

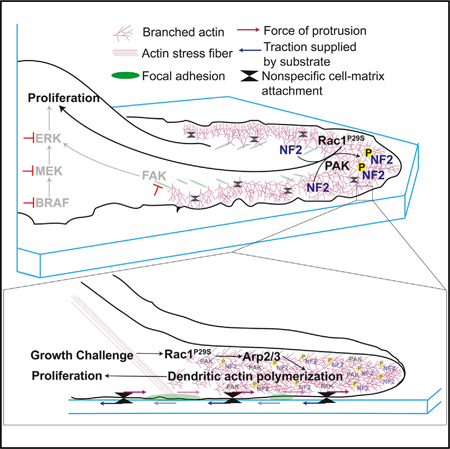
SUMMARY
Actin assembly supplies the structural framework for cell morphology and migration. Beyond structure, this actin framework can also be engaged to drive biochemical signaling programs. Here, we describe how the hyperactivation of Rac1 via the P29S mutation (Rac1P29S) in melanoma hijacks branched actin network assembly to coordinate proliferative cues that facilitate metastasis and drug resistance. Upon growth challenge, Rac1P29S-harboring melanoma cells massively upregulate lamellipodia formation by dendritic actin polymerization. These extended lamellipodia form a signaling microdomain that sequesters and phospho-inactivates the tumor suppressor NF2/Merlin, driving Rac1P29S cell proliferation in growth suppressive conditions. These biochemically active lamellipodia require cell-substrate attachment but not focal adhesion assembly and drive proliferation independently of the ERK/MAPK pathway. These data suggest a critical link between cell morphology and cell signaling and reconcile the dichotomy of Rac1’s regulation of both proliferation and actin assembly by revealing a mutual signaling axis wherein actin assembly drives proliferation in melanoma.
In Brief
The RhoGTPase Rac1 is a regulator of cell morphology and proliferation. Mohan et al. report that these functions converge in Rac1P29S-mutant melanoma cells. Under growth challenge, Rac1P29S cells form extended lamellipodia that sequester and phospho-inactivate NF2/ Merlin, resulting in sustained cell proliferation that is advantageous for metastasis and drug tolerance.
Graphical Abstract
INTRODUCTION
Rac1 is a master regulator of actin polymerization (Ridley et al., 1992). Rac1 is also required for cell cycle progression and oncogenic transformation (Olson et al., 1995; Qiu et al., 1995). This dichotomous regulation of actin assembly and cell proliferation have historically been described as independent functions of Rac1. Mechanisms that link Rac1-mediated cytoskeleton regulation and cell cycle progression remain tenuous (Lamarche et al., 1996; Hall, 1998; Coleman et al., 2004).
Systematic investigation of such mechanisms is increasingly necessary as discoveries of clinically relevant dysregulation of Rac1 in cancers are becoming ever more prevalent. As an effector of Ras, Rac1 is likely dysregulated in the ~30% of all cancers that harbor Ras mutations. Rac1 is required for Rasmediated tumorigenesis and metastasis, further suggesting the importance of this altered signaling (Kissil et al., 2007; Qiu et al., 1995; Malliri et al., 2002; Chow et al., 2012). In a cohort of patients with invasive melanoma, 47% of tumors were found to be immunoreactive for Rac1 activity (Mar et al., 2014), and melanomas refractory to combinatorial treatment with MAPK pathway inhibitors show striking sensitivity to inhibition of the Rac1 effector protein, PAK (Lu et al., 2017; Araiza-Olivera etal., 2018).
Rac1 is also dysregulated through a wide assortment of mutations and expression changes in regulatory proteins including guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). The Rac1-activating GEFs Tiam1, P-REX1, and Vav have oncogenic roles in skin, breast, prostate, renal cell, colon, and pancreatic cancer (Malliri et al., 2002; Sosa et al., 2010; Cook et al., 2014; Wertheimer et al., 2012). In melanoma, overexpression of and activating mutations in the GEFs TIAM-1 and PREX-1,2 are associated with tumor growth and invasion (Cook et al., 2014). These GEFs, along with Vav-1 and Dock 3, are mutated at a cumulative frequency of 89.5% for the set of 469 patient cases listed in The Cancer Genome Atlas’ Skin Cutaneous Melanoma database (cancer.gov/tcga).
Of the ongoing discoveries of hyperactivating point mutations in Rac1 itself, the Rac1P29S mutation is the most common. Rac1P29S was found originally in melanomas (Hodis et al., 2012; Krauthammer et al., 2012), but is emerging also in breast, endometrial, and head and neck cancers (Chang et al., 2016; Kawazu et al., 2013). In line with the phenotypic presentation of other modalities of Rac1 pathway hyperactivation, the Rac1P29S mutation is associated with increased tumor aggressiveness such as increased frequency of nodal metastases and drug resistance (Watson et al., 2014; Chen et al., 2016). As with GEF and GAP modifications, the Rac1P29S mutation preserves the GTP hydrolysis activity of Rac1 (Davis et al., 2013; Kawazu et al., 2013). Located in the Switch I loop of Rac1, the P29S mutation results in fast cycling of GDP/GTP exchange (Davis et al., 2013). This contrasts the constitutively active Rac1G12V and Rac1Q61L mutations, which have been designed in the lab to study properties and function of Rac1 pathways but which are largely absent in tumors (Lin et al., 1997; Olson, 2018).
Despite the clear clinical evidence of Rac1 hyperactivation as an oncogenic event, little insight exists regarding the mechanism by which shifts in Rac1 signaling promote aggressive cancers. To further investigate this mechanism, we used the Rac1P29S point mutation as a pathologically relevant, directly-hyperactivating perturbation of Rac1 that is likely to represent the effects of the broad class of more indirect Rac1 hyperactivating conditions.
Our experiments show that hyperactivation of Rac1 primes cells with the abilityto engage the actin cytoskeleton during growth challenge to coordinate proliferative signaling. Rac1P29S cells enhance dendritic actin polymerization forming dramatic lamellipodial structures that sequester and phospho-inactivate the tumor suppressor NF2/Merlin. Thus, Rac1 regulation of the actin cytoskeleton directs subsequent proliferative signaling. This reconciles Rac1’s dichoto-mous function as a proliferative and cytoskeletal regulator and sheds light on a critical role of cell morphology in signaling.
RESULTS
Rac1P29S Confers Growth Advantage to Melanoma Cells in Metastases and upon MAPKi Treatment
Rac1 activity has strong implications for oncogenic and especially metastasis-promoting cell functions (Machesky, 2008; Etienne-Manneville and Hall, 2002; Bid et al., 2013). To investigate whether hyperactivation of Rac1 through the P29S mutation results in increased metastasis, we used a mouse xenograft tumor protocol previously shown to recapitulate the metastatic potential of tumors in melanoma patients (Quintana et al., 2012). Xenograft tumors were created by injecting A375 melanoma cells exogenously expressing Rac1P29S (+P29S), Rac1WT (+WT), and empty vector (+EV) subcutaneously into NSG mice. To rule out overexpression artifacts and cell line specific effects, a separate cohort of mice were injected with IGR1 melanoma cells that endogenously express the Rac1P29S mutation (IGR1P29S). To generate the appropriate control, we used CRISPR-Cas9 genome editing to introduce a single base-pair change that reverted the Rac1P29S mutation to Rac1WT (IGR1WT) (Figures S1A and S1B). At the endpoint of primary tumor growth (2.5 cm), bioluminescence imaging (BLI) of whole-body organ dissections was applied to detect and quantify both micro-and macro-metastases (Figures 1A and 1B; Data S1A and S1B). These experiments revealed that the overall metastatic burden was significantly increased for mice with xenograft tumors expressing Rac1P29S (Figures 1C and 1D), suggesting that the mutation drives a metastatic advantage in melanoma. Gross observation as well as histological quantification of lung metastases revealed that the size of lung metastases in mice with +P29S xenograft tumors was strikingly larger than those in +EV and +WT controls (Figures 1E and 1F; Data S1C). This suggested that expression of Rac1P29S might confer a growth advantage in metastases.
Figure 1. Rac1P29S Confers Growth Advantage to Melanoma Cells in Metastases.
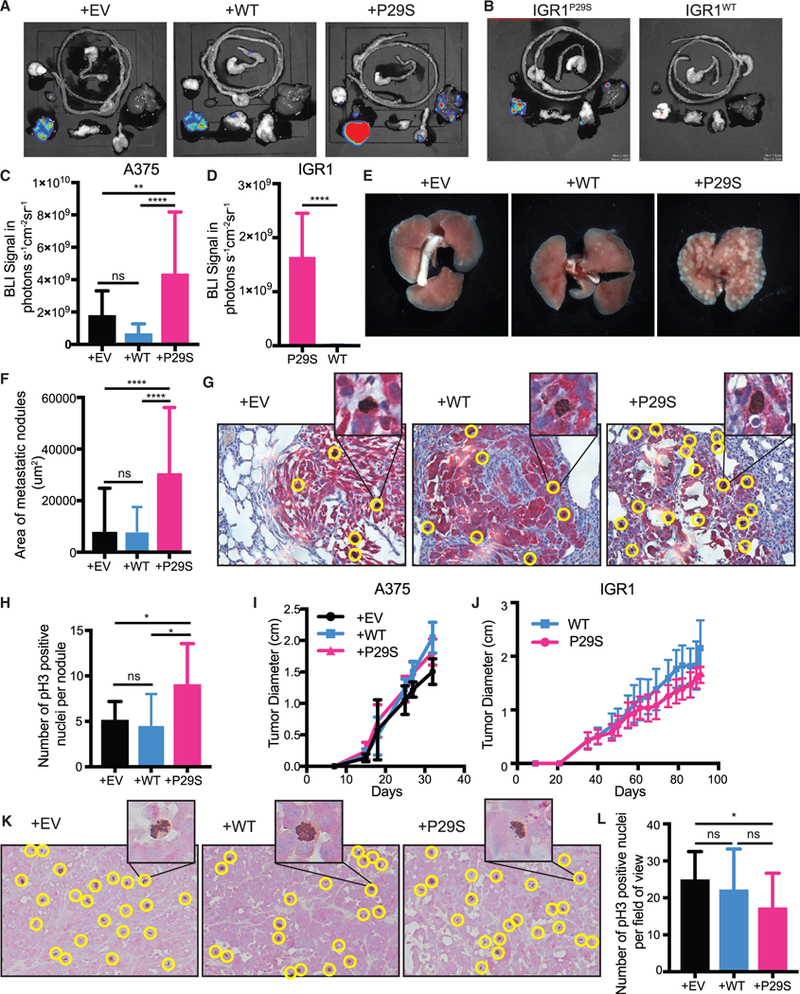
(A-E) Endpoint metastatic burden in mice with tumors that were between 2.0–2.5 cm. A375, n = 20 mice per condition from two independent experiments, IGR1, n = 10 mice per condition, see also Data S1.
(A-D) Bioluminescence imaging (BLI) signal detected in dissected organs.
(E) Macrometastases in gross lungs post fixation in 10% neutral-buffered formalin for 48 h.
(F-H) Paraffin-embedded lungs were sectioned and dual-labeled with anti-dsRed antibody for tumor nodule detection and anti-phospho Histone 3 for detection of mitotic nuclei.
(F) Area of metastatic nodules determined using pixel classification of dsRed staining by Ilastik machine learning software (Sommer et al., 2011).
(G) Representative images of dual staining of metastatic melanoma cells (red) and mitotic nuclei (brown, positive nuclei circled in yellow).
(H) All metastatic lung nodules controlled for size were randomized and hand counted for phospho-Histone 3 positive nuclei to determine number of proliferating cells (n ≥ 9 metastatic lung nodules for each condition).
(I and J) Growth of xenograft tumors in NSG mice from (I) A375 melanoma cells exogenously expressing Rac1P29S (+P29S), Rac1WT(+WT), or empty vector (+EV) (n = 20 mice precondition from two independent experiments) and from (J) IGR1 cells with endogenous Rac1 P29S(P29S) or endogenous Rac1WT(WT), reverted by CRISPR-Cas9 genome editing (n = 10 mice per condition).
(K and L) Paraffin-embedded primary-xenograft tumors were sectioned and dual-labeled with anti-dsRed antibody for tumor cell detection (red) and anti-phospho Histone 3 for detection of mitotic nuclei (brown).
(K) Positive nuclei, circled yellow in representative images, were (L) hand counted across each 20x magnification field of view acquired to determine number of proliferating cells (n ≥ 20 fields of view for each condition). All data represent mean ± SD. Statistical significance was assessed by one-way ANOVA with Tukey’s multiple comparisons test (C, F, H, and L) and unpaired Student’s t-test (D). *p < 0.05, **p < 0.01, ****p < 0.0001.
See also Figure S1.
One of the factors that must be overcome for metastatic growth is proliferation-limiting conditions at the site of colonization (Shibue and Weinberg, 2009). To test if metastases expressing Rac1P29S have a proliferative advantage, we used phospho-Histone 3 as a marker for mitotic nuclei to detect proliferating cells in lung metastases. Metastatic nodules in mice with +P29S primary tumors had a significantly greater number of mitotic nuclei than those in mice with +EV or +WT tumors, suggesting that metastases with Rac1P29S indeed have increased proliferation (Figures 1G and 1H). Strikingly, this growth advantage upon Rac1P29S expression was absent in the primary tumors. The primary tumors expressing Rac1P29S grew at the same or even slower rates than primary tumors expressing Rac1WT (Figures 1I and 1J). Detection of mitotic nuclei in the primary tumors by labeling phospho-Histone 3 revealed no difference in proliferation between tumors expressing Rac1WT and Rac1P29S (Figures 1K and 1L). This suggests that proliferation and growth at the primary injection site are not affected by the Rac1P29S mutation. We reasoned that expression of Rac1P29S might confer a proliferative advantage specifically during metastasis, bestowing melanoma cells with a growth advantage in secondary sites that is not at play in the primary tumor.
Consistent with the insensitivity of the primary tumor growth rates to the Rac1-mutational status in vivo, control cells and cells expressing Rac1P29S exhibit similar proliferation in vitro (Figures 2A and 2B). We reasoned that a growth challenge like the one experienced by metastases in vivo would be necessary to observe the Rac1P29S-driven proliferative advantage with cell cultures in vitro. Inhibition of the MAPK pathway (MAPKi) using drugs that target BRAF and MEK is a common modality to inhibit melanoma cell growth. We tested if growth suppression by MAPKi treatment with the BRAF inhibitor, Dabrafenib, and the MEK inhibitor, Trametinib, would reveal a Rac1P29S proliferative advantage in vitro. Indeed, upon MAPKi treatment, control cells suffer a massive drop in proliferation while cells expressing Rac1P29S exhibit sustained proliferation (Figures 2C–2E). Cell death upon MAPKi treatment was negligible for both control cells and Rac1P29S cells (Figures S2A and S2B). Similarly, while IGR1P29S cells proliferate at a slightly higher rate than IGR1WT cells under baseline conditions (Figure 2F), the most prominent proliferative advantage for IGR1P29S was noted upon MAPKi treatment. IGR1P29S cells exhibit sustained proliferation under both BRAF and MEK inhibition, whereas proliferation in the CRISPR-reverted IGR1WT cells is sensitive to these drugs (Figures 2G and 2H). These data, along with the in vivo results, suggest that Rac1 hyperactivity drives a proliferative mechanism that is highly advantageous to melanoma cells specifically upon growth challenge. Additionally, this proliferative advantage provides an explanation for the recently observed resistance of Rac1P29S cells to MAPKi treatment (Watson et al., 2014) (Figures S2C and S2D).
Figure 2. Rac1P29S Confers a Proliferative Advantage to Melanoma Cells upon MAPKi Treatment.
