

Daptomycin binds to bacterial cell membranes and disrupts essential cell envelope processes, leading to cell death. Bacteria respond to daptomycin by altering their cell envelopes to either decrease antibiotic binding to the membrane or by diverting binding away from septal targets. In Enterococcus faecalis, daptomycin resistance is typically coordinated by the three-component cell envelope stress response system, LiaFSR.
KEYWORDS: Enterococcus, adaptive resistance, drug resistance evolution
ABSTRACT
Daptomycin binds to bacterial cell membranes and disrupts essential cell envelope processes, leading to cell death. Bacteria respond to daptomycin by altering their cell envelopes to either decrease antibiotic binding to the membrane or by diverting binding away from septal targets. In Enterococcus faecalis, daptomycin resistance is typically coordinated by the three-component cell envelope stress response system, LiaFSR. Here, studying a clinical strain of multidrug-resistant Enterococcus faecium containing alleles associated with activation of the LiaFSR signaling pathway, we found that specific environments selected for different evolutionary trajectories, leading to high-level daptomycin resistance. Planktonic environments favored pathways that increased cell surface charge via yvcRS upregulation of dltABCD and mprF, causing a reduction in daptomycin binding. Alternatively, environments favoring complex structured communities, including biofilms, evolved both diversion and repulsion strategies via divIVA and oatA mutations, respectively. Both environments subsequently converged on cardiolipin synthase (cls) mutations, suggesting the importance of membrane modification across strategies. Our findings indicate that E. faecium can evolve diverse evolutionary trajectories to daptomycin resistance that are shaped by the environment to produce a combination of resistance strategies. The accessibility of multiple and different biochemical pathways simultaneously suggests that the outcome of daptomycin exposure results in a polymorphic population of resistant phenotypes, making E. faecium a recalcitrant nosocomial pathogen.
INTRODUCTION
The rise of multidrug-resistant (MDR) pathogens is one of the most pressing biomedical problems of this century. The Centers for Disease Control and Prevention (CDC) reports that 2 million antibiotic-resistant infections, resulting in 23,000 deaths, occur each year (1). Vancomycin-resistant enterococci (VRE) cause approximately 1,300 deaths annually, with the number of infections increasing substantially over the last 15 years (1). The Infectious Disease Society of America has listed Enterococcus faecium among the “no ESKAPE” pathogens (E. faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.), for which there is an urgent need for new therapies (2). E. faecium accounts for a significant number of enterococcal health care-associated infections, particularly in severely immunocompromised patients. E. faecium strains that are resistant to all antienterococcal antibiotics have been widely described (3–6), making these infections untreatable in certain scenarios (1, 3–5, 7, 8).
Daptomycin (DAP) is a bactericidal cyclic lipopeptide antibiotic approved in 2003 and used widely as a “rescue” drug against MDR Gram-positive organisms, such as Staphylococcus aureus, E. faecium, and Enterococcus faecalis (4, 9, 10). While the DAP mechanism of action remains unclear, DAP acts in a calcium-dependent manner, where the DAP:Ca+2 complex binds to the cell membrane in a phosphatidylglycerol-dependent manner with high avidity for division septa. DAP binding has pleiotropic effects in the cell membrane that include mislocalization of key proteins involved in cell wall and cell membrane metabolism and, ultimately, cell death (11–14). Unfortunately, DAP resistance is increasingly observed in the clinic. A clear understanding of the biochemical basis for resistance can potentially identify novel targets, therapeutic approaches, and diagnostic markers for these MDR infections to restore the effectiveness of antibiotics (including DAP) against recalcitrant strains (15).
To date, the manner in which organisms evolve DAP resistance falls into three categories: repulsion from the cell surface (16–18), diversion of DAP binding away from the division septa (13, 15, 16, 18–20), and antibiotic hyperaccumulation in selected cells (seen in streptococci) (21) to presumably protect the bacterial population. S. aureus predominately exploits repulsion, often mediated through gain-of-function mutations in the dltABCD operon and the multiple-peptide resistance factor mprF (16, 18, 20). Mutations in the dltABCD operon, responsible for incorporating d-alanine into lipoteichoic acids (LTA), often result in a more positively charged cell envelope and reduced DAP binding (18, 22, 23). Similarly, MprF catalyzes the transfer of a lysyl group to phosphatidylglycerol, causing a net increase in cell surface charge and reduced DAP binding (18), although it has also been postulated that this reaction affects DAP susceptibility by decreasing the availability of phosphatidylglycerol for DAP binding (24). In contrast, E. faecalis employs a different strategy that involves redistribution of anionic phospholipid microdomains away from the septum to divert DAP from critical septal targets. This mechanism is mediated by the LiaFSR cell envelope stress response system in association with cardiolipin synthase (cls) (15, 25, 26). Our initial mechanistic studies (27) suggested that DAP resistance in E. faecium is not mediated by membrane remodeling but rather involves repulsion of the antibiotic from the cell surface. Nonetheless, the LiaFSR system is involved in the E. faecium DAP response, since deletion of the gene encoding the LiaR response regulator resulted in hypersusceptibility independent of the genetic background of the strain (28).
To deconstruct the potential resistance strategies available to E. faecium, we used specific adaptive environments to evolve the clinical strain E. faecium HOU503, harboring the most common clinical LiaRS substitutions (LiaRW73C and LiaST120A), which predispose system activation and DAP resistance (29). Despite HOU503 containing alleles associated with LiaFSR activation and DAP redistribution, we show that we can use the environment to favor different biochemical resistance strategies. Unlike the evolution of E. faecalis and S. aureus to DAP resistance, our work suggests that E. faecium is able to employ various resistance strategies that may help explain the recalcitrant nature of these infections and the therapeutic challenges they pose (27).
RESULTS
Distinct adaptive environments select for divergent phenotypes and evolutionary trajectories.
To favor distinct evolutionary trajectories that might be associated with the evolution of DAP resistance by E. faecium in the presence of LiaFSR substitutions, we performed experimental evolution using two different techniques that established a very different basis for the selection of adaptive phenotypes. We chose E. faecium HOU503 as the ancestor because it is a vancomycin (VAN)-resistant isolate (MIC of >256 μg/ml) with a DAP MIC of 3 μg/ml, and is poised to make the transition to DAP resistance (8). HOU503 contains mutated alleles within the LiaFSR pathway (LiaRW73C and LiaST120A) that, in combination, increase the strength of LiaFSR signaling (29). Thus, this strain provided the ideal scenario to investigate subsequent and viable evolutionary trajectories upon antibiotic exposures when an initial step has been taken.
First, we evolved five HOU503 populations to DAP resistance using a traditional serial-flask transfer model where planktonic cells were transferred daily to increasing DAP concentrations. The stepwise increase in DAP concentration was performed below the current population MIC to allow the establishment of multiple evolutionary trajectories within the population (25). The populations were passaged for a total of 8 days, with the final populations containing 8 μg/ml DAP (resistant by clinical standards). The environment established by flask-transfer strongly reduces the selection for trajectories that might rely upon biofilm, as cells that adhere to surfaces are less likely to be transferred.
We next evolved two independent HOU503 populations to DAP resistance in a bioreactor where the vessel remained constant and the culture was maintained at its highest growth rate. As in the flask-transfer experiments, the bioreactor population was subjected to stepwise increases in DAP concentration, below the population MIC, to maintain diversity until the final population was growing at 8 μg/ml DAP, within 10 to 12 days. The bioreactor environment is, in many respects, the opposite of the flask-transfer environment, where cells that form biofilms or adhere to surfaces remain within the vessel. The production of long-term biofilms contributes, in part, to the high level of polymorphism found within the bioreactor (30, 31). While the formation of biofilms is one difference between the flask transfer and bioreactor environments, the flask transfer populations also experience fluctuations in population size and spend a significant amount of time in stationary phase. Based on our results, it is clear that these different environments can provide a useful way of identifying and exploring evolutionary trajectories. For further discussion, see the text in the supplemental material.
Following adaptation, two isolates from each of the five flask-transfer populations (10 isolates total) were selected for whole-genome sequencing (WGS) and phenotypic characterization. From each bioreactor-adapted population, 10 and 9 isolates, respectively, with distinct phenotypic properties (DAP MIC, cell density at stationary phase, and the ability to form floc) underwent WGS and further characterization (19 isolates total). Antibiotic cross-sensitivities were also tested and are discussed in the text and Table S1 in the supplemental material.
To confirm that each technique produced a different adaptive environment, we performed a crystal violet assay to quantify endpoint isolate biofilm growth and assayed the growth rates of 16 isolates with diverse genomes from both environments (isolates with identical genomes were excluded from these assays). As shown in Fig. 1A, bioreactor-derived isolates produced up to 10-fold more biofilm than HOU503 and the flask-transfer isolates, consistent with growing in different adaptive environments. In fact, FT5 and FT8 produced less biofilm than the ancestor, suggesting that the production of biofilm was not selected for in these isolates. While the bioreactor-derived isolates typically formed strong biofilms, isolate R2P29 produced little biofilm, consistent with our previous studies showing that bioreactors can favor highly polymorphic populations and maintain planktonic subpopulations (25, 30–33). This isolate is discussed further in the supplemental text.
FIG 1.
