

Soil microbiome composition and functioning determine the outcome of plant-pathogen interactions under natural field conditions.
Abstract
Plant-pathogen interactions are shaped by multiple environmental factors, making it difficult to predict disease dynamics even in relatively simple agricultural monocultures. Here, we explored how variation in the initial soil microbiome predicts future disease outcomes at the level of individual plants. We found that the composition and functioning of the initial soil microbiome predetermined whether the plants survived or succumbed to disease. Surviving plant microbiomes were associated with specific rare taxa, highly pathogen-suppressing Pseudomonas and Bacillus bacteria, and high abundance of genes encoding antimicrobial compounds. Microbiome-mediated plant protection could subsequently be transferred to the next plant generation via soil transplantation. Together, our results suggest that small initial variation in soil microbiome composition and functioning can determine the outcomes of plant-pathogen interactions under natural field conditions.
INTRODUCTION
Crop loss to plant pathogens is an ever-increasing threat for agricultural production. While production demands increase, there is a pressing need to reduce the use of environmentally unfriendly pesticides and agrochemicals (1). To achieve this, manipulation of the plant microbiomes has been suggested as a viable alternative to reduce disease levels by, for example, improving microbe-mediated pathogen suppression (2) and priming the plant immune system (3). However, we still poorly understand how the complex interactions between plants, their pathogens, and soil microbiota determine disease development under natural field conditions. In particular, separating stochastic and deterministic drivers is challenging because disease outcomes are affected by multiple factors including population density dynamics (4), the genetic background of the pathogen (5) and the host (6), temporal and spatial variation in biotic and abiotic conditions (7), and the composition and assembly of the host-associated microbiome (8). Considerable spatial variation in disease dynamics exists even in relatively homogenous agricultural monocultures that are exposed to similar stochastic and temporal environmental variation locally (9, 10). This suggests that local spatial variation within agricultural fields could be important in determining the outcome of plant-pathogen interactions, thereby raising the question “To what extent does the initial microbiome composition and functioning determine variation in disease incidence across plant individuals?”
To examine this question, we used a newly developed rhizobox system that allows for nondestructive repeated sampling of individual plants (tomatoes in this case) at different growth stages under natural field conditions (Fig. 1A and fig. S1A). Although repeated sampling has previously been applied for studying plant-soil interactions (11), nondestructive repeated sampling of plant rhizosphere microbiomes enables sampling of the same plant individuals in time. This time series approach is similar to medical cohort studies, where individuals are repeatedly sampled at distinct time scales throughout their life cycle (12). We used soil-borne plant pathogenic Ralstonia solanacearum bacterium as our focal pathogen and tomato plants as model hosts on a field that had been naturally infected by R. solanacearum for more than15 years. This pathogen is a causal agent of bacterial wilt disease that is able to infect numerous economically important crops and is a very severe pathogen globally and especially in China and Asia (13). Three replicate plots with 16 tomato plants in each were established under natural field conditions, and each plant was grown in a rhizobox (fig. S1B) filled with local soil to retain natural physicochemical conditions and microbial community structure. Three individual rhizosphere samples were nondestructively collected from each plant at every sampling time point (Fig. 1B and movie S1), including initial soil microbiome samples before planting of tomatoes (to assess the local soil species pool including pathogen without plant influence) and spanning the full vegetative period to track changes in microbiome assembly and pathogen growth at the level of individual plants (Fig. 1, C and D). All samples were kept frozen (−80°C), and soil collection was terminated at week 6 after disease dynamics were allowed to stabilize for one additional week. The number of healthy and diseased plants found in different replicate plots varied, and the average disease incidence reached 54% by the end of the experiment (26 wilted, 12 healthy, and 10 latently infected plants; fig. S1, B to D). Bacterial wilt is commonly characterized by symptomless latent infections (14) due to differences in the development of bacterial wilt disease or due to variation in systemic plant resistance induced by soil microbiota (15). In this study, we focused only on clearly determinable disease outcomes and hence randomly selected 12 plants that remained healthy and 12 plants that succumbed to disease for further analysis. We aimed to answer three main questions: (i) to what extent do initial differences in rhizosphere bacterial communities predict the future disease outcomes? (ii) Does bacterial community assembly during the tomato plant growth affect disease development? (iii) What are the underlying taxonomic and functional differences between diseased and healthy rhizosphere microbiomes? To achieve these aims, we analyzed all samples repeatedly collected from these plants throughout the field experiments, and linked final disease outcomes to initial microbiome composition, subsequent bacterial community assembly, and pathogen and bacterial density dynamics. At the beginning of the experiment, we also analyzed the initial physicochemical properties of soil samples in each rhizobox.
Fig. 1. Schematic figure of the rhizobox sampling system and the experimental design.
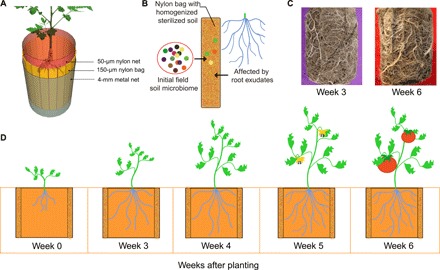
(A) The rhizobox consisted of a three-layer cylinder with a height of 136 mm and a diameter of 110 mm. The inner layer (root compartment) is made of a 50-μm nylon mesh net, which prevents roots from entering into the middle layer, and the outer layer is made of a 4-mm metal mesh to support the rhizobox. (B) The middle sampling layer consisted of 18 individual nylon mesh bags (150 μm nylon mesh; height, 136 mm; width, 18 to 21 mm; thickness, 1 to 2 mm) containing homogenized and sterilized field soil. The soil in the nylon mesh bags of the middle layer was thus in close contact with plant roots and root exudates and was used as a proxy of rhizosphere bacterial community. (C) The central root compartment was densely colonized by plant roots after 3 weeks of transplantation of tomato seedlings to the field (photo credit: Yian Gu, Nanjing Agricultural University). (D) Initial bulk soil was collected when the experiment was conducted (0 weeks), and three middle-layer nylon bags from each rhizobox were randomly collected at every sampling time point (3, 4, 5, and 6 weeks after planting) to sample the rhizosphere soil.
RESULTS
Linking initial differences in microbiomes with future disease outcomes
The initial variation in pathogen abundances, soil physicochemical properties, total bacterial densities, bacterial community diversity (table S1 and fig. S2), or plant location within plots (fig. S3) could not predict whether plants remained healthy or became infected by the pathogen over the course of the experiment. However, the initial soil bacterial community composition clustered into two distinct groups that corresponded well with plants that remained healthy or became diseased later during the experiment [analysis of molecular variance (AMOVA) on unweighted UniFrac, P < 0.001, Fig. 2B]. In particular, future plant survival was associated with a set of rare bacterial taxa present in the initial microbiome (figs. S4 and S5A), and a total of 46 and 53 unique operational taxonomic units (OTUs) were found to be closely associated with plants that remained healthy and became diseased, respectively (figs. S5, B and C, and S6). Of these discriminating OTUs, the initial soils that were later associated with healthy plants had higher abundances of Alphaproteobacteria, Firmicutes, and Cyanobacteria, while initial soils that were later associated with diseased plants had higher abundances of Acidobacteria, Actinobacteria, and Verrucomicrobia (week 0; Fig. 2C and data file S1). The co-occurrence networks of healthy plants were initially much larger, having higher number of nodes and edges, longer average path lengths, and higher modularity compared to the initial networks of diseased plants (Fig. 2D and table S2). On the basis of “NetShift” analysis, we were able to link Massilia, Dyadobacter, Terrabacter, Arachidicoccus, and Dyella genera as potential keystone taxa behind pathogen suppression in the initial microbiomes of healthy plants (Fig. 2E). At the functional level, plants that remained healthy were initially associated with microbiomes with relatively high levels of functional genes (Fig. 2F) linked to nonribosomal peptide and polyketide synthases (Fig. 2G and data file S2). These functions are responsible for the production of potent antimicrobial compounds, which may either directly inhibit the pathogen or contribute to the assembly of a pathogen-suppressing microbiome (16). Bacillus, Pseudomonas, and Actinobacteria represent the best-described producers of nonribosomal antimicrobial peptides and polyketides (17, 18). However, their abundances were not correlated with the relative abundance of these functional genes. To explore potential differences in the production of antimicrobials by these bacteria, we used semiselective media to isolate Bacillus and Pseudomonas from the initial soils that later became associated with healthy and diseased plants. We found that the total abundance of these two bacterial genera did not differ depending on the future plant survival (fig. S7). However, the initial microbiomes of surviving plants were associated with Bacillus and Pseudomonas isolates that were much more effective at inhibiting the pathogen (Fig. 2H). Together, these culture-dependent and culture-independent techniques demonstrate that small functional differences in the initial soil microbiomes can predetermine whether the plants later survive or succumb to bacterial wilt disease.
