

Abstract
Objective
Stress granules (SG) are dynamic cytoplasmic aggregates containing mRNA, RNA-binding proteins and translation factors that form in response to cellular stress. SG have been shown to contribute to the pathogenesis of several human diseases, but their role in vascular diseases is unknown. This study shows SG accumulate in vascular smooth muscle cells (VSMC) and macrophages during atherosclerosis.
Approach and Results
Immunohistochemical analysis of atherosclerotic plaques from LDLR−/− mice revealed an increase in the stress granule-specific markers Ras‐GTPase‐activating protein SH3 domain‐binding protein (G3BP) and Poly-A-Binding Protein (PABP) in intimal macrophages and smooth muscle cells that correlated with disease progression. In vitro, PABP+ and G3BP+ stress granules were rapidly induced in VSMC and bone marrow derived macrophages in response to atherosclerotic stimuli, including oxidized low density lipoprotein and mediators of mitochondrial or oxidative stress. We observed an increase in eIF2α phosphorylation, a requisite for stress granule formation, in cells exposed to these stimuli. Interestingly, SG formation, PABP expression, and eIF2α phosphorylation in VSMCs is reversed by treatment with the anti-inflammatory cytokine interleukin-19. Microtubule inhibitors reduced stress granule accumulation in VSMC, suggesting cytoskeletal regulation of stress granule formation. SG formation in VSMCs was also observed in other vascular disease pathologies, including vascular restenosis. Reduction of SG component G3PB1 by siRNA significantly altered expression profiles of inflammatory, apoptotic, and proliferative genes.
Conclusions
These results indicate that SG formation is a common feature of the vascular response to injury and disease, and that modification of inflammation reduces stress granule formation in VSMC.
Introduction:
Cardiovascular disease is the leading cause of death worldwide, with vascular diseases in particular accounting for two-thirds of all cardiovascular disease-related mortalities1. Many vascular diseases such as atherosclerosis are pro-inflammatory in etiology2. In addition to immune cells, vascular smooth muscle cells (VSMCs) respond to inflammatory stimuli and synthesize pro-inflammatory modulators which further propagate VSMC activation and the recruitment of leukocytes to the atherosclerotic lesion3–6, resulting in localized inflammation and plaque. Indeed, recent studies have suggested that a majority of the cells in atherosclerotic lesions are SMC-derived7. Therefore, a better understanding of mechanisms that modulate the VSMC response to injury is key to development of therapeutics to combat multiple vascular diseases. The recent CANTOS trial provided the first-in-human evidence that targeting inflammation (by inhibiting interleukin-1β) prevented cardiovascular events in patients with residual inflammatory risk, without additional lipid lowering8. This supports the targeting of inflammatory components in vascular disease as a potential area of intervention.
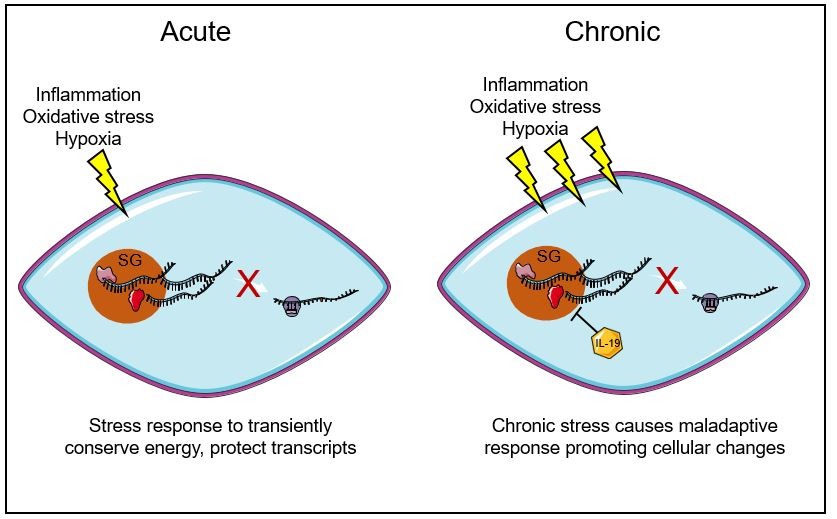
Initiation and resolution of inflammation is a dynamic and tightly regulated process. The post-transcriptional regulation of mRNA stability and translation provides a mechanism by which VSMC can rapidly respond to inflammatory stimuli. RNA binding proteins can bind to AU-rich elements (AREs) in the 3’UTR of mammalian mRNAs to regulate transcript processing9,10. Notably, many inflammatory cytokine mRNAs contain conserved or semi-conserved AU-rich elements in their 3’UTR, imparting target specificity for a potential anti-inflammatory modality9. Under various forms of stress or drug-induced inhibition of translation, mRNAs stalled at a step in translation initiation assemble to form cytoplasmic structures called stress granules (SG). Stress granules are ribonucleoprotein complexes that contain stalled translation initiation complexes as well as mRNA. RNA-binding proteins such as HuR, TIA-1, and TTP, and translation initiation factors such as eIF4E, eIF4G, eIF4A, and eIF4B co-localize in SG11,12. The protein and RNA composition of stress granules suggests that processing of ARE-containing inflammatory transcripts likely occurs there. Controlling mRNA decay allows the cell to fine-tune mRNA abundance and translation for rapid adaptation to environmental conditions, especially inflammation11, and it has been posited that SG formation represents a “decision point” in the cell’s response to stress11. Several studies have suggested that transient SG formation is a cytoprotective response, whereas chronic appearance of SG is indicative of prolonged translational repression, with associated detrimental cellular effects. Indeed, prolonged SG assembly is recognized as a distinguishing cellular feature of Alzheimer’s disease13. Given the chronic nature of many vascular diseases, and atherosclerosis in particular, we hypothesized that SG formation occurs and may participate in the vascular response to injury.
Presently, the role of SG in vascular tissue, and their association with vascular diseases such as atherosclerosis, are poorly understood. Using an unbiased Mass-SPEC-based proteomic approach designed to identify proteins involved in RNA processing in VSMC, our lab identified 15 of 18 candidate proteins as SG components14. We hypothesized that atherogenic stimuli can induce SG formation in VSMC, and that the appearance and abundance of these structures is associated with disease progression. In this study we report that SGs are induced in a time-dependent fashion in VSMC and macrophages in mouse models of vascular disease. SG components are increased in coronary artery from failing compared with non-failing human heart. In vitro modeling of atherosclerotic plaque VSMCs and macrophages showed that SG are rapidly induced in response to oxLDL stimulation and oxidative stress. Notably, stress granule formation in VSMCs could be reversed by treatment with the anti-inflammatory cytokine IL-19, which we had previously identified as a regulator of inflammatory cytokine mRNAs and HuR in VSMCs. Together, these results suggest that stress granule formation represents a vascular response to inflammation, and that modification of inflammation reduces SG formation. Stress granule formation may function as a marker and potentially a cellular decision point in the progression of such diseases.
Material and Methods
The authors declare that all supporting data are available within the article [and its online supplementary files].
VSMC Culture.
Primary human coronary artery vascular smooth muscle cells were obtained as cryopreserved secondary culture from LifeLine Cell Technology from a male individual which are utilized due to consistency with previous studies14–16(Frederick, MD) and maintained as we described17,18. Cells were used from passage 3–5. To induce SG formation, 20μM clotrimazole (Sigma-Aldrich, Allentown, PA) was added in serum-free media directly to VSMCs for 45 minutes, at which time cells were washed with PBS, fixed, and stained as detailed in the immunohistochemistry methods. For OxLDL (Kalen Biomedical, LLC, Germantown, MD) treatment, VSMC were serum-starved in 2% FBS media prior to stimulation with 50μg/ml for 24 hours unless indicated otherwise. For IL-19 (Peprotech, Inc., Rocky Hill, NJ) treatment, we stimulated with 100ng/ml of human recombinant IL-19 for the times indicated. For calpain inhibition, VSMC were pre-treated with 100 μmol/L zLLal (Biomol Research Laboratories, cat# BML-PI116, Farmingdale, NY) for 30 minutes prior to stimulation. For Proliferation, HVSMCs were transfected with G3BP1 siRNA or scrambled control siRNA for 48h. Cells were seeded at 10,000 cells/well and counted at days 3 and 7 as described 17.
PCR Array.
Gene expression in hVSMCs transfected with G3BP1 siRNA or scrambled control was performed using the RT2 Profiler™ PCR Array Human Atherosclerosis, catalog # PAHS-038Z from Qiagen (Hilden, Germany) as described by the manufacturer. The results are graphed as SD from independent experiments. Primer sequences for RT-PCR are presented in Supplemental Materials Table 1.
BMDM isolation and treatment.
Bone marrow derived macrophages were isolated from the tibiae and femorae of C57BL/6J mice, both male and female, and differentiated in DMEM supplemented with 20% L929-conditioned medium, 10% FBS and 1% penicillin-streptomycin for 5 days. BMDMs were incubated with 500 μM sodium arsenite (S7400, Sigma Inc., Cambridge, MA) for 30 minutes, or with 50 μg/mL of OxLDL (J65591, ThermoFisher Scientific, Waltham, MA) or aggregated LDL (J65039, ThermoFisher Scientific, Waltham, MA) for 24 hours.
