

Abstract
Delivery to the proper tissue compartment is a major obstacle hampering the potential of cellular therapeutics for medical conditions. Delivery of cells within biomaterials may improve localization, but traditional and newer void-forming hydrogels must be made in advance with cells being added into the scaffold during the manufacturing process. We recently developed injectable, in situ cross-linking microporous scaffolds that demonstrated a remarkable ability to provide a matrix for cellular proliferation and growth 3-dimensionally in vitro. The ability of these scaffolds to deliver cells in vivo is currently unknown. Herein, we show that mesenchymal stem cells (MSCs) can be co-injected locally with microparticle scaffolds assembled in situ immediately following injection. MSC delivery within a microporous scaffold enhanced MSC retention subcutaneously when compared to cell delivery alone or delivery within traditional in situ cross-linked nanoporous hydrogels. After two weeks, endothelial cells forming blood vessels were recruited to the scaffold and cells retaining the MSC marker CD29 remained viable within the scaffold. These findings highlight the utility of this approach at achieving localized delivery of stem cells through an injectable porous matrix while limiting obstacles of introducing cells within the scaffold manufacturing process.
Keywords: stem cell delivery, injectable microporous scaffolds, hydrogel microparticles, microfluidics, tissue engineering
Graphical Abstarct
Void-forming hydrogels for cell delivery must be made in advance with cells being added into the scaffold during the manufacturing process. Here, mesenchymal stem cells are co-injected locally with microparticle scaffolds that assembled in situ immediately following injection. This approach enhanced MSC retention subcutaneously when compared to cell delivery alone or delivery within traditional in situ cross-linked nanoporous hydrogels.
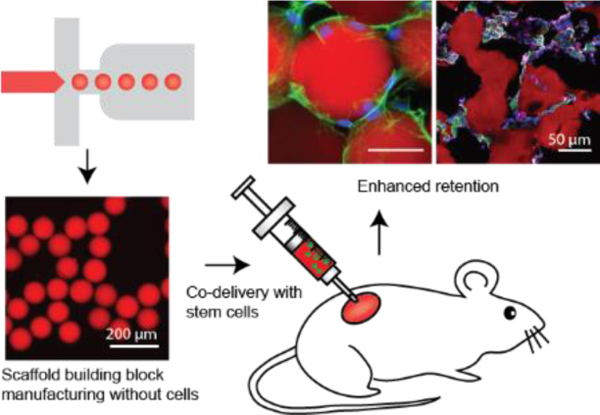
Introduction
Stem cell therapies hold promise for many intractable diseases through a variety of mechanisms. These mechanisms include the promotion of tissue repair, replacement of mutated or missing factors, and modulation of the immune system [1–4]. Mesenchymal stem cells (MSCs) represent a particularly attractive stem cell therapeutic candidate as they are multipotent cells with the capacity to migrate to injured or inflamed organs to induce tissue repair through secreted factors[5] and differentiate into functional tissue[6]. However, clinical translation has been challenging due to poor homing, survival, and engraftment of transplanted stem cells to a disease site [7,8]. Moreover, loss of control by the engrafted cells in a disrupted biological environment limits the ability to harness the stem cells for meaningful therapeutic outcomes [5,9].
Localized delivery of stem cells using biomaterials can mitigate these issues[10–13]. An ideal material should not only enable minimally invasive delivery by injection, and retain cells after transplantation to achieve sustained function, but also create an artificial stem cell niche in situ for higher efficacy and longer maintenance of the therapy [14,15]. Specifically, the biomaterial should provide suitable biophysical and biochemical microenvironmental cues for enhanced control of cell function in vivo [16,17]. However, current injectable biomaterials suffer from ineffective modulation or lack of porosity for mass transport, cell motility, proliferation, cell-cell adhesion, or new tissue formation [14,15,18].
To this end, void forming hydrogels were created to facilitate expansion of cells within the hydrogel coupled with the degradation of porogens in the hydrogel matrix [19]. While these hydrogels display tremendous promise, a major limitation of this approach is that cells need to be supplied at the time of manufacture of the hydrogel, which represents a significant manufacturing and application hurdle as cells need to survive from the time of manufacture to the time delivery. Acute medical conditions that require immediate therapeutics may be less amenable to this approach. Approaches where therapeutics can be mixed with the delivery vehicle immediately before application to form a scaffold in situ within tissue may overcome the manufacturing limitation.
We originally created microporous annealed particle (MAP) scaffolds as a way to combine microporosity and injectability which showed a tremendous potential to heal wounds[20]. We and others with similar technology have shown that in vitro, cells use these microporous scaffolds to expand and proliferate in three dimensions around the microparticles [20–25]. These findings suggested that cells can be mixed with hydrogel immediately before delivery, and this can be used as a cellular delivery platform that can overcome some of the limitations of current hydrogel technologies.
Here, we present data demonstrating delivery of stem cells in a microporous, injectable scaffold enhances their retention in tissue, without requiring the integration of the stem cells during the microparticle building block manufacturing process (Figure 1A). When subcutaneously implanted without cells, we obtained additional evidence that subcutaneous delivery of MAP scaffolds by themselves resulted in tremendous cellular integration, new blood vessel formation, and an absence of foreign body formation around the implant, and collagen deposition and vascularization deep into the scaffold volume. Seeding MAP scaffolds with mesenchymal stem cells (MSCs) immediately before subcutaneous delivery results in enhanced maintenance of MSCs when compared to MSCs delivered in PBS or non-porous hydrogels and allows for MSCs to quickly migrate, adhere, and proliferate, leading to enhanced maintenance of transplanted cells in vivo. Moreover, we show that the material properties can be tuned to promote the maintenance of the stem cell population while integrating with surrounding tissues through vascularization. We anticipate that this approach can be easily translated and generally applied to delivery of other treatment-appropriate therapeutic cells, given cell production can be independent of biomaterial production, and molecularly and biophysically tailored niches can be created.
Figure 1. Mesenchymal stem cell (MSC) delivery and in situ niche creation strategy using annealed monodisperse hydrogel particles.