Fig. 2. The outcome of plant-pathogen interaction is associated with the initial soil microbiome composition and functioning.
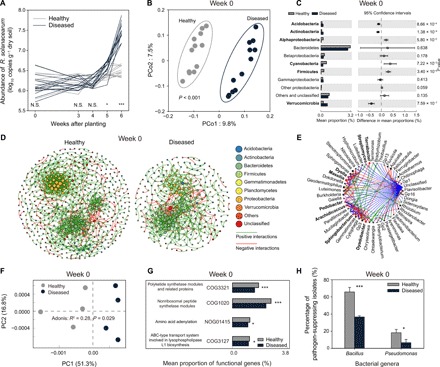
(A) The population dynamics of R. solanacearum bacterial pathogen in the rhizosphere of healthy and diseased plants. (B) The initial soil microbiomes associated with healthy and diseased plants were clearly distinct (F1,22 = 2.3, P < 0.001, AMOVA on unweighted UniFrac); the percentage of explained variation is shown on the x and y axes. (C) Differences in the abundances of rare discriminating OTUs (linear discriminant analysis score ≥ 2, fold change ≥ 2, and significance test P < 0.05) in the initial soils (week 0) that later became associated with healthy and diseased plants. P values were calculated using Student’s t test (P < 0.05), and significantly associated phyla are highlighted in bold. (D) Co-occurrence networks of initial microbiomes that later became associated with healthy (left) and diseased plants (right). (E) Potential “driver taxa” behind pathogen suppression based on bacterial network analysis of initial microbiomes that later became associated with healthy and diseased plants. Node sizes are proportional to their scaled NESH (neighbor shift) score (a score identifying important microbial taxa of microbial association networks), and a node is colored red if its betweenness increases when comparing soil microbiomes associated with diseased to healthy plants. As a result, large red nodes denote particularly important driver taxa behind pathogen suppression, and these taxa names are shown in bold. Line colors indicate node (taxa) connections as follows: association present only in healthy plant microbiomes (red edges), association present only in diseased plant microbiomes (green edges), and association present in both healthy and diseased plant microbiomes (blue edges). (F) Distinct functional gene profiles associated with the initial microbiomes of future healthy and diseased plants. (G) The abundance of representative genes related to secondary metabolism synthesis in the initial soil microbiomes of future healthy and diseased plants. (H) The percentage of R. solanacearum pathogen-suppressing Bacillus and Pseudomonas bacteria isolated from the initial soil microbiomes of healthy and diseased plants (pairwise t test, mean ± SD, n = 3; N.S., nonsignificant; *P < 0.05; ***P < 0.001).
Tracking changes in microbiome composition, diversity, and functioning during plant growth and comparing soil suppressiveness in the second plant generation
We next explored whether the initial differences in the taxonomic and functional soil microbiome composition changed during the plant development (Fig. 3). We found that the healthy and diseased plants harbored distinct microbiomes throughout the experiment (fig. S8), even though the microbiome composition tended to converge toward the end of the experiment (Fig. 3A), possibly due to plant root exudate–induced changes in microbiome composition and modification of plant endosphere (19, 20). Most of the rare taxa associated with the initial soil microbiome of healthy plants were rapidly replaced, while the rare taxa associated with the initial soil microbiome of diseased plants persisted or even increased in abundance toward the end of the experiment (Fig. 3B and figs. S9 and S10). This suggests that initially rare discriminating taxa likely played a transient role in healthy plant microbiomes and a more persistent role in diseased plant microbiomes. Rare taxa associated with healthy plants could have driven priority effects in microbiome assembly during the early stages of plant development (21), either leading to more suppressive bacterial communities (22) or by priming the plant immune system (20). In contrast, the rare taxa associated with diseased plants could have even facilitated the pathogen, thereby potentially promoting infections (23, 24). Further studies are, however, needed to directly test and verify these hypotheses. At later phases of plant development, higher abundances of discriminating Acidobacteria and unclassified bacterial taxa were found in healthy plant microbiomes and higher abundances of discriminating Actinobacteria, Bacteroidetes, and Betaproteobacteria taxa were found in diseased plant microbiomes (week 6; Fig. 3C and data file S3). Similar to initial soil samples, healthy plant microbiomes were still associated with larger co-occurrence networks with higher number of nodes and edges and longer average path lengths compared to diseased plants (Fig. 3D and table S2). However, differences between the networks were less pronounced at the end of the experiment compared to the initial samples. On the basis of NetShift analysis, Gp16 (Acidobacteria), Spartobacteria_genera_incertae_sedis, Dyella, Hyphomicrobium, and Sphingomonas were identified as keystone genera associated with pathogen suppression in the healthy plant microbiomes (Fig. 3E). We also found that healthy and diseased plant microbiomes converged from a perspective of functional genes over the course of the experiment (Fig. 3F and fig. S11). However, some specific functional characteristics, such as the high abundance of polyketide synthases genes, remained higher in healthy plant microbiomes throughout the experiment (Fig. 3G and data file S4), which suggests that they potentially retained their capability to suppress the pathogen.
Fig. 3. Taxonomic and functional differences between healthy and diseased plant microbiomes persist throughout the tomato growth season and can be transferred to the next plant generation via soil transplantation.
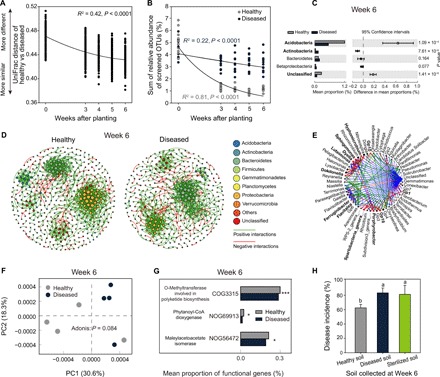
(A) Decay in the phylogenetic distance (unweighted UniFrac distance) between microbiomes associated with healthy and diseased plants. (B) Temporal dynamics of the relative abundance of rare OTUs enriched in the initial microbiomes of healthy and diseased plants. (C) Differences in the abundances of rare discriminating OTUs (linear discriminant analysis score ≥ 2, fold change ≥ 2, and significance test P < 0.05) associated with healthy and diseased plant microbiomes at the end of the experiment (week 6). P values were calculated using Student’s t test (P < 0.05), and significantly associated phyla are highlighted in bold. (D) Co-occurrence networks associated with healthy (left) and diseased plants (right) at the end of the experiment (week 6). (E) Potential driver taxa behind pathogen suppression based on bacterial network analysis of healthy and diseased plant microbiomes at the end of the experiment (week 6). Node sizes are proportional to their scaled NESH score (a score identifying important microbial taxa of microbial association networks), and a node is colored red if its betweenness increases when comparing soil microbiomes associated with diseased to healthy plants. As a result, large red nodes denote particularly important driver taxa behind pathogen suppression, and these taxa names are shown in bold. Line colors indicate node (taxa) connections as follows: association present only in healthy plant microbiomes (red edges), association present only in diseased plant microbiomes (green edges), and association present in both healthy and diseased plant microbiomes (blue edges). (F) Distinct functional gene profiles associated with healthy and diseased plant microbiomes at the end of the experiment (week 6). (G) The abundance of representative genes related to secondary metabolism synthesis of healthy and diseased plant microbiomes at the end of the experiment (week 6). (H) The disease incidence in the second plant generation after transplantation of soil from the first-generation healthy, diseased, or sterilized healthy soil (mean ± SD, n = 4). Different lowercase letters denote significance at P < 0.05 (Duncan’s multiple range test).
