

HNTs/siRIPK4 nanoparticles have been shown to be an effective treatment for bladder cancer.
Abstract
RNA interference (RNAi) technology can specifically silence the expression of a target gene and has emerged as a promising therapeutic method to treat cancer. In the present study, we showed that natural halloysite nanotube (HNT)–assisted delivery of an active small interfering RNA (siRNA) targeting receptor-interacting protein kinase 4 (RIPK4) efficiently silenced its expression to treat bladder cancer. The HNTs/siRNA complex increased the serum stability of the siRNA, increased its circulation lifetime in blood, and promoted the cellular uptake and tumor accumulation of the siRNA. The siRNA markedly down-regulated RIPK4 expression in bladder cancer cells and bladder tumors, thus inhibiting tumorigenesis and progression in three bladder tumor models (a subcutaneous model, an in situ bladder tumor model, and a lung metastasis model), with no adverse effects. Thus, we revealed a simple but effective method to inhibit bladder cancer using RIPK4 silencing, indicating a promising therapeutic method for bladder cancer.
INTRODUCTION
For tumor eradication, targeted gene therapy has received increased research attention. Small interfering RNA (siRNA) has shown potential as a molecular approach to down-regulate specific gene expression in cancer cells (1, 2). To silence genes that inhibit proliferation, promote apoptosis, or prevent multidrug resistance, siRNA delivered using nanocarriers has become a new focus for anticancer therapy (3). Delivery of siRNA to specific tumor cells and tissues is necessary to limit off-target effects that could adversely affect the therapeutic index and the clinical application of siRNA (4). Targeting cancer using nanoparticle drug carriers usually comprises optimizing the biophysical properties of the carrier to enhance nonspecific permeation and increase accumulation in the tumor (5). This is usually achieved by the biochemical targeting of cancer cell–specific, overexpressed surface receptors (6). For example, a phase 1 human trial data reported safe gene silencing in patients with melanoma using nanoparticle-mediated molecular targeting of cancer cells overexpressing the transferrin receptor (7). In the past decade, siRNA-mediated delivery has become more widespread; however, the lack of efficient in vivo delivery vehicles, particularly for the systemic delivery into immune cells, remains a major challenge for translating siRNA into therapeutics (8, 9). Recently, however, nanoparticle siRNA delivery vehicles have been developed that are highly potent, specific, and biocompatible (10–12). However, developing clinical nanosystems to selectively deliver siRNA into both immune cells and cancer cells remains a challenge (13).
Materials such as lipids, polymers, dendrimers, polymeric micelles, and metallic core nanoparticles have been constructed to efficiently deliver siRNA (9, 14, 15). Natural halloysite nanotubes (HNTs; external diameter, 50 to 70 nm; lumenal diameter, 10 to 15 nm; and length, 1 to 1.5 μm), which are often used as nonviral vectors to entrap and release active agents, have shown benefits in the medical and chemical industries. The application of HNTs for nucleic acid molecules could resolve the problems of inefficient use, easy decomposition, and toxic side effects of siRNA. Research activities have focused on appropriate size regulation, smart lumen construction, easy coupling of surface modifications, and enhanced biocompatibility (16–18).
Therefore, in the present study, we used HNTs to deliver therapeutic anticancer siRNA that actively targets the oncogene RIPK4 (receptor-interacting protein kinase 4), which is a serine/threonine kinase of the RIP kinase family that has an apparent molecular weight of 91.6 kDa. RIPK4 is overexpressed in skin, ovarian, cervical, and colorectal cancers (19, 20). Previously, we showed that RIPK4 is highly expressed in bladder cancer tissues and represents an independent prognostic marker for poor survival (21). In addition, marked antitumor effects were observed after RIPK4 knockdown in bladder cancer (21).
In the present study, HNT nanoclusters were developed to efficiently deliver an siRNA targeting RIPK4 (RIPK4-siRNA) to silence RIPK4 expression in bladder cancer, with the aim of inhibiting bladder cancer tumorigenesis. The results showed that the HNT-encapsulated siRNA was more stable in serum, had an increased circulation lifetime in blood, was more readily taken up by bladder cancer cells, and accumulated in bladder cancer tumors. The HNTs/siRNA complex knocked down RIPK4 expression markedly in bladder cancer cells, which suppressed bladder cancer tumorigenesis and progression effectively. Thus, the nanoparticle-mediated delivery system developed in the present study is a simple and effective method to inhibit bladder cancer tumorigenesis as a potential previously unidentified therapy for bladder cancer.
RESULTS
Synthesis and characterization of HNTs
Figure 1A shows the regular morphology and size homogeneity of halloysites in the range of ~500 nm prepared by the combination of high-speed shear homogenization and two-step uniform viscosity centrifugation (Fig. 1A). Nanoparticles within the 500-nm range should be suitable candidates as carriers to deliver siRNA into cells, according to previous studies (22, 23). Oversized or undersized halloysites would probably not be taken up by cells and would show ineffective siRNA loading. The ends of the nanotubes were unblocked, and the lumen of the nanotubes was visible. Negatively charged siRNA [owing to the phosphate group (PO43−)] was absorbed into the positively charged lumen of the halloysites [owing to the aluminol group (Al-OH)] using vacuum impregnation methods (Fig. 1B). After vacuum impregnation, most of the HNTs were filled with siRNA. Time-dependent release of the siRNA occurred under neutral conditions (Fig. 1C). The isoelectric point was approximately 6.5 (Fig. 1D), and the zeta potential ranged from 13.8 mV (pH 2.0) to −29.0 mV (pH 12.0). The pH-dependent charged form will benefit the controlled release of siRNA and improve their resistance to degradation.
Fig. 1. Characterization of the siRNA-loaded HNTs.
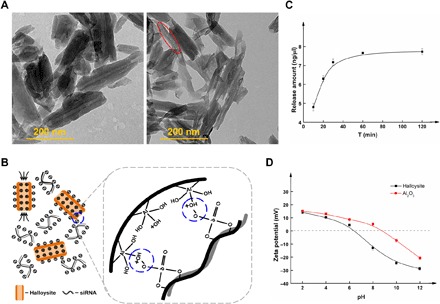
(A) Transmission electron microscopy images of HNTs and HNTs/siRNA. (B) Schematic diagram of HNTs combined with siRNA. (C) siRNA cumulative release from HNTs/siRNA under neutral conditions. (D) Zeta potential of HNTs and Al2O3.
siRNA-binding efficiency and cytotoxicity of HNTs
The efficiency of siRNA binding was determined using electrophoretic mobility shift assays in agarose gels. The ratio of the HNTs to siRNA ranged from 1:1 to 100:1. Figure S1A shows that the mobility of the siRNA through the gel was reduced as increasing amounts of HNTs were used. The siRNA band did not exit the well at a ratio of 50:1, which suggested that the optimal ratio of HNTs to siRNA was 50:1. The cytotoxicity of the HNTs was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The HNTs were used to deliver the negative control siRNA (siNC) into T24 (established from a highly malignant grade III human urinary bladder carcinoma) cells to determine their cytotoxicity. Cells were treated with different concentrations of HNTs/siNC complexes. As shown in fig. S1 (B and C), at a high concentration of HNT complexes of 500 μg/ml, which was 50-fold higher than the concentration required for highly efficient transfection, the HNTs/siNC complex did not markedly affect cell viability. In contrast, Lipofectamine 3000 showed some cytotoxicity at the dose used for efficient transfection according to the manufacturer’s protocol. These results demonstrated that because of their low toxicity, HNTs could be used as an siRNA transfer vector.