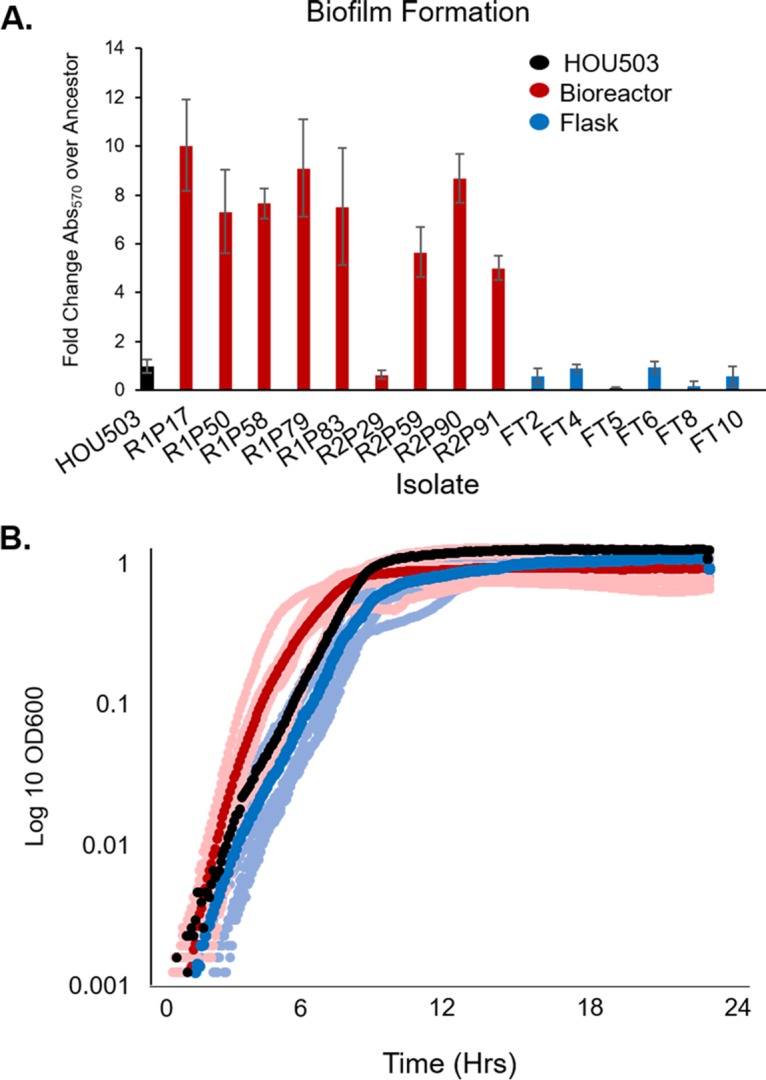
Varying the adaptive environment selects for distinctive and divergent phenotypes. The bioreactor (red) and flask (blue) environments evolve distinctly different phenotypes. (A) A crystal violet assay was used to quantify biofilm formation of 16 endpoint isolates and is reported as the fold change in crystal violet absorbance at 570 nm (Abs570) over the level for the ancestor. All bioreactor isolates (except R2PXXIX) produced more biofilm than the ancestor. Error bars represent standard deviations. (B) Growth rates were performed in a microplate reader in triplicate for all endpoint isolates. The dark red and blue markers indicate the average growth for each adaptive environment, with the lighter shades showing the average growth of individual isolates and the ancestor in black.
In addition to biofilm formation, the growth rates of each endpoint isolate type differed (Fig. 1B). Flask-transfer isolates tended to grow more slowly than both the ancestor and the bioreactor-adapted isolates, with flask-transfer generation times ranging from 0.6 to 1.7 h, while the HOU503 generation time was 0.66 h. The bioreactor-derived isolate generation times were 0.4 to 0.7 h, with only one isolate (R1P83) having a significantly longer generation time of 1.6 h (Fig. 1B). Since the bioreactor is run as a turbidostat without nutrient limitation, the environment selects for both biofilm formation and rapid growth while flask environments select for planktonic cells and experience complex growth dynamics of entering and exiting stationary phase. As shown in Fig. 1, bioreactor-derived lineages have both a strong propensity to form biofilms and faster planktonic growth rates, consistent with the bioreactor selection conditions. Together, these data show that the adaptive environments established by the different experimental evolution approaches favored distinct phenotypes that potentially could reveal different evolutionary trajectories leading to DAP resistance.
Flask-adapted HOU503 repeatedly evolved mutations within yvcRS that led to an increase in dltABCD and mprF transcripts, consistent with the repulsion phenotype.
Comparison of the 10 flask-transfer isolate WGS to those of HOU503 and the closed reference genome HOU503F_del_liaR (the ancestor with a liaR deletion; Sanger sequencing in adapted isolates of liaR showed no mutations) revealed the repeated evolution of mutations in orf_2375 or orf_2376, which are annotated as homologs of the yvcRS system associated with bacitracin resistance in E. faecalis and was recently found to be involved in E. faecalis evolved to DAP resistance without a functional LiaFSR system (Table 1) (34). This sampling does not provide an exhaustive study of all possible DAP resistance alleles evolved in flasks; however, the repeated evolution of yvcRS mutations in multiple populations indicates their importance. Isolate FT9 was evidenced as containing a mixed population and was subsequently excluded from analysis. Below, we report on the genomic mutations, while plasmid dynamics are discussed in the supplemental text. YvcS is a transmembrane permease that senses bacitracin and, in conjunction with YvcR, a cytoplasmic ATPase, transmits the signal to the YxdK sensor kinase and YxdJ response regulator (34). This system shares significant similarity to the VraFG/GraSR system in S. aureus that upregulates both dltABCD and mprF in the presence of cationic antimicrobial peptides (CAMPs) (17). Interestingly, yvcRS in enterococci is located directly upstream of the dltABCD operon. Therefore, we hypothesized that, in E. faecium, YvcRS could regulate dltABCD and possibly mprF.
TABLE 1.
Flask-transfer isolate genomesa

+, gene identified in this study.
To test this hypothesis, we performed qPCR to measure the effect of yvcRS mutations on the transcription of dltA and mprF. We assayed endpoint isolates with diverse genomes and, as such, FT1, FT3, and FT7 were not included in these assays, as their genomes were identical to those of other isolates tested. As shown in Fig. 2, flask-transfer isolates had increases of 2- to 9-fold in dltA transcripts and 2- to 5-fold in mprF transcripts compared to levels for the housekeeping gene gdhIV (glucose-1-dehydrogenase 4). Furthermore, all dlt operon transcripts were upregulated in FT6 (yvcRH203Y, clsA20D), suggesting a link between the dlt operon and YvcRS (Fig. S1).
FIG 2.

Flask-transfer isolates with mutations in yvcRS had upregulated dltA and mprF transcripts. qPCR was used to measure transcript levels of flask-transfer isolates using gdhIV as a reference. (A) dltA transcript levels compared to those of the ancestor. (B) mprF transcript levels compared to those of the ancestor.
The isolates used for the assays shown in Fig. 2 and below contained one additional single-nucleotide polymorphism (SNP) outside yvcRS, making it difficult to assert causality for the mutations in yvcRS and the upregulation of the dltABCD and mprF transcripts. We report the genetic changes associated with DAP resistance within metagenomic analysis of the populations but with the caveat that other mutations can be present within the endpoint isolates. For example, to evaluate the likely effects of yvcS mutations, we characterized isolates FT2 (yvcSG133V, pgsA−62) and FT5 (yvcSS23I, clsR218Q), because the only common mutation between them was present in yvcS, suggesting that the yvcS mutations caused the increase in transcripts. Similarly, FT6 (yvcRH203Y, clsA20D) and FT10 (yvcRG173C, clsA20D) were chosen to study the effects of yvcR mutations, as the secondary mutation clsA20D is in the well-characterized cls gene. Mutations in cls are commonly found in the enterococcal DAP response and are associated with phospholipid redistribution (13, 35). As shown in our later analyses, other lineages containing clsA20D had a strikingly different phenotype, suggesting that clsA20D could not be directly responsible for the phenotype observed here. By comparing strains with different secondary mutations, the basis of the conclusion linking potential upregulation of dltABCD or mprF is one of parsimony rather than direct causality.
Isolates with mutations in yvcRS had a more positively charged cell surface and bound less BDP:DAP than the ancestor.
To determine if isolates containing yvcRS mutations possessed an increase in cell surface charge, likely due to the upregulation of the dltABCD operon and mprF, flask-transfer isolates were incubated with the positively charged molecule poly-l-lysine conjugated to fluorescein isothiocyanate (PLL:FITC), and cell fluorescence was quantified using fluorescence microscopy. Cells binding less PLL:FITC correlates with a more positively charged cell surface (36). All flask-transfer isolates tested bound 30 to 60% less PLL:FITC than the ancestor (P < 0.05 using Mann-Whitney test with post hoc Holm-Bonferroni adjustment), indicating that flask-transfer isolates produced a more positively charged cell surface and that the regulatory changes shown in Fig. 2 likely lead to an increase in cell surface charge (Fig. 3A and Fig. S2). We also examined whether the isolates with mutations in yvcRS bound less bodipy-DAP (BDP:DAP) than the ancestor. BDP:DAP is a conjugation of the fluorophore bodipy-FL with DAP and is used as a proxy for DAP binding (27, 28, 37). After incubation with 32 μg/ml BDP:DAP, bound BDP:DAP was quantified using fluorescence microscopy (Fig. 3B and C). All flask-transfer isolates bound 53 to 70% less BDP:DAP than the ancestor (P < 0.005 using Mann-Whitney test with post hoc Holm-Bonferroni), supporting the hypothesis that mutations in yvcRS are associated with repulsion of DAP from the cell surface. Incubation with the anionic phospholipid dye 10-N-nonyl acridine orange (NAO) (15, 38, 39) revealed no redistribution of phospholipid microdomains (Fig. S3).
FIG 3.
