

Mechanisms of magnesium homeostasis in Mycobacterium tuberculosis are poorly understood. Here, we describe the characterization of a pyrimidinetrione amide scaffold that disrupts magnesium homeostasis in the pathogen by direct binding to the CorA Mg2+/Co2+ transporter. Mutations in domains of CorA that are predicted to regulate the pore opening in response to Mg2+ ions conferred resistance to this scaffold.
KEYWORDS: CorA transporter, Mycobacterium tuberculosis, magnesium homeostasis, pyrimidinetrione amide, structure-activity relationship
ABSTRACT
Mechanisms of magnesium homeostasis in Mycobacterium tuberculosis are poorly understood. Here, we describe the characterization of a pyrimidinetrione amide scaffold that disrupts magnesium homeostasis in the pathogen by direct binding to the CorA Mg2+/Co2+ transporter. Mutations in domains of CorA that are predicted to regulate the pore opening in response to Mg2+ ions conferred resistance to this scaffold. The pyrimidinetrione amides were cidal against the pathogen under both actively replicating and nonreplicating conditions in vitro and were efficacious against the organism during macrophage infection. However, the compound lacked efficacy in infected mice, possibly due to limited exposure. Our results indicate that inhibition of Mg2+ homeostasis by CorA is an attractive target for tuberculosis drug discovery and encourage identification of improved CorA inhibitors.
INTRODUCTION
Short-course chemotherapy for tuberculosis was adopted by the WHO in 1991 for drug-sensitive disease, requiring 6 months of multidrug treatment (1). The risk of drug-related toxicity, as well as noncompliance, increases proportionally over time, which has motivated efforts to reduce the treatment duration (2). The current front-line drug regimen consists of drugs that target RNA synthesis (rifampin) and cell wall biosynthesis (isoniazid/ethambutol) and pyrazinamide (which has an ill-defined mechanism of action) (3, 4). Identification of drugs that target novel pathways in the pathogen has been proposed as a way of developing new drug combinations that could reduce treatment duration (5). To this end, mechanisms that are critical for survival under various growth conditions are predicted to encompass targets that could lead to a rapid decline in bacterial viability in vivo.
Magnesium is an essential cofactor in a plethora of enzymatic reactions. It plays a structural role in the integrity of nucleic acids and proteins, as well as maintaining the integrity of the cell membrane and cell wall (6, 7). In M. tuberculosis, three mutants that exhibited attenuation at low extracellular magnesium concentrations have been described: perM, encoding a membrane protein of unknown function, deletion of which results in in vivo attenuation and inability to grow at low Mg2+ concentrations; phoP, encoding the transcriptional regulator of a two-component system important for complex lipid biosynthesis and virulence; and mgtC, which encodes a P-type ATPase likely acting as an accessory protein for Mg2+ import, deletion of which results in inability to grow at low Mg2+ concentrations at acidic pH and in loss of virulence (8–10). The in vivo attenuation of these mutants in M. tuberculosis, as well as other bacterial pathogens with inactivated Mg2+-dependent processes, has been hypothesized to point to restriction of this essential cation as a possibly host-mediated mechanism to control bacterial pathogens (7–10). Indeed, phagosomal Mg2+ concentrations, at least during Salmonella enterica serovar Typhimurium infection, have been estimated to be in the 10 to 50 μM range (11). The predicted low phagosomal Mg2+ concentrations and the increased Mg2+ requirement of M. tuberculosis at low pH suggest that access to the nutrient could be growth limiting in vivo. Consequently, inhibitors of Mg2+ homeostasis would affect bacterial survival at every stage of host pathogenesis (12). The mechanisms by which M. tuberculosis maintains Mg2+ homeostasis are poorly understood. M. tuberculosis encodes two Mg2+ transporters, CorA and MgtE, neither of which is apparently essential based on saturating genome transposon mutagenesis studies (13, 14).
In this work, we describe the identification of a series of compounds that exert potent growth inhibition on M. tuberculosis and inhibit Mg2+ uptake via direct binding to the CorA transporter. A representative of this inhibitor series lacked in vivo efficacy against M. tuberculosis in mice. This was presumed to be related to insufficient compound exposure in vivo.
RESULTS
Identification of a pyrimidinetrione amide with Gram-positive antibacterial activity.
The series, as represented by compound 1, was identified in a large whole-cell phenotypic screen against M. tuberculosis and was found to have potent antimycobacterial activity on two distinct growth media (Table 1). The pyrimidinetrione amide core is based on barbituric acid. In order to establish some preliminary structure-activity relationship (SAR), analogues were prepared with different amines, including a simple aniline (compound 2), cyclohexylamine (compound 3), butylamine (compound 4), benzylamine (compound 5), and 3-picolylamine (compound 6). Except for compound 2, none showed equivalent antimicrobial activities in both media (Table 1). Although compound 5 showed good potency in glycerol-alanine salts-Tween 80 (GAST) medium (a minimal medium lacking bovine serum albumin [BSA]), the lack of potency in 7H9 medium suggested high protein binding, discouraging further evaluation. The modification of the amide linker to generate either a sulfonylamine (compound 7) or a ketone (compound 8) also resulted in complete loss of antitubercular activity.
TABLE 1.
Antitubercular activities of pyrimidinetrione amide analogues

Compounds 1 to 8 are all known molecules.
MIC of compound against M. tuberculosis H37Rv in Middlebrook 7H9-BSA containing glucose/glycerol/Tween 80 (7H9) or GAS medium with Tween 80 (GAST).
Based on the initial SAR described in Table 1, a series of compounds modified in the aniline moiety were further evaluated (Table 2). Interestingly, irrespective of the substituents in the para position of the aniline, compounds 9 to 14 showed good antitubercular activities. Compounds 15 and 16, which had a substituent in the ortho position, also showed activity, while other analogues with heteroaromatic rings instead of phenyl groups, such as pyridine (compounds 17 to 19) and isoxazole (compound 20), generally had reduced potency. Because the core is related to barbituric acid, we wanted to rule out nonspecific effects due to mitochondrial-membrane depolarization (15). We tested for cytotoxicity against HepG2 cells during growth on either glucose or galactose as a sole carbon source. Growth on galactose forces cells to use mitochondrial respiration to generate ATP rather than through glycolysis, as occurs during growth on glucose (16). This showed that several of the compounds were associated with clear mitochondrial toxicity, as evidenced by the enhanced cytotoxicity of compounds during growth on galactose. Cytotoxicity in galactose medium generally tracked with antimycobacterial potency (see Fig. S1 in the supplemental material). However, compounds having electron-donating alkyl groups, methyl (compound 9) and isopropyl (compound 10), were both selective and had no mitochondrial toxicity based on a lack of inhibition of HepG2 cell growth on either glucose or galactose.,
TABLE 2.
Antitubercular activities and cytotoxicities of pyrimidinetrione amide analogues


Compounds 1, 2, 9 to 11, and 13 are known molecules.
