

SUMMARY
Vacuolar-proton ATPases (V-ATPases) are conserved complexes that couple the hydrolysis of ATP to the pumping of protons across membranes. V-ATPases are known to play diverse roles in cellular physiology. We studied the Toxoplasma gondii V-ATPase complex and discovered a dual role of the pump in protecting parasites against ionic stress and in the maturation of secretory proteins in endosomal-like compartments. Toxoplasma V-ATPase subunits localize to the plasma membrane and to acidic vesicles, and characterization of conditional mutants of the a1 subunit highlighted the functionality of the complex at both locations. Microneme and rhoptry proteins are required for invasion and modulation of host cells, and they traffic via endosome-like compartments in which proteolytic maturation occurs. We show that the V-ATPase supports the maturation of rhoptry and microneme proteins, and their maturases, during their traffic to their corresponding organelles. This work underscores a role for V-ATPases in regulating virulence pathways.
Graphical Abstract
In Brief
Stasic et al. characterize the function of the vacuolar proton ATPase in the life cycle of Toxoplasma gondii, a widespread parasite that infects almost one-third of the world’s population. The work presents molecular evidence of the pump’s role in the synthesis of virulence factors of a highly successful pathogen.
INTRODUCTION
Toxoplasma gondii is an Apicomplexan parasite that infects a wide range of hosts, including humans. T. gondii infections are usually asymptomatic in healthy adults, but in immunosuppressed individuals, infections can cause serious complications and be fatal. The pathogenicity of this obligate intracellular parasite is linked to its lytic cycle, which consists of invasion of mammalian cells, replication inside a parasitophorous vacuole (PV), and egress (Black and Boothroyd, 2000). As T. gondii progresses through its lytic cycle, it encounters dramatic changes in the ionic (Ca2+, Na+, K+, Cl−) and nutrient composition of its surrounding milieu. Sophisticated regulatory mechanisms are in place for the parasite to deal with these changes and also to use these ionic gradients for its own benefit such as filling its intracellular Ca2+ stores (Pace et al., 2014). T. gondii tachyzoites, the fast-growing form, replicate inside host cells, which lyse upon exit of the parasite, a phase responsible for the pathology of Toxoplasma. Our laboratory has previously characterized an organelle termed the plant-like vacuole or PLV (also termed VAC) that becomes prominent in extracellular tachyzoites and fragments shortly after invasion in intracellular parasites. The PLV is thought to protect parasites against ionic and osmotic stress (Miranda et al., 2010; Parussini et al., 2010). The PLV houses lytic enzymes like cathepsins, plant-like pumps like the vacuolar proton pyrophosphatase, and channels like aquaporins, and acts to regulate ions and/or as a post-Golgi sorting compartment for secretory proteins destined to the micronemes, rhoptries, and acidocalcisomes. The PLV plays a central role during the extracellular phase of the parasite not only in resisting environmental stress but also in preparing parasites for subsequent host cell invasion. Most of the functions of this organelle depend on its ability to maintain an acidic gradient at its membrane, which in plants is driven by two distinct electrogenic proton pumps: the H+-translocating inorganic pyrophosphatase (VP1) and the vacuolar-H+-ATPase (V-ATPase).
The V-ATPase is an evolutionarily conserved proton (H+) pump that couples the hydrolysis of ATP with the translocation of H+ across membranes, often into the lumen of a vesicle. Typically, these protein complexes consist of at least 14 different subunits that compose a membrane anchoring V0 domain (a, c, c′, c″, d, and e subunits) and a peripheral V1 domain (subunits A to H) (Forgac, 2007). In this work, we characterized the a1 subunit of the T. gondii V-ATPase complex. Subunit a is a 100-kDa transmembrane protein containing an amino-terminal cytoplasmic domain and a carboxy-terminal hydrophobic domain with eight to nine transmembrane helices (Leng et al., 1999). We investigated the link between the T. gondii V-ATPase and PLV function to gain knowledge of the mechanism by which this organelle protects parasites against ionic stress and its role in sorting and maturation of essential secretory proteins like microneme and rhoptry proteins. Our data showed that the V-ATPase is also functional at the plasma membrane of T. gondii tachyzoites where it pumps H+ out of the parasite. We propose a model for the dual role of this multipurpose pump and its adaptation to the unique needs of intracellular and extracellular tachyzoites and their parasitism.
RESULTS
Genomic Organization and Expression of the T. gondii vha1 Gene
V-ATPases are complexes composed of two domains, V1 and V0 (Figure S1A), which couple the hydrolysis of ATP with transport of H+. Subunit a of the V0 domain is a 100-kDa integral membrane protein that spans both domains of the complex and is involved in its assembly (Forgac, 2007) (Figure S1A). The N-terminal domain connects V1 and V0, and stabilizes the complex during rotary catalysis. The C-terminal domain is membrane-embedded and is involved in proton transport (Wang et al., 2008). T. gondii appears to express two a isoforms, a1 and a2 (TGGT1_232830 and TGGT1_290720, respectively). We chose to characterize a1 because of its central role in translocating H+ and its potential interaction with both domains V0 and V1. Subunit a1 (TGGT1_232830) had a higher BLAST expectant score compared with the yeast Vph1p (Table S2), stronger evidence for expression, and a stronger phenotype score than a2 according to ToxoDB (Sidik et al., 2016). We termed this gene Tgvha1 (vha1 henceforth), which encodes for a predicted protein of 909 amino acids (Vha1) with an N-terminal signal peptide covering the first 26 amino acids. The topology of Vha1 predicts the presence of 7 transmembrane domains (Figure S1B).
The V-ATPase Localizes to the Plasma Membrane and the PLV
To study the localization of the V-ATPase in T. gondii, the vha1 gene was endogenously tagged with a 3xHA at the C terminus (Figure 1A) using the pLIC plasmid approach (Huynh and Carruthers, 2009; Sheiner et al., 2011). The vha1-HA parasites were isolated and insertion of the tag was confirmed by PCR (Figures S1C and S1D) and further evaluated by western blots with αHA antibodies (Figure 1B). Immunofluorescence assays (IFAs) with αHA antibodies showed plasma membrane localization in intracellular (Figure 1Ci) and extracellular parasites and co-localization with the plasma membrane protein, surface antigen 1 or SAG1 (Figures 1Cii and S2A). Specific localization of Vha1 to the plasma membrane was shown by Clostridium septicum alpha-toxin treatment, which induces separation of the plasma membrane away from the inner membrane complex (IMC) (Wichroski et al., 2002). IFAs showed that Vha1 did not co-localize with the IMC marker (Figure S2B). In intracellular parasites, we observed labeling of vesicles (Figures 1C and S2A), but the plasma membrane labeling was the most predominant (Figure 1C). In extracellular parasites, in addition to the plasma membrane, Vha1 labeled a large vacuole (Figures 1Cii–Civ and S2C) that co-localized with cathepsin L (TgCPL or CPL) and the vacuolar proton pyrophosphatase (VP1), which label the PLV and other endosome-like compartments (ELCs) (Parussini et al., 2010) (Miranda et al., 2010) (Figures 1Ciii, Civ, and S2C). Immuno-electron microscopy (immuno-EM) confirmed the presence of Vha1 at the plasma membrane and the PLV (Figure 1D). The localization of Vha1 to the PLV became more noticeable with longer exposure of parasites to the extracellular milieu (Figure S2G). We also showed PLV localization by super-resolution microscopy (Figures 1Ciii and S2C) and confirmed this localization by IFAs with specific antibodies generated in mice against the a1 subunit (Figures S2H and S2I). Western blots of Toxoplasma lysates developed with the affinity-purified mouse serum showed a band corresponding to the correct size of Vha1 (Figure S2H). The traffic of the V-ATPase to the PLV and its underlying mechanism remains to be characterized. We do not believe that it is being turned over or degraded because no degradation products showed in immunoblots (Figure S2H) and our data indicate that the complex is functional at the PLV.
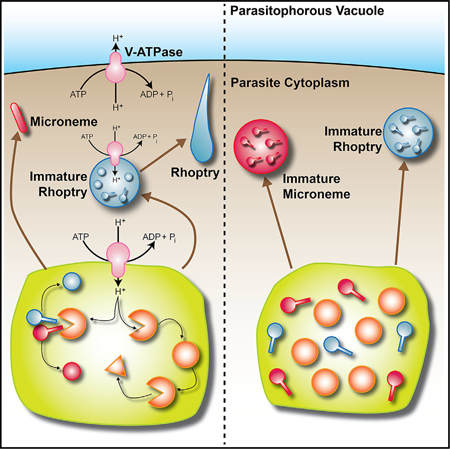
Figure 1. Vha1 Localization.
(A) 3xHA tagging: HA, hemagglutinin; CAT, chloramphenicol acetyltransferase.
(B) Western blots of lysates from vha1-HA (left panel), vhE-HA and vhG-HA (right panel) parasites with αHA antibody.
(C) IFAs of vha1-HA cells with αHA showing (i) localization of Vha1 in intracellular tachyzoites; (ii) co-localization with αSAG1; (iii) super-resolution IFAs showing co-localization with CPL (PLV marker) (dashed lines mark parasites); and (iv) extracellular tachyzoites showing co-localization with VP1.
(D) Immuno-EM with aHA showing (i) labeling at the plasma membrane and PLV and (ii) PLV fusing with vesicles.
(E) vha1-mNeonGreen clones showing co-localization of mNeonGreen with LysoTracker.
(F) IFAs of intracellular vhG-HA tachyzoites with αHA and αVP1.
(G) IFAs of intracellular tachyzoites expressing vhE-HA with αVP1 and αHA. The Mander’s colocalization coefficient of VP1/Vha1 and CPL/Vha1 from three independent trials was 0.33 and 0.62, respectively. Scale bars: 2 μm.
We previously showed ATP-stimulated proton transport in a PLV-enriched fraction (Miranda et al., 2010; see Figure 6B) supporting activity of the V-ATPase in the PLV. We tagged the a1 subunit with the green fluorescent protein mNeonGreen (Sidik et al., 2016), and parasites expressing Vha1-mNeonGreen were loaded with LysoTracker, a lysosomal marker that labels acidic compartments. LysoTracker co-localized with the Vha1-mNeon at the PLV in extracellular tachyzoites, confirming the acidic nature of the PLV and its co-localization with the V-ATPase (Figure 1E).
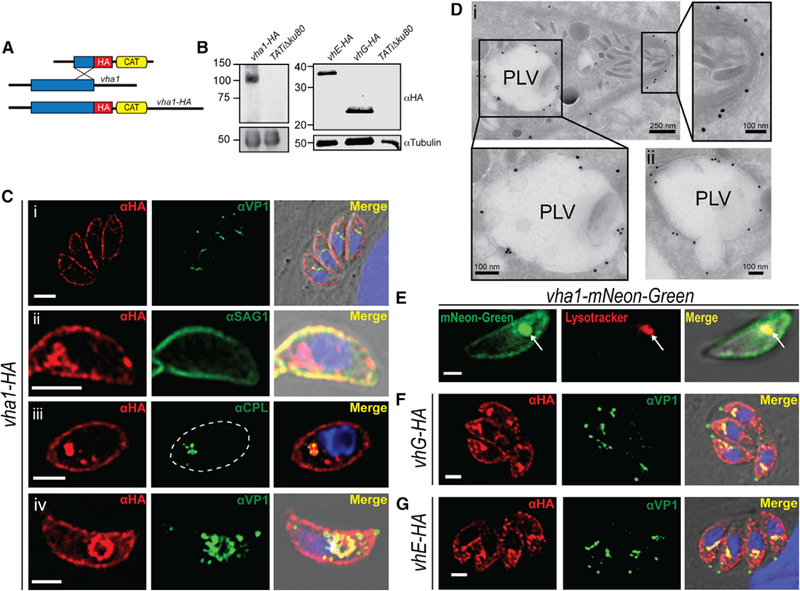
Figure 6. The V-ATPase and Rhoptry Maturation.
(A) Western blots of total lysates of iΔvha1-HA tachyzoites grown + and − ATc (3 days) and probed for rhoptry bulb proteins ROP4, ROP7, or TgCA_RP. Immature forms are indicated with arrowheads, and mature forms with arrows.
(B) Comparison between the percentage of immature ROP4, ROP7, or TgCA_RP between lysates of iΔvha1-HA parasites grown + or − ATc.
(C) IFAs of iΔvha1-HA tachyzoites grown + or − ATc and probed with αIMC (highlights nascent daughter cells in dividing parasites), αproROP4 (labels immature rhoptries), and αCA_RP (rhoptries). These are super-resolution images showing the labeling with αproROP4 of dividing parasites (αIMC1 labels nascent daughters; left panels) and non-dividing parasites (right panels). Scale bars: 2 mm.
(D) Quantification of PVs with parasites expressing immature and mature rhoptries in iΔvha1-HA tachyzoite vacuoles grown + and − ATc. There was a significant increase of αproROP4 labeling in non-dividing parasites with ATc (red columns).
(E) Routine EM of iΔvha1-HA tachyzoites grown −ATc (left, −ATc) showing normal rhoptries and their characteristic rhoptry neck (arrowheads) and bulb regions (arrows). In iΔvha1-HA +ATc (3 days), mature rhoptries are absent, and accumulation of vesicular immature rhoptry structures was seen (arrows).
(F) Quantification of the EM images showing significant reduction in the number of tachyzoites containing mature rhoptries after treatment with ATc. Approximately 52–73 parasites were enumerated from three trials.
(G) Super-resolution IFA showing that Vha1 encircles nascent rhoptries labeled by proROP4 (1:500) and TgCA_RP (1:1,000) in intracellular parasites. Statistical analysis was done using a Student’s t test, where *p < 0.05, **p < 0.01, and ****p < 0.0001; n.s., not significant.