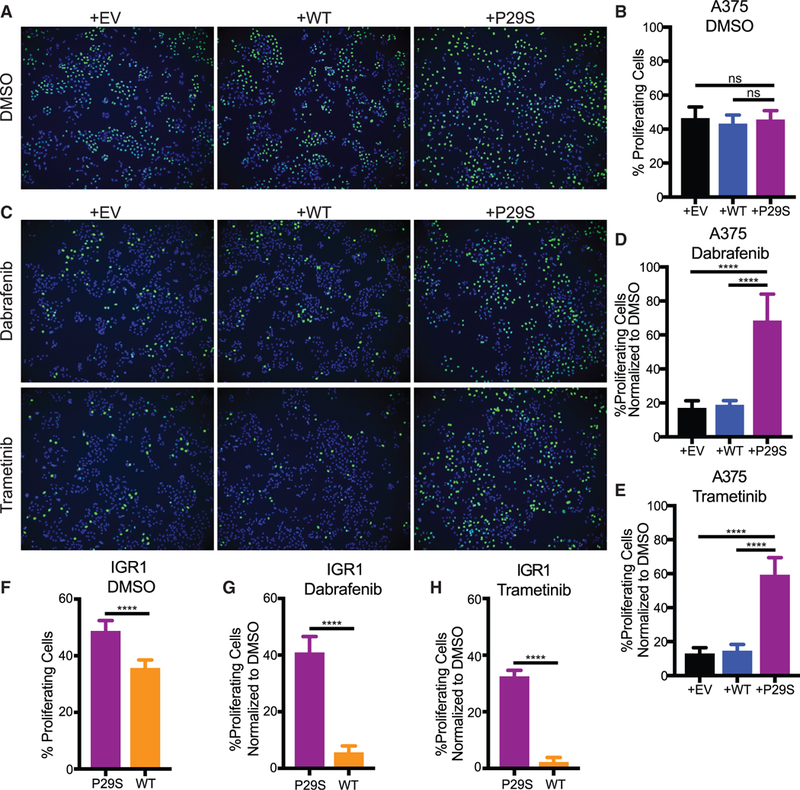
(A-E) Incorporation of fluorescently labeled Edu Into S-phase nuclei of A375 cells exogenously expressing Rac1P29S (+P29S), Rac1WT (+WT), or empty vector (+EV) to detect:
(A and C) proliferating cells (green) and Hoechst-labeled nuclei (blue) to determine fraction of proliferating cells, denoted in %, upon 48 h treatment with (A and B) DMSO (0.003%), (C-E) Dabrafenib (33.3 nM), and Trametinib (3.3 nM).
(F-H) Proliferation assay applied to IGR1 cells endogenously expressing Rac1WT or Rac1P29S upon 48 h treatment with (F) DMSO (0.1%), (G) Dabrafenib (10,000 nM), and (H) Trametinib (6.6 nM).
For all Edu incorporation assays n = 15 images were counted for each condition from three independent experiments. All data represent mean ± SD. Statistical significance was assessed by one-way ANOVA with Tukey’s multiple comparisons test (B, D, and E) and unpaired Student’s t-test (F, G, and H). ****p < 0.0001.
See also Figure S2.
Rac1P29S’s Proliferative Advantage under Growth-Challenging Conditions Is Driven by Enhanced Assembly of a Dendritic Actin Network
When plated on tissue culture dishes, Rac1P29S cells exhibit a distinct morphological phenotype under MAPKi-induced growth challenge (Figure 3A). Rac1WT and Rac1P29S cells have similar cell areas in untreated conditions (Figure 3B). However, upon MAPKi treatment, Rac1P29S cells respond by dramatically enhancing cell spreading (Figures 3B, 3C, and S3). Consistent with previous observations of increased cellular contractility upon MAPKi treatment and our own work showing that shortterm inhibition of ERK abrogates lamellipodial actin polymerization (Mendoza et al., 2015), Rac1WT cells also change in morphology but form long, narrow, spindle-like protrusions (Figures 3A and S3). Thus, the enhancement in the spreading of Rac1P29S cells was unexpected and suggested that the mutation elicits an effect on cell morphology under MAPKi treatment. Three-dimensional light-sheet imaging of fluorescently-labeled actin in control and Rac1P29S cells cultured on a more physiological collagen gel revealed prominent membrane ruffling in Rac1P29S cells (Figure 3D; Video S1). MAPKi treatment of these cells resulted in the formation of large, highly dynamic lamellipodia (Figure 3D; Video S1). On a two-dimensional surface, cryo-EM of control and Rac1P29S cells upon MAPKi treatment also revealed a unique expansion of a fine lamellipodial actin network beyond cortical actin structures in Rac1P29S cells (Figure 3E). To accurately measure the size of dynamic lamellipodial structures, we acquired high-resolution time lapse videos of fluorescently labeled actin in MAPKi-treated cells (Figure 3F; Videos S2 and S3). We then developed an algorithm to determine lamellipo-dia widths, exploiting the feature that in lamellipodia the actin fluorescence fluctuates significantly more over the period of a 10-min video compared to other regions because of rapid actin treadmilling (see STAR Methods) (Figures 3G–3I; Video S4). Using this approach, we found that lamellipodial protrusions in Rac1P29S cells were greatly extended upon MAPKi treatment, whereas changes in control cells were more moderate (Figure 3J). Additionally, the lamellipodia in MAPKi-treated Rac1P29S cells are significantly larger than those in untreated and MAPKi-treated control cells (Figure 3J). This indicated that the morphology change is indeed unique to cells expressing Rac1P29S.
Figure 3. Lamellipodia Formation is Dramatically Enhanced upon MAPKi Treatment of Rac1P29S Cells.
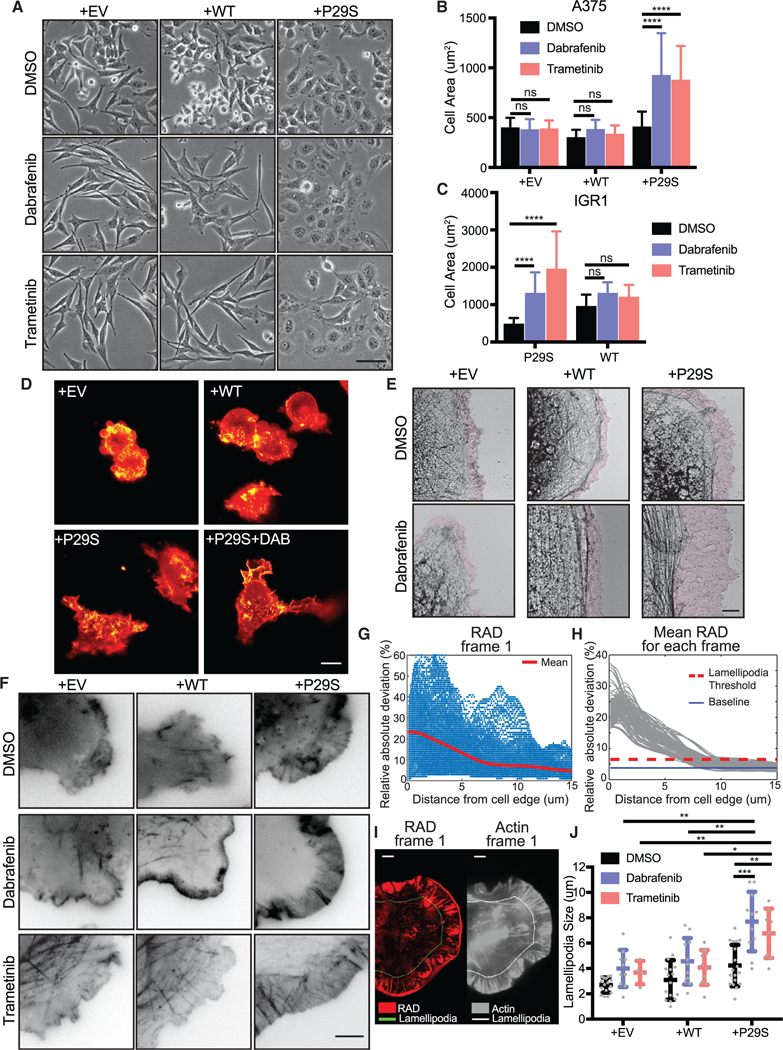
(A) Phase contrast imaging of morphology changes of A375 cells exogenously expressing Rac1P29S (+P29S), Rac1WT (+WT), or empty vector (+EV) upon 48 h treatment with DMSO (0.003%), Dabrafenib (33.3 nM), and Trametinib (3.3 nM) (20x magnification). Scale bar, 50 μm.
(B) Quantification of cell area of +P29S, +WT, and +EV A375 cells upon 48 h drug treatment.
(C) Quantification of cell area of IGR1 cells with endogenous Rac1P29S (P29S) and Rac1WT (WT) upon 48 h drug treatment with DMSO (0.1%), Dabrafenib (10,000 nM), and Trametinib (6.6 nM). Statistical significance was assessed in (B) and (C) using a two-way ANOVA and Tukey’s multiple comparisons test.
(D) Diagonally scanned light sheet microscopy (Dean et al., 2016) of +EV, +WT, and +P29S cells expressing mNeonGreen-Actin-C-35 following cell culture on 2 mg/mL bovine collagen gels for 48 h with or without drug treatment. Scale bar, 10 μm.
(E) Electron micrographs of cell edges of +EV, +WT, and +P29S cells following 48 h drug treatment. Purple colored zones highlight branched actin networks characteristic of lamellipodia regions. Scale bar, 2 μm.
(F) Contrast-inverted TIRF microscopy (60x magnification) of +EV, +WT, and +P29S cells expressing mNeonGreen-Actin-C-35 following 48 h drug treatment. Scale bar, 5 μm.
(G-J) Quantification of lamellipodia size in living cells. Videos of cells expressing mNeonGreen-Actin-C-35 were acquired for 10 min with an imaging interval of 5 sec. The procedure is illustrated for a single representative video of a +P29S cell treated with Dabrafenib (48 h, 33.3 nM):
(G) Relative absolute deviation (RAD) in fluorescence intensity for each pixel plotted as a function of the pixel distance from the cell edge; red line, mean RAD curve for a single frame (Frame 1).
(H) Gray lines, mean RAD curves for all frames of the video. Red line, lamellipodia threshold determined as mean(RAD) + 4*std(RAD). Blue line, min(RAD).
(I) Left, RAD values at each pixel (red color scale) mapped onto the cell mask of frame 1. Right, raw fluorescence intensity of mNeonGreen-Actin-C-35 (gray color scale) in frame 1; the line in both panels indicates the locus where RAD curves, on average, fall below the lamellipodia threshold. Scale bars, 5 μm.
(J) Distributions of lamellipodia widths for all videos (DMSO (0.003%); +EV, n = 16, +WT, n = 16, +P29S, n = 18. Dabrafenib (33.3nM); +EV, n = 10, +WT, n = 11, +P29S, n = 11. Trametinib (3.3 nM); +EV, n = 4, +WT, n = 10, +P29S, n = 8). Statistical significance was assessed using Mann-Whitney tests.
All graphs represent mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S3 and Videos S1, S2, S3, and S4.
To test if the growth challenge-specific proliferation advantage in Rac1P29S cells is dependent on the growth challenge-enhanced lamellipodia, we disrupted lamellipodia formation by pharmacologically targeting the branched actin filament nucleator, Arp2/3 (Pollard and Borisy, 2003), using CK666 and CK689 as active and inactive inhibitory compounds, respectively (Figure S4A). We also disrupted the polymerization of linear actin filaments using the pan-formin inhibitor SMIFH2 (Isogai et al., 2015). Inhibition of either branched actin or linear actin polymerization resulted in a similar decrease in cell proliferation, regardless of Rac1 mutational status (Figure 4A). However, inhibition of branched actin polymerization with the compound CK666 uniquely reduced Rac1P29S cells’ proliferative advantage under MAPKi treatment (Figure 4B). Neither treatment with the inactive control compound CK689 nor inhibition of linear actin polymerization with SMIFH2 increased Rac1P29S cells’ sensitivity to MAPKi treatment (Figures 4C and 4D). Genetic abrogation of Arp2/3 activity using CRISPR/Cas9-mediated genomic knockout of the ARPC1B regulatory domain (Abella et al., 2016) (Figures S4B and S4C) also restored sensitivity to MAPKi treatment in Rac1P29S cells (Figure 4E).
Figure 4. Enhanced Assembly of a Dendritic Actin Network Drives Rac1P29S’s Proliferative Advantage.
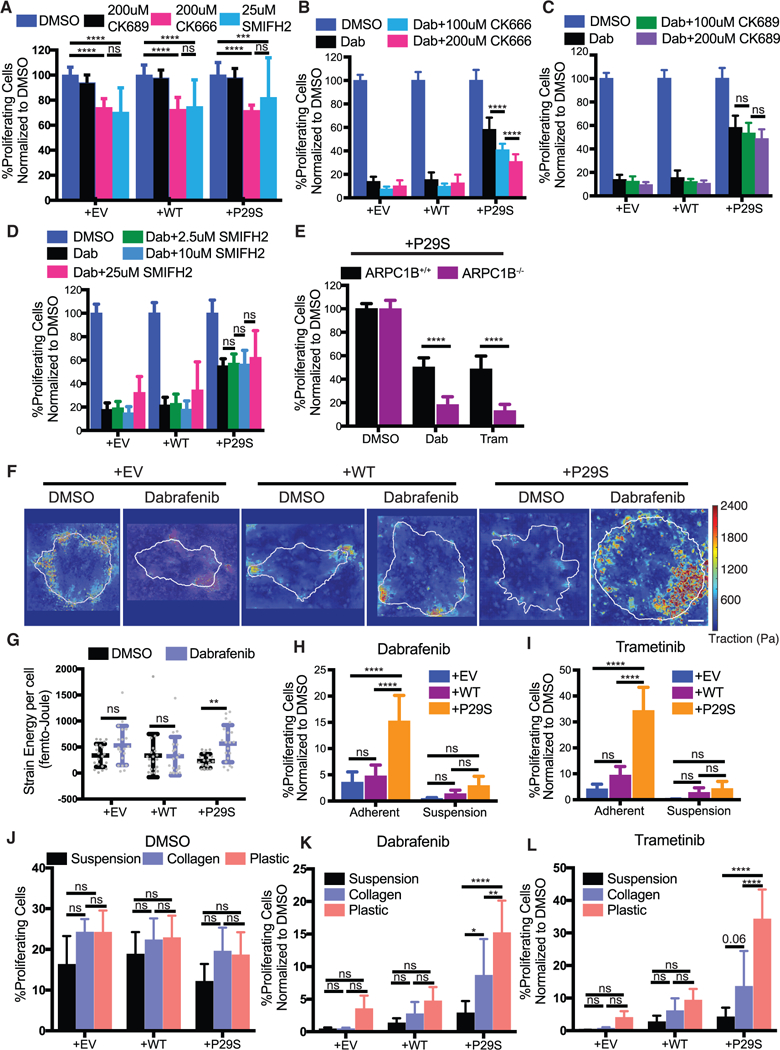
(A) Proliferation assay applied to A375 cells expressing Rac1 P29S(+P29S), Rac1WT(+WT), or empty vector (+EV) treated with DMSO (0.003%), the Arp2/3 complex Inhibitor CK666 (200 μM), the control peptide CK689 (200 μM), and the pan-formin inhibitor SMIFH2 (25 μM). Treatment of cells with CK666 or SMIFH2 alone suppresses proliferation to the same degree and similarly across all cell lines regardless of Rac1 status.
(B-D) Proliferation assay applied to+P29S, +WT, or +EV cells upon 48 h treatment with Dabrafenib (33.3 nM) along with indicated concentrations of (B) CK666, (C) CK689, and (D) SMIFH2.