Intracellular uptake of HNTs/siRNA
To investigate intracellular uptake behavior of HNT nanoparticles to deliver siRNA into cancer cells, the uptake of HNTs/FAM-siRNA complexes by T24 cells was examined under an inverted fluorescence microscope using fluorescein (FAM)-labeled siRNA (FAM-siRNA). Figure 2A shows that after 6 hours of incubation, FAM-siRNA was observed within the T24 cells, indicating that the HNTs were internalized. To determine precisely the effect of the composition of the formulations on the internalization of the siRNA, we incubated T24 cells with HNTs/FAM-siRNA for 6 hours and then used flow cytometry to analyze the cells after quenching the extracellular fluorescence using trypan blue. Figure 2B shows that for the HNTs/FAM-siRNA complexes, the proportion of fluorescent cells increased with increasing doses of FAM-siRNA. Furthermore, at a dose of 200 nM, the cells treated with HNTs/FAM-siRNA complexes showed a comparable transfection efficiency to that achieved using Lipofectamine 3000/FAM-siRNA complexes. At 250 nM, the transfection did not change notably. Therefore, we decided to use 200 nM of siRNA as the optimal dose for HNT transfection in T24 cells. The use of HNTs induced similarly low toxicity to that of Lipofectamine 3000 during siRNA transfection.
Fig. 2. The transfection efficiency and distribution of HNT-delivered siRIPK4 in T24 bladder cancer cells.
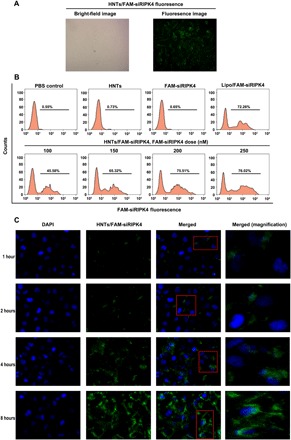
(A) Bright-field and fluorescence microscopy images of HNTs/FAM-siRIPK4 fluorescence at 6 hours after transfection. (B) Flow cytometry analysis of fluorescent cells: representative histograms (upper panels) and means ± SD (lower panels, from three independent experiments). FAM-siRIPK4, fluorescein-labeled siRNA targeting RIPK4. (C) Confocal laser scanning microscopy analysis of the distribution of HNTs/FAM-siRIPK4 in T24 bladder cancer. Fluorescein (green)–labeled siRIPK4. DAPI (4′,6-diamidino-2-phenylindole; blue) was used to stain cell nuclei.
Intracellular distribution and lysosomal escape of FAM-siRNA
To achieve cytoplasmic posttranscriptional gene silencing, siRNA escape from lysosomes after entering the cell is essential (24). To investigate whether the nanoparticle-delivered siRNA could exit the endosomes and/or lysosomes, T24 cells were treated with HNTs/FAM-siRNA for different periods, and the nanoparticles’ cellular localizations were determined. DAPI (4′,6-diamidino-2-phenylindole) was used to counterstain the cell nuclei. As shown in Fig. 2C, initially, HNTs with FAM-siRNA (green) were distributed mainly in the cell membrane. Two hours later, efficient codelivery of the siRNA into the cytoplasm was demonstrated by the punctate distribution in the T24 cell cytoplasm and at the periphery of the nuclei of the FAM-siRNA fluorescence intensity. In addition, at 4 and 8 hours, the fluorescence intensity was significantly enhanced. The results suggested that immediately after transfection, the hybrid nanoparticles are located in the endosomes or early lysosomes. After prolonged incubation, the FAM-siRNA were released from the lysosomes and showed marked fluorescence intensity in the cytoplasm. Subsequently, T24 cells were stained using LysoTracker, and the cells were monitored using confocal microscopy to study the escape of HNTs/siRNA from lysosomes (fig. S2). After 1 hour of incubation, the green (FAM-siRNA) and red (LysoTracker) fluorescence was colocalized in the T24 cells (fig. S2), indicating that the FAM-labeled siRNA targeting RIPK4 (FAM-siRIPK4) was located in the lysosomes. After 4 hours of incubation, the green fluorescence (FAM-siRIPK4) was separated from the red fluorescence (LysoTracker; fig. S2), indicating that the FAM-siRIPK4 had escaped from the lysosomes into the cytoplasm. We then analyzed the colocalization of siRIPK4 with the lysosomes over incubation time quantitatively (fig. S3A), which provided results that were consistent with the observations in fig. S2. In addition, the ratio of the escape of siRIPK4 from the lysosomes increased from 34 to 80.5% from 3 to 6 hours (fig. S3B). Thus, we believe that intact siRIPK4 escaped from the lysosomes into the cytoplasm to perform siRNA-mediated gene silencing.
RIPK4 gene knockdown by HNTs/siRIPK4 complexes in T24 cells
The RIPK4 gene knockdown effect of the HNTs/siRIPK4 complexes was measured in T24 cells. As shown in fig. S4, siRIPK4 reduced the level of the RIPK4 mRNA dose dependently. siRIPK4 at 100, 150, and 200 nM induced 57.4, 87.6, and 97.2% reductions in the RIPK4 mRNA level, respectively, compared with that in the HNTs/siNC complexes group (P < 0.001). The HNTs/siNC complexes group showed no reduction compared with that in the control. As expected, the RIPK4 protein level, as assessed using Western blotting, decreased dose dependently (fig. S4). These results showed that transfection with the HNTs/siRIPK4 complexes efficiently knocked down the expression of RIPK4 at the mRNA and protein levels in T24 cells.
Cell cycle, apoptosis, and cell proliferation after RIPK4 knockdown
The effect of RIPK4 knockdown by HNTs/siRIPK4 on the proliferation of T24 cells in vitro was assessed using the MTT and the plate colony formation assays. The growth and colony formation of T24 cells was significantly decreased after RIPK4 knockdown compared with that in the HNTs/siNC complexes group (P < 0.001; figs. S5 and S6). By contrast, this reduction was not observed in the HNTs/siNC complexes group (figs. S5 and S6). Thus, the proliferation of T24 cells was specifically reduced by the HNTs/siRIPK4 complexes by controlling the in vivo expression of RIPK4. Annexin V–fluorescein isothiocyanate (FITC) and propidium iodide staining were used to determine the effect of HNTs/siRIPK4 on the apoptosis of T24 cells. As shown in fig. S7, the siRIPK4 delivered by the HNTs significantly increased the level of apoptosis compared with that induced in the HNTs/siNC cells (P < 0.001). Moreover, no apoptosis was induced by HNTs/siNC (fig. S7), which demonstrated that RIPK4 down-regulation caused apoptosis. In T24 cells treated with various formulations, the cell cycle of was examined using flow cytometry. As shown in fig. S8, HNTs/siRIPK4 treatment markedly decreased the proliferation index compared with that in the control groups. These results indicated that depletion of RIPK4 by HNTs/siRIPK4 inhibited proliferation and induced apoptosis and cell cycle arrest in T24 cancer cells.