(A) An artificial stem cell niche is created in situ by annealing a suspension of injectable monodisperse hydrogel particles. Highly monodisperse particle building blocks enable the generation of a pore network in a highly controllable manner, which promotes the transport of oxygen and nutrients as well as cell migration. The biophysical properties of building blocks are modulated to enhance the functions of the transplanted MSCs. (B) Monodisperse hydrogel particles or µgels produced by the microfluidic device. Scale bar: 200 µm. (C) Tissue scaffold assembled from monodisperse µgels. Scale bar: 200 µm. (D) Diffusivity of 70 kDa dextran and 0.3 kDa FITC in non-porous scaffolds (N), MAP scaffolds (M) and PBS (n = 4 – 7). Data are presented as average ± s.d. Statistical significance based on one-way ANOVA followed by Tukey’s HSD post hoc test (significance compared to N, ****p < 0.0001). (E) Hydraulic conductivity of PBS through the non-porous scaffold (N) and MAP scaffold (M) at atmospheric pressure (n = 3). Data are presented as average ± s.d. Statistical significance based on standard two-tailed Student t-test (****p < 0.0001). (F) Fluorescent images of MSCs in vitro cultured in microporous scaffolds and non-porous scaffolds at week 2. Blue, nucleus; Green, actin; Red, gel. Scale bar: 50 µm.
Results
Enhanced mass transport through a microporous network
We hypothesized that covalently-linked assemblies of monodisperse hydrogel particles would produce an interconnected pore space beneficial for the transport of oxygen and nutrients and may overcome diffusion limitations of conventional non-porous hydrogels. To test whether this is indeed the case, highly-monodisperse (CV < 5%) microscale hydrogel particles were generated using a microfluidic approach (Figure S1). These particles were enzymatically annealed in vitro to generate MAP scaffolds (M) (Figure 1B, C). To assess transport by diffusion, we performed fluorescence recovery after photobleaching (FRAP) using fluorescein (0.3 kDa) and dextran-conjugated fluorescein (70 kDa) (Figure 1D and Figure S2). The fluorescent intensity recovery was on the order of tens of seconds in MAP scaffolds, resulting in a calculated diffusivity for 70 kDa dextran approximately 50% of that in PBS, while no diffusion was detected into the hydrogel particles in the same timeframe. Given the relatively large hydrodynamic diameter of 70 kDa dextran that is comparable to the nanoscale pores in the gel mesh network, steric hindrance severely limits diffusion within nanoporous hydrogels [26,27]. In fact, the observation of diffusion in macroscale gels revealed that the diffusivity was 50-fold lower than that in PBS (0.42 µm2/s), in agreement with previous reports [28]. For 0.3kDa fluorescein molecules, MAP scaffolds again demonstrated a significantly enhanced diffusivity compared to chemically-matched non-porous gels (Figure 1D).
Higher convective flux of fluid also resulted from the interconnected pore network of the scaffolds. To measure hydraulic conductivity, non-porous scaffolds and MAP scaffolds were placed on top of a membrane with 5µm pores in a custom-designed device which allowed for precise gravity-driven flow (Figure S3). While only limited permeation was observed through the non-porous scaffold resulting in a conductivity of ~1.6 × 10−3 µm/s at atmospheric pressure, the interconnected porosity of MAP scaffolds yielded ~600-fold enhancement of the conductivity (~1 µm/s) (Figure 1E), which was comparable to physiologic convection in the extracellular fluid [29]. This not only indicates that the pores are interconnected throughout the scaffold, but also suggests that nutrients and waste can be transported by convection [30], which may be beneficial for cell survival and proliferation in a macroscale biomaterial formed from MAP gel in vitro and in vivo.
New tissue formation within subcutaneously acellularly-delivered microporous scaffolds
As a proof of concept as to whether microporous scaffold implants can support stem cells in vivo, we first assessed whether scaffolds subcutaneously injected result in a foreign body response that would limit nutrient delivery to cells within the scaffold (Figure S4). To our surprise, not only did the MAP scaffolds display no foreign body response (Figure S4B), they supported the ingrowth of both PECAM (CD31) expressing blood vessels (Figure S4C), and collagen I and III expressing fibroblasts (Figure S4B) through the majority of the scaffold. By contrast, chemically-identical traditional (i.e. nanoporous) hydrogels elicited a significant collagen I-rich fibrotic encapsulation without significant integration within the scaffold (Figure S4A). We considered the findings a positive indication that MAP scaffolds would support nutrient delivery to co-injected stem cells in vivo leading to enhanced survival and proceeded to test this hypothesis.
Enhanced MSC proliferation and survival in microporous scaffolds in vitro and in vivo
Given the increased potential for enhanced nutrient transport and tissue formation in MAP scaffolds, we next tested whether MAP scaffolds do indeed support cell survival. MSCs incorporated in MAP scaffolds showed highly-interconnected and spread morphology through the void spaces between hydrogel building blocks (Figure 1F and 2A). On the other hand, MSCs in non-porous gels possessed a round morphology with limited spreading and connection between cells (Figure 1F and 2A). Although cells can degrade the hydrogel matrix locally to infiltrate, the time course for degradation prevents cells from migrating to occupy the space throughout the scaffold. In fact, multiple cells were confined to a small area (Figure 1F inset).
Figure 2. Controlled microporosity generated by MAP scaffolds facilitates the highest proliferation in vitro and retention in vivo.

(A) Fluorescent images of MSCs growing in MAP scaffolds (M), and non-porous scaffolds (N) following two weeks of in vitro culture. Corresponding heat map of nucleus density in the fields of view. Darker red color indicates a region with a higher number of nuclei. (Blue, nucleus; Green, actin; Red, gel). Scale bar: 200 µm. (B) Cell proliferation of fluorescently transfected MSCs measured by increase in fluorescence intensity over time (n = 4). (C) Histograms of nucleus density for five scaffold conditions (n = 4 scaffolds per condition). The red dashed line indicates the threshold for no nuclei in a region. (D) Representative fluorescence IVIS images of MSCs producing RFP that were subcutaneously injected into C57BL/6 mice with MAP scaffold (M), non-porous scaffold (N) and PBS at 0, 2, 5, 7, 10, and 14 days post-implantation. (E) Integrated fluorescent intensity at each time point (n = 6–11). (F) Comparison of cell retention at day 7 relative to day 0. (G) Comparison of cell area at day 7 relative to day 0. Cell area was defined as an area with radiant efficiency higher than 2×107. Each point represents an individual mouse. All data are presented as average ± s.d. Statistical significance based on one-way ANOVA followed by Tukey’s HSD post hoc test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001); n.s. indicates not significant.
The ability to spread and migrate throughout the MAP scaffold led to more rapid proliferation, with increased void volume in MAP scaffolds leading to significantly higher proliferation rate. To correlate the microscale pore network with cell growth and proliferation, the intensity of red fluorescent protein (RFP) produced by RFP-transfected MSCs (RFP-MSCs) was measured for each scaffold over a two-week period. While the expansion of cells incorporated in non-porous gels yielded only a 2.3-fold increase, the expansion of RFP-MSCs in MAP scaffolds yielded a 17-fold increase (Figure 2B). Enhanced proliferation was confirmed through the analysis of nucleus density, in which much lower number of cell-free regions were observed in MAP scaffolds (Figure 2C).