MICs of compounds against M. tuberculosis H37Rv in Middlebrook 7H9-BSA containing glucose/glycerol/Tween 80 (7H9), GAS medium with Tween 80 (GAST), Middlebrook 7H9-BSA-tyloxapol-butyrate-0.1 mM nitrite, pH 6.0 (Butyrate), and a CorA-E212D mutant M. tuberculosis strain in Middlebrook 7H9-BSA containing glucose-glycerol-Tween 80 [7H9 (corA::E212D)].
Cytotoxicities of compounds tested against HepG2 cells in DMEM-10% FBS supplemented with glucose or galactose.
ND, not done.
The compound series was not selectively antitubercular, as seen from its broader-spectrum activity against Mycobacterium smegmatis, Bacillus subtilis, and Staphylococcus aureus (see Table S1 in the supplemental material). However, the series lacked activity against Gram-negative bacteria, such as Escherichia coli and Pseudomonas aeruginosa (data not shown), suggesting a mechanism of action unique to Gram-positive bacteria.
The pyrimidinetrione amide inhibits magnesium uptake in M. tuberculosis.
In an effort to understand the mechanism of action of the series, mutants resistant to compound 1 were raised. Mutants were obtained at a frequency of resistance of 10−8. Three that were confirmed to be more than 100-fold resistant to the compound were submitted for whole-genome sequencing. Two had independent mutations in corA (Rv1239c), corA::E212D, and corA::A317S; other single nucleotide polymorphisms (SNPs) present showed no evidence of mechanism-of-action-associated resistance (Table 3). The third mutant had an SNP profile identical to that of the second mutant, suggesting that it may have arisen as a sibling.
TABLE 3.
Mutations identified in pyrimidinetrione amide-resistant M. tuberculosis strains
| Strain | Polymorphismsa |
|---|---|
| E2.1 | corA::E212D, 4221182::T → C (30 bp upstream of Rv3776) |
| E2.2 | corA::A317S, Rv0275c::S233S, 3281706 → G (in aa 342/2111 of mas) |
| E2.6 | Same as E2.2 |
aa, amino acids.
The corA gene encodes a putative magnesium and cobalt transporter predicted to be nonessential by genome-wide transposon mutagenesis studies (13, 14). To confirm that SNPs in corA were associated with resistance, we raised strains resistant to compounds 1, 10, 12, 13, and 15 and specifically sequenced their corA genes. All the resistant strains contained point mutations within the corA gene (see Table S2 in the supplemental material). To interpret the effects of these mutations on corA function, an M. tuberculosis CorA (MtCorA) homology model was generated from the crystal structure of Thermotoga maritima CorA (TmCorA) using Phyre2 (17). Unexpectedly, the mutations did not occur at the divalent-cation sensor region, where Mg2+ ions bind. Instead, the homology model predicted that the mutations occurred within the acidic (E212D) and kink (G299S and M300V/L) regions, which are involved in the gating motion of CorA, and the hydrophobic tunnel (A317S), through which Mg2+ ions flow (see Fig. S2 in the supplemental material). The mutation data suggested that the pyrimidinetrione amide scaffold affected the gating mechanism of CorA, resulting in the inhibition of Mg2+ uptake (see Fig. S2). This hypothesis was supported by the Mg2+ dependence of the growth inhibition in M. tuberculosis (Fig. 1A), as well as other Gram-positive bacteria (see Table S2).
FIG 1.

The pyrimidinetrione amide scaffold inhibits Mg2+ uptake by M. tuberculosis. (A) Magnesium dependence of the pyrimidinetrione amide MIC against M. tuberculosis in Mg2+-free medium with the indicated concentrations of Mg2+. (B) Co2+ dependence of the pyrimidinetrione amide MIC against M. tuberculosis in Co2+-free medium with the indicated concentrations of Co2+. (C) Fold changes in intracellular Mg2+ concentrations as measured by ICP-MS compared to vehicle (DMSO) control after 24 h of treatment with moxifloxacin (Mox) or compound 10 at 1- or 10-fold the MIC in 7H9 medium that contained 0.4 mM Mg2+. Significance between groups was determined by Tukey’s multiple-comparison test, with significance set at P values of less than 0.05. *, 0.02; ***, 0.0006; ns, not significant. The error bars indicate standard deviation.
Co2+ was unable to rescue cells from pyrimidinetrione-dependent growth inhibition (Fig. 1B). The inability of Co2+ to rescue growth inhibition was not surprising, since there are no known essential cobalt-dependent enzymes in M. tuberculosis. Magnesium is an essential metal cofactor in many key metabolic enzymes, and the specificity of some of the pyrimidinetrione amides for inhibiting bacterial growth and not HepG2 cell growth suggested that magnesium uptake could be a novel target for drug development.
To confirm that the series inhibited Mg2+ uptake, intracellular concentrations of the cation were measured by inductively coupled plasma mass spectrometry (ICP-MS). This analysis confirmed that compound 10 treatment generated a concentration-dependent decrease in intracellular magnesium levels (Fig. 1C). Moxifloxacin was chosen as a negative control because it was not expected to affect cell wall or membrane integrity, both of which could potentially influence intracellular Mg2+ levels. However, moxifloxacin resulted in an unexpected increase in intracellular magnesium concentrations (Fig. 1C) for reasons that were not clear.
The pyrimidinetrione amide-Mg2+ complex binds to the CorA transporter.
The core of the series shows structural similarity to the pyrimidinedione scaffold associated with Mg2+ binding in drugs such as raltegravir (18). To explore whether this series bound Mg2+ ions, we analyzed the UV-visible (Vis) spectral response of compound 10 upon titration with Mg2+ ions. A clear cation-dependent decrease in absorbance at 280 nm was seen (see Fig. S3 in the supplemental material). The coordination complex of compound 10 with Mg2+ was further investigated by nuclear magnetic resonance (NMR) studies; based on the splitting pattern of the signals of the asymmetric pyrimidinetrione moiety, compound 10 forms the enol tautomer when interacting with Mg2+ (see Fig. S4 in the supplemental material). The coordination of Mg2+ by this scaffold could suggest that the compounds merely act to deplete free Mg2+ ions, with the corA mutations conferring higher affinity for the Mg2+·6H2O complex. However, arguing against this notion, in the absence of compound the corA mutants required a 250-fold higher Mg2+ concentration than the wild-type strain in order to grow (see Table S3 in the supplemental material).
In order to test whether the Mg2+-pyrimidinetrione amide complex directly bound to CorA, we determined the thermal stability of recombinantly expressed CorA in the presence of Mg2+ and compound 10 (Fig. 2). This showed that all these ligands increased the thermal stability of the protein, indicating binding to the transporter (Fig. 2 and Table 4). The apparent dissociation constant of CorA with metal ions and compound 10 was determined using fluorescence change based on tryptophan quenching (Table 5; see Fig. S5 in the supplemental material). The results indicated that Co2+ was poorly bound to CorA compared to Mg2+, whereas compound 10 bound with 2-fold higher affinity than Mg2+ alone, irrespective of the presence of Mg2+. Although this result showed that the Mg2+-pyrimidinetrione amide complex bound and stabilized the CorA structure, it was still unclear how this event affected intracellular Mg2+ homeostasis, resulting in bactericidality. We predicted that mutant strains, such as the corA::E212D strain, could survive exposure to compound 10 as a result of lower binding affinity of the CorA::E212D protein to the Mg2+-compound 10 complex, thereby overcoming the disruption of Mg2+ homeostasis. Therefore, we performed similar thermal-stability assays and fluorescence shift assays using one of the mutant CorA proteins, the purified recombinant CorA::E212D, in the presence of Mg2+, compound 10, or compound 10 with Mg2+. The assay result showed that the Mg2+-pyrimidinetrione amide complex could also bind to the mutated CorA::E212D (see Tables S4 and S5 in the supplemental material). This result suggested that the mutations observed on CorA do not have a measurable effect on the binding affinity of the Mg2+-pyrimidinetrione amide complex for the transporter (bearing in mind the limited analytical sensitivity of these assays).