Mice.
All mouse experiments were approved by the Institutional Animal Care and Use Committees of Temple University and New York University Medical Center. For all animals, food and water access is ad libitum, the facility uses a 12-hour light/12-hour dark cycle, and the cage bedding is Bed-o’-cob 1/8” made by Anderson’s Lab Bedding. LDLR−/− mice on the C57BL/6J background were purchased from The Jackson Laboratory (Stock number 002207) and both male and female mice were used. Ligated mouse carotid arteries, aortic arch plaques from LDLR−/− mice fed western diet for 0, 5, 10, and 14 weeks, and human coronary arteries were collected as part of studies described previously19,20. Briefly, mice were housed in an AAALAC-approved facility and maintained on a normal laboratory diet until study commencement. Mice of both sexes were entered in the study in accordance with Robinet et al21., and at 8 weeks of age normal laboratory diet (TestDiet’s “PicoLab Rodent Diet 20, composed of 20% protein, catalog #5053 from Animal Specialties and Provisions) was replaced with a Western diet (42% fat, 0.2% cholesterol, Harlan Western diet TD.88137) as part of a study described in Ellison et. al and Ray et. al16,19. The partial ligation model of injury was performed as part of a study we described in Ellison et. al22. Briefly, mice were anesthetized by injection of ketamine and xylazine. The left common carotid artery was dissected and ligated near the bifurcation. After 28 days, mice were euthanized and tissue was prepared for immunohistochemistry (IHC) and morphological analysis. Severity of hyperplasia is known to be strain dependent, and the C57BL/6 develops a more limited, intimal hyperplasia in response to carotid ligation23,24. For analysis of atherosclerosis in the aortic root, LDLR−/− mice were fed western diet (21% [wt/wt] fat, 0.3% cholesterol; Research Diets) for 8, 12, or 16 weeks. At sacrifice, mice were anaesthetized with isoflurane and exsanguinated by cardiac puncture, perfused with PBS, followed by 10% sucrose in PBS. Aortic roots were embedded in OCT medium and frozen immediately for subsequent sectioning and staining. All animal models of vascular disease adhered to the guidelines for experimental atherosclerosis as described in Daugherty et al25.
Atherosclerosis analysis.
Hearts embedded in OCT were sectioned through the aortic root (6 μm) and stained with Oil Red O for lesion quantification using 6 sections per mouse, spanning the entire aortic root26. Lesion area was quantified by digital imaging system using ImageJ software. Immunofluorescence staining for CD68 (MCA1957, rat anti-mouse Bio-Rad Laboratories Inc., Hercules, CA), smooth muscle cell α-actin (53–9760-82, ThermoFisher Scientific, Waltham, MA) and G3BP (ab181150, Abcam Inc., Cambridge, MA) were performed on frozen atherosclerotic sections as previously described27. Briefly, after 4% PFA fixation, sections were incubated with a serum-free protein blocker solution (Agilent Technologies, Santa Clara, CA) for 1 hour, followed by an overnight incubation with primary antibodies in 1% BSA and 5% goat serum in PBS. Tissues were incubated with appropriate fluorescent labeled secondary antibodies, followed by DAPI (D9542, Sigma Inc., Cambridge, MA) nuclear stain. The sections were imaged on a Leica SP5 microscope and the ImageJ plug-in Analyze Particles was used to count the number of SGs in the aortic root. Measurements were conducted on 6 mice per group under blinded conditions.
Immunofluorescent staining and SG quantitation.
SGs were measured by the program StarSearch (Java program from University of Pennsylvania) and ImageJ as described in previous studies28,29. For cultured cells, the number of SG-bearing cells was quantified using the ImageJ software. IF images were randomly taken with a 20× and 40× objective lens in 10–20 different fields. The ImageJ plug-in “Analyze particles” was used to count the number of SGs in 40–50 cells per condition. Additionally, we used Star Search to confirm ImageJ results. Positive identification of SG formation was considered to be cytoplasmic punctate immunoreactivity for accepted SG marker proteins, including PABP and G3BP112. Immunostaining procedure was followed as outlined in Kedersha and Anderson, 200712. Immunofluorescent staining was performed on formalin fixed paraffin embedded tissue. Briefly, tissue was fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned at 5um, deparaffinized in xylene and rehydrated through graded ethanols. Heat induced antigen unmasking was performed. Tissue sections were blocked with 5% donkey serum followed by primary antibodies; FXR1, PABP, G3BP1, smooth muscle cell α actin (SMC α actin), and HUR (all Abcam Inc., Cambridge, MA). Then tissues were incubated with appropriate fluorescently labeled (ALEXA FLUOR 488 – green and 568 – red) secondary antibodies, followed by DAPI (all Life Technologies Corp., Carlsbad, CA) nuclear stain.
Western blotting and protein determination.
Human VSMC extracts were prepared as described 17,19,30. Membranes were incubated with a 1:5000–9000 dilution of primary antibody, and a 1:5000 dilution of secondary antibody. Antibodies to G3BP1, eIF2a, p-eIF2a from AbCam. HSP70, HSP90 and tubulin were from Santa Cruz Biotechnology (Dallas, TX, USA). Reactive proteins were visualized using enhanced chemiluminescence (Amersham, Piscataway, NJ, USA) according to manufacturer’s instructions. Relative intensity of bands was quantitated by scanning image analysis and the Image J densitometry program and normalized to internal control proteins (HSP90 or tubulin).
Statistical Analysis.
The normality of distribution was tested with the D’Agostino-Pearson (omnibus K2) and Kolmogorov-Smirnov tests implemented in Prism statistical software (GraphPad, La Jolla, CA). Statistical significance between two groups was evaluated with a two-tailed Student’s t-test with Welch’s correction to determine equal variance. For multiple group comparisons, one-way analysis of variance (ANOVA) followed by Dunn’s multiple comparison test for non-parametric data and Tukey’s multiple comparison test for parametric data were performed. Results are expressed as mean ± SEM. Significance was accepted at the level of P ≤ 0.05. All experiments using cultured cells were performed in triplicate, from at least three independent experiments.
Results
Stress granule formation correlates with atherosclerosis progression.
Atherosclerosis results from chronic inflammation in the artery wall, and several forms of cellular stress can drive its progression, including oxidative, mitochondrial, endoplasmic reticular and hemodynamic stresses. To determine whether stress granules accumulate in cells in the developing atherosclerotic lesion, we fed atherosclerosis-prone LDLR−/− mice a western diet (WD) for 0, 8, 12, and 16 weeks. We performed co-immunostaining for the stress granule-specific marker Ras‐GTPase‐activating protein SH3 domain‐binding protein (G3BP1) and smooth muscle cell (alpha actin) and macrophage (CD68) markers in cross-sections of the aortic root. While there was little G3BP1 expression in the healthy aortic root, we found that G3BP1 accumulated in VSMC and macrophages in atherosclerotic plaques (Figure 1A). G3BP1 expression increased with plaque progression and was significantly upregulated after 12 and 16 weeks of western diet feeding (Figure 1B–C). To examine atherosclerosis in other sites in the aorta, we stained plaques in the aortic arch of LDLR−/− mice fed WD for up to 14 weeks. Similar to our findings in aortic root plaques, we observed cumulative staining of the stress granule-specific marker Poly-A Binding Protein (PABP) with atherosclerosis progression (Supplemental Data Figure I and II). Co-localization of PABP and smooth muscle cells showed that PABP expression was increased specifically in the synthetic intima and cap layer of smooth muscle cells juxtaposed to the plaque, as compared to the quiescent and contractile medial smooth muscle cells in these lesions. Together, these data indicate that stress granule formation correlates with atherosclerotic vascular disease progression.
Figure 1.