In vivo biodistribution via intratumoral injection
Delivering therapeutic siRNA to the target tissue directly (i.e., localized siRNA delivery) has the potential to reduce the adverse effects usually induced by systemic administration. In various mouse xenograft models, direct intratumoral injection of siRNA to achieve local delivery has been reported. Polyethyleneimine loaded with siRNA inhibited tumor growth after intratumoral injection into mice bearing glioblastoma xenografts (25). To prevent the recurrence of superficial bladder cancer postoperatively without inducing serious side effects induced by systemic administration, intravesical instillation of chemotherapy drugs (e.g., Calmette-Guerin, pirarubicin, and gemcitabine) is the most widely accepted treatment option. Therefore, the challenges associated with the systemic delivery of siRNA may be avoided in bladder cancer. To assess whether direct tumor injection in a mouse bladder cancer model could achieve an even distribution of siRNA throughout the tumor, HNTs/siRIPK4 were injected intratumorally into subcutaneous mouse xenografts. The distribution of FAM-siRIPK4 was assessed using in vivo imaging. Naked FAM-siRIPK4, HNTs, and phosphate-buffered saline (PBS) were used as controls. Figure 3A shows that 0.5 hour after injection with HNTs/FAM-siRIPK4, more intense fluorescence at the tumor tissue was observed compared with that of the naked FAM-siRIPK4 group. Consistently, the HNTs/FAM-siRIPK4–treated group had a significantly larger fluorescence distribution area than the naked FAM-siRIPK4–treated group. This suggested excellent tissue penetration of HNTs carrying FAM-siRIPK4 into the tumor sites and the subsequent distribution over a larger tumor area compared with that of the naked FAM-siRIPK4 group. At 16 hours after injection, the HNTs/FAM-siRIPK4 group still showed fluorescence at the tumor site, whereas no visible fluorescence was seen in the naked FAM-siRIPK4–, HNT-, or PBS-treated groups. In the tumor tissues, we speculated that the HNT-packaged FAM-siRIPK4 prevented aggregation and nonspecific protein adsorption of the nanoparticles; thus, HNTs/FAM-siRIPK4 remained longer at the tumor site than did the naked FAM-siRIPK4.
Fig. 3. Tumor transfection in vivo and inhibition of tumor growth by HNTs/siRIPK4.
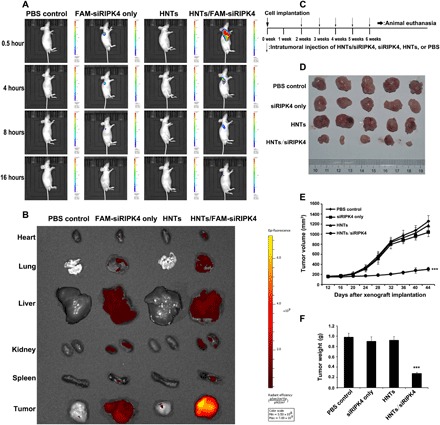
(A) In vivo imaging of T24 xenograft–bearing mice after intratumoral injection of HNTs/FAM-siRIPK4, free FAM-siRIPK4, HNTs, or PBS (n = 3 mice in each group). (B) In vivo imaging of T24 xenograft–bearing mice after tail vein injection of HNTs/FAM-siRIPK4, free FAM-siRIPK4, HNTs, or PBS (n = 3 mice in each group). (C) Timeline for the assessment of the antitumor activities of the HNTs/siRIPK4 complexes in the in vivo subcutaneous xenograft model. (D) Images of the tumors dissected from tumor-bearing mice receiving various treatments (n = 5 mice in each group). (E) Relative tumor volume in BALB/c nude mice with HNTs/siRIPK4 xenografts over time. (F) Weight of tumors dissected from mice after treatment. ***P < 0.001.
In vivo biodistribution via tail vein injection
We wondered if HNT nanoparticle–coated siRIPK4 could pass through the bloodstream to reach tumor sites, and whether they are stable in circulation. Mice were injected via the tail vein with PBS, free FAM-siRIPK4, HNTs, or HNTs/FAM-siRIPK4. The biodistribution of the complexes in mice was detected by the Xenogen IVIS Lumina system after 6 hours of injection. As shown in Fig. 3B, the HNTs/FAM-siRIPK4–injected group showed the accumulation of the HNTs/FAM-siRIPK4 complexes in the heart, lung, liver, kidneys, spleen, and, most importantly, tumor, whereas only the background of fluorescence in the tumor can be observed in the FAM-siRIPK4–injected group, suggesting that siRNA may be quickly cleared by the bloodstream in vivo in the absence of nanoparticle coats. Our data suggest that HNTs/FAM-siRIPK4 is stable in circulation and could be efficiently delivered to the tumor sites via tail vein injection in vivo.
Antitumor efficiency
Next, the gene knockdown and antitumor effects of the HNTs/siRIPK4 complexes were investigated in three bladder tumor models, including a subcutaneous model, an in situ bladder tumor model, and a tail vein injection model of lung metastasis.
Antitumor effect in a subcutaneous model
The antitumor growth effect of HNTs/siRIPK4 was compared with that of naked siRIPK4. Athymic mice bearing T24 cell xenografts were injected intratumorally with HNTs/siRIPK4 at 20 μg per injection once a week for five times in total (Fig. 3C). The HNTs/siRIPK4 treatment suppressed tumor growth most significantly among the treatments, whereas the free siRIPK4–induced tumor growth suppression was not significant compared with that in the control group (Fig. 3D). The tumor volumes in mice treated with HNTs/siRIPK4 were reduced by 77.4% compared with that of the control group (Fig. 3E). In contrast, the free siRIPK4 treatment had no significant effect on tumor growth (Fig. 3, D and E). The tumor weight in the HNTs/siRIPK4 group was also the lowest among all the groups (Fig. 3F). Moreover, suppression of the RIPK4 protein levels was only observed in the HNTs/siRIPK4 xenografts, as assessed by Western blotting and immunohistochemistry (IHC) (figs. S9 and S10). Thereafter, the IHC was used to analyze the expression of the proliferation-related gene Ki-67 to evaluate whether down-regulation of RIPK4 inhibited tumor tissue cell proliferation. Treatment with HNTs/siRIPK4 reduced the levels of the Ki-67 protein (fig. S11). To determine tumor cell apoptosis, the terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay was used. Tumors treated with HNTs/siRIPK4 had significantly increased numbers of apoptotic cells (fig. S11). Therefore, the delivery of siRIPK4 via HNTs specifically inhibited the growth of bladder cancer xenografts.
Antitumor effect in an in situ bladder tumor model
To test the clinical significance of the HNTs/siRIPK4 complexes, the tumor suppression effect of HNTs/siRIPK4 was studied in an in situ bladder cancer model, because urinary bladder instillation chemotherapy is one of the main treatments for bladder cancer. The rat in situ bladder cancer model was established successfully. Thereafter, the antitumor effects of HNTs/siRIPK4, HNTs, free siRIPK4, and PBS were determined. The weights of the rat bladders and the number of rat bladder lesions differed among the treatment groups (table S1). The observed differences in bladder weight and the histopathological changes among the different treatment groups were significant (P < 0.05). After treatment with HNTs/siRIPK4, most of the bladder tumors were in a lower stage (stage pT1/Ta), while in the PBS control group, most of them were in higher tumor stages (>pT2). This suggested that the therapeutic effect of HNTs/siRIPK4 was significantly better compared with that of PBS. This conclusion was confirmed histologically in excised bladders and in hematoxylin and eosin (H&E)–stained tissue slices (Fig. 4).
Fig. 4. In vivo siRIPK4 delivered by HNTs inhibits tumor promotion of T24 bladder cancer cells in an in situ model of bladder cancer (n = 12 rats in each group).
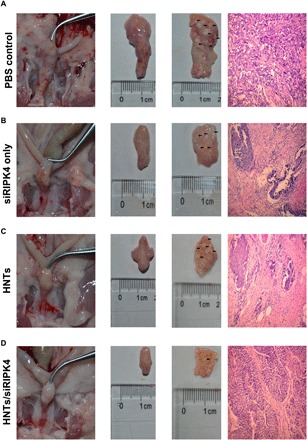
(A) Representative excised bladders and H&E-stained tissue slices (muscle-invasive bladder cancer: ≥stage pT2) treated with PBS. (B) Representative excised bladders and H&E-stained tissue slices (muscle-invasive bladder cancer: ≥stage pT2) treated with siRIPK4. (C) Representative excised bladders and H&E-stained tissue slices (muscle-invasive bladder cancer: ≥stage pT2) treated with HNTs. (D) Representative excised bladders and H&E-stained tissue slices (noninvasive papillary carcinoma: stage pTa) treated with HNTs/siRIPK4. (Photo credit: Jianye Liu and Ke Cao, The Third Xiangya Hospital of Central South University).