We also studied the localization of subunits E (TGGT1_305290; henceforth vhE) and G (TGGT1_246560; henceforth vhG) following a similar genetic strategy as described. Subunits E (VhE) and G (VhG) form part of the peripheral V1 domain and are involved in establishing the stator of the V-ATPase complex and interact with the regulator of ATPase of vacuoles and endosomes (RAVE) complex (Smardon et al., 2002). Both VhG and VhE localized to the plasma membrane and co-localized with VP1 in intracellular and extracellular tachyzoites (Figures 1F, 1G, S2E, and S2F). We also tagged vhG with a Ty tag in parasites expressing vha1-HA (Figure S2D) to study co-localization. IFAs with αTy and αHA, showed co-localization of both subunits supporting their role within the complex (Figure S2D). Our results established that the T. gondii V-ATPase localizes to the plasma membrane and the PLV, which becomes more prominent in extracellular parasites.
vha1 Can Partially Rescue Growth of a Subunit-Deficient Yeast
The specific function of the vha1 gene product as part of the V-ATPase complex was investigated by expressing the T. gondii vha1 gene in Saccharomyces cerevisiae vph1Δstv1Δ mutants (Perzov et al., 2002). These mutants do not express either of their a subunits (Vph1p and Stv1p, which localize to the yeast vacuole or Golgi, respectively) and are unable to grow at pH ≥7 due to deficient acidification of the yeast vacuole (Manolson et al., 1994). The T. gondii vha1-HA gene was cloned in the galactose inducible expression plasmid, pYES2/NT C, and transformed in the vph1Δstv1Δ yeast strain (Figure 2A). The vha-HA gene was expressed in the vph1Δstv1Δ strain in CSM-ura media with 2% galactose, while no expression was seen in 2% glucose (Figure 2B). The vph1Δstv1Δ strain does not grow at pH 7, but expression of the vha-HA gene partially restored growth in this medium (Figures 2C and S3A). Quantification of the slopes during exponential growth (4–16 h for wild-type [WT] and 8–32 h for vph1Δstv1Δ yeast) showed that vph1Δstv1Δ expressing vha1-HA grew significantly better than vph1Δstv1Δ yeast transformed with the empty vector (EV) (Figure 2C). Using a similar strategy, the gene of the subunit a2 (vha2) was expressed in the vph1Δstv1Δ strain (Figures S3A), and it also partially complemented their growth. Expression of GFP-Vha1 showed localization of Vha1 to the yeast vacuole (and other internal compartments) (Figure S3B). GFP-Vph1 was also expressed as control, which localized mostly to the yeast vacuole (Figure S3B). These results showed that the T. gondii a1 subunit can function as part of the V-ATPase complex, providing genetic evidence of its conserved biochemical function.
Figure 2. Functional Analysis of Vha1 in Saccharomyces cerevisiae.
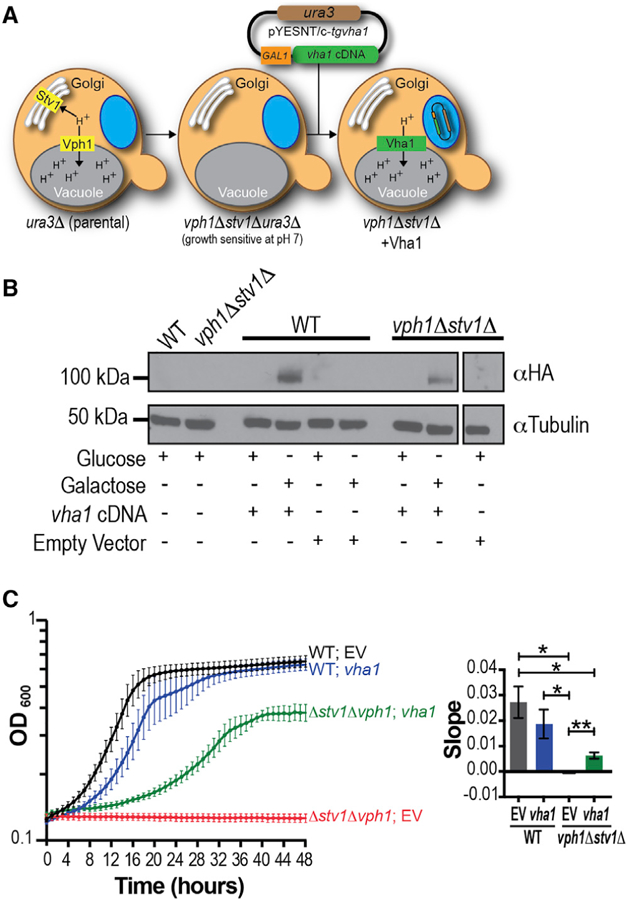
(A) Strategy used for complementation of vph1Δstv1Δ S. cerevisiae with the vha1 gene. GAL1, galactose inducible promoter; ura3, metabolic marker that contains the ura3 gene.
(B) Western blot of vph1Δstv1Δ lysates grown on glucose or galactose (pH 5.5) transformed with pYES2 containing the vha1-HA cDNA. Control strains were transformed with an empty pYES2/NT C vector or no vector.
(C) Growth of WT and vph1Δstv1Δ yeast harboring vha1 cDNA (vha1) or empty vector (EV) grown in CSM-ura pH 7.0 with 2% galactose for 48 h. Data are from three independent trials done in triplicate. Quantification of the slopes during exponential growth (4–16 h in WT yeast and 8–32 h in vph1Δstv1Δ yeast) using one-way ANOVA test, where *p < 0.05 and **p < 0.01.
Vha1 and the Lytic Cycle of T. gondii
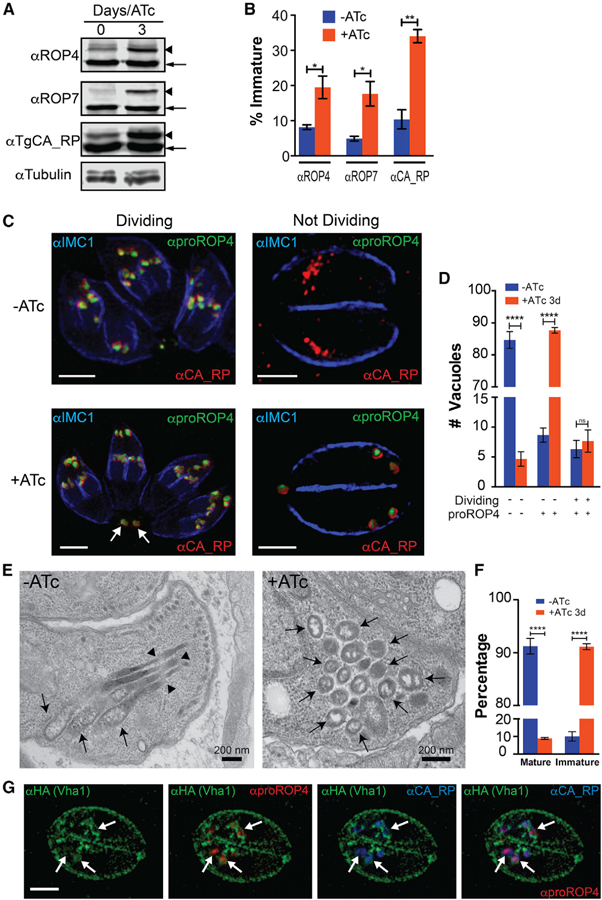
We generated conditional iΔvha1-HA mutants by using a tetracycline-regulated transactivator system and the parental line TATiΔku80, which combines regulated gene expression (Sheiner et al., 2011) with high efficiency of homologous recombination (Fox et al., 2009) (Figures 3A and S4A). Proper insertion and orientation of the promoter was shown by PCR (Figure S4B), and western blots confirmed regulation of Vha1 expression. Vha1 was not evident by western after 24 h of growth with anhydrotetracyclin (ATc) (Figure 3A), and IFAs did not detect the Vha1 signal after 3 days with ATc (Figure S4C, +ATc). We also created a complemented strain, iΔvha1-HA-CM, that constitutively expressed vha1 from a tubulin promoter at the uprt gene locus (Donald and Roos, 1995) (Figures 3B and S4D) for control experiments. Note that the western in Figure 3B was done using αHA (left panel) and the polyclonal antibody αVha1 generated in this work (right panel), which shows that the complemented vha1 is expressed even in the presence of ATc (Figure S2H).
Figure 3. The V-ATPase and the T. gondii Lytic Cycle.
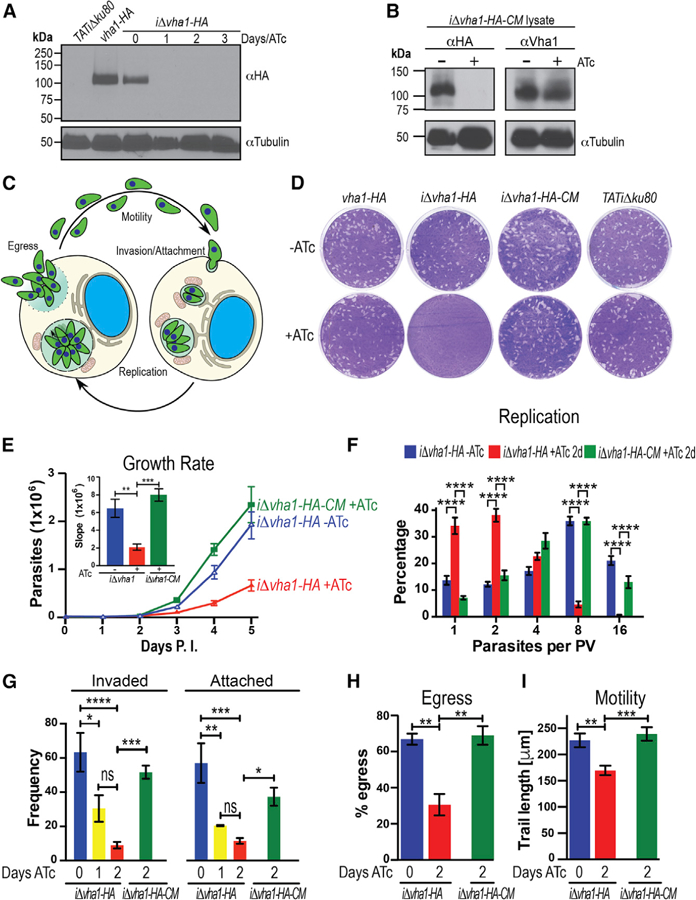
(A) Western blots of lysates from parental (TATiΔku80), vha1-HA, and iΔvha1-HA grown + or ATc parasites. Immunoblots were developed with rat αHA (1:200). Anti-tubulin was the loading control.
(B) Western blots of lysates of iΔvha1-HA-CM parasites showing no expression of the endogenous copy of vha1 in the presence of ATc, while the extra copy of Vha1 was constitutively expressed as evident with the mouse aVha1 antibody (1:500) (see Figure S2H for more details on the antibody).
(C) Overview of the lytic cycle. Adapted from Hortua Triana et al. (2018).
(D) Plaque assays of vha1-HA, iΔvha1-HA, iΔvha1-HA-CM, and TATiΔku80 clones grown + or ATc (0.5 mg/mL). Each well was infected with 200 parasites and grown for 8 days, fixed, and stained with crystal violet.
(E) 4,000 tdTomato-expressing parasites were grown in hTERT cells for 5 days. Data are presented as the average of three independent trials done in triplicate. Inset: slopes from days 2–5.
(F) Growth kinetics of iΔvha1-HA and iΔvha1-HA-CM + or ATc. Parasites from 115 to 150 PVs were counted per clone in three independent trials.
(G) Red-green assay for quantification of attachment and invasion of T. gondii. Data were from three to four independent experiments counting 10 random fields per clone.
(H) Egress triggered with 10 mM nigericin for 30 min at 37°C. Data were from three independent experiments counting 10 random fields per clone and comparing them to a DMSO control. Only PVs with two or more parasites were enumerated.
(I) tdTomato-expressing parasites were re-suspended in Ringer’s buffer without Ca2+, and motility was stimulated with 1.8 mM Ca2+. The average length (microns) parasites traveled is reported from five to six individual parasites from three independent trails. Values are means ± SEM.
(E) and (F) were compared with two-way ANOVA test and (G), (H), and (I) were compared with one-way ANOVA test, where *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
The pathogenicity of T. gondii stems from its lytic cycle, which is initiated by active invasion of host cells, replication, and egress (Figure 3C). To establish whether Vha1 is important for parasite growth, we performed plaque assays of vha1-HA, iΔvha1-HA, iΔvha1-HA-CM, and the parental strain TATiΔku80 under identical conditions with (+) and without (−) ATc (Figure 3D). iΔvha1-HA+ATc could not form plaques demonstrating that the expression of vha1 was critical for at least one major step of the lytic cycle. We introduced a cytosolic tdTomato into the iΔvha1-HA and iΔvha1-HA-CM cells to analyze growth kinetics. To determine the impact of ATc on growth, tdTomato-expressing parasites were preincubated with ATc, and the drug was removed at the indicated times (Figure S5A). We observed that 4 days with ATc blocked growth completely while some growth was seen after 1–2 days +ATc (Figure S5A). Based on these results, we studied a variety of phenotypic features with 1, 2, or 3 days of ATc treatment. We reasoned that longer exposure to ATc would likely result in stronger phenotypic characteristics but could be the result of non-specific effects. iΔvha1-HA parasites grown +ATc for 3 days were tested for viability using the trypan blue exclusion assay to rule out that they were dead or dying (Figure S5B). We evaluated the daily growth rate and found that iΔvha1-HA+ATc parasites grew significantly slower than the controls (iΔvha1-HA-ATc) and complemented (iΔvha1-HA-CM+ATc) lines (Figures 3E and S5C). To assay for replication, parasites were pre-incubated (+ATc) or not (−ATc) with ATc for 24 h and passaged to new fibroblast cells + or −ATc, respectively. After 24 h of exposure to ATc, followed by another 24 h in a new passage (48-h +ATc), there was a significant reduction in the ability of iΔvha1-HA+ATc to replicate as shown by the low number of parasites per PV counted (Figure 3F, red columns).
We next analyzed each step of the lytic cycle and found that 2 days +ATc reduced invasion of host cells 7-fold and attachment 5-fold (Figure 3G); these defects were ameliorated in the iΔvha1-HA-CM line. We studied egress by stimulating intracellular tachyzoites with nigericin or saponin plus calcium (Borges-Pereira et al., 2015), and the iΔvha1-HA+ATc mutants showed a significant delay in their response to these inducers of egress (Figures 3H and S5D). There was a significant difference in the percentage of lysed PVs after 30 min of exposing intracellular parasites to 10 mM nigericin (66.9% and 68.9% for the iΔvha1-HA-ATc and iΔvha1-HA-CM+ATc, respectively, and 30.5% for iΔvha1-HA+ATc) (Figure 3H). We also measured time to egress after induction with 2 mM Ca2+ in the presence of 0.01% saponin to permeabilize host cells. iΔvha1-HA-ATc and iΔvha1-HA-CM+ATc parasites took 303 ± 35 and 342 ± 80 s, respectively, to exit host cells, while iΔvha1-HA+ATc took 527 ± 45 s (Figure S5D). We also evaluated motility, an essential part of the lytic cycle of T. gondii and found that the mutants +ATc traveled 57 mm less than the controls −ATc and 69 μm less than the complemented strains +ATc (Figure 3I). In summary, these results show that the activity of the V-ATPase impacts every step of the lytic cycle of T. gondii.
