer initiation be fully assessed? PRDM14’s role in tumour initiation, metastasis and relapse has largely been ignored based on gene expression and exon sequencing. PRDM14’s mis-expression and mutation status is often associated with genomic amplification or epigenetic changes that can be detected using alternative sequencing strategies.
Are PRDM14-expressing CSCs transient or are they resident in the tumour? PRDM14 is not normally expressed in adult cells, so a small amount of expression in a rare progenitor would make detection difficult in the tumour bulk. The tumour may become independent of PRDM14 due to clonal evolution.
Is PRDM14’s role in initiating many cancer subtypes dependent upon the expression of ETO family members alone, or are other binding partners present in different cell types? PRDM14’s ability to initiate tumours in progenitors rather than fully differentiated cells implies that partners present only in progenitor cells are required. These interactions may be reasonable entry points for therapy.
How and why does DNA damage occur in ETO- and PRDM14-expressing tumours? It is likely that a DNA repair component is missing or mis-appropriated in adult cells, allowing PRDM14 to catalyze genomic rearrangements that enable cancer development. The identification of missing factors could prevent a large proportion of PRDM14-induced cancers.
Are pluripotency oncogenes such as PRDM14 responsible for genetic changes that occur during tumour evolution, metastasis and relapse? A PRDM14-expressing CSC that is resident in a tumour could produce subclones, which may out-compete the original CSCs to carry different translocations and genetic alterations from the original tumour post chemotherapy, which could initiate cancer relapse. A CSC that that sustains self-renewal while promoting genomic instability is deadly.
Trends Box.
The epigenetic regulator PRDM14, which establishes and maintains pluripotency in embryonic cells, can mimic this function in adult progenitor cells, reprogramming them to a pluripotent stem cell-like state to establish cancer stem cells.
The involvement of PRDM14 in initiating human cancers may be underestimated because its expression is difficult to detect and it is altered primarily by copy number variation and epigenetic changes, rather than intragenic mutations.
Mis-expression of PRDM14 expands progenitor cells that have genomic instability, allowing for rapid growth of cancer subclones that lead to tumour heterogeneity and uncontrolled growth.
PRDM14 requires a protein partner in progenitor cells to initiate cancer, providing avenues to eliminate cancer stem cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Lab website: http://lab.research.sickkids.ca/justice/
REFERENCES
- 1.Lapidot T et al. (1994) A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367 (6464), 645–8. [DOI] [PubMed] [Google Scholar]
- 2.Bakhshinyan D et al. (2018) Introduction to Cancer Stem Cells: Past, Present, and Future. Methods Mol Biol 1692, 1–16. [DOI] [PubMed] [Google Scholar]
- 3.Wang H and Unternaehrer JJ (2019) Epithelial-mesenchymal Transition and Cancer Stem Cells: At the Crossroads of Differentiation and Dedifferentiation. Dev Dyn 248 (1), 10–20. [DOI] [PubMed] [Google Scholar]
- 4.Prager BC et al. (2019) Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 24 (1), 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reya T et al. (2001) Stem cells, cancer, and cancer stem cells. Nature 414 (6859), 105–11. [DOI] [PubMed] [Google Scholar]
- 6.Kreso A and Dick JE (2014) Evolution of the cancer stem cell model. Cell Stem Cell 14 (3), 275–91. [DOI] [PubMed] [Google Scholar]
- 7.Batlle E and Clevers H (2017) Cancer stem cells revisited. Nat Med 23 (10), 1124–1134. [DOI] [PubMed] [Google Scholar]
- 8.Vogelstein B et al. (2013) Cancer genome landscapes. Science 339 (6127), 1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garraway LA and Lander ES (2013) Lessons from the cancer genome. Cell 153 (1), 17–37. [DOI] [PubMed] [Google Scholar]