Antitumor effect in a tail vein injection model of lung metastasis
We injected the T24 cells into the tail veins of BALB/c nude mice to form metastatic lung tumors. Pulmonary metastasis nodules were counted in the excised lungs of each group (Fig. 5A). In the HNTs/siRIPK4–treated group, the formation of macroscopic lung tumor metastases was almost completely abrogated, which contrasted with the results for the PBS control and other groups. Compared with the PBS control, free siRIPK4, and HNTs groups, in mice treated with HNTs/siRIPK4, the numbers of pulmonary metastatic nodules decreased by 77.8, 76.5, and 76.9%, respectively. Moreover, the results of the H&E staining of lung sections demonstrated the superior suppressive effect of HNTs/siRIPK4 on macro- and micrometastatic foci formation in the lungs compared with those of the other groups (Fig. 5A). The occurrence of tumor invasion and metastasis could be promoted by the rapid growth and proliferation of tumor cells (26); thus, the remarkable antitumor effect of HNTs/siRIPK4 could account for its marked antimetastatic efficacy. Together, these results showed that in vivo, HNTs/siRIPK4 showed greater antitumor efficiency and antimetastatic effects than free siRIPK4 or the other treatments, suggesting the potential clinical application of this nanoparticle-based delivery system.
Fig. 5. In vivo inhibits tumor metastasis and toxicity of HNTs/siRIPK4 by tail vein injection of HNTs/siRIPK4 (n = 5 mice in each group).
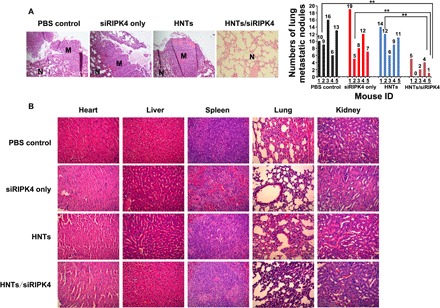
(A) In vivo siRIPK4 delivered by HNTs inhibits tumor metastasis in the tail vein injection of the lung metastasis model. N, normal lung; M, lung metastatic tumor. Separated lungs from mice receiving PBS, siRIPK4, HNTs, or HNTs/siRIPK4 were stained with H&E (left). Quantitative assessment of pulmonary metastatic nodules in the T24 tumor–bearing mice (right). **P < 0.01. (B) Twenty-four hours after the final intravenous injection of PBS, siRIPK4, HNTs, and HNTs/siRIPK4, H&E staining of tissue sections for histopathological analysis.
In vivo toxicity evaluation
The mammalian innate immune system is potently activated by free siRNA duplexes, leading to systemic in vivo inflammation resulting from the induction of high levels of inflammatory cytokines (27). This inflammatory response to siRNA could result in significant in vivo toxicity. We used the HNTs/siRIPK4 complexes to reduce the level of free siRIPK4–induced immunostimulation. We investigated the in vivo toxicity of the siRIPK4 complexes at various doses. H&E-stained sections of the liver, lung, heart, spleen, and kidney showed no marked histological changes between the HNTs/siRIPK4 group and the other groups (Fig. 5B). Thus, the HNTs/siRIPK4 complexes displayed no apparent organ toxicity. The liver and kidney functional parameters were within the normal range in the HNTs/siRIPK4 group (table S2). However, relatively high levels of alanine amino transferase (ALT) and aspartate amino transferase (AST) were induced in the free siRNA group (table S2), indicating that the free siRNA induced hepatic dysfunction (28). Moreover, therapeutic doses of HNTs/siRIPK4 induced no significant immune response, as the levels of serum cytokines, including interferon-α (IFN-α), interleukin-1β (IL-1β), IL-6, and tumor necrosis factor–α (TNF-α), were similar to those of the control group (fig. S12). By contrast, relatively high levels of IFN-α, IL-1β, IL-6, and TNF-α were induced in the free siRNA group (fig. S12) (27). Together, these results indicated that in vivo, the HNTs/siRIPK4 complexes are safer and less toxic than free siRNA.
Expression level of the nuclear factor κB signal pathway in tumor tissues
Next, we explored the downstream signaling pathway involved in RIPK4’s regulation of bladder cancer cell tumorigenesis and progression. In various diffuse large B cell lymphoma cell lines, up-regulation of RIPK4 activated nuclear factor κB (NF-κB), whereas inactivation of RIPK4 had the opposite effect (29). Consistently, overexpression of RIPK4 caused dose-dependent activation of NF-κB in 293T cells (30). In our previous study, we also observed that RIPK4 activated the NF-κB pathway in bladder cancer (21). Consequently, we used Western blotting and IHC assays to verify the HNTs/siRIPK4 complexes’ in vivo RNA interference (RNAi) mechanism after the animals were euthanized. Western blotting analyses of the levels of NF-κB–related proteins in each tumor mass showed significant reductions in mice treated with HNTs/siRIPK4 (fig. S9). However, in mice treated with unloaded HNTs or free siRIPK4, the NF-κB protein levels showed no significant decrease compared with that in the PBS control. Similar changes to those observed by Western blotting were seen upon immunohistochemical staining of subcutaneous tumor tissues (fig. S10). Thus, RIPK4’s regulation of the proliferation and progression of bladder cancer might involve NF-κB signaling as the downstream pathway.
DISCUSSION
In the present study, we constructed an siRNA-based gene silencing complex, termed HNTs/siRIPK4, to inhibit bladder cancer proliferation and progression effectively by depleting RIPK4 levels. HNTs/siRIPK4 down-regulated RIPK4 expression successfully via the siRNA/RNA-induced silencing complex pathway (Fig. 6). Analysis of the in vivo RNAi-mediated mechanism showed that the NF-κB signaling pathway could be the downstream signal pathway involved in RIPK4’s regulation of the proliferation and progression of bladder cancer.
Fig. 6. Delivery of HNTs/siRIPK4 complexes for RIPK4 silencing and bladder cancer therapy.
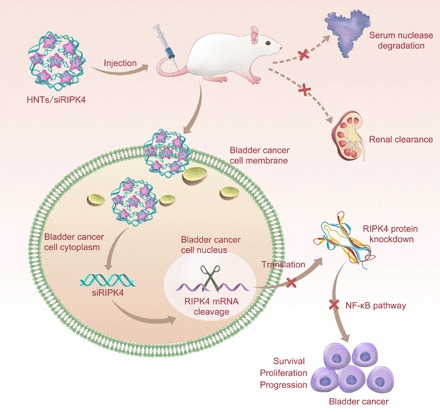
The HNTs/siRIPK4 complex protects the RIPK4 siRNA from serum degradation by nucleases and clearance through the kidneys, and promotes RIPK4 siRNA accumulation in tumor cells, thereby silencing RIPK4 in bladder tumors. Down-regulated RIPK4 expression inhibits bladder cancer proliferation and progression.
Gene silencing has been proposed as a treatment for human diseases, with siRNA as the most promising silencing mechanism. However, the intracellular delivery of siRNA to the specific tissues and organs expressing the target gene is the major challenge to their use in humans (7). Two practical considerations must be taken into account: the siRNA itself (stability and specificity) and its delivery system (31). In the bloodstream, serum nucleases degrade naked RNA, which can lead to stimulation of the innate immune system (32). In the present study, we showed that HNT-based delivery of siRNA increased their serum stability and decreased their off-target effects while maintaining their silencing ability. The application of HNTs to deliver nucleic acid molecules could resolve the problems of inefficient use, easy decomposition, and toxic side effects (16).
