

Abstract
Objective:
Fatty acid uptake and oxidation characterize the metabolism of alternatively activated (AA) macrophage polarization in vitro, but the in vivo biology is less clear. We assessed the roles of LpL-mediated lipid uptake in macrophage polarization in vitro and in several important tissues in vivo.
Approach and Results:
We created mice with both global and myeloid-cell specific LpL deficiency. LpL deficiency in the presence of VLDL altered gene expression of bone marrow derived macrophages and led to reduced lipid uptake but an increase in some anti- and some pro-inflammatory markers. However, LpL deficiency did not alter lipid accumulation or gene expression in circulating monocytes nor did it change the ratio of Ly6Chigh/Ly6Clow. In adipose, less macrophage lipid accumulation was found with global but not myeloid-specific LpL deficiency. Neither deletion affected the expression of inflammatory genes. Global LpL deficiency also reduced the numbers of elicited peritoneal macrophages. Finally, we assessed gene expression in macrophages from atherosclerotic lesions during regression; LpL deficiency did not affect the polarity of plaque macrophages.
Conclusions:
The phenotypic changes observed in macrophages upon deletion of Lpl in vitro is not mimicked in tissue macrophages.
Keywords: lipoprotein lipase lipids, fatty acid, lipids, macrophage polarization, inflammation
Graphical Abstract
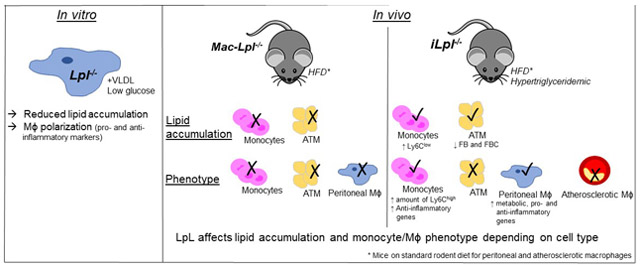
Introduction
Macrophages (Mϕs) are indispensable for maintaining tissue homeostasis and defending against multiple threats as members of the innate immune system. They have essential developmental roles and regulate normal physiology as “first responders” by communicating with the host’s adaptive immune system 1. Although the role and phenotype of different tissue Mϕs are diverse and environment-specific, the Mϕ phenotype is often oversimplified as a dichotomy of classically activated Mϕs (CAMϕs or M1) and alternatively activated Mϕs (AAMϕs or M2). Key determinants of these two phenotypes are thought to include differences in the metabolic pathways used by these cells, and a number of studies, predominantly in vitro, suggest that metabolic substrate use controls Mϕ polarization 2, 3.
These two forms of Mϕs defend against different pathogens. CAMϕs are implicated in acute bacterial infection; they are inflammatory, and their metabolic phenotype is glycolytic and hypoxia inducible factor 1α-dependent 3. AAMϕs play a significant role in parasitic infection. They utilize fatty acid oxidation (FAO) and play roles in resolving inflammation, including experimental atherosclerosis 4, and in wound healing. Canonical induction of M2 polarization is mediated by signal transducer and activator of transcription 6, which transcriptionally upregulates both peroxisome proliferator-activated receptors (PPARs; PPARδ, PPARγ) and PPARγ coactivator-1 beta (PGC1β) 3. PPARs and PGC1β activation induce anti-inflammatory markers (Arg1, Ym/Chi3l3, and Fizz1/Relma), FAO-related genes (Cpt1a, Aox) 3, 5, and fatty acid (FA) uptake genes such as cluster of differentiation 36 (Cd36) and lipoprotein lipase (Lpl) 5.
As noted above, AAMϕs perform oxidative phosphorylation of FAs and also of glucose 6 and glutamine 7, with FAO being the dominant source of energy. Uptake and use of FAs have shown to be critical to conversion of Mϕs to AAMϕs in vitro 5, but the in vivo biology is less clear. LpL is the rate-limiting enzyme for the conversion of circulating triglycerides (TGs), within chylomicrons and very low-density lipoproteins (VLDL), to free FAs (FFAs). It is also responsible, in part, for regulating plasma TG levels. Further, LpL increases the association of lipoproteins with proteoglycans, often referred to as bridging, and hydrolyses circulating lipoprotein, which is critical for lipid uptake in skeletal muscle, heart, and brown adipose tissue (AT) 8-10. LpL deletion reduces cellular uptake of both FFA and VLDL that occurs via non-receptor (bridging) pathways 11; in vitro studies have documented the importance of this process in monocytes and Mϕs 12-15. Notably, macrophage LpL deficiency does not affect receptor-mediated endocytosis16. Several in vitro studies have shown that LpL-mediated VLDL lipid accumulation in monocytes induces expression of inflammatory genes such as interleukin-1 beta (Il1β) and tumor necrosis factor alpha (Tnfα) 17, 18. This indicates that VLDL-derived lipid uptake is inflammatory in monocytes. In contrast, LpL-mediated VLDL lipid uptake has been proposed to suppress Mϕ inflammation via PPARδ activation 19, 20, suggesting that LpL-mediated lipid uptake promotes an AA phenotype. In contrast to these purported roles in driving Mϕs to a less inflammatory and more reparative phenotype, Mϕ-derived LpL is atherogenic 21. These data imply that pathways of lipid uptake and their effects on Mϕ polarization found in vitro may differ from what occurs in vivo.
Thus, we asked whether the influences of lipid uptake pathways on Mϕ polarity are microenvironment-specific. To answer this, we assessed the roles of LpL in Mϕ polarization in vitro and in several important depots in vivo, including AT, the peritoneal cavity, and atherosclerotic plaques. Our data indicate that LpL affects both lipid uptake and Mϕ polarity. The situation in vivo is more complicated as complete LpL deficiency also causes marked hypertriglyceridemia. However, this issue was controlled for, in part, by also studying mice with a myeloid-specific LpL deletion. Finally, our data show that LpL exerts distinct effects in each tissue depot in vivo, an observation that cannot be easily modeled in vitro.
Materials and Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
Animals:
Global inducible Lpl knockout (iLpl−/−) mice were generated by crossing Lplfl/fl mice with TgCreER transgenic mice (The Jackson Laboratory) 22, which harbor the tamoxifen-inducible Cre recombinase driven by the chicken beta actin promoter/enhancer coupled with the cytomegalovirus (CMV) immediate-early enhancer (β-actin-MerCreMer). Myeloid cell-specific Lpl knockout (Mac-Lpl−/−) mice 21 were generated by crossing Lplfl/fl mice with LysMCre mice (The Jackson Laboratory).
Cell culture:
Bone marrow (BM) cells were isolated by flushing cells from the femurs or tibiae of mice. Cells were differentiated into BM-derived Mϕs (BMDMs) in normal (5 mM) glucose DMEM with 10% Fetal Bovine Serum (FBS), 1% penicillin/streptomycin, and murine M-CSF (10 ng/mL; PeproTech) for 7 days. The BMDMs were cultured in low (1 mM) glucose or normal (5 mM) glucose or high (25 mM) glucose DMEM for 24 hours with or without murine IL-4 (20 ng/mL; PeproTech) and/or human VLDL (100 μg/mL; Alfar Aesar) plus 5% FBS for 24 hours.
Plasma Lipid Measurement:
Total triglyceride (TG) and total cholesterol (TC) were measured by using Infinity Total Triglyceride Reagent (Thermo Fisher Scientific; #TR22421) and Infinity Total Cholesterol Reagent (Thermo Fisher Scientific; #TR13421). Non-esterified FAs (NEFAs) were measured by using Wako Diagnostic NEFA reagents (Wako Life Sciences, Inc).
Lipoprotein Fractionation:
Equal amounts of plasma (70 μl-100 μl) were used for sequential density ultracentrifugation to separate VLDL (d<1.006 g/mL), low-density lipoprotein (LDL) (d=1.006-1.063 g/mL), high-density lipoprotein (HDL) (d=1.063-1.21 g/mL) in a TLA 100 rotor (Beckmann Instruments). Fractions were used to measure TC and TG as described above.
Glucose and Insulin Measurement:
Blood glucose was measured by using OneTouch Ultra2 meter (One Touch). Plasma insulin was measured by using Mouse Ultrasensitive Insulin ELISA kit (Alpco; #80-ISMSU-E01).
White blood cell counts:
Total while blood cell counts in freshly collected mouse blood were obtained using a hematology cell counter (Oxford Science Inc.).
Blood Leukocytes:
Monocytes (total and subsets) and neutrophils were identified from whole blood as previously described 23.
Hematopoietic stem cells:
Hematopoietic stem and progenitor cells from the BM and spleen were analyzed by flow cytometry as previously described 23.
Adipose tissue macrophage (ATM) isolation:
ATMs were isolated as previously described 24.
Peritoneal macrophage isolation:
Peritoneal exudate cells were harvested by peritoneal lavage with FACS buffer (PBS+ 0.2%BSA+ 5mM EDTA) at a volume of 10 mL per mouse. Total peritoneal exudate cells were sorted by using FACS AriaII (BD Bioscience). The gating strategy to sort large peritoneal Mϕs (LPMs) and small peritoneal Mϕs (SPMs) was as previously described 25 and is shown in Supplemental Figure 6.
Seahorse extracellular flux analysis:
BMDMs from Lplfl/fl and iLpl−/− were seeded (300,000 cells per well) in 5mM glucose supplemented with 2%FBS into XF24 cell culture microplates (Seahorse Bioscience) and stimulated with IL-4 as described above. After 24 hours, oxygen consumption rates (OCR) were measured using an XFe24 instrument (Seahorse Bioscience). For cellular mitochondrial oxidation assessments, the XF Cell Mito Stress Test Kit was used according to the manufacturer’s protocol.
Neutral lipid content measurement:
Adipo-Red (Lonza) was used according to the manufacturer’s protocol to measure intracellular lipid content in BMDMs. Freshly isolated monocytes, ATMs, and peritoneal Mϕs were stained with Bodipy (1:500 dilution; Sigma) for 30 mins in dark on ice to fluorescently label intracellular lipid accumulation. Total neutral lipid content was quantified by using flow cytometry (LSRII) and analyzed by using FlowJo software.
Aortic Transplant.
The aortic transplant model has been described before 4, 26-29 A plaque burden aortic arch from Ldlr−/− mice was transplanted into the recipient mice (iLpl−/− or Lplfl/fl mice), which was inter-positioned with the abdominal aorta. During the transplantation, blood flow was directed through the graft. At time of the transplant, all recipient mice were approximately 22 weeks old and maintained on a normal rodent diet. All mice were sacrificed 14 days after the aortic arch transplantation. AHA guidelines for experimental atherosclerosis studies have been followed 30.
Laser capture microdissection and quantitative Real Time PCR.
CD68+ cells were selected from atherosclerotic plaques by laser capture microdissection. All laser capture microdissection procedures were performed under RNase-free conditions. Aortic root sections were stained with hematoxylin-eosin as previously described 31, 32.
Immunohistochemistry and plaque assessment.
Grafted arches were removed after perfusion with 10% sucrose in saline, and embedded in optimum cutting temperature (OCT, Tissue-Tek; Sakura Finetek USA) block and freezed. Serial sections at (6 μm thick) were cut and stained for CD68 (Bio-Rad MCA1957) and CD206, also known as Mannose Receptor (MR) (Bio-Rad MCA2235). Negative controls were performed using an isotype control (Rat IgG2a) instead of the primary antibody (Abcam ab18450). ImagePro Plus 7.0 software was used to determine CD68+ and MR+ areas. Overlay was done using Adobe Photoshop CC2018.
Statistical Analyses:
Data are presented as mean ± SD. Normality test (Shapiro-Wilk normality test (∝=0.05) and visual inspection of Q-Q plots) and homogeneity of variance (Brown-Forsythe test) were performed. Depending on the results, parametric or non-parametric tests were chosen as indicated in the figure legends. *p<0.05; **p<0.01. All calculations were performed using Microsoft Excel and Graphpad Prism 7.