- 10.Shlush LI et al. (2014) Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506 (7488), 328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishikawa N et al. (2007) Gene amplification and overexpression of PRDM14 in breast cancers. Cancer Res 67 (20), 9649–57. [DOI] [PubMed] [Google Scholar]
- 12.Moelans CB et al. (2010) Molecular profiling of invasive breast cancer by multiplex ligation-dependent probe amplification-based copy number analysis of tumor suppressor and oncogenes. Mod Pathol [DOI] [PubMed]
- 13.Taniguchi H et al. (2017) Silencing PRDM14 expression by an innovative RNAi therapy inhibits stemness, tumorigenicity, and metastasis of breast cancer. Oncotarget 8 (29), 46856–46874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moriya C et al. (2018) PRDM14 is overexpressed in chronic pancreatitis prior to pancreatic cancer. FEBS Open Bio 8 (10), 1733–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moriya C et al. (2017) Inhibition of PRDM14 expression in pancreatic cancer suppresses cancer stem-like properties and liver metastasis in mice. Carcinogenesis 38 (6), 638–648. [DOI] [PubMed] [Google Scholar]
- 16.Dettman EJ and Justice MJ (2008) The Zinc Finger SET Domain Gene Prdm14 is Overexpressed in Lymphoblastic Lymphomas with Retroviral Insertions at Evi32. PLoS ONE 3 (11), e3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dettman EJ et al. (2011) Prdm14 initiates lymphoblastic leukemia after expanding a population of cells resembling common lymphoid progenitors. Oncogene 30 (25), 2859–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burton A et al. (2013) Single-cell profiling of epigenetic modifiers identifies PRDM14 as an inducer of cell fate in the mammalian embryo. Cell Rep 5 (3), 687–701. [DOI] [PubMed] [Google Scholar]
- 19.Grabole N et al. (2013) Prdm14 promotes germline fate and naive pluripotency by repressing FGF signalling and DNA methylation. EMBO Rep 14 (7), 629–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okashita N et al. (2014) PRDM14 promotes active DNA demethylation through the ten-eleven translocation (TET)-mediated base excision repair pathway in embryonic stem cells. Development 141 (2), 269–80. [DOI] [PubMed] [Google Scholar]
- 21.Hackett JA et al. (2013) Synergistic mechanisms of DNA demethylation during transition to ground-state pluripotency. Stem Cell Reports 1 (6), 518–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tesar PJ et al. (2007) New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 448 (7150), 196–9. [DOI] [PubMed] [Google Scholar]
- 23.Chia NY et al. (2010) A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature 468 (7321), 316–20. [DOI] [PubMed] [Google Scholar]
- 24.Ma Z et al. (2011) Sequence-specific regulator Prdm14 safeguards mouse ESCs from entering extraembryonic endoderm fates. Nat Struct Mol Biol 18 (2), 120–7. [DOI] [PubMed] [Google Scholar]
- 25.Okashita N et al. (2016) PRDM14 Drives OCT3/4 Recruitment via Active Demethylation in the Transition from Primed to Naive Pluripotency. Stem Cell Reports 7 (6), 1072–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohinata Y et al. (2005) Blimp1 is a critical determinant of the germ cell lineage in mice. Nature 436 (7048), 207–13. [DOI] [PubMed] [Google Scholar]
- 27.Yamaji M et al. (2008) Critical function of Prdm14 for the establishment of the germ cell lineage in mice. Nat Genet 40 (8), 1016–22. [DOI] [PubMed] [Google Scholar]
- 28.Magnusdottir E et al. (2013) A tripartite transcription factor network regulates primordial germ cell specification in mice. Nat Cell Biol 15 (8), 905–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayashi K et al. (2011) Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 146 (4), 519–32. [DOI] [PubMed] [Google Scholar]
- 30.Nakaki F et al. (2013) Induction of mouse germ-cell fate by transcription factors in vitro. Nature 501 (7466), 222–6. [DOI] [PubMed] [Google Scholar]
- 31.Chan YS et al. (2013) A PRC2-dependent repressive role of PRDM14 in human embryonic stem cells and induced pluripotent stem cell reprogramming. Stem Cells 31 (4), 682–92. [DOI] [PubMed] [Google Scholar]