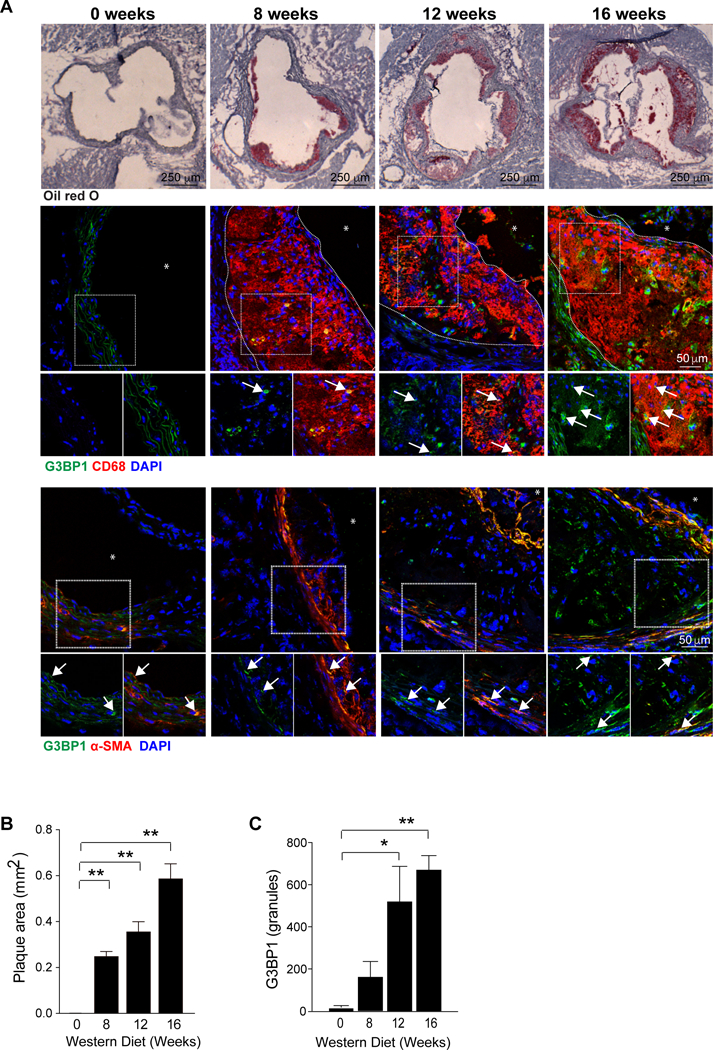
Stress granules are induced during atherogenesis and increase with disease progression. (A) Representative images of atherosclerotic plaques in the aortic root of LDLR−/− mice fed western diet for 0, 8, 12 and 16 weeks, and stained with oil red O stain to identify neutral lipid (top panel, 40X), the macrophage marker CD68 (red) and G3BP (green) (middle panel, 400X; * = lumen), and the smooth muscle cell marker alpha actin (red) and G3BP (green) (bottom panel, 40X; * = lumen). Arrows indicate co-stained cells. Plaque area has been outlined with a dotted line. (B-C) Quantification of plaque size (B) and G3BP1 fluorescence intensity in plaques (C). Data are the mean ± s.e.m. of n=6 mice per time point. Significance was determined by one-way analysis of variance (ANOVA) followed by post-test of Dunn. *P≤0.05, **P≤0.005.
Stress granules form in VSMC in response to mitochondrial and oxidative stress
To understand the mechanisms of stress granule formation in VSMCs during atherosclerosis, we measured the accumulation of stress granule components including PABP and the RBPs HuR and FXR112 in response to various stimuli. In unstimulated human VSMC, we observed diffuse, cytoplasmic PABP and FXR1 staining, and nuclear HuR staining (Figure 2A), consistent with the appearance of VSMC in the healthy aorta. Treatment of VSMCs with a known inducer of mitochondrial stress, clotrimazole, induced the co-localization of PABP, HuR, and FXR1 in cytoplasmic punctae (Fig 2A). Quantification of PABP+ punctae revealed that clotrimazole induced a 4-fold increase in the number of stress granules per cell, compared to control VSMC (Fig 2B). The SG inducer arsenite also induced SG formation in VSMC (Supplemental Data IIIB). As an independent measure of stress granules, we assayed phosphorylation of eIF2α, which signals ER stress and correlates with stress granule formation. Consistent with the increase in PABP+ puncta in VSMC treated with clotrimazole, we also observed an increase in eIF2α phosphorylation in these cells by Western blotting (Fig. 2C). Compared to control VSMC, there was a significant increase in the ratio to phospho-eIF2α to total eIF2α protein in clotrimazole-treated VSMC (Figure 2C).
Figure 2.
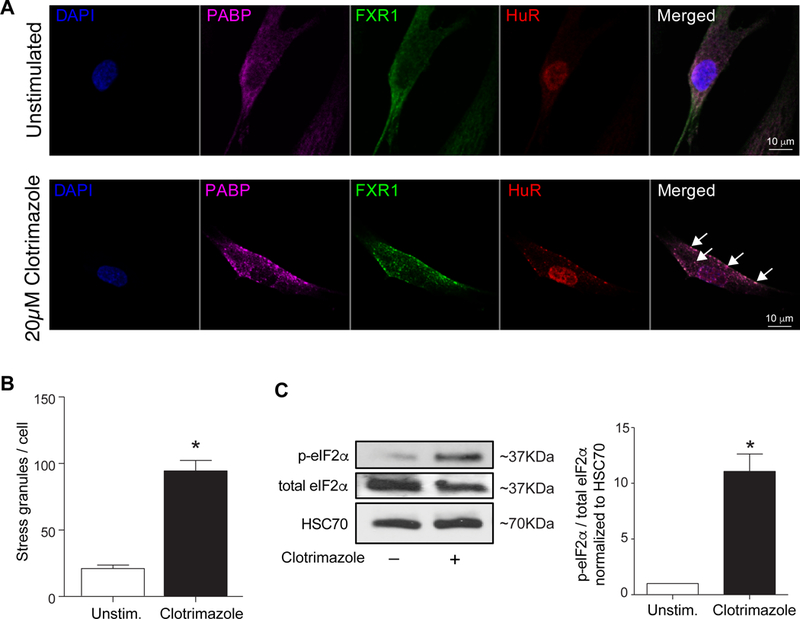
Primary human vascular cells develop stress granules in response to mitochondrial stress. (A) Representative images of human VSMC either unstimulated or treated with the mitochondrial stressor clotrimazole (20 μM) and stained for the stress granule markers, PABP (purple), FXR1 (green), HuR (red). Cell nuclei were stained with DAPI (blue). (B) Quantification of punctate immunoreactivity in unstimulated and clotrimazole-treated human VSMC. Data are the mean ± s.d. of 50 cells per condition. (C) Western blot analysis of total and phospho-eIF-2α in unstimulated and clotrimazole-treated human VSMC. Quantification of the ratio of p-eIF2α:total eIF2α is shown below. Data are the mean ± s.d. of 3 experiments. *P≤0.05.
As oxidized forms of LDL (oxLDL) accumulate in the artery wall during atherosclerosis, we next tested whether oxLDL could stimulate the formation of stress granules in human VSMCs. While not as robust as clotrimazole, oxLDL treatment induced the accumulation of both PABP (Fig. 3A–B) and G3BP1 (Fig. 3C) in VSMCs, as measured by immunostaining and western blotting, respectively. Interestingly, in oxLDL-stimulated VSMC, stress granule localization was predominantly perinuclear or in cytoplasmic lamellae, whereas clotrimazole induced both perinuclear and cytoplasmic punctate staining. Furthermore, consistent with the observed increase in PABP+ granules and G3BP1 accumulation in VSMC exposed to oxLDL, we observed an increase in the ratio to phospho-eIF2α to total eIF2α protein in oxLDL-treated VSMC compared to control VSMC, as measured by western blotting (Fig. 3D). Previous studies from our laboratory have shown that the anti-inflammatory cytokine IL-19 has important inhibitory effects on development of atherosclerosis, restenosis, and the VSMC response to inflammatory stimuli14–16,19,31. IL-19 significantly reduced both oxLDL-driven SG formation and eIF2α phosphorylation in VSMC (Figure 3E). This suggests a link between SG formation and atherogenic stimuli.
Figure 3.
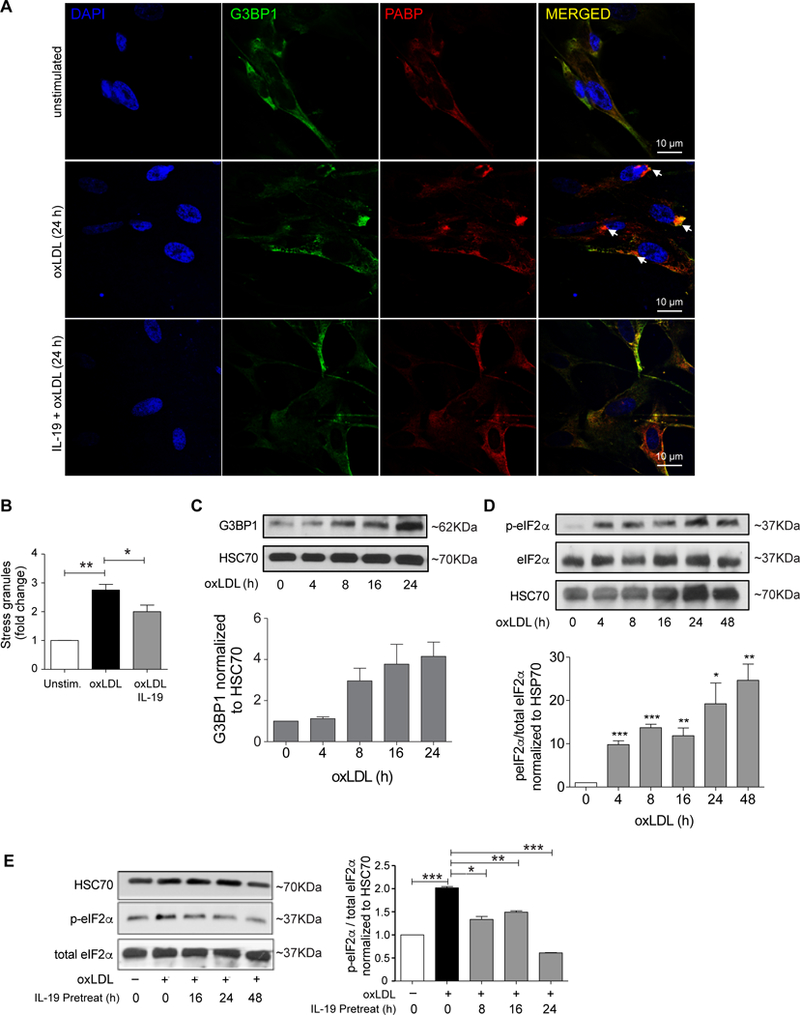
OxLDL induces stress granule formation in human VSMCs. (A) Representative images of human VSMC either unstimulated or treated with oxLDL (50 μg/mL) and stained for the stress granule marker PABP (red) or alpha actin (green). Cell nuclei were stained with DAPI (blue). Arrows indicate PABP+ puncta. (B) Quantification of punctate immunoreactivity in unstimulated and oxLDL-treated human VSMC. (C) Western blot analysis of G3BP1 and HSC70 protein levels in human VSMCs treated with oxLDL for the indicated times. Densitometry analysis of G3BP1 protein relative to HSC70 is shown below. Data are the mean of 3 independent experiments. (D) Western blot analysis of total and phospho-eIF-2α in unstimulated and oxLDL-treated human VSMC, densitometry of phospho-to-total protein ratios are shown below. (E) Western blot analysis of total and phospho-eIF-2α in VSMCs untreated or pretreated with IL-19 prior to stimulation with oxLDL. Data are representative of 3 independent experiments. *P≤0.05.