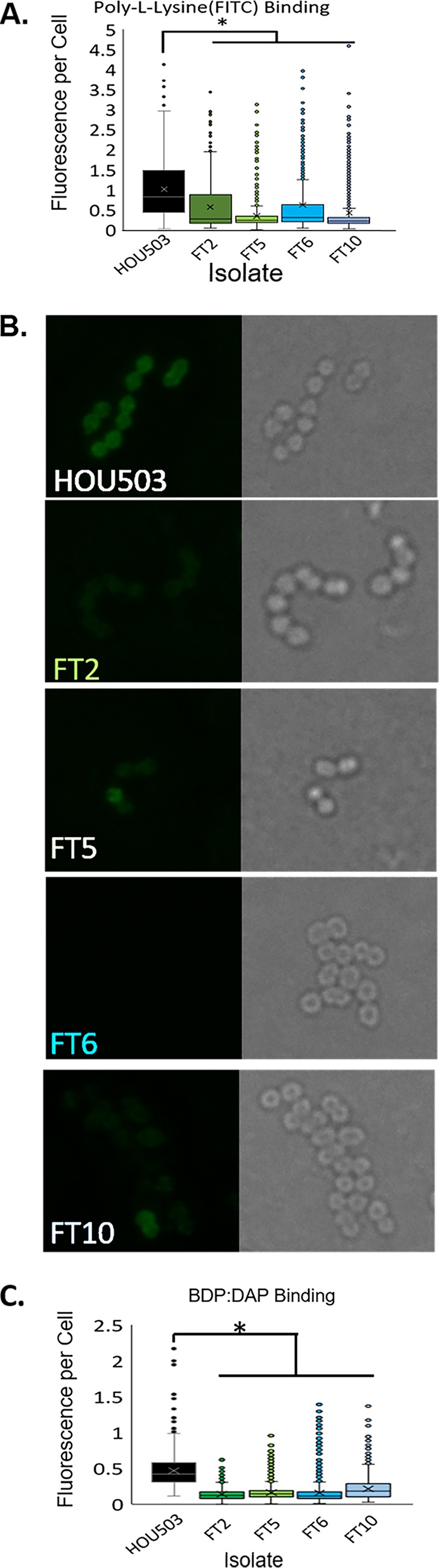
Isolates with mutations in yvcRS had a more positively charged cell surface and bound less BDP:DAP than the ancestor, HOU503. (A) The relative cell surface charge was determined by incubation with PLL:FITC. Cells that bind less PLL:FITC have a more positive cell surface charge. An asterisks indicates statistical significance (P < 0.05) using the Mann-Whitney test with post hoc Holm-Bonferroni adjustment. ImageJ was used for quantification. Physical images can be viewed in Fig. S2. (B) Isolates were incubated with BDP:DAP to determine DAP binding patterns. (C) Quantification of BDP:DAP binding per cell. An asterisks indicates statistical significance (P < 0.05) using the Mann-Whitney test with post hoc Holm-Bonferroni adjustment. ImageJ was used for quantification.
Adaptation in a bioreactor produced two main DAP resistance trajectories via mutations in divIVA or oatA.
Using methods described previously (25, 30–33) and more extensively in the text in the supplemental material, two independent HOU503 populations were evolved to DAP resistance within 12 and 10 days (population 1 and population 2, respectively) in a bioreactor system that favors polymorphic populations and biofilm formation (25, 31–33). To identify the frequency of each mutation over time and the likely order of mutations, a polymorphic sample was taken daily for metagenomic deep sequencing (Fig. S4). Afterwards, 10 and 9 nonrandom, phenotypically diverse endpoint isolates from populations 1 and 2, respectively, underwent WGS to identify the linkages between mutations (Tables 2 and 3). These endpoint isolates were selected based upon phenotypic diversity to increase the chance of identifying diverse genotypes; therefore, the percentage of endpoint isolates carrying any particular mutation does not represent the frequency of that mutation within the entire population. By combining the daily frequencies with the genetic linkages from endpoint isolates, adaptive timelines were created, detailing the likely sequence of events that resulted in DAP resistance (Fig. 4). Note that these trajectories do not include plasmid-associated mutations. Plasmids are easily transferred horizontally; therefor, pinpointing a plasmid-encoded acquisition of mutations in different lineages is difficult (see the supplemental text) (40). Similarly, this timeline was constructed with the assumption that homologous recombination and recurrent evolution of identical alleles was not occurring. Interestingly, several identical mutations were present on day 1 of both populations (rpoBG32G, purAL409S, ansP−288, tagBR330S, orf_280C84C, and orf_2338−276), suggesting heterogeneity at these loci in the ancestor. Because these mutations were present at high frequencies on day 1, without DAP present, and their frequency fell upon the addition of DAP, it is likely that these mutations were not the result of DAP adaptation and were hitchhiker mutations. Thus, any mutations identified prior to the addition of DAP were removed from subsequent analysis.
TABLE 2.
Bioreactor-derived population 1 endpoint isolate genomes

TABLE 3.
Bioreactor-derived population 2 endpoint isolate genomes

FIG 4.

Adaption within a bioreactor environment favoring rapid growth and biofilm formation produced two predominant evolutionary trajectories. Combining the WGS data from endpoint isolates that identified genetic linkage with the metagenomic frequency data over time established the likely sequence of events that resulted in DAP-resistant trajectories. Dashed lines indicate that the frequency of the subsequent mutation(s) identified in specific endpoint isolates were below the level of detection (<3%) in the overall bioreactor population. The low frequency of these mutations within the population suggests that they were acquired toward the end of adaptation. The DAP concentration each day of the experiment is across the top in gray. Final DAP MICs of each trajectory are denoted in red at the right. (A) Population 1 evolved 2 main trajectories opening with either a mutation in oatA or Δ1915, followed by additional mutations, including in cls. (B) Population 2 evolved one main trajectory with a mutation in divIVA followed by mutations in cls.
Because mutations that arise early and at high frequency are those more likely to contribute to DAP resistance (25, 30, 31, 33), we identified divIVA and oatA as candidate genes for further study (Fig. 4). An evolutionary trajectory that reached high frequency within population 1 was Δorf_2427-2429, divIVA, a deletion of 1,915 bp (here referred to as Δ1915) that resulted in the truncation of the C terminus of a putative sepF gene (residues 107 to 200), deletions of an S4 domain-containing gene and a yygT superfamily gene, and the N-terminal deletion of divIVA (residues 1 to 75). SepF forms ring structures at the division septum and directly interacts with FtsZ in Bacillus subtilis (41, 42), while DivIVA acts as a scaffold in the septum formation complex and aids in chromosomal segregation by providing a scaffold at the cellular poles (43). In population 2, a SNP in divIVA (Q75K) was the prominent allele, found in 69% of the population, suggesting the importance of changes in divIVA toward DAP resistance. Alternatively, mutations in oatA were observed in four endpoint isolates in population 1 and were prominent alleles, based on daily frequency data, during early adaptation in population 2. In the absence of genetic experiments to determine causality of these alleles, the experiments described below can only draw the most parsimonious arguments to link these genotypes to phenotypes.
After the acquisition of the primary mutations described above, mutations in cls were acquired and found in 13/19 endpoint isolates, comprising 36% and 61% of the final populations. The emergence of mutations in cls closely mimics E. faecalis adaptation to DAP, corroborating the important role of Cls in the evolution of DAP resistance but only after an initial set of mutations establish a biochemical basis for their acquisition (25). Our extensive sampling of the bioreactor and deep sequencing of daily populations provides no evidence of yvcRS mutations occurring in this environment, indicating a difference between adaptation in flasks and a bioreactor. See the supplemental text for further discussion of alleles.
Bioreactor-derived isolates containing divIVA-associated mutations produced abnormal division septa.
In total, 11/19 bioreactor-derived isolates contained a mutation in divIVA that comprised 41% and 69% of the final populations (Tables 1 and 2, and Fig. S4). In population 1, Δ1915 affected four genes, all of which had predicted functions involved in cell division, including the N-terminal deletion of divIVA. The mutation observed in population 2 (Q75K) was in a predicted loop region of divIVA between the first two predicted helices of the N terminus. Because the deletion affecting divIVA would result in a loss of function, we speculate that divIVAQ75K had reduced function, although this remains untested. Interestingly, divIVAQ75K was also identified in population 1, although it was less successful than Δ1915 (Fig. S4A). To understand how Δ1915 affects DAP resistance, we evaluated isolate R1P79 (Δ1915, clsR211L), as the only additional genomic mutation was clsR211L, which was also observed in a separate trajectory not containing mutations within divIVA, R1P50. To study divIVAQ75K, we used isolate R2P90, containing divIVAQ75K and clsA20D (the cls allele assessed in FT6 and FT10).
Using transmission electron microscopy (TEM), we found that the Δ1915- and divIVAQ75K-containing isolates had increased abnormal septation events and abnormal clustering or chaining (Fig. 5). We measured the number of cells with abnormal septation events compared to those of the ancestor to provide a quantitative metric and found that R1P79 (Δ1915, clsR211L) exhibited an increase in abnormal septation events from 9% (ancestor) to 82%, whereas R2P90 (divIVAQ75K, clsA20D) produced 50% abnormal septation events (Fig. 5D). This suggests that Δ1915 produced a more severe phenotype. This could be due to losing two additional genes associated with cell division and the truncation of the cell division allele sepF in addition to the loss of divIVA. Alternatively, this more severe phenotype may result from the loss of divIVA alone. It is important to note that isolates containing clsA20D and clsR211L did not have obvious septal defects in the presence of yvcRS mutations (Fig. 3), supporting the observation that changes in divIVA are associated with septal abnormalities.