Building off of these in vitro results and acellular MAP scaffold support of robust endogenous cell ingrowth in vivo (Figure S4), we hypothesized that MAP scaffolds would enhance the retention of MSCs in tissue when compared with PBS or non-porous scaffolds in vivo (Figure 2D, E). MSCs expressing RFP (RFP-MSCs) were injected subcutaneously in C57BL/6 mice, an immunocompetent mouse strain, to recapitulate MSC survival in the presence of a functional immune system. The fluorescent intensity was measured over a two-week period. RFP fluorescence intensity remained the highest for cells co-delivered in MAP scaffolds compared to PBS and non-porous scaffolds at the end of two weeks (Figure 2F). Likewise, the cell area, which was defined by the area above a radiant efficiency of 2×107, was also significantly higher in MAP scaffolds (Figure 2G). The half-life of MSCs for each case calculated from the RFP intensity decay over time was 6.13 days for MAP scaffolds, 2.35 days for non-porous gel and 1.91 days for PBS. Combined, these results support that the formation of a microporous scaffold in situ promotes cell proliferation and survival in vivo, perhaps due to enhanced transport, cell distribution and connectivity throughout the scaffold, as identified in vitro.
Physical and chemical properties of MAP scaffolds modulate MSC behavior
Since the in vivo fluorescence studies suggest that MAP scaffolds improved the retention of the subcutaneously delivered MSCs, we next wished to confirm the retention of MSCs within the MAP hydrogel. We also wished to test whether MSC retention in the injected scaffold could be improved through modulation of material properties of the building blocks, such as degradability, stiffness, and cell-binding motif amount, as these parameters were shown to play a critical role in cell delivery with other biomaterials [31–33]. To arrive at a final set of microgel parameters for in vivo experiments, different weight percent, stoichiometry, crosslinker types and cell binding motif (RGD) concentrations were screened (Figure 3A, Table S1). Two soft building blocks were designed to have storage moduli of 500 Pa with enzymatically-degradable (SoD1; corresponding to the same/original formulation of MAP hydrogel used in Figure 2) and non-degradable (SoN1) crosslinkers and a standard RGD concentration (0.5 mM) to isolate the effect of MMP-triggered degradability. Stiff building blocks were designed using the non-degradable formulation, decoupling degradability from stiffness. We avoided simultaneous modulation of stiffness with MMP-degradable crosslinkers since we observed that stiffness and degradability were difficult to independently control. For example, doubling the crosslinking concentration of degradable crosslinkers resulted in higher stiffness but also resulted in a significant loss of degradability (Figure S5A). In fact, the degree of cell spreading through local degradation is reduced for cells encapsulated in 10wt% degradable gel (Figure S5B), which contrasts with increasing spread cell morphology on a stiffer 2D substrate [34]. In our system, the crosslinking density of the stiffer microgel building blocks was increased to achieve a storage modulus of 2,500 Pa while maintaining RGD concentration (StN1) such that the effect of stiffness could be investigated independently from degradability or adhesive ligand concentration. Importantly, due to in situ assembly of cells into micropores formed within the annealed MAP scaffolds, increasing the stiffness of hydrogel building blocks did not result in changes in confinement of cells. To investigate the effect of cell binding motif concentration, the RGD concentration within the stiff non-enzymatically-degradable hydrogel formulation was increased five-fold (StN5), without changing the storage modulus (Figure S6A). Although these four distinctive compositions (SoN1, StN1, StN5, SoD1; Figure 3A) had different swelling ratios in an aqueous buffer, we used microfluidic droplet generation to tune the pre-swollen building block size, such that each microgel type was similar in size after swelling (Figure S6B). This was important to preserve the microporous structure of the MAP scaffolds and decouple potential effects of material properties from microporosity. By manufacturing microgel particles with well-controlled material properties we could create MAP scaffolds with orthogonally-controlled properties to study the effect of microenvironmental cues on stem cells.
Figure 3. Modulating MAP scaffold material properties affect MSC function.

(A) Four MAP gel formulations manufactured to study the effect of degradability, stiffness and binding motif concentration. (B) Relative cell expansion in vitro at 7 days (n = 5). (C) Representative fluorescent images of MSC growth in vitro following two weeks (Blue, nucleus; Green, actin). Scale bar: 50 µm. (D) Average actin area normalized by the number of cells for in vitro culture (n = 5). (E) Representative fluorescent images of GFP-producing MSCs in MAP scaffolds subcutaneously injected into C57BL/6 mice following two weeks (Red, gel; Blue, nucleus; Green, GFP; Magenta, CD29). Scale bar: 50 µm. (F) CD29+ cells per area in scaffolds at week 2 (n = 5). (G) Ratio of CD29+ cells to all cells in the scaffolds at week 2 (n = 5). Each dot in the plots represents an individual mouse. All data are presented as average ± s.d. Statistical significance based on one-way ANOVA followed by Tukey’s HSD post hoc test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Indeed, all three material properties tested affected in vitro MSC proliferation (Figure 3B). SoD1 showed a higher expansion rate than SoN1, indicating that material degradability plays a role when material stiffness is low. Stiff gel building blocks also led to higher proliferation, which is in agreement with previous work that indicated that MSCs spread more and proliferate more readily on stiffer 2D substrates [24,35]. MSC expansion was further enhanced on these stiff scaffolds by incorporating higher RGD concentration. For the enzymatically-non-degradable gel conditions, more RGD sites on the surface may be beneficial to promote growth as cells have more difficulty in degrading and revealing new RGD binding sites. In fact, StN5 was observed to promote slightly larger actin spread area per cell than StN1, indicating that the high RGD enhanced binding of cells to the substrate and spreading (Figure 3C, D).