However, delivery issues still inhibit the production of safe, robust, and efficient siRNA-based treatments (33). Despite their small size, siRNA transit across the cell membrane is hampered by their negative charge and hydrophilicity. Furthermore, siRNA are rapidly eliminated from circulation before reaching the diseased tissue (34, 35). Thus, modifications and delivery systems for siRNA can have intended effects (targeted gene silencing) and unintended effects, such as immune stimulation, toxicity, and off-target silencing (35). For in vivo delivery, many materials have been explored, including lipids, polymers antibodies, peptides, aptamers, and small molecules.
To minimize the side effects of conventional nontargeted chemo- and small RNA therapeutics, we hypothesized that the physiochemical properties of the materials alone could endow selective delivery to specific cells. We identified selective HNTs that efficiently transfected bladder cancer cells in vitro and in vivo. We demonstrated that they could be used to develop cancer cell–specific drug carriers. The present study established the feasibility of this approach to increase on-target drug activity. It also showed that HNT carriers exhibit cell type–dependent delivery.
We showed that HNTs could deliver siRNA efficiently and specifically into bladder cancer cells to achieve targeted gene silencing. RIPK4 gene selective knockdown by HNTs/siRIPK4 inhibited in vitro and in vivo bladder cancer growth and progression by inhibiting proliferation and inducing apoptosis. The efficiency of siRNA uptake and gene knockdown using fusion protein complexes could be affected by the affinity of the antibody, the cell surface variations in the density of the receptor, the efficiency of receptor internalization and endocytosis, and their subsequent release from endosomes. Further study is needed to determine the mechanisms governing the release of the complexed siRNA from the endosomes, their entry into the cytoplasm, and the cellular location of RNA-induced silencing complex formation and activity, which are crucial bottlenecks for intracellular delivery (33). Generally, in early-phase clinical studies, liposomes and polymer-based particles for the systemic delivery of anticancer siRNA are retained in the liver or excreted through kidney filtration, and thus do not concentrate in tumors (35). By contrast, in vivo, siRIPK4 delivered by HNTs was targeted to, and accumulated in, the tumors. We hypothesized that the diameters of the siRIPK4 and HNT particles were small enough such that the complex can cross the hepatic sinusoidal endothelium and glomerular filtration barrier to access the malignant tissue.
The innate immune system expresses many pattern recognition receptors, like Toll-like receptors 3, 7, and 8 (TLR3, TLR7, and TLR8, respectively), double-stranded RNA (dsRNA)–dependent protein kinase (PKR), and the RNA helicase protein retinoic acid-inducible gene 1 protein (RIG-I), which recognize different aspects of RNA structure, and thus may affect siRNA delivery (36). siRNA-induced immune activation may cause transient increases in cytokine levels between 1 and 24 hours after siRNA administration (37). However, no marked changes in cytokines were detected at any time point after treatment, which contrasted with the increased IFN-α and IL-6 levels observed at 6 hours after the injection siRNA delivered via polyconjugates (38). The results of the present study showed that the antitumor activities of HNTs/siRIPK4 are not the result of siRNA-mediated nonspecific activation of the innate immune response. In addition, no treatment-related or dose-dependent toxicities of HNTs/siRIPK4s were observed, and they were very well tolerated.
Our results showed that HNTs/siRIPK4s have marked potential to treat bladder cancer; however, some challenges must be resolved before their clinical application. First, the purification process should be improved to eliminate potential contaminants, such as endotoxins. Second, optimization of the active targeting antibody, siRNA, and dosing regimen is required in rodents and nonhuman primates. Third, the trafficking pathway allowing the siRNA to exit the endosomes and enter the cytoplasm should be determined. These optimization and mechanistic studies should increase the antitumor activity of the HNTs/siRIPK4. The significance of the present study lies in the selective delivery of siRIPK4 to bladder tumors. Our previous finding that most of the bladder cancer cases express RIPK4 (21) indicated the therapeutic potential of targeting RIPK4. The results demonstrate the potential of siRNA to treat patients with bladder cancer.
In the present study, biocompatible HNTs were developed as a delivery platform for siRNA (HNTs/siRNA complex) to target RIPK4 for bladder cancer treatment via RIPK4 depletion. The HNTs/siRNA complex not only provides protection against the harsh biological environment but also passively targets tumor tissues. The HNTs/siRNA complex enhanced the specific in vitro and in vivo knockdown of RIPK4 in bladder cancer cells and bladder tumors, which effectively suppressed tumor growth and progression in three bladder tumor models with no adverse effects or toxicity. HNT-assisted delivery of siRNA targeting RIPK4 holds promise as a therapeutic approach to treat bladder cancer.
MATERIALS AND METHODS
Materials, siRNA, and cell lines
Tumor cell line T24 (human bladder cells; American Type Culture Collection) was grown in Dulbecco’s modified Eagle’s medium (DMEM; CellGro) with 10% heat-inactivated fetal bovine serum (FBS) (CellGro) in 5% CO2 at 37°C. The oligoribonucleotide sequence of the siNC and human RIPK4 siRNA (siRIPK4) were as follows: 5′-GACACCAGCAAACUGAUGATT-3′ (sense) and 5′-UCAUCAGUUUGCUGGUGUCCC-3′ (antisense) (Shanghai GenePharma Co. Ltd., Shanghai, China). Fluorescein-tagged siRNA (FAM-siRNA) was synthesized using fluorescein modification of the 3′-end of the sense strand. Mouse antibodies recognizing RIPK4 and β-actin were obtained from Abcam (Cambridge, UK). Rabbit antibodies recognizing marker of proliferation Ki-67 (Ki-67), inhibitor of NF-κB (IκB) kinase subunit β (IKK), phospho (p)–IKK, IκB, p-IκB, and NF-κB p65 subunit (NF-κB-p65) antibodies were purchased from Cell Signaling Technology (Denver, MA, USA).
HNTs/siRNA preparation and characterization
HNT aqueous solution preparation: 1.0 g of halloysite ore was processed for 90 s using high-speed crushing, 400-mesh sieving, 25-min ultrasound, and differential centrifugation (5 min at 3000 rpm and 10 min at 9000 rpm), and then centrifuged for 5 min at 10,000 rpm. The microstructure of the HNTs was characterized using scanning electron microscopy (TESCAN MIRA3 LMU), transmission electron microscopy (JEOL JEM-200CX), x-ray diffraction (DX-2700), and Fourier transform infrared spectra (Nexus 670).
siRNA loading: 30 μl of HNT aqueous solution (3 mg/ml) was mixed with 12 μl of siRNA solution [1 OD unit of siRNA was dispersed into 125 μl of DEPC (diethyl pyrocarbonate) water], vacuum impregnated for 3 hours, and then centrifuged for 5 min at 10,000 rpm. The siRNA loading efficiency was evaluated by gel electrophoresis of the supernatant.
Gel electrophoresis
Agarose gel electrophoresis (1%) was used to assess complex formation between HNTs and siRNA, using mass ratios of HNTs to siRNAs of 100:1, 50:1, 30:1, 20:1, 10:1, and 1:1. The resulting HNTs/siRNA complexes were left for 30 min to form complexes at room temperature, after which the samples were subjected to centrifugation at 5000 rpm for 5 min. The suspensions were reserved for analysis. The pellets were electrophoresed through 1% agarose gels and with ethidium bromide (0.1 μg/ml) at 100 V for 40 min in tris-acetate (TAE) buffer (0.045 M TAE; 0.001 M EDTA); the bands were visualized on an ultraviolet transilluminator.
In vitro transfection and the distribution of HNTs/FAM-siRIPK4
Briefly, in 24-well plates, trypsinized cells were seeded (n = 3) at 5 × 104 cells per well and incubated overnight until they reached 60 to 80% confluence, after which they were transfected with various formulations. FAM-siRNA uptake by the cells was assessed after 6 hours using an inverted fluorescence microscope (Olympus, IX71, Japan). In addition, cells were trypsinized, centrifuged (2000g, 3 min), resuspended in PBS, and analyzed using flow cytometry (Beckman Coulter).