Stress granules form in macrophages in response to cholesterol loading and oxidative stress
During atherogenesis, monocytes are recruited into the arterial intima to clear accumulated lipoproteins. Macrophage uptake of oxLDL is thought to be a key event in the formation of cholesterol-loaded macrophage foam cells in the plaque. To test whether oxLDL induces stress granule formation in macrophages as well as VSMC, we treated bone marrow derived macrophages (BMDM) with oxLDL for 24h. While there was little detectable staining for G3BP1 in untreated BMDM, upon treatment with oxLDL G3BP1 accumulated in cytoplasmic puncta (Fig. 4A). Furthermore, oxLDL treatment of BMDM induced an increase in G3BP1 protein as measured by western blotting (Fig. 4B). To understand whether the observed increase in stress granule formation was result of oxidative stress induced by prolonged exposure to oxLDL or a result of cholesterol loading of the macrophages, we treated BMDMs with arsenite as an inducer of oxidative stress or aggregated LDL as an alternative means of cholesterol loading. Importantly, both arsenite and aggregated LDL treatment of macrophages increased the accumulation of G3BP1 punctae as measured by immunostaining (Fig. 4A). Furthermore, western blotting showed that oxLDL and aggregated LDL increased macrophage G3BP1 protein levels to a similar extent (Fig. 4B–C). These data suggest that multiple cellular stresses present in atherosclerotic plaques, including oxidative stress and cholesterol loading, may contribute to stress granule formation in macrophage foam cells.
Figure 4.
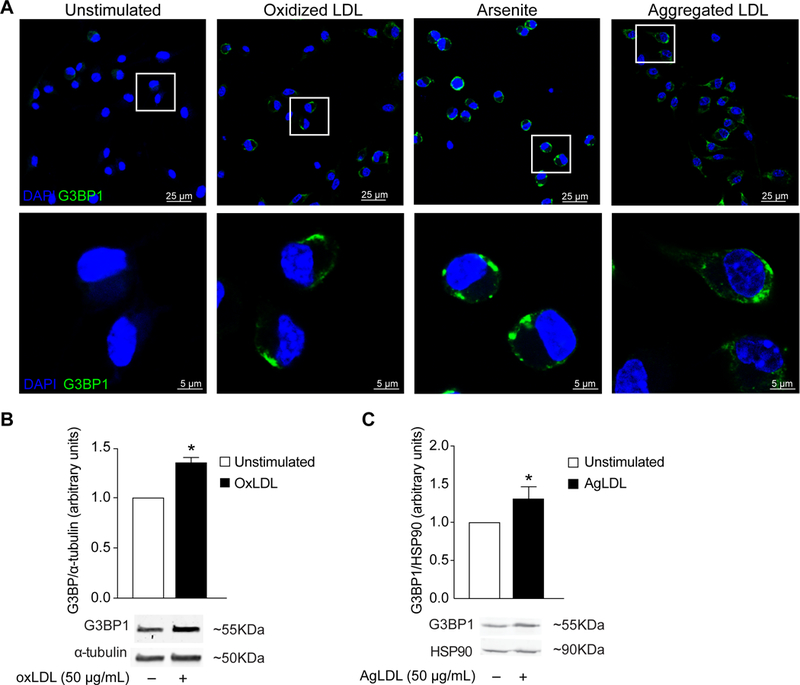
Cholesterol loading and oxidative stress induce stress granules in macrophages. (A) Representative images of bone marrow derived macrophages (BMDMs) treated with oxidized or aggregated LDL (50 μg/mL) for 24 hours, or with the oxidative stressor sodium arsenite (500 μM) for 30 minutes, and stained for the stress granule marker G3BP1 (green). Cell nuclei were stained with DAPI (blue). (B-C) Western blot and densitometry analysis of G3BP1 protein levels in BMDMs treated with (B) oxLDL or (C) aggregated LDL. Data are the mean of 3 independent experiments. *P≤0.05.
SG are increased in VSMC in vascular proliferative syndromes
To understand whether stress granules are formed in VSMC in other vascular pathologies, we next examined vascular restenosis, a disease of VSMC proliferation and migration that results from a maladaptive response to mechanical injury and cellular stress. We used a mouse carotid artery ligation model, which leads to VSMC migration and proliferation, and results in a neointima consisting of synthetic VSMCs22. Consistent with our findings in the healthy aortic arch and aortic root (Figure 1, Supplemental Figures. I, II), we observed little PABP immunoreactivity in unligated control mouse carotid arteries (Figure 5A). In contrast, following carotid artery ligation, there was a marked increase in PABP staining that localized to alpha actin-expressing VSMCs (Figure 5A–B). Importantly, PABP immunoreactivity was almost exclusively found in synthetic, neointimal VSMC compared to the medial, quiescent VSMCs (Figure 5A), suggesting that stress granule formation is associated with the VSMC response to injury.
Figure 5.
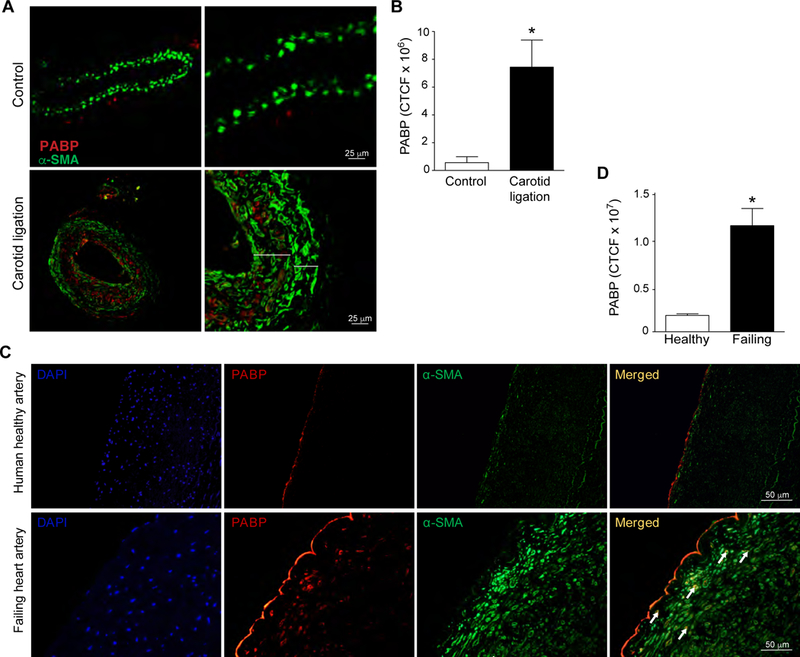
Enhanced PABP expression in injured mouse and human arteries. (A) Representative images of control and ligated mouse carotid arteries stained for PABP (red) and alpha smooth muscle actin (green) Larger bracket indicates neointima, smaller bracket is the media. (B) Quantification of PABP immunoreactivity in control or ligated mouse carotid arteries. N=4 mice/group. (C) Representative images of normal human coronary artery and coronary artery from a failing human heart stained for PABP (red), alpha smooth muscle actin (green), and DAPI (blue). (D) Quantification of PABP immunoreactivity in human artery samples expressed as corrected total cellular fluorescence (CTCF). n=3 control, n=4 failing heart.
To translate these findings to human disease states, we stained coronary arteries from control and failing human hearts for PABP and alpha smooth muscle actin. A small, but detectable amount of PABP staining was observed in coronary arteries from non-diseased human hearts (Figure 5C). In contrast, coronary artery neointimal SMC from failing human hearts demonstrated significantly more PABP that co-localized with alpha smooth muscle actin staining (Figures 5C,D). Although it is difficult to detect punctate staining in formalin-fixed, paraffin embedded tissue, the increase in PABP accumulation in VSMC is consistent with stress granule formation in the failing heart, and is in agreement with our findings of stress granule formation in response to vascular injury in mouse models of atherosclerosis and restenosis.