Results
LpL deficiency in Mϕs affects their polarization and lipid accumulation in vitro:
LpL mediates the hydrolysis of VLDL-derived TG and activates PPARδ 14, which suppresses inflammatory pathways in cultured Mϕs 20. To assess whether LpL deficiency affects VLDL-dependent lipid uptake and alternative activation (AA) under various culture conditions, BMDMs from wild type and Mϕ-specific LpL deficient mice (Lplflfl; LysMCre, Mac-Lpl−/−) were cultured with 100 μg/mL of human VLDL plus 5% FBS for 24 hours in low-glucose (1 mM) containing media. LpL actions enhance Mϕs phagocytic function only in low glucose media 16 when FAs are likely to be a more critical source of energy substrate. Efficient Lpl deletion in BMDMs is shown in Figure 1A. As expected, LpL deficiency led to a marked reduction in FFA in the VLDL-containing culture system (Figure 1B). Loss of LpL in Mϕs significantly reduced intracellular neutral lipids as measured by bodipy and Nile-Red staining (Figure 1C, 1D). To assess whether lack of LpL alters known PPARδ-targeted genes, we measured a lipid droplet-related gene, perilipin 2 (Plin2) and a FAO-related gene, carnitine palmitoyltransferase 1A (Cpt1a). Consistent with the intracellular lipid droplet results, Plin2 and Cpt1a gene expression were decreased in Mac-Lpl−/− BMDMs (Figure 1E, 1F). In order to examine whether lack of LpL induces other metabolic-related gene expression as a compensatory mechanism, we measured glucose transporter 1 (Glut1/Slc2a1) and FA synthase (Fasn) mRNA levels. Lpl deficiency increased both Glut1 and Fasn mRNA expression upon VLDL treatment (Figure 1G, 1H), suggesting that lack of LpL-mediated FA uptake increases glucose uptake and de novo FA synthesis. These results demonstrate that Mϕ-derived LpL is indispensable for VLDL-mediated FA uptake under a low glucose culture condition.
Figure 1. LpL is required for VLDL-derived lipid uptake in macrophages and regulates both inflammatory and anti-inflammatory genes in the presence of VLDL containing low glucose media in vitro.
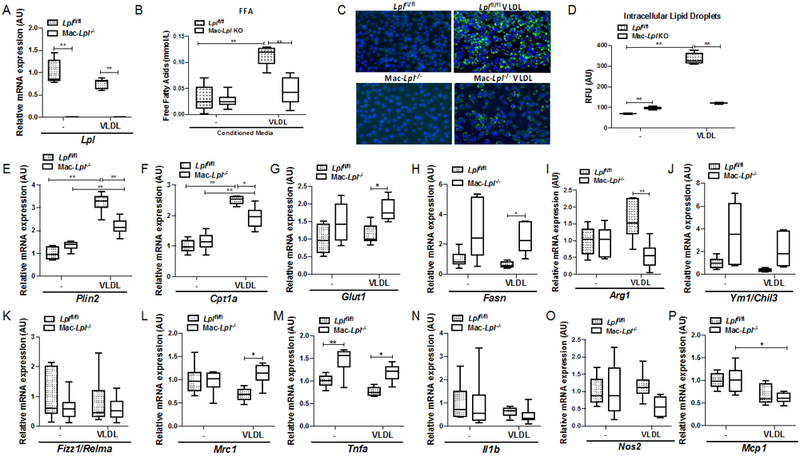
BMDMs from Lplfl/fl and Mac-Lpl−/− mice were used to measure metabolism- related and M1/M2-related gene expression in the presence of human VLDL (100ug/mL). (A) The gene expression of Lpl in BMDMs. (B) Conditioned media FFAs concentration from cultured BMDMs from Lplfl/fl and Mac-Lpl−/− mice. (C) Fluorescent staining of nuclei by DAPI and neutral lipid by bodipy dye in VLDL treated BMDMs (Blue: nuclei, Green: neutral lipids). (D) Quantification of neutral lipid content measured by Nile-red stain in VLDL treated BMDMs (RFU: Relative Fluorescent Unit). (E-F) The expression of PPARδ-targeted genes (Plin2, Cpt1a) in BMDMs. (G-H) The expression of Glut1 and Fasn in Mac-Lpl−/− BMDMs. (I-L) The gene expression of anti-inflammatory genes (Arg1, Ym1, Fizz1, Mrc1) in VLDL treated BMDMs. (M-P) The expression of inflammatory genes (Tnfa, I1b, Nos2, Mcp1) in VLDL treated BMDMs. N=6/group.*p<0.05, **p<0.01. Results are represented as median with 25th and 75th percentiles, capped bars indicate 10th and 90th percentile and compared using Two-way ANOVA, Sidak’s multiple comparison.
Next, we investigated whether loss of LpL-mediated VLDL FA uptake impairs M2 polarization and/or augments M1 polarization. Although Arg1 gene expression was decreased, as had been suggested by Chawla et al. 5, mRNA levels of other M2 markers, Ym1 and Mrc1, were increased in Mac-Lpl−/− BMDMs in the presence of VLDL, as compared to Lplfl/fl BMDMs (Figure 1I, 1J, 1L). Among inflammatory genes, Tnfα was increased in Mac-Lpl−/− BMDMs (Figure 1M), but Nos2 gene expression showed a trend to decreased expression in Mac-Lpl−/− BMDMs in the presence of VLDL(Figure 1O). These results show that lack of LpL in Mϕs reduces lipid uptake and alters both inflammatory- and anti-inflammatory genes in VLDL-enriched culture conditions. However, the mixed phenotypic responses indicate a level of complexity in Mϕ polarization due to different transcriptional regulation within both the inflammatory and AA pathways.
To investigate whether LpL regulates Mϕ polarity in response to the standard inducer of AAMs in vitro, we cultured Lplfl/fl and Mac-Lpl−/− BMDMs with IL-4 (20 ng/mL) and IL-4 plus VLDL for 24 hours under low glucose conditions. Surprisingly, IL-4 stimuli alone increased Plin2, Cpt1a, and Fasn mRNA levels in Mac-Lpl−/− BMDMs, as compared to Lplfl/fl BMDMs (Figure 2A, 2B, and 2D). Since IL-4 induces FAO 5, lack of FA uptake perhaps stimulated cells to increase de novo FA synthesis. Culturing Mϕs with IL-4 plus VLDL led to a significant decrease in Plin2 and Cpt1a, but a trend of increase in Glut1 and a significant ∼ 5-fold increase in Fasn gene expression in Mac-Lpl−/− BMDMs (Figure 2A, 2B, 2C, and 2D), similar to in vitro findings in Figure 1 E-H. This suggests that in M2-like Mϕs, LpL-mediated VLDL-derived lipid uptake regulates PPARδ-targeted genes, but lack of LpL seem to induce metabolic compensations via de novo FA synthesis.
Figure 2. LpL is necessary for VLDL-derived FA uptake, but it is not anti-inflammatory under low-glucose condition in vitro.
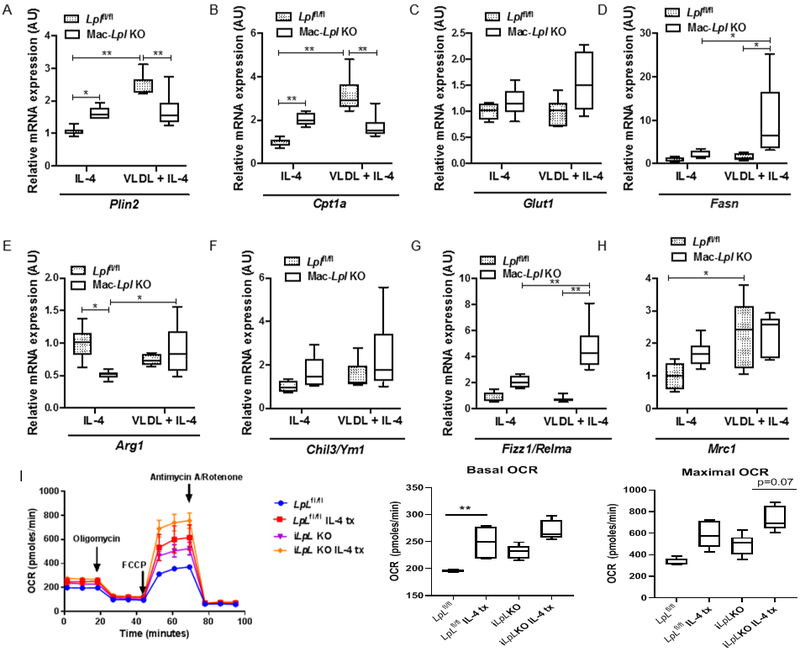
BMDMs from Lplfl/fl and Mac-Lpl−/− mice were used to measure metabolism-related and M2-related genes in the presence of IL-4 (20ng/mL) or IL-4 plus human VLDL. (A) The expression of a lipid droplet-related protein, Plin2, gene in BMDMs. (B) The expression of a fatty acid oxidation-related gene, Cpt1a in BMDMs. (C) The expression of a glucose transporter, Glut1 (Slc2a1), gene in BMDMs. (D)The expression of fatty acid synthase gene in BMDMs. (E-H) The expression of canonical M2 (anti-inflammatory) genes in BMDMs. (I) Oxygen consumption rate comparing IL-4 treatment in Lplfl/fl and Mac-Lpl−/−. N=3/group. *p<0.05, **p<0.01. Results are represented as median with 25th and 75th percentiles, capped bars indicate 10th and 90th percentile and compared compared using Two-way ANOVA with Sidak’s multiple comparison test.
As expected, upon IL-4 treatment alone, lack of LpL prevented up-regulation of Arg1 (Figure 2E). Deletion of LpL did not affect Ym1, Fizz1 and Mrc1 gene expression (Figure 2F-H). Upon IL-4 and VLDL treatment, Lpl deficiency did not affect Arg1, Ym1, and Mrc1 but dramatically increased Fizz1/Relma expression. Huang et al. demonstrated that glucose-mediated FA synthesis is necessary for M2 activation as measured by Fizz1/Relma and programmed death-ligand 1 expression 6. Thus, up-regulation of Fizz1 expression is probably due to glucose-mediated FA synthesis, as we found an increase in both Glut1 and Fasn mRNA levels in Mac-Lpl−/− BMDMs. To test the metabolic status of Mϕs in LpL depleted conditions, we performed OCR experiments in cultured Lplfl/fl and Mac-Lpl−/− BMDMs incubated with IL-4 (20 ng/mL) (Figure 2 I-J). The results showed that OCR is increased when LpL is depleted, being in accord with the changes we found in Cpt1a gene expression. Overall, our data show that in AA Mϕs, LpL is necessary for FA uptake, but it is not anti-inflammatory under the low glucose condition in vitro. Further, increase in Glut1 mRNA level suggests that LpL deficient Mϕs switch to a greater use of glucose.
To test whether FFAs not derived from LpL-hydrolysis alter the phenotype of Mac-Lpl−/− BMDMs, we incubated BMDMs with or without oleic acid (OA) (300 μM) for 24 hours. OA significantly decreased Lpl expression, but increased Cpt1a gene expression in Lplfl/fl BMDMs (Supplemental Figure 1). OA treatment also significantly lowered Mrc1 gene expression in Mac-Lpl−/− BMDMs with a trend towards decreased expression in Lplfl/fl BMDMs. Unexpectedly, we did not find altered expression of other anti-inflammatory related-genes such as Ym1/Chil3 or Fizz1/Relma, whereas inflammatory genes increased upon OA treatment (Supplemental Figure 1). Although OA treatment is generally known to induce M2 genes (Arg1, Mrc1) 33-35, our data suggest that OA-dependent M2 activation can be time- or dose-dependent. The pattern of gene expression in response to OA treatment was similar in Lplfl/fl compared to Mac-Lpl−/− BMDMs, suggesting that FA uptake does regulate anti-inflammatory or inflammatory gene expression independent of LpL expression, whereas VLDL-derived FA requires LpL expression to invoke an inflammatory response.