The Role of Vha1 in Intracellular pH
Intracellular pH (pHi) must be strictly controlled because of the narrow optimum pH of most enzymatic processes (Demaurex, 2002). Because of this, cells regulate their pHi by active transport of H+ across membranes. We showed that the maintenance of the cytoplasmic pH of T. gondii tachyzoites was sensitive to bafilomycin A1, a specific inhibitor of V-ATPases (Moreno et al., 1998), and a role for the V-ATPase in pHi regulation was deduced. We therefore tested pHi in the iΔvha1-HA+ATc mutants (extracellulars) by loading them with the pH indicator BCECF-AM (Figures 4A and 4B). pHi of iΔvha1-HA+ATc tachyzoites appeared to vary in a similar way to the pHi of iΔvha1-HA-ATc parasites when exposed to extracellular pHs between 5.5 and 8.5 (Figure 4B). Apparently under these conditions, iΔvha1-HA+ATc mutants are still able to regulate pHi, which is reasonable considering the importance of maintaining physiological pHi levels within a normal range. To study the function of the V-ATPase at the plasma membrane, we measured H+ extrusion using the free acid form of BCECF in a weakly buffered solution (Figure 4C) (Pace et al., 2011). This protocol measures changes in extracellular pH (pHe) that result from H+ extruded from the cell into the extracellular milieu. Addition of glucose, which stimulates glycolytic activity, resulted in medium acidification (Figure 4C; see green and blue slopes). Changes in pHe of −0.40 to −0.46 pH units were observed with the control ( iΔvha1-HA-ATc) or the complemented ( iΔvha1-HA-CM+ATc) parasites. However, mutant iΔvha1-HA+ATc tachyzoites produced a change in pHe of only 0.19 pH units (Figure 4C and inset). The H+ extrusion activity was blocked by bafilomycin A1 for all three cell lines, iΔvha1-HA–ATc, iΔvha1-HA+ATc, and iΔvha1-HA-CM+ATc (Figure 4D). These data highlighted the role for the T. gondii V-ATPase in extrusion of protons, which could be stimulated with glucose.
Figure 4. Function of Vha1 in T. gondii.
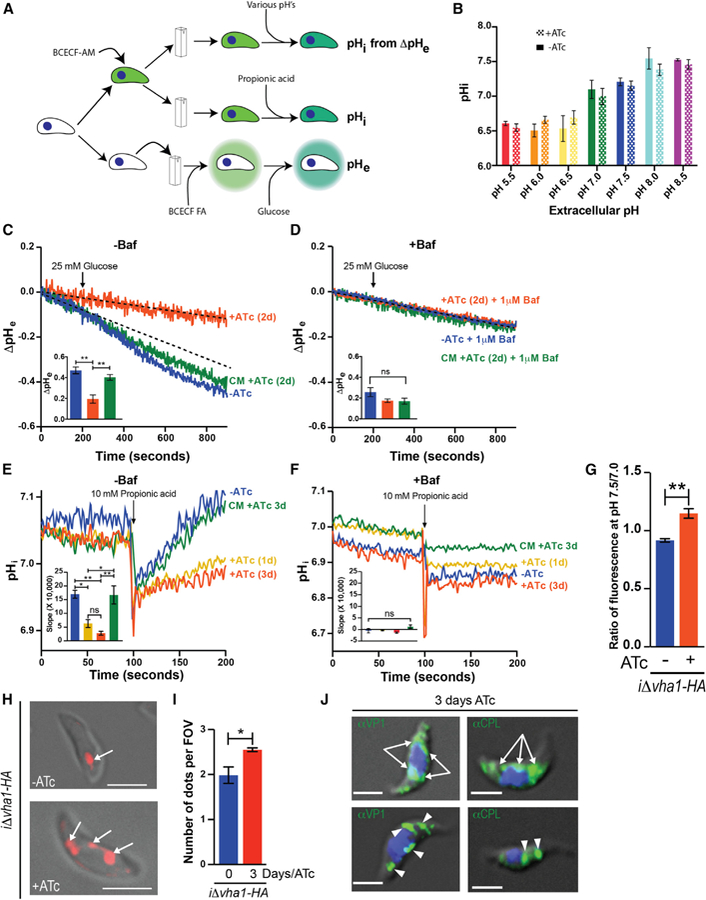
(A) Protocol used to measure intracellular pH (pHi), extracellular pH (ΔpHe), and proton extrusion (pHe).
(B) pHi measurements of iΔvha1-HA cells at various pHe values. Parasites were loaded with the pH indicator BCECF-AM, and fluorescence measurements were done as described in STAR Methods. Data were from three independent trials.
(C) Representative tracings of proton extrusion of iΔvha1-HA + or −ATc after adding 5 mM glucose. BCECF free acid in a weakly buffered solution was used. Changes in pH were followed as described in STAR Methods. Inset shows analysis from three to six independent trials. The dashed line represents the –ATc slope before adding glucose.
(D) Same as (C), but cells were pretreated with 1 μM bafilomycin for 3 min. Insets show quantifications of slopes.
(E) Acid load and pH recovery of parasites loaded with BCECF-AM. 10 mM propionic acid (PA) was added where indicated to suspensions of iΔvha1-HA and iΔvha1-HA-CM (3 days ATc) grown +ATc for 0, 1, and 3 days. Quantifications are from three to four independent trials.
(F) Same as (E) but cells were pretreated with 1 μM bafilomycin for 3 min. Insets show the quantification of slopes from 105 to 200 s from three independent trials.
(G) Membrane potential measurements of iΔvha1-HA cells grown + or −ATc for 2 days and incubated with bisoxanol. Fluorescence measurements at pH 7.5 to 7.0 were compared and are from three independent trials.
(H) iΔvha1-HA parasites + or −ATc (0 or 3 days, respectively) and loaded with 10 μM LysoTracker red for 30 min at 37°C in BAG.
(I) Quantification of compartments stained by LysoTracker. 5–15 cells per field of view were counted and a minimum of eight fields. Graph shows quantifications of puncta in iΔvha1-HA parasites + and −ATc.
(J) IFA of iΔvha1-HA+ATc (3 days) parasites and probed with αVP1 or αCPL. The arrows point to ER localization, and the arrowheads point to discrete puncta of VP1 or CPL staining.
Statistical analysis was performed using a Student’s t test (G and I) or one-way ANOVA (B and C–F), where *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001; ns, not significant.
Exposing iΔvha1-HA-ATc (controls) tachyzoites to 10 mM propionic acid caused transient acidification of the cytosolic pH, which is quickly recovered to normal values within 50 s of the acid pulse (Figure 4E, blue tracing). Recovery of iΔvha1-HA+ATc parasites after acid treatment was very slow, and at 100 s post-treatment, their pHi was still low, while the pH of control parasites was completely recovered (Figure 4E, yellow and red tracings). The complemented line, iΔvha1-HA-CM+ATc, was able to restore its pHi, almost as fast and efficiently as parental parasites (Figure 4E, green tracing). Bafilomycin A1 blocked recovery of pHi in all lines (+ or ATc and complemented strain) (Figure 4F), showing that the V-ATPase functions to protect T. gondii from acid stress by pumping protons out of the parasite cytosol.
Deficient H+ pumping at the plasma membrane may also impact membrane potential. We used the membrane potential-sensitive fluorescent probe bisoxonol (Moreno et al., 1998) and found that the iΔvha1-HA+ATc parasites were modestly depolarized (higher fluorescence ratio) compared to their controls (Figure 4G). These data indicated that the proton pumping activity of the V-ATPase generated a H+ gradient at the plasma membrane that contributes partially to the buildup of the membrane potential. Likely, other mechanisms are at play and prevent cells from becoming completely depolarized.
Co-localization of LysoTracker with Vha1 in the PLV (Figure 1E) of iΔvha1-HA-ATc cells supports the acidic nature of the PLV. When we loaded iΔvha1-HA+ATc parasites with LysoTracker instead of a single vacuole labeled, we observed several puncta showing fragmentation of the PLV (Figures 4H and 4I), which could result from defective biogenesis. We next performed IFAs of iΔvha1-HA+ATc parasites with the PLV markers VP1 and CPL, and we also observed labeling of both markers in vesicles distributed through the cell (Figure 4J, arrowheads) including some labeling of the endoplasmic reticulum (ER) (Figure 4J, arrows). It is possible that the PLV is not forming properly in extracellular iΔvha1-HA+ATc tachyzoites, probably due to defective vesicle fusion (Figures 1Ciii and 1Civ). The labeling with αVP1 was similar to the one observed with LysoTracker, indicating that the defective formation of the PLV was likely leading to an accumulation of VP1 vesicles, which were acidic but failed to fuse. We were unable to evaluate the difference in acidification because of the non-quantitative nature of Lyso-Tracker. We expressed a genetically encoded pH indicator (mCherry-SEpHluorin) in the PLV (see Video S1) of control iΔvha1-HA cells, but because of the fragmented nature of the PLV, it was not possible to estimate pH in the vesicles formed in iΔvha1-HA+ATc mutants (data not shown). Our data support a role for the V-ATPase in trafficking of vesicles to or from the PLV.
Vha1 Is Important for the Localization, Maturation, and Secretion of Micronemes
Micronemes are secretory organelles involved in the invasion of host cells by tachyzoites (Carruthers et al., 1999). Several microneme proteins undergo proteolytic maturation while traveling to the micronemes, and a role for endosomal compartments in this maturation has been shown (Harper et al., 2006). In extracellular tachyzoites, the V-ATPase translocates from the plasma membrane to the PLV, which harbors microneme maturase activity (Parussini et al., 2010). Parasite attachment and invasion are associated with the secretion of micronemes (Carruthers et al., 1999), and since iΔvha1-HA+ATc parasites display defects in attachment and invasion, we studied microneme secretion. We tested the release of MIC2 into the excreted/secreted antigen fraction collected from supernatants of extracellular tachyzoites without stimulation (Figure 5A, constitutive) or after induction by ethanol (Figure 5A, 1% ethanol). Mutants iΔvha1-HA+ATc parasites were not able to secrete micronemes constitutively, and they responded poorly to stimulation by ethanol (Figure 5A). The amount of total MIC2 protein in the iΔvha1-HA lysate was similar to that of parental and complemented strains. Figure 5B shows the quantification of three independent experiments using GRA1 secretion as control. We found that, after 3 days with ATc, iΔvha1-HA+ATc tachyzoites are deficient at secreting MIC2, while secretion of GRA1 was normal (indicating viable parasites) (Figure 5B). The complemented line, iΔvha1-HA–CM, showed normal secretion of MIC2 even after 3 days +ATc. We also tested the secretion of the apical membrane antigen 1 (TgAMA1), a transmembrane protein that localizes to the micronemes (Hehl et al., 2000). Secretion of TgAMA1 was also significantly reduced in the iΔvha1-HA+ATc line (Figures S6A and S6B).
Figure 5. V-ATPase and Microneme Secretion.
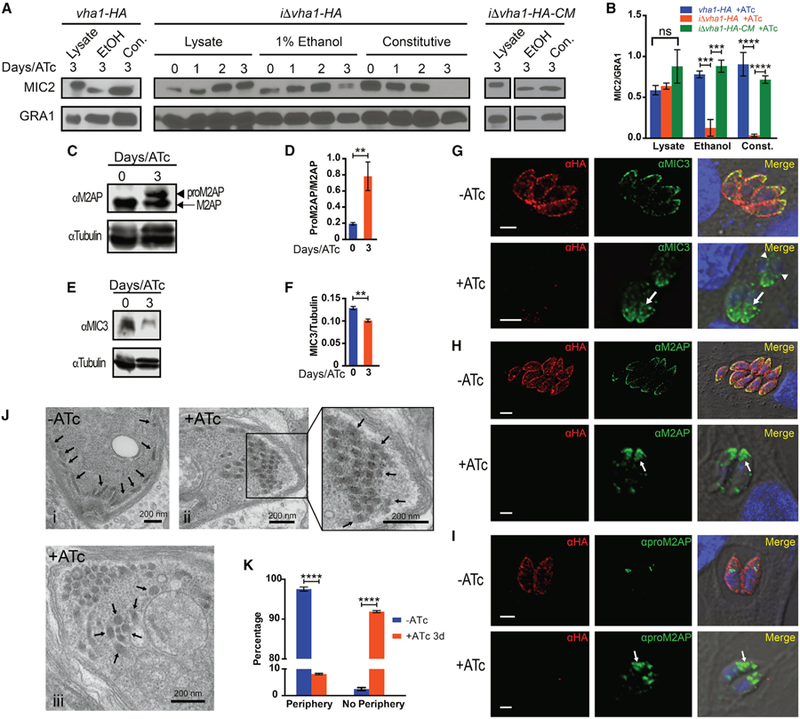
(A) Microneme secretion of vha1-HA, iΔvha1-HA, and iΔvha1-HA-CM parasites. Parasites were incubated in invasion media alone for constitutive expression (con.) or in the presence of 1% ethanol (EtOH). Lysate samples were also included. Supernatants were analyzed by western blots with anti-MIC2 or anti-GRA1.
(B) Quantification of MIC2 secretion by pixel density from ImageJ (ratio of MIC2/GRA1) from four to five independent trials. Values are means ± SEM.
(C) Western blots of total lysates of iΔvha1-HA + and ATc showing the increase in the signal for proM2AP (arrowhead) compared to mature M2AP (arrow).
(D) Ratio of the signal intensity of proM2AP and M2AP. Bar graphs shows quantification of at least three independent trials.
(E) Western blots of total lysates of iΔvha1-HA parasites +ATc for 3 days or ATc (0) probed with αMIC3.
(F) Quantification of the ratio of intensity of the bands for MIC3/tubulin from three independent trials.
(G) IFAs with aMIC3 and αHA showing localization of MIC3 in iΔvha1-HA parasites grown + and ATc. Arrowheads point to perinuclear labeling and arrows point staining not at the periphery.
(H) IFAs with aM2AP and αHA showing the localization of M2AP in iΔvha1-HA parasites grown + and ATc.
(I) IFAs with aproM2AP and αHA in iΔvha1-HA parasites grown + and ATc.
(J) Transmission EM of iΔvha1-HA parasites grown ATc (i) or +ATc for 3 days (ii and iii). (i) Arrows point toward peripheral micronemes and apical end; (ii) micronemes are indicated by the arrows at the apical end in parasites +ATc for 3 days; (iii) micronemes are more centrally located in parasites incubated +ATc for 3 days.
(K) Quantification of EM images from three independent preparations comparing the localization of micronemes of iΔvha1-HA grown + and ATc for 3 days. The STAR Methods explain how was done the quantification. Approximately 59–72 parasites were enumerated from three independent trials.
(D) and (F) were quantified by pixel density using Image Studio from three independent trials. Graph error bars are SEM. Statistical analysis was performed using a one-way ANOVA, where **p < 0.01; ***p < 0.001, and ****p < 0.0001; ns, not significant.