Next, T24 cells were seeded in a 35-mm glass-bottom culture dish (MatTek Corp.) at 5 × 104 cells per well and incubated at 37°C in 5% CO2 for 24 hours. The cell culture medium was then substituted by HNTs/FAM-siRIPK4 complexes in 500 μl of serum-free DMEM in each well. At various time points, the cell culture medium was removed, and the cells were washed three times with PBS. DAPI (Sigma-Aldrich) was then used to stain the nuclei for 5 min. The cells were directly observed using confocal laser scanning microscopy with an Olympus FLUOVIEW confocal microscope and analyzed using the FV10-ASW viewer software (Olympus, Tokyo, Japan).
Lysosomal escape of siRNA
To study the escape of the siRNAs from lysosomes, T24 cells (1 × 105 cells per well) were seeded into a confocal dish and incubated for 24 hours. LysoTracker Red (75 nM, 1 hour, Invitrogen) was then used to stain the T24 cells. Thereafter, HNTs/FAM-siRNA and free FAM-siRNA (200 nM FAM-siRNA equivalent) were incubated with the T24 cells at 37°C for 1 hour. The cells were then rinsed and incubated in fresh medium for 1 to 6 hours. Samples were taken at different time points, the cells were fixed using 4% paraformaldehyde (PFA), and observed using confocal microscopy. LysoTracker Red and FAM-siRNA were detected at excitation wavelengths of 550 and 480 nm, respectively.
RIPK4 expression analysis
Only HNTs/siRIPK4 (100, 150, or 200 nM siRIPK4 at a mass ratio of 50:1), HNTs, HNTs/siNC (200 nM siNC), or PBS was used to transfect T24 cells (5 × 104) in 24-well plates (n = 3). RNA and proteins were harvested from the cells after 24 or 48 hours.
TRIzol reagent (Invitrogen, Carlsbad, CA, USA) was used to isolate total RNA, and a PrimeScript RT Reagent Kit (Promega, Madison, WI, USA) was used to synthesize cDNA. An ABI 7900HT fast real-time polymerase chain reaction (PCR) system (Applied Biosystems, Foster City, CA, USA) was used to perform real-time quantitative PCR (qPCR). The reaction comprised 30 cycles of 30 s at 94°C, 30 s at 55°C, and 60 s at 72°C. The qPCR primers for RIPK4 and ACTB (encoding β-actin) were as follows: RIPK4-specific primer, 5′-GCCCACTACCACGTCAAGAT-3′ (forward) and 5′-TTCACCATGATGTGCAGGAT-3′ (reverse); β-actin–specific primer, 5′-CATTAAGGAGAAGCTGTGCT-3′ (forward) and 5′-GTTGAAGGTAGTTTCGTGGA-3′ (reverse). SDS–polyacrylamide gel electrophoresis was used to resolve equal amounts of whole-cell lysates. The separated proteins were transferred electrophoretically onto a polyvinylidene difluoride membrane (Pall Corp., Port Washington, NY). Western blotting analysis was then carried out using standard protocols.
MTT assay
The T24 cells were plated in 96-well plates at 3000 cells per well in 100 μl of DMEM supplemented with 10% FBS. HNTs (10, 50, 100, 250, and 500 μg/ml) carrying 50 nM siNC, or Lipofectamine 3000 conjugated with the same amount siNC, or the siNC only, or PBS control was used to treat the cells for 6 hours. The cell culture medium was replaced with fresh medium, and the cells were incubated for another 72 hours. Following the addition of 20 μl of MTT reagent (5 mg/ml) to each well and incubation for 4 hours, the medium was replaced with 200 μl of dimethylsulfoxide (Sigma, St. Louis, MO, USA). The absorbance was then measured using a microplate reader at a wavelength of 490 nm.
Measurement of LDH in culture media by Enzyme-Linked Immunosorbent Assay
Cells grown in 10-cm dishes were treated at the indicated time as described in the figure legends. The culture media were collected, and the concentration of lactate dehydrogenase (LDH) was determined with an LDH ELISA (enzyme-linked immunosorbent assay) kit (Abcam) according to the manufacturer’s protocols.
Colony formation assays
After treatment with the indicated formulations for 48 hours, T24 cells in six-well plates (1 × 103 cells per well) were cultured for 10 to 14 days. After a clone had formed, the cells were washed twice using PBS, fixed with PFA, and stained using crystal violet.
Apoptosis analysis using flow cytometry
T24 cells were plated at a density of 3 × 105 per well in six-well plates. Forty-eight hours later, various formulations were added to the medium. Thereafter, an Annexin V–FITC Apoptosis Detection Kit (Medical & Biological Laboratories, Nagoya, Japan) was used to assess the apoptosis rate according to the manufacturer’s protocol.
Cell cycle assays
For cell cycle analysis, T24 cells were plated at a density of 5 × 104 cells per well in 24-well plates. At 48 hours after transfection with HNTs/siRIPK4, HNTs/siNC, unloaded HNTs, or PBS, cells in each well were trypsinized, fixed with 70% ethanol, and then stained using a Coulter DNA-Prep Reagents Kit (Beckman Coulter, Fullerton, CA). Flow cytometry was then used to detect the DNA content in the cells of each sample (Becton Dickinson, San Jose, CA, USA).
In vivo fluorescence imaging
We purchased female BALB/c-nu/nu athymic mice (4 to 5 weeks old) from the Shanghai Slac Laboratory Animal Co. Ltd. (Shanghai, China). The Institute Research Medical Ethics Committee of The Third Xiangya Hospital of Central South University approved the animal care and experimental protocols used in this study.
T24 cells (4 × 107) in 100 μl of PBS were injected subcutaneously to grow tumors. When the tumors developed, the mice were randomly assigned to four treatment groups (n = 3 mice), which were injected intratumorally or via the tail intravenously with 400 μl of HNTs/FAM-siRIPK4, or an equal dose of free FAM-siRIPK4, HNTs only, or PBS solution, respectively. The mice were placed onto a prewarmed stage inside an IVIS light-tight chamber after intratumoral injection. The mice were kept under anesthesia using 2.5% isoflurane. The Xenogen IVIS Lumina system (Caliper Life Sciences, USA) was used to acquire images, and Living Image 3.1 software (Caliper Life Sciences) was used to analyze the results. For in vivo distribution of the complex, mice were euthanized 6 hours after tail intravenous injection. The heart, lung, liver, kidney, spleen, and tumor tissues were harvested and analyzed by the Xenogen IVIS Lumina system and Living Image 3.1 software.
In vivo antitumor activity in a model of subcutaneous bladder cancer
T24 cells were allowed to form a monolayer, after which trypsin digestion was used for form a single-cell suspension. The cell density was adjusted to 4 × 107 in 100 μl of PBS and then subcutaneously injected into the right flank of each mouse. Once the T24 cells developed a tumor (tumor volume about 100 mm3), the mice were randomly assigned to four treatment groups (n = 5 mice in each group). The tumors of the mice in each group were injected directly with HNTs/siRIPK4 (20 μg of siRIPK4 per injection, 50:1 mass ratio) or an equal amount of unloaded HNTs, siRIPK4 only, or PBS, respectively. The injections were repeated once a week for 5 weeks. The long and short axial lengths of the tumors were measured every other day. The tumor xenografts were harvested, weighed, and snap frozen for cryosectioning or formalin fixed for paraffin sectioning. H&E was used to stain the histological sections of the tumors. Tumor apoptosis was examined histologically using the TUNEL assay on primary tumor sections using a Colorimetric TUNEL Apoptosis Assay Kit (Beyotime, Haimen, China) according to the manufacturer’s protocol. A Dako Real Envision Kit (K5007, Dako) was then used for immunohistochemical staining with primary antibodies to detect the protein levels.