(E) Proliferation assay applied to +P29S cells with CRISPR/Cas9-mediated knockout oftheArp2/3 regulatory component ARPC1B (ARPC1B−/−) and with control levels ofARPC1B (ARPC1B+/+). Both cell types were treated for48 h with DMSO (0.003%), Dabrafenib (33.3 nM), and Trametinib (3.3 nM). An unpaired Student’s t-test was used to determine statistical significance.
(F and G) Traction Force Microscopy (TFM) of +P29S, +WT, or +EV cells cultured on TFM substrates for 48 h with DMSO (0.003%) or Dabrafenib (33.3 nM) treatment.
(F) Representative traction force maps. Scale bar, 10 μm.
(G) Strain energy percell determined byTFM analysis increases upon MAPKi treatmentfor+P29S, but notfor+EVor+WTcontrol cells. A Mann-Whitneytestwas used to determine statistical significance because of non-normal distribution of the data determined using D’Agostino-Pearson omnibus normality test.
(H and I) Proliferation assay applied to+P29S, +WT, and +EV cells grown ontissue culture coated plastictofacilitate adhesion orin low-adhesion dishesto inhibit adhesion following 48 h treatment with (H) Dabrafenib (33.3 nM) and (I) Trametinib (3.3 nM).
(J-L) Proliferation assay assessedfor+P29S, +WT, or+EVcellscultured using indicated substratesandtreated with (J) DMSO (0.003%), (K) Dabrafenib(33.3 nM), or (L) Trametinib (3.3 nM) for 48 h.
Statistical significance was determined using a two-way ANOVA and Tukey’s multiple comparisons test for (A-D and H-L). All graphs represent mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S4.
Together, these data demonstrate that the upregulation of a specific actin network assembly in lamellipodia of cells harboring Rac1P29S is directly related to enhanced cell proliferation. Rac1P29S drives the proliferative advantage via Arp2/3-mediated dendritic actin polymerization. This is distinct from linear actin polymerization. Thus, Rac1 drives cell cycle progression and oncogenic proliferation through its function as a cytoskeletal regulator, reconciling these previously divergent arms of Rac1 signaling.
The Rac1P29S-Driven Proliferative Advantage Is Dependent on Cell-Matrix Attachment
Extended lamellipodia formation and cell spreading are expected to require enhanced traction provided by a substrate in order to balance the increase in membrane tension during cell protrusion (Ji et al., 2008). Using high-resolution traction force microscopy (Han et al., 2015), we found indeed that cells expressing Rac1P29S specifically generated more traction strain energy upon MAPKi treatment (Figures 4F and 4G). Inhibition of lamellipodia formation during MAPKi treatment prevented this increase in traction strain energy (Figure S4D). We conjectured that removing the mechanical grip by culturing cells in suspension would inhibit the formation of lamellipodia, and thus, the proliferative advantage of Rac1P29S cells, even though actin polymerization machinery (i.e., Arp2/3 activity) would remain intact. To test this, we compared the response in proliferation upon MAPKi treatment between cells cultured in suspension and cells cultured on adherent surfaces. Without MAPKi treatment both Rac1P29S cells and control cells grown in suspension were resistant to anoikis and proliferated readily like attached cells (Figure S4E). However, upon MAPKi treatment, Rac1P29S cells grown in suspension lost their proliferative advantage and were equally sensitive to growth inhibition as control cells (Figures 4H and 4I). We also noted that by culturing cells in soft collagen, which partially restores the substrate grip, Rac1P29S driven proliferation is rescued compared to cells cultured in suspension. Specifically, upon MAPKi treatment, Rac1P29S cells exhibited enhanced proliferation on ~500 Pa collagen gels (Bordeleau et al., 2017) versus in suspension, albeit not to the extent observed on stiff glass (2–4 GPa) (Butcher et al., 2009) (Figures 4J–4L). Thus, the Rac1P29S-mediated activation boost in lamellipodia formation upon growth challenge requires minimal substrate traction to drive cell proliferation. Furthermore, these modulations of substrate traction provide physical perturbations of lamellipodia formation that complement our chemical and genetic perturbations (Figures 4A–4E) in demonstrating the dependence of Rac1P29S-driven proliferation on the robust assembly of lamellipodial machinery.
Lamellipodia Formation in Rac1P29S Cells Drives Proliferative Advantage Independently of Focal Adhesion Signaling
One modality of cell-substrate attachment is mediated via focal adhesion (FA) complexes. These complexes are mechanosensitive (i.e., stronger forces promote larger adhesions) and have been described as active signaling centers that drive cell proliferation and drug resistance in cancer (Paszek et al., 2005; Hirata et al., 2015). Thus, we hypothesized that the enhanced traction observed upon MAPKi treatment of Rac1P29S cells would be synonymous with enhanced FA assembly and signaling. To test this, we imaged by total internal reflection fluorescence (TIRF) microscopy immunofluorescently labeled Paxillin as a marker of FAs. Indeed, we observed that exogenous expression of Rac1P29S in A375 cells resulted in the formation of large FAs (Figures 5A and S5A; Video S5) that increased in size upon MAPKi treatment (Figures S5B and S5C). IGR1P29S cells endogenously harboring Rac1P29S also had a striking phenotype in adhesion assembly, although in these cells the number of adhesions and distribution across the entire ventral surface of the cell, rather than the adhesion size, were most striking (Figure 5B). Reversion of endogenous Rac1P29S to Rac1WT resulted in cells with a more stereotypical peripheral distribution of FAs. This suggested that expression of Rac1P29S yields specific alterations in FA assembly (Figure 5B). Additionally, we found that on substrates coated with collagen, laminin, and fibronectin, cells expressing Rac1P29S attached more readily than control cells (Figures 5C and 5D). Surprisingly however, we noted that Rac1P29S cells also exhibited increased attachment to poly-lysine substrates (Figures 5C and 5D) (Lawson and Burridge, 2014; Mazia et al., 1975). This suggested that the attachment of Rac1P29S cells is facilitated non-specifically, possibly through the increased electrostatic interactions that result from wider cell spreading. Upon growth challenge, Rac1P29S cells plated on poly-lysine substrates continued to exhibit sustained proliferation compared to control cells (Figure S5D). This suggests that a non-specific method of cell attachment is sufficient for Rac1P29S cell attachment and lamellipodia formation to drive proliferation.
Figure 5. Lamellipodia Formation in Rac1P29S Cells Drives Proliferative Advantage Independently of Focal Adhesion Signaling.
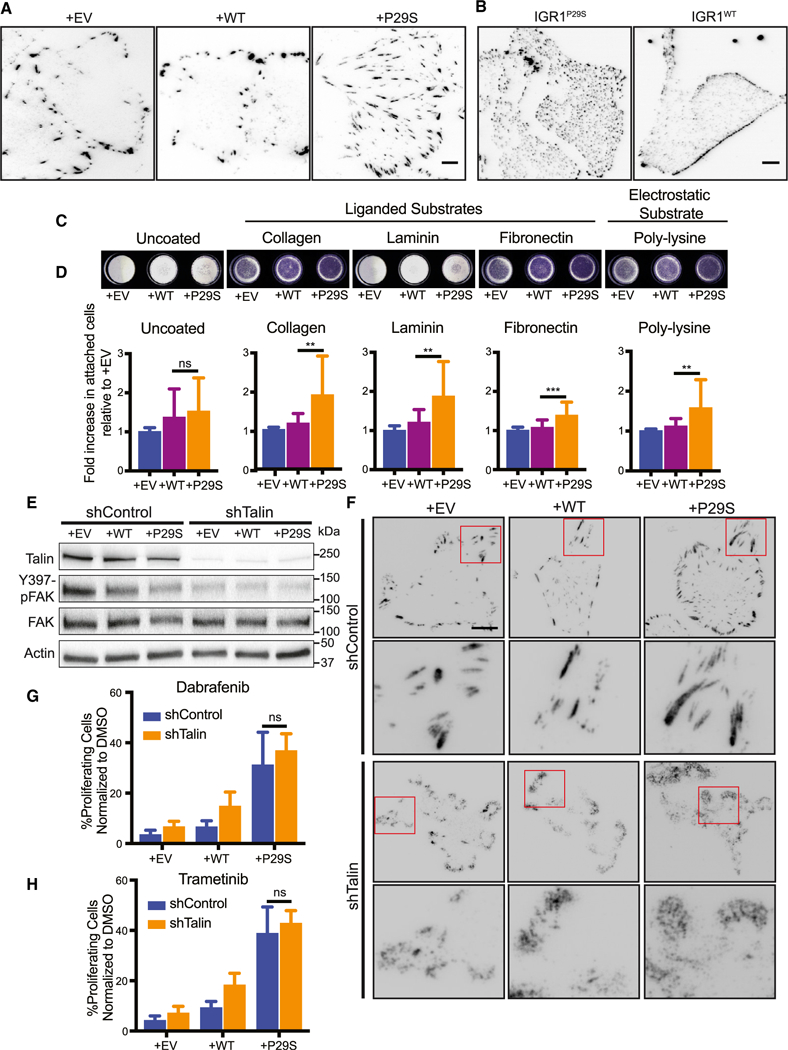
(A and B) Differences in focal adhesions assessed by paxillin immunofluorescence for (A) A375 cells expressing Rac1P29S (+P29S), Rac1WT (+WT), or empty vector (+EV) and (B) IGR1 cells endogenously expressing Rac1P29S or Rac1WT.
(C and D) Attachment of +P29S, +WT, or +EV cells to dishes that are uncoated or coated with poly-L-lysine (0.001%), Type 1 Collagen (2 μg/mL), Laminin (10 μg/mL), and Fibronectin (10 μg/mL) following 15 min incubation after seeding.
(C) Cells that attached in 15 min to coated wells stained with Crystal Violet.
(D) Calorimetric quantification of attached cells normalized to +EV control. Statistical significance was assessed using a one-way ANOVA and Tukey’s multiple comparisons test. The p value for +WT versus +P29S cell attachment to uncoated wells is 0.7786.
(E and F) Disruption of focal adhesion complexes by stable expression of pGIPZ-TLN-V2LHS-56643 (shTalin) to achieve Talin knockdown or pGIPZ-control-V2LHS (shControl).
(E) Immunoblot showing Talin expression and FAK activity upon shTalin and shControl expression across all A375 cells lines, +P29S, +WT, and +EV.
(F) Immunofluorescencedetection of focal adhesion disruption following expression of shTalin or shControl using anti-paxillin antibody in +P29S, +WT, and +EV cells. Scale bar, 10 μm.
(G and H) Proliferation assay applied to A375 cells expressing shControl versus shTalin upon 48 h treatment with (G) Dabrafenib (33.3 nM) and (H) Trametinib (3.3 nM). An unpaired Student’s t-test was performed to compare proliferation between Rac1P29S cells expressing shControl versus shTalin when treated with Dabrafenib (p = 0.146) and Trametinib (p = 0.184).
All bar graphs represent mean ± SD. **p < 0.01, ***p < 0.001. See also Figure S5 and Video S5.
We tested if bona fide FAs that interact with the extracellular matrix via integrins still contribute to the Rac1 P29S-driven proliferative advantage. Neither focal adhesion kinase (FAK) nor Paxillin, canonical markers of FA signaling, showed heightened activity in Rac1P29S cells in the presence or absence of MAPKi treatment (Figure S5E). Moreover, while FA assembly and signaling are reciprocally reinforced by RhoA activity and actomyosin contractility (Paszek et al., 2005), neither RhoA nor myosin revealed enhanced activity (Figure S5F). In fact, Rac1P29S cells exhibited reduced RhoA activity upon drug treatment (Figure S5F). Likewise, while MAPKi treatment led to an expected overall elevation in myosin phosphorylation (Kim et al., 2016), this effect was not unique to cells expressing Rac1P29S (Figure S5F). Additionally, MAPKi-treated Rac1P29S cells maintained their drug resistance compared to control cells upon inhibition of actomyosin contractility with Blebbistatin and ROCK inhibitortreatment (Figures S5G–S5I). This suggested that the Rac1 P29S-driven proliferative advantage is independent of FA signaling and actomyosin contractility.
To more specifically test that Rac1P29S’s attachment-dependent proliferative signaling is autonomous from FA signals, we disrupted FA formation by stable knockdown of Talin (Figure 5E). Talin is an adhesion scaffolding protein that serves as the main molecular linker between integrins and the actomyosin network whose contractility elicits adhesion maturation and signaling (Hytonen and Wehrle-Haller, 2016). As expected, stable knockdown of Talin resulted in reduced activation of FAK (Figure 5E). In cells expressing intact Talin, immunofluorescent labeling of Paxillin showed well-defined focal adhesions, whereas in cells with Talin knockdown Paxillin localized to the cell edge in diffuse, amorphous clouds (Figure 5F). This suggests substantial destabilization of focal adhesions. Nonetheless, the proliferative advantage conferred by Rac1P29S was completely unperturbed by Talin knockdown (Figures 5G, 5H, and S5J). Thus, lamellipo-dia in Rac1P29S cells drive a proliferative advantage independently of FA assembly and signaling. Non-specific cell-substrate attachment is sufficient for this Rac1 P29S-driven enhancement of lamellipodia formation and proliferation.
Rac1P29S-Driven Proliferation via Dendritic Actin Polymerization Is Independent of the MAPK Pathway and JNK, YAP, p38MAPK, and PI3K
To elucidate how Rac1P29S cells relay enhanced dendritic actin polymerization into biochemical signals that drive proliferation, we first tested if Rac1P29S drives proliferation directly through ERK activity. Previous studies have reasoned that Rac1P29S confers drug resistance via ERK activation by noting increased ERK signaling in cells expressing Rac1P29S (Krauthammer et al., 2012; Watson et al., 2014; Araiza-Olivera et al., 2018; Mar et al., 2014). While we also found that exogenous expression of Rac1P29S resulted in some upregulation of ERK activity (Figure 6A), Dabrafenib and Trametinib treatment successfully suppressed phospho-MEKand phospho-ERK levels in Rac1P29S cells (Figure 6A). The expression of cyclin D1, a marker of G-|/S cell cycle progression, was highly sensitive to MEK and ERK suppression in control cells (Figure 6A). However, in Rac1P29S cells cyclin D1 expression was sustained even though MEK and ERK activity were suppressed (Figure 6A). This suggested that MEK and ERK do not mediate the sustained proliferation we observed in Rac1P29S cells (Figures 2C–2E). To rule out that the residual ERK activity observed upon Dabrafenib and Trametinib treatment of Rac1P29S cells (Figure 6A) contributes to the sustained cyclin D1 expression and proliferation, we directly inhibited ERK activity using SCH772984. Treatment with SCH772984 completely ablated ERK phosphorylation in both Rac1P29S cells and control cells (Figure 6B), but cyclin D1 levels remained elevated in Rac1P29S cells (Figure 6B). Most critically, Rac1P29S cells under direct inhibition of ERK activity with SCH772984 proliferated on par with Rac1P29S cells treated with Dabrafenib. This demonstrates that Rac1P29S-driven proliferation is uncoupled from ERK signaling and is likely not mediated by ERK reactivation through mechanisms like paradoxical BRAF activation (Hatzivassiliou et al., 2010; Poulikakos et al., 2010). Indeed, proliferation was also sustained in Rac1P29S cells upon inhibition with the pan-RAF inhibitor LY3009120 that inhibits paradoxical RAF activation (Peng et al., 2015) (Figure S6A). Thus, we concluded that sustained proliferation–and consequently drug resistance–in cells expressing Rac1P29S is mediated independently of MAPK or ERK signaling (Figure S6B).
Figure 6. Rac1P29S Drives Proliferation Independent of MAPK Signaling via NF2/Merlin Phospho-inactivation.