In order to determine whether increasing glucose concentration from low glucose culture conditions (1 mM) to normal (5 mM) affects Mϕ polarity in vitro, we performed the same experiment with both media. In the normal glucose condition, Lpl deletion increased Plin2, Cpt1a, and Fasn mRNA levels (Supplemental Figure 2A, 2B and 2E) but did not alter Glut1 expression in the presence of VLDL (Supplemental Figure 2D). These data indicate that under the normal glucose condition, in vitro, Mϕs mainly utilize glucose as the energy fuel as they do in vivo 36; thus, lack of LpL-mediated FA uptake does not impact Glut1 expression. With regards to Mϕ polarity, similar to low glucose culture conditions, Arg1 was significantly decreased in Mac-Lpl−/− BMDMs, whereas other typical M2 genes such as Ym1 and Fizz1 were up-regulated in the presence of VLDL (Supplemental Figure 2F, 2G, and 2H). Upon IL-4 stimulation in the normal glucose condition, we found a similar pattern to the low glucose conditions, suggesting that increasing glucose concentration from 1 mM to 5 mM does not significantly impact Lpl deficient Mϕ polarity. The changes that we observe only matter to M2-induced conditions using VLDL and/or IL-4, as culture conditions without substrate added to the enzyme and no induction for anti-inflammation served as baseline conditions.
Since our results show that Lpl deficiency up-regulates Glut1 and Fasn gene expression under low glucose and up-regulates Fasn gene expression under the normal glucose culture condition, we hypothesized that glucose-mediated FA synthesis is responsible for increasing some M2-related genes (Ym1 and Fizz1). Thus, we performed the same in vitro experiment under high (25 mM) glucose conditions to examine whether an excess amount of glucose impacts both metabolic- and M2-related genes. Unlike low and normal glucose culture conditions, we found no significant difference between Lpl expressing and non-expressing cells in all M2-related genes (Arg1, Ym1, Fizz1, Mrc1) in high glucose containing media (Supplemental Figure 3G, 3H, 3I and 3J). These data indicate that with excess glucose, Mϕs mainly use glucose as their energy source; thus, ablating LpL-mediated FA uptake does not affect either metabolism or polarization.
The impact of myeloid-specific Lpl ablation on circulating monocytes in obese mice:
While we could reproduce many of the in vitro findings of previously reported studies 14, 19, we found that these effects varied by culture condition. For this reason, we next investigated the role of LpL in monocytes and Mϕs in a lipid-rich microenvironment in vivo. In order to induce a hyperlipidemic condition, we used mice fed a HFD. Obesity increases circulating levels of myeloid-derived cells 37. This diet also enables us to examine ATMs, which are limited in animals fed a normal rodent diet. Thus, we sought to determine whether myeloid cell-derived LpL regulates monocyte subpopulations (Ly6Chi and Ly6Clow monocytes), lipid accumulation, and phenotype of monocytes. The detailed experimental design is shown in Figure 3A. We confirmed that Lpl was efficiently deleted in monocytes in Mac-Lpl−/− mice (Figure 3B). Ablation of Lpl in myeloid-derived cells with HFD did not affect plasma glucose levels (Figure 3C). As expected, levels of plasma TG and TC in VLDL, LDL, and HDL in Mac-Lpl−/− mice were not significantly different from TG and TC levels in Lplfl/fl mice (Figure 3D, 3E). Lack of LpL in monocytes with HFD did not affect circulating levels of Ly6Chi- and Ly6Clow-monocytes (Figure 3F). BM progenitors (common myeloid progenitors and granulocyte-Mϕ progenitors) in Mac-Lpl−/− mice were not significantly different from Lplfl/fl mice (Supplemental Figure 4A), as circulating levels of monocytes were unaltered. Thus, differences in BM progenitors and circulating monocytes noted with normal rodent diets 38 were abrogated by the HFD, which is known to stimulate BM progenitors 37. Ablation of Lpl did not alter intracellular lipid content as measured by percent of bodipy+ cells from each monocyte population (Figure 3G, 3H) and bodipy+ geometric mean fluorescent intensity (gMFI) (Figure 3I). Consistent with the intracellular lipid content, expression of Plin2 and other lipid-related genes were unaltered in Mac-Lpl−/− mice (Figure 3J). Also, Lpl deficiency in monocytes did not lead to an increase in inflammatory gene expression (Figure 3K) and did not affect expression of anti-inflammatory-related genes (Figure 3L). These results indicate that myeloid cell-derived LpL in vivo is not required in monocytes for lipid accumulation and is not inflammatory in these cells.
Figure 3. Myeloid-cell derived LpL does not affect systemic metabolism and circulating levels of monocytes, and is not required for lipid accumulation in monocytes in vivo.
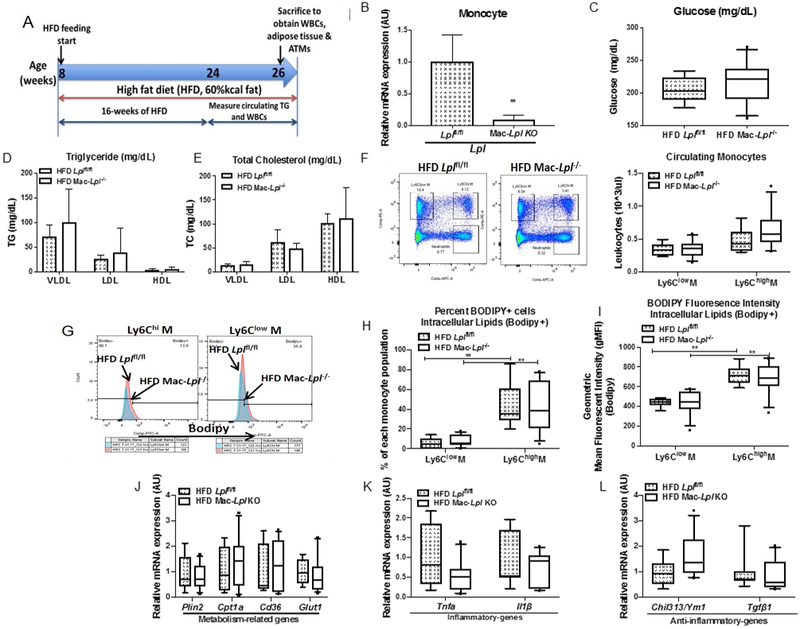
Littermate controls (Lplfl/fl) and Mac-Lpl−/− (Lplfl/fl;LysMCre) mice were studied for plasma metabolic parameters, circulating levels of monocytes, and lipid content and phenotype of monocytes. (A) Experimental design: Lplfl/fl and Mac-Lpl−/− mice were fed with a HFD (60%kcal fat) from 8 weeks old of age until sacrifice (26 weeks old of age); Body weight and plasma parameters were measured between 24 weeks of age and 25 weeks of age; Circulating monocytes (Ly6Chi and Ly6Clow), adipose tissue (PGAT, SCAT, and BAT), and adipose tissue macrophages (CD45+F4/80+CD11b+ and CD45+F4/80+CD11b+CD11c+) were obtained at 26 weeks old of age. (B) The expression of Lpl in monocytes (CD45+CD115+) that were isolated by fluorescence-activated cell sorting (FACS) method. (C) Plasma fasting glucose level. (D) Levels of plasma triglyceride in VLDL, LDL, and HDL fraction. (E) Levels of plasma total cholesterol in VLDL, LDL, and HDL fraction. (F) Representative flow cytometry plots of blood leukocytes and quantified number of circulating Ly6Chi and Ly6Clow monocyte. (G) Circulating Ly6Chi and Ly6Clow monocytes from HFD Lplfl/fl and HFD Mac-Lpl−/− mice were analyzed using flow cytometry for neutral lipid content (BODIPY); Representative flow cytometry histogram plots are shown for BODIPY fluorescence. (H) Quantified percentage of BODIPY fluorescence in Ly6Chi and Ly6Clow monocytes. (I) Quantification of BODIPY geometric mean fluorescence intensity (gMFI) in Ly6Chi and Ly6Clow monocytes. (J) The expression of metabolism-related genes (Plin2, Cpt1a, Cd36, Glut1) in total monocytes. (K) The expression of inflammatory genes (Tnfa, Il1b) in total monocytes. (L) The expression of anti-inflammatory genes (Ym1 and Tgfbi) in total monocytes. N=6/group. *p<0.05, **p<0.01. Results are represented as mean ± SD (B, D, E) and median with 25th and 75th percentiles, capped bars indicate 10th and 90th percentile (F-L) and Lplfl/fl and Mac-Lpl−/− compared using unpaired t-test (B-E, J-L). Two-way ANOVA with Sidak’s multiple comparison test was used for (F-I).
The effect of Lpl deletion on ATMs:
Next, we investigated whether deletion of LpL in myeloid-derived cells affects ATM lipid uptake and polarity. Visceral AT-specific acute silencing of Lpl in ATMs using glucan-encapsulated siRNA particles showed that Lpl deficiency in ATMs slightly decreased ATM neutral lipid content in leptin deficient (ob/ob) mice 39. However, it is less clear whether LpL affects subpopulations and polarization. Because acute Lpl silencing affected lipid content in ATMs in vivo37 and we found a significant reduction in lipid content of BMDMs in vitro (Figure 1), we postulated that LpL affects the phenotype of Mϕs residing in a high-fat environment. For that reason, we first investigated the role of LpL in ATMs of obese mice.
ATMs are thought to consist of two major subpopulations: F4/80+CD11b (FBs) and F4/80+CD11b+CD11c+ (FBCs). Although several studies described that FBs are less inflammatory, while FBCs are more inflammatory 40, Xu et al. 24 demonstrated that FBCs have more neutral lipid content, increased transcriptional profile of lysosomal-dependent lipid metabolism, and are not more inflammatory than FBs. So, the data are inconsistent. In our experiments, ablation of LpL in myeloid-derived cells did not alter total ATM (F4/80+) content (Figure 4A). Unexpectedly, we found that Lpl deficiency in ATMs did not lead to fewer Cd11c+ ATMs (Figure 4B, 4C) or less lipid accumulation measured by percent of bodipy+ cells (Figure 4D, 4E) and bodipy+ intensity as measured by bodipy gMFI (Figure 4E). Upon Lpl deletion, Lpl deficient ATMs showed approximately 20% higher Glut1 mRNA expression than control Mϕ s (Figure 4G), suggesting a slight metabolic shift from FA uptake to glucose uptake in ATMs. Inflammatory and anti-inflammatory gene expression did not change (Figure 4H, 4I). Our findings are similar to a previous report showing that myeloid cell-specific GLUT1 overexpression did not alter inflammatory phenotype in vivo 36. Here, we illustrate that LpL is not necessary for the development of more lipid-laden ATMs (FBCs), intracellular lipid accumulation, and ATM polarization.
Figure 4. Myeloid-cell derived LpL is not required for more lipid-laden ATM development (FBCs), intracellular lipid accumulation, and ATM polarization.