FIG 5.
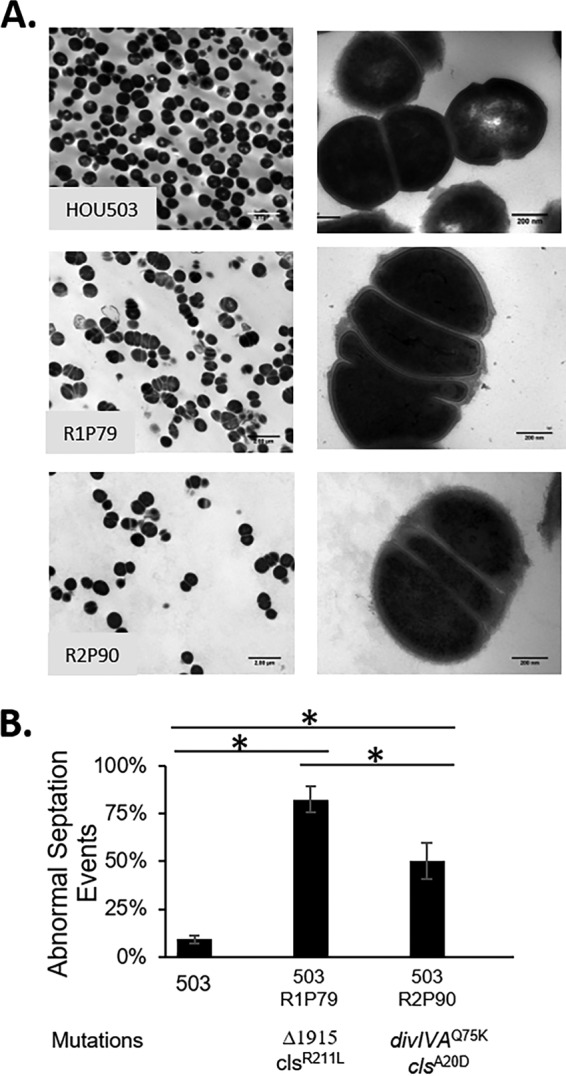
Bioreactor isolates containing divIVA-associated mutations produced abnormal septa. (A) TEM was used at 5,000× (left) and 50,000× (right) to observe cellular morphology of endpoint isolates containing mutations in divIVA. (B) The percentage of abnormal septal events was determined. An asterisks indicates statistical significance (P < 0.05) using the Student t test.
Bioreactor-derived isolates containing divIVA-associated mutations produced differing DAP resistance mechanisms.
We assayed PLL:FITC, BDP:DAP, and NAO binding to determine how mutations in divIVA resulted in DAP resistance. E. faecalis S613 acts as a control to show uniform DAP and normal NAO staining, while E. faecalis R712 showcases the DAP and anionic phospholipid redistribution phenotype. We found that R1P79 (Δ1915, clsR211L) bound PLL:FITC similarly to the ancestor yet bound BDP:DAP in a speckled manner, similar to that observed in the DAP-resistant E. faecalis strain R712 (44). Incubation with NAO also revealed a speckled phenotype, suggesting that Δ1915 resulted in a redistribution of lipid microdomains and DAP binding (Fig. 6 and Fig. S2). Interestingly, R2P90 (divIVAQ75K, clsA20D), while producing aberrant septa, showed different staining patterns: binding less PLL:FITC (suggesting a more positive surface; P < 0.05 using Mann-Whitney test with post hoc Holm-Bonferroni), showing no NAO redistribution, and binding BDP:DAP in a strikingly bimodal manner, reminiscent of streptococci (Fig. 6) (21). Only a subpopulation (approximately 10%) of cells bound significantly more BDP:DAP than the ancestor, whereas the remaining population did not have any discernible difference in drug binding (Fig. 6C). Thus, mechanistically, in R2P90 (divIVAQ75K, clsA20D), DAP resistance appears to have been achieved by a combination of cell envelope changes associated with altered cell division and modest changes in cell surface charge, resulting in hyperaccumulation of DAP in a subset of cells.
FIG 6.

Bioreactor isolates with divIVA-associated mutations produced more complex DAP resistance phenotypes. (A) The relative cell surface charge was determined by incubating the isolates with PLL:FITC. Cells that bind less PLL:FITC have a more positive cell surface charge. R2PXC bound significantly less PLL:FITC than the ancestor, indicating a more positively charged cell surface. An asterisks indicates statistical significance (P < 0.05) using the Mann-Whitney test with post hoc Holm-Bonferroni adjustment. Physical images can be viewed in Fig. S2. (B) Quantification of BDP:DAP binding per cell using ImageJ. An asterisks indicates statistical significance (P < 0.05) using the Mann-Whitney test with post hoc Holm-Bonferroni adjustment. (C) Isolates were incubated with BDP:DAP to determine DAP binding patterns. E. faecalis R712 acts as a control to show the redistribution of binding phenotype. (D) Isolates were incubated with NAO to determine phospholipid microdomain patterning. E. faecalis R712 acts as a control to show the redistribution phenotype, while E. faecalis S613 shows normal staining.
Bioreactor-derived isolates with mutations in oatA had decreased peptidoglycan O-acetylation and increased cell surface charge, consistent with reduced BDP:DAP binding.
Seven different oatA mutations were observed between bioreactor population 1 and population 2, six of which resulted in a truncation of the catalytic C-terminal domain (predicted residues 460 to 628). OatA catalyzes the acetylation of the C-6 hydroxyl group of N-acetylmuramic acid (MurNAc), which contributes to lysozyme resistance and is linked to increased pathogenesis in many species (45–47). Here, two different mutations were identified in endpoint isolates: E480* and E460* (Table 2). All seven different oatA mutations combined comprised 29% of the population 1 on the final day (Fig. S4). While oatA mutations were not observed in population 2 endpoint isolates, two separate oatA mutations (E598* and E460*) were prominent throughout the population 2 experiment (Fig. S4). Their combined presence remained at over 50% of the population between days 5 and 7 but fell to 8% on the final day, as the mutation divIVAQ75K found greater success.
The isolates containing mutations in oatA had several additional mutations. We selected R1P50 (oatAE480*, clsR211L, rpoBG32G, purAL409S, ansP−288, tagBR330S, orf_280C84C, orf_2338−276, and orf_30G25S) and R1P83 (oatAE460*, recAA304S, orf_2207V218A, orf_2597Q74*, and orf_2689G232V) for further analysis, as the only shared mutations between these two endpoint isolates were truncations of oatA. Isolates with either oatA mutation had increased sensitivity to lysozyme, as tested by monitoring cell lysis after incubation with lysozyme, suggesting a loss in catalytic activity and a decrease in peptidoglycan O-acetylation (Fig. 7A). Additionally, we found that R1P50 and R1P83 bound 10% and 40% less PLL:FITC on average, respectively (P < 0.05 using Mann-Whitney test with post hoc Holm-Bonferroni adjustment) (Fig. 7B and Fig. S2), and bound less BDP:DAP (39% and 33%, respectively) than the ancestor (P < 0.05 using the Mann-Whitney with post hoc Holm-Bonferroni adjustment) (Fig. 7C and D). This suggests that the trajectories with oatA mutations conferred increased DAP resistance via repulsion of DAP from the cell surface. Incubation with NAO revealed no evidence of phospholipid redistribution (Fig. S3). Thus, in both flask and bioreactor environments, E. faecium was able to repulse DAP.
FIG 7.

Bioreactor-derived isolates with mutations in oatA had decreased peptidoglycan O-acetylation and increased cell surface charge, consistent with reduced BDP:DAP binding. (A) Lysozyme sensitivity was determined by incubation with or without 0.5 mg/ml lysozyme for 30 min at 37°C. Lane 1, EZ-RUN prestained protein ladder; lanes 2 to 4, incubation with no lysozyme; lanes 2, 5, 8, and 11, HOU503; lanes 3, 6, 9, and 12, R1P50; lanes 4, 7, 10, and 13, R1P83. (B) The relative cell surface charge was determined by incubating the isolates with PLL:FITC. Cells that bind less PLL:FITC have a more positive cell surface charge. An asterisks indicates statistical significance (P < 0.05) using the Mann-Whitney test with post hoc Holm-Bonferroni adjustment. ImageJ was used for quantification. Physical images can be viewed in Fig. S2. (C) Isolates were incubated with BDP:DAP to determine DAP binding patterns. (D) Quantification of BDP:DAP binding per cell. An asterisks indicates statistical significance (P < 0.05) using the Mann-Whitney test with post hoc Holm-Bonferroni adjustment. ImageJ was used for quantification. E. faecalis R712 acts as a control to show the redistribution phenotype, while E. faecalis S613 shows normal staining.
After initial changes to establish either repulsion or redistribution, all evolutionary trajectories converged on alleles linked to membrane homeostasis.