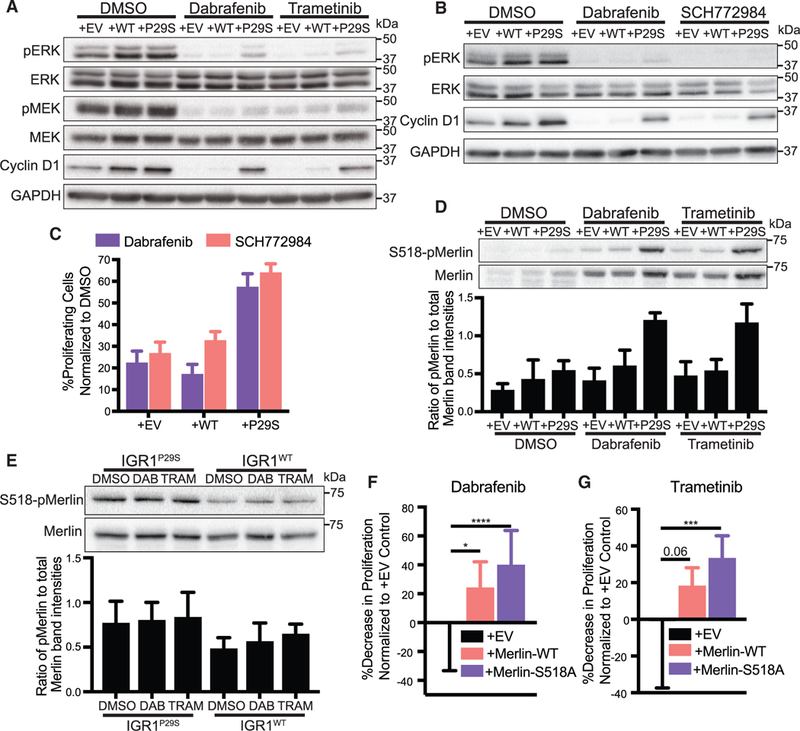
(A) Immunoblot of MAPK signaling represented by phospho-ERK and phospho-MEK and immunoblot of cyclin D1 in A375 melanoma cells expressing Rac1P29S (+P29S), Rac1WT(+WT), or empty vector (+EV) upon 48 h treatment with DMSO (0.003%), Dabrafenib (33.3 nM), and Trametinib (3.3 nM). Data are representative of two independent experiments.
(B) Immunoblot of MAPK signaling represented by phospho-ERK and immunoblot of cyclin D1 in +P29S, +WT, and +EV cells upon 48 h treatment with DMSO (0.003%), Dabrafenib (33.3 nM), and SCH772984 (100 nM). Data are representative of two independent experiments.
(C) Proliferation assay applied to +P29S, +WT, and +EV cells upon 48 h treatment with DMSO (0.003%), Dabrafenib (33.3 nM), and SCH772984 (100 nM). Statistical significance was assessed using an unpaired Student’s t-test.
(D and E) Immunoblot of endogenous levels of total Merlin and inactive S518-phospho-Merlin in (D) +P29S, +WT, and +EV cells upon 48 h treatment with DMSO (0.003%), Dabrafenib (33.3 nM), and Trametinib (3.3 nM) and (E) IGR1 cells endogenously expressing Rac1P29S (IGR1P29S) or Rac1WT (IGR1WT) upon 48 h drug treatment with DMSO (0.1%), Dabrafenib (10000 nM), and Trametinib (6.6 nM). Data are representative of three independent experiments for which quantification by densitometry is the pixel area under the curve of band intensities.
(F and G) Proliferation assay applied to IGR1P29S cells exogenously expressing empty vector (+EV), wild type Merlin (+Merlin-WT), and mutant phospho-deficient Merlin (+Merlin-S518A) upon 48 h treatment with (F) Dabrafenib (10000 nM) and (G) Trametinib (6.6 nM). Data are normalized to DMSO (0.1%) conditions and show the relative decrease in proliferative advantage (expressed in %) to +EV control. Statistical significance was assessed using a one-way ANOVA and Tukey’s multiple comparisons test.
Bar graphs represent mean ± SD. *p < 0.05, ***p < 0.001, ****p < 0.0001. See also Figures S6 and S7.
We next explored numerous alternative mechanisms to explain the dependence of the Rac1P29S-driven proliferative advantage on substrate attachment and robust actin polymerization. Specifically, we tested if the proliferative signaling of pathways including JNK, YAP, p38MAPK, and PI3K, which are all known to be Rac1 regulated, might account for the proliferative advantage of Rac1P29S cells (Dupont et al., 2011). Perturbation of the PI3K pathway did not affect Rac1P29S drug resistance (Figure S6C). YAP nuclear localization was reduced upon MAPKi treatment similarly across control and Rac1P29S cells (Figure S6D). Moreover, perturbations of the YAP/TAZ pathway and the p38 MAPK pathway did not uniquely alter Rac1P29S cells’ proliferation under MAPK inhibition (Figures S6E and S6F). And JNK signaling was unaffected in control and Rac1P29S cells upon MAPKi treatment (Figure S6G). Thus, we concluded that Rac1P29S drives proliferation independently of these pathways.
Rac1P29S-Driven Dendritic Actin Polymerization Sustains Melanoma Cell Proliferation by Sequestration and Phospho-Inactivation of the Tumor Suppressor Merlin
Rac1 activity negatively regulates Merlin, a member of the ERM family of proteins that functions as a membrane-cytoskeletal linker and thus, localizes to the cell edge in regions of dendritic actin polymerization (Mani et al., 2011; Kissil et al., 2002). As the protein product of the NF2 gene that downregulates cyclin D1 and triggers growth arrest, Merlin also functions as a tumor suppressor (Shaw et al., 2001; Xiao et al., 2002). We hypothesized that hyperactive Rac1P29S sustains proliferation by negative regulation of the tumor suppressor Merlin through enhanced dendritic actin polymerization. We observed that Merlin was expressed in both control and Rac1P29S melanoma cells upon MAPKi treatment, however cells expressing Rac1P29S uniquely exhibited increased S518-phospho-inactivation of Merlin (Figures 6D, 6E, S7A, and S7B). To test if enhanced Merlin phosphorylation in Rac1P29S cells rescues these cells from proliferative suppression, we exogenously expressed phospho-deficient S518A-Merlin in Rac1P29S cells. We found that melanoma cell sensitivity to MAPKi treatment was significantly restored in Rac1P29S cells expressing phospho-deficient Merlin (Figures 6F, 6G, and S7C).
We next tested the dependence of Merlin phospho-inactivation on the Rac1P29S-enhanced assembly of a dendritic actin network. Upon MAPKi treatment, phosphorylated, inactivated Merlin localized within 2 mm of the cell edge, characteristic of lamellipodia and branched actin polymerization (Figure 7A). This is consistent with a mechanism in which hyperactive Rac1 drives dendritic actin polymerization resulting in the phosphorylation and inactivation of Merlin. Merlin is thought to be phospho-inactivated at S518 by PAK upon Rac1 binding (Xiao et al., 2002; Kissil et al., 2002). In agreement with a model in which dendritic actin polymerization results in phospho-inactivation of Merlin, we found that PAK also localized to the cell edge upon MAPKi treatment (Figures 7B and 7C). Additionally, co-inhibition of PAK with the compound PF3758309 restored the sensitivity of Rac1P29S cells to MAPKi treatment in a dose-dependent fashion (Figure 7D). At 200 nM this PAK inhibitor fully abrogated the resistance of Rac1P29S cells to MAPKi treatment (Figure 7E). From these data we inferred that phospho-inactivation of Merlin depends on two mechanisms evoked in a Rac1P29S background: (1) Activation of PAK and (2) sequestration of PAK and NF2 at the cell edge via dendritic actin polymerization. To test if phosphorylation and inactivation of Merlin in Rac1P29S cells were directly dependent on Rac1P29S-driven enhanced dendritic actin polymerization, we measured Merlin phosphorylation in Rac1P29S cells upon Arp2/3 inhibition with CK666. Treatment with CK666 abrogated Merlin localization to the lamellipodia (Figures 7F–7H). Most critically, treatment with CK666 also abrogated Merlin phosphorylation (Figures 7I and S7D). This shows that while PAK activity is necessary, it is not sufficient for Merlin phospho-inactivation without the sequestration of Merlin to the cell edge by enhanced dendritic actin polymerization. Thus, Rac1P29S downregulates Merlin’s tumor suppressor activity to drive proliferation through enhanced dendritic actin polymerization that is required for both concentration and PAK-induced phospho-inactivation of Merlin in a lamellipodial signaling microdomain.
Figure 7. Enhanced Dendritic Actin Network in Rac1P29S Cells Sequesters and Inactivates Tumor Suppressor NF2/Merlin in Lamellipodia.
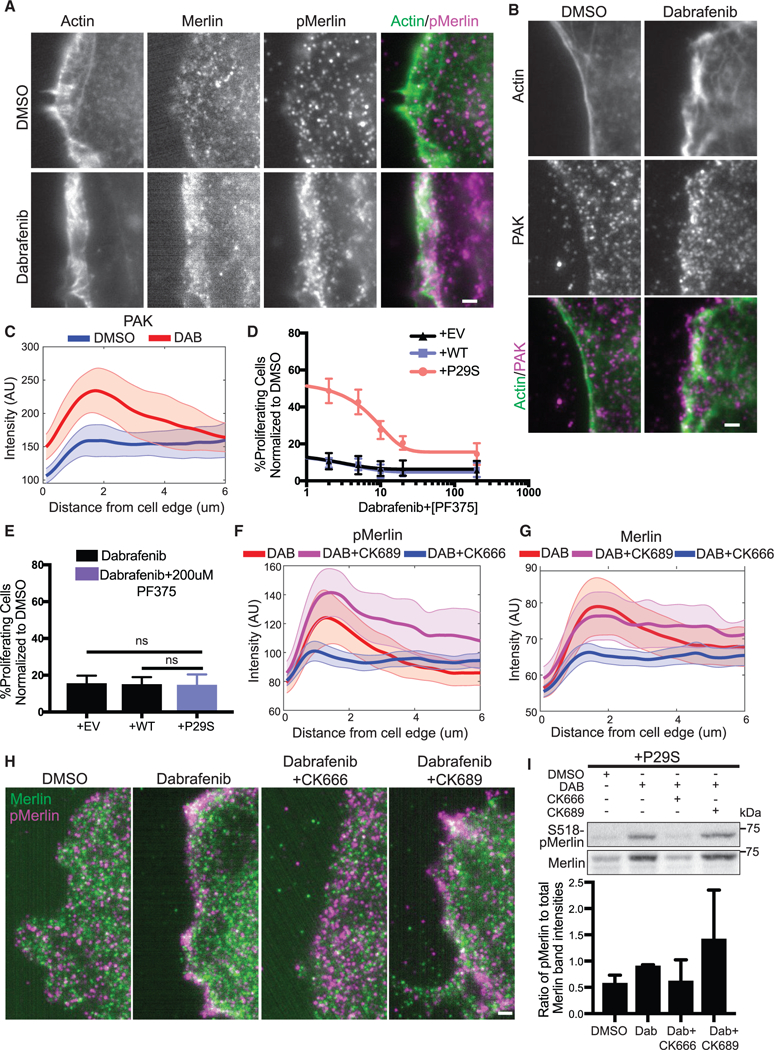
(Aand B) Fluorescent protein detection ofmNeonGreen-Actin-C-35and immunofluorescencedetection of(A) endogenoustotal Merlin and S518-phospho-Merlin and (B) endogenous PAK in A375 melanoma cells expressing Rac1P29S following 48 h treatment with DMSO (0.003%) and Dabrafenib (33.3nM). Scale bars, 2 μm.
(C) Averaged intensity curves of PAK as a function of a distance from the cell edge (0.1~6 μm); n = 12 (DMSO), n = 10 (DAB). Shaded areas indicate 95% confidence intervals at each distance.
(D) Proliferation dose response to increasing concentration ofthe PAK inhibitor PF3758309 (2.0, 5.0,10.0, 20.0, and 200.0 nM) when added in combination with Dabrafenib treatment (33.3 nM) for 48 h in A375 melanoma cells expressing Rac1P29S (+P29S), Rac1WT (+WT), or empty vector (+EV). Data are normalized to DMSO (0.003%) control.
(E) Combined Dabrafenib (33.3 nM) and PF3758309 (200 mM) treatment restores +P29S cells’ proliferative sensitivity to the level of control +EV and +WT cells treated with Dabrafenib (33.3 nM). Statistical significance was assessed using a using a one-way ANOVA and Tukey’s multiple comparisons test.
(F and G) Endogenous(F) S518-phospho-Merlin localization and (G)total Merlin localization in +P29S cellsasafunctionofdistancefromthecell edge(0.1~6 μm); averages are calculated first in distance bands per cell and then compiled over multiple cells upon three treatments; n = 9 (DAB, 33.3 nM), n = 11 (DAB, 33.3 nM+CK689, 200 μM), n = 15 (DAB, 33.3 nM+CK666, 200 μM). Shaded area indicates 95% confidence intervals at each distance.
(H) Immunofluorescence detection of endogenous total Merlin (green) S518-phospho-Merlin (magenta) in +P29S cellstreated as described in (F) and (G). Scale bar, 2 μm.
(I) Immunoblot of endogenous levels of total Merlin and inactive S518-phospho-Merlin in +P29S cells upon 48 h treatment with DMSO (0.003%), Dabrafenib (33.3 nM), and Dabrafenib (33.3 nM) in combination with CK666 (200 μM) or CK689 (200 μM). Data are representative ofthree independent experimentsforwhich quantification by densitometry is the pixel area under the curve of band intensities.
Bar graphs represent mean ± SD. See also Figure S7.
DISCUSSION
The actin cytoskeleton supplies critical mechanical machinery for cancer invasion and metastasis (Machesky, 2008). Our data reveal that, beyond its structural roles, the actin cytoskeleton is also actively involved in regulation of cell signaling. We find that lamellipodia are biochemical regulators of cellular proliferation and that they can be engaged by hyperactive Rac1-primed melanoma cells to drive oncogenic programs. The convergence of actin polymerization and proliferation into one biochemical signaling axis reconciles the dichotomous functions of Rac1 as a regulator of two processes that have hitherto been considered disparate. Moreover, our data establish a critical role of cellular morphology in dictating cellular signaling.
While Rac1 and actin are ubiquitously expressed, the engagement of the specific actin structure that drives oncogenic proliferation in melanoma cells requires three additional conditions: first, Rac1 hyperactivity, second, a growth challenge, and third, a minimal level of substrate traction. In this study, we use the point mutation P29S to achieve Rac1 hyperactivation because this mutation is naturally occurring, clinically relevant, and biochemically well characterized (Krauthammer et al., 2012, Hodis et al., 2012; Davis et al., 2013). However, Rac1 itself is highly regulated, and dysregulation upstream of Rac1 has been observed to result in enhanced cell spreading and increased membrane ruffles (Vial et al., 2003). Thus, we postulate that the numerous modalities of Rac1 dysregulation observed in cancer, including Ras activation and the frequent mutations in GEFs and GAPs, would also provide the level of Rac1 pathway stimulation to drive this mechanism (Malliri et al., 2002; Sosa et al., 2010; Cook et al., 2014; Wertheimer et al., 2012).
Despite the observation of increased membrane ruffling in cells with hyperactive Rac1, whether by the P29S mutation or other mechanisms, the quantitatively significant increase in cell spreading and dramatic enlargement of lamellipodia was only observed upon drug-induced growth challenge. We observed this morphological shift through three different methods: (1) Cryo-EM revealed dramatic extension of lamellipodia in Rac1P29S cells beyond lamellipodia in control cells. (2) Using high-resolution live-cell imaging of fluorescently-labeled actin, quantification of actin flow demonstrated that these dynamic structures are significantly larger than pre-treatment lamellipodia and those of control cells. (3) Light sheet microcopy of growth-challenged Rac1P29S cells cultured on collagen gels showed striking lamellipodia formation in 3D, confirming the relevance of this morphological shift beyond glass. The engagement of this pathway specifically under growth challenge was also evident in vivo. Rac1P29S has no effect on primary tumor growth but offers a significant growth advantage at metastatic sites, where a small number of cells must overcome the challenges to proliferation imposed by foreign microenvironments (Shibue and Weinberg, 2009). This regulatability in the engagement of the Rac1P29S pathway suggests the requirement of either a disinhibition or activation step that is achieved by growth challenge. While this regulatability warrants further study especially in the context of drug targeting, one possibility is that MAPKi-regulation of GEF activity either directly (Ryan et al., 2016) or via loss of feedback inhibition (Smith and Wellbrock, 2016) might favor specific GEF-Rac1P29S binding events and thus favor effector functions (Marei and Malliri, 2017) specific to that GEF-Rac1P29S interaction.