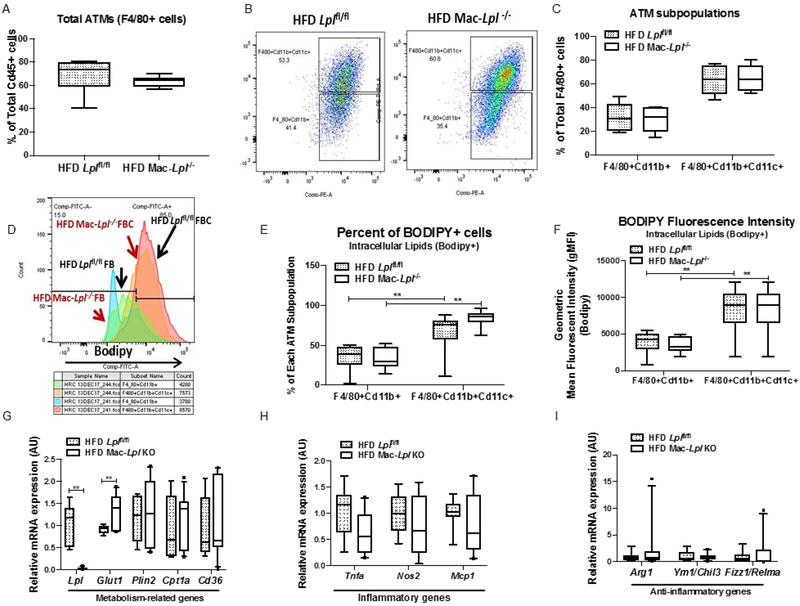
HFD fed littermate controls (Lplfl/fl) and Mac-Lpl−/− (Lplfl/fl;LysMCre) mice were studied for total ATM (CD45+F4/80+) content, ATM subpopulation (CD45+F4/80+Cd11b+ and CD45+F4/80+CD11b+CD11c+), and ATM polarity. (A) Flow cytometry analysis of total ATMs (CD45+F4/80+). (B) Representative flow cytometry plots for ATM subpopulations; FBs (CD45+F4/80+Cd11b+) and FBCs (CD45+F4/80+CD11b+CD11c+) and quantified FBs and FBCs. (C) Quantified percentage of FBs and FBCs from CD45+F4/80+ cells. (D) Representative flow cytometry histogram plots are shown for BODIPY fluorescence in FBs and FBCs from HFD Lplfl/fl and HFD Mac-Lpl−/− mice. (E) Quantified percentage of BODIPY fluorescence in FBs and FBCs. (F) Quantification of BODIPY geometric mean fluorescence intensity (gMFI) in FBs and FBCs. (G) The expression of metabolism-related genes (Lpl, Glut1, Plin2, Cpt1a, Cd36) in FBCs. (H) The expression of inflammatory genes (Tnfa, Nos2, Mcp1) in FBCs. (I) The expression of anti-inflammatory genes (Arg1, Ym1, Fizz1) in total FBCs. N=6/group. *p<0.05, **p<0.01. Results are represented as median with 25th and 75th percentiles, capped bars at 10th and 90th percentile and compared using Two-Way ANOVA, Sidak’s multiple comparison test (A-F) or unpaired t-test, or Mann-Whitney Test (Glut1 Plin2, Cd36, Arg1, Ym1/Chil3, Fizz1/Relma) (G-I).
These results would not be expected if LpL-mediated lipolysis was needed to supply lipids and to activate PPARs, as occurs in other cells and tissues, such as the heart and skeletal muscle 41. Therefore, lipid supply to circulating monocytes and Mϕs must either come from FFAs, uptake of remnant lipoproteins that were partially hydrolyzed by LpL in other tissues, or endocytosis of TG-rich lipoproteins followed by intracellular lipolysis or de novo lipogenesis.
The impact of inducible total body Lpl deletion in myeloid-derived cells in obesity:
To determine whether deletion of LpL in myeloid-derived cells is compensated by LpL in other tissues, we analyzed monocytes and ATMs in iLpl−/− and Lplfl/fl mice. To induce obesity, we fed control Lplfl/fl mice and iLpl−/− mice a HFD for 16 weeks. Tamoxifen was injected intraperitoneally into both groups, globally ablating Lpl in the experimental (iLpl−/−) group (Figure 5A) 22, 42. We confirmed efficient Lpl deletion by measuring Lpl expression in AT (Figure 5B).
Figure 5. Inducible Lpl deletion increased plasma triglyceride level, circulating Ly6Chi monocytes level, and anti-inflammatory genes in monocyte despite more lipid accumulation in monocytes.
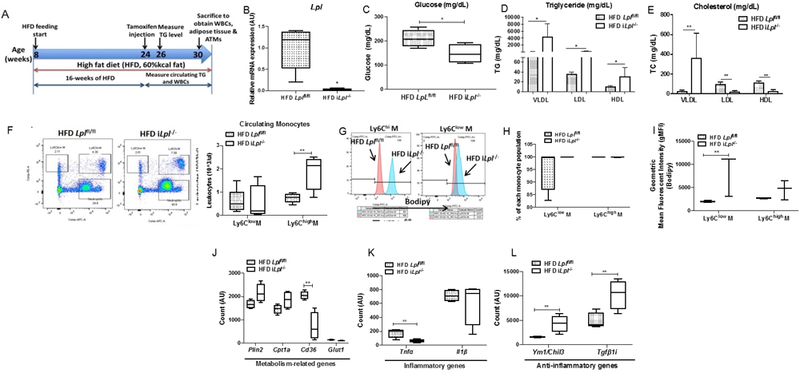
HFD fed littermate controls (Lplfl/fl) and iLpl−/− (Lplfl/fl;β-actin-MerCreMer) mice were studied for plasma metabolic parameters, circulating levels of monocytes, and lipid content and phenotype of monocytes. (A) Experimental design: Lplfl/fl and iLpl−/− mice were fed with a HFD from 8 weeks old of age for until sacrifice (30 weeks old of age); Body weight and plasma parameters were measured between 24 weeks of age and 30 weeks of age; Circulating monocytes (Ly6Chi and Ly6Clow), adipose tissue (PGAT, SCAT, and BAT), and adipose tissue macrophages (CD45+F4/80+CD11b+ and CD45+F4/80+CD11b+CD11c+) were obtained at 30 weeks old of age. (B) The expression of Lpl in adipose tissue (N=4-5/group). (C) Plasma fasting glucose level (N=11-17/group). (D) Levels of plasma triglyceride in VLDL, LDL, and HDL fraction (N=4-5/group). (E) Levels of plasma total cholesterol in VLDL, LDL, and HDL fraction (N=4-5/group). (F) Representative flow cytometry plots of blood leukocytes and quantified number of circulating Ly6Chi and Ly6Clow monocyte (N=11-17/group). (G) Circulating Ly6Chi and Ly6Clow monocytes from HFD Lplfl/fl and HFD iLpl−/− mice were analyzed using flow cytometry for neutral lipid content (BODIPY); Representative flow cytometry histogram plots are shown for BODIPY fluorescence (N=3-5/group). (H) Quantified percentage of BODIPY fluorescence in Ly6Chi and Ly6Clow monocytes (N=3-5/group). (I) Quantification of BODIPY geometric mean fluorescence intensity (gMFI) in Ly6Chi and Ly6Clow monocytes (N=3-5/group). (J) The expression of metabolism-related genes (Plin2, Cpt1a, Cd36, Glut1) in total monocytes (N=4/group). (K) The expression of inflammatory genes (Tnfa, Il1b) in total monocytes (N=4/group). (L) The expression of anti-inflammatory genes (Ym1 and Tgfbi) in total monocytes (N=4/group). *p<0.05, **p<0.01. Results are represented as median with 25th and 75th percentiles, capped bars at 10th and 90th percentile and compared between HFD Lplfl/fl and HFD iLpl−/− using unpaired t-test.
The circulating glucose level was decreased in HFD iLpl−/− mice (Figure 5C), suggesting that lack of LpL-mediated lipid uptake causes an increase in glucose uptake into tissues as was found with neonatal LpL deficiency 43. As expected, global Lpl deletion also increased VLDL-derived TG levels from 88 mg/dL to 4000 mg/mL as well as LDL- and HDL-TG (Figure 5D). VLDL-derived TC levels significantly increased, whereas LDL-TC and HDL-TC decreased in iLpl−/− mice (Figure 5E).
Induced loss of LpL also altered circulating white bloods cells in these HFD-fed mice. Ly6Chi circulating monocytes were significantly increased in HFD iLpL−/− mice relative to HFD Lplfl/fl mice (Figure 5F). The increase in levels of circulating Ly6Chi monocytes occurred with increased granulocyte-Mϕ progenitors in bone marrow (Supplemental Figure 4D). Although percentage of bodipy+ cells in both Ly6Clow- and Ly6Chigh- monocytes was unaltered in HFD iLpl−/− mice (Figure 5G, 5H), we found that bodipy+ gMFI was significantly higher in Ly6Clow monocytes as compared to those in Lplfl/fl mice (Figure 5I). These intracellular lipid content results indicate that although monocytes from both HFD Lplfl/fl and iLpL−/− mice are fully loaded with lipid droplets, lipid droplet size and number are greater in monocytes in HFD iLpl−/− mice. Thus, these lipids are obtained via a non-LpL-mediated lipid uptake.
To study the transcriptional profile and determine the role of LpL in monocytes in HFD iLpl−/− mice, we used RNA sequencing as an unbiased approach. As described previously, we analyzed inflammatory- and anti-inflammatory genes, as well as metabolism-related genes. An inflammatory gene, Tnfα, was significantly decreased, but Il1β was unaltered in HFD iLpl−/− mice (Figure 5K), while anti-inflammatory genes were up-regulated (Tgfbi, Ym1/Chi313) in HFD iLpl−/− mice (Figure 5L). CD36 mRNA levels were significantly downregulated in HFD iLpl−/− mice, whereas other metabolism-related genes showed a trend towards increased expression (Plin2, Cpt1a) or were unaffected (Figure 5J). However, KEGG pathway analysis showed that lack of Lpl increased oxidative phosphorylation and lysosome-related pathways (Supplemental Figure 7), suggesting that Lpl deficiency leads to a metabolic reprogramming by increasing endocytic TG-rich uptake for more oxidative phosphorylation. These data show that hypertriglyceridemia caused by global Lpl deletion with HFD induces BM progenitors to produce more Ly6Chi monocytes. Also, hypertriglyceridemia in HFD iLpl−/− mice leads to an increase of lipid content in Ly6Clow monocytes, which induces M2 gene expression but decreases a M1 gene, Tnfα, in monocytes.
The effect of inducible Lpl deficiency in adipose tissue macrophages in obesity:
Because myeloid cell-derived-specific Lpl deficiency did not alter ATM phenotype, we postulated that this is due to developmental compensation and early action of LysMCre. We used iLpl−/− mice to determine whether deleting Lpl after inducing obesity affects ATM subpopulation, lipid content, and phenotype. ATM Lpl expression was decreased 90% in iLpl−/− mice, however, total ATM (F4/80+) content did not differ between control and HFD iLpl−/− mice (Figure 6A). With regard to subpopulations, percent of FBs from total F4/80+ cells was significantly higher, whereas FBCs from total F4/80+ cells was significantly lower in HFD iLpl−/− mice as compared to HFD Lplfl/fl mice (Figure 6B, 6C). This suggests that LpL is necessary for lipid accumulation in ATMs and important for FBC ATM development in obesity. To further examine whether Lpl deletion affects lipid content in ATMs, we measured neutral lipid content in both FBs and FBCs. Intracellular neutral lipid accumulation measured by percentage of bodipy+ cells and bodipy+ gMFI in FBs and FBCs in HFD iLpl−/− mice tended to be lower in HFD iLpl−/− mice despite the marked hypertriglyceridemia in these mice (Figure 6D, 6E, and 6F). These data show that lack of Lpl in adipocytes decreases quantity of FBCs and prevents lipid accumulation in ATMs.
Figure 6. Inducible Lpl ablation decreases CD11c+ ATMs (FBCs) content, ATM lipid accumulation, but does not affect ATM polarity despite a dramatic increase in Glut1 expression.