To investigate the in vivo response to the four types of MAP scaffolds, MSCs expressing GFP (GFP-MSCs) were subcutaneously injected in C57BL/6 mice along with the different hydrogel formulations and scaffolds were excised two weeks after implantation. Since MSCs expresses the stem cell marker CD29 [36,37], we examined whether GFP-MSCs retaining CD29, and any recruited myeloid cells were present within the different MAP scaffolds. In all MAP scaffolds, injected MSCs were identified by the colocalization of CD29 and GFP (Figure 3E) and negative staining for CD11b. The number of cells with CD29 staining in the scaffold were counted to calculate the density and fraction of cells retaining stem cell markers (Figure S7). The density of CD29+ cells was the highest in StN5 scaffolds (Figure 3F), corresponding to the results of our in vitro cell proliferation experiments. The lower in vivo preservation of MSCs in SoD1, degradable gels, than in vitro may be due to the degradable formulation losing physical integrity more rapidly once implanted and exposed to proteases. Stiffer gels also resulted in CD29+ cells occupying a larger fraction of the total cells present in scaffolds (Figure 3G). For the highest performing scaffold, StN5, we also observed GFP-producing MSCs in tissue cross-sections after subcutaneous implantation for a longer time period of 56 days (Figure S8). Among cells extensively occupying the scaffold, GFP-positive cells that survived for two months were identified. Over this two month period, the scaffold also maintained an interconnected porous structure without significant degradation. The intensity of fluorophores conjugated to the gels did not significantly change from day 14 to day 56 (Figure S9), indicating that the gel matrix or fluorophores were not degraded. Therefore, the scaffold is expected to maintain its physical properties for 56 days. Overall, the stiffer gels with higher RGD (StN5) retained the implanted stem cell population at higher levels compared to other formulations, and cells and scaffold were both maintained up to 56 days after implantation.
Tissue ingrowth and vascularization into the MSC-containing MAP scaffolds
Microporosity of the implanted MSC-containing MAP scaffolds also induced cell migration, in-growth of host tissue, and vascularization important for improved clinical function. The integration of the MAP scaffolds was evaluated by the number of cells near the boundary between the surrounding tissue and MAP scaffold (Figure 4A, B). The density of cells within a 200 µm region outside of and neighboring the scaffold was about 1,580 cells/mm2. Within a region ~100 µm deep into the scaffold boundary, the cell density was similar to the surrounding tissue, indicating that all four types of MAP scaffolds integrated well with the tissue. Cell density gradually decreased in all MAP scaffolds up to a distance of 1.5 mm, with SoN1 showing the lowest cell density in the scaffold core. The other formulations remained cellular (~ 500 cells/mm2) throughout the scaffold, even at depths of 1.5 mm from the implant interface. This value is above the initial seeding density (~ 125 cells/mm2 given that the tissue sectioning was ~25 µm in thickness). Since MSC retention cannot account for the increase in cell numbers and MSCs produce factors that recruit other cell types [38], this indicates that MAP scaffolds likely induced the migration of endogenous cells deep into the scaffold and maintained these cells over weeks overcoming transport limitations of conventional hydrogels. The SoD1 formulation resulted in a greater number of cells in the periphery of the scaffold, suggesting more cell migration and tissue ingrowth into the scaffold corresponding to scaffold degradation over time.
Figure 4. Tissue ingrowth and vascularization into microparticle scaffolds.

(A) Representative images of tissue slices following two weeks of implantation of MSCs with corresponding building block materials. The dotted line indicates a boundary with the surrounding tissue. Scale bar: 200 µm. (B) Number of cells per area as a function of the distance from the interface between the MAP scaffold and the tissue (n = 5). Data presented as average ± s.e.m. Statistical significance performed using one-way ANOVA with a Dunnett post hoc multiple comparison test (*p < 0.05). (C) Representative images of platelet endothelial cell adhesion molecule (PECAM-1) and GFP immunostaining of the scaffolds following two weeks indicating the presence of implanted GFP-MSCs and endothelial cells growing in from surrounding tissue. Scale bar: 50 µm. (D) CD31+ cells per area in scaffolds at week 2 (n = 5). (E) Ratio of CD31+ cells to all cells in the scaffolds at week 2 (n = 5). Each dot in the plots in (D), (E) represents an individual mouse. Data are presented as average ± s.d. Statistical significance based on one-way ANOVA followed by Tukey’s HSD post hoc test (*p < 0.05, **p < 0.01).
Next, we examined the recruitment of specific cell types into the different formulations of MAP hydrogel. Since MSCs produce factors that actively recruit myeloid cells [39], we examined whether modulating different hydrogel parameters resulted in a differential ability for MSCs to attract myeloid cells. Indeed, MSCs implanted in all of the hydrogel scaffolds recruited CD11b myeloid cells, without significant differences between the different formulations (Figure S10). While biomaterial-induced immune responses often lead to foreign body encapsulation [40], we examined the location of the CD11b+ cells and whether they were forming multinucleated/fused cells consistent with foreign body encapsulation. While the majority of the CD11b+ myeloid cells were present within the peripheral aspects of the hydrogel, neither multinucleation nor encapsulation were evident to any of the formulations. These findings suggest that the scaffold-MSC implants were similarly able to recruit myeloid cells, without resulting in a foreign body response that often results in material rejection.
Since we observed blood vessel structures within subcutaneously implanted MAP scaffolds without MSCs (Fig S4), we next wished to evaluate whether different formulations of MAP scaffolds with MSCs resulted in differential formation of blood vessels by examining the endothelial cell markers (PECAM-1). Indeed, we found cells expressing PECAM-1within all four types of MAP scaffolds (Figure 4C). There were significantly higher numbers of PECAM-1-expressing cells within StN1, StN5 and SoD1 scaffolds compared to the SoN1 scaffolds (Figure 4D, E), which may correspond to the ability of these same scaffolds to retain the injected CD29+ cells at higher levels as MSCs present in the scaffold may secrete angiogenic factors such as vascular endothelial growth factor (VEGF)[41,42].
Discussion
The application of stem cell-based therapies has been limited by low retention rate of transplanted cells and lack of efficient control of the cell state and function. Biomaterial-enhanced delivery of stem cell therapies offers a potential solution to overcome this, but non-porous or nanoporous materials do not allow for sufficient incorporation of cells into the hydrogel, requiring significant degradation for cell proliferation and migration, and yielding low transport of nutrients which may limit incorporated cell survival. Void forming hydrogels offer an advantage, as they provide a scaffold that can be rapidly degraded while cells proliferate and expand. However, these hydrogels require the cellular therapeutics to be populated into the scaffold during manufacture, representing a major hurdle to their use as this could affect stem cell function, viability, and long-term storage, requiring great care during manufacture and deployment [19], ultimately resulting in higher costs for therapies. This study demonstrates increased in vivo maintenance of mesenchymal stem cells (MSCs) co-injected with a modular microporous scaffold made through the in situ crosslinking of flowable monodisperse hydrogel particles. The stem cells can be mixed with hydrogel particle building blocks immediately before injection/deployment, decoupling the manufacturing of scaffolds from the culture of therapeutic cells. Assemblies of spherical hydrogel building blocks created interconnected microscale void spaces which enhanced nutrient transport, and promoted cell migration and cell-cell connections, inducing more than ~7-times higher proliferation than chemically identical non-porous hydrogels in vitro and ~8-times enhanced MSC retention in a subcutaneous murine implantation model. Furthermore, the possibility of independent modulation of material properties of the building blocks, including stiffness and adhesive ligand composition, can be performed with different cell types, allowing tuning of conditions appropriate for many different populations of therapeutic cells for specific downstream applications in vivo (e.g. delivery to heart, bone, skin, nerves, etc). MSCs represent a promising therapeutic cell population for tissue/organ regeneration, with local therapy showing some positive effects in human disease[43]. Deployment of a high concentration of MSCs within a regenerative scaffold material immediately before delivery to tissue may improve their efficacy and overcome a manufacturing hurdle of other scaffold materials that require stem cells to be incorporated during scaffold synthesis. Overall, we show that cell delivery in conjunction with an in situ-formed microporous niche promotes the survival of delivered cells and should enhance cell therapies in regenerative medicine and immunologic applications.