Regardless of adaptive technique or DAP resistance mechanism, after initial mutations were acquired, changes were made to genes affecting lipid/membrane homeostasis. The most commonly affected gene was cls, affecting three flask-transfer isolates and 13 bioreactor-derived isolates and comprising 36% and 61% of the populations 1 and 2, respectively, on the final days. In flask-transfer isolates without a mutation in cls, mutations in glycerophosphoryl diester phosphodiesterase (gdpD), phosphatidylglycerol synthase (pgsA), or orf_1482 (upstream of a different putative pgsA, denoted orf_1481) were observed, suggesting that while mutations to cls were more common, there were alternative evolutionary trajectories that affect membrane phospholipids that may produce the same biological outcome. GdpD is a component of cell membrane phospholipid metabolism, and gdpD mutations have been reported in clinical DAP-resistant isolates E. faecium R494 and E. faecalis R712 (I283P and ΔI170, respectively) as well as in one flask-adapted population here (gdpDH29R) (3, 39). A single G→A mutation was observed 62 bp upstream of pgsA (pgsA−62) in FT1 and FT2. PgsA catalyzes the formation of phosphatidylglycerol-3-P from CDP-diacylglycerol, which is then converted to phosphatidylglycerol by PgpABC. DAP adaptive mutations in pgsA have previously been reported in streptococci, S. aureus, and B. subtilis, resulting in a loss of catalytic function or decrease in expression, causing a potential decrease in available phosphatidylglycerol for DAP binding (48). Here, we found that isolates containing mutations upstream of pgsA and the putative pgsA (mutation in orf_1482 affecting orf_1481 expression) produced a very modest decrease in both transcripts (Fig. S5). It is possible that these transcript reductions result in less phosphatidylglycerol and contribute to DAP resistance.
DISCUSSION
As MDR bacteria spread, physicians are increasingly forced to administer drugs of last resort, such as DAP. It is predicted that the ascent of pan-resistant strains will result in a postantibiotic era in which many aspects of modern medicine would be threatened. In this work, we have mapped out the DAP resistance trajectories available to E. faecium poised to exploit liaFSR-mediated resistance (the most common pathway observed in clinical practice), showing how the environment impacts their acquisition and revealing the multilayered nature of the resistance phenotype in this organism. Our results provide clarity to the complex and seemingly contradictory sets of observations surrounding the acquisition of DAP resistance in E. faecium and allow us to make important distinctions from E. faecalis (5, 27, 39).
Previously, it was found that E. faecalis evolves DAP resistance through mutations in the liaFSR envelope stress response system and cls that divert DAP binding away from the division septum of cells via lipid remodeling. Mutations in these systems are seen in the clinic and in in vitro studies using both flask transfers and bioreactors, suggesting that E. faecalis evolves DAP resistance largely through phospholipid redistribution (25, 28, 44, 49, 50). Mutations within E. faecium liaFSR have also been observed, but strains have not shown evidence of phospholipid redistribution (27). For example, E. faecium HOU503, used here, contains two alleles linked to increased DAP resistance (liaRW73C and liaST120A) and exhibits tolerance to DAP, but, as shown in Fig. 3B, 6C, and 7C and Fig. S3, neither BDP:DAP nor NAO was redistributed away from the division septa. While liaR73C and liaS120A may provide HOU503 with an ability to tolerate DAP, additional mutations were required for HOU503 to survive at higher DAP concentrations. Perhaps surprisingly, none of these additional mutations were present in yycFG, a highly conserved two-component system in which mutations have been observed in clinically derived DAP-resistant isolates of E. faecium and S. aureus (26, 39). This suggests that adapting cells can increase resistance via liaFSR or yycFG mutations but not both, implying that the systems are redundant or engage in cross talk that produces the same net outputs in signaling. The mutations reported here, after committing to liaFSR-associated resistance, predominately resulted in repulsion of DAP from the cell surface. However, in the bioreactor environment of long-term biofilm and rapid growth, HOU503 also diverted DAP binding from septal areas. This combination of resistance strategies, resulting from different adaptive environments, marks a major difference between these two closely related species.
In support of the hypothesis that repulsion is a critical driver for DAP resistance in E. faecium, flask-adapted HOU503 repeatedly evolved mutations in the yvcRS multicomponent system that resulted in the upregulation of the dltABCD operon and mprF, an increase in cell surface charge, and a reduction in DAP binding. These data suggest that YvcRS in enterococci is analogous to the VraFG system in S. aureus, which senses bacitracin and cyclic AMPs in conjunction with GraXSR (potentially analogous to YxdJK in enterococci) to upregulate dltABCD and mprF and mediate the repulsion phenotype (17, 34, 51, 52). Demonstrating that the adaptive mutations in yvcRS are responsible for upregulation of the dltABCD and mprF operons will require genetic studies.
Notably, our recent study that evolved E. faecalis lacking the liaR response regulator to DAP resistance in flasks to identify liaFSR-independent DAP resistance mechanisms found that the two endpoint isolates derived from that experiment contained a mutation in either yvcR or yxdK (53). These E. faecalis isolates containing mutations in the yxdJK-yvcRS system did not exhibit an increase in cell surface charge, did not have a reduction in DAP binding, and did not redistribute DAP binding. This phenomenon marks a potential difference between how yvcRS functions in enterococcal species, but more importantly, it highlights that when the LiaFSR pathway is disabled, alternate evolutionary trajectories can emerge in E. faecalis, as is seen here with the multitude of DAP resistance strategies employed by E. faecium. This adaptability also suggests that diverse signaling pathways that respond to environmental stress are able to act indirectly to compensate for the loss or damage to other systems. Radeck and coworkers noted a similar layered compensatory network in the B. subtilis response to bacitracin (54).
In addition to yvcRS-mediated repulsion, we also found that changes in MurNAc acetylation through oatA mutations contributed to DAP resistance. It is important to note that loss of OatA function may be illustrative of how repulsion can be achieved, but it may be much less likely to occur in a clinical setting. The loss of acetylation leads to lysozyme sensitivity and would likely dramatically decrease pathogenicity, as the host innate immune system would be more effective at clearing this infection (46, 55).
While repulsion was the favored DAP resistance mechanism, redistribution of DAP binding was still observed in a bioreactor environment through the 1,915-bp deletion of cell division-associated genes, including divIVA. The isolate containing this mutation had a dramatic increase in abnormal septation events, redistributed phospholipid microdomains, and diverted DAP binding. The frequent formation of division septa could have the net effect of decreasing the efficiency of DAP in disrupting division by simply increasing the number of potential targets for the antibiotic, ultimately diluting the DAP concentration within the membrane. Interestingly, abnormal septal defects are a common phenotype in DAP-resistant isolates of both E. faecalis and E. faecium (3, 53).
The role of the mutation divIVAQ75K in DAP resistance appears more complex and less clear than that of other trajectories. While these cells have a higher frequency of aberrant septa than the ancestor, there was no diversion phenotype. Of note, divIVAQ75K was the only genotype that produced a strongly bimodal DAP binding phenotype. BDP:DAP-stained cells showed a very distinct subpopulation that hyperaccumulated DAP binding in a uniform manner, similar to what has been seen in Streptococcus mitis/oralis, where it was suggested that such subpopulations act as “superbinders” to bind significantly more DAP and thereby protect the surrounding population (21).
After acquiring the mutations that led to either repulsion or DAP diversion, mutations were subsequently acquired in cls or other genes associated with lipid/membrane chemistry. cls mutations have been reported in DAP-resistant isolates of both E. faecium and E. faecalis (3, 25, 27). Here, we found cls mutations in both adaptive environments, showing their importance in contributing to DAP resistance. While a variety of mutations in cls were observed in the bioreactor (six), two of these mutations were also found in flask-adapted endpoint isolates. Both H215R and R218Q have been found in clinical DAP-resistant isolates of E. faecium (3). Previous work found that these cls mutations increase catalytic activity, creating more cardiolipin, but are neither necessary nor sufficient to confer DAP resistance (35, 39). Furthermore, we found that cls mutations were acquired after initial mutations in the liaFSR operon during E. faecalis DAP adaptation (25). During E. faecium adaptation to DAP in the bioreactor, we again observed that changes to cls were acquired at a later stage, suggesting an ordered and epistatic pathway, regardless of whether repulsion or diversion was being employed as the initial step (25). While not directly conferring DAP resistance, it is clear that cls mutations play an important role in the enterococcal counterattack against DAP.
In summary, we have shown here that the environment influences how E. faecium evolves DAP resistance, even with the presence of alleles that are associated with DAP diversion. Flask environments select for repulsion-mediated resistance via mutations to the yvcRS system that seem to play a role in dltABCD and mprF regulation. Conversely, environments that favor biofilm and more complex structured communities produce both repulsion and DAP diversion mechanisms, although DAP diversion appears less frequently. It is possible that these selective environments are representative of distinct E. faecium infection environments and may predict how different infections (i.e., bacteremia versus colonization of catheters/stents) will evolve DAP resistance. The unifying theme across resistance strategies, adaptive environments, and enterococcal species was mutations affecting membrane architecture, specifically in cls, which may have utility as a DAP resistance marker for clinicians.
MATERIALS AND METHODS
Flask-transfer adaptation.
Five populations of HOU503 were adapted to DAP resistance using 100-fold dilutions each day. To start, five different colonies were used to inoculate each of the five populations containing brain heart infusion (BHI) and Ca+ (50 mg/liter CaCl2). The following day, the populations were transferred to fresh tubes containing 1.5 μg/ml DAP (half the initial DAP MIC). After this, the populations were transferred to two tubes, containing either 1.5× or 2× the current DAP concentration. The tube with the best growth was then propagated into two new tubes. This model was followed until the populations were growing in 8 μg/ml DAP. At the end of adaptation, each population was serially diluted onto nonselective BHI. Two isolates from each population were selected at random for WGS and further analysis.
Directed evolution of E. faecium in a bioreactor.