Inhibiting VSMC cytoskeletal regulation and inflammation reduces stress granule formation
Studies have revealed a key role for microtubules in active transport and localization of stress granule components upon stressful stimuli, as well as participation in the nucleation and growth of the granule32. Calpain expression and function in particular are associated with vascular degenerative diseases, and calpain inhibitors are known to disturb microtubular dynamics and inhibition of calpain has anti-inflammatory effects in cardiovascular diseases33,34. To test whether cytoskeletal function may be critical for the formation of functional stress granules in VSMCs, we treated human VSMCs with the calpain inhibitor zLLal prior to treatment with clotrimazole. Compared to VSMCs treated with clotrimazole alone, cells treated with zLLal + clotrimazole showed a marked reduction in the number of stress granules per cell (Figure 6A and Supplemental Figure IIIA). Similar results were obtained with the SG inducer arsenite (Supplemental Figure IIIB). These findings point to a critical role of cytoskeletal regulation in the formation of stress granules in VSMCs. Considering clotrimazole is a potent SG inducer, we next treated human VSMCs with IL-19 prior to addition of clotrimazole. Pretreatment with IL-19 for 8 or 24 hours significantly reduced clotrimazole-induced accumulation of PABP, HuR and FXR1 staining in cytoplasmic puncta (Figure 6B,C). Quantification of PABP puncta showed a time-dependent effect of IL-19 treatment on reduction of stress granule formation in VSMC (Figure 6C). Furthermore, IL-19 pre-treatment significantly reduced the phosphorylation of eIF2α in VSMCs treated with clotrimazole as determined by western blotting (Figures 6D). Together, these data suggest that SG formation is linked with the inflammatory state of the cell, and the anti-inflammatory cytokine IL-19 can reduce VSMC stress granule formation.
Figure 6.
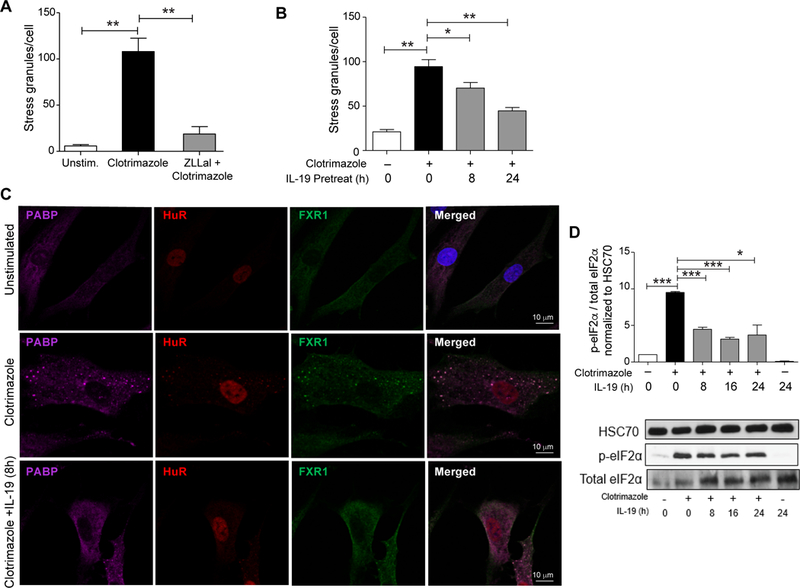
Calpain inhibition and IL-19 reduce stress granule formation in human VSMCs. (A) Quantification of stress granules in human VSMCs unstimulated or pretreated with the calpain inhibitor zLLal prior to stimulation with clotrimazole. (B) Quantification of stress granules in human VSMCs untreated or pretreated with IL-19 prior to stimulation with clotrimazole (n=50). (C) Immunostaining for PABP (purple), HuR (red), and FXR1 (green) in human VSMCs untreated or pretreated with IL-19 prior to stimulation with clotrimazole. (D) Western blot analysis of total and phospho-eIF-2α in VSMCs untreated or pretreated with IL-19 prior to stimulation with clotrimazole. Data are the mean of 3 independent experiments. (E) Quantification of stress granules in human VSMCs untreated or pretreated with IL-19 prior to stimulation with oxLDL (n=50). (F) Western blot analysis of total and phospho-eIF-2α in VSMCs untreated or pretreated with IL-19 prior to stimulation with oxLDL. Data are the mean of 3 independent experiments. *P≤0.05, ** P≤0.01, or *** P≤0.001.
The loss of G3BP1 in hVSMC reduces SG formation and alters the VSMC stress response.
G3BP1 is considered a required component for normal stress granule assembly and importantly, recently identified as a master regulatory gene for CAD35,36. G3BP1 has been studied in numerous stress granule induction systems, but nothing has been reported regarding G3BP1 function in VSMC. To determine a functional role for SG in VSMC, human VSMC were transfected with G3BP1 or scrambled control siRNA, then challenged with clotrimazole. SG were quantitated by punctate PABP immunohistochemistry and G3BP1 western blotting. Figure 7A,B shows that the loss of G3BP1 significantly reduced stress granule formation in response to clotrimazole. Loss of G3BP1 was validated by qRT-PCR and western blot (Figure 7C, D). Because of SG involvement in mRNA processing, we next tested the possibility that loss of G3BP would alter gene expression in response to an atherogenic stimuli. Human VSMC were transfected with G3BP1 siRNA or scrambled control, challenged with oxLDL, and mRNA abundance measured by quantitative PCR array biased for genes involved in atherosclerosis. Figure 7D shows that human VSMCs lacking G3BP1 had significant differences in abundance of transcripts associated with inflammation, adhesion, apoptosis, coagulation and circulation, lipid metabolism, and cell growth and proliferation. This approach was repeated and abundance of selected inflammatory transcripts as well as others not present on the PCR array were quantitated by qRT-PCR. Supplemental Data Figure IV shows that inflammatory and proliferative transcripts were reduced in VSMC lacking G3BP1. Importantly, abundance of HuR, an mRNA stability protein known to be attenuated by IL-19, was significantly reduced, suggesting that IL-19 and G3BP1 have common mRNA targets15,16,19. Finally, G3BP1 knockdown did not significantly change VSMC proliferation (Supplemental Data Figure V ). Together, these data show that loss of G3BP1 significantly alters the VSMC ability to form SG and respond to atherogenic stimuli, suggesting a critical role for SG components in vascular disease progression.
Figure 7.
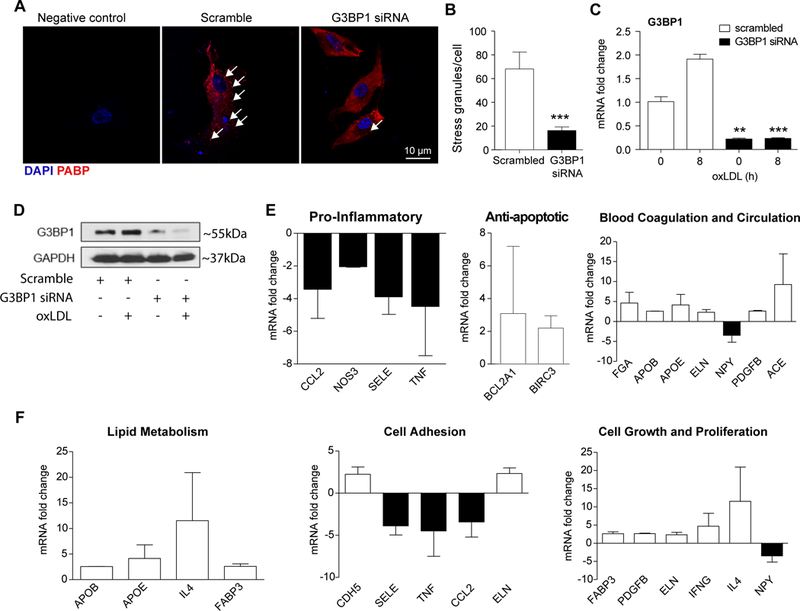
Loss of G3BP1 alters hVSMC response to SG formation and inflammation. HVSMCs transfected with siRNA targeting G3BP1 or scrambled control were treated with clotrimazole and stained for PABP (red) (A), Arrows indicate punctate staining. SG formation was quantified using ImageJ and StarSearch software and graphed in (B), n=40. (C), qRT-PCR and (D) Western blot analysis verified G3BP1 knockdown in hVSMCs. (E) HVSMCs transfected with siRNA targeting G3BP1 or scrambled control were serum-starved and stimulated with oxLDL for 16 hours, RNA collected, and expression of atherosclerosis-related transcripts were quantitated by PCR Array. The results are graphed as SD from two independent experiments. ** P≤0.01, or *** P≤0.001.