FIG 2.
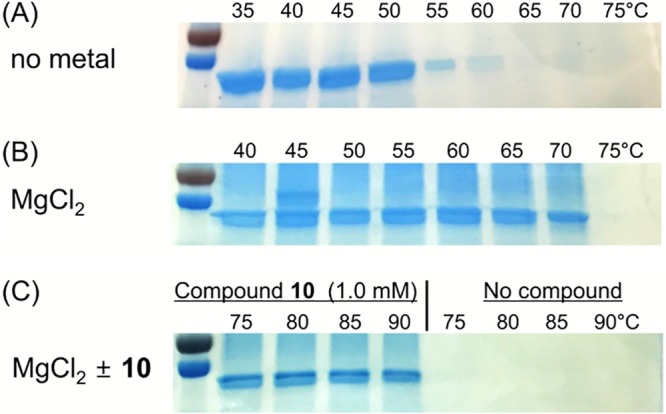
Magnesium and the pyrimidinetrione scaffold bind to the CorA transporter protein. The thermostability of recombinantly expressed CorA from M. tuberculosis was measured with no ligand (A), 0.125 mM MgCl2 (B), and 0.125 mM MgCl2 with or without 1.0 mM compound 10 (C). The protein was preincubated at room temperature with the indicated ligands before heating to the indicated temperatures in a thermocycler. Protein precipitates were removed by filtration, and soluble protein was analyzed by SDS-PAGE.
TABLE 4.
Thermostability of CorA in the presence of metal cations, EDTA, and compound 10
| CorA additive | Melting temp (°C) |
|---|---|
| None | 55 |
| 0.125 mM MgCl2 | 70 |
| 1 mM compound 10 | ≥95 |
| 1 mM compound 10/0.125 mM MgCl2 | ≥95 |
| 1 mM EDTA/0.125 mM MgCl2 | ≥95 |
TABLE 5.
Kd(app) of CorA with metal cations and compound 10
| CorA additive in fluorescence shift assay | Kd(app) (μM) |
|---|---|
| Mg2+ | 47.5 ± 3.0 |
| Co2+ | 205 ± 18 |
| Compound 10 | 20.0 ± 4.3 |
| Compound 10/1 mM Mg2+ | 24.1 ± 6.3 |
| Compound 10/1 mM Co2+ | 21.8 ± 4.1 |
Pyrimidinetrione amide decreases M. tuberculosis viability in vitro and ex vivo but lacks apparent in vivo efficacy.
The pyrimidinetrione amide compound 10 was cidal against actively growing M. tuberculosis cells, showing a 2-log-unit kill over a week of exposure, even at concentrations as low as 2 times the MIC value. Higher concentrations resulted in a more rapid cidal effect with no detectable viable cells remaining, as determined by colony counts on solid agar (Fig. 3A). We next sought to investigate the importance of Mg2+ uptake under nonreplicating conditions using nonreplicating persistence induced by adaptation to hypoxia with subsequent exposure of cells to compounds under anaerobic conditions. This showed that Mg2+ uptake was critical under these conditions, as evidenced by the greater than 3-log-unit kill after a week of compound exposure (Fig. 3B).
FIG 3.

The pyrimidinetrione amide scaffold is cidal against replicating, as well as nonreplicating, M. tuberculosis in vitro and ex vivo. (A) Kill kinetics of compound 10 against M. tuberculosis during active replication in vitro compared to negative (DMSO) and positive (5 μg/ml rifampin [RIF]) controls. (B) Anaerobic cidal activities of compounds 9, 10, and 20 compared to negative (untreated, DMSO, and 1 μg/ml isoniazid [INH]) and positive (100 μM metronidazole) controls after 7 days of compound exposure. M, metronidazole; U, untreated (control); D, DMSO; 10×, 10-fold MIC concentration isoniazid. (C) Efficacies of compounds 1, 9, 10, and 16 during growth of M. tuberculosis in J774 macrophages compared to the negative (DMSO) and positive (0.5 and 5 μg/ml RIF) controls. The horizontal bar indicates inoculum (day 0). The error bars indicate SD.
The cidal consequence of disrupting Mg2+ homeostasis during active growth, as well as nonreplicating persistence, prompted us to explore the antitubercular effect during parasitism of host macrophages. We selected compounds that had no general cytotoxic or mitochondrial toxic effect (compounds 9 and 10); no general cytotoxic effect yet with limited mitochondrial toxicity, giving a 10-fold selectivity window (compound 16); or limited general cytotoxicity and higher mitochondrial toxicity that still gave us a greater than 10-fold selectivity window (compound 1) (Table 1). All of these compounds exerted time- and dose-dependent intracellular (ex vivo) killing of M. tuberculosis growing in macrophages (Fig. 3C).
The promising activity of compound 10 against M. tuberculosis during growth in macrophages encouraged us to explore its in vivo efficacy. Preliminary pharmacokinetic (PK) analysis indicated the half-life of compound 10 was 2.5 ± 0.4 h when dosed once daily, showing that at least twice-per-day (BID) dosing was needed. Unfortunately, the compound was not tolerable by mice at high (100 mg/kg of body weight) doses. At 30 mg/kg, dosed BID by oral gavage, the compound achieved an area under the concentration-time curve (AUC) of 56 μg · h/ml, which gave an AUC/MIC ratio of 250 and plasma concentrations that were almost 10-fold above MICs at the first trough and just above the MIC at 24 h (0.3 μg/ml ± 0.16 [standard deviation {SD}]) (Fig. 4A). This dosing schedule was tolerable in naive mice. Subsequently, M. tuberculosis -infected mice were treated with 30 or 15 mg/kg BID; however, after multiple BID doses, 30 mg/kg was intolerable for the infected mice, necessitating a de-escalation to 30 mg/kg once per day for that group for the remaining dosing period. Due to adverse effects, compound 10 could not be administered at higher concentrations, and at the highest tolerable concentration, it lacked efficacy after 2 and 4 weeks of treatment in infected mouse lungs (Fig. 4B) and spleens (results not shown).
FIG 4.

The pyrimidinetrione amide lacks in vivo efficacy. (A) PK profile of compound 10 after twice-daily administration at 30 mg/kg. (B) M. tuberculosis bacterial burdens in infected mice after 2 and 4 weeks of once-daily or twice-daily treatment at 30 and 15 mg/kg, respectively. Treatment was initiated at 2 weeks after aerosol infection of the mice. The error bars indicate SD. Q, once daily; B, twice daily.