Clinical isolate E. faecium HOU503 was adapted to DAP resistance in two replicate runs in BHI with supplemented calcium (50 mg/liter CaCl2). Experiments were completed in a Sartorius Stedum Biostat B Plus 1-liter vessel. A 200-ml culture volume was maintained which received an airflow of 0.2 liters/min and was stirred at 100 rpm. The bioreactor was run as a turbidostat, maintaining constant cell density. However, due to the prevalence of biofilm, optical density (OD) probes were rendered useless. To circumvent this problem, CO2 was measured by a Magellan Tandem Pro gas analyzer and used as a proxy to monitor cell density and maintain logarithmic growth as described previously (25, 31–33). Manual OD measurements were taken periodically to ensure appropriate cell density. For inoculation, an overnight (ON) culture was grown from a single colony on nonselective media. One milliliter of this ON culture was then used for inoculating the vessel. The culture was initially grown in the absence of DAP to allow the cells to acclimate to the vessel. DAP was then added at half the initial MIC (1.5 μg/ml). Every 2 days, MIC testing via 2-fold broth dilution was performed on a sample taken from the bioreactor to determine the subsequent DAP concentration. The DAP concentration was only increased in the vessel if the sample culture grew equally well in the higher DAP concentration, as was observed with the current working DAP concentration. By maintaining the population at subinhibitory levels of DAP, there is less selective pressure acting on the population, preventing a bottleneck and allowing for more polymorphism within the population. Samples were taken daily and plated onto BHI and bile esculin agar (BEA) to ensure that the vessel was not contaminated. Three 15-ml samples were taken daily and stored as pellets at −80°C alongside their corresponding supernatants and a glycerol stock. At the end of each run, the population within the vessel was serially diluted and plated onto nonselective BHI agar. To identify phenotypic differences embodying potential different genetic trajectories, 90 isolates were chosen at random and underwent three phenotypic screens: (i) DAP MICs were determined via broth dilution, (ii) cell densities at stationary phase were measured, and (iii) propensity to grow as floc in broth was noted. Based on these three characteristics, 10 or 9 isolates from each run with diverse characteristics were selected for further characterization and WGS. For further details, see the text in the supplemental material.
Crystal violet biofilm assay.
ON culturess were used to inoculate fresh Trypticase soy broth (TSB) and outgrown to an OD at 600 nm (OD600) of 0.5. These cultures were then used to inoculate TSB and grown for 16 h at 37°C in a 96-well plate with shaking. Planktonic cells were aspirated, and the remaining biofilm was fixed with 99% methanol. Plates were washed three times with phosphate-buffered saline (PBS) and then air dried. The biofilm was stained with 0.2% crystal violet and incubated at room temperature for 15 min. The crystal violet was removed, followed by three additional PBS washings, and allowed to air dry. Bound crystal violet was solubilized in 33% acetic acid, and absorbance was measured at A570. The assay was performed in triplicate.
Growth rates.
ON cultures were normalized to an OD600 of 0.05 and used to inoculate fresh BHI in a 96-well plate. Cells were grown within a BioTek EpochII microplate reader with orbital shaking at 37°C. Measurements were taken every 5 min for 24 h. The assay was performed in biological triplicates.
Isolating gDNA and library preparation.
The UltraClean microbial DNA isolation kit (MoBio) was used for isolating gDNA from both endpoint isolates and the daily population samples. Each endpoint isolate was grown ON in 10 ml BHI and pelleted. Alternatively, the pellets collected each day from the bioreactor and stored at −80°C were thawed and immediately used for genomic DNA (gDNA) extraction to eliminate the possible effects of freeze/thaw on the outgrown population. In addition to the published protocol, 5 μl of 5 U/ml mutanolysin and 12.5 μl of 200 mg/ml lysozyme were added to the sample suspended in the microbead solution and incubated at 37°C for 1 h. The Nextera XT kit was used for the generation of paired-end libraries using 2.5 μl gDNA and extending the tagmentation step to 9 min at 55°C. Libraries were sequenced by Genewiz on Hiseq with 2 × 150-bp reads. End-point isolates were sequenced with a minimum 100× coverage, and metagenomic sequences were sequenced with at least 300× coverage.
Analyzing genomic sequencing.
Illumina short reads were aligned to the ancestor, HOU503, and the closed reference genome HOU503_del_LiaR (BioProject accession no. PRJNA544687) using the Breseq-0.29.0 pipeline. HOU503_del_LiaR is the ancestor HOU503 with a liaR deletion, so Sanger sequencing was performed on liaR in HOU503 endpoint isolates to confirm the lack of mutations in this gene. Daily samples were analyzed utilizing the polymorphism command (-p) to identify the frequency of each mutation on a given day. Alleles that reached a minimum of 5% on any day were manually examined to ensure accurate mutation calling. All sequences can be found under BioProject accession no. PRJNA522390.
qPCR.
Total RNA was extracted in accordance with the published Qiagen RNeasy Mini protocol, with the addition of a 30-min incubation at 37°C with mutanolysin and lysozyme to help break open the cells. Samples were DNase I treated in accordance with the Invitrogen protocol, using Taq PCR to confirm the removal of contaminating DNA. cDNA was synthesized using Invitrogen SuperScript III in accordance with the manufacturer’s instructions. qPCR was performed using BioRad iQ Sybr green in accordance with the manufacturer’s instructions on a Bio-Rad CFX connect real-time system. gdhIV was used as the housekeeping gene. Changes in expression were calculated using the 2-ΔΔCT method. Experiments were performed in biological and technical triplicate.
Poly-l-lysine-FITC assay.
Isolates were grown overnight in BHI and then used to inoculate fresh tubes containing BHI. Cells were grown until they reached an OD600 of 0.5 and then washed 3 times in HEPES (20 mM, pH 7.0). Cells were then resuspended in HEPES to an OD600 of 0.1 and incubated with 10 μg/ml PLL:FITC, with shaking at room temperature for 10 min. The culture was then washed once with HEPES to remove unbound PLL:FITC. Cells were resuspended in VectaShield and imaged on a Keyence BZ-Z710. Fluorescence per cell was calculated in ImageJ. Experiments were completed in duplicate on separate days.
BDP:DAP.
The conjugation of the fluorophore Bodipy-Fl to DAP was performed as described previously (37). In brief, DAP and BDP were incubated with shaking at room temperature in 0.2 M sodium carbonate buffer, pH 8.5, for 1 h, followed by extensive dialysis against distilled H20 at 4°C. BDP:DAP was then incubated with different enterococcal isolates with known different BDP:DAP binding patterns at different concentrations to confirm appropriate labeling. To determine BDP:DAP binding patterns, we used methods described previously (11, 12, 44, 56). Briefly, overnight cultures of each isolate were used to inoculate fresh BHI containing Ca+ and grown to an OD600 of 0.5. Cells were then incubated with 32 μg/ml BDP:DAP for 20 min in the dark at 37°C with shaking. Cells were washed once with HEPES (20 mM, pH 7.0). The labeled pellet was resuspended in Vectashield and mounted onto poly-l-lysine-coated coverslips and imaged on a Keyence BZ-Z710 using a standard FITC filter. Experiments were completed in duplicate on two separate days. Fluorescence per cell was calculated using ImageJ.
NAO staining.
NAO has been shown to preferentially bind anionic phospholipids in cell membranes and has been used previously to show phenotypes of phospholipid redistribution (15, 38, 44). Isolates were grown to early exponential phase (OD600 of 0.2) in TSB and then incubated with 500 nM NAO at 37°C with shaking in the dark for 3.5 h. Cells were then washed three times in 0.9% saline, resuspended in Vectashield, immobilized on poly-l-lysine-coated coverslips, and visualized on the Keyence BZ-Z710 microscope.
Transmission electron microscopy.
Selected isolates were grown ON in BHI. One milliliter of culture was pelleted and washed 3 times in 0.1 M Millonig’s phosphate buffer. The pellet was then resuspended in 1 ml glutaraldehyde in Millonig’s phosphate buffer and further processed by the University of Texas Health Science Center Electron Microscopy Core. Imaging was performed on a JEOL JEM 1200 EX electron microscope. To quantify abnormal septation, a minimum of 25 events were selected in a field of view at 5,000× and deemed normal or abnormal in appearance. This was repeated for 6 fields of view, resulting in the characterization of at least 125 events.
Antibiotic cross-sensitivity.
All MICs were determined in triplicate via 2-fold broth dilution. ON cultures were grown at 37°C with shaking at 225 rpm. ON cultures were normalized to an OD600 of 0.05, and 5 μl was used to inoculate 0.5 ml BHI with different concentrations of antibiotics and grown ON with shaking at 37°C. The lowest concentration with no visible bacterial growth was considered the MIC.
Lysozyme sensitivity.
ON cultures were used to inoculate fresh BHI and outgrown to an OD600 of 0.5. These cultures were then pelleted and washed three times in Tris-EDTA (TE). Cells were resuspended in TE and normalized to an OD600 of 0.3. Samples were then incubated with or without 0.5 mg/ml lysozyme at 37°C for 30 min. The supernatants were then run on an SDS-PAGE gel to visualize the degree to which cells were lysed.
Data availability.
All genomic sequences were submitted under BioProject accession numbers PRJNA522390 and PRJNA544687.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health, National Institute of Allergy and Infectious Diseases, grants R01AI to Y.S., K08 AI135093 to W.R.M., K24-AI121296 and R01-AI134637 to C.A.A., and K08-AI113317 to T.T. Funding agencies did not play a role in experimental design, performance, or analysis. C.A.A. has received grants from Merck, MeMEd Diagnostics, and Entasis Therapeutics. W.R.M. has received a grant from Merck and honoraria from Achaogen and Shionogi. T.T. has received a grant from Merck.
A.G.P., H.M., Y.S., and C.A.A. contributed to experimental design and conceptualization. A.G.P., H.M., and A.J.K. completed experiments. W.R.M. and T.T. aided in analysis and data acquisition. A.G.P., C.A.A., and Y.S. contributed to writing the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00790-19.