Discussion
Our current understanding of stress granules has been that they are formed in response to stress to preserve specific endogenous mRNA from degradation, storing and perhaps protecting these transcripts, rather than allowing re-initiation of translation or degradation37. Literature has focused on SG formation in neuron and glial cells, but they are not specific to those cells. Recent studies have shown that SGs may participate in the pathogenesis of neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease38,39. We hypothesized that SG may be involved in other chronic inflammatory diseases, such as atherosclerosis, and in the cellular response to vascular injury. The present study demonstrates that SG are actively formed in response to atherosclerotic inflammation and accumulate in intimal macrophages and VSMC with disease progression. This work identifies a number of pro-atherosclerotic stimuli that may stimulate stress granule formation in plaque macrophages and VSMC, including modified forms of LDL and inducers of mitochondrial and oxidative stress. Moreover, we also observed increased stress granule formation in other vascular pathologies in mice and humans, including restenosis and heart failure, respectively. Interestingly, we show that stress granule formation in VSMC can be significantly reduced by treatment with IL-19, an anti-inflammatory cytokine that we previously showed could inhibit atherosclerosis and the VSMC response to inflammatory stimuli. Together, these data suggest that stress granule formation may be a common response to vascular injury and/or inflammation, which allows VSMC and other cell types that participate in vascular inflammation to sequester mRNA transcripts and halt their translation.
Our findings are consistent with a previous report that oxidized LDL and high-fat diet can induce stress granule formation in mouse macrophages and liver40. Our study extends these findings by showing that multiple stressors present in atherosclerotic plaques may contribute to the formation of stress granules in lesional macrophages, including aggregated forms of LDL that can mediate cholesterol loading of the macrophage, and mediators of oxidative stress. Moreover, our data indicate that the formation of stress granules is not limited to macrophages in atherosclerosis, and that these RNP granules readily accumulate in VSMC during atherogenesis and in response to vascular injury. As we observed in macrophages, oxLDL triggered stress granule formation in VSMC, although not to the same extent as chemical inducers of mitochondrial and oxidative stress (e.g., clotrimazole and sodium arsenite), which may activate different, or more potent stress response pathways12. Consistent with stress granule formation in other cell types41,42, oxLDL induced punctate staining and perinuclear clustering of PABP and G3BP1, and increased phosphorylation of eIF2α. However, in vivo, it is likely that oxLDL acts in concert with other pro-inflammatory factors to induce the formation of stress granules in both macrophages and VSMC in the plaque. For example, previous studies have shown that IL-1β can induce SGs in other cell types43, and considerable literature points to a role for this inflammatory cytokine in the development of atherosclerosis. Furthermore, VSMCs experience turbulent hemodynamics and inflammation, which may trigger the stress response pathways in the cells44, and this aspect remains to be further investigated.
VSMC are highly plastic, and as atherosclerosis progresses, normally quiescent medial VSMC respond to chronic local inflammation by modulating their phenotype to a synthetic, proliferative, and migratory de-differentiated myofibroid cell type44. Murine carotid artery ligation is a commonly used model to induce VSMC migration, proliferation, and de-differentiation in vivo. In both atherosclerotic arteries and after carotid ligation, we observed significantly enhanced PABP immunoreactivity in the activated neointimal VSMCs compared with medial VSMC, suggesting these synthetic cells respond to stress by SG formation more so than the quiescent VSMCs. Similarly, significantly increased PABP immunoreactivity was detected in coronary artery neointimal SMC from failing human hearts compared with non-diseased hearts. As in animal models, these neointimal VSMCs are more synthetic than medial VSMCs, and as such, increased expression of SG components likely reflects the vascular transcriptomic response to localized inflammation. Stress granule formation has not been previously reported in this type of vascular injury, and these findings emphasize the response to vascular stress that cells in failing hearts are exposed to. Our finding of SG formation in VSMC in multiple vascular pathologies suggest that this may be a common mechanism by which the cell fine-tunes its transcriptional response to the mechanical and inflammatory stresses the vasculature must respond to.
Actin polymerization and cytoskeletal dynamics play a major role in the regulation of active tension development in smooth muscle45. Interestingly, previous studies have shown that many cytoskeletal-associated proteins localize to SGs and participate in SG dynamics46, suggesting that the cytoskeleton of VSMCs may be critical in modulating SG formation and the stress response. Calpains are activated by various pathophysiological stressors associated with vascular disorders (e.g., hypoxia, oxidized lipids, and inflammation47,48) and are expressed in atherosclerotic plaques of LDLR−/− mice34,49. Work by Thiemermann indicated that acute calpain inhibition significantly ameliorated cardiovascular dysfunction by reduction of acute and chronic inflammation34. Moreover, cells expressing the Calpain inhibitor calpastatin fail to extend lamellipodia, as well as abnormal filopodia, extensions, and retractions, suggesting that calpain may regulate SG dynamics50. Indeed we showed that the calpain inhibitor N-benzyloxycarbonyl-L-leucyl-L-leucinal (zLLal), which reversibly blocks the calcium-dependent neutral cysteine protease calpain I51, significantly reduced clotrimazole-induced SG formation. This is notable in that a recent proteomic analysis of atherosclerotic coronary artery VSMC implied that dysregulation of the cytoskeleton may explain the switch between the contractile and synthetic phenotype of VSMCs52. In this regard, cytoskeletal changes observed in plaque and neointimal VSMC may have major implications on SG formation in VSMC during the development and progression of atherosclerosis and restenosis. A precise mechanistic link between calpain activity and SG dynamics remains to be elucidated.
Previous studies from our group identified potent anti-atherosclerotic and restenotic effects of the cytokine IL-19. In particular, we showed that IL-19 can reduce the stability of pro-inflammatory mRNA transcripts in hVSMC14,15. We demonstrate in this study that IL-19 reduces SG formation in VSMC. Indeed, IL-19 was effective at reducing the assembly of PABP+ and HuR+ puncta in both clotrimazole- and oxLDL-treated VSMC, and also reduced phosphorylation of eIF2α in these cells. These data identify a novel mechanism by which IL-19 may function to decrease vascular inflammation by reducing stress granule formation.
SGs can be difficult to characterize in vivo and their role may be better determined by examination of key components. In HeLa and 293T cell lines, G3BP1 is required for arsenate-induced SG formation, while overexpression of G3BP1 has been shown to induce SGs, even in the absence of stress35,53. A viable mouse knockout of G3BP1 revealed major alteration of the CNS, including impaired motor coordination and dysfunctions of synaptic transmission, suggesting a critical role for G3BP1 and SGs in neuronal function54. G3BP1 is implicated as a master regulatory gene for development of CAD36. In the present study, when knocked down in VSMC, not only was SG formation significantly reduced, but the VSMC response to atherogenic stimuli was altered, as the abundance of mRNA for such crucial functions as adhesion, cell proliferation, inflammation, lipid metabolism were significantly altered. This is a novel role for the SG component G3BP1 in any cell type, and points to a previously uncharacterized mechanism where SGs mediate the VSMC response to inflammatory stimuli. The role we observed for G3BP1 is congruent with our hypothesis that long-term expression of SGs or SG components may be maladaptive, such as the chronic inflammation in VSMCs that occurs in atherosclerosis. Addition of IL-19 to VSMC reduces HuR abundance, and targeted deletion of IL-19 in LDLR−/− mice increased levels of inflammatory cytokine mRNAs and the mRNA stability protein HuR in BMDMs and VSMCs16, that, similar to SGs, suggested a potential regulatory role for this cytokine in mRNA processing. Importantly, in addition to inflammatory transcripts, when G3BP1 is knocked down, HuR mRNA abundance was significantly reduced as well, suggesting a molecular link between IL-19 activity, SG formation, and VSMC response to inflammatory stimuli. Additional work is needed to define a precise IL-19/SG/HuR pathway in the response to inflammatory stress.
There remain several interesting questions to be addressed. First, does atherogenic stress induction of SG formation contribute to vascular disease? It is known that RNA binding proteins target mRNAs with AU-rich elements (AREs) in their 3’UTR and recruit these transcripts to stress granules, and our study suggests that these structures may mediate the VSMC response to atherogenic stimuli. Whether atherogenic stimuli induce a specific subset of RNA binding proteins to mediate this process is not known. Second, what is the nature of the mRNAs being sequestered in stress granules? Previous studies have suggested that many housekeeping and cytokine mRNAs are present in stress granules, and defining the stress granule transcriptome of VSMC and macrophages will be of interest to determine which mRNAs are being sequestered in these structures. Finally, what are the consequences of stress granule assembly or disassembly in VSMC and immune cells in vascular pathologies? Although stress granules have been predominantly labeled as protective to prevent further cellular damage, the ramifications chronic SG formation and function in chronic vascular diseases remains to be fully characterized.
Supplementary Material
{kind=link}
Supplemental Figure I. Expression of SG markers in atherosclerotic plaque. (A) G3BP1 costaining with macrophage and VSMC markers in plaque from LDLR−/− mice fed a western diet for 14 weeks. (B)Poly-A Binding Protein (PABP) expression in atherosclerotic plaque. Very little PABP was detected in LDLR−/− mice fed a normal laboratory diet, but large amount of PABP is detected in plaque from LDLR−/− mice fed a western diet for 14 weeks. Magnification 400X. (C) Colocalization PABP and a-smooth muscle actin in atherosclerotic plaque from LDLR−/− mice fed a western diet for 14 weeks. Co-localization of PABP and smooth muscle cells showed that PABP expression was increased specifically in the synthetic intima and cap layer of smooth muscle cells juxtaposed to the plaque, as compared to the quiescent and contractile medial smooth muscle cells in these lesions. Magnification 600X.