To confirm this, we tested whether the microbial communities isolated at the end of the experiment could provide protection against the pathogen in the next plant generation by conducting “soil transplantation” experiments. We found that soils derived from the “first-generation” healthy plants had 20.8% lower disease incidence compared to soils derived from the first-generation diseased plants (P = 0.002, t = −5, df = 6, Student’s t test; Fig. 3H). No significant difference (P = 0.78, t = 0.29, df = 6, Student’s t test; Fig. 3H) in disease incidence was observed between soils derived from the first-generation diseased plants (75 to 91.7% of disease incidence) and healthy plant soils that had their microbiome removed by sterilization (75 to 100% of disease incidence). Together, these results suggest that the initial differences in soil microbiome composition and functioning persisted throughout the plant development and could be carried over to subsequent plant generations.
DISCUSSION
Host-associated microbiomes have repeatedly been observed to vary between healthy and diseased individuals with plants and humans (25, 26). It has, however, remained unclear whether these differences are the cause or consequence of disease development. Previous studies on mammals have proposed that early-life microbiomes may be connected with the future health of individuals (12). However, direct, causal evidence is still scarce (27, 28). By using a repeated sampling of individual tomato plants that either remained healthy or got infected by soil-borne plant pathogenic R. solanacearum bacterium, we demonstrate that initial differences in the local bacterial species pool can predetermine the outcome of host-pathogen interactions long before the onset of disease outbreaks.
We found no difference in the soil physicochemical properties between initial microbiomes that later became associated with healthy and diseased plants. This suggests that local variation in abiotic environmental conditions within replicate plots was an unlikely driver of differential disease outcomes. Initial community assembly could also have been affected by priority effects during early stages of the experiment when rhizoboxes were transferred to the field (29). However, we did not find phylum-level differences between the initial bulk soil samples and 5-day-old microbiome samples, which originally contained soil sterilized by gamma radiation (fig. S12A). This suggests that rhizoboxes were rapidly colonized by bacteria from the surrounding local environment. While priority effects were not observed to play an important role for the initial microbiome assembly, the microbiomes of 5-day-old samples clustered between soils that later became associated with healthy and diseased plants (fig. S12B), indicative of substantial and strong initial differences. Together, these results suggest that bacterial wilt disease development was highly predictable based on initial differences in bacterial community composition and functional capacity even under temporally varying field conditions.
Even though no clear differences were found at the phylum level, we found that certain taxa could clearly discriminate between plant microbiomes that later became associated with healthy and diseased plants. Specifically, previous studies have linked these taxa to pathogen suppression (7, 30) and, for example, Actinobacteria has been shown to be linked to bacteria-mediated suppression of Fusarium wilt at a continental scale (31). One potential mechanism behind the suppression could be the production of antibiotics, and in support of this, we found that initial microbiomes associated with healthy plants had high levels of functional genes linked to nonribosomal peptide and polyketide synthases. While the abundance of the genes was not significantly associated with any specific taxa, they were linked to more antagonistic Bacillus and Pseudomonas bacteria isolated from the initial microbiome samples that were later associated with healthy plants. We also found that certain other bacterial taxa (Massilia, Dyadobacter, Terrabacter, Arachidicoccus, and Dyella genera) could have played a key role in driving the infection in the rhizosphere. Even though we could not isolate these taxa, it has previously been shown that Massilia and Dyadobacter have been associated with soils that can suppress Fusarium wilt (32, 33), while Terrabacter and Dyella have been linked to the suppression of wheat diseases (34, 35). These clear differences in microbiome composition and functioning were not correlated with differences in the initial pathogen abundances, which became clearer only toward the very end of the experiment. One explanation for this finding is that bacteria were less active in the beginning of the experiment when the tomatoes were still relatively small, providing only a small amount of root exudate for bacterial growth and secondary metabolism (36).
We found that initial differences in the composition of microbiomes associated with healthy and diseased plants persisted throughout the field experiment. However, we did observe slight convergence in community composition and functioning between healthy and diseased plants, which was likely due to similar effects imposed on microbiota by the same tomato cultivars. Discriminating bacterial taxa that could initially separate healthy and diseased microbiomes was lost during the tomato growth, and the taxa associated with healthy and diseased plants changed considerably. Even though the healthy soils were still associated with increased number of functional genes linked to antibiotic production, only the Dyella genus was still associated as a potential driver taxon behind R. solanacearum suppression based on our network analyses. Instead, Gp16 (Acidobacteria), Spartobacteria_genera_incertae_sedis, Hyphomicrobium, and Sphingomonas were associated as potential driver taxa behind pathogen suppression in the healthy plant microbiomes at the end of the experiment. Of these taxa, the Sphingomonadaceae and Spartobacteria have previously been linked to R. solanacearum–driven bacterial wilt infections (37), while Hyphomicrobium has been linked to suppression of black shank (38). Together, these results suggest that pathogen suppression was potentially driven by different taxa in the beginning and at the end of the field experiment, indicative of functional redundancy present in rhizosphere microbiomes.
While initial bacterial community composition could well predict future disease outcomes, we also found that community diversity became significantly different between healthy and diseased plant microbiomes at weeks 5 and 6. Specifically, the microbiomes associated with healthy plants became less diverse, while no clear change was observed in the diseased plant microbiomes. While it is difficult to disentangle the potential drivers behind this, the fact that the discriminating taxa initially associated with healthy plants disappeared during the experiment suggests that it was driven by differences in bacterial community assembly during the experiment. The species co-occurrence networks also differed substantially between the initial and final microbiomes associated with healthy and diseased plants. In general, the networks associated with diseased plants had fewer associated species, fewer connections, shorter average path lengths, and fewer modular structures. Similar to changes in community composition, diversity, and functioning, network differences were initially much clearer and converged between healthy and diseased plants toward the end of the experiment. Together, these results suggest that bacteria were potentially interacting more often and more predictably in healthy versus diseased plant microbiomes, which could have increased the community stability against pathogen invasions (39). In addition, the higher modularity observed in healthy plant microbiomes suggests that certain bacteria were stably associated with each other, indicative of potentially stabilizing mutualistic or commensal interactions (40, 41). While these hypotheses need to be confirmed experimentally in the future, they suggest that in addition to specific taxa, the interactions between certain groups of bacteria are also likely to be important for constraining R. solanacearum infections in the rhizosphere (39, 42, 43).