MIC2 forms a heterohexameric complex with M2AP (MIC2-associated protein), and the complex MIC2-M2AP plays fundamental roles in gliding motility and invasion. M2AP is initially translated with a propeptide that is removed in an endosomal compartment, and blocking the removal of the propeptide affects trafficking of the M2AP-MIC2 complex to the periphery. (Harper et al., 2006). We studied processing and trafficking of M2AP in the iΔvha1-HA+ATc cells (3 days) and observed a significant accumulation of the immature form (Figures 5C and 5D). MIC3 is a homodimeric adhesin that is synthesized as a precursor that is proteolytically processed during its traffic through the secretory pathway on its way to the micronemes. This processing is important for the binding function of the protein (Cérède et al., 2002). Mature MIC3 abundance was significantly reduced in iΔvha1-HA+ATc mutants when compared to controls (Figures 5E and 5F). Parental lines (TATiΔku80+ATc) were tested for M2AP and MIC3 maturation in the presence of ATc and is shown in Figures S6C–S6E. ATc did not affect maturation of these proteins in parental lines.
We next looked at the distribution of microneme proteins by IFAs of iΔvha1-HA+ATc mutants (Figures 5G–5I). There was a notable difference in the distribution of MIC3 in the iΔvha1-HA+ATc mutants. MIC3 labeling was increased in perinuclear areas and accumulated in internal vesicles likely destined to the secretory pathway (Figure 5G, +ATc, arrows). This was distinct from the neatly distributed labeling of MIC3 at the periphery of parasites that express normal levels of Vha1 ( iΔvha1-HA-ATc) (Figure 5G, −ATc). It was previously reported that unprocessed MIC3 builds up in perinuclear compartments such as the ER and/or Golgi compartments and accumulates in the secretory pathway during parasite division (El Hajj et al., 2008). Our data point toward retention of MIC3 in the ER and secretory pathway because of its defective processing, a consequence of inactive V-ATPase (Figure 5G, arrowheads). MIC3 and M2AP are synthesized almost simultaneously, but pro-M2AP was mainly observed in endosome-like compartments while pro-MIC3 was observed in both the ER-Golgi and endosome-like compartments (El Hajj et al., 2008). We studied the localization of M2AP in the iΔvha1-HA+ATc mutants (Figure 5H) and saw a remarkable difference in their distribution. M2AP labeling in the controls iΔvha1-HA-ATc showed the typical peripheral distribution, while labeling in the iΔvha1-HA+ATc mutants was clustered within the central region of the extreme apical end (Figure 5H, compare and +ATc). proM2AP labeling was increased in the iΔvha1-HA+ATc mutants (Figure 5I). Disruption of M2AP (Harper et al., 2006) results in secretory retention of MIC2, leading to reduced MIC2 secretion from the micronemes and impaired invasion. MIC2 localization was also altered, and it accumulated in vesicles with intense labeling toward the apical end of the cell close to the plasma membrane (Figure S6F).
Transmission EM of iΔvha1-HA+ATc showed that their micronemes are not able to dock or fuse at the periphery, instead presenting a more central location in the apical region of the tachyzoite (Figure 5J). These micronemes also exhibited a rounded morphology rather than the typical cigar shape (Tomavo et al., 2013) (Figure 5J). We found significantly fewer micronemes at the periphery in the iΔvha1-HA+ATc mutants (Figure 5K). These results showed that the V-ATPase plays a significant role in microneme maturation, function, and organelle distribution.
Vha1 and Maturation of Rhoptries
The majority of rhoptry bulb proteins are initially translated with an ER signal peptide and a propeptide that acts as a trafficking signal for the rhoptries (Hoppe et al., 2000). Because pro-peptides are proteolytically cleaved within endosomal compartments (Ngô et al., 2004), we questioned whether the activity of the V-ATPase would impact maturation of rhoptries. Immunoblots of lysates from iΔvha1-HA-ATc and iΔvha1-HA+ATc tachyzoites revealed that the maturation of the rhoptry proteins ROP4, ROP7, and TgCA_RP (Chasen et al., 2017) were altered, as demonstrated by a significant increase in the percentage of immature protein (Figures 6A and 6B). The effect of ATc on the maturation of Rhoptries and maturases of parental cell lines is shown in Figure S7. Rhoptry maturation was tested in Figures S7A and S7B.
We next compared IFAs of iΔvha1-HA-ATc and iΔvha1-HA+ATc tachyzoites with antibodies against the propeptide of ROP4 (αproROP4), which labels immature rhoptries (Carey et al., 2004). We found a significant increase in the number of iΔvha1-HA+ATc tachyzoites that contained immature rhoptries. Immature rhoptries are usually seen in dividing tachyzoites, and we observed them in tachyzoites that were not in the division stage of their cell cycle, as determined by IMC labeling (Figure 6C, not dividing). When tachyzoites were dividing (following the IMC labeling), αproROP4 and αCA_RP labeling appeared indistinguishable between iΔvha1-HA-ATc and iΔvha1-HA+ATc tachyzoites, with the only observation of an increased number of nascent rhoptries in the residual body of iΔvha1-HA+ATc vacuoles (Figure 6C, dividing). Trafficking from the ER appeared to be normal as indicated by the localization of rhoptry bulb proteins. When the parasites had completed division, we still saw labeling of aproROP4 in the iΔvha1-HA+ATc mutants. We quantified the number of PVs containing tachyzoites with immature rhoptries and we saw a significant increase (>80%) of aproROP4 labeling in the iΔvha1-HA+ATc (3 days) mutants (Figure 6D). The number of tachyzoites containing daughter cells (labeled with aIMC1) was not significantly different in iΔvha1-HA+ATc mutants, suggesting that this step of endodyogeny is not affected. Routine EM of iΔvha1-HA+ATc parasites showed a striking phenotype where there is no evidence of mature rhoptries (Figures 6E and 6F). Our data support a role for the V-ATPase in the maturation of rhoptry proteins and the morphological changes associated with the formation of the characteristic elongated mature rhoptries.
We performed IFAs with αCA_RP (rhoptries) αproROP4 (immature rhoptries), and αHA in iΔvha1-HA parasites (no ATc) using super-resolution microscopy. We saw labeling of Vha1 in vesicles that surround immature rhoptries (Figures 6G, arrows, and S8A), but none in mature rhoptries (Figure S8B). With the aim to confirm that this localization is consistent for the entire V-ATPase complex, we performed IFAs on vhE-HA and vhG-HA and saw that both E and G encircled immature rhoptries, supporting the presence of the whole complex at this location (Figures S8C and S8D). Immature rhoptries were previously shown to be acidic (pH 3.5–5.5) (Shaw et al., 1998), and our data show the potential mechanism of acidification. It remains to be determined how the V-ATPase-labeled vesicles split allowing the mature rhoptry proteins to traffic to the rhoptries while the V-ATPase follows a different path to other endosomal compartments.
The V-ATPase and Proteolytic Activities
We investigated the role of the V-ATPase on the maturation of known endosomal proteases. CPL, an enzyme present in the PLV and shown to play a role in the maturation of M2AP and MIC3 (Parussini et al., 2010), is proteolytically processed and acidic pH is required for both its maturation and its function. CPL maturation was affected in iΔvha1-HA+ATc mutants as accumulation of immature CPL was seen 48 h after treatment with ATc (Figures 7A and 7B), while ATc had no effect on the maturation of CPL in parental lines (TATiDku80) (Figures S7C and S7D).
Figure 7. The V-ATPase and Protease Maturation.
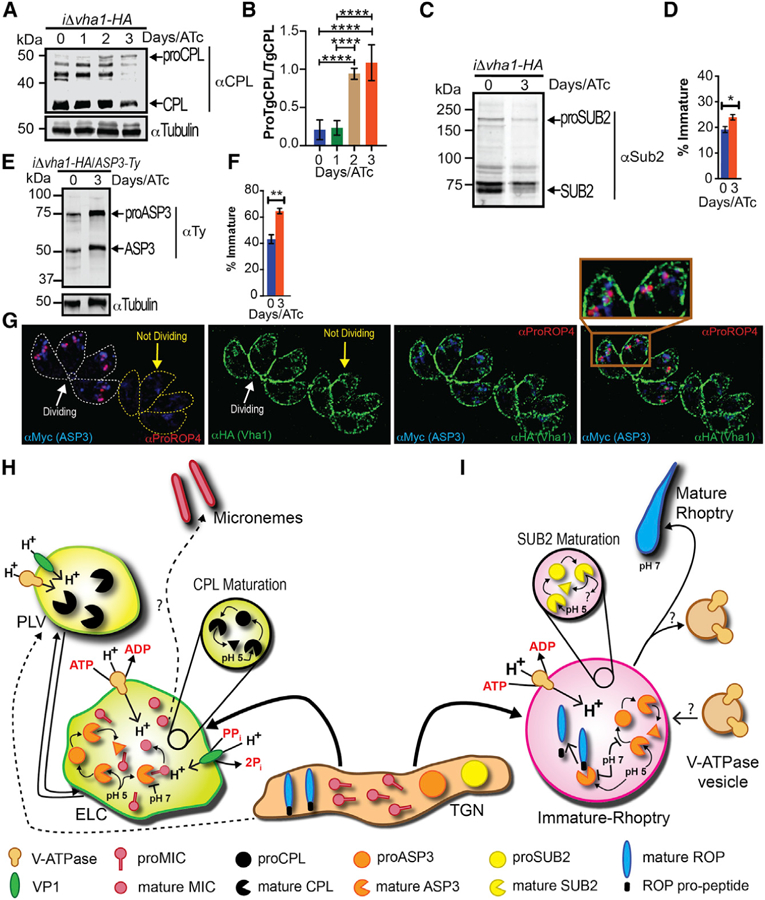
(A) Western blots with αCPL (1:500) of lysates from iΔvha1-HA +ATc parasites showing accumulation of proCPL.
(B) Quantification of bands shown in (A) from four to five trials. Experiments were standardized to tubulin and the ratio of proCPL/CPL was determined by ImageJ.
(C) Western blots of lysates from iΔvha1-HA tachyzoites grown + and ATc (3 days) and probed with αTgSUB2 (1:500).
(D) Quantification of immature SUB2 as percentage of total SUB2. Quantification was from lysates of iΔvha1-HA grown + and ATc.
(E) Western blots with αTy of iΔvha1-HA-ASP3-Ty parasites. Parasites were grown +ATc for 0 or 3 days.
(F) Quantification of the maturation of ASP3 as percentage of immature form from total ASP3.
(G) Super-resolution images showing ASP3 co-localization with proROP4 and V-ATPase, which surrounds ASP3 and proROP4 in dividing cells (lines outline parasites). Statistical analysis was done using a one-way ANOVA or Student’s t test, where *p < 0.05, **p < 0.01, and ****p < 0.0001; ns, not significant.
(H) Proposed maturation of micronemes: Newly synthesized MICs are transported from the Golgi to the trans-Golgi network and then to the ELC. The ELC is acidified by both VP1 and V-ATPase. The PLV is mainly acidified by the V-ATPase, and we propose that this compartment would be more acidic and most likely would not contain MICs. Maturation of CPL occurs by self-cleavage. Maturation of ASP3 occurs in the ELCs. In the V-ATPase acidified endosome, proMICs encounter mature ASP3 and/or other lytic enzymes. Cleavage of pro-peptides may occur at this point, and once the late endosome is matured, processed MICs would be transported to the micronemes.
(I) Rhoptry biogenesis: immature ROP proteins, immature SUB2, and ASP3 traffic through the TGN. The V-ATPase associates with the forming rhoptry creating the acidic environment that is important for SUB2 and ASP3 maturation. In pro-rhoptries, APS3 would be active and able to process immature ROPs. According to this model, the activity of SUB2 would likely be decreased at low pH. The V-ATPase separates by an unknown mechanism allowing the pH of the mature rhoptry to become more neutral and rhoptry organelles to mature.
A rhoptry localized subtilisin-like serine protease, TgSUB2 was proposed to be involved in maturation of ROP proteins (Miller et al., 2003), although TgSUB2 knockouts did not show ROP maturation defects (Dogga et al., 2017). It has been reported that some subtilisin proteases require pH below 7 for their own maturation (Anderson et al., 1997; Gawlik et al., 2009), but this information is not known for TgSUB2. The activity of subtilisin proteases on their substrate, however, may need neutral or slightly alkaline pH (Shinde et al., 1999). We tested the maturation of TgSUB2 in the iΔvha1-HA+ATc mutants and found a significant difference in the ratio between mature and immature forms of TgSUB2 as compared with the same ratio present in the controls, iΔvha1-HA-ATc (Figures 7C and 7D). These results demonstrate that matured SUB2 is reduced in the iΔvha1-HA+ATc mutants; however, the effect on the proteolytic function of TgSUB2 remains to be seen.
Aspartic proteases require low pH for maturation and activity (Szecsi, 1992), and it was reported that aspartic protease 3 (ASP3) was the maturase for microneme and rhoptry proteins in T. gondii (Dogga et al., 2017). To investigate whether maturation of ASP3 was affected in iΔvha1-HA cells, we tagged ASP3 using the 50UPRT-pTub8-Asp3–3Ty-3′UPRT plasmid in the iΔvha1-HA line. We exposed these parasites ( iΔvha1-HA-ASP3-Ty) to ATc to reduce the expression of vha1 and performed westerns and observed an increase in immature ASP3 (and decrease of mature form) compared to the level present in iΔvha1-HA-ATc cells (Figures 7E and 7F). Both lines showed a noticeable level of immature and mature forms (Figure 7E). ASP3 was localized to the ELC (Dogga et al., 2017), so we performed co-localization IFAs with αHA in the vha1-HA line. In dividing intracellular tachyzoites, we observed that ASP3 colocalized with proROP4 and Vha1 labeling encircled the signal of proROP4 (Figure 7G, dividing PV). Our results indicate that the H+ gradient generated by the V-ATPase in immature rhoptries is likely responsible for the generation of the ideal environment required for ASP3 maturation to its proteolytically active product (Figure 7G, dividing PV). Our results support a role for the V-ATPase in regulating pH of endosomal compartments where important maturation of critical secretory proteins occurs.
DISCUSSION
The function of the V-ATPase complex is to pump H+ against a concentration gradient, and this activity impacts the physiology of every eukaryotic cell (Saroussi and Nelson, 2009). The gradient generated by the activity of the V-ATPases in organelles and in membranes of eukaryotic cells is used as a driving force for a number of secondary transport processes (Beyenbach and Wieczorek, 2006). V-ATPases are ATP-dependent H+ pumps that localize to a variety of cellular membranes in eukaryotic cells (Toei et al., 2010), including endosomes, lysosomes, Golgi-derived vesicles, secretory vesicles, and, in some cells, also the plasma membrane (Forgac, 2007). The activity of V-ATPases in the endocytic pathway results in a pH gradient that decreases from pH 6.0 in early endosomes to pH 5.0–5.5 in lysosomes (Hurtado-Lorenzo et al., 2006). Intra-endosomal acidification is required for the enzymatic activity of hydrolytic enzymes (Nishi and Forgac, 2002) among other functions.