Experimental Section
Microfluidic device fabrication:
Microfluidic devices were fabricated using soft lithography as previously described [20]. Briefly, master molds were fabricated on mechanical grade silicon wafers (University wafer) using KMPR 1050 photoresist (Microchem). Devices were molded from the masters using poly(dimethyl)siloxane (PDMS) Sylgard 184 kit (Dow Corning). The base and crosslinker were mixed at a 10:1 mass ratio, poured over the mold, and degassed prior to curing for overnight at 65 °C. Channels were sealed by treating the PDMS mold and a glass microscope slide (VWR) with oxygen plasma at 500 mTorr and 80W for 30 seconds. The channels were functionalized by injecting 100μL of Aquapel (88625–47100, Aquapel) and reacting for 30 seconds until washed by Novec 7500 (9802122937, 3M). The channels were dried by air suction and kept in the oven at 65 °C until used.
Microgel production:
Monodisperse microgels were produced as follows. Two aqueous solutions were prepared: (i) 4 Arm-PEG VS MW 20,000 (PTE-200VS, NOF) at 8, 10, 20 and 24 % (w/v) in 0.3 M triethyloamine (TEOA) pH 8.25, pre-reacted with K-peptide (Ac-FKGGERCG-NH2), Q-peptide (Ac-NQEQVSPLGGERCG-NH2) at a final concentration of 250 µM and with RGD peptide (Ac-RGDSPGERCG-NH2) at a final concentration of 500 µM or 2.5mM and (ii) an 8mM di-cysteine modified Matrix Metallo-protease (MMP) (Ac-GCRDGPQGIWGQDRCG-NH2) (Genscript) or 6 mM, 18 mM or 20 mM poly(ethylene glycol) dithiol MW 1,000 (717142–1G, Sigma-Aldrich) pre-reacted with 10 µM Alexa-fluor 488 or 568-maleimide (Life Technologies). Please see the Table S1 for the composition according to MAP gel types. These pre-gel solutions were sterile-filtered through a 0.2 µm Polyethersulfone (PES) membrane in a leur-lok syringe filter, injected into the microfluidic device and pinched off by oil phase (0.1% Pico-Surf in Novec 7500, SF-000149, Sphere Fluidics) (Figure S1A). The flow rate for aqueous solutions was 1 ~ 12 µL/min and for oil solutions was 4 ~ 400 µL/min to fine-tune the size of droplets (Figure S1B). Gels were collected from the device into a tube in oil phase, incubated overnight at room temperature in dark. Microgels in oil phase were vortexed with 20% 1H,1H,2H,2H-Perfluoro-1-octanol (PFO) (370533–25G, Sigma-Aldrich) in Novec 7500 for 10 seconds. Microgels were then mixed with 1:1 mixture of HEPES buffer (100 mM HEPES, 40 mM NaCl pH 7.4) and hexane followed by centrifugation at 10,000 rpm to separate microgels from oil for five times. Microgels were incubated in sterile-filtered 70% ethanol solution at 4 °C at least overnight for sterilization. Before in vivo or in vitro experiment, microgels were washed with HEPES buffer with 10 mM CaCl2 for five times.
Annealing of microgels:
Equal volumes of two microgel solutions were incubated in HEPES-buffered saline (pH 7.4) containing FXIII (10 U/mL) or thrombin (2U/mL) respectively at 4°C overnight. The two solutions were centrifuged at 10,000 rpm for 5 minutes and supernatants were removed to concentrate the microgels. These concentrated solutions were thoroughly mixed with each other by pipetting up and down, pipetted into a desired location and kept at 37°C for 90 minutes to anneal the microgels into a MAP scaffold.
Rheology techniques for measuring the storage modulus of MAP building blocks:
We measured the storage modulus of an 8-mm disc gel using an Anton paar physica mcr 301 Rheometer. 40 µL of pre-gel solutions (20 µL of PEG with peptides, 20 µL of crosslinker) were pipetted onto sterile siliconized (Sigmacote; SL2–25ML, Sigma-Aldrich) slide glass, covered with another glass with 1 mm spacer and incubated at 37 °C for two hours. Disc gels were swollen to equilibrium in PBS overnight before being measured. We performed an amplitude sweep (0.01–10% strain) to find the linear amplitude range for each. An amplitude within the linear range was chosen to run a frequency sweep (0.5–5 Hz). At least, four disc-gels were measure for each condition.
Diffusion measurement using photobleaching in MAP scaffold:
MAP gels were incubated with 100 µM 70 kDa dextran-FITC (FD70S-100MG, Sigma-Aldrich) solution in PBS or a 100 nM fluorescein solution in PBS. 20µL of microgels were pipetted and annealed in a 3-mm diameter PDMS well on a glass coverslip to form a MAP scaffold. Fluorescence recovery after photobleaching (FRAP) was conducted using a Leica TCS SP5 confocal microscope. A 20x dry objective and argon laser were used for bleaching and imaging. For pore diffusivity measurements, bleaching was performed with 30% laser power and 100% transmission, with imaging at 15% transmission to limit additional bleaching. For the single-phase bleaching measurements in non-porous hydrogel and PBS, 70% laser power and 100% transmission were used for bleaching, with 6% transmission used for imaging. After bleaching for 8 seconds, at least 50 images were taken with the interval of 390 ms (Figure S2). A circle of 100 μm diameter centered on the bleach spot was taken as the analysis region of interest (ROI) in all cases using ImageJ. The diffusivity was calculated via the approach of Soumpasis [44] (1):
| (1) |
Where is the ROI radius, is the halftime calculated by fitting the mean intensity of the ROI in time to an exponential equation (2):
| (2) |
Where and were obtained from the fitted curve.