REFERENCES
- 1.CDC. 2013. Antibiotic resistance threats in the United States, 2013. Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- 2.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 3.Arias CA, Panesso D, McGrath DM, Qin X, Mojica MF, Miller C, Diaz L, Tran TT, Rincon S, Barbu EM, Reyes J, Roh JH, Lobos E, Sodergren E, Pasqualini R, Arap W, Quinn JP, Shamoo Y, Murray BE, Weinstock GM. 2011. Genetic basis for in vivo daptomycin resistance in enterococci. N Engl J Med 365:892–900. doi: 10.1056/NEJMoa1011138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arias CA, Murray BE. 2012. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol 10:266. doi: 10.1038/nrmicro2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hollenbeck BL, Rice LB. 2012. Intrinsic and acquired resistance mechanisms in enterococcus. Virulence 3:421–433. doi: 10.4161/viru.21282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilmore MS, Lebreton F, van Schaik W. 2013. Genomic transition of enterococci from gut commensals to leading causes of multidrug-resistant hospital infection in the antibiotic era. Curr Opin Microbiol 16:10–16. doi: 10.1016/j.mib.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Werth BJ, Barber KE, Ireland CE, Rybak MJ. 2014. Evaluation of ceftaroline, vancomycin, daptomycin, or ceftaroline plus daptomycin against daptomycin-nonsusceptible methicillin-resistant Staphylococcus aureus in an in vitro pharmacokinetic/pharmacodynamic model of simulated endocardial vegetations. Antimicrob Agents Chemother 58:3177–3181. doi: 10.1128/AAC.00088-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munita JM, Mishra NN, Alvarez D, Tran TT, Diaz L, Panesso D, Reyes J, Murray BE, Adachi JA, Bayer AS, Arias CA. 2014. Failure of high-dose daptomycin for bacteremia caused by daptomycin-susceptible Enterococcus faecium harboring LiaSR substitutions. Clin Infect Dis 59:1277–1280. doi: 10.1093/cid/ciu642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.FDA. 2003. Center for drug evaluation and research approval package for: application number 21-572. Food and Drug Administration, Rockville, MD. [Google Scholar]
- 10.Miller WR, Murray BE, Rice LB, Arias CA. 2016. Vancomycin-resistant enterococci: therapeutic challenges in the 21st century. Infect Dis Clin North Am 30:415–439. doi: 10.1016/j.idc.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 11.Hachmann A-B, Angert ER, Helmann JD. 2009. Genetic analysis of factors affecting susceptibility of Bacillus subtilis to daptomycin. Antimicrob Agents Chemother 53:1598–1609. doi: 10.1128/AAC.01329-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pogliano J, Pogliano N, Silverman JA. 2012. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J Bacteriol 194:4494–4504. doi: 10.1128/JB.00011-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tran TT, Munita JM, Arias CA. 2015. Mechanisms of drug resistance: daptomycin resistance. Ann N Y Acad Sci 1354:32–53. doi: 10.1111/nyas.12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Müller A, Wenzel M, Strahl H, Grein F, Saaki TV, Kohl B, Siersma T, Bandow JE, Sahl H-G, Schneider T, Hamoen LW. 2016. Daptomycin inhibits cell envelope synthesis by interfering with fluid membrane microdomains. Proc Natl Acad Sci U S A 113:E7077–E7086. doi: 10.1073/pnas.1611173113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reyes J, Panesso D, Tran TT, Mishra NN, Cruz MR, Munita JM, Singh KV, Yeaman MR, Murray BE, Shamoo Y, Garsin D, Bayer AS, Arias CA. 2014. A liaR deletion restores susceptibility to daptomycin and antimicrobial peptides in multidrug-resistant Enterococcus faecalis. J Infect Dis 211:1317–13125. doi: 10.1093/infdis/jiu602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ernst CM, Staubitz P, Mishra NN, Yang S-J, Hornig G, Kalbacher H, Bayer AS, Kraus D, Peschel A. 2009. The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS Pathog 5:e1000660. doi: 10.1371/journal.ppat.1000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Falord M, Karimova G, Hiron A, Msadek T. 2012. GraXSR proteins interact with the VraFG ABC transporter to form a five-component system required for cationic antimicrobial peptide sensing and resistance in Staphylococcus aureus. Antimicrob Agents Chemother 56:1047–1058. doi: 10.1128/AAC.05054-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mishra NN, Yang SJ, Sawa A, Rubio A, Nast CC, Yeaman MR, Bayer AS. 2009. Analysis of cell membrane characteristics of in vitro-selected daptomycin-resistant strains of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 53:2312–2318. doi: 10.1128/AAC.01682-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mishra NN, Bayer AS, Weidenmaier C, Grau T, Wanner S, Stefani A, Cafiso V, Bertuccio T, Yeaman MR, Nast CC, Yang SJ. 2014. Phenotypic and genotypic characterization of daptomycin-resistant methicillin-resistant Staphylococcus aureus strains: relative roles of mprF and dlt operons. PLoS One 9:e107426-18. doi: 10.1371/journal.pone.0107426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones T, Yeaman MR, Sakoulas G, Yang SJ, Proctor RA, Sahl HG, Schrenzel J, Xiong YQ, Bayer AS. 2008. Failures in clinical treatment of Staphylococcus aureus infection with daptomycin are associated with alterations in surface charge, membrane phospholipid asymmetry, and drug binding. Antimicrob Agents Chemother 52:269–278. doi: 10.1128/AAC.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mishra NN, Tran TT, Seepersaud R, Garcia-de-la-Maria C, Faull K, Yoon A, Proctor R, Miro JM, Rybak MJ, Bayer AS, Arias CA. 2017. Perturbations of phosphatidate cytidylyltransferase (CdsA) mediate daptomycin resistance in Streptococcus mitis/oralis by a novel mechanism. Antimicrob Agents Chemother 61:e02435-16. doi: 10.1128/AAC.02435-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang S, Kreiswirth BN, Sakoulas G, Yeaman MR, Yan Q, Sawa A, Bayer AS. 2009. Enhanced expression of dltABCD is associated with development of daptomycin nonsusceptibility in a clinical endocarditis isolate of Staphylococcus aureus. J Infect Dis 200:1916–1920. doi: 10.1086/648473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bayer AS, Schneider T, Sahl H-G. 2013. Mechanisms of daptomycin resistance in Staphylococcus aureus: role of the cell membrane and cell wall. Ann N Y Acad Sci 1277:139–158. doi: 10.1111/j.1749-6632.2012.06819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khatib TO, Stevenson H, Yeaman MR, Bayer AS, Pokorny A. 2016. Binding of daptomycin to anionic lipid vesicles is reduced in the presence of lysyl-phosphatidylglycerol. Antimicrob Agents Chemother 60:5051–5053. doi: 10.1128/AAC.00744-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller C, Kong J, Tran TT, Arias CA, Saxer G, Shamoo Y. 2013. Adaptation of Enterococcus faecalis to daptomycin reveals an ordered progression to resistance. Antimicrob Agents Chemother 57:5373–5383. doi: 10.1128/AAC.01473-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Werth BJ, Steed ME, Ireland CE, Tran TT, Nonejuie P, Murray BE, Rose WE, Sakoulas G, Pogliano J, Arias CA, Rybak MJ. 2014. Defining daptomycin resistance prevention exposures in vancomycin-resistant Enterococcus faecium and E. faecalis. Antimicrob Agents Chemother 58:5253–5261. doi: 10.1128/AAC.00098-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diaz L, Tran TT, Munita JM, Miller WR, Rincon S, Carvajal LP, Wollam A, Reyes J, Panesso D, Rojas NL, Shamoo Y, Murray BE, Weinstock GM, Arias CA. 2014. Whole-genome analyses of Enterococcus faecium isolates with diverse daptomycin MICs. Antimicrob Agents Chemother 58:4527–4534. doi: 10.1128/AAC.02686-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panesso D, Reyes J, Gaston E, Deal M, Londoño A, Nigo M, Munita JM, Miller W, Shamoo Y, Tran TT, Arias CA. 2015. Deletion of liaR reverses daptomycin resistance in Enterococcus faecium independent of the genetic background. Antimicrob Agents Chemother 59:7327–7334. doi: 10.1128/AAC.01073-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davlieva M, Wu C, Zhou Y, Arias CA, Shamoo Y. 2018. Two mutations commonly associated with daptomycin resistance in Enterococcus faecium LiaST120A and LiaRW73C appear to function epistatically in LiaFSR signaling. Biochemistry 57:6797–6805. doi: 10.1021/acs.biochem.8b01072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mehta HH, Weng J, Prater AG, Elworth RAL, Han X, Shamoo Y. 2018. Pathogenic Nocardia cyriacigeorgica and Nocardia nova evolve to resist trimethoprim-sulfamethoxazole by both expected and unexpected pathways. Antimicrob Agents Chemother 62:e00364-18. doi: 10.1128/AAC.00364-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehta HH, Prater AG, Shamoo Y. 2017. Using experimental evolution to identify druggable targets that could inhibit the evolution of antimicrobial resistance. J Antibiot 71:279–286. doi: 10.1038/ja.2017.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammerstrom TG, Beabout K, Clements TP, Saxer G, Shamoo Y. 2015. Acinetobacter baumannii repeatedly evolves a hypermutator phenotype in response to tigecycline that effectively surveys evolutionary trajectories to resistance. PLoS One 10:e0140489. doi: 10.1371/journal.pone.0140489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beabout K, Hammerstrom TG, Wang TT, Bhatt M, Christie PJ, Saxer G, Shamoo Y. 2015. Rampant parasexuality evolves in a hospital pathogen during antibiotic selection. Mol Biol Evol 32:2585–2597. doi: 10.1093/molbev/msv133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gebhard S, Fang C, Shaaly A, Leslie DJ, Weimar MR, Kalamorz F, Carne A, Cook GM. 2014. Identification and characterization of a bacitracin resistance network in Enterococcus faecalis. Antimicrob Agents Chemother 58:1425–1433. doi: 10.1128/AAC.02111-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davlieva M, Zhang W, Arias CA, Shamoo Y. 2013. Biochemical characterization of cardiolipin synthase mutations associated with daptomycin resistance in enterococci. Antimicrob Agents Chemother 57:289–296. doi: 10.1128/AAC.01743-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hartmann W, Galla H-J. 1978. Binding pf polylysine to charged bilayer membranes molecular organization of a lipid:peptide complex. North Holl Biomed Press 509:474–490. doi: 10.1016/0005-2736(78)90241-9. [DOI] [PubMed] [Google Scholar]