The Toxoplasma genome shows evidence for the presence of most subunits of the V-ATPase with two a subunit isoforms, a1 and a2 (TgGT1_232830 and TgGT1_290720). In this work, we characterized the a1 subunit. Expression of the Toxoplasma vha1 gene partially complemented the growth of the mutant strain S. cerevisiae vph1Δstv1Δ at pH 7 (Manolson et al., 1994). This result showed that Vha1 functions as part of the V-ATPase complex, and the function of the V-ATPase complex could be studied by manipulating the expression of the vha1 gene. The expression of the a2 gene in the same strain of S. cerevisiae also partially complemented their growth, indicating that a2 may also function as part of the complex. We focused on the characterization of a1 because it had higher homology to other a subunits of V-ATPases of yeast, plants, and mammalian cells and also because the sequence of a2 contained an extra domain of 12–15 kDa at the C terminus, the domain important for H+ translocation, which is not found in yeast, plants, or mammalian cells.
The V-ATPase of T. gondii showed H+-pumping activity at the plasma membrane, which acted in the recovery of normal cytosolic pH from acid loads. The active H+ extrusion into the surrounding media shows that the V-ATPase is fully functional at the plasma membrane and protons generated from normal metabolic functions, or artificially induced with acid load, are pumped outside the cell. It is likely that other mechanisms are functional, like a P-type ATPase or a sodium/proton exchanger (Arrizabalaga et al., 2004), thus explaining why parasites are still alive at 48 h even though the absence of Vha1 expression.
The H+ gradient generated by the activity of the V-ATPase at the plasma membrane contributed to the generation of a membrane potential, and mutant parasites were slightly depolarized (Moreno et al., 1998). This depolarization is modest because, most likely, parasites would not survive if their membranes were considerably depolarized. Changes in membrane potential are often linked with ion influx or efflux (Åkerman, 1978).
The expression of the vha1 gene could be regulated with ATc in the iΔvha1-HA mutants. These parasites showed a strong growth defect and strong phenotypic differences, already observed 48 h after ATc treatment, and even stronger differences were observed when cells were grown longer with ATc. All of the major steps of the lytic cycle were defective, including maturation and secretion of micronemes, invasion, motility, and egress, supporting an essential role for the V-ATPase. The invasion defect was evident 2 days after ATc treatment, earlier than the microneme secretion defect, which is only evident 3 days after ATc treatment. The H+ pumping activity is already non-functional at day 2 of ATc, so it is likely that the invasion decrease at this stage is due to a defect in signaling, which is linked to H+ gradients generated by the V-ATPase (Roiko et al., 2014).
Intracellular replication was affected in the iΔvha1-HA+ATc mutants, and our interpretation is that metabolically active intra-cellular parasites produce large quantities of acid, which needs to be extruded by the pumps at the plasma membrane. We measured pHi in extracellular parasites and that did not appear to be affected when cells were exposed to regular changes in pHe. However, when the parasite cytosol was challenged with propionic acid, the recovery response observed was defective in the mutants. This supports an important function for the pump in protecting cytoplasmic pH and, when the pump is defective, would cause alterations of pHi, disrupting cell fitness and replication.
Secretory organelles, like rhoptries and micronemes, are made de novo in daughter parasites during the process of endodyogeny, the mechanism by which Toxoplasma replicates (Black and Boothroyd, 2000). A large body of evidence indicates that the biogenesis of rhoptries and micronemes occurs at the Golgi (Tomavo et al., 2013). Trafficking of cargo to the micronemes involves the participation of endosome-like compartments, and their proteolytic maturation involves the participation of specific maturases (Dogga et al., 2017; Parussini et al., 2010). pro-MICs have been observed in structures bearing late endosomal markers (Harper et al., 2006), and nascent micronemes have been observed in close proximity to the PLV. Specific inhibitors of the V-ATPase, like bafilomycin A1, reduced the maturation of CPL, an enzyme that localizes to the PLV, shown to be self-processed (Dou et al., 2013) and to be involved in the maturation of microneme proteins (Parussini et al., 2010).
Disruption of the activity of the V-ATPase negatively impacted the maturation of MIC3 and M2AP, two microneme proteins that are processed before storage in the microneme compartment (Soldati et al., 2001). Defective maturation of M2AP leads to mistargeting of MIC2 as it was shown that proteolytic stabilization of the MIC2-M2AP complex within the micronemes is important for the correct packaging within micronemes favoring its rapid secretion onto the parasite surface (Harper et al., 2006). The localization of M2AP, MIC3, proM2AP, and MIC2 were altered in iΔvha1-HA+ATc mutant parasites, and microneme organelles were not correctly oriented. We proposed a model for the role of the V-H+-ATPase in the maturation of micronemes (Figure 7H). Two potential endosome acidic compartments, one expressing higher levels of the VP1 (VP1 compartment as in Harper et al., 2006, and Liu et al. 2014) and a second one where the V-ATPase predominates (PLV/VAC). Acidification of these organelles could be important for either the maturation process of relevant maturases and/or for their specific proteolytic activity on their substrates, as for example essential adhesins secreted for invasion. The presence of more than one of these compartments could represent the means by which lytic activity is regulated by limiting and/or allowing contact with substrates. Vha1 depletion disrupted the biogenesis of the PLV, and this resulted in mis-targeting of important secretory proteins.
Rhoptries are club-shaped secretory organelles with a discrete neck and bulb region uniquely present in Apicomplexan parasites. Mature rhoptries become club-shaped when mature, and they secrete proteins during the process of host cell invasion, an essential function for parasite virulence (Dubremetz, 2007). A number of rhoptry proteins are secreted during host cell invasion and participate in the building of the PV. In addition, some rhoptry proteins are targeted to the host cell nucleus to control host functions (Dubremetz, 2007). In intracellular para-sites, the V-ATPase labeled immature rhoptries, which have been previously shown to be acidic (pH 3.5–5) in comparison to the more neutral mature rhoptries (pH 5–7) (Shaw et al., 1998). Pro-rhoptries form transiently between the Golgi and the apical area just prior to cytokinesis (Dubremetz, 2007), but the mechanism by which these vesicles form is not known (Ngô et al., 2003). How proteins are delivered to the rhoptries is also not clear; however, adaptins have been implicated in this trafficking (Ngô et al., 2003; Venugopal et al., 2017).
Many of the proteins destined to the bulb of the rhoptries (ROPs) contain an N-terminal ER signal peptide and an N-terminal pro-peptide that is cleaved during rhoptry maturation. It was proposed that the N-terminal ROP pro-peptides could be cleaved by subtilisin-like proteases, like TgSUB2, at the SΦX(E/D) motif first identified in ROP1 (Miller et al., 2003; Turetzky et al., 2010). Subtilisin-like proteases typically undergo autocatalytic cleavage of a pro-peptide that initially helps with folding but must be cleaved before proteolytic activity can occur on substrate proteins (Ikemura and Inouye, 1988). The autocatalytic cleavage and substrate protease activity of subtilisin-like proteases typically have different pH optimums. In the case of subtilisin E from Bacillus subtilis, the optimal pH for autocatalytic cleavage of the pro-peptide is pH 7.0, whereas the optimal pro-teolytic activity on substrate proteins is pH 8.5 (Shinde et al., 1999). TgSUB2 has an atypical cleavage site, which occurs after an acidic residue instead of a basic residue, raising the possibility that the optimal conditions for its cleavage activity are also atypical. There is no experimental evidence to support TgSUB2 maturation in the neutral environment of the ER (Miller et al., 2003).
Aspartic protease 3 (ASP3), an aspartyl protease of the ELC, was critical for invasion and egress of Toxoplasma, albeit it did not appear to be implicated in parasite replication, gliding motility, and attachment (Dogga et al., 2017). It was also proposed that the activity of ASP3 superseded the previously proposed roles for SUB2 (Miller et al., 2003) and CPL (Parussini et al., 2010) for the maturation of rhoptries and microneme proteins, respectively. The acidification role of the V-ATPase is relevant for the efficient maturation of SUB2 and also ASP3. In this model, SUB2 autocatalytic cleavage occurs in the immature rhoptries at low pH and acidification of immature rhoptries would affect maturation of SUB2, but the role of this enzyme in ROP proteins secretion and maturation is less clear. Maturation and/or activity of ASP3 in pro-rhoptries would be impacted by the acidification role of the V-ATPase explaining the delay in ASP3 maturation and the defective rhoptries in the iΔvha1-HA+ATc mutants. It is evident that the phenotype of iΔvha1-HA+ATc parasites partly mimics the phenotypes of iDASP3(+ATc) on maturation of rhoptries and micronemes. However, iΔvha1-HA+ATc parasites also present other phenotypic features probably linked to the role of the pump at the plasma membrane. See our model for the role of the V-H+-ATPase in maturation of rhoptries (Figure 7I).
This work presents a characterization of the V-ATPase protein complex at the molecular level in Toxoplasma gondii. The work revealed interesting and significant features of this complex such as its distinct localization in intracellular and extracellular parasites and its functionality at the plasma membrane and the endosomal system. The V-ATPase proton pump activity supports the processing/synthesis of microneme and rhoptry proteins, which are critical for the lytic cycle of Toxoplasma, a central feature of the pathogenesis of the parasite. Especially intriguing is its localization with immature rhoptries and downstream dissociation from rhoptries likely to allow the maturation of the organelle. The traffic of Vha1 to the PLV, soon after egress of tachyzoites and its functional role at the lysosomal-like organelle where it acts in the acidification and maturation of secreted proteins, is also an intriguing feature to adjust to the needs of extra-cellular tachyzoites.
In summary, our findings highlight the function of this pump in higher-order physiological processes essential for Toxoplasma parasitism. In doing so, the activity of the pump impacts the processing/synthesis of virulence factors. Disruption of these delicate mechanisms will alter the most essential aspect of the parasitological cycle of Toxoplasma. Our work directly connects the proton-pumping activity of the V-ATPase complex to processes necessary for Toxoplasma virulence.
STAR☆METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Silvia Moreno (smoreno@uga.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Parasites and Host Cell Culture
T. gondii tachyzoites were maintained at 37 C in either immortal or mortal human foreskin fibroblasts (hTERT, ATCC® CRL-4001; HFF, ATCC® SCRC-1041) monolayers cultured in Dulbecco’s modified Eagle’s medium with high glucose supplemented with 1% HyClone fetal bovine serum (GE Healthcare Life Sciences). Parasites were regularly passed to new flasks containing new host cells when approximately 50%–80% of the parasites have lysed out of the old culture. Flasks were maintained at 37° C with 5% CO2. All strains and host cell lines were determined to be mycoplasma negative with the MycoScope kit (Genlantis). All parasite lines and host cells used in this study are listed in the key resource table.
Parasite mutants were passed under similar conditions but the media was supplemented with the appropriate antibiotic used for selection.
Yeast growth
Yeast were grown on yeast peptone dextrose (YPD) agar plates or in liquid YPD at 30° C. Yeast were also grown on CSM-ura media on agar plates or broth as described below. For transformations, 1 mL of cultures was used to inoculate 10–15 mL of fresh YPD media at 30° C. Yeast were grown until the OD600 reached 1.5–2.0.
METHOD DETAILS
Epitope-tagging
Carboxy-terminus tagging was done in the parental line RHTatiDku80 (TatiΔku80) (Sheiner et al., 2011) a parasite line that contains the tetracycline-regulated transactivator system that allows conditional expression of genes and also in which the ku80 gene was deleted increasing efficiency of homologous recombination. Primers 1–6 (Table S1) were used to create C-terminal insert fragments of genes vha1 (TgGT1_232830), vhE (TgGT1_305290), and vhG (TgGT1_ 246560) (Table S2) that were suitable for cloning into a pLIC-3XHA, pLIC-mNeonGreen, or pLIC-GFP plasmids. Linearized plasmids were transfected into TATiΔku80 cells followed by drug selection. Upstream gene locus primers 7–9 (Table S1) and a pLIC reverse primer (primer 10) were used to verify proper insertion in the correct gene locus. Western blot analyses with rat αHA (1:200) confirmed the presence of the 3xHA tagging. The cell lines isolated were termed according to the tagged gene and the tag used as vha1-HA, vhE-HA, vhG-HA, vha1-mNeonGreen, vha1-GFP. For tdTomato expressing cell lines, parasites were transfected with a tdTomato plasmid (Chtanova et al., 2008) (a gift from Boris Striepen, University of Georgia), enriched using a Bio-Rad S3 cell sorter, and subcloned. C-terminal Ty1 or myc tagging of aspartic protease 3 (ASP3; TgGT1_246550) was performed in the iΔvha1-HA strain by co-transfecting a plasmid that contained CAS9 and a protospacer against the UPRT gene locus along with a plasmid that contained the ASP3–3Ty1 cDNA or a pLIC plasmid with a C-terminal myc tag, both plasmids were generous gifts from Dominique Soldati-Favre (Dogga et al., 2017). Correct ASP3 C-terminal tags were confirmed by PCR (Primers 26, 27, and/or 10) and western blots with anti-Ty1 or anti-myc.
Inducible Knockdown
For vha1 knockdowns, a tetracycline-regulatable element (Sheiner et al., 2011) was inserted upstream of the translational start codon via double homologous recombination. Primers 11–14 (Table S1) were used to generate the upstream UTR and gene fragments respectively for promoter insertion. Primes 15–18 were used to confirm insertion in the correct genetic locus. Repression of vha1 was accomplished with 0.5 mg/mL anhydrotetracycline. The clonal line isolated after transfection, selection, and subcloning was named iΔvha1-HA.
Complementation of iΔvha1-HA Parasites
Complementation was achieved by cloning the cDNA of the vha1 gene, using primers 19 and 20 (Table S1), into a UPRT cDNA shuttle vector, which contains the 5° and 3° UTR’s of the UPRT gene and a tubulin promoter. The plasmid was transfected into iΔvha1-HA cells expressing tdTomato and selected using 5 µM 5-fluorodeoxyuridine (FUDR). Primers 18 and 21 (Table S1) were used to verify if vha1 cDNA was inserted into the UPRT gene. The line isolated was named iΔvha1-HA-CM.
Immunofluorescence Assays
For IFAs of intracellular tachyzoites, hTERT or HFF monolayers were grown on 18 mm glass coverslips for 24 h and at this time infected with 5 X 105 parasites and allowed to grow for another 24 h. After 18–24 hours, parasites were fixed in 3% paraformaldehyde for 15 min.