In vivo antitumor activity in a model of in situ bladder cancer
Female Sprague-Dawley rats (aged 6 to 7 weeks; 120 to 150 g) were purchased from Shanghai Slac Laboratory Animal Co. Ltd. Ether inhalation was used to anesthetize the rats. N-methyl-nitrosurea solution (0.2 ml at 10 mg/ml; Sigma, St. Louis, MO, USA) was used to infuse the rat bladders via a 22-gauge angiocatheter every other week for five times. The animals remained anesthetized for approximately 45 min after catheterization to avoid spontaneous micturition (39).
Tumors were successfully induced at 16 weeks. Thereafter, 48 rats were assigned to four groups with 12 rats in each group. From the 16th week, and after anesthetizing the rats, their bladders were instilled with 500 μl of HNTs/siRIPK4, an equal dose of free HNTs, siRIPK4 only, or PBS solution. After instillation, the animals were kept under anesthesia for about 45 min to avoid spontaneous micturition. The treatments were repeated once a week for 5 weeks. Two days after the end of therapy, the animals were euthanized. Their bladders were removed, weighed, fixed in 4% PFA for 24 hours, embedded in paraffin, and analyzed histopathologically. Transverse sections were cut through the midportion of the bladder and subjected to H&E staining.
In vivo antitumor activity in a lung metastasis model
BALB/c nude mice were divided randomly into three groups (n = 5 per group) and treated through the tail vein with bladder cancer cells (2.5 × 106) that had been treated with HNTs/siRIPK4, an equal dose of free HNTs, siRIPK4 only, or PBS solution every 7 days for five times, respectively. Fort-eight hours after cessation of therapy, the animals were euthanized. The mouse lungs were excised, photographed, and examined histologically. In each lung, the numbers of macroscopic metastatic nodules were counted.
Toxicity assay in mice
At 24 hours after the final intravenous injection, the major organs (heart, liver, spleen, lungs, and kidney) of the mice were excised, fixed, sectioned, and stained with H&E for histological analysis. The tissue slices were subjected to microscopic observation (Leica DMI 6000B). Before organ collection, an electronic balance was used to determine the body weights of the mice. At 24 hours after the final intravenous injection, blood was sampled from the venous plexus of the mice’s eyes. The blood samples were subjected to immediate centrifugation at 3000g for 5 min at 4°C. Hematological analysis was then performed on the supernatant. The blood biochemical analysis comprised determining the ALT, AST, blood urea nitrogen, and creatinine levels using Modular Analytics (Roche, Germany). ALT and AST were used as indicators of hepatic functions, and blood urea nitrogen and creatinine were used as indicators of renal functions. To assess the immunotoxicity of the various formulations, we determined cytokines present in the serum using ELISA kits for IL-1β, IL-6, IFN-α, and TNF-α (Abcam).
Statistical analysis
Data are presented as means ± SD. For comparisons between two groups, the unpaired Student’s t test (two tailed) was used. For multiple group analysis, one-way analysis of variance (ANOVA) with Bonferroni tests was used. Statistical significance was considered at a P value less than 0.05.
Supplementary Material
Acknowledgments
We thank the Hunan Key Laboratory of Pharmacodynamics and Safety Evaluation of New Drugs for performing the animal experiment. Funding: This work was supported by the National Natural Science Foundation of China (81802556 and 81874137), the Natural Science Foundation of Hunan Province (2019JJ30039), the New Xiangya Talent Projects of The Third Xiangya Hospital of Central South University (JY201615), and the Scientific Projects of Health and Family Planning Commission of Hunan Province (B2017034). Author contributions: K.C. and J.L. designed the experiments. J.L., Y.Z., Q.Z., H.Z., H.X., and H.B. performed the experiments. J.L., XY.L., L.H., Z.L., and M.X. analyzed the data. K.C., J.L., YX.Z., Q.Z., P.W., XM.L., and Y.Z. wrote the paper. All the authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaaw6499/DC1
Fig. S1. Efficiency and cytotoxicity of the siRNA binding to HNTs.
Fig. S2. Lysosomal escape of HNTs/siRIPK4 in T24 bladder cancer cells.
Fig. S3. Intracellular trafficking and lysosomal escape of HNTs/siRIPK4.
Fig. S4. qRT-PCR of RIPK4 mRNA and Western blotting analysis of the RIPK4 protein in T24 cells after HNTs/siRIPK4 transfection.
Fig. S5. HNTs/siRIPK4 inhibited the growth of T24 cells in vitro by MTT assay and LDH assay.
Fig. S6. HNTs/siRIPK4 inhibited the growth of T24 cells in vitro (colony formation).
Fig. S7. Annexin V and propidium iodide staining of T24 cells treated with HNTs/siRIPK4 complexes and other formulations, and analysis of apoptosis using flow cytometry.
Fig. S8. After HNT-mediated RIPK4 knockdown in T24 cells, FACS was used to analyze the cell cycle.
Fig. S9. Tumor tissues analyzed using Western blotting from the subcutaneous xenograft model.
Fig. S10. Tumor tissues analyzed using IHC from the subcutaneous xenograft model.
Fig. S11. HNTs/siRIPK4 reduced Ki-67 expression and induced apoptosis of mice bearing T24 xenografts.
Fig. S12. Immunotoxicity analysis of blood of mice at 24 hours after PBS, siRIPK4, HNTs, and HNTs/siRIPK4 injection.
Table S1. Tumor suppression effect of different treatment on bladder weight and histopathologic changes in SD rat bladders of different groups.
Table S2. Biochemical parameters of blood from the mice at 24 h after last injection of various drugs.