Finally, our data showed a minimal requirement of the pathway for cell-substrate attachment. We interpret this requirement as the necessity of the extended lamellipodial branched actin network assembly under Rac1-hyperactivation to experience sustained mechanical resistance against the gradually increasing plasma membrane tension (Ji et al., 2008): first, Rac1 P29S-driven proliferation is completely abrogated in suspended cells, where no such resistance can arise. Second, for cells plated on stiffer substrates, where the plasma membrane-cortex system is less compliant to actin polymerization than on softer substrates like a collagen gel, the proliferative advantage conferred by the pathway is enhanced. Consistent with this interpretation, under growth challenge by MAPK inhibitors, the engagement of the Rac1P29S mediated pathway increases cell traction force, a direct measure of the force level generated by actin polymerization against the plasma membrane-cortex system (Lee et al., 2015; Ji et al., 2008). Given that Rac1P29S cells exhibit increased attachment even on poly-lysine coated substrates compared to control cells, and that their proliferative advantage is maintained, we note that non-specific cell-substrate interactions such as electrostatically primed surfaces are sufficient for lamellipodia formation and the dependent proliferation-promoting signaling.
Intriguingly, focal adhesion formation is also upregulated in cells expressing Rac1P29S. While all previous studies to our knowledge that discuss the role of actin in cancer signaling implicate mechanical engagement of acto-myosin stress fibers and converge exclusively on FAK signaling (Paszeketal., 2005; Hirata et al., 2015; Baeetal., 2014), we noted that proliferation driven by Rac1-enhanced lamellipodia is independent of this pathway. We found FAK activity unaffected by Rac1P29S expression and drug challenge, and, most compellingly, disruption of focal adhesions by knocking down the essential mechanical linker component, Talin, has no effect on Rac1P29S driven proliferation. The uncoupling of Rho and myosin signaling from Rac1 P29S’s growth advantage suggests that signaling via branched actin polymerization is independent of actin stress fibers. This is inline with the independence of Rac1P29S signaling from linear actin polymerization, and lends the idea that unique actin network architectures–stress fibers, branched actin networks, linear actin bundles–can coordinate unique signaling cues.
Lamellipodia-extension in Rac1P29S cells leads to a signaling microdomain that spatially coordinates and locally amplifies a biochemical cue that has the potency to drive cell proliferation. This adds the case of a morphologically driven oncogenic signaling cue to an emerging body of literature that highlights the importance of cell shape in signaling control (Ron et al., 2017; Rangamani et al., 2013; Kuo et al., 2012). We find that the Rac1P29S-enhanced lamellipodial branched actin network is necessary for the sequestration and phospho-inactivation of Merlin. Merlin is a tumor suppressor known to downregulate cyclin D1 and cell cycle progression (Xiao et al., 2005; Mani et al., 2011). Merlin has been shown to be phosphorylated and inactivated at Serine-518 by PAK (Xiao et al., 2002; Kissil et al., 2002). Upon MAPKi drug challenge, both Merlin and PAK localize to the lamellipodial cell edge, and Merlin phosphorylation is uniquely enhanced in Rac1P29S cells. Introducing the phospho-deficient Merlin mutation, S518A, into Rac1P29S cells restores sensitivity of these cells to MAPKi treatment. Consistently, inhibition of PAK activity also restores sensitivity of cells expressing Rac1P29S to growth suppression. Critically however, PAK activity alone, even in a Rac1-hyperactivated system, is insufficient for Merlin phospho-inactivation without lamellipodia formation. Upon inhibition of branched actin polymerization, phosphorylation of Merlin in Rac1P29S cells is abrogated. Additionally, both inhibition of branched actin polymerization and suspension and soft collagen culture specifically diminish lamellipodia formation, resulting in a pronounced suppression of Rac1 P29S-driven proliferation. Thus, lamellipodia formation is a necessary driver in the phospho-inactivation of Merlin that allows Rac1P29S cells to subvert growth challenge. We note that unlike a large number of melanoma drug resistance pathways that converge on ERK reactivation (Van Allen et al., 2014; Shi et al., 2014), this lamellipodial coordination of Merlin phosphorylation is independent of ERK signaling and thus unveils an ERK-independent drug resistance mechanism.
In conclusion, the pathway driven by hyperactive Rac1P29S confers a proliferative advantage that potentiates drug resistance and metastasis, two of the most critical factors in determining prognosis of cancer patients. The conditional engagement of biochemically active lamellipodia and the subsequent proliferative signaling suggests that Rac1P29S’s function must undergo an activation or disinhibition step that is provided by growth challenges under drug treatment and in metastases. This regulation of oncogenic function is unique in the context of conventional oncogenic transformations such as those elicited by constitutively active mutations such as BRAFV600E and KRASG12V. The regulatability of this pathway, along with its ERK independence and the frequent alterations in Rac1-activating proteins in melanoma as well as other forms of cancer (Kazanietzand Caloca, 2017), present an arm of targetable signaling that can be pursued independently of targets in adhesion and MAPK signaling and provides the opportunity to address the clinically reported failure of MAPK inhibitors in these cancers. Moreover, Rac1 is a highly regulated driver of proliferation even in normal cells. Therefore, this study unveils a morphology-driven mechanism of cell growth that may be active in a wide range of other scenarios, from development to neoplasm to homeostasis of challenged tissue.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Gaudenz Danuser (gaudenz.danuser@utsouthwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS Cell
Cell Lines
A375 cells (human malignant melanoma cells harvested from a 54-year-old female; CRL-1619, ATCC, RRID: CVCL_0132) and IGR1 cells (human malignant melanoma cells harvested from a 42-year-old male; ACC236, DSMZ, RRID: CVCL_1303) were cultured at 37°C, 5% CO2 using DMEM (11995, Gibco) supplemented with 10% FBS and 0.2% antibiotic-antimycotic (15240062, Gibco).
Mouse Studies
All xenograft tumor studies were conducted using four-to eight-week old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (The Jackson Laboratory, RRID: IMSR_JAX:005557) that weighed 20–25g. The mice were housed in a level two barrier facility to minimize opportunistic infections. Five mice were housed in each cage, and the mice were fed a standard diet consisting of irradiated food and autoclaved water. Xenograft experiments were performed according to the protocol approved by the UT Southwestern Institutional Animal Care and Use Committee (protocol 2016–101360). Mice were allocated to experiments randomly and samples processed in an arbitrary order, but formal randomization techniques were not used.
METHOD DETAILS
Constructs and Reagents
Rac1 mutant and wild type viral vectors were generated from a pcDNA3-GFP-Rac1 wild type (Rac1WT) construct (Cell BioLabs). The QuikChange Lightning Mutagenesis Kit (210515, Agilent) was used to introduce a single-base pair 85C>T transition in the pcDNA3-GFP-Rac1WT coding sequence resulting in the proline-to-serine mutation at amino acid 29 (Rac1P29S). Primers for the reaction were designed using the Agilent online tool (www.agilent.com/genomics/qcpd): CTGATCAGTTACACAACCAATGCATTTTCTGGAGAA TATATCCC (forward) GGGATATATTCTCCAGAAAATGCATTGGTTGTGTAACTGATCAG (reverse)
Coding sequences of Rac1P29S and Rac1WT were cut out of the resulting pcDNA3-GFP-Rac1P29S construct and the original pcDNA3-GFP-Rac1WT construct, respectively using the enzymes EcoR1 and Xho1. These coding sequences were ligated into a pLVX-IRES-puromycin lentiviral construct (pLVX-puro) that was cut with the same enzymes to create pLVX-puro-Rac1P29S and pLVX-puro-Rac1WT constructs. Undigested pLVX-puro construct was included in subsequent steps to serve as an appropriate empty-vector negative control to rule out effects due to expression of the backbone vector. HEK293T cells were transfected using PEI at 3ul/ug total DNA concentration with these constructs (5ug) and VSV envelope vector pspax2 (7ug) and pmd2g viral packaging construct (5 ug). Viral media was harvested, filtered (0.45 um), and mixed with 1ul/ml dilution of polybrene.
To generate A375 cells lines that exogenously express Rac1WT (+WT), Rac1P29S (+P29S), and empty pLVX-puro vector (+EV), parent A375 cells were spin-infected for 1hr at 3000 rpm. Viral media was replaced with fresh media 24 hrs following infection. Cells transduced with pLVX-puro-Rac1WT, pLVX-puro-Rac1P29S, and undigested pLVX-puro (empty vector) were selected with 2ug/ml puromycin.
To titrate expression of fluorescent protein-tagged constructs and avoid overexpression artifacts, a series of truncated CMV promoters were created using the pLVX-shRNA2 backbone (Clontech). Briefly, the U6 promoter, multiple cloning site, and ZsGreen reporter gene were removed and replaced with iteratively increasing 100 basepair increments of the CMV promoter starting with the 5’ end. Thus, 6 constructs were prepared, with the first 100,200,300, 400, 500, and 600 base pairs of the promoter region (available on Addgene: IDs 110718, 110719, 110720, 110721, 110722, 110723), fluorescent protein fusions were cloned downstream using the restriction enzymes SpeI andXhoI. To label adhesions, mNeonGreen-Paxillin-22 (Allele Biotechnology), a C-terminal fluorescent protein fusion with a 22 amino acid linker, was cloned into the 100 base pair variant. To label the actin cytoskeleton, mNeonGreen-Actin-C-35 (Allele Biotechnology), an N-terminal fluorescent protein fusion with a 35 amino acid linker, was cloned into the 300 base pair variant. Lentiviral particles for both constructs were prepared according to the manufacturers recommendations, and infected A375 cells were enriched with fluorescence activated cell sorting (FACS) using a FACSAria system (Children’s Research Institute Flow Cytometry Facility).
To generate cell lines that exogenously express wild type and mutant NF2/Merlin, NF2/Merlin wild type, S518A, and S518D constructs were cloned out of pcDNA vectors generously provided by the lab of Dr. Duojia Pan and into pLVX-IRES-puromycin lentiviral vectors. Virus was prepared from these constructs as described above. IGR1WT and IGR1P29S cells were transduced and selected using 0.2ug/ml puromycin.
Drugs were acquired from the following sources: Dabrafenib, Trametinib, SB203580, and YAP-TEAD Inhibitor 1 (Peptide 17) from Selleckchem, SMIFH2 from Tocris, CK666, CK689, Blebbistatin, and Y27632 ROCK Inhibitor from Sigma, and SCH772984 from ChemieTek.
CRISPR/Cas9 Genome Editing
For the single-base pair change of endogenous Rac1P29S in IGR1 cells to Rac1WT, CRISPR/Cas9 mediated genome-editing was used, and homologous recombination was achieved using a single-stranded DNA oligonucleotide (ssODN) repair template by following the protocol described by the Zhang Lab (Ran et al., 2013). The sgRNA guide sequences 5’-TACACAACCAATGCATTTTC-3’ and 5’-ATATTCTCCAGAAAATGCAT-3’ were designed using the CRISPR Design Tool (http://tools.genome-engineering.org) and the ssODN repair sequences AAGA TACTTACACAGTAGGGATATATTCTCCAGGAAATGCATTGGTTGTGTAACTGATCAGTAGGCAAGT and AAAACTTGCCTACTGATC AGTTACACAACCAATGCATTTCCTGGAGAATATATCCCTACTGTGTAAGTAT
corresponding to each guide, respectively, were designed using the construct visualization tool Benchling (Benchling.com). Guide sequences were cloned into the pSpCas9(BB)-2A-GFP (PX458) vector (48138, Addgene) (Ran et al., 2013). IGR1 cells plated in 10cm dishes were transfected with both guides independently following 30 min treatment of cells with 0.05 uM of SCR7 pyrazine (SML1546, Sigma) using 15 ug of CRISPR/Cas9-guide construct DNA, 30 ul of ssODN reconstituted to 10 uM and 30 ul of Lipofect-amine LTX Reagent (15338030, Thermo). GFP-positive cells were then plated as single cells using FACS into 96-well plates containing IGR1-pre-conditioned media. Colonies were expanded in 24 well plates and screened for successful genome-editing using gDNA extraction using QuickExtract (QE09050 , Epicentre) and Sanger sequencing.
For CRISPR/Cas9-mediated knockout of ARPC1B, selected DNA target sequences from exon 3 of ARPC1B were pasted into the CRISPR design tool CRISPOR (http://crispor.tefor.net). Resulting potential target sites with a high efficiency score were used for designing the sgRNA constructs (20 nucleotides), which were cloned into pSpCas9(BB)-2A-GFP (pX458; Addgene plasmid ID: 48138) using BbsI and sequence verified (Ran et al., 2013). To disrupt ARPC1B gene expression, a blasticidin selection cassette was inserted at the cut site employing a self-cleaving donor vector. The sequence for the sgRNA used to target ARPC1B is as follows: 5’-CTCGTGCACCTTGGTCCATT-3’. ARPC1B knockout and the Arp 2/3 complex were evaluated with western blots using the following antibodies: anti-ARP3 1:500 (sc-48344), anti-ARPC2 1:500 (sc-515754), and anti-ARPC1B 1:500 (sc-137125) all from Santa Cruz and anti-ARPC1A 1:1000 (HPA004334, Sigma).
Xenograft Tumor Model and Immunohistochemistry
Viral supernatant prepared from a dsRed2-P2A-Luc lentiviral construct was provided generously by the Morrison lab to generate dsRed-luciferase positive cells for xenograft tumor injections and subsequent bioluminescence imaging to detect metastases (Pis-kounova et al., 2015). Following infection, dsRed positive cells were enriched using flow cytometry and expanded. 100 cells were counted and prepared in 25% Matrigel as described in (Piskounova et al., 2015) for subcutaneous injection into the flank region of NSG mice. Tumor growth was measured using a Marathon CO030150 Digital Caliper until tumors reached around 2.0 cm but no more than 2.5 cm before mice were euthanized for end-point analysis of metastasis (UT Southwestern IACUC protocol 2016101360). Bioluminescence imaging of dissected organs and analysis were performed as described in (Piskounova et al., 2015). Dissected lungs were fixed in 10% neutral-buffered formalin for 48 hrs on a rotator at room temperature then transferred to PBS. Tissues were paraffin embedded and sectioned by the UT Southwestern Histo Pathology Core.