Despite a slight convergence in microbiome composition during plant development, the beneficial effects of healthy plant microbiota could be transferred to future plant generations in a separate soil transplantation experiment highlighting the importance of functional microbiomes for disease resistance. This raises an important question: Do healthy plant-associated microbiomes have something extra or do diseased plant-associated microbiomes lack something? To fully answer this question would require first defining the core root microbiome of tomato as a baseline control. While being out of scope of this study, such investigations should be an integral part of future plant microbiome research (44, 45) to elucidate how taxonomic and functional differences in microbiomes causally determine plant disease outcomes. However, diseased plant microbiomes were also associated with certain rare taxa that were absent in healthy plant microbiomes and could thus have promoted pathogen infection (24). Moreover, differences in the pathogen densities between healthy and diseased microbiomes became evident only toward the end of the experiment, suggesting that the effects of initial microbiomes were potentially magnified during the community assembly. Development of dysbiosis, a microbial imbalance associated with the increased dominance of bacterial pathogen (46, 47), could thus also be important in explaining the bacterial wilt disease dynamics (9). While further experiments are needed to unequivocally separate these potential explanations, our results suggest that both initial and developmental differences in microbiome composition might play an important role for bacterial wilt outbreaks.
On the basis of our results, we propose a rethinking of current approaches used to manage plant diseases in agricultural systems. First, instead of a pathogen-focused view, better solutions for controlling plant disease outbreaks may be achieved by managing the composition of soil microbiome as a whole. For example, instead of attempting to directly reduce pathogen densities, increasing the relative abundance of naturally protective bacteria using organic fertilization might offer a better strategy. Second, the discriminating power of certain bacterial taxa suggests that the rare microbiome may represent an untapped pool of beneficial microorganisms (48) that could be selected and enriched by plant breeding, direct microbial inoculation along with the seedlings (49), or soil management. For example, certain agricultural management practices, such as organic fertilization, might promote physicochemical and biological properties linked to soil suppressiveness. In a wider perspective, our results suggest that initial microbiome composition plays a crucial role in predetermining the disease dynamics and future health of associated host organisms. These results might be important for understanding potential beneficial effects of crop rotation for pathogen suppression. It is known that different crop species harbor specific core microbiomes that can suppress pathogens (50). Rotating or intercropping certain plant species in combination could thus potentially be used to shift and modify the composition and suppressiveness of natural soil microbiomes without the need for pesticides. Moreover, our results have potentially important implications for managing diseases also in medical contexts. For example, if minor changes in the composition of an individual’s early life microbiome cascade into differential health outcomes later in life, then many diseases could be potentially prevented by ensuring the development of an initial “healthy” microbiome. We conclude that despite the often-stochastic nature of disease variability in natural environments, plant disease dynamics can also be driven by deterministic processes linked to within-field spatial differences in taxonomic and functional properties of soil microbiomes.
MATERIALS AND METHODS
Rhizobox system and repeated sampling design
We designed a semi-open mesocosm system (“rhizobox”) to repeatedly collect rhizosphere soil from each individual plant without damaging the root system. This method allowed a reconstruction of time series ranging from initial soil samples throughout plant development and disease onset and quantifying changes in pathogen density dynamics, physicochemical soil properties, and microbiome assembly. The rhizobox consisted of a three-layer cylinder (Fig. 1A and fig. S1A) with a height of 136 mm and a diameter of 110 mm. The outer layer was made of a 4-mm metal mesh to support the rhizobox. The inner layer (root compartment) was made of a 50-μm nylon mesh net, which prevents roots from entering into the middle layer. It consisted of 18 separate nylon mesh bags that could be removed individually during sampling (height, 136 mm; width, 18 to 21 mm; thickness, 1 to 2 mm; mesh size, 150 μm). Each bag was filled with 4 g of homogenized and sterilized field soil dug out from the same location where the rhizoboxes were placed during the field experiment. To this end, we used soil from a tomato field in Qilin, Nanjing, China, that has been naturally infected by R. solanacearum for decades (50). After removing plant debris, the soil was sieved (<2 mm), homogenized, divided into nylon bags, and sterilized by gamma irradiation (50 kilograys). Each bag was sprayed with sterile distilled water, gently squeezed to 1- to 2-mm thickness, and arranged between the outer and the inner layers of the rhizobox to create the middle sampling layer. The inner layer was filled with natural soil dug out from the location where the rhizoboxes were buried during the field experiment. The soil in the nylon mesh bags of the middle layer could thus be colonized by bacteria from the surrounding natural soil and affected by the development of plant roots and secretion of plant root exudates (Fig. 1B). As a result, the soil in the nylon mesh bags of the middle layer initially represented microbial communities present in the bulk soil (fig. S12) before the inner compartment of rhizobox became densely filled with roots after 3 weeks of planting in the field (Fig. 1C).
The design and sampling of the field experiment
Tomato seeds (Solanum lycopersicum cv. “Jipin”) were surface-sterilized with NaClO (3%; v:v) and germinated on moist filter paper for 2 days before sowing in sterilized nursery substrate (commercially available from Huaian Agricultural Technology Development Ltd., Huaian, Jiangsu, China). Tomato seedlings were incubated in the greenhouse at 30° ± 3°C for 3 weeks before planting in the field in rhizoboxes.
The field experiment was conducted in the same field described above (Qilin, Nanjing, China). The study site had been under a tomato monoculture system with two crop seasons per year for 19 years. The spring crop season typically lasts from February to June and the autumn crop season lasts from July to November. The nitrogen fertilizer was provided at 103.1 kg ha−1, phosphate was provided at 31.3 kg ha−1, and potassium was provided at 70.3 kg ha−1 per crop season; irrigation was applied three times per crop season; and pesticides were applied when necessary. The study field was tilled conventionally after every crop season. We conducted the field experiment in 2015 during the late-spring crop season between May and June. A soil corer was used to create 136-mm-deep holes suitable for the rhizobox. After removing the plant debris and rocks, the soil was placed right back into the root compartment of the rhizobox, which was immediately buried back into the hole. Triplicate soil samples were collected from the inner and outer sides of the middle sampling layer using a soil corer (diameter, 1 cm), and homogenized and pooled soil samples were regarded as the initial bulk soil samples (week 0). A subset of initial soil samples was mixed with 30% (v:v) glycerol and stored at −80°C for further processing. One tomato seedling was planted to the root compartment of each rhizobox, and rhizoboxes were placed 30 cm apart from each other. Three experimental plots (1 m apart from each other) were set up with a total of 16 rhizoboxes per plot (fig. S1B).
Five days later, three nylon mesh bags were randomly removed from rhizoboxes to explore the primary effects of microbial colonization from surrounding bulk soil to the middle sampling layer as, at this stage, plant roots were too small to exert any influence on the microbiome. Sampling of rhizoboxes was started 3 weeks after planting of tomatoes (Fig. 1, C and D), and from week 3 onward, three nylon bags were randomly collected from each rhizobox on a weekly basis up until week 6. These samples were considered as rhizosphere samples (Fig. 1, B and C) and stored at −80°C for further analyses after homogenization of replicate samples per sampling time point of each plant. More than half of the tomato plants showed disease symptoms after 6 weeks of planting. The field experiment was maintained for another week (total of 7 weeks) to ensure that disease incidence had reached its maximum peak and that none of the healthy plants developed any disease symptoms (fig. S1C). Seven weeks after planting of tomatoes, R. solanacearum densities present in the plant stem crowns were measured for all the 48 plants by macerating 5 g of stem crown tissue in 45 ml of sterile distilled water followed by serial dilution plating on R. solanacearum semiselective media (SMSA) (51). Individual plants that showed no wilt symptoms and were negative for pathogen isolation were classified as healthy plants (fig. S1D). Plants that showed wilt symptoms and were positive for pathogen isolation were classified as diseased plants. Plants that did not show wilt symptoms but were positive for pathogen isolation were considered as latently infected plants (52) and were not included in the further analyses in this study. The repeated sampling strategy allowed us to go back in time after observing the final health condition (diseased versus healthy) of individual plants. To this end, we randomly chose 12 healthy and 12 diseased plants and traced back the changes in microbiome composition, physicochemical soil properties, and pathogen densities for healthy and diseased plants individually. We also collected soil samples from healthy and diseased plants at the end of the experiment (week 7) to test soil suppressiveness in a separate greenhouse with a second generation of tomato plants (described in detail later).