Diffusion measurement in non-porous hydrogel using fluorescent intensity profile:
8-mm disc gels were prepared as previously described in the rheology technique section. Gels were swollen in PBS overnight and placed between two slide glasses in PBS with 100 µM 70 kDa dextran-FITC (FD70S-100MG, Sigma-Aldrich). The fluorescent images of gels (FITC) were taken every day and the intensity profiles over time were used to calculate the diffusivity using Fick’s law.
Gel degradation experiment:
10 µL of pre-gel solutions (5 µL of PEG with peptides, 5 µL of crosslinker pre-reacted with 10 µM Alexa-fluor 488-maleimide) were pipetted onto sterile siliconized (Sigmacote; SL2–25ML, Sigma-Aldrich) slide glass, covered with another glass with 1 mm spacer and incubated at 37°C for two hours. The final concentrations of PEG and crosslinkers were 5wt% 4-Arm PEG-VS with 4 mM PEG-dithiol (5%ND), 5wt% 4-Arm PEG-VS with 4 mM MMP-cleavable dithiol (5%D) and 10wt% 4-Arm PEG-VS with 8 mM MMP-cleavable dithiol (10%D). Gels were swollen to equilibrium in PBS overnight before being transferred to a 24-well insert with fluorescent-blocking membrane (08–772-147, Fisher Scientific); one gel per a well. The insert was inserted into a 24 well-plate with 1 mL of 1% TryPLE (12604013, Gibco) in PBS. The fluorescent level at the bottom of the plate was measured by the plate-reader (BioTek Cytation5) at 37°C for 12 hrs.
Hydraulic conductivity measurement in the scaffold:
A custom-designed device was designed using Autodesk Inventor 3D CAD software, and printed in Watershed XC 11122 Normal-Resolution Stereolithography build in 0.004” layers from Proto Labs, Inc. (Figure S3). For the MAP scaffold, 25 µL of microgel building blocks (5wt% crosslinked with MMP-cleavable dithiol) was casted on top of a 5 µm pore size cellulose membrane (SMWP01300, Fisher Scientific) in the bottom plane of the device and annealed followed by the overnight incubation in PBS. For the non-porous scaffold, 10 µL of pre-gel solution (5 wt% PEG with 4 mM MMP-cleavable dithiol) was casted on top of the membrane in the device and incubated at 37 °C for two hours followed by the overnight incubation in PBS. Then 1 mL of PBS with blue food dye was injected into the device and the permeated volume over time was measured. The hydraulic conductivity was calculated based on Darcy’s law [45]:
| (3) |
where, is the inner cross-sectional area of the graduated tube (cm2), is the test sample thickness (cm), is the test sample cross-sectional area (cm2). is the time elapsed between the initial head and the final head (s), is the initial head across the test specimen (cm), and is the final head across the test specimen (cm).
Mesenchymal stem cell culture in flasks:
Strain C57BL/6 Mouse Mesenchymal Stem Cells with RFP (MUBMX-01201, Cyagen Biosciences) and strain C57BL/6 Mouse Mesenchymal Stem Cells with GFP (MUBMX-01101, Cyagen Biosciences) were maintained in Mouse Mesenchymal Stem Cell Growth Medium (MUXMX-90011, Cyagen Biosciences) according to manufacturer’s specifications to retain stemness. Cells between passages 4–6 were used. Cells were maintained at lower than 80% confluency in culture.
Mesenchymal stem cell in vitro culture on the MAP scaffold:
MSCs labelled with RFP (MUBMX-01201, Cyagen Biosciences) were dissociated by trypsin and centrifuged down to remove medium. 60 µL of MAP in HEPES-buffered saline (pH 7.4) containing FXIII (10 U/mL) and 10 mM CaCl2 was combined as mixed thoroughly with 60 µL of MAP microgel building blocks in HEPES-buffered saline (pH 7.4) containing thrombin (2 U/mL) with a positive displacement pipette (MICROMAN, Gilson, Inc.). Cells were resuspended and spiked in MAP building blocks at 1,000 cells/µl concentration. These MAP gels were kept on ice to prevent annealing as well as maintaining the MSC viability. 20 µL of MAP gels with MSCs at 1,000 cells/µL was pipetted into silicone isolators (GBL664206–25EA, Sigma-Aldrich) in a tissue culture plate (08–772-50, Thermo Fisher Scientific) and incubated for 90 mins at 37 °C followed by adding a complete cell culture medium. Cells were grown in 5 % CO2 and 37 °C and 1 mL of medium was changed every 3–4 days. The fluorescent intensity from RFP was detected using a plate-reader (BioTek Cytation5). In each sample, at least 6 points of RFP intensity were measured (Ex: 545, Em: 605) using the area scanning function. At day 14, samples were gently washed with PBS twice, fixed with 4% PFA overnight at 4 °C followed by Hoechst (1/500) (Thermo Fisher Scientific) and phalloidin 647 (1/500) (Thermo Fisher Scientific) staining at room temperature for 4 hours. Then the scaffolds were gently washed twice with PBS and kept at 4 °C until imaged.
Mesenchymal stem cell in vitro culture on the non-porous scaffold:
PEG-VS scaffolds (5wt% r=0.8 MMP-1 crosslinker, 250 µM K, 250 µM Q, 500 µM RGD) were used to encapsulate MSCs (1,000 cells/µL). Gels were formed for 15 minutes (TEOA 0.3M, pH 8.25) before being placed into appropriate media. The fluorescent intensity detection and staining was performed as for MAP scaffold in vitro experiments.
In Vivo Imaging System (IVIS) experiments:
Strain C57BL/6J mice (The Jackson Laboratory) were anesthetized using continuous application of aerosolized isofluorane (3 vol%) throughout the duration of the procedure. 60 µL of MAP in HEPES-buffered saline (pH 7.4) containing FXIII (10 U/mL) and 10 mM CaCl2 was combined as mixed thoroughly with 60 µL of MAP building blocks in HEPES-buffered saline (pH 7.4) containing thrombin (2 U/mL) with a positive displacement pipette (MICROMAN, Gilson, Inc.). 100 µL of MAP building blocks with one million MSCs expressing RFP (MUBMX-01201, Cyagen Biosciences) was injected subcutaneously into mice via 25-gauge needles. As controls, 100 µL of PBS with one million MSCs expressing RFP and 40 µL of pre-gel solution (5wt% PEG with 4mM MMP-cleavable dithiol, swelling ratio 2.5) with one million MSCs expressing RFP were injected using the same method. Two injection sites on opposing sides of the back per mouse were used to avoid potential signal overlap during imaging. To monitor cell viability and distribution, the RFP fluorescence was measured using a Perkin Elmer IVIS Lumina II on days 0, 2, 5, 7, 10, 14. Before imaging, mice were anesthetized with 3% isofluorane/air. For each image acquisition, a gray scale body surface image was collected, followed by an overlay of the fluorescent (Ex: 535, Em: 600) and their radiant efficiency were quantified using Living Image software (Caliper LifeSciences). All animal experiments were performed according to established animal protocols.