Immunohistochemistry was performed to dual label mouse lung tissues for melanoma metastases using rabbit anti-RFP antibody (Rockland) to recognize dsRed-luciferase positive disseminated human melanoma cells and mouse anti-S10-phospho-Histone H3 (9706, Cell Signaling) antibody to recognize mitotic cells. Briefly, sections were deparaffinized for 5 min in a xylene bath, done three times. Sections were hydrated in sequential ethanol baths: 100% ethanol, 5 min, 90% ethanol, 2 min, 80% ethanol, 2 min, 70% ethanol, 2 min, and 50% ethanol, 2 min. Sections were left under a gentle stream of running water for 5 min. Antigen presentation was achieved by cooking sections in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) inside a heated pressure cooker for 5 min. Following cool down, slides were rinsed with water and removed to 1x TBST. To block endogenous peroxidase, slides were incubated with a 3% H2O2 solution for 15 min. Sections were rinsed for 5 min twice with 1xTBST and Biotin-Avidin blocking was performed (SP-2001, Vector Laboratories). The Vector M.O.M Peroxidase-based Immunodetection Kit (Vector Laboratories) was followed for additional blocking, mouse anti-S10-phospho-Histone H3 primary antibody incubation (1:300 dilution, 4°C, overnight), and secondary antibody incubation. Phospho-Histone H3 positive tissue was visualized using the ImmPACT DAB Peroxidase Substrate Kit (SK-4105, Vector Laboratories). Sections were washed with 1xTBST then blocked and incubated in rabbit anti-RFP antibody (Rockland) according to the ImmPRESS-AP Reagent Anti-Rabbit IgG alkaline phosphatase-based staining kit (MP-5401, Vector Laboratories). Metastatic melanoma cells were visualized using ImmPACT Vector Red alkaline phosphatase substrate (SK-5105, Vector Laboratories). Sections were then hematoxylin stained (15 sec, H3404, Vector Laboratories).
Cell Culture and Proliferation Assays
The fraction of proliferating cells was determined using the Click-iT Plus EdU Alexa Fluor Imaging Kit (Molecular Probes). Briefly, 15,000–30,000 A375 cells or 70,000 IGR1 cells were seeded into each well of a 12-well plate and drug treated for 48hrs. Cells were incubated with 10uM of EdU for 3hrs prior to fixation with 4% PFA and permeabilization with 0.5% Triton. Edu-positive nuclei were labeled according to manufacturer protocol followed by labeling of all nuclei with Hoechst (10mg/ml) diluted 1:2000. An inverted phase contrast and fluorescence Nikon Ti-Eclipse microscope equipped with a Zyla sCMOS camera, SOLA solid state white-light excitation system and a motorized filter turret with filters for DAPI, FITC and TRITC along with Nikon Elements image acquisition software was used to image Hoechst and Edu-positive nuclei at 10x magnification. For all Edu incorporation assays, three biological replicates were performed on separate days. Around five images, sampling multiple regions of the well, were acquired for each condition (n≈15).
For experiments testing proliferation sensitivity to drug for cells cultured in low adhesion or soft collagen, percent proliferation was determined using the Click-iT Plus Edu Alexa Fluor Flow Cytometry Kit (C10632, Molecular Probes). For low adhesion experiments, 300,000 cells were seeded into tissue culture coated 6 well plates or ultra-low attachment 6 well plates (Corning) to keep cells in suspension. For soft collagen experiments, 300,000 cells were counted and resuspended in 2 mg/mL collagen (Advanced Biomatrix) (1 mL cocktail of pre-warmed reagents: 100 uL 10x PBS, 10uL 1M NaOH, 250 uL H2O, 640 uL 3mg/mL collagen). 100 mM ribose (R9629, Sigma) was supplemented in the collagen cocktail to increase rigidity by enhancing crosslinking (Bordeleau et al., 2017). Cells were plated in dishes pre-warmed to 37°C. Following drug treatment and Edu incubation, cells were either trypsinized from plastic dishes, collected from suspension dishes by aspiration, or released from collagen gels by incubation in collagenase (1mg/ml in PBS, added 1:1 collagen to collagenase volume) for 3 hrs at 37oC (5030, Advanced Biomatrix) prior to neutralization with media then fixation and permeabilization according to manufacturer’s protocol. Labeled cells were analyzed using a FACSAria II Cell Sorter (BD Biosciences, San Jose, CA) and FlowJo v10 analysis software.
Electron Microscopy
Cells were incubated in the presence of DMSO or Dabrafenib for 48 hours, after which the cells were trypsinized and re-plated with DMSO or Dabrafenib treatment onto glow-discharged carbon-film gold EM grids (EMS CF200-Au). After 12–16 hours, the grids were processed as follows (Vinzenz et al., 2012): grids were washed by dipping into 4 droplets of ‘cytoskeleton buffer’ (CB) (Mueller et al., 2017), after wicking off the last droplet, the grids were lightly fixed and extracted in CB containing 0.25% glutaraldehyde and 0.5% triton-X 100 for 1 minute. Grids were washed by dipping into 1 droplet of CB. The grids were then fixed for 10 minutes in CB containing 2% glutaraldehyde. After washing in 3 water droplets for 2 minutes each, the grids were stained with 1% uranyl acetate for 2 minutes; washed twice in water droplets for 2 minutes each, then air dried. Imaging was done on a JEM-1400 Plus transmission electron microscope equipped with a LaB6 source operated at 120 kV using an AMT-BioSprint 16M CCD camera; supported by NIH grant 1S10OD021685–01A1.
Cell Adhesion Assay
For cell adhesion assays, cells were trypsinized and washed with DMEM containing no serum. 100,000 cells in 100 ml serum-free DMEM were seeded into 96-well plates coated with 0.001% poly-L-lysine, 2 μg/ml collagen type-I, 10 mg/ml fibronectin, 10 μg/ml laminin or no coating, and blocked with blocking buffer (DMEM supplemented with 0.5% BSA). Cells were incubated for 15 min at 37°C and non-adherent cells were removed by three washes using washing buffer (DMEM supplemented with 0.1% BSA). Attached cells were fixed with 4% paraformaldehyde (PFA) for 10 min, washed twice and stained with Crystal Violet (5 mg/ml in 2% ethanol) for 10 min. Fixed cells were extensively washed with water and lysed with 2% SDS for 30 min. Absorbance was measured at 590 nm using a plate reader.
Western Blotting and Activity Assays
Cells were washed twice in chilled Hank’s Balanced Salt Solution containing calcium and magnesium (HBSS, 14025092, Gibco) and lysed in chilled 1x JS buffer (2x JS: 100mM Hepes, pH 7.5, 300mM NaCl, 10mM EGTA, 3mM MgCl2, 2% Glycerol, 2% Triton X-100, supplemented with protease and phosphatase inhibitors). Protein concentrations were determined using Pierce BCA Assay kit (Thermo). Samples were run on 4–20% Mini-PROTEAN pre-cast gels (Biorad) or either 10% or 15% homemade gels and transferred to PVDF membranes (Thermo). For the Rho activity assay GST-RBD (Rhotekin) was purified as previously described (Isogai et al., 2015 JCS). The RBD assay was carried out by incubating 500 μg of total cell lysate (lysis buffer: 50mM Tris pH 7.5, 150 mM NaCl, 10mM MgCl2, 1% Triton X-100, 1mM DTT, supplemented with protease and phosphatase inhibitors) with 30 μg of GST-RBD and GST-CRIB, respectively, as previously described (Isogai et al., 2015 JCS). Antibodies used and dilutions are as follows: T202/Y204-pERK 1:500 (E10, Cell Signaling), ERK 1:1000 (sc-93, Santa Cruz), pMEK1/2 1:1000(9121, Cell Signaling), MEK1/2 1:1000 (9122, Cell Signaling), Cyclin D1 1:500 (sc-8396, Santa Cruz), GAPDH 1:5000 (G9545, Sigma), Y397-pFAK 1:500 (D20B1, Cell Signaling), FAK 1:1000 (D2R2E, Cell Signaling), Y118-pPaxillin 1:1000 (Cell Signaling), Y31-pPaxillin 1:1000 (R&D Systems), Paxillin 1:10,000 (ab32084, Abcam), Talin 1:1000 (Sigma), β-Actin 1:100,000 (AC15 Sigma), RhoA 1:500 (Cell Signaling), T18/S19-pMYLC2 1:1000 (Cell Signaling), MYLC2 1:1000 (D18E2, Cell Signaling), JNK 1:500 (sc7345, Santa Cruz), pJNK 1:1000 (sc6254, Santa Cruz), S518-pNF2 1:1000 (9163, Cell Signaling), NF2 1:1000 (D3S3W, Cell Signaling). Western blot quantification was done using the gel densitometry application in ImageJ.
Immunofluorescence
Paxillin in cell adhesions was visualized using immunofluorescence at least 24 hrs following cell plating on #1.5 acid-etched cover-glasses in 6-well plates or #1.5 glass-bottom chamber slides (155382, nunc). Nuclear YAP localization was visualized using immunofluorescence 48 hours after seeding and drug treatment in #1.5 glass-bottom chamber slides (155382, nunc). Cells were washed three times in HBSS. For Paxillin, cells were then incubated for 2 min in a 2% paraformaldehyde/0.5% Triton X-100 solution followed by 4% paraformaldehyde for 30 min. For YAP localization, cells were incubated in 4.0% paraformaldehyde in PBS for 15 min, washed with PBS, then incubated in 0.1% Triton X-100 in PBS for 10min. Samples were then washed three times in PBS. Quenching-PBS (Q-PBS, 225 ml PBS, 22.5 mg saponin, 4.5 g BSA, 225 mg lysine, pH 7.4) was added to samples for 30 min to block non-specific binding sites. Rabbit anti-paxillin antibody 1:500 (ab32084, Abcam) and rabbit anti-YAP antibody 1:500 (abcam, ab205270) were prepared in Q-PBS and added to samples for 1 hr then washed for 5 min, 6 times in Q-PBS. Alexa Fluor-conjugated secondary antibodies (Molecular Probes) were prepared at 1:500 dilutions in Q-PBS. Samples were incubated for 1hr and washed for 5 min, 6 times in PBS. Samples were post-fixed for 10 min in 4% paraformaldehyde and washed thrice in PBS. Paxillin-labeled adhesions were visualized with total internal reflection fluorescence microscopy (TIRF) using a Nikon Ti-Eclipse inverted microscope equipped with a 100× 1.49 NA Apo TIRF objective, a Diskovery Platform (Andor Technology) for TIRF illumination, and an sCMOS camera. YAP localization was visualized by collecting a z-stack (15 sections at 4.2um thickness) by epifluorescence illumination using a DeltaVision OMX at 60x magnification.
Localization of total NF2, PAK, and phospho-NF2 were visualized using immunofluorescence 48 hours after seeding and drug treatment in #1.5 glass-bottom chamber slides (155382, nunc). Cells were washed three times in HBSS and fixed with 4% paraformaldehyde in CPB (Mueller et al., 2017; Leyton Puig et al., 2016) for 15 min. Cells were then covered in ice-cold 100% methanol and left at −20°C for 15 min. Following washes, samples were blocked and treated with antibodies as described above. Primary antibodies were prepared in Q-PBS using the following dilutions: S518-pNF2 1:50 (9163, Cell Signaling), NF2 1:100 (B-12, sc-55575), PAK 1:100 (abcam, ab154284). Alexa Fluor-conjugated secondary antibodies (Molecular Probes) were prepared at 1:500 dilutions in Q-PBS. For NF2, PAK, and phospho-NF2, localization was visualized with TIR-FM using a DeltaVision OMX at 60x magnification.
Live Cell Imaging
Cells were plated in 35 mm WillCo dishes on #1.5 cover glasses coated with thin (~30 μm) layers of soft silicone gels with high refractive indices, making the substrates both deformable under cell traction forces and compatible with TIRF microscopy (Gutierrez et al., 2011). Surfaces of the gels were decorated with covalently bonded 40 nm dark red (660/680 nm) fluorescent beads (ThermoFisher), which served as tracer particles, enabling traction force microscopy (TFM). Silicone gel substrates were coated with fibronectin by incubating them for 30 minutes under a solution of 20 ul of a 10 mg/mL 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) solution, 30 ul of a 5 mg/mL fibronectin solution, and 2 mL of Ca2+ and Mg2+ containing Dulbeccos Phosphate Buffered Saline (DPBS, 14040117, Gibco). Following coating, substrates were rinsed multiple times with DPBS followed by a final rinse in DMEM and incubated at 37°C prior to cell plating and drug treatment (0.003% DMSO, 33.3nM Dabrafenib). High-resolution imaging was performed 48 hrs later using a DeltaVision OMX.
Fortraction-force microscopy, the microscope was operated in a ring-TIRF illumination condition, which permits more homogeneous illumination of the basal surface of the cell. Images were acquired at a 60x magnification, providing an 80 nm pixel size and Nyquist sampling. Paxillin and the fluorescent tracer particles were imaged with 488 and 640 nm lasers, respectively, and focus offsets were introduced for each spectral channel to maximize image focus. Following imaging, cells were removed rapidly by adding mL of 30% bleach to the 2 mL of cell media, and the tracer particles on the substrate at each cell position were imaged under the relaxed condition.
For actin imaging, cells were plated in #1.5 glass-bottom chamber slides (155382, nunc), and the DeltaVision OMX microscope was operated in an oblique illumination mode, thus minimizing unnecessary illumination of the cells. Images were acquired every 5 seconds, which is sufficient to Nyquist sample actin polymerization, protrusion, and retraction events.
For 3D imaging, +EV, +WT, and +P29S cells expressing mNeonGreen-Actin-C-35 were cultured on 2mg/ml bovine collagen gels for 48hrs with or without drug treatment and imaged with a high numerical aperture variant of diagonally scanned light sheet microscopy75. Briefly, 80 timepoints were acquired with 488 nm illumination and a 35 ms camera exposure time, resulting in a 3.19 sec interval to image a single volume. The lateral and axial pixel sizes were 104 and 500 nm, respectively, for a field of view of 106 × 106 × 45 microns. Images were deconvolved with a synthetic point-spread function using 40 iterations of the Richardson-Lucy algorithm implemented with Microvolution, and rendered using Arivis or ImageJ.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of Immunohistochemistry
In order to quantify proliferation of melanoma metastases in the lung, a few key issues had to be addressed: first, metastases needed to be distinguished from normal lung tissue; second, the area of melanoma cells within metastases needed to be determined since metastases were heterogenous in terms of packing of cells, nodularity versus infiltration or dispersion with the normal tissue, and the presence of stromal or extracellular matrix deposition; and third, area of pH3-labeled dividing nuclei relative to area of metastasis was not a viable metric to determine fraction of cell proliferation because nuclei size and shape were variable based on tissue sectioning and mitotic stage (e.g. mitotic nuclei in prophase undergoing chromosome condensation are largerthan mitotic nuclei in metaphase).
Thus, we developed a strategy to distinguish metastases and determine the area of cancer cells within metastases with high accuracy. We then used this information to normalize metastases by area and then performed hand counting to determine frequency of proliferation within metastases. To ensure blinded, unbiased hand counting, histology data representing each condition were pooled and assigned a random number and renamed using MATLAB. The key was maintained by an independent author not involved in the data analysis. Since cells in our xenograft tumor model (described above) stably expressed the dsRed2-P2A-Luc lentiviral construct, melanoma cells in metastases were labeled by antibody staining for RFP. We then employed Ilastik machine learning software (https://www.ilastik.org/) for pixel classification to determine pixel area of all metastatic nodules positively labeled for dsRed expression (stained red). All metastatic nodules with pixel areas between 20,000 and 40,000 um2 were de-identified, randomized and phospho-Histone 3 positive nuclei were hand counted to determine number of proliferating cells (n ≥ 9 metastatic lung nodules for each condition).
Quantification of Cell Proliferation
Nuclei were counted automatically in a custom pipeline for image processing, quality control and condition analysis developed in MATLAB (The MathWorks, Inc.). The detection algorithm uses the 99th percentile of the background noise to estimate an adaptive threshold for significant nuclei signal. In order to discriminate nuclei in close proximity in the mask of significance, local intensity maxima are detected on the band-passed filtered image before watershed segmentation. The band-pass filter allows for selectivity in scale of the object of interest. The scales used for the band-pass filter are 8 pixels and 15 pixels for low-pass and high-pass filter, respectively. Since we didn’t observe variations in nuclei size across experiments, this algorithm provides us with an unbiased method to compare nuclei accounts across conditions. We then used the u-trackGUI (Jaqaman et al., 2008) to allow for a systematic review of the stationarity of imaging condition. The measurements associated to each nuclei detection (position, intensity, channel and time) are then grouped by condition and stored in a spreadsheet for convenient plotting.