- 32.Seki Y (2018) PRDM14 Is a Unique Epigenetic Regulator Stabilizing Transcriptional Networks for Pluripotency. Front Cell Dev Biol 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nady N et al. (2015) ETO family protein Mtgr1 mediates Prdm14 functions in stem cell maintenance and primordial germ cell formation. Elife 4, e10150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tu S et al. (2016) Co-repressor CBFA2T2 regulates pluripotency and germline development. Nature 534 (7607), 387–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iacono G et al. (2018) Increased H3K9 methylation and impaired expression of Protocadherins are associated with the cognitive dysfunctions of the Kleefstra syndrome. Nucleic Acids Res 46 (10), 4950–4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.da Rocha ST et al. (2008) Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet 24 (6), 306–16. [DOI] [PubMed] [Google Scholar]
- 37.Carofino BL and Justice MJ (2015) Tissue-Specific Regulation of Oncogene Expression Using Cre-Inducible ROSA26 Knock-In Transgenic Mice. Curr Protoc Mouse Biol 5 (2), 187–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carofino BL et al. (2013) A mouse model for inducible overexpression of Prdm14 results in rapid-onset and highly penetrant T-cell acute lymphoblastic leukemia (T-ALL). Dis Model Mech 6 (6), 1494–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carofino BL et al. (2016) PRDM14 promotes RAG-dependent Notch1 driver mutations in mouse T-ALL. Biol Open 5 (5), 645–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tracey LJ et al. (2019) The pluripotency regulator PRDM14 requires hematopoietic regulator CBFA2T3 to initiate leukemia in mice. Molecular Cancer Research In press. [DOI] [PMC free article] [PubMed]
- 41.Chyla BJ et al. (2008) Deletion of Mtg16, a target of t(16;21), alters hematopoietic progenitor cell proliferation and lineage allocation. Mol Cell Biol 28 (20), 6234–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Engel ME et al. (2010) Myeloid translocation gene 16 (MTG16) interacts with Notch transcription complex components to integrate Notch signaling in hematopoietic cell fate specification. Mol Cell Biol 30 (7), 1852–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cai Y et al. (2009) Eto2/MTG16 and MTGR1 are heteromeric corepressors of the TAL1/SCL transcription factor in murine erythroid progenitors. Biochem Biophys Res Commun 390 (2), 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hunt A et al. (2011) Mtg16/Eto2 contributes to murine T-cell development. Mol Cell Biol 31 (13), 2544–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schuh AH et al. (2005) ETO-2 associates with SCL in erythroid cells and megakaryocytes and provides repressor functions in erythropoiesis. Mol Cell Biol 25 (23), 10235–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steinauer N et al. (2017) Emerging Roles of MTG16 in Cell-Fate Control of Hematopoietic Stem Cells and Cancer. Stem Cells Int 2017, 6301385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hug BA and Lazar MA (2004) ETO interacting proteins. Oncogene 23 (24), 4270–4. [DOI] [PubMed] [Google Scholar]
- 48.Sood R et al. (2017) Role of RUNX1 in hematological malignancies. Blood 129 (15), 2070–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lindberg SR et al. (2003) Interactions between the leukaemia-associated ETO homologues of nuclear repressor proteins. Eur J Haematol 71 (6), 439–47. [DOI] [PubMed] [Google Scholar]
- 50.Kawaguchi M et al. (2019) Co-option of the PRDM14-CBFA2T complex from motor neurons to pluripotent cells during vertebrate evolution. Development 146 (2). [DOI] [PubMed] [Google Scholar]
- 51.Lindberg SR et al. (2005) The Leukemia-associated ETO homologues are differently expressed during hematopoietic differentiation. Exp Hematol 33 (2), 189–98. [DOI] [PubMed] [Google Scholar]
- 52.Heng TS et al. (2008) The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9 (10), 1091–4. [DOI] [PubMed] [Google Scholar]