For extracellular parasites, freshly lysed tachyzoites were collected, centrifuged and washed with Buffer A with glucose (BAG) (116 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 50 mM HEPES, and 5.5 mM dextrose). Extracellular parasites were affixed to pre-coated coverslips with polylysine and fixed in 3% paraformaldehyde for 15 min. After fixation, both intracellular and extracellular were treated identically. Parasites were permeabilized with 0.25% Triton X-100 at room temperature for 10 minutes. Blocking followed with 3% bovine serum albumin (BSA) in PBS at pH 8.0 for 30 min at room temperature. Primary and secondary antibodies were prepared in PBS at pH 8.0. Coverslips with parasites were washed 5 times after each antibody incubation and mounted on glass slides. IFA images were taken with an Olympus IX-71 inverted fluorescence microscope with a Photometrix CoolSnapHQ CCD camera driven by DeltaVision software or with a Zeiss ELYRA S1 (SR-SIM) super-resolution microscope. Images were deconvolved using Applied Precision’s Softworx imaging suite using 10 cycles of enhanced ratio deconvolution or with ZEN 2011 software with SIM analysis module for SR-SIM images. For all IFAs, a control of TATiΔKu80 incubated with secondary antibody alone were used to subtract non-specific background.
The antibodies used for IFAs were: rat αHA (1:25); mouse αTy: (1:1,000), mouse aCPL (1:100), mouse aMIC2 (1:500), mouse aMIC3(1:400), rabbit aM2AP (1:500), rabbit aproM2AP (1:250), mouse αSAG1 (1:100), mouse aMyc (1:100), mouse aVha1(1:100), and rabbit αVP1 (1:4,000). The secondaries were all used at a concentration of 1:1,000.
For the experiment looking at pro-rhoptry labeling the specific antibody against immature rhoptry α-proROP4 (UVT-70, 1:500) (a gift from Gary Ward), the rhoptry bulb antibody α-TgCA_RP (Guinea Pig 1:1000) and an inner membrane complex marker that labels the IMC (α-IMC1 Mouse, 1:500), and allows to identification of dividing tachyzoites were used. For the quantification of this result, 100 vacuoles (containing 2–8 tachyzoites) were examined for the presence or absence of tachyzoites containing nascent rhoptries (α-proROP4 labeling) and/or undergoing cytokinesis (α-IMC1 labeling of daughter cells).
Electron Microscopy
Routine electron microscopy was performed at the Georgia Electron Microcopy Center. Parasites were fixed in 2.5% glutaraldehyde and 2% paraformaldehyde and post-fixed in 2% osmium tetroxide in 0.1 M cacodylate buffer (pH 7.2). Samples were then ‘‘En bloc’’ stained in 0.5% uranyl acetate and dehydrated in an ascending ethanol series and embedded in Spurr’s plastic (Spurr, 1969). Cells were polymerized in an Eppendorf tube at 70° C for 12 hours. Embedded cells were sectioned on grids post-stained with uranyl acetate and lead nitrate. Grids were viewed in a JEOL JEM-1011 transmission electron microscope operated at 80 kV. The most apical point of the plasma membrane was defined, and then a line was drawn from this most apical end extending into the cytosol of the parasite 400 nm. Anything within this zone, was defined as apical micronemes and not peripheral. Anything within 100 nm of the plasma membrane and out of the apical zone was defined as periphery. Micronemes were identified by their higher electron density relative to other structures and organelles of the parasite. Structures with greater or equivalent electron density like dense granules, rhoptry necks, and acidocalcisomes are significantly larger and different in shape to be easily distinguishable from micronemes. Rhoptries were considered mature if any visible portion of the rhoptry was longer than 200 nm (in any direction). Rhoptries were identified by either their bulbs or necks, with necks having characteristically tubular morphology with high relative electron density and rhoptry bulbs with much less electron density than the necks and the cytosol. Parasites with a minimum of 1 microneme or rhoptry structure were enumerated.
Immuno-electron Microscopy
Immuno-electron microscopy was done using Vha1-HA or Vha1-GFP tagged parasites. Parasites were manually lysed through a 25 gauge needle and incubated in DMEM media with serum for 0 or 1.5 hours at 37° C shaking incubator. Cells were pelleted, washed with PBS, then fixed with 4% paraformaldehyde and 0.05% glutaraldehyde for 1 hour on ice and washed once with phosphate buffered saline once. Sample preparation and images were performed by Dr. Wandy Beatty at the Department of Molecular Microbiology, Washington University School of Medicine, St. Louis, MO 63110.
Generation of mouse anti-Vha1 serum
Primers 22 and 23 (Table S1) were used to clone the first 327 amino acids of the vha1 gene into the bacterial inducible expression plasmid pQE-80L. Expression was induced with 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) for 2 hours at 37° C. Before and after the addition of IPTG, a sample corresponding to 1 OD600 was taken. The cells were sonicated, and the supernatant and pellet fractions were run on a 10% SDS-PAGE gel. Induction of expression with IPTG resulted in a significant band corresponding to the correct size of 36 kDa. Recombinant protein was purified from the bacteria lysate with a Thermo scientific HisPur Ni-NTA chromatography cartridge. Mice were inoculated intraperitoneally with 100 µg of purified and sterilized peptide solution mixed with Freund’s complete adjuvant and boosted twice every 2 weeks with 50 µg of peptide and Freund’s incomplete adjuvant. Antigen-adjuvant mixture was injected intraperitoneally (0.1 mL/injection) and blood was collected every two weeks to determine antibody titers. At the end of the immunization period mice were anesthetized, and blood was collected by cardiac puncture. This serum was tested against parasite lysates and compared with the αHA labeling of vha1-HA lysates to confirm correct size and antibody purity (~37 kDa). Parasite lysates were run on a 10% SDS-PAGE gel and transferred to a nitrocellulose membrane for developing with αHA and the αVha1 serum. The observed bands were around 100 kDa. The serum was affinity purified and the aVha1 antibody was used for IFAs and westerns. Work with mice was carried out in strict accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals and Association for the Assessment and Accreditation of Laboratory Animal Care guidelines. The animal protocol was approved by the University of Georgia’s Committee on the Use and Care of Animals (protocol A2015 02–025-R2). All efforts were made to humanely euthanize the mice after collecting blood.
Plaque Assays
Plaque assays were performed with confluent hTERT host cells grown in six-well plates. Parasites obtained from T-25 flasks were pelleted, washed with PBS, counted using a hemocytometer and diluted in DMEM-HG for infection of each well with 200 parasites. Plates were incubated with or without ATc at 37° C. After 8 days of undisturbed growth, wells were washed twice with PBS, fixed with 100% ethanol for 5 minutes, and washed twice with PBS. Washed wells were stained with 5X crystal violet, washed twice with PBS, and allowed to dry for 4 hours before being photographed.
Red/Green Invasion Assay
The Red/Green invasion assay was performed as described (Kafsack et al., 2004) with some modifications. HFF fibroblasts were grown in a 12 well plate containing 18 mm coverslips for 24 h. The ideal monolayer should be at 70%–80% confluency for this experiment. The plate was placed on ice prior to the experiment. Freshly harvested and purified parasites were resuspended in ice-cold invasion media (3% serum and 10 mM HEPES in DMEM) at a concentration of 2.0 x parasites per mL. The media from the pre-chilled 12 well plate was aspirated and replaced with 1 mL of media plus 1 mL of the parasite suspension (2 × 107 parasites in 2 mL) and the plate incubated on ice for 20 min. At this point, the 12 well plate is transferred to a 37° C water bath and parasites are allowed to invade host cells for 5 min. Invasion is stopped by washing each well with PBS twice, immediately fixing with 3% paraformaldehyde, and blocked with 10% fetal bovine serum (FBS) for 20 min. Extracellular parasites are labeled with Rabbit αSAG1 polyclonal antibody (1:1000) (a generous gift from John Boothroyd) for 60 min at room temperature. Wells were washed with PBS 5 times to remove anti-body, permeabilizated with 1% Triton X-100 in PBS for 10 minutes, and washed 3 times. Parasites were then probed with anti-SAG1 monoclonal antibody (1:500) (Thermo Fisher) for 60 min at room temperature. Cells were washed with PBS 5 times to remove anti-body. Secondaries against rabbit (red; 1,1000) and mouse (green; 1:500) were used to distinguish invasion from attachment. Secondaries were incubated for 60 minutes at room temperature and washed with PBS 5 times. Counting of red and green labeled parasites was compiled from three independent experiments by counting ten fields of view selected at random.
Replication and Growth Assays
Parasite strains were transfected with a plasmid that overexpresses the tdTomato gene. FACS sorting was performed to select a stable tdTomato expressing line. tdTomato-expressing parasites were incubated with or without ATc for 24 hours. These parasites (1 × 105) were used to infect (37° C for 30 min) sub-confluent HFF cells previously grown on coverslips. Following this incubation, parasites were washed twice to remove any extracellular parasites and incubated with or without ATc for an additional 24 hours. After 48 hours of total ATc incubation, Toxoplasma-infected cells were washed twice with PBS and fixed with 3% paraformaldehyde for 10 min. Between 115–150 parasitophorous vacuoles (PV) per condition were counted and the number of parasites inside each PV enumerated. These experiments were repeated a minimum of three times.
Growth was assayed by infecting confluent hTERT cells in 96 well plates with 4,000 tdTomato-expressing parasites with or without ATc in the media. Parasites were allowed to grow in DMEM media without phenol red for 6 days. Each day a fluorescensce reading was recorded on a BioTek Synergy H1 hybrid plate reader. A standard curve was used to correlate parasite numbers, (1 × 106 to 1 × 101) with fluorescence values.
Egress and Motility Assays
For egress experiments, tdTomato-expressing parasites were grown with ATc for 2 days on hTERT host cells grown on 18 mm coverslips. Two coverslips per cell line of interest (one to serve as the experimental and the other as a control) were infected with 2 × 105 parasites in DMEM media and incubated for 24 hours at 37° C. Coverslips were then washed with PBS three times with pre-warmed PBS (pH 7.4) to 37° C. Egress was triggered with 10 µM nigericin from a stock solution made in DMSO and compared with a coverslip treated with DMSO alone as control. Coverslips were incubated at 37° C for 30 minutes. After the incubation, coverslips were fixed with 3.5% paraformaldehyde for 20 min at room temperature. Coverslips were then washed once with PBS and then mounted on a slide. For each sample, the number of intact parasitophorous vacuoles in 10 randomly selected fields was recorded. To determine % of egress the following equation was used:
Only PV’s with 2 or more parasites were counted.
An additional egress assay was performed using 0.01% saponin. HFF cells were seeded onto 35 mm MatTek culture dishes 24 hours prior to use. tdTomato-expressing parasites were pre-grown for 24 hours with ATc in hTERTs host cells, harvested, and used to infect the HFFs previously grown in MatTek culture dishes. After an additional 24 hours (48 hours total with ATc), parasites were assayed using time-lapse microscopy. This was done by removing the DMEM-HA media and replacing it with 2 mL of Ringer buffer with 2 mM calcium. Parasites were equilibrated for 2 minutes before adding 20 µL of a 0.01% saponin solution to the dish. A minimum of 6 PV’s per field of view were assayed and the time for each PV to egress was enumerated. If no PV’s egressed after 20 minutes from the time of saponin addition, those PVs were assigned an egress time of 1,200 s.
Motility was assayed by resuspending parasites in Ringer buffer without calcium (155 mM NaCl, 3 mM KCl, 1 mM MgCl2, 3 mM NaH2PO4, and 10 mM HEPES, and 10 mM dextrose) with 100 µM EGTA. tdTomato-expressing parasites incubated with or without ATc were tested. 24 hours prior to the experiment, 35 mm MatTek dishes were treated with 10% FBS to provide sufficient protein to form a surface conducive to motility. MatTek dishes were washed once with PBS and loaded with 2 mL of Ringer without Ca2+. MatTek dishes were chilled on ice and 5 × 105 parasites were added and allowed to adhere to the surface for 1.5 min. Dishes were then placed in a Zeiss LSM 710 Confocal Microscope environmental chamber set to 37° C. 1.8 mM Ca2+ was added at the indicated time and the motility assay was recorded via time lapse microscopy. Length of trials was manually traced in ImageJ using the MTrackJ plugin. For each trial, the motility of 5 parasites from 4 independent trials per strain (iΔvha1-HA-ATc, iΔvha1-HA +ATc, and iΔvha1-HA-CM+ATc) was quantified and averaged. The length of parasite movement is reported in µm.
Yeast Transformation
A colony of yeast grown on yeast peptone dextrose (YPD) agar plates was inoculated in 10 mL of YPD liquid media overnight at 30° C. 1 mL of the overnight culture was used to inoculate 10–15 mL of fresh YPD media at 30° C. Yeast were grown until the OD600 reached 1.5–2.0. Yeast were centrifuged and the pellet resuspended in 200 µL of 0.1 M lithium acetate in water for 15 s and centrifuged. Yeast were resuspended in 80 µL of 0.1 M lithium acetate and the volume adjusted to 100 µl, which was split into two 50 µL tubes. Each 50 µL tube was centrifuged and the pellets resuspended in the following recipes:
| Experimental Tube | Control Tube |
|---|---|
| 240 μL 50% sterile PEG3350 | 240 μL 50% sterile PEG3350 |
| 36 μL 1 M Lithium Acetate | 36 μL 1 M Lithium Acetate |
| 50 μL Salmon sperm DNA (2 mg/mL) | 50 μL Salmon sperm DNA (2 mg/mL) |
| DNA (100–500 ng) plus water to 34 μL | 34 μL water (no DNA) |
Tubes were vortexed for 1 min and incubated first at 30° C for 30 min and then at 42° C for 30 min. Yeast were plated on Complete Supplement Mixture medium lacking uracil (CSM-ura) plates at 30° C for 3 days.
Yeast complementation
The yeast mutant vph1Δstv1Δ, that does not express either of the V-ATPase a subunits was used for functional complementation studies because these mutants are unable to grow a neutral pH. Primers 24–25 and 28–29 (Table S1) were used to clone vha1-HA or vha2 cDNA, respectively, into the pYES2NT/c yeast galactose inducible plasmid by restriction digest. Plasmids were transformed into WT and vph1Δstv1Δ yeast and plated on CSM-ura pH 6.5 agar plates. Colonies were analyzed via PCR for the presence of correct inserts. For inducible expression analysis, yeasts were grown in CSM-ura media at pH 5.5 with 2% galactose or glucose.