Quantification of Cell Area
Cell areas were measured following 48 hr drug treatment with DMSO (0.003%), Dabrafenib (33.3 nM), and Trametinib (3.3 nM). Images were acquired using live cell phase contrast imaging at 20x magnification and a Retiga R3 CCD camera (QImaging). ROIs were carefully hand drawn for n ≥ 20 cells randomly selected for each cell line (A375+EV, A375+WT, A375+P29S, IGR1P29S, and IGR1WT) and for each drug condition (DMSO, Dabrafenib, and Trametinib). ImageJ was used to measure cell area.
Traction Force Analysis
Traction force was analyzed using a MATLAB-based algorithm that uses L1 regularization (Han et al., 2015). The regularization parameter was determined by optimal regularization selection criteria-an inflection point in the L-curve, which is a plot between the residual norm vs. the self-norm of the traction solution. The strain energy (1/2 * displacement * traction), which represents the overall mechanical work done by a cell on the soft gel, was quantified from traction maps and displacement maps of individual cells, integrated over an entire, segmented cell area. Strain energy distributions of A375 cells expressing empty vector, Rac1WT, or Rac1P29S were compared following treatment for 48hrs with either 0.003% DMSO or 33.3nM Dabrafenib.
Quantification of Focal Adhesions
Focal adhesion (FA) segmentation was performed as described previously (Han et al., 2015), based on a combination of Otsu and Rosin thresholding after preprocessing images with noise removal and background subtraction. Based on the segmentation, statistics for FA area were determined and compared for A375 cells expressing empty vector, Rac1WT, or Rac1P29S following treatment with either 0.003% DMSO or 33.3nM Dabrafenib.
Quantification of Immunofluorescence
We analyzed the intensities of immunofluorescently-labeled phospho-NF2 and total NF2 in images acquired by TIRF microscopy to determine localization and signaling of NF2 in A375 cells lines treated for 48hrs with DMSO, Dabrafenib or Dabrafenib combined with the Arp2/3 inhibitor CK666 or the inactive peptide CK689. The cell boundaries were segmented from the pNF2 images by intensity thresholding, and the distances from the cell edge were computed for each pixel within each cell. For individual cells, we collected the pixel-by-pixel distances from the edge and intensities of pNF2 and NF2. We computed mean intensities at a given distance for the range 0.1 – 6 um using Nadaraya-Watson kernel regression and a custom-built MATLAB function. The obtained mean intensity functions of multiple cells were averaged and 95% confidence intervals were computed at each distance to quantify cell-to-cell variation. The same analysis pipeline was applied to determine PAK localization following 48hrs of treatment with DMSO and Dabrafenib.
To determine the corrected total nuclear YAP fluorescence, sum intensity projections (SIPs) were generated using ImageJ from z-stacks (15 sections at 4.2um thickness) acquired with a DeltaVision OMX at 60x magnification with epifluorescence illumination. The DAPI nuclear labeling was used to draw ROIs for all nuclei in each SIP image, and ImageJ was used to measure nuclear area and the integrated density (IntDen) of YAP signal in the nucleus. Mean background intensity was determined by selecting ROIs with no fluorescence signal from the YAP channel of each image. Nuclear YAP fluorescence was determined using the equation for corrected total fluorescence: Corrected Total Fluorescence = Integrated Density - (Area of nuclei x Mean fluorescence of background readings) (Martin Fitzpatrick, University of Birmingham, United Kingdom, The Open Lab Book).
Determination of Lamellipodia Widths
To measure the size of lamellipodial structures in time lapse images of fluorescently labeled actin we utilized a similar approach as for the quantification of PAK and pNF2 localization described above. However, raw actin intensities do not delineate lamellipodia boundaries, because transversal arcs and stress-fibers outside the lamellipodia display intensity values similar or higher than the dense lamellipodial actin brushes. The fundamental difference between lamellipodial and non-lamellipodial actin structures is the dynamics. Actin treadmilling in the lamellipodium yields a visually obvious flow that is reflected by high temporal intensity variation over a ~10-minute movie, whereas transversal arcs and stress fibers generate a more stationary intensity signal. To capture this behavior we transformed each actin image into an image that captures the relative absolute deviation (RAD) from the temporal mean intensity at each pixel within the cell mask:
RAD images showed high signals in lamellipodia and low signals in static structures. Averaged RAD values decreased as a function of the distance from the cell edge. At the distance characteristic for the lamellipodia width the averaged RAD curves reacheda baseline level. We used the temporal mean and standard deviation (std) of the baseline RAD values for an entire movie to determine a threshold mean(Rad) + 4*std(RAD). Using the threshold, we computed for each frame of the movie the distance of the high RAD region, which reflects the spatially averaged lamellipodium width for the particular frame. To systematically compare lamellipodia extensions between experimental conditions we assigned to each movie the median distance of the high RAD region over all frames as a robust measurement of the overall lamellipodium width.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism software and details including statistical test used, value of n, what n represents, and dispersion and precision measures accompany respective figures in the figure legend. All biological replicates were performed on separate days. P-value cut offs were assigned as follows: *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-phospho-p44/42 MAPK (Erk1/2) (T202/Y204) | Cell Signaling | E10; Cat#9106; RRID: AB_331768 |
| Rabbit polyclonal anti-ERK 1 | Santa Cruz | C-16; Cat#sc-93; RRID: AB_631453 |
| Rabbit polyclonal anti-phospho-MEK 1/2 (S217/S221) | Cell Signaling | Cat#9121; RRID: AB_331648 |
| Rabbit polyclonal anti-MEK 1/2 | Cell Signaling | Cat#9122; RRID: AB_823567 |
| Mouse monoclonal anti-Cyclin D1 | Santa Cruz | A-12; Cat#sc-8396; RRID: AB_627344 |
| Rabbit polyclonal anti-GAPDH | Sigma-Aldrich | Cat#G9545; RRID: AB_796208 |
| Rabbit monoclonal anti-phospho-FAK (Y397) | Cell Signaling | D20B1; Cat#8556; RRID: AB_10891442 |
| Rabbit monoclonal anti-FAK | Cell Signaling | D2R2E; Cat#13009; RRID: AB_2798086 |
| Rabbit polyclonal anti-phospho-Paxillin (Y118) | Cell Signaling | Cat#2541; RRID: AB_2174466 |
| Mouse monoclonal anti-phospho-Paxillin (Y31) | R&D Systems | Cat# MAB61641; RRID: AB_10972644 |
| Rabbit monoclonal anti-Paxillin | Abcam | Y113; ab32084; RRID: AB_779033 |
| Mouse monoclonal anti-Talin | Sigma-Aldrich | Cat# T3287; RRID: AB_477572 |
| Mouse monoclonal anti-β-Actin | Sigma-Aldrich | AC-15; Cat#A1978; RRID: AB_476692 |
| Rabbit monoclonal anti-RhoA | Cell Signaling | 67B9; Cat#2117; RRID: AB_10693922 |
| Rabbit polyclonal anti-phospho-Myosin Light Chain 2 (T18/S19) | Cell Signaling | Cat#3674; RRID: AB_2147464 |
| Rabbit monoclonal anti-Myosin Light Chain 2 | Cell Signaling | D18E2; Cat#8505; RRID: AB_2728760 |
| Mouse monoclonal anti-JNK | Santa Cruz | D-2; sc-7345; RRID: AB_675864 |
| Mouse monoclonal anti-phospho-JNK | Santa Cruz | G-7; sc-6254; RRID: AB_628232 |
| Rabbit polyclonal anti-phospho-Merlin (S518) | Cell Signaling | Cat#9163; RRID: AB_2149793 |
| Rabbit monoclonal anti-Merlin | Cell Signaling | D3S3W; Cat#12888; RRID: AB_2650551 |
| Mouse monoclonal anti-NF2 | Santa Cruz | B-12; Cat# sc-55575; RRID: AB_831599 |
| Rabbit polyclonal anti-RFP | Rockland | Cat# 600–401-379; RRID: AB_2209751 |
| Mouse monoclonal anti-phospho-Histone H3 (S10) | Cell Signaling | 6G3; Cat#9706; RRID: AB_331748 |
| Rabbit polyclonal anti-PAK 1 | abcam | Cat#ab154284 |
| Goat polyclonal anti-Rabbit IgG, Alexa Fluor 488-conjugated secondary | ThermoFisher | Cat#A11034; RRID: AB_2576217 |
| Donkey polyclonal anti-Rabbit IgG, Alexa Fluor 647-conjugated secondary | ThermoFisher | Cat# A31573; RRID: AB_2536183 |
| Donkey polyclonal anti-Mouse IgG, Alexa Fluor 568-conjugated secondary | ThermoFisher | Cat# A10037; RRID: AB_2534013 |
| Mouse monoclonal anti-ARP3 | Santa Cruz | A-1; sc-48344; RRID: AB_626700 |
| Mouse monoclonal anti-p34-ARC (ARPC2) | Santa Cruz | F-5; sc-515754 |
| Mouse monoclonal anti-p41-ARCb (ARPC1B) | Santa Cruz | C-3; sc-137125; RRID: AB_2289927 |
| Rabbit polyclonal anti-ARPC1A | Sigma-Aldrich | HPA004334; RRID: AB_1078215 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| mNeonGreen-Paxillin-22 | Allele Biotechnology | http://www.allelebiotech.com/mneongreen-fusions-and-constructs/ |
| mNeonGreen-Actin-C-35 | Allele Biotechnology | http://www.allelebiotech.com/mneongreen-fusions-and-constructs/ |
| Dabrafenib | Selleckchem | S2807 |
| Trametinib | Selleckchem | S2673 |
| SB203580 | Selleckchem | S1076 |
| YAP-TEAD Inhibitor 1 (Peptide 17) | Selleckchem | S8164 |
| SMIFH2 | Tocris | S4826 |
| CK666 | Sigma-Aldrich | 182515 |
| CK689 | Sigma-Aldrich | 182517 |
| (−)-Blebbistatin | Sigma-Aldrich | B0560 |
| ROCK Inhibitor | Sigma-Aldrich | Y-27632; SCM075 |
| SCH772984 | ChemieTek | CT-SCH772 |
| Bovine collagen, Type 1 | Advanced Biomatrix | 5133 |
| Collagenase | Advanced Biomatrix | 5030 |
| Poly-L-Lysine | Sigma-Aldrich | P8920 |
| Fibronectin | Sigma-Aldrich | F1141 |
| Laminin | Santa Cruz | sc-29012 |
| Critical Commercial Assays | ||
| QuikChange Lightning Mutagenesis Kit | Agilent | 210515 |
| Biotin-Avidin Blocking Kit | Vector Laboratories | SP-2001, |
| Vector M.O.M Peroxidase-based Immunodetection Kit | Vector Laboratories | N/A |
| ImmPACT DAB Peroxidase Substrate Kit | Vector Laboratories | SK-4105 |
| ImmPRESS-AP Reagent Anti-Rabbit IgG alkaline phosphatase-based staining kit | Vector Laboratories | MP-5401 |
| ImmPACT Vector Red alkaline phosphatase substrate | Vector Laboratories | SK-5105 |
| Click-iT Plus EdU Alexa Fluor Imaging Kits (555 and 488) | Molecular Probes | C10638 (555) and C10637 (488) |
| Click-iT Plus Edu Alexa Fluor Flow Cytometry Kit | Molecular Probes | C10632 |
| Experimental Models: Cell Lines | ||
| Human melanoma: A375 | ATCC | Cat# CRL-1619; RRID: CVCL_0132 |
| Human melanoma: IGR1 | DSMZ | Cat# ACC-236; RRID: CVCL_1303 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Adult female NOD/SCID IL2Rγnull mice | S. Morrison (Quintana et al., 2012) | https://www.jax.org/strain/005557; RRID: IMSR_JAX:005557 |
| Oligonucleotides | ||
| Rac1P29S QuikChange primer (forward): CTGATCAGTTACACAACCAATGCATTTTCTGGAGAATATATCCC | This paper | N/A |
| Rac1P29S QuikChange primer (reverse): GGGATATATTCTCCAGAAAATGCATTGGTTGTGTAACTGATCAG | This paper | N/A |
| CRISPR editing Rac1P29S to Rac1WT: sgRNA guide sequence 1: 5’- TACACAACCAATGC ATTTTC-3’ | This paper | N/A |
| CRISPR editing Rac1P29S to Rac1WT: sgRNA guide sequence 2: 5’- ATATTCTCCAGAAAA TGCAT-3’ | This paper | N/A |
| CRISPR editing Rac1P29S to Rac1WT: ssODN repair sequence 1: AAGATACTTACACAGTAGGGATATATTCTCCAGGAAATGCATTGGTTGTGTAACTGATCAGTAGGCAAGT | This paper | N/A |
| CRISPR editing Rac1P29S to Rac1WT: ssODN repair sequence 2: AAAACTTGCCTACTGATCAGTTACACAACCAATGCATTTCCTGGAGAATATATCCCTACTGTGTAAGTAT | This paper | N/A |
| CRISPR knockout of ARPC1B: sgRNA guide sequence: 5’-CTCGTGCACCTTGGTCCATT-3’ | This paper | N/A |
| Recombinant DNA | ||
| pcDNA3-GFP-Rac1 (Wild Type) | Cell Biolabs, inc. | STA-450, 345001 |
| pcDNA3-GFP-Rac1 P29S | This paper | N/A |
| pLVX-Puro | Clontech | 632164 |
| pLVX-Puro-Rac1WT | This paper | N/A |
| pLVX-Puro-RaclP29S | This paper | N/A |
| pLVX-shRNA2 | Clontech | 632179 |
| pLVX-CMV100-mNeonGreen-Paxillin-22 | This paper | N/A |
| pLVX-CMV300-mNeonGreen-Actin-C-35 | This paper | N/A |
| pcDNA-NF2 (Wild Type) | D. Pan | N/A |
| pcDNA-NF2-S518A | D. Pan | N/A |
| pLVX-Puro-NF2 (Wild Type) | This paper | N/A |
| pLVX-Puro-NF2-S518A | This paper | N/A |
| dsRed2-P2A-Luc | S. Morrison (Piskounova et al., 2015) | N/A |
| pSpCas9(BB)-2A-GFP (PX458) | Ran et al., 2013. | Addgene ID: 48138, RRID: Addgene_48138 |
| Software and Algorithms | ||
| Ilastik | https://www.ilastik.org/ | RRID:SCR_015246 |
| Matlab | The MathWorks, Inc.; https://www.mathworks.com/products/matlab.html | RRID:SCR_001622 |
| Fiji | http://fiji.sc/ | RRID:SCR_002285 |
| GraphPad Prism | https://www.graphpad.com/ | RRID:SCR_002798 |
| U-track Particle Tracking (Version 2.2.1) | Danuser Lab (Jaqaman et al., 2008) | http://www.utsouthwestern.edu/labs/danuser/software/ |
| Traction Force Analysis (Version 1.1.3) | Danuser Lab (Han et al., 2015) | http://www.utsouthwestern.edu/labs/danuser/software/ |
| Focal Adhesion Segmentation | Danuser Lab (Han et al., 2015) | http://www.utsouthwestern.edu/labs/danuser/software/ |
| Immunofluorescence Localization Analysis | This paper | N/A |
| Lamellipodia Widths Analysis | This paper | N/A |
| CRISPR Design Tool | http://tools.genome-engineering.org | |
| CRISPOR | http://crispor.tefor.net | |
| Benchling | https://benchling.com/ | RRID:SCR_013955 |
Highlights.