DNA extraction, quantitative PCR, and 16S rRNA amplicon sequencing
Soil DNA was isolated from 500 mg of soil using the PowerSoil DNA Isolation Kit (MoBio, Carlsbad, CA, USA) according to the manufacturer’s instructions. DNA extracts were quantified using a NanoDrop spectrophotometer (ND2000, Thermo Scientific, DE, USA). Aliquots of DNA extract were used for quantitative polymerase chain reaction (qPCR) analyses, and the other was used for PCR amplification before 16S ribosomal RNA (rRNA) amplicon sequencing.
The total bacterial and R. solanacearum pathogen abundances were determined using qPCR with primer sets Eub338/Eub518 (53) and Rsol_fliC (54), respectively, using SYBR Premix Ex Taq Kit (Takara, Dalian, China) according to the manufacturer’s instructions. Each individual sample was measured in triplicate using a 7500 Fast Real-Time PCR System (Applied Biosystems, CA, USA). A plasmid standard (pMD 19-T vector; Takara, Dalian, China) was generated from cloned target genes (fliC and 16S rRNA) from R. solanacearum strain QL-Rs1115 (50), which is the dominant pathogen genotype in the field described above (Qilin, Nanjing, China).
The V4 regions of the bacterial 16S rRNA gene were PCR-amplified using the primer pairs 563F and 802R (55) with an Illumina adaptor (Illumina, CA, USA) sequence and index sequences following a previously described PCR method (56). The PCR products were purified (AxyPrep PCR Clean-up Kit, Axygen Biosciences, CA, USA) before performing agarose gel electrophoresis. The concentrations of the purified amplicon products were determined using QuantiFluor-ST (Promega, WI, USA) and sequenced on an Illumina MiSeq platform at Shanghai Biozeron Biological Technology Co. Ltd.
The sequence data were processed with the UPARSE pipeline (57). Briefly, read pairs from each sample were assembled, and sequences were screened with a maximal expected error of 0.25 and a minimum length of 200 base pairs (bp). Singletons were discarded, and sequence reads were then clustered into OTUs at 3% dissimilarity. Chimeras were removed using UCHIME (58). The representative sequences and OTU table obtained were then analyzed using mothur (59). All the samples were rarefied to the depth of the smallest sample (25,316 reads). The taxonomies of each OTU were annotated using the RDP 16S rRNA classifier (60) with a confidence threshold of 80%.
Metagenomic analysis
In addition to taxonomic composition, we performed a metagenomic analysis to gain insights into the functional differences between healthy and diseased microbiomes. A total of four healthy and four diseased samples were randomly selected at both the initial (week 0) and the final (week 6) week of the field experiment for metagenomic analysis. Metagenomic shotgun sequencing libraries were prepared and sequenced at Shanghai Biozeron Biological Technology Co. Ltd. For each sample, 1 mg of genomic DNA was used with Illumina’s TruSeq for library preparation. Libraries were sequenced using the Illumina HiSeq 4000, 50-bp paired-end technology. Raw FASTQ sequence files were filter-trimmed using Trimmomatic to remove adapter sequences (61). Reads were quality-filtered using fastq_quality_filter from the FASTX toolkit at default settings. Functional annotation was performed by comparison of quality-filtered reads to annotated ones using COG and eggNOG databases.
Culture-dependent assessment of antimicrobial activity of Bacillus and Pseudomonas bacteria against R. solanacearum
As a validation of the metagenomics data, we performed an additional screening of antimicrobial activity of initial soil bacteria associated with healthy and diseased plants. Four initial soil replicates were randomly pooled into one, resulting in three replicates for healthy and diseased plants. We focused on two common soil bacterial genera, Bacillus and Pseudomonas, that are well-characterized bacterial groups known to produce a broad range of antibiotics and linked to pathogen suppression in several previous studies (7, 62). For isolation of Bacillus and Pseudomonas, bacteria from the initial soil, two semiselective media were used: V8 juice agar for generic members of Bacillus (326 ml of V8 juice, 33 g of NaCl, and 0.8 g of glucose in 490 ml of distilled water at pH 5.2 containing filter-sterilized solutions of 45 mg of actidione and 22.5 mg of polymyxin B) and CFC for pseudomonads (63). Soil suspensions were serially diluted and plated on the semiselective media (soil suspensions used for isolation of Bacillus strains were pretreated at 80°C for 10 min to enrich heat-resistant Bacillus spores). After 2 days of incubation at 30°C, 40 purified Bacillus and Pseudomonas isolates per replicate (n = 3) were randomly isolated from the selective plates based on colony morphology, resulting in a total of 120 Bacillus and Pseudomonas isolates for both future healthy and diseased plants. The inhibition effect on R. solanacearum QL-Rs1115 pathogen was carried out by using a spot-spraying method described previously (50).
Soil transplantation experiment linking the suppressiveness of healthy and diseased plant microbiomes between plant generations
To test whether the initial differences in healthy and diseased plant microbiomes translated into long-term suppression of the pathogen during successive plant generation, we directly tested how soils derived from first-generation plants constrained pathogen growth in the second plant generation using a separate greenhouse experiment. To this end, we collected soil from the root compartment of rhizoboxes at the end of the experiment (week 7, one week after the last rhizosphere soil sampling). Three root compartment soil replicates were randomly pooled into one, resulting in four soil replicates for both healthy and diseased plants. Pathogen abundance in the root compartment soil was determined using serial dilution plating on SMSA media after total pathogen abundances were adjusted to 3 × 107 colony-forming units per gram of soil for all samples using a pure culture of R. solanacearum QL-Rs1115 strain isolated from the same field (50). Three treatments were set up: (i) healthy soil originating from healthy plants, (ii) diseased soil originating from diseased plants, and (iii) sterilized soil from the healthy plants (incubated at 80°C for 3 hours). With all treatments, tomato seeds (cv. Jipin) were surface-sterilized (3% NaClO), germinated (30°C for 2 days), and sown in pots (height, 135 mm; diameter, 120 mm) containing the different soils (dry weight, 400 g). Each pot with 12 tomato plants was regarded as one replicate, and each treatment had four replicate pots. The pots were incubated in growth chambers with a 16-hour light/8-hour dark photoperiod at 26°C and replenished with sterile distilled water to equal weight to maintain identical soil moisture between treatments. Disease incidence was recorded as the percentage of wilted tomato plants per replicate after 3 weeks of sowing.
Statistical analyses
The soil microbiome composition was ordinated by principal coordinates analysis (PCoA) using unweighted UniFrac distance. Differences between microbiome composition of healthy and diseased plants were calculated by using AMOVA and mothur. Unweighted UniFrac distances are sensitive to rare OTUs and thus emphasize differences in the presence or absence of taxa (64). Initial soil physicochemical properties were ordinated by principal components analysis [R package FactoMineR (65)]. Linear discriminant analysis and a significance test were used to explore the most discriminating OTUs between health conditions using LEfSe (66) and DESeq2 (67), respectively. Three screening criteria were used: (i) linear discriminant analysis with a score of ≥2 (health condition relative to diseased condition), (ii) fold change ≥2 (health condition relative to diseased condition), and (iii) significance test with P < 0.05. We further validated this approach using the random forest approach (68) and found that random forest analysis yielded very similar results, identifying 10 discriminating OTUs that were all also identified by our discrimination criteria at week 0, and 10 discriminating OTUs, of which 6 were also identified by our discrimination criteria at week 6 (fig. S6). Ralstonia OTUs were not included in the analyses exploring the role of discriminating OTUs during plant and microbiome development. Co-occurrence network analysis was performed following Molecular Ecological Network Analyses Pipeline (69). The top 1000 OTUs per sampling time point were retained for analysis, and the number of sequences was log-transformed and analyzed using a random matrix theory–based approach (70). The edges (i.e., connections between taxa as OTUs) correspond to a strong and significant (positive or negative) correlation between nodes (i.e., taxa as OTUs) (71). The network was graphed using Gephi (72). We also used the NetShift method to identify potential keystone driver taxa based on differences in network interactions between healthy and diseased plant microbiomes (https://web.rniapps.net/netshift). This method allows one to quantify the directional changes in the individual node interactions (73) by exploring whether there is a significant overall change in community patterns between healthy and diseased samples, whether there are major changes in associations of each constituent node (taxon) in healthy and diseased samples, whether specific nodes (taxa) have been important members of the community, and whether there is an increase in their importance in the diseased samples. Temporal dynamics of phylogenetic similarity between soil microbiomes associated with healthy and diseased plants were analyzed with linear regression as a function of time (quadratic term). The correlation between the relative abundance of screened rare OTUs enriched in healthy and diseased plants and sampling time was assessed with an exponential decay model. Student’s t test and one-way analysis of variance (Duncan’s multiple range test) were used to compare mean differences between the treatments using SPSS (v. 19) and STAMP (74). All amplicon sequencing data have been deposited in the DDBJ SRA under the accession number SRP090147. The metagenomics-derived gene catalogs used for the current analysis are publicly available (SRA database accession number PRJNA492172).