DISCUSSION
We have identified a pyrimidinetrione amide scaffold with potent in vitro activity against M. tuberculosis. Resistant mutants mapped to the CorA transporter, with Mg2+ supplementation abrogating the growth inhibition in a concentration-dependent fashion. Using thermostability assays and fluorescence shift assays, we were able to demonstrate that the inhibitor binds to CorA in the presence and absence of Mg2+. The ligand binding site for the inhibitor remains to be identified, but we were also able to show that resistance-conferring amino acid mutations had no effect on ligand binding. Instead, the mutations mapped to the acidic and kink regions of the cytoplasmic domain of the CorA transporter, which are involved in the gating motion of the pore, indicating that modes of resistance were not due to changes in CorA-ligand interaction but likely the results of perturbation of the gating motion in response to Mg2+ dissociation from the protein. Low cytosolic Mg2+ concentrations result in cation dissociation, with the resultant channel opening driven by this gating mechanism. In line with a loss of regulation of the pore opening of the cation channel, the corA::E212D and corA::A317 mutants required a 250-fold higher Mg2+ concentration in the medium than parental wild-type cells for growth (results not shown). During the preparation of this article, a report was published that found mutations in CorA similar to those of a pyrimidinetrione, as well as a 4-hydroxyquinoline scaffold (19). These mutations were predicted to result in channel opening, resulting in increased cytosolic Mg2+ concentrations. Similar to our work, cytosolic concentrations of both free and bound Mg2+ decreased, complementing our data showing that these inhibitors directly influence the influx/efflux of the cation.
The observation that the metabolic consequences of CorA inhibition are both growth arrest and death was surprising, since there is an alternative Mg2+ transporter encoded by mgtE in the bacterium. The finding that mutations in only corA could confer resistance suggests that regulation of Mg2+ homeostasis by the cation is predominantly conferred by the CorA transporter, where changes in MgtE levels do not occur rapidly enough to overcome this dysregulation of Mg2+ homeostasis. In contrast, during genetic inactivation of corA, compensatory genetic changes could occur to alter levels of MgtE as cells are gradually depleted of CorA protein. The finding that corA, but not mgtC or mgtE, is conserved in Mycobacterium leprae, a mycobacterium with a minimal genome, points to the conserved nature of Mg2+ homeostasis by the transporter. Despite this, there is no evidence for the in vivo essentiality of M. tuberculosis corA during mouse pathogenesis (20). Intriguingly, these compounds were active only against the Gram-positive bacteria tested and not Gram-negative bacteria. CorA is widely distributed in bacteria, including E. coli and P. aeruginosa (6). Lack of activity against these Gram-negative microbes could be due to lack of binding to their CorA counterpart, different mechanisms of Mg2+ homeostasis, lack of transport of the pyrimidinetrione amides across the outer membrane, or their efflux by multidrug efflux pumps.
Our results emphasize the importance of Mg2+ homeostasis during active replication, as well as nonreplicating persistence in vitro and also during parasitism of host macrophages. Free Mg2+ concentrations in the lung were determined to be 0.67 ± 0.08 mM (data not shown), a value similar to reported concentrations in plasma (21), which is below the level that overcomes the growth inhibition by the pyrimidinetrione amides. The lack of in vivo efficacy was disappointing, although the pharmacokinetic studies suggested that blood serum drug concentrations were only sufficiently above the MIC for half of the dosing period when extrapolated from the PK parameters (Fig. 4A). Furthermore, the concentration of compound in the cellular lesions may be well below the MIC level during this dosing period. In addition, despite the lack of cytotoxicity of the compound, clinical signs of toxicity were evident when mice were dosed at 100 mg/kg (results not shown), prohibiting additional studies to probe the in vivo efficacy of the compound.
In conclusion, our results demonstrate the critical role of CorA in maintaining Mg2+ homeostasis and its attractiveness as a drug target. Despite the in vitro and ex vivo efficacy of the pyrimidinetrione amides, our data suggest that an alternative scaffold that inhibits CorA function would need to be identified in order to perform the proof of concept experiments that establish the validity of this target in vivo.
MATERIALS AND METHODS
Chemistry.
All chemicals and reagents were purchased from Aldrich (Sigma-Aldrich, St. Louis, MO, USA) and Lancaster (Alfa Aesar, Johnson Matthey Company, Ward Hill, MA, USA) and were used without further purification. Commercially available analogues of pyrimidinetrione amide were purchased from Enamine LLC (Monmouth Junction, NJ, USA), Bionet America, Inc. (Tustin, CA, USA), and MicroCombiChem GmbH (Wiesbaden, Germany). Reactions were monitored by thin-layer chromatography (TLC) performed on silica gel glass plates containing 60 GF-254 (Millipore Sigma, MO, USA), and visualization on TLC was achieved by UV light or iodine indicator. Column chromatography was performed with Merck 60- to 120-mesh silica gel. 1H nuclear magnetic resonance (NMR) spectra were recorded on a Varian Mercury-300 NMR spectrometer, and chemical shifts are reported in parts per million downfield from an internal trimethylsilane (TMS) standard. Routine mass analyses (low-resolution mass spectrometry [LRMS]) were performed on an HP Agilent liquid chromatography-mass spectrometry (LC-MS) series 1100 system equipped with a reverse-phase column (Agilent Poroshell 120 EC-C18; 2.7 μm; 50 by 2.1 mm) and a photodiode array detector using electrospray ionization (ESI). All the reported compounds were pure and >95%.
Synthesis of compounds 12 and 14 to 20 (22).
To a solution of 1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (1.0 eq) in dichloromethane (DCM) (0.1 M) were added pyridine (1.2 eq) and 4-dimethylaminopyridine (0.1 eq) at 0°C. Ethylcarbonochloridate (1.0 eq) was added dropwise to the reaction mixture at the same temperature over 1 h. After stirring for 12 h at 0°C, the reaction mixture was allowed to warm to room temperature and then diluted with DCM and washed with water. The organic layer was dried over Na2SO4 and evaporated in vacuo. The crude mixture was recrystallized from ethanol to get ethyl 1,3-dimethyl-2,4,6-trioxohexahydropyrimidine-5-carboxylate as a white solid (1.09 g; 75%). 1H NMR (DMSO [dimethyl sulfoxide]-d6, 300 MHz) δ 4.28 (qt, J = 6.9 Hz, 2-H), 3.16 (s, 3-H), 1.27 to 1.23 (t, J = 6.9 Hz, 3-H); LCMS (ESI) m/z 229 [M + H]+.
To a solution of ethyl 1,3-dimethyl-2,4,6-trioxohexahydropyrimidine-5-carboxylate (1.0 eq) in toluene (0.5 M) was added each amine (1.0 eq) and triethylamine (TEA) (1.2 eq). The reaction mixture was refluxed for about 12 h. After cooling down to room temperature, the reaction mixture was diluted with ethyl acetate and then washed with water. The organic layer was dried over Na2SO4 and evaporated in vacuo. The crude mixture was recrystallized from acetonitrile to get the desired products, compounds 12 and 14 to 20.