Subcutaneous MSC injection model:
Strain C57BL/6J mice (The Jackson Laboratory) were anesthetized using continuous application of aerosolized isofluorane (3 vol%) throughout the duration of the procedure. 60 µL of MAP in HEPES-buffered saline (pH 7.4) containing FXIII (10 U/mL) and 10 mM CaCl2 was combined as mixed thoroughly with 60 µL of MAP building blocks in HEPES-buffered saline (pH 7.4) containing thrombin (2 U/mL) with a positive displacement pipette (MICROMAN, Gilson, Inc.). 2.4 µl of MSC suspension was then added and mixed thoroughly. 100 µL of MAP building blocks with MSC GFP (MUBMX-01101, Cyagen Biosciences) at 5,000 cells/µL was injected subcutaneously into mice via 25-gauge needles. Four injection sites on the back per mouse were used to accommodate all four test conditions (SoD1, SoN1, StN1 and StN5). Mice were sacrificed at week 2 and MAP scaffolds were excised and immediately fixed in 4% PFA for two hours before flash frozen in OCT compound (Tissue-Tek). These OCT tissue blocks were kept at −80 °C until sectioned at 25 µm thickness using cryostat (Leica CM1950) and collected onto 25 × 75 mm charged slides (1358W, Globe Scientific). Sections were dried at room temperature overnight and kept at −20 °C until stained. All animal experiments were performed according to established animal protocols.
Tissue section immunofluorescence:
Slides containing tissue sections were washed and blocked using 10% normal goat serum (50062Z, Invitrogen) for two hours at room temperature and then stained with primary antibodies overnight at 4 °C. Primary antibody dilutions were prepared as in the Table S2. Sections were incubated in secondary antibodies with 1/500 diluted Hoechst for 2 hours at room temperature, and subsequently washed with PBS. Secondary antibody dilutions were prepared as in the Table S2. Sections were mounted in Antifade Gold mounting medium (9071S, Cell Signaling Technology) and imaged using Leica Confocal SP-5 with 63x water immersion objective lens or Nikon Ti Eclipse with 10x objective lens.
Computational analysis of tissue images:
Images were analyzed using a MATLAB code. Briefly, the code defines the region of an injected MAP scaffold region using the TRITC channel. It divides the region into two: a periphery region of interest (ROI) (200 μm into and out of the scaffold) and core ROI (inside of the periphery region) (Figure S7). For ROI regions, the code reads the Cy5 channel intensity profiles and defines regions of protein expression using a threshold (identical for all sample images). The ratio of cells with protein expression was calculated by the ratio of cells having a nuclear stain also in the protein expression region for each ROI. The density of cells was calculated by the total number of cells under the protein expression mask divided by the area of the ROI. The cell density as a function of the distance from the interface was calculated by counting the number of cells in a subregion of the scaffold that was divided by distance (Figure S7E). We verified that the quantification by this method is insensitive to imaging conditions: exposure time and number of scans, although we kept the imaging condition the same for all samples (Figure S11). Tissue slice areas with physical defects from imperfect tissue sectioning were excluded manually in the analysis.
Statistical analysis:
All values are depicted as mean ± standard deviation unless stated. Statistical comparisons were performed using GraphPad Prism 8.0 or MATLAB version R2016b (MathWorks, Inc.). The significance between two groups was analyzed by a two-tailed Student t-test. The significance between multiple groups was analyzed by a one-way ANOVA. Values were considered to be significantly different when p < 0.05.
Supplementary Material
Acknowledgements
This work was supported by the Presidential Early Career Award for Scientists and Engineers (N00014-16-1-2997). J.K. was supported by the Kwanjeong Educational Foundation, Korea. P.O.S. was supported by the Mentored Clinical Scientist Development Award (K08-AR066545) through NIAMS. The authors would like to thank Weikun Xiao and Dr. Stephanie Seidlits for technical assistance and comments, Dr. Jason Lee at the Crump Institute in California NanoSystems Institute (CNSI) for discussions and assistant for IVIS, Dr. Matt Schibler and Dr. Laurent Bentolila at Advanced Light Microscopy/Spectroscopy in CNSI for confocal imaging consultation, Dr. Westbrook Weaver, Joseph de Rutte, Dr. Maria Antfolk and Wei Yang for helpful discussions. J.K., D.R.G., T.S., P.O.S., D.D.C. conceived the study and designed the experiment. J.K., M.A. performed in vitro studies, J.K., D.R.G., A.F., P.O.S. performed in vivo studies. J.K., T.H., M.M., D.Z. characterized the MAP scaffold. J.K. designed and fabricated the microfluidic device. J.K., D.D.C. analyzed and interpreted the data. J.K., P.O.S. and D.D.C. wrote the manuscript with input from all the authors.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Jaekyung Koh, Department of Bioengineering, University of California, Los Angeles, CA, 90024, USA..
Donald R. Griffin, Department of Biomedical Engineering, University of Virginia, Charlottesville, VA, 22904, USA.
Maani M. Archang, Department of Bioengineering, University of California, Los Angeles, CA, 90024, USA.
An-Chieh Feng, Department of Microbiology, Immunology, and Molecular Genetics, David Geffen School of Medicine, University of California, Los Angeles, CA, 90024, USA..
Thomas Horn, Department of Bioengineering, University of California, Los Angeles, CA, 90024, USA..
Michael Margolis, Department of Bioengineering, University of California, Los Angeles, CA, 90024, USA..
David Zalazar, Department of Bioengineering, University of California, Los Angeles, CA, 90024, USA..
Tatiana Segura, Department of Biomedical Engineering, Neurology, Dermatology, Duke University, Durham, NC, 27708, USA..
Philip O. Scumpia, Division of Dermatology, Department of Medicine, David Geffen School of Medicine, University of California, Los Angeles, CA, 90024, USA. Department of Dermatology, VA Greater Los Angeles Healthcare System, Los Angeles, CA, 90073, USA.
Dino Di Carlo, Department of Bioengineering, University of California, Los Angeles, CA, 90024, USA. Department of Mechanical Engineering, Mechanical and Aerospace Engineering, University of California, Los Angeles, CA, 90024, USA. California NanoSystems Institute (CNSI), University of California, Los Angeles, CA, 90024, USA. Jonsson Comprehensive Cancer Center, University of California, Los Angeles, CA, 90024, USA..