- 37.Pader V, Hakim S, Painter KL, Wigneshweraraj S, Clarke TB, Edwards AM. 2016. Staphylococcus aureus inactivates daptomycin by releasing membrane phospholipids. Nat Microbiol 2:16194. doi: 10.1038/nmicrobiol.2016.194. [DOI] [PubMed] [Google Scholar]
- 38.Barák I, Muchová K. 2013. The role of lipid domains in bacterial cell processes. Int J Mol Sci 14:4050–4065. doi: 10.3390/ijms14024050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tran TT, Panesso D, Gao H, Roh JH, Munita JM, Reyes J, Diaz L, Lobos EA, Shamoo Y, Mishra NN, Bayer AS, Murray BE, Weinstock GM, Arias CA. 2013. Whole-genome analysis of a daptomycin-susceptible Enterococcus faecium strain and its daptomycin-resistant variant arising during therapy. Antimicrob Agents Chemother 57:261–268. doi: 10.1128/AAC.01454-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palmer KL, Kos VN, Gilmore MS. 2010. Horizontal gene transfer and the genomics of enterococcal antibiotic resistance. Curr Opin Microbiol 13:632–639. doi: 10.1016/j.mib.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamoen LW, Meile J, De Jong W, Noirot P, Errington J. 2006. SepF, a novel FtsZ-interacting protein required for a late step in cell division. Mol Microbiol 59:989–999. doi: 10.1111/j.1365-2958.2005.04987.x. [DOI] [PubMed] [Google Scholar]
- 42.Gundogdu ME, Kawai Y, Pavlendova N, Ogasawara N, Errington J, Scheffers D. 2011. Large ring polymers align FtsZ polymers for normal septum formation. EMBO J 30:617–626. doi: 10.1038/emboj.2010.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pinho MG, Kjos M, Veening J. 2013. How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nat Rev Microbiol 11:601–614. doi: 10.1038/nrmicro3088. [DOI] [PubMed] [Google Scholar]
- 44.Tran TT, Panesso D, Mishra NN, Mileykovskaya E, Guan Z, Munita JM, Reyes J, Diaz L, Weinstock GM, Murray BE, Shamoo Y, Dowhan W, Bayer AS, Arias CA. 2013. Daptomycin-resistant Enterococcus faecalis diverts the antibiotic molecule from the division septum and remodels cell membrane. mBio 4:1–10. doi: 10.1128/mBio.00281-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sychantha D, Jones CS, Little DJ, Moynihan PJ, Robinson H, Galley NF, Roper DI, Dowson CG, Howell PL, Clarke AJ. 2017. In vitro characterization of the antivirulence target of Gram-positive pathogens, peptidoglycan O-acetyltransferase A (OatA). PLoS Pathog 13:e1006667. doi: 10.1371/journal.ppat.1006667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bera A, Herbert S, Jakob A, Vollmer W, Götz F. 2005. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol Microbiol 55:778–787. doi: 10.1111/j.1365-2958.2004.04446.x. [DOI] [PubMed] [Google Scholar]
- 47.Sychantha D, Clarke AJ. 2018. Peptidoglycan modification by the catalytic domain of Streptococcus pneumoniae OatA follows a ping-pong bi-bi mechanism of action. Biochemistry 57:2394–2401. doi: 10.1021/acs.biochem.8b00301. [DOI] [PubMed] [Google Scholar]
- 48.Peleg AY, Miyakis S, Ward DV, Earl AM, Rubio A, Cameron DR, Pillai S, Moellering RC, Eliopoulos GM. 2012. Whole genome characterization of the mechanisms of daptomycin resistance in clinical and laboratory derived isolates of staphylococcus aureus. PLoS One 7:e28316. doi: 10.1371/journal.pone.0028316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davlieva M, Shi Y, Leonard PG, Johnson TA, Zianni MR, Arias CA, Ladbury JE, Shamoo Y. 2015. A variable DNA recognition site organization establishes the LiaR-mediated cell envelope stress response of enterococci to daptomycin. Nucleic Acids Res 43:4758–4773. doi: 10.1093/nar/gkv321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palmer KL, Daniel A, Hardy C, Silverman J, Gilmore MS, Al P. 2011. Genetic basis for daptomycin resistance in Enterococci. Antimicrob Agents Chemother 55:3345–3356. doi: 10.1128/AAC.00207-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meehl M, Herbert S, Go F, Cheung A. 2007. Interaction of the GraRS two-component system with the VraFG ABC transporter to support vancomycin-intermediate resistance in Staphylococcus aureus. Antimicrob Agents Chemother 51:2679–2689. doi: 10.1128/AAC.00209-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dintner S, Heermann R, Fang C, Jung K, Gebhard S. 2014. A sensory complex consisting of an ATP-binding cassette transporter and a two-component regulatory system controls bacitracin resistance in Bacillus subtilis. J Biol Chem 289:27899–27910. doi: 10.1074/jbc.M114.596221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller WR, Tran TT, Diaz L, Rios R, Khan A, Reyes J, Prater AG, Panesso D, Shamoo Y, Arias CA. 2019. LiaR-independent pathways to daptomycin resistance in Enterococcus faecalis reveal a multilayer defense against cell envelope antibiotics. Mol Microbiol 111:811–824. doi: 10.1111/mmi.14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Radeck J, Gebhard S, Orchard PS, Kirchner M, Bauer S, Mascher T, Fritz G. 2016. Anatomy of the bacitracin resistance network in Bacillus subtilis. Mol Microbiol 100:607–620. doi: 10.1111/mmi.13336. [DOI] [PubMed] [Google Scholar]
- 55.Herbert S, Bera A, Nerz C, Kraus D, Peschel A, Goerke C, Meehl M, Cheung A, Götz F. 2007. Molecular basis of resistance to muramidase and cationic antimicrobial peptide activity of lysozyme in staphylococci. PLoS Pathog 3:e102. doi: 10.1371/journal.ppat.0030102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hachmann AB, Sevim E, Gaballa A, Popham DL, Antelmann H, Helmann JD. 2011. Reduction in membrane phosphatidylglycerol content leads to daptomycin resistance in Bacillus subtilis. Antimicrob Agents Chemother 55:4326–4337. doi: 10.1128/AAC.01819-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller WR, Bayer AS, Arias CA. 2017. Mechanism of action and resistance to daptomycin in staphylococcus aureus and enterococci. Cold Spring Harb Perspect Med 6:a026997. doi: 10.1101/cshperspect.a026997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Munoz-Price LS, Lolans K, Quinn JP. 2005. Emergence of resistance to daptomycin during treatment of vancomycin-resistant Enterococcus faecalis infection. Clin Infect Dis 41:565–566. doi: 10.1086/432121. [DOI] [PubMed] [Google Scholar]
- 59.Bæk KT, Thøgersen L, Mogenssen RG, Mellergaard M, Thomsen LE, Petersen A, Skov S, Cameron DR, Peleg AY, Frees D. 2015. Stepwise decrease in daptomycin susceptibility in clinical Staphylococcus aureus isolates associated with an initial mutation in rpoB and a compensatory inactivation of the clpX gene. Antimicrob Agents Chemother 59:6983–6991. doi: 10.1128/AAC.01303-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cui L, Isii T, Fukuda M, Ochiai T, Neoh H-M, Camargo IL, Watanabe Y, Shoji M, Hishinuma T, Hiramatsu K. 2010. An RpoB mutation confers dual heteroresistance to daptomycin and vancomycin in Staphylococcus aureus. Antimicrob Agents Chemother 54:5222–5233. doi: 10.1128/AAC.00437-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fischer A, Yang SJ, Bayer AS, Vaezzadeh AR, Herzig S, Stenz L, Girard M, Sakoulas G, Scherl A, Yeaman MR, Proctor RA, Schrenzel J, François P. 2011. Daptomycin resistance mechanisms in clinically derived staphylococcus aureus strains assessed by a combined transcriptomics and proteomics approach. J Antimicrob Chemother 66:1696–1711. doi: 10.1093/jac/dkr195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mishra NN, McKinnell J, Yeaman MR, Rubio A, Nast CC, Chen L, Kreiswirth BN, Bayer AS. 2011. In vitro cross-resistance to daptomycin and host defense cationic antimicrobial peptides in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob Agents Chemother 55:4012–4018. doi: 10.1128/AAC.00223-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Howden BP, Davies JK, Johnson PDR, Stinear TP, Grayson ML. 2010. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev 23:99–139. doi: 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomic sequences were submitted under BioProject accession numbers PRJNA522390 and PRJNA544687.