(i) 1,3-Dimethyl-2,4,6-trioxo-N-(4-phenoxyphenyl)hexahydropyrimidine-5 carboxamide (compound 12). White solid (75%): 1H NMR (CDCl3, 300 MHz) δ 11.92 (s, 1-H), 7.46 (d, J = 6.9 Hz, 2-H), 7.35 (t, J = 7.2 Hz, 2-H), 7.12 (t, J = 7.5 Hz, 1-H), 7.03 to 7.00 (m, 4-H), 3.41 (s, 3-H), 3.38 (s, 3-H); LC-MS (ESI) m/z 368 [M + H]+.
(ii) N-(4-acetylphenyl)-1,3-dimethyl-2,4,6 trioxohexahydropyrimidine-5-carboxamide (compound 14). White solid (79%): 1H NMR (CDCl3, 300 MHz) δ 12.10 (s, 1-H), 7.97 (d, J = 8.7 Hz, 2-H), 7.63 (d, J = 8.7 Hz, 2-H), 3.41 (s, 3-H), 3.37 (s, 3-H), 2.59 (s, 3-H); LC-MS (ESI) m/z 318 [M + H]+.
(iii) N-(4-bromo-2-fluorophenyl)-1,3-dimethyl-2,4,6-trioxohexahydropyrimidine-5-carboxamide (compound 15). White solid (46%): 1H NMR (CDCl3, 300 MHz) δ 12.04 (s, 1H), 7.98 (t, J = 8.5 Hz, 1-H), 7.38 to 7.26 (m, 2-H), 3.42 (s, 3-H), 3.38 (s, 3-H); LC-MS (ESI) m/z 370 [M − H]−.
(iv) 1,3-Dimethyl-2,4,6-trioxo-N-(2,4,6-trichlorophenyl)hexahydropyrimidine-5-carboxamide (compound 16). White solid (48%): 1H NMR (CDCl3, 300 MHz) δ 11.35 (s, 1-H), 7.44 (s, 2-H), 3.40 (s, 3-H), 3.39 (s, 3-H); LC-MS (ESI) m/z 378 [M + H]+.
(v) N-(6-chloropyridin-3-yl)-1,3-dimethyl-2,4,6 trioxohexahydropyrimidine-5-carboxamide (compound 17). White solid (88%): 1H NMR (CDCl3, 300 MHz) δ 11.94 (s, 1-H), 8.52 (d, J = 2.4 Hz, 1-H) 8.01 to 7.96 (dd, J = 2.7 and 8.7 Hz, 1-H), 7.35 (d, J = 8.4 Hz, 1-H), 3.41 (s, 3-H), 3.37 (s, 3-H); LC-MS (ESI) m/z 311 [M + H]+.
(vi) 1,3-dimethyl-N-(6-methylpyridin-2-yl)-2,4,6-trioxohexahydropyrimidine-5-carboxamide (compound 18). White solid (71%): 1H NMR (CDCl3, 300 MHz) δ 12.13 (s, 1-H), 7.70 (d, J = 7 Hz, 1-H), 7.63 (t, J = 7.8 Hz, 1-H), 6.98 (d, J = 7.5 Hz, 1-H), 3.39 (s, 6-H), 2.50 (s, 3-H); LC-MS (ESI) m/z 291 [M + H]+.
(vii) 1,3-Dimethyl-2,4,6-trioxo-N-(4(trifluoromethyl)pyridin-2-yl)hexahydropyrimidine-5-carboxamide) (compound 19). White solid (73%): 1H NMR (CDCl3, 300 MHz) δ 12.18 (s, 1-H), 8.55 (d, J = 5.1 Hz, 1-H), 8.27 (s, 1-H), 7.32 (d, J = 4.8 Hz, 1-H), 3.43 (s, 3-H), 3.39 (s, 3-H); LC-MS (ESI) m/z 345 [M + H]+.
(viii) 1,3-Dimethyl-N-(5-methylisoxazol-3-yl)-2,4,6-trioxohexahydropyrimidine-5-carboxamide (compound 20). White solid (69%): 1H NMR (CDCl3, 300 MHz) δ 12.04 (s, 1-H), 6.52 (s, 1-H), 3.41 (s, 3-H), 3.36 (s, 3-H), 2.43 (s,3-H); 13C NMR (CDCl3, 75 MHz) δ 169.1, 167.8, 162.8, 161.7, 158.4, 91.7, 81.6, 28.5, 12.1; LC-MS (ESI) m/z 281 [M + H]+.
Strains, media, and materials.
M. tuberculosis H37Rv (ATCC 27294) was used for all experiments. To determine the antimicrobial spectrum of the compounds, B. subtilis and S. aureus were used as Gram-positive strains and E. coli and P. aeruginosa were used as Gram-negative strains. For liquid cultures, M. tuberculosis was grown in 7H9 medium, which consists of Middlebrook 7H9 (Becton, Dickinson) broth base supplemented with albumin (50 g/liter)-dextrose (20 g/liter)-NaCl (8.1 g/liter) (ADC), 0.2% glycerol, and 0.05% Tween 80. Solid agar medium (7H11-OADC) for M. tuberculosis growth consisted of Middlebrook 7H11 (Becton, Dickinson) agar base supplemented with OADC (ADC with 0.06% oleic acid). GAST was used as a defined minimal liquid growth medium for M. tuberculosis (23). Mg2+-free GAST medium was made by omitting MgCl2·6H2O from the medium composition. The low-pH nitrosative-stress medium contained bovine serum albumin fraction V (5 g/liter), butyric acid (2.5 mM), NaNO2 (6.9 mg/ml), NaCl (0.81 g/liter), and 0.05% tyloxapol pH adjusted to 6.0. MIC determination was performed as previously described (23). Luria-Bertani (LB) broth (Difco) was used to determine the MICs of Gram-positive and -negative strains. The screening conditions under which the original pyrimidinetrione hits were identified have been described previously; in brief, they consisted of 7H9 medium with readout of M. tuberculosis growth after 3 days of compound exposure (24).
Generation and characterization of pyrimidinetrione-resistant mutants.
To generate mutants spontaneously resistant to the compounds, 50 ml of M. tuberculosis was grown to an optical density at 650 nm (OD650) of 0.2. Harvested cells were resuspended in 500 μl of medium, and 100-μl aliquots (109 cells) were plated on 7H11-OADC containing compound 1 at concentrations corresponding to 5×, 10×, and 20× MIC. Appropriate dilutions of the cell suspensions were also plated on drug-free plates to calculate bacterial numbers in the suspensions for determination of frequencies (the resistance frequency is the number of resistant mutants obtained divided by the number of bacteria plated). The agar plates were incubated at 37°C for 4 weeks. After 4 weeks, 4 colonies growing on 7H11-OADC containing 10× MIC of compound 1 were inoculated in 7H9 medium, and resistance was confirmed by MIC determination. Genomic DNA of the mutants was isolated by the CTAB (cetyltrimethylammonium bromide) method (25). Whole-genome sequencing was performed and analyzed as described previously (26).
Cytotoxicity in HepG2 cells.