Supplementary Material
Acknowledgments
Funding: This research was financially supported by the National Key Basic Research Program of China (2015CB150503), the National Key Research and Development Program of China (2018YFD1000800), the National Natural Science Foundation of China (31801952 and 41922053), the Natural Science Foundation of Jiangsu Province (BK20181068 and BK20170085), the 111 project (B12009), the Natural Science Research Program of Huaian (HAB201829), the Fundamental Research Funds for the Central Universities (KY2201719 and KYT201802), and Key S&T Special Projects of China National Tobacco Corporation [110201601025 (LS-05)]. A.J. was supported by the Netherlands Organisation for Scientific Research (NWO) project ALW.870.15.050 and the Koninklijke Nederlandse Akademie van Wetenschappen (KNAW) project 530-5CDP18. V.-P.F. was supported by the Wellcome Trust (reference number 105624) through the Centre for Chronic Diseases and Disorders (C2D2) and Royal Society Research Grants (RSG\R1\180213 and CHL\R1\180031) at the University of York. Author contributions: All authors wrote the manuscript. Y.X., Y.G., and Z.W. developed the ideas and designed the experimental plans. Y.G. and Z.W. performed the experiments. Y.G., A.J., and Z.W. analyzed the data. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the corresponding authors Qirong Shen (shenqirong@njau.edu.cn) or Yangchun Xu (ycxu@njau.edu.cn).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaaw0759/DC1
Fig. S1. The placement of rhizoboxes and observed disease dynamics during the field experiment.
Fig. S2. Differences in the physicochemical soil properties, total bacterial abundances, and bacterial diversity between healthy and diseased plants during the field experiment.
Fig. S3. Relationships between physical distance and bacterial phylogenetic distance (unweighted UniFrac distance) between rhizoboxes within three replicated plots.
Fig. S4. Differences in the abundances of bacterial phyla in the initial soils (week 0) that later became associated with healthy and diseased plants (week 6).
Fig. S5. Rare OTUs discriminate the initial soil microbiomes that later become associated with healthy and diseased plants.
Fig. S6. Ten best discriminant OTUs linked to future plant disease outcomes based on random forest analysis.
Fig. S7. The total abundance of Bacillus and Pseudomonas bacteria and their ability to inhibit the growth of Ralstonia solanacearum.
Fig. S8. Differences in bacterial community composition associated with healthy and diseased plants persisted throughout the field experiment.
Fig. S9. Temporal changes in discriminating rare OTUs observed in the initial microbiomes associated with healthy and diseased plants.
Fig. S10. Heatmaps showing the dynamics of discriminating rare OTUs associated with healthy and diseased plants during the field experiment.
Fig. S11. PCoA of Bray-Curtis distances of bacterial functional gene profiles in the beginning (week 0) and at the end of the field experiment (week 6).
Fig. S12. Comparison of bacterial community between initial bulk soil and 5-day old nylon bag samples.
Table S1. Physicochemical properties of initial soils that later became associated with healthy and diseased plants.
Table S2. Topological properties of networks associated with healthy and diseased plant microbiomes at weeks 0 and 6.
Data file S1. Screened OTUs enriched in initial soil microbiome associated with later healthy and diseased plants.
Data file S2. Functional genes significantly different in initial soil microbiome associated with later healthy and diseased plants.
Data file S3. Screened OTUs enriched in soil microbiome associated with later healthy and diseased plants at 6 week after planting.
Data file S4. Functional genes significantly different in soil microbiome associated with later healthy and diseased plants at 6 weeks after planting.
Movie S1. Method of sampling middle-layer nylon bags from rhizobox.
REFERENCES AND NOTES
- 1.D. Pennock, N. McKenzie, L. Montanarella, Status of the World's Soil Resources. Technical Summary FAO, Rome, Italy (2015).
- 2.Fierer N., Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 15, 579–590 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Raaijmakers J. M., Mazzola M., Soil immune responses. Science 352, 1392–1393 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Campbell C. L., Noe J. P., The spatial analysis of soilborne pathogens and root diseases. Annu. Rev. Phytopathol. 23, 129–148 (1985). [Google Scholar]
- 5.Wang J.-F., Olivier J., Thoquet P., Mangin B., Sauviac L., Grimsley N. H., Resistance of tomato line Hawaii7996 to Ralstonia solanacearum Pss4 in Taiwan is controlled mainly by a major strain-specific locus. Mol. Plant-Microbe Interact. 13, 6–13 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Dodds P. N., Rathjen J. P., Plant immunity: Towards an integrated view of plant–pathogen interactions. Nat. Rev. Genet. 11, 539–548 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Mendes R., Kruijt M., de Bruijn I., Dekkers E., van der Voort M., Schneider J. H. M., Piceno Y. M., De Santis T. Z., Andersen G. L., Bakker P. A. H. M., Raaijmakers J. M., Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 332, 1097–1100 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Kwak M.-J., Kong H. G., Choi K., Kwon S.-K., Song J. Y., Lee J., Lee P. A., Choi S. Y., Seo M., Lee H. J., Jung E. J., Park H., Roy N., Kim H., Lee M. M., Rubin E. M., Lee S.-W., Kim J. F., Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 36, 1100–1109 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Wei Z., Hu J., Gu Y., Yin S., Xu Y., Jousset A., Shen Q., Friman V.-P., Ralstonia solanacearum pathogen disrupts bacterial rhizosphere microbiome during an invasion. Soil Biol. Biochem. 118, 8–17 (2018). [Google Scholar]
- 10.Wei Z., Huang J.-F., Hu J., Gu Y.-A., Yang C.-L., Mei X.-L., Shen Q.-R., Xu Y.-C., Friman V.-P., Altering transplantation time to avoid periods of high temperature can efficiently reduce bacterial wilt disease incidence with tomato. PLOS ONE 10, e0139313 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinbeiss S., Beßler H., Engels C., Temperton V. M., Buchmann N., Roscher C., Kreutziger Y., Baade J., Habekost M., Gleixner G., Plant diversity positively affects short-term soil carbon storage in experimental grasslands. Glob. Chang. Biol. 14, 2937–2949 (2008). [Google Scholar]
- 12.Tamburini S., Shen N., Wu H. C., Clemente J. C., The microbiome in early life: Implications for health outcomes. Nat. Med. 22, 713–722 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Hayward A. C., Biology and epidemiology of bacterial wilt caused by Pseudomonas solanacearum. Annu. Rev. Phytopathol. 29, 65–87 (1991). [DOI] [PubMed] [Google Scholar]