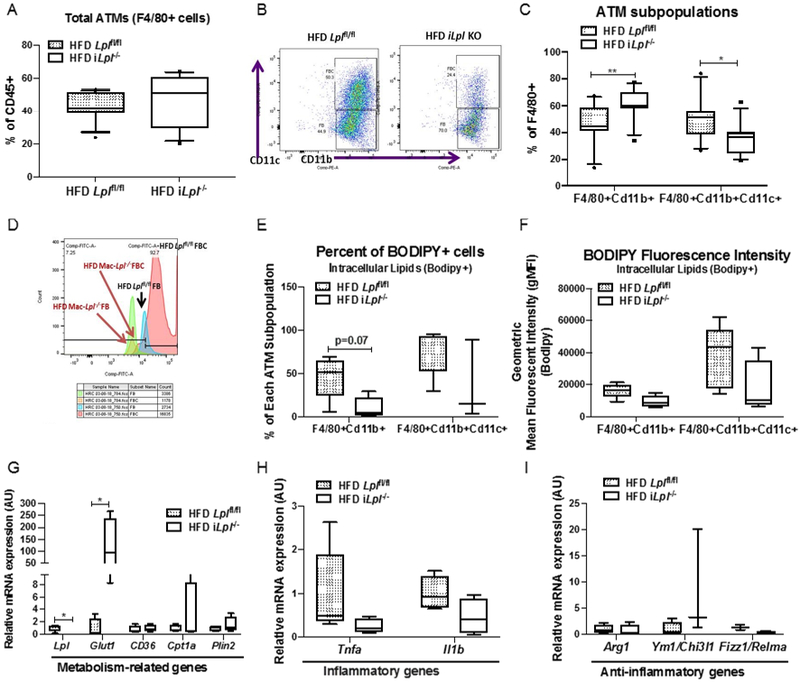
HFD fed littermate controls (Lplfl/fl) and iLpl−/− (Lplfl/fl;β-actin-MerCreMer) mice were studied for total ATM (CD45+F4/80+) content, ATM subpopulation (CD45+F4/80+Cd11b+ and CD45+F4/80+CD11b+CD11c+), and ATM polarity. (A) Flow cytometry analysis of total ATMs (CD45+F4/80+). (B) Representative flow cytometry plots for ATM subpopulations; FBs (CD45+F4/80+Cd11b+) and FBCs (CD45+F4/80+CD11b+CD11c+) and quantified FBs and FBCs (N=11-17/group). (C) Quantified percentage of FBs and FBCs from CD45+F4/80+ cells. (D) Representative flow cytometry histogram plots are shown for BODIPY fluorescence in FBs and FBCs from HFD Lplfl/fl and HFD iLpl−/− mice (N=3-5/group). (E) Quantified percentage of BODIPY fluorescence in FBs and FBCs (N=3-5/group). (F) Quantification of BODIPY geometric mean fluorescence intensity (gMFI) in FBs and FBCs (N=3-5/group). (G) The expression of metabolism-related genes (Lpl, Glut1, Cd36, Cpt1a, Plin2) in ATMs (N=4-5/group). (H) The expression of inflammatory genes (Tnfa, Il1b) in ATMs (N=4-5/group). (I) The expression of anti-inflammatory genes (Arg1, Ym1, Fizz1) in ATMs (N=4-5/group). *p<0.05, **p<0.01. Results are represented as median with 25th and 75th percentiles, capped bars at 10th and 90th percentile and compared between Lplfl/fl and iLpl−/− using unpaired t-test (A-F, CD36, Tnfa, Il1b) or Mann-Whitney Test (LpL, Glut1, Cpt1a, Plin2).
To examine whether LpL affects ATM phenotype, we measured both inflammatory and anti-inflammatory gene expression. Although Glut1 gene expression was dramatically increased in total ATMs of HFD iLpl−/− mice (Figure 6G), mRNA levels of inflammatory genes (Il1β, Tnfa) tended to be lower (Figure 6H). One anti-inflammatory gene, Ym-1, increased, but other M2 genes (Arg-1 and Fizz-1) were unaltered (Figure 5E and 6I) in total ATMs. A FAO-related gene, Cpt1a, and other lipid-related genes (Cd36, Plin2) were not affected (Figure 6G). Overall, our data show that although lack of LpL reduced the number of lipid-laden ATMs and appeared to shift Mϕs to a more glucose-oxidizing phenotype (increased Glut1 mRNA level), this phenomenon did not lead to M1 phenotype in ATMs.
The impact of myeloid cell-specific Lpl deletion in peritoneal Mϕs:
Tissue-specific Mϕs have distinct phenotypes and functions, depending on the tissue microenvironment. A previous report demonstrated that VLDL-derived FA uptake induces inflammation in peritoneal Mϕs in vitro, suggesting that LpL-dependent lipid uptake is inflammatory 13. We next assessed whether LpL affects peritoneal Mϕ number and polarity, both in resident (F4/80hiMHCIIlow) and recruited (F4/80lowMHCIIhigh) populations. We induced Mϕ recruitment using zymosan A (100 μg/mouse; Sigma) in mice fed a normal rodent diet, as shown in Figure 7A. We found that lack of LpL did not affect Mϕ accumulation in the peritoneal cavity (Figure 7B, 7C, and 7D). Glut1 mRNA expression was also significantly up-regulated in Mac-Lpl−/− LPMs as compared to Lplfl/fl LPMs (Figure 8B), indicating that genetic ablation of LpL may lead to a metabolic reprogramming. However, despite an increase in Glut1, both inflammatory and anti-inflammatory genes were not significantly different in LPMs between Mac-Lpl−/− and Lplfl/fl mice (Figure 8E-8I). These results show that independent of hyperlipidemic conditions, LpL deficiency seems to shift the energy source from FA to glucose, as measured by Glut1 expression, this metabolic reprogramming does not impact M1/M2 phenotype of tissue Mϕs both in AT and the peritoneal cavity.
Figure 7. Myeloid-cell derived Lpl deficiency does not affect number of peritoneal macrophages.
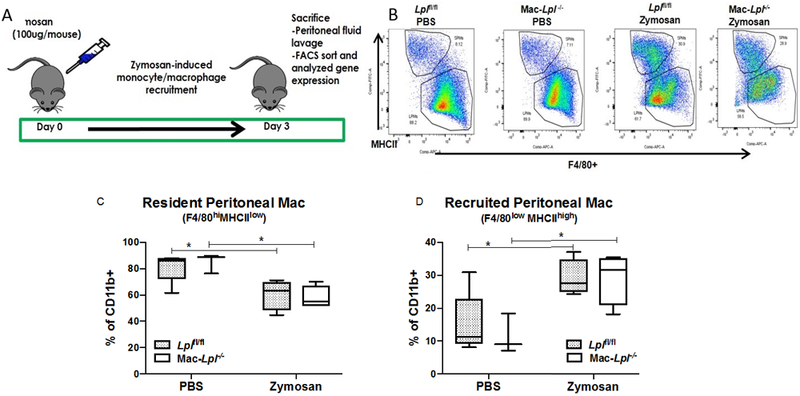
Lean littermate controls (Lplfl/fl) and Mac-Lpl−/− (Lplfl/fl;LysMCre) mice were studied for both resident peritoneal macrophages (F4/80highMHCIIlow) and recruited peritoneal macrophages (F4/80lowMHCIIhigh) under zymosan-induced peritonitis. (A) Experimental design: Zymosan (100 μg/mouse) was i.p injected into 12-week-old lean Lplfl/fl and Mac-Lpl−/−mice on day 0 to induce monocyte/macrophage recruitment to peritoneal cavity. All mice were sacrificed on day 3 for peritoneal macrophage analysis using flow cytometry. (B) Representative flow cytometry plots for peritoneal macrophages subpopulations. (C) Flow cytometry analysis of resident peritoneal macrophages. (D) Flow cytometry analysis of recruited peritoneal macrophages. N=3-5/group. *p<0.05. Results are represented as median with 25th and 75th percentiles, capped bars at 10th and 90th percentile and compared using Two-Way ANOVA with Sidak’s multiple comparison test.
Figure 8. Myeloid-cell derived Lpl deficiency increases metabolic genes, but does not profoundly affect canonical inflammatory and anti-inflammatory genes in resident peritoneal macrophages under zymosan stimuli.
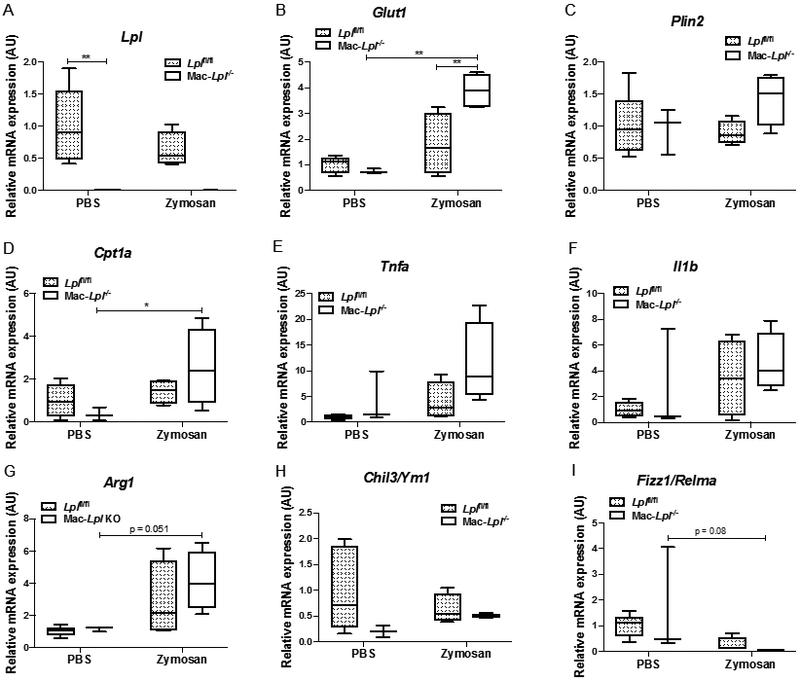
(A) The expression of Lpl in resident peritoneal macrophages (Cd45+F4/80highMHCIIlow). (B) The expression of Glut1 in resident peritoneal macrophages. (C) The expression of Plin2 in resident peritoneal macrophages. (D) The expression of Cpt1a in resident peritoneal macrophages. (E) The expression of Tnfa in resident peritoneal macrophages. (F) The expression of Il1b in resident peritoneal macrophages. (G) The expression of Arg1 in resident peritoneal macrophages. (H) The expression of Ym1 in resident peritoneal macrophages. (I) The expression of Fizz1 in resident peritoneal macrophages. N=3-5/group. *p<0.05, **p<0.01. Results are represented as median with 25th and 75th percentiles, capped bars at 10th and 90th percentile and compared using Two-Way ANOVA with Sidak’s multiple comparison test.
The effect of inducible Lpl deficiency in peritoneal Mϕs:
In contrast to Mac-Lpl−/− mice, there was more than a 60% decrease in both resident and recruited peritoneal Mϕs in iLpl−/− mice as compared to those in Lplf/fl mice (Figure 9B, 9C, 9D). The dramatic reduction in the resident population was evident even in the control (PBS treatment) group, showing that LpL is required for maintaining the tissue resident Mϕs in the cavity. These results are parallel to those found with angiopoietin-like protein 4 deficiency where greater LpL activity is associated with severe peritoneal inflammation 44. The decrease in recruited population illustrates that LpL is also important in monocyte recruitment into the peritoneal cavity upon interaction with inflammatory stimuli such as zymosan. While we were analyzing the M1/M2 gene expression profile from the resident and recruited peritoneal Mϕs, we were unable to obtain an optimum amount of RNA to measure those genes due to the low number of resident peritoneal Mϕs (LPMS). The gene expression profile from the recruited population (SPMs), however, was analyzed. We found a pattern of increase in Glut1 expression (Figure 10B) along with significant Tnfα and Il1b expression (Figure 10E) in iLpl−/− mice relative to Lplf/fl mice. When we analyzed M2 genes, Arg1 was significantly upregulated in iLpl−/− mice compared to Lplf/fl mice, but not Ym1 and Fizz1 (Figure 10G, 10H, 10I). Because we measured the gene expression in the recruited population, it is difficult to make a parallel comparison with the results from the Mac-Lpl−/− mice. However, the gene expression profile from total peritoneal Mϕs was identical to the gene expression from the recruited population (Supplemental Figure 9). These data show that LpL derived from non-myeloid cells is required for Mϕ development, recruitment, and polarization in peritoneal cavity.