Cytotoxicity in HepG2 cells was measured by CellTiter-Glo luminescent cell viability assay (Promega). For glucose medium, 20,000 HepG2 cells were seeded in 96-well white flat-bottom plates (Corning Inc.) in Dulbecco’s modified Eagle’s medium (DMEM) GlutaMax (Gibco) supplemented with 10% fetal bovine serum, 20 mM HEPES, and 0.5 mM sodium pyruvate. For galactose medium, HepG2 cells were precultured in galactose DMEM and seeded in the same plate with glucose medium. Galactose DMEM consists of glucose-free DMEM (Gibco) supplemented with 10% fetal bovine serum, 10 mM d-galactose, 2 mM l-glutamine, 5 mM HEPES, and 0.5 mM sodium pyruvate. The plates were incubated at 37°C with 5% CO2. The following day, 2-fold serial dilutions of compound up to 100 μM were added to the respective growth medium in duplicate, and the cells were incubated for 24 h. After this, 20 μl of CellTiter-Glo reagent was added to each well, followed by incubation at 37°C with 5% CO2 for 10 min. Cytotoxicity was determined by measuring the luminescence using Fluostar Optima FL (BMG Labtech).
In silico analysis.
The amino acid sequence of MtCorA (Rv1239c) was retrieved from the Mycobrowser database (https://mycobrowser.epfl.ch/). The FASTA format of the MtCorA protein sequence was submitted to the PHYRE2 protein fold recognition server, and 99 sequence alignment hits were obtained. The sequence of TmCorA was selected as the single highest-scoring template, and 332 residues (91% of the MtCorA sequence) were modeled with 100% confidence by the structure of TmCorA.
Magnesium and cobalt concentration-dependent MIC.
We used Mg2+-free GAST medium to test the magnesium dependence of the MICs of the compounds. Two-fold-concentrated GAST generated without the addition of magnesium chloride hexahydrate was prepared. An equal volume of magnesium chloride in sterile distilled water was added to the GAST medium to give final concentrations ranging from 0.12 mM up to 240 mM. MIC determinations for the compounds were set up in duplicate as previously described for each MgCl2 concentration (27). Briefly, 50-ul aliquots of GAST medium at the defined MgCl2 concentration with compound serially diluted in duplicate from 50 to 0.024 μM were added to all wells of a round-bottom 96-well plate (Nunclon). M. tuberculosis culture in GAST at an OD650 of 0.2 was diluted 1:500 in 2× Mg2+-free GAST, and 50 μl was added to each well of the 96-well plates. The plates were incubated under 5% CO2 at 37°C. Growth was measured using an inverted enlarging mirror at 1, 2, and 3 weeks, with the MIC taken to be the concentration that completely inhibited all visible growth. For the cobalt-dependent MIC test, CoCl2 was added to GAST medium at concentrations from 0.001 to 5 mM, and MICs were determined as described above.
Magnesium concentration measurement by ICP-MS.
We measured magnesium concentrations in M. tuberculosis cells treated by ICP-MS. Two liters of M. tuberculosis was grown to an OD650 of 0.6 in 7H9. Five hundred-milliliter culture volumes were incubated with 1× and 10× MIC of compound 10, 10× MIC of moxifloxacin, or an equivalent volume of DMSO vehicle control (0.1% [vol/vol]) for 24 h in 2-liter-capacity rolling bottles. Cells were harvested and washed three times with 1/10 volume phosphate-buffered saline containing 0.05% Tween 80 (PBST). The wet weight of M. tuberculosis cell pellets was recorded to normalize the magnesium concentration prior to autoclaving. ICP-MS analysis service was provided by Elementary Analysis Inc. (Lexington, KY, USA).
UV spectrum scanning to test magnesium binding of pyrimidinetrione.
One milliliter of 100 μM compound 10 dissolved in 100 mM HEPES buffer was put in 1-cm-path-length quartz crystal cuvettes for UV scanning. The absorbance of the compound 10 solution was scanned at 200-nm to 400-nm wavelengths with a Varian Cary 300 Bio UV-Vis spectrophotometer. We sequentially added 10 μl of 1 mM MgSO4 to the compound solution to observe changes in the absorbance spectrum. A standard curve was plotted with A280s of 5, 10, 50, and 100 μM compound 10. Absorbance at 280 nm was used to calculate the concentration of compound 10 after correcting for changes in volume, and the values were plotted in a graph.
Minimum magnesium requirement test.
M. tuberculosis H37Rv, corA::E21D, and corA::A317S strains were cultured in GAST to an OD650 of 0.2 at 37°C. Fifty microliters of Mg2+-free GAST was distributed to every well of a round-bottom 96-well plate (Nunclon). Fifty microliters of 240 mM MgCl2 dissolved in Mg2+-free GAST was added to the wells of the first column of the plate, and 50-μl volumes were transferred from column 1 to column 12, generating a 2-fold serial dilution series of MgCl2 across the plate. M. tuberculosis cultures were diluted 1:500 in Mg2+-free GAST, and 50 μl was added to each well of the 96-well plates. Growth was measured using an inverted enlarging mirror at 1, 2, and 3 weeks, with the minimum Mg2+ concentration to support growth taken to be the lowest concentration that supported any visible evidence of growth.
Cloning, expression, and purification of MtCorA.
The M. tuberculosis corA gene (rv1239c), was cloned into the pET28a vector. The gene was amplified by PCR using Herculase II Fusion DNA polymerase (Agilent) and the following primers: forward, 5′-CGCGGCAGCCATATGTTCCCAGGGTTT-3′, and reverse, 5′-TGCGGCCGCAAGCTTCTAGAGCCAGTTTCT-3′.The PCR-amplified gene was inserted between two restriction sites, NdeI and HindIII, using In-Fusion HD Cloning Plus (TaKaRa). Site-directed mutagenesis was performed to generate the E212D mutation on corA. MtCorA contained an N-terminal polyhistidine tag. The plasmids carrying the wild-type and mutant corA genes were transformed into OverExpress C41(DE3) (Lucigen). The cells were grown in LB medium at 37°C. The induction was initiated with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at an OD600 of 0.7 to 0.8, and after 6 h, the cells were harvested by centrifugation at 3,500 rpm for 20 min. The cell pellet was resuspended in lysis buffer containing 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5 mM dithiothreitol (DTT), and SigmaFast protease inhibitor cocktail tablets, EDTA free (Sigma-Aldrich). After sonication, the lysed cells were pelleted by centrifugation at 20,000 × g for 60 min, and the membrane was isolated from the supernatant by ultracentrifugation at 142,000 × g for 60 min. The isolated membrane was then solubilized in resuspension buffer, 50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 20 mM imidazole, 0.5 mM DTT, with 1% (wt/vol) dodecyl maltoside (DDM) (Anatrace). The resulting supernatant was loaded onto an Ni-nitrilotriacetic acid (NTA) column. The column was washed with wash buffer consisting of 50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 40 mM imidazole, 0.5 mM DTT, with 0.1% (wt/vol) DDM. The CorA or CorA mutant protein was eluted with elution buffer (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 300 mM imidazole, 0.5 mM DTT, with 0.05% [wt/vol] DDM), and the resulting supernatant fractions containing CorA were identified using SDS-PAGE, pooled, and dialyzed against dialysis buffer (50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 0.5 mM DTT, with 0.05% [wt/vol] DDM). The protein was finally purified on a Superdex 200 16/60 (GE Healthcare) in 50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 0.5 mM DTT, with 0.05% (wt/vol) DDM.