Supplemental Figure II. PABP expression correlates with atherogenesis in LDLR−/− mice. Cumulative staining of the stress granule-specific marker Poly-A Binding Protein (PABP) with atherosclerosis progression. (A) Representative images of aortic arch sections from LDLR−/− mice fed western diet for 0, 5, 10, and 14 weeks that were immunostained for PABP (red), alpha smooth muscle actin (green), and DAPI (blue). (B) Quantification of PABP staining of aortic arch sections by corrected total cell fluorescence (CTCF). *P≤0.05, ** P≤0.01, or *** P≤0.001.
Supplemental Figure III. Immunostaining for PABP (red), G3BP (green) and DAPI (blue) in human VSMCs untreated or pretreated with the calpain inhibitor zLLal prior to stimulation with clotrimazole (A), and Arsenite (B). Data are representative of 3 independent experiments.
Supplemental Figure IV. HVSMCs transfected with siRNA targeting G3BP1 or scrambled control were serum-starved and stimulated with oxLDL for 16 hours, RNA collected, and expression of atherosclerosis-related transcripts were quantitated by PCR. *P≤0.05, ** P≤0.01, or *** P≤0.001.
Supplemental Figure V. G3BP1 knockdown does not affect VSMC proliferation. HVSMCs were transfected with G3BP1 siRNA or scrambled control siRNA for 48h. Cells were seeded at 10,000 cells/well and counted at days 3 and 7.
Highlights.
Stress granules are induced in vascular disease.
Stress granules form in cultured VSMC in response to atherogenic stimuli
Stress granule formation can be inhibited by anti-inflammatory IL-19
Reduction in stress granule formation alters the VSMC response to atherogenic stimuli
Acknowledgments
Sources of Funding
This work was supported by grants from the National Heart Lung, and Blood Institute of the National Institutes of Health (HL141108 and HL117724 to MVA; R35HL135799, P01HL131481 and P01HL131478 to KJM) and grant 13GRNT1685003 from the American Heart Association to MVA. MR, AH, and CV were supported by American Heart Association pre-doctoral fellowships 16PRE31220005, 17PRE33670798, and 18PRE34080191.
Non-standard Abbreviations
- ARE
AU-rich element
- G3PB1
GTPase Activating Protein (SH3 Domain) Binding Protein 1
- FXR1
Fragile X-Related 1
- HuR
Human antigen R
- oxLDL
oxidized LDL
- PABP
Poly A Binding Protein
- SG
Stress Granules
- VSMC
Vascular Smooth Muscle Cell
Footnotes
Disclosures
We have nothing to disclose
REFERENCES
- 1.Roth GA, Johnson C, Abajobir A, Abd-Allah F, Abera SF, Abyu G, Ahmed M, Aksut B, Alam T, Alam K, Alla F, Alvis-Guzman N, Amrock S, Ansari H, Ärnlöv J, et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. Journal of the American College of Cardiology. 2017;70:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ross R Atherosclerosis--an inflammatory disease. The New England Journal of Medicine. 1999;340:115–126. [DOI] [PubMed] [Google Scholar]
- 3.Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28:812–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hansson GK, Robertson A-KL, Söderberg-Nauclér C. Inflammation and atherosclerosis. Annual Review of Pathology. 2006;1:297–329. [DOI] [PubMed] [Google Scholar]
- 5.Singer CA, Salinthone S, Baker KJ, Gerthoffer WT. Synthesis of immune modulators by smooth muscles. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology. 2004;26:646–655. [DOI] [PubMed] [Google Scholar]
- 6.Libby P, Geng YJ, Sukhova GK, Simon DI, Lee RT. Molecular determinants of atherosclerotic plaque vulnerability. Annals of the New York Academy of Sciences. 1997;811:134–142; discussion 142–145. [DOI] [PubMed] [Google Scholar]
- 7.Albarrán-Juárez J, Kaur H, Grimm M, Offermanns S, Wettschureck N. Lineage tracing of cells involved in atherosclerosis. Atherosclerosis. 2016;251:445–453. [DOI] [PubMed] [Google Scholar]
- 8.Weber C, von Hundelshausen P. CANTOS Trial Validates the Inflammatory Pathogenesis of Atherosclerosis: Setting the Stage for a New Chapter in Therapeutic Targeting. Circulation Research. 2017;121:1119–1121. [DOI] [PubMed] [Google Scholar]
- 9.Gillis P, Malter JS. The adenosine-uridine binding factor recognizes the AU-rich elements of cytokine, lymphokine, and oncogene mRNAs. Journal of Biological Chemistry. 1991;266:3172–3177. [PubMed] [Google Scholar]
- 10.Bakheet T, Williams BRG, Khabar KSA. ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Research. 2006;34:D111–D114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buchan JR, Parker R. Eukaryotic stress granules: the ins and outs of translation. Molecular Cell. 2009;36:932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kedersha N, Anderson P. Mammalian Stress Granules and Processing Bodies In: Methods in Enzymology.Vol 431 Elsevier; 2007:61–81. [DOI] [PubMed] [Google Scholar]
- 13.Ash PEA, Vanderweyde TE, Youmans KL, Apicco DJ, Wolozin B. Pathological stress granules in Alzheimer’s disease. Brain Research. 2014;1584:52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herman AB, Vrakas CN, Ray M, Kelemen SE, Sweredoski MJ, Moradian A, Haines DS, Autieri MV. FXR1 Is an IL-19-Responsive RNA-Binding Protein that Destabilizes Pro-inflammatory Transcripts in Vascular Smooth Muscle Cells. Cell Reports. 2018;24:1176–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuneo AA, Herrick D, Autieri MV. Il-19 reduces VSMC activation by regulation of mRNA regulatory factor HuR and reduction of mRNA stability. Journal of Molecular and Cellular Cardiology. 2010;49:647–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ray M, Gabunia K, Vrakas CN, Herman AB, Kako F, Kelemen SE, Grisanti LA, Autieri MV. Genetic Deletion of IL-19 (Interleukin-19) Exacerbates Atherogenesis in Il19−/−×LDLR−/− Double Knockout Mice by Dysregulation of mRNA Stability Protein HuR (Human Antigen R). Arteriosclerosis, Thrombosis, and Vascular Biology. 2018;38:1297–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gabunia K, Herman AB, Ray M, Kelemen SE, England RN, DeLa Cadena R, Foster WJ, Elliott KJ, Eguchi S, Autieri MV. Induction of MiR133a expression by IL-19 targets LDLRAP1 and reduces oxLDL uptake in VSMC. Journal of Molecular and Cellular Cardiology. 2017;105:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tian Y, Sommerville LJ, Cuneo A, Kelemen SE, Autieri MV. Expression and suppressive effects of interleukin-19 on vascular smooth muscle cell pathophysiology and development of intimal hyperplasia. The American Journal of Pathology. 2008;173:901–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellison S, Gabunia K, Kelemen SE, England RN, Scalia R, Richards JM, Orr AW, Orr W, Traylor JG, Rogers T, Cornwell W, Berglund LM, Goncalves I, Gomez MF, Autieri MV. Attenuation of experimental atherosclerosis by interleukin-19. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33:2316–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ellison S, Gabunia K, Richards JM, Kelemen SE, England RN, Rudic D, Azuma Y-T, Munroy MA, Eguchi S, Autieri MV. IL-19 Reduces Ligation-Mediated Neointimal Hyperplasia by Reducing Vascular Smooth Muscle Cell Activation. The American Journal of Pathology. 2014;184:2134–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD. Consideration of Sex Differences in Design and Reporting of Experimental Arterial Pathology Studies-Statement From ATVB Council. Arteriosclerosis, Thrombosis, and Vascular Biology. 2018;38:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellison S, Gabunia K, Richards JM, Kelemen SE, England RN, Rudic D, Azuma Y-T, Munroy MA, Eguchi S, Autieri MV. IL-19 reduces ligation-mediated neointimal hyperplasia by reducing vascular smooth muscle cell activation. The American Journal of Pathology. 2014;184:2134–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harmon KJ, Couper LL, Lindner V. Strain-dependent vascular remodeling phenotypes in inbred mice. The American Journal of Pathology. 2000;156:1741–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuhel DG, Zhu B, Witte DP, Hui DY. Distinction in genetic determinants for injury-induced neointimal hyperplasia and diet-induced atherosclerosis in inbred mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22:955–960. [DOI] [PubMed] [Google Scholar]
- 25.Daugherty A, Tall AR, Daemen MJAP, Falk E, Fisher EA, García-Cardeña G, Lusis AJ, Owens AP, Rosenfeld ME, Virmani R. American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology; and Council on Basic Cardiovascular Sciences. Recommendation on Design, Execution, and Reporting of Animal Atherosclerosis Studies: A Scientific Statement From the American Heart Association. Arteriosclerosis, Thrombosis, and Vascular Biology. 2017;37:e131–e157. [DOI] [PubMed] [Google Scholar]