Figure 9. Inducible Lpl deletion dramatically decreases both resident and recruited peritoneal macrophages content upon zymosan stimuli.
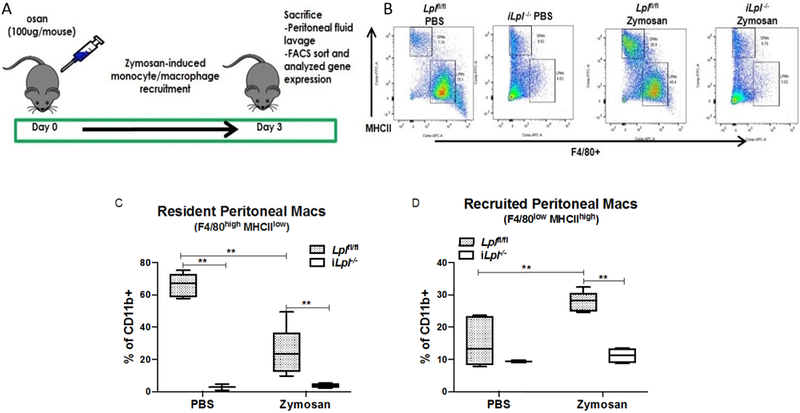
Lean littermate controls (Lplfl/fl) and iLpl−/− (Lplfl/fl;β-actin-MerCreMer) mice were studied for both resident peritoneal macrophages (F4/80highMHCIIlow) and recruited peritoneal macrophages (F4/80lowMHCIIhigh) under zymosan-induced peritonitis. (A) Experimental design: Zymosan (100 μg/mouse) was i.p injected into 12-week-old lean Lplfl/fl and Mac-Lpl−/−mice on day 0 to induce monocyte/macrophage recruitment to peritoneal cavity. All mice were sacrificed on day 3 for peritoneal macrophage analysis using flow cytometry. (B) Representative flow cytometry plots for peritoneal macrophages subpopulations. (C) Flow cytometry analysis of resident peritoneal macrophages. (D) Flow cytometry analysis of recruited peritoneal macrophages. N=3-5/group. *p<0.05. Results are represented as median with 25th and 75th percentiles, capped bars at 10th and 90th percentile and compared using Two-Way ANOVA with Sidak’s multiple comparison test.
Figure 10. Inducible Lpl ablation increases metabolic-, inflammatory-, and anti-inflammatory genes in recruited peritoneal macrophages upon zymosan stimuli.
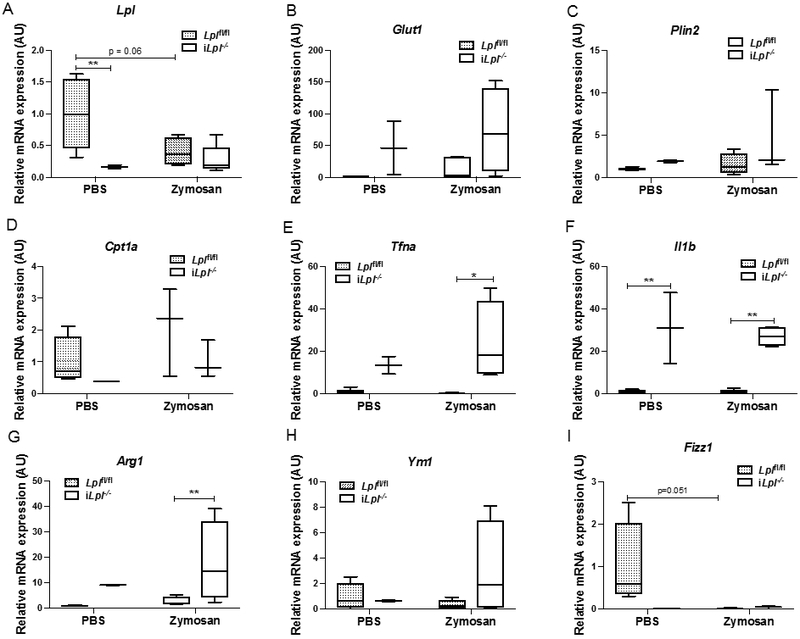
(A) The expression of Lpl in recruited peritoneal macrophages (Cd45+F4/80lowMHCIIhigh). (B) The expression of Glut1 in resident peritoneal macrophages. (C) The expression of Plin2 in resident peritoneal macrophages. (D) The expression of Cpt1a in resident peritoneal macrophages. (E) The expression of Tnfa in resident peritoneal macrophages. (F) The expression of Il1b in resident peritoneal macrophages. (G) The expression of Arg1 in resident peritoneal macrophages. (H) The expression of Ym1 in resident peritoneal macrophages. (I) The expression of Fizz1 in resident peritoneal macrophages. N=3-5/group. *p<0.05, **p<0.01. Results are represented as median with 25th and 75th percentiles, capped bars at 10th and 90th percentile and compared using Two-Way ANOVA with Sidak’s multiple comparison test.
The impact of inducible Lpl deletion on atherosclerosis plaque Mϕs in the context of regression:
We hypothesized that lack of Mϕ-derived LpL would prevent the polarization of newly recruited Mϕs to M2 and negatively affect regression. To test our hypothesis, we utilized a well-established aortic transplant methodology 28, 29 to create a plaque regression microenvironment in Lplfl/fl and iLpl−/− mice as described in Figure 11A. Two weeks after inducing regression, we isolated CD68+ Mϕs from plaques using laser capture microdissection 26, 45 and measured gene expression. Consistent with the other tissue Mϕs, Lpl deletion increased Glut1 mRNA levels (Figure 11B; p=0.08). Similar to in vivo ATMs, the expression of Plin2, Cd36, and Cpt1a genes were not altered in iLpl−/− mice. Although we found a pattern of decrease in Mcp1 mRNA level in iLpl−/− mice as compared to Lplfl/fl mice, unaltered expression of other typical inflammatory genes (Tnfa, Ilb and Nos2) was observed (Figure 11C). In contrast to our hypothesis, the anti-inflammatory genes (Mrc1, Fizz1, Il10) were not significantly decreased upon deletion of Lpl (Figure 11D). We confirmed our gene expression results for mannose receptor Mrc1/CD206, a representative M2 marker in mouse and human plaque Mϕs 4, by CD206 immunostaining in the regressing plaques. The quantified area of mannose receptor in iLpl−/− mice was comparable with Lplfl/fl mice (Figure 11E), indicating that LpL does not affect plaque Mϕ M2 polarization in regression. As expected, Lpl deficiency had no impact on regressing plaque area (Figure 11F). Of note, despite plaque area not having changed in the regression groups compared to baseline, amount of CD68+ cells were reduced, representing atherosclerosis regression (data not shown). Overall, we found that LpL is dispensable for M2 polarization in plaque Mϕs and resolution of atherosclerosis.
Figure 11. Inducible Lpl ablation does not profoundly affect plaque area and inflammatory/anti-inflammatory genes in plaque macrophages in the context of atherosclerosis regression.
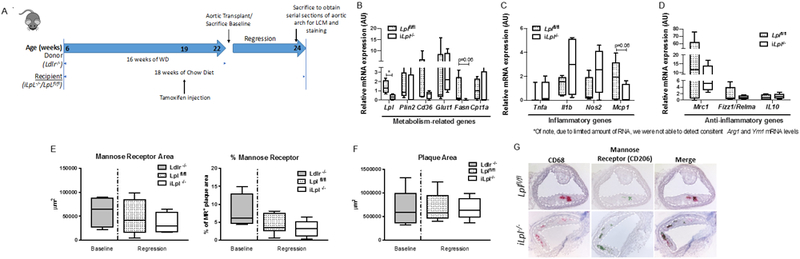
(A) Experimental design: A plaque burden aortic arch from Ldlr−/− mice was transplanted into the 22-week-old recipient mice (iLpl−/− or Lplfl/fl mice, maintained on a standard laboratory diet), inter-positioned with the abdominal aorta, and blood flow was directed through the graft. All mice were sacrificed 14 days after the aortic arch transplantation. (B) The expression of metabolism-related genes (Lpl, Glut1, Plin2, Cd36, Fsn, Cpt1a) in plaque macrophages. (C) The expression of inflammatory genes (Tnfa, Il1b, Nos2, Mcp1[Ccl2]) in plaque macrophages. (D) The expression of anti-inflammatory genes (Mrc1, Fizz1, Il10) in plaque macrophages. (E) Quantification of mannose receptor (Cd206) staining in the atherosclerotic plaque (as shown with area unit, um2, and percent of the plaque size per field). (F) Quantification of plaque area. (G) Representative images for the co-localization of CD68+ and CD206+ staining and the quantification of double-positive (CD206+CD68+) cells. N=4-7/group (baseline n=4, Lplfl/fl n=7, iLpl−/− n=7). *p<0.05. Results are represented as median with 25th and 75th percentiles, capped bars at 10th and 90th percentile and compared using unpaired t-test (Lpl, Plin2, Glut1, Cd36, Nos2, Mcp1, Fizz1/Relma, IL-10), Mann-Whitney test (Fasn, Cpta1, Mrc1), Welch’s t-test (Il1b, Tnfa), or One-way ANOVA with Tukey’s multiple comparison (E, F).
Discussion
Genetic ablation of Lpl in vitro and/or in vivo demonstrated distinct effects on Mϕ phenotype. It is widely accepted that Mϕ phenotype is associated with, and likely depends on, cellular differences in lipid and glucose metabolism 3, 46. Moreover, several in vitro studies indicate that LpL is a primary regulator of Mϕ lipid uptake 14 and therefore a modulator of Mϕ polarity 5, 19. By comparing the role of this enzyme both in vitro and in in vivo, our data challenge these assumptions. We show that deleting myeloid cell-derived LpL does not alter Mϕ polarity in obese AT, peritoneal cavity, and regressing atherosclerotic plaques. In all depots, cellular lipid metabolism does not correlate with Mϕ phenotype, and the results support a model in which tissue environment plays the central role in determining how subsets of Mϕs respond to changes in ability to produce FFAs from TGs.
Although we were able to create a TG-rich environment both in the circulation and in AT by feeding the mice a HFD, we also created a robust inflammatory microenvironment with high concentrations of glucose, insulin, and FFAs–so called metabolic chronic inflammation. In a human monocyte cell line (THP-1), LpL-mediated VLDL uptake induces both intracellular lipid accumulation and inflammation, as measured by Il1β and Tnfα mRNA expression 17, 18. However, in vivo monocytes, markers of metabolism and phenotype were unchanged in Mac-Lpl−/− mice, indicating that the inflammatory microenvironment induced by a HFD in vivo adapted to compensate for the lack of LpL in monocytes. In ATMs, local FFA concentrations are likely higher than FFAs in systemic circulation due to the close proximity to adipocyte lipolysis. Thus, our findings in ATMs in Mac-Lpl−/− mice–unaltered ATM subpopulation, lipid accumulation, and phenotype–are likely due to FFA uptake via FA transporters or the flip-flop pathway, endocytic TG-rich lipoprotein uptake, or accumulation of adipocyte-derived lipid vesicles 47. This may explain the lack of changes in inflammatory gene expression phenotype in Mac-Lpl−/− mice despite increased Glut1 mRNA expression in ATMs.