REFERENCES AND NOTES
- 1.Davidson B. L., McCray P. B. Jr., Current prospects for RNA interference-based therapies. Nat. Rev. Genet. 12, 329–340 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grimm D., Small silencing RNAs: State-of-the-art. Adv. Drug Deliv. Rev. 61, 672–703 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Kanasty R. L., Whitehead K. A., Vegas A. J., Anderson D. G., Action and reaction: The biological response to siRNA and its delivery vehicles. Mol. Ther. 20, 513–524 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burnett J. C., Rossi J. J., RNA-based therapeutics: Current progress and future prospects. Chem. Biol. 19, 60–71 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsumura Y., Maeda H., A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 46, 6387–6392 (1986). [PubMed] [Google Scholar]
- 6.Narayanan S., Binulal N. S., Mony U., Manzoor K., Nair S., Menon D., Folate targeted polymeric ‘green’ nanotherapy for cancer. Nanotechnology 21, 285107 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Davis M. E., Zuckerman J. E., Choi C. H., Seligson D., Tolcher A., Alabi C. A., Yen Y., Heidel J. D., Ribas A., Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 464, 1067–1070 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitehead K. A., Langer R., Anderson D. G., Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 8, 129–138 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanasty R., Dorkin J. R., Vegas A., Anderson D., Delivery materials for siRNA therapeutics. Nat. Mater. 12, 967–977 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Goldberg M. S., Xing D., Ren Y., Orsulic S., Bhatia S. N., Sharp P. A., Nanoparticle-mediated delivery of siRNA targeting Parp1 extends survival of mice bearing tumors derived from Brca1-deficient ovarian cancer cells. Proc. Natl. Acad. Sci. U.S.A. 108, 745–750 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conde J., Edelman E. R., Artzi N., Target-responsive DNA/RNA nanomaterials for microRNA sensing and inhibition: The jack-of-all-trades in cancer nanotheranostics? Adv. Drug Deliv. Rev. 81, 169–183 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Khan O. F., Kowalski P. S., Doloff J. C., Tsosie J. K., Bakthavatchalu V., Winn C. B., Haupt J., Jamiel M., Langer R., Anderson D. G., Endothelial siRNA delivery in nonhuman primates using ionizable low–molecular weight polymeric nanoparticles. Sci. Adv. 4, eaar8409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinstein S., Peer D., RNAi nanomedicines: Challenges and opportunities within the immune system. Nanotechnology 21, 232001 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Guo P., Coban O., Snead N. M., Trebley J., Hoeprich S., Guo S., Shu Y., Engineering RNA for targeted siRNA delivery and medical application. Adv. Drug Deliv. Rev. 62, 650–666 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williford J. M., Wu J., Ren Y., Archang M. M., Leong K. W., Mao H. Q., Recent advances in nanoparticle-mediated siRNA delivery. Annu. Rev. Biomed. Eng. 16, 347–370 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Lvov Y., Wang W., Zhang L., Fakhrullin R., Halloysite clay nanotubes for loading and sustained release of functional compounds. Adv. Mater. 28, 1227–1250 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Xue J., Niu Y., Gong M., Shi R., Chen D., Zhang L., Lvov Y., Electrospun microfiber membranes embedded with drug-loaded clay nanotubes for sustained antimicrobial protection. ACS Nano 9, 1600–1612 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y., Tang A., Yang H., Ouyang J., Applications and interfaces of halloysite nanocomposites. Appl Clay Sci. 119, 8–17 (2016). [Google Scholar]
- 19.Huang X., McGann J. C., Liu B. Y., Hannoush R. N., Lill J. R., Pham V., Newton K., Kakunda M., Liu J., Yu C., Hymowitz S. G., Hongo J.-A., Wynshaw-Boris A., Polakis P., Harland R. M., Dixit V. M., Phosphorylation of Dishevelled by protein kinase RIPK4 regulates Wnt signaling. Science 339, 1441–1445 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu D.-Q., Li F.-F., Zhang J.-B., Zhou T.-J., Xue W.-Q., Zheng X.-H., Chen Y.-B., Liao X.-Y., Zhang L., Zhang S.-D., Hu Y.-Z., Jia W.-H., Increased RIPK4 expression is associated with progression and poor prognosis in cervical squamous cell carcinoma patients. Sci. Rep. 5, 11955 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu J.-Y., Zeng Q.-H., Cao P.-G., Xie D., Chen X., Yang F., He L.-Y., Dai Y.-B., Li J.-J., Liu X.-M., Zeng H.-L., Zhu Y.-X., Gong L., Cheng Y., Zhou J.-D., Hu J., Bo H., Xu Z.-Z., Cao K., RIPK4 promotes bladder urothelial carcinoma cell aggressiveness by upregulating VEGF-A through the NF-κB pathway. Br. J. Cancer 118, 1617–1627 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H., Zhang W., Zhu G., Xie J., Chen X., Rethinking cancer nanotheranostics. Nat. Rev. Mater. 2, 17024 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun T., Zhang Y. S., Pang B., Hyun D. C., Yang M., Xia Y., Engineered nanoparticles for drug delivery in cancer therapy. Angew. Chem. Int. Ed. Engl. 53, 12320–12364 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Ma G., Ren Y., Wang K., He J., SRC-3 has a role in cancer other than as a nuclear receptor coactivator. Int. J. Biol. Sci. 7, 664–672 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niu X. Y., Peng Z. L., Duan W. Q., Wang H., Wang P., Inhibition of HPV 16 E6 oncogene expression by RNA interference in vitro and in vivo. Int. J. Gynecol. Cancer 16, 743–751 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Koontongkaew S., The tumor microenvironment contribution to development, growth, invasion and metastasis of head and neck squamous cell carcinomas. J. Cancer 4, 66–83 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Judge A. D., Sood V., Shaw J. R., Fang D., McClintock K., MacLachlan I., Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 23, 457–462 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Wang J., Zhou G., Chen C., Yu H., Wang T., Ma Y., Jia G., Gao Y., Li B., Sun J., Li Y., Jiao F., Zhao Y., Chai Z., Acute toxicity and biodistribution of different sized titanium dioxide particles in mice after oral administration. Toxicol. Lett. 168, 176–185 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Kim S.-W., Oleksyn D. W., Rossi R. M., Jordan C. T., Sanz I., Chen L., Zhao J., Protein kinase C–associated kinase is required for NF-κB signaling and survival in diffuse large B-cell lymphoma cells. Blood 111, 1644–1653 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Meylan E., Martinon F., Thome M., Gschwendt M., Tschopp J., RIP4 (DIK/PKK), a novel member of the RIP kinase family, activates NF-κB and is processed during apoptosis. EMBO Rep. 3, 1201–1208 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee S. J., Son S., Yhee J. Y., Choi K., Kwon I. C., Kim S. H., Kim K., Structural modification of siRNA for efficient gene silencing. Biotechnol. Adv. 31, 491–503 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Jackson A. L., Linsley P. S., Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 9, 57–67 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Nguyen J., Szoka F. C., Nucleic acid delivery: The missing pieces of the puzzle? Acc. Chem. Res. 45, 1153–1162 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ashihara E., Kawata E., Maekawa T., Future prospect of RNA interference for cancer therapies. Curr. Drug Targets 11, 345–360 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Oh Y. K., Park T. G., siRNA delivery systems for cancer treatment. Adv. Drug Deliv. Rev. 61, 850–862 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Whitehead K. A., Dahlman J. E., Langer R. S., Anderson D. G., Silencing or stimulation? siRNA delivery and the immune system. Annu. Rev. Chem. Biomol. Eng. 2, 77–96 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Robbins M., Judge A., MacLachlan I., siRNA and innate immunity. Oligonucleotides 19, 89–102 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Rozema D. B., Lewis D. L., Wakefield D. H., Wong S. C., Klein J. J., Roesch P. L., Bertin S. L., Reppen T. W., Chu Q., Blokhin A. V., Hagstrom J. E., Wolff J. A., Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc. Natl. Acad. Sci. U.S.A. 104, 12982–12987 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steinberg G. D., Brendler C. B., Ichikawa T., Squire R. A., Isaacs J. T., Characterization of an N-methyl-N-nitrosourea-induced autochthonous rat bladder cancer model. Cancer Res. 50, 6668–6674 (1990). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaaw6499/DC1
Fig. S1. Efficiency and cytotoxicity of the siRNA binding to HNTs.
Fig. S2. Lysosomal escape of HNTs/siRIPK4 in T24 bladder cancer cells.
Fig. S3. Intracellular trafficking and lysosomal escape of HNTs/siRIPK4.
Fig. S4. qRT-PCR of RIPK4 mRNA and Western blotting analysis of the RIPK4 protein in T24 cells after HNTs/siRIPK4 transfection.
Fig. S5. HNTs/siRIPK4 inhibited the growth of T24 cells in vitro by MTT assay and LDH assay.
Fig. S6. HNTs/siRIPK4 inhibited the growth of T24 cells in vitro (colony formation).
Fig. S7. Annexin V and propidium iodide staining of T24 cells treated with HNTs/siRIPK4 complexes and other formulations, and analysis of apoptosis using flow cytometry.
Fig. S8. After HNT-mediated RIPK4 knockdown in T24 cells, FACS was used to analyze the cell cycle.
Fig. S9. Tumor tissues analyzed using Western blotting from the subcutaneous xenograft model.
Fig. S10. Tumor tissues analyzed using IHC from the subcutaneous xenograft model.
Fig. S11. HNTs/siRIPK4 reduced Ki-67 expression and induced apoptosis of mice bearing T24 xenografts.
Fig. S12. Immunotoxicity analysis of blood of mice at 24 hours after PBS, siRIPK4, HNTs, and HNTs/siRIPK4 injection.
Table S1. Tumor suppression effect of different treatment on bladder weight and histopathologic changes in SD rat bladders of different groups.
Table S2. Biochemical parameters of blood from the mice at 24 h after last injection of various drugs.