- 53.Chambers SM et al. (2007) Hematopoietic fingerprints: an expression database of stem cells and their progeny. Cell Stem Cell 1 (5), 578–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kochetkova M et al. (2002) CBFA2T3 (MTG16) is a putative breast tumor suppressor gene from the breast cancer loss of heterozygosity region at 16q24.3. Cancer Res 62 (16), 4599–604. [PubMed] [Google Scholar]
- 55.Amann JM et al. (2005) Mtgr1 is a transcriptional corepressor that is required for maintenance of the secretory cell lineage in the small intestine. Mol Cell Biol 25 (21), 9576–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rocha CR et al. (2013) The role of DNA repair in the pluripotency and differentiation of human stem cells. Mutat Res 752 (1), 25–35. [DOI] [PubMed] [Google Scholar]
- 57.Henry MP et al. (2018) The Genomic Health of Human Pluripotent Stem Cells: Genomic Instability and the Consequences on Nuclear Organization. Front Genet 9, 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simko SJ et al. (2012) Mouse Lymphoblastic Leukemias Induced by Aberrant Prdm14 Expression Demonstrate Widespread Copy Number Alterations Also Found in Human ALL. Cancers (Basel) 4 (4), 1050–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van der Kouwe E and Staber PB (2019) RUNX1-ETO: Attacking the Epigenome for Genomic Instable Leukemia. Int J Mol Sci 20 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grey C et al. (2018) PRDM9, a driver of the genetic map. PLoS Genet 14 (8), e1007479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Teng G et al. (2015) RAG Represents a Widespread Threat to the Lymphocyte Genome. Cell 162 (4), 751–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davi F et al. (1996) Lymphocytic progenitor cell origin and clonal evolution of human B-lineage acute lymphoblastic leukemia. Blood 88 (2), 609–21. [PubMed] [Google Scholar]
- 63.Davidsson J et al. (2010) Relapsed childhood high hyperdiploid acute lymphoblastic leukemia: presence of preleukemic ancestral clones and the secondary nature of microdeletions and RTK-RAS mutations. Leukemia 24 (5), 924–31. [DOI] [PubMed] [Google Scholar]
- 64.Notta F et al. (2011) Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 469 (7330), 362–7. [DOI] [PubMed] [Google Scholar]
- 65.Ou M et al. (2018) PRDM14: A Potential Target for Cancer Therapy. Curr Cancer Drug Targets 18 (10), 945–956. [DOI] [PubMed] [Google Scholar]
- 66.Leitch HG et al. (2013) Primordial germ-cell development and epigenetic reprogramming in mammals. Curr Top Dev Biol 104, 149–87. [DOI] [PubMed] [Google Scholar]
- 67.Harikumar A and Meshorer E (2015) Chromatin remodeling and bivalent histone modifications in embryonic stem cells. EMBO Rep 16 (12), 1609–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hayashi K et al. (2008) Dynamic equilibrium and heterogeneity of mouse pluripotent stem cells with distinct functional and epigenetic states. Cell Stem Cell 3 (4), 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamaji M et al. (2013) PRDM14 ensures naive pluripotency through dual regulation of signaling and epigenetic pathways in mouse embryonic stem cells. Cell Stem Cell 12 (3), 368–82. [DOI] [PubMed] [Google Scholar]
- 70.Leitch HG et al. (2013) Naive pluripotency is associated with global DNA hypomethylation. Nat Struct Mol Biol 20 (3), 311–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Takahashi K et al. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131 (5), 861–72. [DOI] [PubMed] [Google Scholar]
- 72.Mzoughi S et al. (2016) The role of PRDMs in cancer: one family, two sides. Curr Opin Genet Dev 36, 83–91. [DOI] [PubMed] [Google Scholar]
- 73.Huang S et al. (1998) The PR domain of the Rb-binding zinc finger protein RIZ1 is a protein binding interface and is related to the SET domain functioning in chromatin-mediated gene expression. J Biol Chem 273 (26), 15933–9. [DOI] [PubMed] [Google Scholar]
- 74.Krivtsov AV and Armstrong SA (2007) MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 7 (11), 823–33. [DOI] [PubMed] [Google Scholar]