Yeast were grown in YPD broth or CSM-ura media and normalized to OD600. Yeast grown in media were pelleted to remove old media and then resuspended in 200 µL lysis buffer (0.1 M NaOH, 0.05 M EDTA, 2% SDS, with 2% 2-Mercaptoethanol) and boiled for 10 min. After boiling, 5 µL of 4 M acetic acid was added and the suspension was vortexed for 30 s. Lysed yeast were boiled again for 10 minutes and 50 µL of loading buffer was added (0.25 M Tris-HCl, 50% glycerol, 0.05% Bromophenolblue). 20 µg (quantified via nanodrop) of lysate was run on a 10% SDS-PAGE gel for western analysis with rat αHA (1:200) antibodies. To test for complementation of growth, strains were grown on CSM-ura media at pH 7.0 with 2% galactose for 48 (liquid) or 96 h (plates) at 30° C. For growth in liquid CSM-ura, yeast strains were normalized to an OD600 of 1 and diluted 1:10 in liquid media in triplicate. Plates were read in a BioTek Synergy H1 hybrid tester every hour for 48 h under high orbital shaking incubated at 37° C. For growth on agar plates, yeast strains first grown in suspension were normalized to an OD600 of 1 and serially diluted 1:10 on the plates and the experiment repeated three times. Plates were photographed 96 h after inoculation. For the localization of vha1 in yeast, primers 28–31 were used to amplify vha1 or vph1 cDNA to clone via yeast gap repair into the pGO36 plasmid (O’Brien et al., 2015) which will fuse GFP to the gene. Yeast were grown on CSM-ura plates for 4 days and colonies were isolated.
pHi Measurements
Intracellular pH (pHi) of iΔvha1-HA and iΔvha1-HA–CM grown plus and minus ATc was determined fluorometrically by loading parasites with 2′,7′-Bis(2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester (BCECF-AM) (Thermo Fisher Scientific). Purified extracellular tachyzoites were resuspended at a final density of 1 × 109/ mL and loaded with 9 µM BCECF-AM in BAG containing 1.5% sucrose at 37° C for 20 min. At the end of the loading time parasites were washed twice with BAG and resuspended to a final concentration of 1 X 109 cells/mL in the same buffer and kept on ice protected from light. For fluorescence measurements, a 50 µL aliquot of the cell suspension was diluted into 2.45 mL of standard buffer (135 mM NaCl, 5 mM KCl, 1 mM MgSO4, 1 mM CaCl2, 5 mM glucose and 10 mM HEPES/Tris, pH 7.4) to a final density of 2 × 107 cells/mL. The cell suspension was allowed to equilibrate for 2.5 min. in a cuvette before being placed in an Hitachi F-7000 fluorescence spectrophotometer (Hitachi High Technologies). Fluorescence ratios were calculated with excitations at 505 and 440 nm and emission at 530 nm. A standard curve was created using parasites in high potassium standard buffer (140 mM KCl, 1 mM MgSO4, 1 mM CaCl2, 5 mM glucose and 10 mM HEPES/Tris, pH 7.4) at pH’s ranging from 5.5 to 8 (in 0.5 pH increments) and by adding 5.2 µM of nigericin to the cell suspension. The pH of the buffer and the fluorescence value reached after the stabilization of the trace was used for building the standard curve. The standard curve was then used to determine pHis. To determine the effect of changing extracellular pH (pHe) on pHi, iΔvha1-HA + and ATc loaded with BCECF-AM were incubated in standard buffer at the indicated varying pHe. For the recovery experiments, loaded parasites in suspension were exposed to 10 mM propionic acid, which was added to the cuvette at the indicated time.
Proton Extrusion Measurements
Proton extrusion was determined fluorometrically by suspending parasites in a weakly buffered solution containing the free form of BCECF (Thermo Fisher Scientific). A 100 µL aliquot of a cell suspension (at 1 × 109 cell/mL) was diluted into 2.45 mL of a weak buffer (135 mM NaCl, 5 mM KCl, 1 mM MgSO4, 1 mM CaCl2, 0.1 mM HEPES, and 0.1 mM Tris-Base) with 0.38 µM BCECF free acid. The cell suspension was added to a cuvette and placed in a Hitachi F-7000 fluorescence spectrophotometer. 25 mM glucose was added to stimulate metabolic activity and proton extrusion. A standard curve of known pH’s was used to determine the change of extracellular pH.
Membrane Potential Measurements
Membrane potential was measured fluorometrically with bisoxonol. Bisoxonol (0.2 µM) was added to 5 × 107 cells suspended in 2.5 mL of standard buffer at pH 7.5 or 7.0 at 37° C and the fluorescence excitation was set at 540 nm and emission at 580 nm. The cell suspension was added to a cuvette and placed in a Hitachi F-7000 fluorescence spectrophotometer. The last 100 s of the tracing were averaged at each pH and the ratio of pH 7.5/7.0 determined.
Lysotracker Measurements
Purified parasites (at a concentration of 1 × 108) were loaded with 10 mM Lysotracker red by incubating them for 30 min at 37° C in BAG, washed twice with BAG, and placed in 35 mm MatTek dishes in Ringer buffer. For quantification of Lysotracker stained compartments, the number of vacuoles from 10 fields of view with at least 5 parasites was tallied. To minimize bias, counting was performed double blinded.
Microneme Secretion
Parasites were collected from a < 10% lysed culture, passed through a 23 gauge needle, filtered through 3 µm nucleopore membrane, pelleted, and resuspended in ice cold invasion media (20 mM HEPES in DMEM-HG without serum). For constitutive expression, 4 × 108 parasites were incubated at 37° C in a shaking water bath for 30 min. Parasites were pelleted and the supernatant was collected and used for western analysis. For induced expression, 4 × 108 parasites were incubated in microneme secretion buffer (9.8 mL of invasion media with 200 mL of 10% ethanol), incubated in a shaking 37° C water bath for 2 minutes, and the supernatant was collected. The supernatants and lysate were run on a 12.5% SDS-PAGE gel for 45 minutes at 100 V. Immunoblots were probed with αMIC2 (1:4,000), αAMA1 (1:500), and αGRA1 (1:1,000).
Microneme Maturation
M2AP and MIC3 maturation were measured by using an antibody created against the propeptide part of M2AP (Harper et al., 2006) (1:1,000) or against MIC3 (Céréde et al., 2002) (1:40). iΔvha1-HA parasites were incubated with ATc for 0 to 3 days, parasite lysate was generated, and run on a 10% SDS-PAGE gel and probed with αM2AP, αMIC3 or αTubulin (1:30,000). The intensity of the bands obtained in the immunoblots were analyzed using pixel density by ImageJ or Image Studio (where indicated). Quantification was determined either by a ratio of immature/mature or mature/tubulin respectively.
Maturation of proteases
Maturation of CPL was performed using an antibody against CPL, which recognizes both the mature and immature forms. iΔvha1-HA parasites were grown with ATc for 0 to 3 days, lysed, run on an 10% SDS-PAGE gel, which was transferred to a nitrocellulose membrane for western analysis. αCPL at 1:500 was used to identify proCPL from mature CPL. Determination of the mature from immature was done based on the sizes described in (Parussini et al., 2010). For TgSUB2 maturation, aTgSUB2 (1:500) (Miller et al., 2003) was used for westerns as described above. Determination of the mature from immature was done based on sizes described in (Miller et al., 2003). For aspartic protease 3 (ASP3) maturation, the plasmid 5′UPRT-pTub8-Asp3–3Ty-3′UPRT from Dominique Soldati-Favre’s lab (Dogga et al., 2017) was transfected into iΔvha1-HA parasites. αTy1 (1:1,000) was used for westerns of membranes containing lysates generated as above. Determination of the mature from immature was based on sizes described in (Dogga et al., 2017). For all quantifications, pixel densities generated using ImageJ of the propeptide and mature peptide was measured to determine maturation.
Maturation of Rhoptry Proteins
Maturation of rhoptry proteins was analyzed using α-ROP4 (UVT-68, Rabbit 1:500), α-ROP7 (1:1,000), and α-TgCA_RP (Guinea Pig 1:1,000) via western blots which were done similarly to the ones described above for microneme and protease maturations. Quantification of band intensity was performed using signal intensity determined by LI-COR Image Studio Software. Percentage of immature protein was determined by dividing the band intensity of the immature band by the total combined intensity of mature and immature bands.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using GraphPad Prism 6. Unless otherwise noted, all error bars are presented as the standard error of the mean (SEM) and from a minimum of three independent trials. Significant differences were only considered if P values were < 0.05, where *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001. NS designates when the comparison is not statistically significant. Experiment-specific statistical information is provided in the figure legends or associated method details including trials (n), standard error of the mean SEM, and statistical test performed.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| αMIC2 | Vern Carruthers | Harper et al., 2006 |
| αMIC3 | Maryse Lebrun | Céréde et al., 2002 |
| αM2AP | Vern Carruthers | Harper et al., 2006 |
| αproM2AP | Vern Carruthers | Harper et al., 2006 |
| αGRA1 | Vern Carruthers | Huynh and Carruthers, 2006 |
| αCPL | Vern Carruthers | Parussini et al., 2010 |
| αVha1 | This Paper | TgVha1 |
| αTgSUB2 | Kami Kim | Miller et al., 2003 |
| αHA High Affinity, from rat IgG1 | Sigma-Aldrich | Cat#11867423001 |
| αTy1 | Drew Etheridge | Lab Stock |
| αTubulin | Sigma-Aldrich | Cat#T5168 |
| αVP1 | Silvia Moreno | Miranda et al., 2010 |
| αSAG1 | John Boothroyd and Fisher | N/A |
| αROP4 (UVT-68) | Gary Ward | Carey et al., 2004 |
| αROP7 | Peter Bradley | Chasen et al., 2017 |
| αCA_RP | Silvia Moreno | Chasen et al., 2017 |
| αMyc | Sigma-Aldrich | Cat#M5546 |
| Alexa Fluor 350 Antibody | Thermo Fisher Scientific | Cat#A20180 |
| Alexa Fluor 488 Antibody | Thermo Fisher Scientific | Cat#A20181 |
| Alexa Fluor 546 Antibody | Thermo Fisher Scientific | Cat#A20183 |
| α-AMA1 | Dominique Soldati-Favre | Hehl et al., 2000 |
| α-IMC1 | Drew Etheridge | N/A |
| Bacterial and Virus Strains | ||
| Mix and Go | Zymo Research | Cat#T3007 |
| BL21-CodonPlus (DE3)-RILP | VWR | Cat#76193–370 |
| Biological Samples | ||
| TATiDKu80 Toxoplasma gondii | Boris Striepen | Sheiner et al., 2011 |
| vha1-HA, TATiDKu80 Toxoplasma gondii | This work | N/A |
| vhE-HA, TATiDKu80 Toxoplasma gondii | This work | N/A |
| vhG-HA, TATiDKu80 Toxoplasma gondii | This work | N/A |
| vha1-HA, vhG-Ty1; TATiDKu80 Toxoplasma gondii | This work | N/A |
| iΔvha1-HA; TATiDKu80 Toxoplasma gondii | This paper | N/A |
| iΔvha1-HA-CM; TATiDKu80 Toxoplasma gondii | This work | N/A |
| iΔvha1-mNeonGreen; TATiDKu80 Toxoplasma gondii | This work | N/A |
| iΔvha1-GFP; TATiDKu80 Toxoplasma gondii | This work | N/A |
| vph1Δstv1Δ | This work | N/A |
| WT Yeast | Vincent Starai | BY47472 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2h, 7h-bis-(2-carboxyethyl)-5(6)-carboxyfluorescein (BCECF) | Thermo Fisher Scientific | Cat#B1151 |
| 2h, 7h-bis-(2-carboxyethyl)-5(6)-carboxyfluorescein-acetoxymethyl (BCECF-AM) | Thermo Fisher Scientific | Cat#B1170 |
| Anhydrotetracycline (ATc) | Sigma-Aldrich | Cat#1035708 |
| 5-fluorodeoxyuridine (FUDR) | Sigma-Aldrich | Cat#F0503 |
| isopropyl β-D-1-thiogalactopyranoside (IPTG) | Sigma-Aldrich | Cat#I5502 |
| Bafilomycin A1 | Sigma-Aldrich | Cat#B1793 |
| Saponin | Sigma-Aldrich | Cat#47036 |
| Nigericin | Sigma-Aldrich | Cat#N7143 |
| Dulbecco’s modified Eagle medium-High Glucose | Sigma-Aldrich | Cat#D5796 |
| Freund’s Complete Adjuvant | Thermo Fisher Scientific | Cat#77140 |
| Freund’s incomplete adjuvant | Thermo Fisher Scientific | Cat#77145 |
| HisPur Ni-NTA chromatography cartridge | Thermo Fisher Scientific | Cat#90098 |
| 16% Paraformaldehyde | Electron Microscopy Sciences | Cat#15700 |
| Bovine Serum (Calf Serum) | Thermo Fisher Scientific | Cat#16030074 |
| Complete Supplement Mixture medium lacking uracil (CSM-ura) | Sunrise Science | Cat#1004–100 |
| Galactose | Sigma-Aldrich | Cat#B1793 |
| LysoTracker Red DND-99 | Thermo Fisher Scientific | Cat#47036 |
| DiSBAC2(3) (Bis-(1,3-Diethylthiobarbituric Acid)Trimethine Oxonol)(bisoxonol) | Thermo Fisher Scientific | Cat#N7143 |
| Experimental Models: Cell Lines | ||
| Human Foreskin Fibroblast immortalized with hTERT | ATCC | BJ-5ta ATCC® CRL-4001 |
| Human Foreskin Fibroblast (HFF) | ATCC | HFF-1 ATCC® SCRC-1041 |
| Oligonucleotides | ||
| The list of oligonucleotides sequence is shown in Table S1 | N/A | N/A |
| Recombinant DNA | ||
| pLIC-3XHA | Boris Striepen | Sheiner et al., 2011 |
| pLIC-mNeon-Green | This work | N/A |
| pLIC-GFP | This work | N/A |
| ptdTomato overexpression plasmid | Boris Striepen | Chtanova et al., 2008 |
| UPRT Vha1 cDNA shuttle vector | This work | This paper |
| 5′UPRT-pTub8-Asp3–3Ty-3′UPRT | Dominique Soldati-Favre | Dogga et al., 2017 |
| pSAG1-CAS9-U6-sgUPRT | David Sibley | Addgene plasmid #54467 |
| promoter insertion plasmid | Boris Striepen | Sheiner et al., 2011 |
| pGO-GFP | Vincent Starai | OBrien et al., 2015 |
| Software and Algorithms | ||
| FIJI (imageJ) | https://fiji.sc | Version 2.0.0-rc-69/1.52i |
| Graphpad | https://www.graphpad.com | Version 6 |
| Microsoft excel | Microsoft.com | Version 16.22 |
| Microsoft word | Microsoft.com | Version 16.22 |
| Adobe Illustrator | Adobe.com | Version CS6 |
| Adobe Photoshop | Adobe.com | Version CS6 |
| Image Studio | Li-Cor.com | Version 5 |
Highlights.