Thermostability test of CorA with pyrimidinetriones.
Purified MtCorA samples were diluted to a concentration of 0.5 mg/ml. The solution was aliquoted into 30-μl volumes, with 1 μl of a stock solution of different Mg2+ concentrations added to the aliquot. Samples were incubated for 20 min at room temperature and then further incubated for 10 min at every increment of 5°C up to 95°C in a thermocycler. The samples were transferred to a 96-well filter plate (0.45 μm; Millipore) to remove precipitated proteins. The yield of the filtered protein was analyzed by SDS-PAGE.
Substrate binding measured by fluorescence quenching.
The dissociation constants between CorA and its ligands, Mg2+, Co2+, and compound 10, were determined by a ligand dose-dependent fluorescence-quenching assay (Cary Varian Eclipse fluorescence spectrophotometer). CorA was titrated with increasing concentrations of ligands, and fluorescence emission was measured at ∼350 nm following excitation at 283 nm (bandwidths, 5 nm). The fluorescence quenching (ΔF) versus ligand concentration ([S]) was plotted, and the Kd(app) (the apparent dissociation constant of CorA to ligands) values were calculated by fitting the data to an equation, ΔF = (F0 − F) = (ΔFmax × [S])/{Kd(app) + [S]}, where F0 and F are the fluorescence intensities in the absence and presence of substrates, respectively.
Efficacy study in vitro, ex vivo, and in vivo.
The in vitro efficacies of the compounds against M. tuberculosis were measured during aerobic growth, as well as during anaerobic, nonreplicating persistence. Time-kill kinetics were measured under aerobic conditions as follows. Logarithmically growing M. tuberculosis (OD650 = 0.2) was diluted 1,000-fold in 7H9 medium, and 1-ml volumes were exposed to 1×, 2×, 5×, 10×, and 20× MIC of compound 10 for up to 7 days in duplicate. Samples were collected and plated on 7H11-OADC plates for CFU enumeration at 1, 2, 4, and 7 days of treatment. For anaerobic conditions, M. tuberculosis was cultured in the self-generated oxygen depletion model as previously described (27). One-milliliter volumes of anaerobic M. tuberculosis culture were exposed for 1 week to 1×, 5×, 10×, 20×, and 50× MICs of compounds 9, 10, and 20 in an anaerobic chamber (Microbiology International), followed by CFU enumeration as described above.
For ex vivo efficacy testing, J774 cells (6.6 × 104 cells/well) were seeded in flat-bottom 24-well plates (Corning Inc.) in J774 growth medium consisting of DMEM GlutaMax (Gibco) supplemented with 10% fetal bovine serum, 20 mM HEPES, and 0.5 mM sodium pyruvate. M. tuberculosis H37Rv with confirmed high levels of phthiocerol dimycocerosate was grown in 7H9 to an OD650 of 0.5 and filtered through a 5-μm sterile filter to ensure a single-cell suspension, cell density was confirmed at OD650, and the suspension was diluted in J774 growth medium to 4.4 × 105 CFU/ml. Aliquots of 0.1 ml of the M. tuberculosis suspension were added to each well of the 24-well plate, giving a multiplicity of infection (MOI) of 1:1.5. After overnight incubation, the medium was aspirated and monolayers were washed twice in prewarmed PBS (pH 7.4) twice. At this point, four wells were used for CFU enumeration as described below. To each of the remaining wells, 1 ml J774 growth medium containing compound or vehicle (DMSO) control was added. Each compound was tested at 1×, 5×, and 10× its MIC in duplicate wells for each time point and concentration. Rifampin (0.5 and 5 μg/ml) was used as a positive control. Cells were incubated at 37°C, 95% humidity, 5% CO2 for 3 and 7 days. The medium was replenished at days 3 and 5, taking care not to remove any macrophages, since the pyrimidinetrione amides resulted in loss of adherence of macrophages over time. After 3 or 7 days of incubation, medium was removed, taking care not to remove macrophages, and 1 ml of 7H9 medium containing 0.1% SDS was added to each well to ensure macrophage lysis. After 5 min, the lysate was rapidly mixed to shear eukaryotic DNA, diluted in 7H9, and plated on 7H11-OADC plates. Colonies were enumerated after 4 weeks of incubation at 37°C.
Mouse studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health under animal study protocol number LCIM 4E. Pharmacokinetic analyses on 20 C57BL/6J mice were done by administering 30 mg/kg compound 10 suspended in 10% (2-hydroxypropyl)-β-cyclodextrin with 50 mM tricine (pH 8.2) twice per day (with a separation of 7 h) by oral gavage, followed by blood sampling at 0.5, 1, 2, 4, 8, 12, and 24 h; 0.1 ml of blood was collected twice from each animal during the experiment in Li-Heparin Microvette tubes (Sardtedt AG & Co., Numbrecht, Germany) and centrifuged to prepare plasma. The tolerability of compound 10 was tested by dosing 5 naive mice twice per day with 100 or 30 mg/kg for 7 days with observation for 7 subsequent days. For evaluation of in vivo efficacy, C57BL/6J mice were infected by the aerosol route, as previously described (28). After 14 days, groups of 10 mice were dosed with compound 10 given by oral gavage of either 15 mg/kg or 30 mg/kg twice a day. The higher-dose group developed signs of distress after 1 week, so the dose to that group was de-escalated to 30 mg/kg once a day. Control groups were dosed with vehicle control [10% (2-hydroxypropyl)-β-cyclodextrin] with 50 mM tricine (pH 8.2) or 10 mg/kg rifampin. After 2 and 4 weeks of treatment, groups of 5 mice were euthanized, and appropriate dilutions in 7H9 medium of lung and spleen homogenates were plated on 7H11-OADC plates for CFU enumeration.
Pharmacokinetic analysis of pyrimidinetriones.
Pharmacokinetic plasma samples were analyzed with an Agilent 1200 Infinity high-performance liquid chromatography (HPLC) instrument with an Agilent 6460C triple-quadrupole mass selective detector (LC-QqQ) utilizing ESI. Samples were prepared by mixing 50 μl serum with 50 μl labetalol internal standard (IS), 20 μl water, or spike solution, followed by 400 μl acetonitrile-methanol (3:1) for protein precipitation. Samples were centrifuged at 13,000 rpm for 5 min, and 5 μl supernatant was injected into a 2.1- by 50-mm Eclipse Plus C18 1.8-μm column utilizing 0.1% formic acid in water (solvent A) and acetonitrile with 0.1% formic acid (solvent B). The solvent gradient program was as follows: 8% B, hold for 0.5 min, increase to 95% B in 4.5 min, with a flow rate of 0.8 ml/min.