- 14.Swanson J. K., Yao J., Tans-Kersten J., Allen C., Behavior of Ralstonia solanacearum race 3 biovar 2 during latent and active infection of geranium. Phytopathology 95, 136–143 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Berendsen R. L., Pieterse C. M. J., Bakker P. A. H. M., The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Mazurier S., Corberand T., Lemanceau P., Raaijmakers J. M., Phenazine antibiotics produced by fluorescent pseudomonads contribute to natural soil suppressiveness to Fusarium wilt. ISME J. 3, 977–991 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Bais H. P., Fall R., Vivanco J. M., Biocontrol of Bacillus subtilis against infection of Arabidopsis roots by Pseudomonas syringae is facilitated by biofilm formation and surfactin production. Plant Physiol. 134, 307–319 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rangaswamy V., Jiralerspong S., Parry R. J., Bender C. L., Biosynthesis of the Pseudomonas polyketide coronafacic acid requires monofunctional and multifunctional polyketide synthase proteins. Proc. Natl. Acad. Sci. U.S.A. 95, 15469–15474 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mendes L. W., Kuramae E. E., Navarrete A. A., van Veen J. A., Tsai S. M., Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 8, 1577–1587 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castrillo G., Teixeira P. J. P. L., Paredes S. H., Law T. F., de Lorenzo L., Feltcher M. E., Finkel O. M., Breakfield N. W., Mieczkowski P., Jones C. D., Paz-Ares J., Dangl J. L., Root microbiota drive direct integration of phosphate stress and immunity. Nature 543, 513–518 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sprockett D., Fukami T., Relman D. A., Role of priority effects in the early-life assembly of the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 15, 197–205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pagaling E., Strathdee F., Spears B. M., Cates M. E., Allen R. J., Free A., Community history affects the predictability of microbial ecosystem development. ISME J. 8, 19–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abdullah A. S., Moffat C. S., Lopez-Ruiz F. J., Gibberd M. R., Hamblin J., Zerihun A., Host-multi-pathogen warfare: Pathogen interactions in co-infected plants. Front. Plant Sci. 8, 1806 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li M., Wei Z., Wang J., Jousset A., Friman V.-P., Xu Y., Shen Q., Pommier T., Facilitation promotes invasion in plant-associated microbial communities. Ecol. Lett. 22, 149–158 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Cho I., Blaser M. J., The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 13, 260–270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trivedi P., He Z., Van Nostrand J. D., Albrigo G., Zhou J., Wang N., Huanglongbing alters the structure and functional diversity of microbial communities associated with citrus rhizosphere. ISME J. 6, 363–383 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Falony G., Joossens M., Vieira-Silva S., Wang J., Darzi Y., Faust K., Kurilshikov A., Bonder M. J., Valles-Colomer M., Vandeputte D., Tito R. Y., Chaffron S., Rymenans L., Verspecht C., De Sutter L., Lima-Mendez G., D’hoe K., Jonckheere K., Homola D., Garcia R., Tigchelaar E. F., Eeckhaudt L., Fu J., Henckaerts L., Zhernakova A., Wijmenga C., Raes J., Population-level analysis of gut microbiome variation. Science 352, 560–564 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Gerber G. K., The dynamic microbiome. FEBS Lett. 588, 4131–4139 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Hiscox J., Savoury M., Müller C. T., Lindahl B. D., Rogers H. J., Boddy L., Priority effects during fungal community establishment in beech wood. ISME J. 9, 2246–2260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kyselková M., Kopecký J., Frapolli M., Défago G., Ságová-Marečková M., Grundmann G. L., Moënne-Loccoz Y., Comparison of rhizobacterial community composition in soil suppressive or conducive to tobacco black root rot disease. ISME J. 3, 1127–1138 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Trivedi P., Delgado-Baquerizo M., Trivedi C., Hamonts K., Anderson I. C., Singh B. K., Keystone microbial taxa regulate the invasion of a fungal pathogen in agro-ecosystems. Soil Biol. Biochem. 111, 10–14 (2017). [Google Scholar]
- 32.Fu L., Penton C. R., Ruan Y., Shen Z., Xue C., Li R., Shen Q., Inducing the rhizosphere microbiome by biofertilizer application to suppress banana Fusarium wilt disease. Soil Biol. Biochem. 104, 39–48 (2017). [Google Scholar]
- 33.Siegel-Hertz K., Edel-Hermann V., Chapelle E., Terrat S., Raaijmakers J. M., Steinberg C., Comparative microbiome analysis of a Fusarium wilt suppressive soil and a Fusarium wilt conducive soil from the Chateaurenard region. Front. Microbiol. 9, 568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chng S., Cromey M. G., Dodd S. L., Stewart A., Butler R. C., Jaspers M. V., Take-all decline in New Zealand wheat soils and the microorganisms associated with the potential mechanisms of disease suppression. Plant Soil 397, 239–259 (2015). [Google Scholar]
- 35.Yin C., Hulbert S. H., Schroeder K. L., Mavrodi O., Mavrodi D., Dhingra A., Schillinger W. F., Paulitz T. C., Role of bacterial communities in the natural suppression of Rhizoctonia solani bare patch disease of wheat (Triticum aestivum L.). Appl. Environ. Microbiol. 79, 7428–7438 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamilova F., Kravchenko L. V., Shaposhnikov A. I., Azarova T., Makarova N., Lugtenberg B., Organic acids, sugars, and l-tryptophane in exudates of vegetables growing on stonewool and their effects on activities of rhizosphere bacteria. Mol. Plant-Microbe Interact. 19, 250–256 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Lee C. G., Iida T., Inoue Y., Muramoto Y., Watanabe H., Nakaho K., Ohkuma M., Prokaryotic communities at different depths between soils with and without tomato bacterial wilt but pathogen-present in a single greenhouse. Microbes Environ. 32, 118–124 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ros M., Raut I., Santisima-Trinidad A. B., Pascual J. A., Relationship of microbial communities and suppressiveness of Trichoderma fortified composts for pepper seedlings infected by Phytophthora nicotianae. PLOS ONE 12, e0174069 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei Z., Yang T., Friman V.-P., Xu Y., Shen Q., Jousset A., Trophic network architecture of root-associated bacterial communities determines pathogen invasion and plant health. Nat. Commun. 6, 8413 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou J., Deng Y., Luo F., He Z., Yang Y., Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. MBio 2, e00122-11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newman M. E., Modularity and community structure in networks. Proc. Natl. Acad. Sci. U.S.A. 103, 8577–8582 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li M., Wang J., Jousset A., Friman V.-P., Xu Y., Shen Q., Pommier T., Facilitation promotes invasions in plant-associated microbial communities. Ecol. Lett. 22, 149–158 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Hu J., Wei Z., Friman V.-P., Gu S.-h., Wang X.-f., Eisenhauer N., Yang T.-j., Ma J., Shen Q.-r., Xu Y.-c., Jousset A., Probiotic diversity enhances rhizosphere microbiome function and plant disease suppression. MBio 7, e01790-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toju H., Peay K. G., Yamamichi M., Narisawa K., Hiruma K., Naito K., Fukuda S., Ushio M., Nakaoka S., Onoda Y., Yoshida K., Schlaeppi K., Bai Y., Sugiura R., Ichihashi Y., Minamisawa K., Kiers E. T., Core microbiomes for sustainable agroecosystems. Nat. Plants 4, 247–257 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Lundberg D. S., Lebeis S. L., Paredes S. H., Yourstone S., Gehring J., Malfatti S., Tremblay J., Engelbrektson A., Kunin V., del Rio T. G., Edgar R. C., Eickhorst T., Ley R. E., Hugenholtz P., Tringe S. G., Dangl J. L., Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–90 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tamboli C., Neut C., Desreumaux P., Colombel J. F., Dysbiosis in inflammatory bowel disease. Gut 53, 1–4 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fröhlich E. E., Farzi A., Mayerhofer R., Reichmann F., Jačan A., Wagner B., Zinser E., Bordag N., Magnes C., Fröhlich E., Kashofer K., Gorkiewicz G., Holzer P., Cognitive impairment by antibiotic-induced gut dysbiosis: Analysis of gut microbiota-brain communication. Brain Behav. Immun. 56, 140–155 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jousset A., Bienhold C., Chatzinotas A., Gallien L., Gobet A., Kurm V., Küsel K., Rillig M. C., Rivett D. W., Salles J. F., van der Heijden M. G. A., Youssef N. H., Zhang X., Wei Z., Hol W. H. G., Where less may be more: How the rare biosphere pulls ecosystems strings. ISME J. 11, 853–862 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santhanam R., Luu V. T., Weinhold A., Goldberg J., Oh Y., Baldwin I. T., Native root-associated bacteria rescue a plant from a sudden-wilt disease that emerged during continuous cropping. Proc. Natl. Acad. Sci. U.S.A. 112, E5013–E5020 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei Z., Yang X., Yin S., Shen Q., Ran W., Xu Y., Efficacy of Bacillus-fortified organic fertiliser in controlling bacterial wilt of tomato in the field. Appl. Soil Ecol. 48, 152–159 (2011). [Google Scholar]