Rac1P29S confers proliferative advantage during melanoma metastasis and drug treatment
Rac1P29S sustains proliferation in drug-challenged cells by elongation of lamellipodia
Rac1P29S-driven proliferation needs matrix attachment but not focal adhesion signaling
Elongated lamellipodia in Rac1P29S cells sequester and phospho-inactivate NF2/Merlin
ACKNOWLEDGMENTS
We would like to thank Dr. Sean Morrison for providing guidance for in vivo experiments and for critically reading the manuscript and Dr. Duojia Pan for providing NF2 mutant and wild-type constructs. We are grateful for assistance from John Shelton and the UT Southwestern Histo Pathology Core for providing guidance and services for tissue handling and also generous access to supplies and equipment, Dr. Katherine Luby-Phelps and the Electron Microscopy Core for access to the JEM-1400 Plus transmission electron microscope (NIH 1S100D021685–01A1), and the BioHPC team for covering the significant needs of this project in computing and storage infrastructure. We are also grateful to the Shay/Wright Lab for both resources and guidance: Dr. Andrew Ludlow for input on CRISPR/Cas9 genome editing, Crystal Corneliusfor managing and sharing the pGIPZ shRNA construct library, and Krishna Luteal for his immunohistochemistry protocol. We would also like to thank Michael Abrams and the Alto Lab for guidance on CRISPR/Cas9 genome editing and Dr. Marcel Mettlen in the Schmid Lab for his immunofluorescence protocol. This work was supported by funding from the following grants: NIH F30 CA206399 (A.S.M.), NIH F32 GM117793 (K.M.D.), Human Frontier Science Program LT000954/2015(P.R.), NIH K25 K25CA204526(E.S.W.),Welch Foundation I-1840 (G.D.), and NIH R01 GM071868 (G.D.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.devcel.2019.04.007.
DATA AND SOFTWARE AVAILABILITY
The images and data generated during this study are available from the corresponding author upon reasonable request. Custom code used for data analysis include the current version of u-track particle tracking software (Version 2.2.1) and traction force microscopy software (Version 1.1.3), which are both freely available for download from the Danuser Lab website (http://www.utsouthwestern.edu/labs/danuser/software/). The Matlab functions for lamellipodia width analysis can be requested from the corresponding author. Contrary to the aforementioned packages, these functions are not maintained.
REFERENCES
- Abella JVG, Galloni C, Pernier J, Barry DJ, Kjaer S, Carlier MF, and Way M (2016). Isoform diversity in the Arp2/3 complex determines actin filament dynamics. Nat. Cell Biol. 18, 76–86. [DOI] [PubMed] [Google Scholar]
- Araiza-Olivera D, Feng Y, Semenova G, Prudnikova TY, Rhodes J, and Chernoff J (2018). Suppression of RAC1-driven malignant melanoma by group A PAK inhibitors. Oncogene 37, 944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae YH, Mui KL, Hsu BY, Liu SL, Cretu A, Razinia Z, Xu T, Puré E, and Assoian RK (2014). A FAK-cas-Rac-Lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci. Signal. 7, ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bid HK, Roberts RD, Manchanda PK, and Houghton PJ (2013). RAC1: an emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol. Cancer Ther 12, 1925–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordeleau F, Mason BN, Lollis EM, Mazzola M, Zanotelli MR, Somasegar S, Califano JP, Montague C, Lavalley DJ, Huynh J, et al. (2017). Matrix stiffening promotes a tumor vasculature phenotype. Proc. Natl. Acad. Sci. USA 114, 492–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher DT, Alliston T, and Weaver VM (2009). A tense situation: forcing tumour progression. Nat. Rev. Cancer 9, 108–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, Gao J, Socci ND, Solit DB, Olshen AB, et al. (2016). Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol 34, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Mcquade JL, Panka DJ, Hudgens CW, Amin-Mansour A, Mu XJ, Bahl S, Jané-Valbuena J, Wani KM, Reuben A, et al. (2016). Clinical, molecular, and immune analysis of dabrafenib-trametinib combination treatment for braf inhibitor-refractory metastatic melanoma: a phase 2 clinical trial. JAMA Oncol 2, 1056–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow HY, Jubb AM, Koch JN, Jaffer ZM, Stepanova D, Campbell DA, Duron SG, O’farrell M, Cai KQ, Klein-Szanto AJ, et al. (2012). P21-activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res. 72, 5966–5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman ML, Marshall CJ, and Olson MF (2004). RAS and Rho GTPases in G1-phase cell-cycle regulation. Nat. Rev. Mol. Cell Biol. 5, 355–366. [DOI] [PubMed] [Google Scholar]
- Cook DR, Rossman KL, and Der CJ (2014). Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene 33, 4021–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Ha BH, Holman EC, Halaban R, Schlessinger J, and Boggon TJ (2013). RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc. Natl. Acad. Sci. 110, 912–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean KM, Roudot P, Reis CR, Welf ES, Mettlen M, and Fiolka R (2016). Diagonally scanned light-sheet microscopy for fast volumetric imaging of adherent cells. Biophys. J. 110, 1456–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. (2011). Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, and Hall A (2002). Rho GTPases in cell biology. Nature 420, 629–635. [DOI] [PubMed] [Google Scholar]
- Gutierrez E, Tkachenko E, Besser A, Sundd P, Ley K, Danuser G, Ginsberg MH, and Groisman A (2011). High refractive index silicone gels for simultaneous total internal reflection fluorescence and traction force microscopy of adherent cells. PLoS One 6, e23807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A (1998). Rho GTPases and the actin cytoskeleton. Science 279, 509–514. [DOI] [PubMed] [Google Scholar]
- Han SJ, Oak Y, Groisman A, and Danuser G (2015). Traction microscopy to identify force modulation in subresolution adhesions. Nat. Methods 12, 653–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJC, Stokoe D, Gloor SL, Vigers G, et al. (2010). RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464, 431–435. [DOI] [PubMed] [Google Scholar]
- Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, Larkin J, Marais R, and Sahai E (2015). Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell 27, 574–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodis E,Watson I.r., Kryukov G.v., Arold S.t., Imielinski M,Theurillat J-P, Nickerson E, Auclair D, Li L, Place C, et al. (2012). A landscape of driver mutations in melanoma. Cell 750, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hytönen VP, and Wehrle-Haller B (2016). Mechanosensing in cell-matrix adhesions-converting tension into chemical signals. Exp. Cell Res. 343, 35–41. [DOI] [PubMed] [Google Scholar]
- Isogai T, Van Der Kammen R, and Innocenti M (2015). SMIFH2 has effects on formins and p53 that perturb the cell cytoskeleton. Sci. Rep 5, 9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, and Danuser G (2008). Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods 5, 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji L, Lim J, and Danuser G (2008). Fluctuations of intracellular forces during cell protrusion. Nat. Cell Biol. 70, 1393–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawazu M, Ueno T, Kontani K, Ogita Y, Ando M, Fukumura K, Yamato A, Soda M, Takeuchi K, Miki Y, et al. (2013). Transforming mutations of RAC guanosine triphosphatases in human cancers. Proc. Natl. Acad. Sci. USA 770, 3029–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanietz MG, and Caloca MJ (2017). The Rac GTPase in cancer: from old concepts to new paradigms. Cancer Res. 77, 5445–5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Kim J, Hong H, Lee SH, Lee JK, Jung E, and Kim J (2016). Actin remodeling confers BRAF inhibitor resistance to melanoma cellsthrough YAP/TAZ activation. EMBO J 35, 462–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissil JL, Johnson KC, Eckman MS, and Jacks T (2002). Merlin phosphorylation by p21-activated kinase2and effects ofphosphorylation on merlin localization. J. Biol. Chem. 277, 10394–10399. [DOI] [PubMed] [Google Scholar]
- Kissil JL, Walmsley MJ, Hanlon L, Haigis KM, Bender Kim CF, Sweet-Cordero A, Eckman MS, Tuveson DA, Capobianco AJ, Tybulewicz VLJ, et al. (2007). Requirement for Rac1 in a K-ras-induced lung cancer in the mouse. Cancer Res. 67, 8089–8094. [DOI] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, Mccusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al. (2012). Exomesequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 44, 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo PL, Lee H, Bray MA, Geisse NA, Huang YT, Adams WJ, Sheehy SP, and Parker KK (2012). Myocyte shape regulates lateral registry of sarcomeres and contractility. Am. J. Pathol 787, 2030–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarche N, Tapon N, Stowers L, Burbelo PD, Aspenström P, Bridges T, Chant J, and Hall A (1996). Rac and Cdc42 induce actin polymerization and G1 cell cycle progression independently of p65PAK and the JNK/SAPK MAP kinase cascade. Cell 87, 519–529. [DOI] [PubMed] [Google Scholar]
- Lawson CD, and Burridge K (2014). The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 5, e27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Elliott HL, Oak Y, Zee CT, Groisman A, Tytell JD, and Danuser G (2015). Functional hierarchy of redundant actin assembly factors revealed by fine-grained registration of intrinsic image fluctuations. Cell Syst 7, 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyton Puig D, Kedziora KM, Isogai T, Van Den Broek B, Jalink K, and Innocenti M (2016). Tips and tricks for artifact-free PFA-based fixation of the actin cytoskeleton and its regulatory proteins for single molecule localization super-resolution microscopy. Protoc. Exch 70, 1038. [Google Scholar]
- Lin R, Bagrodia S, Cerione R, and Manor D (1997). A novel Cdc42Hs mutant induces cellular transformation. Curr. Biol. 7, 794–797. [DOI] [PubMed] [Google Scholar]
- Lu H, Liu S, Zhang G, Bin Wu B, Zhu Y, Frederick DT, Hu Y, Zhong W, Randell S, Sadek N, et al. (2017). PAK signaling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature 550, 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky LM (2008). Lamellipodia and filopodia in metastasisand invasion. FEBS Lett. 582, 2102–2111. [DOI] [PubMed] [Google Scholar]
- Malliri A, Van DerKammen RA, Clark K, Van DerValk M, Michiels F, and Collard JG (2002). Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature 477, 867–871. [DOI] [PubMed] [Google Scholar]
- Mani T, Hennigan RF, Foster LA, Conrady DG, Herr AB, and Ip W (2011). FERM domain phosphoinositide binding targets merlin to the membrane and is essential for its growth-suppressive function. Mol. Cell. Biol. 37, 1983–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mar VJ, Wong SQ, Logan A, Nguyen T, Cebon J, Kelly JW, Wolfe R, Dobrovic A, Mclean C, and Mcarthur GA (2014). Clinical and pathological associations of the activating RAC1 P29S mutation in primary cutaneous melanoma. Pigment Cell Melanoma Res. 27, 1117–1125. [DOI] [PubMed] [Google Scholar]
- Marei H, and Malliri A (2017). GEFs: dual regulation of Rac1 signaling. Small GTPases 8, 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazia D, Schatten G, and Sale W (1975). Adhesion of cells to surfaces coated with polylysine. Applications to electron microscopy. J. Cell Biol. 66, 198–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza MC,Vilela M, Juarez JE, Blenis J, and Danuser G (2015). ERK reinforces actin polymerization to power persistent edge protrusion during motility. Sci. Signal 8, ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller J, Szep G, Nemethova M, De Vries I, Lieber AD, Winkler C, Kruse K, Small JV, Schmeiser C, Keren K, et al. (2017). Load adaptation of lamellipodial actin networks. Cell 777, 188–200. [DOI] [PubMed] [Google Scholar]
- Olson MF (2018). Rho GTPases, their post-translational modifications, disease-associated mutations and pharmacological inhibitors. Small GTPases 9, 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MF, Ashworth A, and Hall A (1995). An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G. Science 269, 1270–1272. [DOI] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. (2005). Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241–254. [DOI] [PubMed] [Google Scholar]
- Peng S-B, Henry J.r., Kaufman M.d., Lu W-P, Smith B.d., Vogeti S, Rutkoski T.j., Wise S, Chun L, Zhang Y, et al. (2015). Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell 28, 384–398. [DOI] [PubMed] [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, Deberardinis RJ, and Morrison SJ (2015). Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, and Borisy GG (2003). Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465. [DOI] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, and Rosen N (2010). RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu RG, Chen J, Kirn D, Mccormick F, and Symons M (1995). An essential role for Rac in Ras transformation. Nature 374, 457–459. [DOI] [PubMed] [Google Scholar]
- Quintana E, Piskounova E, Shackleton M, Weinberg D, Eskiocak U, Fullen DR, Johnson TM, and Morrison SJ (2012). Human melanoma metastasis in NSG mice correlates with clinical outcome in patients. Sci. Transl. Med 4, 159ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J,Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangamani P, Lipshtat A, Azeloglu EU, Calizo RC, Hu M, Ghassemi S, Hone J, Scarlata S, Neves SR, and Iyengar R (2013). Decoding information in cell shape. Cell 154, 1356–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, and Hall A (1992). The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70, 401–410. [DOI] [PubMed] [Google Scholar]
- Ron A, Azeloglu EU, Calizo RC, Hu M, Bhattacharya S, Chen Y, Jayaraman G, Lee S, Neves-Zaph SR, Li H, et al. (2017). Cell shape information is transduced through tension-independent mechanisms. Nat. Commun 8, 2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MB, Finn AJ, Pedone KH, Thomas NE, Der CJ, and Cox AD (2016). ERK/MAPK signaling drives overexpression of the Rac-GEF, PREX1, in BRAF-and NRAS-mutant melanoma. Mol. Cancer Res. 14, 1009–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Paez JG, Curto M, Yaktine A, Pruitt WM, Saotome I, O’bryan JP, Gupta V, Ratner N, Der CJ, et al. (2001). The Nf2 tumor suppressor, merlin, functions in Rac-dependent signaling. Dev. Cell 1, 63–72. [DOI] [PubMed] [Google Scholar]
- Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al. (2014). Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 4, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibue T, and Weinberg RA (2009). Integrin β1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc. Natl. Acad. Sci. USA 706, 10290–10295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MP, and Wellbrock C (2016). Molecular pathways: maintaining MAPK inhibitor sensitivity by targeting nonmutational tolerance. Clin. Cancer Res. 22, 5966–5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C, Straehle C, Köthe U, and Hamprecht FA (2011). In Ilastik: interactive learning and segmentation toolkit-IEEE International Symposium on Biomedical Imaging (ISBI) 2011, 230–233. [Google Scholar]
- Sosa MS, Lopez-Haber C, Yang C, Wang H, Lemmon MA, Busillo JM, Luo J, Benovic JL, Klein-Szanto A, Yagi H, et al. (2010). Identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol. Cell 40, 877–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, et al. (2014). The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 4, 94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vial E, Sahai E, and Marshall CJ (2003). ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell 4, 67–79. [DOI] [PubMed] [Google Scholar]
- Vinzenz M, Nemethova M, Schur F, Mueller J, Narita A, Urban E, Winkler C, Schmeiser C, Koestler SA, Rottner K, et al. (2012). Actin branching in the initiation and maintenance of lamellipodia. J. Cell Sci. 125, 2775–2785. [DOI] [PubMed] [Google Scholar]
- Watson IR, Li L, Cabeceiras PK, Mahdavi M, Gutschner T, Genovese G, Wang G, Fang Z, Tepper JM, Stemke-Hale K, et al. (2014). The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res. 74, 4845–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheimer E, Gutierrez-Uzquiza A, Rosemblit C, Lopez-Haber C, Sosa MS, and Kazanietz MG (2012). Rac signaling in breast cancer: a tale of GEFs and GAPs. Cell. Signal 24, 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao GH, Beeser A, Chernoff J, and Testa JR (2002). P21-activated kinase links Rac/Cdc42 signaling to merlin. J. Biol. Chem. 277, 883–886. [DOI] [PubMed] [Google Scholar]
- Xiao GH, Gallagher R, Shetler J, Skele K, Altomare DA, Pestell RG, Jhanwar S, and Testa JR (2005). The NF2 tumor suppressor gene product, merlin, inhibits cell proliferation and cell cycle progression by repressing cyclin D1 expression. Mol. Cell. Biol. 25, 2384–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.