References
- [1].Parekkadan B, Milwid JM, Annu. Rev. Biomed. Eng 2010, 12, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Trounson A, McDonald C, Cell Stem Cell 2015, 17, 11. [DOI] [PubMed] [Google Scholar]
- [3].Tran C, Damaser MS, Adv. Drug Deliv. Rev 2015, 82, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ankrum J, Karp JM, Trends Mol. Med 2010, 16, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Galipeau J, Sensébé L, Cell Stem Cell 2018, 22, 824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mishra PJ, Banerjee D, Methods Mol. Biol 2017, 1554, 201. [DOI] [PubMed] [Google Scholar]
- [7].Eggenhofer E, Benseler V, Kroemer A, Popp FC, Geissler EK, Schlitt HJ, Baan CC, Dahlke MH, Hoogduijn MJ, Front. Immunol 2012, 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Elman JS, Murray RM, Wang F, Shen K, Gao S, Conway KE, Yarmush ML, Tannous BA, Weissleder R, Parekkadan B, PLoS One 2014, 9, e89882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ranganath SH, Levy O, Inamdar MS, Karp JM, Cell Stem Cell 2012, 10, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Madl CM, Heilshorn SC, Blau HM, Nature 2018, 557, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Burdick JA, Mauck RL, Gerecht S, Cell Stem Cell 2016, 18, 13. [DOI] [PubMed] [Google Scholar]
- [12].Lane SW, a Williams D, Watt FM, Nat. Biotechnol 2014, 32, 795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cai L, Dewi RE, Heilshorn SC, Adv. Funct. Mater 2015, 25, 1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mitrousis N, Fokina A, Shoichet MS, Nat. Rev. Mater 2018, 3, 441. [Google Scholar]
- [15].Dimatteo R, Darling NJ, Segura T, Adv. Drug Deliv. Rev 2018, 127, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shin JW, Mooney DJ, Cell Stem Cell 2016, 18, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lutolf MP, Gilbert PM, Blau HM, Nature 2009, 462, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tong X, Yang F, Adv. Healthc. Mater 2018, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Huebsch N, Lippens E, Lee K, Mehta M, Koshy ST, Darnell MC, Desai R, Madl CM, Xu M, Zhao X, Chaudhuri O, Verbeke C, Kim WS, Alim K, Nat. Mater 2015, 14, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Griffin DR, Weaver WM, Scumpia PO, Di Carlo D, Segura T, Nat Mater 2015, 14, 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sideris E, Griffin DR, Ding Y, Li S, Weaver WM, Di Carlo D, Hsiai T, Segura T, ACS Biomater. Sci. Eng 2016, 2, 2034. [DOI] [PubMed] [Google Scholar]
- [22].Nih LR, Sideris E, Carmichael ST, Segura T, Adv. Mater 2017, 29, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Darling NJ, Sideris E, Hamada N, Carmichael ST, Segura T, Adv. Sci 2018, 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].de Rutte JM, Koh J, Di Carlo D, Adv. Funct. Mater 2019, 29, 1970174. [Google Scholar]
- [25].Caldwell AS, Campbell GT, Shekiro KMT, Anseth KS, Adv. Healthc. Mater 2017, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kumar NN, Gautam M, Lochhead JJ, Wolak DJ, Ithapu V, Singh V, Thorne RG, Sci. Rep 2016, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li J, Mooney DJ, Nat. Rev. Mater 2016, 1, 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wong AP, Perez-Castillejos R, Christopher Love J, Whitesides GM, Biomaterials 2008, 29, 1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wood A, Biocontrol Sci. Technol 2006, 16, 495. [Google Scholar]
- [30].Sannino A, Netti PA, Madaghiele M, Coccoli V, Luciani A, Maffezzoli A, Nicolais L, J. Biomed. Mater. Res. Part A 2006, 79A, 229. [DOI] [PubMed] [Google Scholar]
- [31].Keung AJ, Kumar S, Schaffer DV, Annu. Rev. Cell Dev. Biol 2010, 26, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sun Y, Chen CS, Fu J, Annu. Rev. Biophys 2012, 41, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gattazzo F, Urciuolo A, Bonaldo P, Biochim. Biophys. Acta - Gen. Subj 2014, 1840, 2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Guo M, Pegoraro AF, Mao A, Zhou EH, Arany PR, Han Y, Burnette DT, Jensen MH, Kasza KE, Moore JR, Mackintosh FC, Fredberg JJ, Mooney DJ, Lippincott-Schwartz J, Weitz DA, Proc. Natl. Acad. Sci 2017, c, 201705179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lee J, Abdeen AA, Kilian KA, Sci. Rep 2014, 4, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ma S, Xie N, Li W, Yuan B, Shi Y, Wang Y, Cell Death Differ 2014, 21, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jurewicz M, Yang S, Augello A, Godwin JG, Moore RF, Azzi J, Fiorina P, Atkinson M, Sayegh MH, Abdi R, Diabetes 2010, 59, 3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].da Silva Meirelles L, Fontes AM, Covas DT, Caplan AI, Cytokine Growth Factor Rev 2009, 20, 419. [DOI] [PubMed] [Google Scholar]
- [39].Romieu-Mourez R, Francois M, Boivin M-N, Bouchentouf M, Spaner DE, Galipeau J, J. Immunol 2009, 182, 7963. [DOI] [PubMed] [Google Scholar]
- [40].Veiseh O, Doloff JC, Ma M, Vegas AJ, Tam HH, Bader AR, Li J, Langan E, Wyckoff J, Loo WS, Jhunjhunwala S, Chiu A, Siebert S, Tang K, Hollister-Lock J, Aresta-Dasilva S, Bochenek M, Mendoza-Elias J, Wang Y, Qi M, Lavin DM, Chen M, Dholakia N, Thakrar R, Lacík I, Weir GC, Oberholzer J, Greiner DL, Langer R, Anderson DG, Nat. Mater 2015, 14, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rustad KC, Wong VW, Sorkin M, Glotzbach JP, Major MR, Rajadas J, Longaker MT, Gurtner GC, Biomaterials 2012, 33, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kim JS, Jung Y, Kim SH, Shin JS, Kim SH, Park CG, Xenotransplantation 2019, 26, 1. [DOI] [PubMed] [Google Scholar]
- [43].Chen Z, Chen L, Zeng C, Wang WE, Stem Cells Int 2018, 2018, 7045245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Soumpasis DM, Biophys. J 1983, 41, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Aboufoul M, Garcia A, Mater. Struct. Constr 2017, 50, 1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.