Another possible interpretation is that since LysM-Cre is effective starting from the development stage, ablation of LpL may have induced a compensatory mechanism in the early developmental stages of mice. We tested the latter hypothesis by using HFD iLpl−/− mice. We found a significant increase in levels of circulating Ly6Chi monocytes with a significant increase in lipid accumulation within Ly6Clow monocytes. Because we did not find increased LDL receptor (Ldlr), VLDL receptor (Vldlr) (data not shown), or Glut1 gene expression level, higher neutral lipid content in Ly6Clow monocytes in HFD iLpl−/− mice is perhaps due to LpL-independent lipid uptake pathway such as FFA uptake via the flip-flop pathway, as serum FFA concentration was higher in HFD iLpl−/− mice than in HFD Lplfl/fl mice (Supplementary Figure 4G). Interestingly, although levels of circulating Ly6Chi monocytes were higher in HFD iLpl−/− mice, a typical inflammatory gene, Tnfα gene expression, was lower, but anti-inflammatory genes were higher as compared to HFD Lplfl/fl mice. These results show that, as previously shown by Fisher and Pearce groups 4, 48, Ly6Chi monocytes have potential to become M2 Mϕs.
Even though inducible LpL deletion in obese mice did not affect total ATM content (F4/80+ cells), it shifted the FB:FBC ratio. Obesity is associated with FBC populations as they accumulate more neutral lipids 24, however, deleting Lpl in obese mice decreased FBCs and increased FBs. There was a pattern of a reduction in intracellular lipid content in FBCs and FBs (p=0.07) in HFD iLpl−/− mice, as compared to ATMs in HFD Lplfl/fl mice. In contrast, ATMs in HFD Mac-Lpl−/− mice and HFD Lplfl/fl mice showed no difference in intracellular lipid content. One explanation for this is that lipid accumulation in Mϕs is dependent on adipocyte-derived LpL. Another explanation is that uptake requires systemic initial lipolysis of TG-rich lipoproteins by LpL to allow their internalization by the ATMs, e.g. via lipoprotein receptors. Lipoprotein receptor uptake of nascent TG-rich lipoproteins requires their partial lipolysis to remnant or intermediate density lipoproteins to allow optimal uptake via lipoprotein receptors. Total deficiency of LpL will eliminate these particles and might be the reason that systemic rather than Mϕ specific LpL deficiency reduced Mϕ lipid droplets. Such a conclusion would contrast with in vitro studies showing Mϕ lipid accumulation from chylomicrons via LpL actions.
Although Glut1 mRNA expression was dramatically increased in ATMs in HFD iLpl−/− mice, this did not affect the M1/M2 phenotype in ATMs, contradicting the current dogma of Mϕ polarization in vitro 2. The dogma is that greater glucose utilization creates inflammatory Mϕs. Our studies do not support this association as causative, as increased Glut1 mRNA did not create a more inflammatory phenotype in LpL deficient Mϕs. Our data are consistent with the Glut1 overexpression mouse model phenotype 36, which did not show more inflammation in Mϕs. Moreover, our data support a recent finding where deletion of a key enzyme involved in FAO, carnitine palmitoyltransferase II (Cpt2), showed no effect on M2 conversion in both BMDMs and ATMs 49. Overall, our data demonstrate that myeloid cell-derived LpL is dispensable for lipid uptake and polarity, however, LpL derived from other cells/tissues are perhaps necessary for ATM lipid accumulation.
The peritoneal cavity is a unique tissue compartment in both mice and humans. In addition to its distinctive anatomical structure, the peritoneal cavity contains many types of immune cells, such as lymphocytes, Mϕs, granulocytes, and mesothelial cells 50, suggesting a complex cytokine profile in peritoneal fluid. Under normal physiological conditions, 91% of total myeloid cells (Cd11b+) are resident Mϕs (LPMs), whereas 9% of total myeloid cells (Cd11b+) are recruited Mϕs (SPMs) 51. Upon a 3-day period of zymosan stimulation, the LPM:SPM ratio shifts from 91:9 to approximately 60:40. We sought to determine the role of LpL-mediated lipid uptake in both LPMs and SPMs in basal and zymosan-stimulated states. We found an increase in Glut1 and Plin2 mRNA levels in Mac-Lpl−/− mice under zymosan-stimuli in the resident population. These changes in metabolic-related gene expression are similar to the Mac-Lpl−/− BMDMs cultured under low glucose media (Figure 1). When we measured glucose concentration in peritoneal cavity, it was low (below detection threshold of the glucometer), and TG concentrations were also lower than in the circulation (Supplemental Figure 8). Although the exact concentration of glucose in peritoneal cavity is unknown, our data suggest that the peritoneal cavity microenvironment is a low glucose milieu and Lpl deficiency in peritoneal Mϕs induces metabolic compensation. However, these changes did not profoundly affect the inflammatory or anti-inflammatory state of the cells.
Of the tissue Mϕs we studied, plaque Mϕs are perhaps the most appropriate type to evaluate in vivo the role of LpL in both lipid metabolism and polarity, as the atherosclerotic lesion microenvironment offers immediate contact between Mϕs and lipid particles. Mϕ-expression of LpL is atherogenic and associated with greater vascular inflammation 21. The role of Mϕ-derived LpL in regression, however, is yet to be defined. A recent study demonstrated that continued recruitment of inflammatory Ly6Chi monocytes and their polarization to the M2 state are required for the resolution of atherosclerotic inflammation and plaque regression 4. This finding led us to question whether LpL has an impact on M2 polarization in plaque Mϕs during plaque regression. We speculated that altering the most upstream pathway of lipid uptake by deleting LpL would affect alternative activation and resolution of atherosclerosis. However, our data show that ablation of LpL does not affect M2 conversion in plaque Mϕs and thus does not change the plaque area. In fact, Lpl deficient Mϕs from the regressing plaques had no change in the typical M2-related genes, including Mrc1 and Fizz1. This result is surprising because we observed a trend towards increased Glut1 mRNA levels in the plaque Mϕs, as we did in other in vivo Mϕs and in vitro BMDMs. These findings again indicate a metabolic shift to glucose metabolism at least at the transcriptional level, but it neither led to an expression increase of inflammatory genes nor a decrease of anti-inflammatory genes. The lack of impact on M2 conversion of plaque Mϕs is perhaps due to multiple types of Mϕs within the lesion area. Since atherosclerosis regression is a dynamic condition with constant recruitment of Ly6Chi monocytes and retention of foam cells (well known as inflammatory), the baseline of regression condition is most likely comprised of a mix of M1/M2 Mϕs, as observed in human plaques 52. Thus, deleting LpL may have affected both types of cells and led to no effect on the total plaque Mϕs phenotype. It should be noted that, although we see a significant decrease in LpL expressing macrophages in iLpl−/− recipient mice, suggesting that the majority of LpL expressing macrophages from the donor has been replaced by recruited Lpl−/− macrophages, we cannot exclude that there are LpL expressing macrophages left in the atherosclerosis lesions also affecting macrophage phenotype.
Overall, we discovered that, unlike previous in vitro studies, LpL-mediated FAs (PPARδ ligands) are not required for AA of Mϕs in vivo. Our data clearly show that monocyte/tissue Mϕ-derived LpL minimally regulates lipid accumulation in those cells in vivo and does not affect polarization. More importantly, LpL in other cells including myocytes, adipocytes, and endothelial cells 53 distinctly affects lipid accumulation and the phenotypes of circulating monocytes and ATMs. Hypertriglyceridemia caused by global Lpl deficiency increases lipid accumulation in monocytes and shifts them to a more M2 phenotype. Global, but not Mϕ-specific, Lpl deficiency in AT decreases lipid accumulation in ATMs and reduces FBC population without affecting M1/M2 polarity. Whether these findings were due to the global knockout causing changes in circulating TG-rich lipoproteins that made them reliant on Mϕ LpL or due to a requirement for adipose LpL in the Mϕ-lipid uptake process is unclear. Most likely, it suggests that multiple sources of lipids are responsible for creation of lipid-rich ATMs. Furthermore, our data support several recent and novel observations related to LpL and cellular lipid metabolism. Most importantly, a LpL-rich cluster of cells has been found in the bone marrow using single cells RNA sequencing 54. Thus, it is possible that LpL directs cellular phenotyping in ways that are exclusive of Mϕ lipid uptake. Within the AT, our data show that Mϕ LpL expression does not alter Mϕ lipid content and is not likely to be a major modulator of lipid uptake of the recently described adipose-derived exosomes 47.
In conclusion, our current findings and the previous in vivo studies demonstrate that the dogma of M1/M2 dichotomy is only applicable in certain tissue culture conditions and clearly not in vivo physiology. The in vivo tissue microenvironment is far more complicated than culture conditions because it involves dynamic interactions between different cell types, energy sources, and many other factors.
Supplementary Material
Highlights.
LpL regulates both inflammatory and anti-inflammatory gene expression in cultured macrophages.
Myeloid LpL is dispensable for lipid accumulation and macrophage polarization in vivo.
Global LpL deficiency reduces adipose macrophage lipid content and the number of induced peritoneal macrophages
The presence or absence of LpL does not affect gene expression of macrophages within regressing atherosclerotic plaques.
Acknowledgements
HRC conducted all in vivo and in vitro experiments and analyzed the data presented in Figures 1-10. She primarily directed the project and wrote the manuscript. TJ performed the experiments and the data analyses for Figure 11, and she wrote the sections describing the methods related to those results. Both HRC and TJ revised the manuscript. DS, YH, LG, and TB assisted with the in vivo mouse studies. NG, SC, JG, and SB contributed to parts of the in vivo and in vitro experiments and performed qRT-PCR and analyzed the data.