- 51.Elphinstone J. G., Hennessy J., Wilson J. K., Stead D. E., Sensitivity of different methods for the detection of Ralstonia solanacearum in potato tuber extracts. EPPO Bull. 26, 663–678 (1996). [Google Scholar]
- 52.Swanson J. K., Montes L., Mejia L., Allen C., Detection of latent Infections of Ralstonia solanacearum race 3 biovar 2 in geranium. Plant Dis. 91, 828–834 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Fierer N., Jackson J. A., Vilgalys R., Jackson R. B., Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Appl. Environ. Microbiol. 71, 4117–4120 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schonfeld J., Heuer H., van Elsas J. D., Smalla K., Specific and sensitive detection of Ralstonia solanacearum in soil on the basis of PCR amplification of fliC fragments. Appl. Environ. Microbiol. 69, 7248–7256 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cardenas E., Wu W.-M., Leigh M. B., Carley J., Carroll S., Gentry T., Luo J., Watson D., Gu B., Ginder-Vogel M., Kitanidis P. K., Jardine P. M., Zhou J., Criddle C. S., Marsh T. L., Tiedje J. M., Significant association between sulfate-reducing bacteria and uranium-reducing microbial communities as revealed by a combined massively parallel sequencing-indicator species approach. Appl. Environ. Microbiol. 76, 6778–6786 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gu Y., Wei Z., Wang X., Friman V.-P., Huang J., Wang X., Mei X., Xu Y., Shen Q., Jousset A., Pathogen invasion indirectly changes the composition of soil microbiome via shifts in root exudation profile. Biol. Fertil. Soils 52, 997–1005 (2016). [Google Scholar]
- 57.Edgar R. C., UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Edgar R. C., Haas B. J., Clemente J. C., Quince C., Knight R., UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schloss P. D., Westcott S. L., Ryabin T., Hall J. R., Hartmann M., Hollister E. B., Lesniewski R. A., Oakley B. B., Parks D. H., Robinson C. J., Sahl J. W., Stres B., Thallinger G. G., Van Horn D. J., Weber C. F., Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Q., Garrity G. M., Tiedje J. M., Cole J. R., Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bolger A., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garbeva P., Postma J., van Veen J. A., van Elsas J. D., Effect of above-ground plant species on soil microbial community structure and its impact on suppression of Rhizoctonia solani AG3. Environ. Microbiol. 8, 233–246 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Mead G. C., Enumeration of pseudomonads using cephaloridine-fucidin-cetrimide agar (CFC). Int. J. Food Microbiol. 2, 21–26 (1985). [Google Scholar]
- 64.Peiffer J. A., Spor A., Koren O., Jin Z., Tringe S. G., Dangl J. L., Buckler E. S., Ley R. E., Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. U.S.A. 110, 6548–6553 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lê S., Josse J., Husson F., FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 25, 1–18 (2008). [Google Scholar]
- 66.Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W. S., Huttenhower C., Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Breiman L., Random forests. Mach. Learn. 45, 5–32 (2001). [Google Scholar]
- 69.Deng Y., Jiang Y.-H., Yang Y., He Z., Luo F., Zhou J., Molecular ecological network analyses. BMC Bioinformatics 13, 113 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou J., Deng Y., Luo F., He Z., Tu Q., Zhi X., Functional molecular ecological networks. MBio 1, e00169-10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dini-Andreote F., de Cássia Pereira e Silva M., Triadó-Margarit X., Casamayor E. O., van Elsas J. D., Salles J. F., Dynamics of bacterial community succession in a salt marsh chronosequence: Evidences for temporal niche partitioning. ISME J. 8, 1989–2001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.M. Bastian, S. Heymann, M. Jacomy, Gephi: an open source software for exploring and manipulating networks, E. Adar, M. Hurst, T. Finin, N. Glance, N. Nicolov, B. Tseng, eds. Proceedings of the International Conference on Weblogs and Social Media (The AAAI Press, Menlo Park, California, 2009). [Google Scholar]
- 73.Kuntal B. K., Chandrakar P., Sadhu S., Mande S. S., ‘NetShift’: A methodology for understanding ‘driver microbes’ from healthy and disease microbiome datasets. ISME J. 13, 442–454 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Parks D. H., Tyson G. W., Hugenholtz P., Beiko R. G., STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaaw0759/DC1
Fig. S1. The placement of rhizoboxes and observed disease dynamics during the field experiment.
Fig. S2. Differences in the physicochemical soil properties, total bacterial abundances, and bacterial diversity between healthy and diseased plants during the field experiment.
Fig. S3. Relationships between physical distance and bacterial phylogenetic distance (unweighted UniFrac distance) between rhizoboxes within three replicated plots.
Fig. S4. Differences in the abundances of bacterial phyla in the initial soils (week 0) that later became associated with healthy and diseased plants (week 6).
Fig. S5. Rare OTUs discriminate the initial soil microbiomes that later become associated with healthy and diseased plants.
Fig. S6. Ten best discriminant OTUs linked to future plant disease outcomes based on random forest analysis.
Fig. S7. The total abundance of Bacillus and Pseudomonas bacteria and their ability to inhibit the growth of Ralstonia solanacearum.
Fig. S8. Differences in bacterial community composition associated with healthy and diseased plants persisted throughout the field experiment.
Fig. S9. Temporal changes in discriminating rare OTUs observed in the initial microbiomes associated with healthy and diseased plants.
Fig. S10. Heatmaps showing the dynamics of discriminating rare OTUs associated with healthy and diseased plants during the field experiment.
Fig. S11. PCoA of Bray-Curtis distances of bacterial functional gene profiles in the beginning (week 0) and at the end of the field experiment (week 6).
Fig. S12. Comparison of bacterial community between initial bulk soil and 5-day old nylon bag samples.
Table S1. Physicochemical properties of initial soils that later became associated with healthy and diseased plants.
Table S2. Topological properties of networks associated with healthy and diseased plant microbiomes at weeks 0 and 6.
Data file S1. Screened OTUs enriched in initial soil microbiome associated with later healthy and diseased plants.
Data file S2. Functional genes significantly different in initial soil microbiome associated with later healthy and diseased plants.
Data file S3. Screened OTUs enriched in soil microbiome associated with later healthy and diseased plants at 6 week after planting.
Data file S4. Functional genes significantly different in soil microbiome associated with later healthy and diseased plants at 6 weeks after planting.
Movie S1. Method of sampling middle-layer nylon bags from rhizobox.