- 26.Moore KJ, Kunjathoor VV, Koehn SL, Manning JJ, Tseng AA, Silver JM, McKee M, Freeman MW. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. The Journal of Clinical Investigation. 2005;115:2192–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Distel E, Barrett TJ, Chung K, Girgis NM, Parathath S, Essau CC, Murphy AJ, Moore KJ, Fisher EA. miR33 inhibition overcomes deleterious effects of diabetes mellitus on atherosclerosis plaque regression in mice. Circulation Research. 2014;115:759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aulas A, Fay MM, Szaflarski W, Kedersha N, Anderson P, Ivanov P. Methods to Classify Cytoplasmic Foci as Mammalian Stress Granules. Journal of Visualized Experiments: JoVE. 2017;123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Somasekharan SP, El-Naggar A, Leprivier G, Cheng H, Hajee S, Grunewald TGP, Zhang F, Ng T, Delattre O, Evdokimova V, Wang Y, Gleave M, Sorensen PH. YB-1 regulates stress granule formation and tumor progression by translationally activating G3BP1. The Journal of Cell Biology. 2015;208:913–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.England RN, Preston KJ, Scalia R, Autieri MV. Interleukin-19 decreases leukocyte-endothelial cell interactions by reduction in endothelial cell adhesion molecule mRNA stability. AJP: Cell Physiology. 2013;305:C255–C265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gabunia K, Ellison S, Kelemen S, Kako F, Cornwell WD, Rogers TJ, Datta PK, Ouimet M, Moore KJ, Autieri MV. IL-19 Halts Progression of Atherosclerotic Plaque, Polarizes, and Increases Cholesterol Uptake and Efflux in Macrophages. The American Journal of Pathology. 2016;186:1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chernov KG, Barbet A, Hamon L, Ovchinnikov LP, Curmi PA, Pastré D. Role of microtubules in stress granule assembly: microtubule dynamical instability favors the formation of micrometric stress granules in cells. The Journal of biological chemistry. 2009;284:36569–36580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miyazaki T, Miyazaki A. Dysregulation of Calpain Proteolytic Systems Underlies Degenerative Vascular Disorders. Journal of Atherosclerosis and Thrombosis. 2018;25:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cuzzocrea S, McDonald MC, Mazzon E, Siriwardena D, Serraino I, Dugo L, Britti D, Mazzullo G, Caputi AP, Thiemermann C. Calpain inhibitor I reduces the development of acute and chronic inflammation. The American Journal of Pathology. 2000;157:2065–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsuki H, Takahashi M, Higuchi M, Makokha GN, Oie M, Fujii M. Both G3BP1 and G3BP2 contribute to stress granule formation. Genes to Cells: Devoted to Molecular & Cellular Mechanisms. 2013;18:135–146. [DOI] [PubMed] [Google Scholar]
- 36.Foroughi Asl H, Talukdar HA, Kindt ASD, Jain RK, Ermel R, Ruusalepp A, Nguyen K-DH, Dobrin R, Reilly DF, Schunkert H, Samani NJ, Braenne I, Erdmann J, Melander O, Qi J, et al. Expression Quantitative Trait Loci Acting Across Multiple Tissues Are Enriched in Inherited Risk for Coronary Artery Disease. Circulation: Cardiovascular Genetics. 2015;8:305–315. [DOI] [PubMed] [Google Scholar]
- 37.Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends in Biochemical Sciences. 2008;33:141–150. [DOI] [PubMed] [Google Scholar]
- 38.Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. The Journal of Cell Biology. 2013;201(3):361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strong MJ. The evidence for altered RNA metabolism in amyotrophic lateral sclerosis (ALS). Journal of the Neurological Sciences. 2010;288:1–12. [DOI] [PubMed] [Google Scholar]
- 40.Bai Y, Dong Z, Shang Q, Zhao H, Wang L, Guo C, Gao F, Zhang L, Wang Q. Pdcd4 Is Involved in the Formation of Stress Granule in Response to Oxidized Low-Density Lipoprotein or High-Fat Diet. PloS One. 2016;11:e0159568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matthews JD, Frey TK. Analysis of subcellular G3BP redistribution during rubella virus infection. Journal of General Virology. 2012;93:267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gray NK, Hrabalkova L, Scanlon JP, Smith RWP. Poly(A)-binding proteins and mRNA localization: who rules the roost? Biochemical Society Transactions. 2015;43:1277–1284. [DOI] [PubMed] [Google Scholar]
- 43.Ansari MY, Haqqi TM. Interleukin-1β induced Stress Granules Sequester COX-2 mRNA and Regulates its Stability and Translation in Human OA Chondrocytes. Scientific Reports. 2016;6:27611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Denes L, Entz L, Jancsik V. Restenosis and therapy. International Journal of Vascular Medicine. 2012;2012:406236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gunst SJ, Zhang W. Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. American Journal of Physiology. Cell Physiology. 2008;295:C576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bartoli KM, Bishop DL, Saunders WS. The Role of Molecular Microtubule Motors and the Microtubule Cytoskeleton in Stress Granule Dynamics. International Journal of Cell Biology. 2011;2011:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miyazaki T, Koya T, Kigawa Y, Oguchi T, Lei X-F, Kim-Kaneyama J, Miyazaki A. Calpain and atherosclerosis. Journal of Atherosclerosis and Thrombosis. 2013;20:228–237. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y, Liu NM, Wang Y, Youn JY, Cai H. Endothelial cell calpain as a critical modulator of angiogenesis. Biochimica Et Biophysica Acta. Molecular Basis of Disease. 2017;1863:1326–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miyazaki T, Taketomi Y, Takimoto M, Lei X-F, Arita S, Kim-Kaneyama J, Arata S, Ohata H, Ota H, Murakami M, Miyazaki A. m-Calpain induction in vascular endothelial cells on human and mouse atheromas and its roles in VE-cadherin disorganization and atherosclerosis. Circulation. 2011;124:2522–2532. [DOI] [PubMed] [Google Scholar]
- 50.Potter DA, Tirnauer JS, Janssen R, Croall DE, Hughes CN, Fiacco KA, Mier JW, Maki M, Herman IM. Calpain regulates actin remodeling during cell spreading. The Journal of Cell Biology. 1998;141:647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohkawa K, Asakura T, Takada K, Sawai T, Hashizume Y, Okawa Y, Yanaihara N. Calpain inhibitor causes accumulation of ubiquitinated P-glycoprotein at the cell surface: possible role of calpain in P-glycoprotein turnover. International Journal of Oncology. 1999;15:677–686. [DOI] [PubMed] [Google Scholar]
- 52.de la Cuesta F, Zubiri I, Maroto AS, Posada M, Padial LR, Vivanco F, Alvarez-Llamas G, Barderas MG. Deregulation of smooth muscle cell cytoskeleton within the human atherosclerotic coronary media layer. Journal of Proteomics. 2013;82:155–165. [DOI] [PubMed] [Google Scholar]
- 53.Tourrière H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, Tazi J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. The Journal of Cell Biology. 2003;160:823–831. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Martin S, Zekri L, Metz A, Maurice T, Chebli K, Vignes M, Tazi J. Deficiency of G3BP1, the stress granules assembly factor, results in abnormal synaptic plasticity and calcium homeostasis in neurons. Journal of Neurochemistry. 2013;125:175–184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure I. Expression of SG markers in atherosclerotic plaque. (A) G3BP1 costaining with macrophage and VSMC markers in plaque from LDLR−/− mice fed a western diet for 14 weeks. (B)Poly-A Binding Protein (PABP) expression in atherosclerotic plaque. Very little PABP was detected in LDLR−/− mice fed a normal laboratory diet, but large amount of PABP is detected in plaque from LDLR−/− mice fed a western diet for 14 weeks. Magnification 400X. (C) Colocalization PABP and a-smooth muscle actin in atherosclerotic plaque from LDLR−/− mice fed a western diet for 14 weeks. Co-localization of PABP and smooth muscle cells showed that PABP expression was increased specifically in the synthetic intima and cap layer of smooth muscle cells juxtaposed to the plaque, as compared to the quiescent and contractile medial smooth muscle cells in these lesions. Magnification 600X.
Supplemental Figure II. PABP expression correlates with atherogenesis in LDLR−/− mice. Cumulative staining of the stress granule-specific marker Poly-A Binding Protein (PABP) with atherosclerosis progression. (A) Representative images of aortic arch sections from LDLR−/− mice fed western diet for 0, 5, 10, and 14 weeks that were immunostained for PABP (red), alpha smooth muscle actin (green), and DAPI (blue). (B) Quantification of PABP staining of aortic arch sections by corrected total cell fluorescence (CTCF). *P≤0.05, ** P≤0.01, or *** P≤0.001.
Supplemental Figure III. Immunostaining for PABP (red), G3BP (green) and DAPI (blue) in human VSMCs untreated or pretreated with the calpain inhibitor zLLal prior to stimulation with clotrimazole (A), and Arsenite (B). Data are representative of 3 independent experiments.
Supplemental Figure IV. HVSMCs transfected with siRNA targeting G3BP1 or scrambled control were serum-starved and stimulated with oxLDL for 16 hours, RNA collected, and expression of atherosclerosis-related transcripts were quantitated by PCR. *P≤0.05, ** P≤0.01, or *** P≤0.001.
Supplemental Figure V. G3BP1 knockdown does not affect VSMC proliferation. HVSMCs were transfected with G3BP1 siRNA or scrambled control siRNA for 48h. Cells were seeded at 10,000 cells/well and counted at days 3 and 7.