Sources of Funding
R01 HL135987, P01 HL092969, NYU Clinical & Translational Science Awards Grant 1TL1 TR001447 from NIH, AHA Predoctoral Fellowship (18PRE33990436)
Abbreviations
- Mϕ
macrophage
- AAMϕ
alternatively activated macrophage
- CAMϕ
classically activated macrophages
- FAO
fatty acid oxidation
- PPAR
peroxisome proliferator-activated receptor
- FA
fatty acid
- LpL
lipoprotein lipase
- CD36
cluster of differentiation 36
- FFA
free fatty acids
- BMDM
bone marrow-derived macrophage
- ATM
adipose tissue macrophage
- LPM
large peritoneal macrophage
- SPM
small peritoneal macrophages
- OA
oleic acid
- OCR
oxygen consumption rate
- ECAR
extracellular acidification rate
Footnotes
Disclosures
None
References
- 1.Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9:259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ganeshan K, Chawla A. Metabolic regulation of immune responses. Annu Rev Immunol. 2014;32:609–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, Gusarova V, Gromada J, Weinstock A, Moore KJ, Loke P, Fisher EA. Inflammatory ly6chi monocytes and their conversion to m2 macrophages drive atherosclerosis regression. J Clin Invest. 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A. Oxidative metabolism and pgc-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4:13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, Pearce EJ. Metabolic reprogramming mediated by the mtorc2-irf4 signaling axis is essential for macrophage alternative activation. Immunity. 2016;45:817–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, Pearce EJ, Driggers EM, Artyomov MN. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42:419–430 [DOI] [PubMed] [Google Scholar]
- 8.Trent CM, Yu S, Hu Y, Skoller N, Huggins LA, Homma S, Goldberg IJ. Lipoprotein lipase activity is required for cardiac lipid droplet production. J Lipid Res. 2014;55:645–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Arcos I, Hiyama Y, Drosatos K, Bharadwaj KG, Hu Y, Son NH, O’Byrne SM, Chang CL, Deckelbaum RJ, Takahashi M, Westerterp M, Obunike JC, Jiang H, Yagyu H, Blaner WS, Goldberg IJ. Adipose-specific lipoprotein lipase deficiency more profoundly affects brown than white fat biology. J Biol Chem. 2013;288:14046–14058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Augustus A, Yagyu H, Haemmerle G, Bensadoun A, Vikramadithyan RK, Park SY, Kim JK, Zechner R, Goldberg IJ. Cardiac-specific knock-out of lipoprotein lipase alters plasma lipoprotein triglyceride metabolism and cardiac gene expression. J Biol Chem. 2004;279:25050–25057 [DOI] [PubMed] [Google Scholar]
- 11.Obunike JC, Edwards IJ, Rumsey SC, Curtiss LK, Wagner WD, Deckelbaum RJ, Goldberg IJ. Cellular differences in lipoprotein lipase-mediated uptake of low density lipoproteins. J Biol Chem. 1994;269:13129–13135 [PubMed] [Google Scholar]
- 12.den Hartigh LJ, Connolly-Rohrbach JE, Fore S, Huser TR, Rutledge JC. Fatty acids from very low-density lipoprotein lipolysis products induce lipid droplet accumulation in human monocytes. J Immunol. 2010;184:3927–3936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saraswathi V, Hasty AH. The role of lipolysis in mediating the proinflammatory effects of very low density lipoproteins in mouse peritoneal macrophages. J Lipid Res. 2006;47:1406–1415 [DOI] [PubMed] [Google Scholar]
- 14.Chawla A, Lee CH, Barak Y, He W, Rosenfeld J, Liao D, Han J, Kang H, Evans RM. Ppardelta is a very low-density lipoprotein sensor in macrophages. Proc Natl Acad Sci U S A. 2003;100:1268–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu C, Jin X, Tsueng G, Afrasiabi C, Su AI. Biogps: Building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res. 2016;44:D313–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin B, Loike JD, Kako Y, Weinstock PH, Breslow JL, Silverstein SC, Goldberg IJ. Lipoprotein lipase regulates fc receptor-mediated phagocytosis by macrophages maintained in glucose-deficient medium. J Clin Invest. 1997;100:649–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.den Hartigh LJ, Altman R, Norman JE, Rutledge JC. Postprandial vldl lipolysis products increase monocyte adhesion and lipid droplet formation via activation of erk2 and nfkappab. Am J Physiol Heart Circ Physiol. 2014;306:H109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bojic LA, Sawyez CG, Telford DE, Edwards JY, Hegele RA, Huff MW. Activation of peroxisome proliferator-activated receptor delta inhibits human macrophage foam cell formation and the inflammatory response induced by very low-density lipoprotein. Arterioscler Thromb Vasc Biol. 2012;32:2919–2928 [DOI] [PubMed] [Google Scholar]
- 19.Lee CH, Kang K, Mehl IR, Nofsinger R, Alaynick WA, Chong LW, Rosenfeld JM, Evans RM. Peroxisome proliferator-activated receptor delta promotes very low-density lipoprotein-derived fatty acid catabolism in the macrophage. Proc Natl Acad Sci U S A. 2006;103:2434–2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barish GD, Atkins AR, Downes M, Olson P, Chong LW, Nelson M, Zou Y, Hwang H, Kang H, Curtiss L, Evans RM, Lee CH. Ppardelta regulates multiple proinflammatory pathways to suppress atherosclerosis. Proc Natl Acad Sci U S A. 2008;105:4271–4276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi M, Yagyu H, Tazoe F, Nagashima S, Ohshiro T, Okada K, Osuga J, Goldberg IJ, Ishibashi S. Macrophage lipoprotein lipase modulates the development of atherosclerosis but not adiposity. J Lipid Res. 2013;54:1124–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bharadwaj KG, Hiyama Y, Hu Y, Huggins LA, Ramakrishnan R, Abumrad NA, Shulman GI, Blaner WS, Goldberg IJ. Chylomicron- and vldl-derived lipids enter the heart through different pathways: In vivo evidence for receptor- and non-receptor-mediated fatty acid uptake. J Biol Chem. 2010;285:37976–37986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, Ramkhelawon B, Distel E, Westerterp M, Huang LS, Schmidt AM, Orchard TJ, Fisher EA, Tall AR, Goldberg IJ. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013;17:695–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW Jr. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 2013;18:816–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell. 2014;157:832–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trogan E, Choudhury RP, Dansky HM, Rong JX, Breslow JL, Fisher EA. Laser capture microdissection analysis of gene expression in macrophages from atherosclerotic lesions of apolipoprotein e-deficient mice. Proc Natl Acad Sci U S A. 2002;99:2234–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trogan E, Fayad ZA, Itskovich VV, Aguinaldo JG, Mani V, Fallon JT, Chereshnev I, Fisher EA. Serial studies of mouse atherosclerosis by in vivo magnetic resonance imaging detect lesion regression after correction of dyslipidemia. Arterioscler Thromb Vasc Biol. 2004;24:1714–1719 [DOI] [PubMed] [Google Scholar]
- 28.Reis ED, Li J, Fayad ZA, Rong JX, Hansoty D, Aguinaldo JG, Fallon JT, Fisher EA. Dramatic remodeling of advanced atherosclerotic plaques of the apolipoprotein e-deficient mouse in a novel transplantation model. J Vasc Surg. 2001;34:541–547 [DOI] [PubMed] [Google Scholar]
- 29.Chereshnev I, Trogan E, Omerhodzic S, Itskovich V, Aguinaldo JG, Fayad ZA, Fisher EA, Reis ED. Mouse model of heterotopic aortic arch transplantation. J Surg Res. 2003;111:171–176 [DOI] [PubMed] [Google Scholar]
- 30.Daugherty A, Tall AR, Daemen M, Falk E, Fisher EA, Garcia-Cardena G, Lusis AJ, Owens AP 3rd, Rosenfeld ME, Virmani R, American Heart Association Council on Arteriosclerosis T, Vascular B, Council on Basic Cardiovascular S. Recommendation on design, execution, and reporting of animal atherosclerosis studies: A scientific statement from the american heart association. Arterioscler Thromb Vasc Biol. 2017;37:e131–e157 [DOI] [PubMed] [Google Scholar]
- 31.Nunnari JJ, Zand T, Joris I, Majno G. Quantitation of oil red o staining of the aorta in hypercholesterolemic rats. Exp Mol Pathol. 1989;51:1–8 [DOI] [PubMed] [Google Scholar]
- 32.Feng B, Zhang D, Kuriakose G, Devlin CM, Kockx M, Tabas I. Niemann-pick c heterozygosity confers resistance to lesional necrosis and macrophage apoptosis in murine atherosclerosis. Proc Natl Acad Sci U S A. 2003;100:10423–10428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Camell C, Smith CW. Dietary oleic acid increases m2 macrophages in the mesenteric adipose tissue. PloS one. 2013;8:e75147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pardo V, Gonzalez-Rodriguez A, Guijas C, Balsinde J, Valverde AM. Opposite cross-talk by oleate and palmitate on insulin signaling in hepatocytes through macrophage activation. J Biol Chem. 2015;290:11663–11677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, Subramanian V, Mukundan L, Ferrante AW, Chawla A. Alternative m2 activation of kupffer cells by ppardelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7:496–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishizawa T, Kanter JE, Kramer F, Barnhart S, Shen X, Vivekanandan-Giri A, Wall VZ, Kowitz J, Devaraj S, O’Brien KD, Pennathur S, Tang J, Miyaoka RS, Raines EW, Bornfeldt KE. Testing the role of myeloid cell glucose flux in inflammation and atherosclerosis. Cell Rep. 2014;7:356–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, Dragoljevic D, Hong ES, Abdel-Latif A, Smyth SS, Choi SH, Korner J, Bornfeldt KE, Fisher EA, Dixit VD, Tall AR, Goldberg IJ, Murphy AJ. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 2014;19:821–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang CL, Garcia-Arcos I, Nyren R, Olivecrona G, Kim JY, Hu Y, Agrawal RR, Murphy AJ, Goldberg IJ, Deckelbaum RJ. Lipoprotein lipase deficiency impairs bone marrow myelopoiesis and reduces circulating monocyte levels. Arterioscler Thromb Vasc Biol. 2018;38:509–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aouadi M, Vangala P, Yawe JC, Tencerova M, Nicoloro SM, Cohen JL, Shen Y, Czech MP. Lipid storage by adipose tissue macrophages regulates systemic glucose tolerance. Am J Physiol Endocrinol Metab. 2014;307:E374–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muoio DM, MacLean PS, Lang DB, Li S, Houmard JA, Way JM, Winegar DA, Corton JC, Dohm GL, Kraus WE. Fatty acid homeostasis and induction of lipid regulatory genes in skeletal muscles of peroxisome proliferator-activated receptor (ppar) alpha knock-out mice. Evidence for compensatory regulation by ppar delta. J Biol Chem. 2002;277:26089–26097 [DOI] [PubMed] [Google Scholar]
- 42.Gordts PL, Nock R, Son NH, Ramms B, Lew I, Gonzales JC, Thacker BE, Basu D, Lee RG, Mullick AE, Graham MJ, Goldberg IJ, Crooke RM, Witztum JL, Esko JD. Apoc-iii inhibits clearance of triglyceride-rich lipoproteins through ldl family receptors. J Clin Invest. 2016;126:2855–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merkel M, Weinstock PH, Chajek-Shaul T, Radner H, Yin B, Breslow JL, Goldberg IJ. Lipoprotein lipase expression exclusively in liver. A mouse model for metabolism in the neonatal period and during cachexia. J Clin Invest. 1998;102:893–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lichtenstein L, Mattijssen F, de Wit NJ, Georgiadi A, Hooiveld GJ, van der Meer R, He Y, Qi L, Koster A, Tamsma JT, Tan NS, Muller M, Kersten S. Angptl4 protects against severe proinflammatory effects of saturated fat by inhibiting fatty acid uptake into mesenteric lymph node macrophages. Cell Metab. 2010;12:580–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feig JE, Fisher EA. Laser capture microdissection for analysis of macrophage gene expression from atherosclerotic lesions. Methods Mol Biol. 2013;1027:123–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gordon S, Martinez FO. Alternative activation of macrophages: Mechanism and functions. Immunity. 2010;32:593–604 [DOI] [PubMed] [Google Scholar]
- 47.Flaherty SE 3rd, Grijalva A, Xu X, Ables E, Nomani A, Ferrante AW Jr. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science. 2019;363:989–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nascimento M, Huang SC, Smith A, Everts B, Lam W, Bassity E, Gautier EL, Randolph GJ, Pearce EJ. Ly6chi monocyte recruitment is responsible for th2 associated host-protective macrophage accumulation in liver inflammation due to schistosomiasis. PLoS Pathog. 2014;10:e1004282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gonzalez-Hurtado E, Lee J, Choi J, Selen Alpergin ES, Collins SL, Horton MR, Wolfgang MJ. Loss of macrophage fatty acid oxidation does not potentiate systemic metabolic dysfunction. Am J Physiol Endocrinol Metab. 2017;312:E381–E393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kitayama J, Emoto S, Yamaguchi H, Ishigami H, Watanabe T. Cd90+ mesothelial-like cells in peritoneal fluid promote peritoneal metastasis by forming a tumor permissive microenvironment. PloS one. 2014;9:e86516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghosn EE, Cassado AA, Govoni GR, Fukuhara T, Yang Y, Monack DM, Bortoluci KR, Almeida SR, Herzenberg LA, Herzenberg LA. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc Natl Acad Sci U S A. 2010;107:2568–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015;12:10–17 [DOI] [PubMed] [Google Scholar]
- 53.Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: Lipoprotein lipase- and cd36-mediated pathways. J Lipid Res. 2009;50 Suppl:S86–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Dominguez A, Pinho S, Akhmetzyanova I, Gao J, Witkowski M, Guillamot M, Gutkin MC, Zhang Y, Marier C, Diefenbach C, Kousteni S, Heguy A, Zhong H, Fooksman DR, Butler JM, Economides A, Frenette PS, Adams RH, Satija R, Tsirigos A, Aifantis I. The bone marrow microenvironment at single-cell resolution. Nature. 2019;569:222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.