The V-ATPase localizes to the plasma membrane, PLV, and pro-rhoptries
The V-ATPase supports the maturation of rhoptry and microneme proteins
V-ATPase deficiency leads to accumulation of immature rhoptry and microneme proteins
The function of the V-ATPase impacts every step of the Toxoplasma lytic cycle
ACKNOWLEDGMENTS
We thank Vern Carruthers for anti-MIC2, proM2AP, and CPL antibodies; Maryse Lebrun for anti-MIC3; and Kami Kim for anti-TgSUB2. We thank Dominique Soldati-Favre for the plasmids for C-terminal tagging of ASP3. Melissa Storey generated the Vha1 antibody. We thank the Georgia Electron Microcopy Center for assisting with the EM images, the Biomedical Microscopy Core, Coverdell Center, for the use of their microscopes, and Dr. Kandasamy for assistance. Wandy Betty from Washington University in St. Louis performed the immuno-EM of Figure 1. We thank Zhicheng Dou, Drew Etheridge, and Vern Carruthers for suggestions and reading the manuscript. Funding for this work was provided by the NIH (AI-096836 and AI-128356 to S.N.J.M. and AI-100913 to V.J.S.). N.M.C. was supported by a pre-doctoral fellowship from the American Heart Association (14PRE19100003). A.J.S. was partly funded through a UGA OVPR fellowship as part of T32 Training Grant T32AI060546.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.04.038.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Åkerman KEO (1978). Changes in membrane potential during calcium ion influx and efflux across the mitochondrial membrane. Biochim. Biophys. Acta 502, 359–366. [DOI] [PubMed] [Google Scholar]
- Anderson ED, VanSlyke JK, Thulin CD, Jean F, and Thomas G (1997). Activation of the furin endoprotease is a multiple-step process: requirements for acidification and internal propeptide cleavage. EMBO J 16, 1508–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrizabalaga G, Ruiz F, Moreno S, and Boothroyd JC (2004). Ionophore-resistant mutant of Toxoplasma gondii reveals involvement of a sodium/hydrogen exchanger in calcium regulation. J. Cell Biol 165, 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyenbach KW, and Wieczorek H (2006). The V-type H+ ATPase: molecular structure and function, physiological roles and regulation. J. Exp. Biol 209, 577–589. [DOI] [PubMed] [Google Scholar]
- Black MW, and Boothroyd JC (2000). Lytic cycle of Toxoplasma gondii. Microbiol. Mol. Biol. Rev 64, 607–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges-Pereira L, Budu A, McKnight CA, Moore CA, Vella SA, Hortua Triana MA, Liu J, Garcia CR, Pace DA, and Moreno SN (2015). Calcium signaling throughout the Toxoplasma gondii lytic cycle: a study using genetically encoded calcium indicators. J. Biol. Chem 290, 26914–26926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey KL, Jongco AM, Kim K, and Ward GE (2004). The Toxoplasma gondii rhoptry protein ROP4 is secreted into the parasitophorous vacuole and becomes phosphorylated in infected cells. Eukaryot. Cell 3, 1320–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carruthers VB, Giddings OK, and Sibley LD (1999). Secretion of micronemal proteins is associated with toxoplasma invasion of host cells. Cell. Microbiol 1, 225–235. [DOI] [PubMed] [Google Scholar]
- Céréde O, Dubremetz JF, Bout D, and Lebrun M (2002). The Toxoplasma gondii protein MIC3 requires pro-peptide cleavage and dimerization to function as adhesin. EMBO J 21, 2526–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chasen NM, Asady B, Lemgruber L, Vommaro RC, Kissinger JC, Coppens I, and Moreno SNJ (2017). A glycosylphosphatidylinositol-anchored carbonic anhydrase-related protein of Toxoplasma gondii is important for rhoptry biogenesis and virulence. MSphere 2, e00027–e00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chtanova T, Schaeffer M, Han S-J, van Dooren GG, Nollmann M, Herzmark P, Chan SW, Satija H, Camfield K, Aaron H, et al. (2008). Dynamics of neutrophil migration in lymph nodes during infection. Immunity 29, 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaurex N (2002). pH Homeostasis of cellular organelles. News Physiol. Sci 17, 1–5. [DOI] [PubMed] [Google Scholar]
- Dogga SK, Mukherjee B, Jacot D, Kockmann T, Molino L, Hammoudi P-M, Hartkoorn RC, Hehl AB, and Soldati-Favre D (2017). A druggable secretory protein maturase of Toxoplasma essential for invasion and egress. eLife 6, e27480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald RG, and Roos DS (1995). Insertional mutagenesis and marker rescue in a protozoan parasite: cloning of the uracil phosphoribosyltransferase locus from Toxoplasma gondii. Proc. Natl. Acad. Sci. USA 92, 5749–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Coppens I, and Carruthers VB (2013). Non-canonical maturation of two papain-family proteases in Toxoplasma gondii. J. Biol. Chem 288, 3523–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubremetz JF (2007). Rhoptries are major players in Toxoplasma gondii invasion and host cell interaction. Cell. Microbiol 9, 841–848. [DOI] [PubMed] [Google Scholar]
- El Hajj H, Papoin J, Céréde O, Garcia-Réguet N, Soête M, Dubremetz JF, and Lebrun M (2008). Molecular signals in the trafficking of Toxoplasma gondii protein MIC3 to the micronemes. Eukaryot. Cell 7, 1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgac M (2007). Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol 8, 917–929. [DOI] [PubMed] [Google Scholar]
- Fox BA, Ristuccia JG, Gigley JP, and Bzik DJ (2009). Efficient gene replacements in Toxoplasma gondii strains deficient for nonhomologous end joining. Eukaryot. Cell 8, 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawlik K, Shiryaev SA, Zhu W, Motamedchaboki K, Desjardins R, Day R, Remacle AG, Stec B, and Strongin AY (2009). Autocatalytic activation of the furin zymogen requires removal of the emerging enzyme’s N-terminus from the active site. PLoS ONE 4, e5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JM, Huynh MH, Coppens I, Parussini F, Moreno S, and Carruthers VB (2006). A cleavable propeptide influences Toxoplasma infection by facilitating the trafficking and secretion of the TgMIC2-M2AP invasion complex. Mol. Biol. Cell 17, 4551–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehl AB, Lekutis C, Grigg ME, Bradley PJ, Dubremetz J-F, Ortega-Barria E, and Boothroyd JC (2000). Toxoplasma gondii homologue of plasmodium apical membrane antigen 1 is involved in invasion of host cells. Infect. Immun 68, 7078–7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe HC, Ngô HM, Yang M, and Joiner KA (2000). Targeting to rhop-try organelles of Toxoplasma gondii involves evolutionarily conserved mechanisms. Nat. Cell Biol 2, 449–456. [DOI] [PubMed] [Google Scholar]
- Hortua Triana MA, Márquez-Nogueras KM, Vella SA, and Moreno SNJ (2018). Calcium signaling and the lytic cycle of the Apicomplexan parasite Toxoplasma gondii. BBA Mol. Cell Res 1865, 1846–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado-Lorenzo A, Skinner M, El Annan J, Futai M, Sun-Wada G-H, Bourgoin S, Casanova J, Wildeman A, Bechoua S, Ausiello DA, et al. (2006). V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat. Cell Biol 8, 124–136. [DOI] [PubMed] [Google Scholar]
- Huynh MH, and Carruthers VB (2006). Toxoplasma MIC2 is a major determinant of invasion and virulence. PLoS Pathog 2, e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh MH, and Carruthers VB (2009). Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot. Cell 8, 530–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura H, and Inouye M (1988). In vitro processing of pro-subtilisin produced in Escherichia coli. J. Biol. Chem 263, 12959–12963. [PubMed] [Google Scholar]
- Kafsack BF, Beckers C, and Carruthers VB (2004). Synchronous invasion of host cells by Toxoplasma gondii. Mol. Biochem. Parasitol 136, 309–311. [DOI] [PubMed] [Google Scholar]
- Koivusalo M, Welch C, Hayashi H, Scott CC, Kim M, Alexander T, Touret N, Hahn KM, and Grinstein S (2010). Amiloride inhibits macropino-cytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol 188, 547–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng XH, Nishi T, and Forgac M (1999). Transmembrane topography of the 100-kDa a subunit (Vph1p) of the yeast vacuolar proton-translocating ATPase. J. Biol. Chem 274, 14655–14661. [DOI] [PubMed] [Google Scholar]
- Liu J, Pace D, Dou Z, King TP, Guidot D, Li Z-H, Carruthers VB, and Moreno SNJ (2014). A vacuolar-H+-pyrophosphatase (TgVP1) is required for microneme secretion, host cell invasion, and extracellular survival of Toxo-plasma gondii. Mol. Microbiol 93, 698–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolson MF, Wu B, Proteau D, Taillon BE, Roberts BT, Hoyt MA, and Jones EW (1994). STV1 gene encodes functional homologue of 95-kDa yeast vacuolar H+-ATPase subunit Vph1p. J. Biol. Chem 269, 14064–14074. [PubMed] [Google Scholar]
- Miller SA, Thathy V, Ajioka JW, Blackman MJ, and Kim K (2003). TgSUB2 is a Toxoplasma gondii rhoptry organelle processing proteinase. Mol. Microbiol 49, 883–894. [DOI] [PubMed] [Google Scholar]
- Miranda K, Pace DA, Cintron R, Rodrigues JCF, Fang J, Smith A, Rohloff P, Coelho E, de Haas F, de Souza W, et al. (2010). Characterization of a novel organelle in Toxoplasma gondii with similar composition and function to the plant vacuole. Mol. Microbiol 76, 1358–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno SN, Zhong L, Lu H-G, Souza WDE, and Benchimol M (1998). Vacuolar-type H+-ATPase regulates cytoplasmic pH in Toxoplasma gondii tachyzoites. Biochem. J 330, 853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngô HM, Yang M, Paprotka K, Pypaert M, Hoppe H, and Joiner KA (2003). AP-1 in Toxoplasma gondii mediates biogenesis of the rhoptry secretory organelle from a post-Golgi compartment. J. Biol. Chem 278, 5343–5352. [DOI] [PubMed] [Google Scholar]
- Ngô HM, Yang M, and Joiner KA (2004). Are rhoptries in Apicomplexan parasites secretory granules or secretory lysosomal granules? Mol. Microbiol 52, 1531–1541. [DOI] [PubMed] [Google Scholar]
- Nishi T, and Forgac M (2002). The vacuolar H+-ATPases—nature’s most versatile proton pumps. Nat. Rev. Mol. Cell Biol 3, 94–103. [DOI] [PubMed] [Google Scholar]
- O’Brien KM, Lindsay EL, and Starai VJ (2015). The Legionella pneumophila effector protein, LegC7, alters yeast endosomal trafficking. PLoS ONE 10, e0116824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace DA, Fang J, Cintrón R, Docampo MD, and Moreno SNJ (2011). Overexpression of a cytosolic pyrophosphatase (TgPPase) reveals a regulatory role of PP(i) in glycolysis for Toxoplasma gondii. Biochem. J 440, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace DA, McKnight CA, Liu J, Jimenez V, and Moreno SNJ (2014). Calcium entry in Toxoplasma gondii and its enhancing effect of invasion-linked traits. J. Biol. Chem 289, 19637–19647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parussini F, Coppens I, Shah PP, Diamond SL, and Carruthers VB (2010). Cathepsin L occupies a vacuolar compartment and is a protein maturase within the endo/exocytic system of Toxoplasma gondii. Mol. Microbiol 76, 1340–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perzov N, Padler-Karavani V, Nelson H, and Nelson N (2002). Characterization of yeast V-ATPase mutants lacking Vph1p or Stv1p and the effect on endocytosis. J. Exp. Biol 205, 1209–1219. [DOI] [PubMed] [Google Scholar]
- Roiko MS, Svezhova N, and Carruthers VB (2014). Acidification activates Toxoplasma gondii motility and egress by enhancing protein secretion and cytolytic activity. PLoS Pathog 10, e1004488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saroussi S, and Nelson N (2009). Vacuolar H+-ATPase—an enzyme for all seasons. Pflugers Arch 457, 581–587. [DOI] [PubMed] [Google Scholar]
- Shaw MK, Roos DS, and Tilney LG (1998). Acidic compartments and rhoptry formation in Toxoplasma gondii. Parasitology 117, 435–443. [DOI] [PubMed] [Google Scholar]
- Sheiner L, Demerly JL, Poulsen N, Beatty WL, Lucas O, Behnke MS, White MW, and Striepen B (2011). A systematic screen to discover and analyze apicoplast proteins identifies a conserved and essential protein import factor. PLoS Pathog 7, e1002392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde U, Fu X, and Inouye M (1999). A pathway for conformational diversity in proteins mediated by intramolecular chaperones. J. Biol. Chem 274, 15615–15621. [DOI] [PubMed] [Google Scholar]
- Sidik SM, Huet D, Ganesan SM, Huynh M-H, Wang T, Nasamu AS, Thiru P, Saeij JPJ, Carruthers VB, Niles JC, and Lourido S (2016). A genome-wide CRISPR screen in Toxoplasma identifies essential apicomplexan genes. Cell 166, 1423–1435.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smardon AM, Tarsio M, and Kane PM (2002). The RAVE complex is essential for stable assembly of the yeast V-ATPase. J. Biol. Chem 277, 13831–13839. [DOI] [PubMed] [Google Scholar]
- Soldati D, Dubremetz JF, and Lebrun M (2001). Microneme proteins: structural and functional requirements to promote adhesion and invasion by the api-complexan parasite Toxoplasma gondii. Int. J. Parasitol 31, 1293–1302. [DOI] [PubMed] [Google Scholar]
- Spurr AR (1969). A low-viscosity epoxy resin embedding medium for electron microscopy. J. Ultrastruct. Res 26, 31–43. [DOI] [PubMed] [Google Scholar]
- Szecsi PB (1992). The aspartic proteases. Scand. J. Clin. Lab. Invest. Suppl 210, 5–22. [PubMed] [Google Scholar]
- Toei M, Saum R, and Forgac M (2010). Regulation and isoform function of the V-ATPases. Biochemistry 49, 4715–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomavo S, Slomianny C, Meissner M, and Carruthers VB (2013). Protein trafficking through the endosomal system prepares intracellular parasites for a home invasion. PLoS Pathog 9, e1003629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turetzky JM, Chu DK, Hajagos BE, and Bradley PJ (2010). Processing and secretion of ROP13: a unique Toxoplasma effector protein. Int. J. Parasitol 40, 1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal K, Werkmeister E, Barois N, Saliou J-M, Poncet A, Huot L, Sindikubwabo F, Hakimi MA, Langsley G, Lafont F, and Marion S (2017). Dual role of the Toxoplasma gondii clathrin adaptor AP1 in the sorting of rhoptry and microneme proteins and in parasite division. PLoS Pathog 13, e1006331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Toei M, and Forgac M (2008). Analysis of the membrane topology of transmembrane segments in the C-terminal hydrophobic domain of the yeast vacuolar ATPase subunit a (Vph1p) by chemical modification. J. Biol. Chem 283, 20696–20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichroski MJ, Melton JA, Donahue CG, Tweten RK, and Ward GE (2002). Clostridium septicum alpha-toxin is active against the parasitic protozoan Toxoplasma gondii and targets members of the SAG family of glycosylphosphatidylinositol-anchored surface proteins. Infect. Immun 70, 4353–4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.