- 75.Moritz LE and Trievel RC (2018) Structure, mechanism, and regulation of polycomb-repressive complex 2. J Biol Chem 293 (36), 13805–13814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fog CK et al. (2012) PRDM proteins: important players in differentiation and disease. Bioessays 34 (1), 50–60. [DOI] [PubMed] [Google Scholar]
- 77.Lin Y et al. (1997) Repression of c-myc transcription by Blimp-1, an inducer of terminal B cell differentiation. Science 276 (5312), 596–9. [DOI] [PubMed] [Google Scholar]
- 78.Boi M et al. (2015) PRDM1/BLIMP1: a tumor suppressor gene in B and T cell lymphomas. Leuk Lymphoma 56 (5), 1223–8. [DOI] [PubMed] [Google Scholar]
- 79.Steele-Perkins G et al. (2001) Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein-methyltransferase superfamily. Genes Dev 15 (17), 2250–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Glass C et al. (2014) The role of EVI1 in myeloid malignancies. Blood Cells Mol Dis 53 (1–2), 67–76. [DOI] [PubMed] [Google Scholar]
- 81.Zhang Y et al. (2011) PR-domain-containing Mds1-Evi1 is critical for long-term hematopoietic stem cell function. Blood 118 (14), 3853–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chuikov S et al. (2010) Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol 12 (10), 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chi J and Cohen P (2016) The Multifaceted Roles of PRDM16: Adipose Biology and Beyond. Trends Endocrinol Metab 27 (1), 11–23. [DOI] [PubMed] [Google Scholar]
- 84.Moelans CB et al. (2010) Molecular differences between ductal carcinoma in situ and adjacent invasive breast carcinoma: a multiplex ligation-dependent probe amplification study. Anal Cell Pathol (Amst) 33 (3), 165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moelans CB et al. (2014) Genomic evolution from primary breast carcinoma to distant metastasis: Few copy number changes of breast cancer related genes. Cancer Lett 344 (1), 138–146. [DOI] [PubMed] [Google Scholar]
- 86.Kitchen MO et al. (2016) Quantitative genome-wide methylation analysis of high-grade non-muscle invasive bladder cancer. Epigenetics 11 (3), 237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Steenbergen RD et al. (2013) Methylation-specific digital karyotyping of HPV16E6E7-expressing human keratinocytes identifies novel methylation events in cervical carcinogenesis. J Pathol 231 (1), 53–62. [DOI] [PubMed] [Google Scholar]
- 88.Ashktorab H et al. (2016) Reduced Representation Bisulfite Sequencing Determination of Distinctive DNA Hypermethylated Genes in the Progression to Colon Cancer in African Americans. Gastroenterol Res Pract 2016, 2102674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Terashima K et al. (2014) Genome-wide analysis of DNA copy number alterations and loss of heterozygosity in intracranial germ cell tumors. Pediatr Blood Cancer 61 (4), 593–600. [DOI] [PubMed] [Google Scholar]
- 90.Gell JJ et al. (2018) PRDM14 is expressed in germ cell tumors with constitutive overexpression altering human germline differentiation and proliferation. Stem Cell Res 27, 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ruark E et al. (2013) Identification of nine new susceptibility loci for testicular cancer, including variants near DAZL and PRDM14. Nat Genet 45 (6), 686–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Baltaci E et al. (2016) Analysis of gene copy number changes in head and neck cancer. Clin Otolaryngol 43 (4): 1004–1009. [DOI] [PubMed] [Google Scholar]
- 93.Zhang T et al. (2013) High expression of PRDM14 correlates with cell differentiation and is a novel prognostic marker in resected non-small cell lung cancer. Med Oncol 30 (3), 605. [DOI] [PubMed] [Google Scholar]
- 94.Baykara O et al. (2015) Amplification of chromosome 8 genes in lung cancer. J Cancer 6 (3), 270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Su Y et al. (2016) Integrating DNA methylation and microRNA biomarkers in sputum for lung cancer detection. Clin Epigenetics 8, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]