Multiple reaction monitoring (MRM) transition for compound 10 was detected as [M − H]− precursor (Q1), collision energy voltage (CEV) 24 V, and 155 product ion (Q2) negative mode, and IS was detected as [M + H]+ precursor (Q1), CEV 8V, and 311.2 product ion (Q2) positive mode. PK parameters were calculated using PK macros in Excel.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded in part by the Intramural Research Program of NIAID (AI000693-25) and by grants from the Foundation for the National Institutes of Health (BARRY11HTB0) with support from the Bill and Melinda Gates Foundation (OPP1024021). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We gratefully acknowledge the assistance of Danielle Weiner in animal efficacy experiments and Aashish Srivastava for preparing samples for sequencing.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01006-19.
REFERENCES
- 1.Leung CC, Lange C, Zhang Y. 2013. Tuberculosis: current state of knowledge: an epilogue. Respirology 18:1047–1055. doi: 10.1111/resp.12156. [DOI] [PubMed] [Google Scholar]
- 2.Horsburgh CR Jr, Barry CE III, Lange C. 2015. Treatment of tuberculosis. N Engl J Med 373:2149–2160. doi: 10.1056/NEJMra1413919. [DOI] [PubMed] [Google Scholar]
- 3.Barry CE. 2011. Lessons from seven decades of antituberculosis drug discovery. Curr Top Med Chem 11:1216–1225. doi: 10.2174/156802611795429158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gopal P, Dick T. 2014. Reactive dirty fragments: implications for tuberculosis drug discovery. Curr Opin Microbiol 21:7–12. doi: 10.1016/j.mib.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 5.Libardo JMD, Boshoff HI, Barry CE III, 2018. The present state of the tuberculosis drug development pipeline. Curr Opin Pharmacol 42:81–94. doi: 10.1016/j.coph.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maguire ME. 2006. Magnesium transporters: properties, regulation and structure. Front Biosci 11:3149–3163. doi: 10.2741/2039. [DOI] [PubMed] [Google Scholar]
- 7.Groisman EA, Hollands K, Kriner MA, Lee EJ, Park SY, Pontes MH. 2013. Bacterial Mg2+ homeostasis, transport, and virulence. Annu Rev Genet 47:625–646. doi: 10.1146/annurev-genet-051313-051025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchmeier N, Blanc-Potard A, Ehrt S, Piddington D, Riley L, Groisman EA. 2000. A parallel intraphagosomal survival strategy shared by Mycobacterium tuberculosis and Salmonella enterica. Mol Microbiol 35:1375–1382. doi: 10.1046/j.1365-2958.2000.01797.x. [DOI] [PubMed] [Google Scholar]
- 9.Goodsmith N, Guo XV, Vandal OH, Vaubourgeix J, Wang R, Botella H, Song S, Bhatt K, Liba A, Salgame P, Schnappinger D, Ehrt S. 2015. Disruption of an M. tuberculosis membrane protein causes a magnesium-dependent cell division defect and failure to persist in mice. PLoS Pathog 11:e1004645. doi: 10.1371/journal.ppat.1004645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walters SB, Dubnau E, Kolesnikova I, Laval F, Daffe M, Smith I. 2006. The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol Microbiol 60:312–330. doi: 10.1111/j.1365-2958.2006.05102.x. [DOI] [PubMed] [Google Scholar]
- 11.Garcia-del Portillo F, Foster JW, Maguire ME, Finlay BB. 1992. Characterization of the micro-environment of Salmonella typhimurium-containing vacuoles within MDCK epithelial cells. Mol Microbiol 6:3289–3297. doi: 10.1111/j.1365-2958.1992.tb02197.x. [DOI] [PubMed] [Google Scholar]
- 12.Piddington DL, Kashkouli A, Buchmeier NA. 2000. Growth of Mycobacterium tuberculosis in a defined medium is very restricted by acid pH and Mg(2+) levels. Infect Immun 68:4518–4522. doi: 10.1128/IAI.68.8.4518-4522.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 14.Lamichhane G, Zignol M, Blades NJ, Geiman DE, Dougherty A, Grosset J, Broman KW, Bishai WR. 2003. A postgenomic method for predicting essential genes at subsaturation levels of mutagenesis: application to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 100:7213–7218. doi: 10.1073/pnas.1231432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson CM, Norquist BA, Vesce S, Nicholls DG, Soine WH, Duan S, Swanson RA. 2002. Barbiturates induce mitochondrial depolarization and potentiate excitotoxic neuronal death. J Neurosci 22:9203–9209. doi: 10.1523/JNEUROSCI.22-21-09203.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. 2007. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol Sci 97:539–547. doi: 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- 17.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mouscadet JF, Tchertanov L. 2009. Raltegravir: molecular basis of its mechanism of action. Eur J Med Res 14Suppl 3:5–16. doi: 10.1186/2047-783x-14-s3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopez Quezada L, Silve S, Kelinske M, Liba A, Diaz Gonzalez C, Kotev M, Goullieux L, Sans S, Roubert C, Lagrange S, Bacqué E, Couturier C, Pellet A, Blanc I, Ferron M, Debu F, Li K, Aubé J, Roberts J, Little D, Ling Y, Zhang J, Gold B, Nathan C. 2019. Bactericidal disruption of magnesium metallostasis in Mycobacterium tuberculosis is counteracted by mutations in the metal ion transporter CorA. mBio 10:e01405-19. doi: 10.1128/mBio.01405-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang YJ, Reddy MC, Ioerger TR, Rothchild AC, Dartois V, Schuster BM, Trauner A, Wallis D, Galaviz S, Huttenhower C, Sacchettini JC, Behar SM, Rubin EJ. 2013. Tryptophan biosynthesis protects mycobacteria from CD4 T-cell-mediated killing. Cell 155:1296–1308. doi: 10.1016/j.cell.2013.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laarakker MC, van Lith HA, Ohl F. 2011. Behavioral characterization of A/J and C57BL/6J mice using a multidimensional test: association between blood plasma and brain magnesium-ion concentration with anxiety. Physiol Behav 102:205–219. doi: 10.1016/j.physbeh.2010.10.019. [DOI] [PubMed] [Google Scholar]
- 22.Wang D, Zhang Z, Lu X, Feng Y, Luo K, Gan J, Yingxue L, Wan J, Li X, Zhang F, Tu Z, Cai Q, Ren X, Ding K, Ding K. 2011. Hybrid compounds as new Bcr/Abl inhibitors. Bioorg Med Chem Lett 21:1965–1968. doi: 10.1016/j.bmcl.2011.02.029. [DOI] [PubMed] [Google Scholar]
- 23.Duckworth BP, Wilson DJ, Nelson KM, Boshoff HI, Barry CE, Aldrich CC. 2012. Development of a selective activity-based probe for adenylating enzymes: profiling MbtA involved in siderophore biosynthesis from Mycobacterium tuberculosis. ACS Chem Biol 7:1653–1658. doi: 10.1021/cb300112x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oh S, Park Y, Engelhart CA, Wallach JB, Schnappinger D, Arora K, Manikkam M, Gac B, Wang H, Murgolo N, Olsen DB, Goodwin M, Sutphin M, Weiner DM, Via LE, Boshoff HIM, Barry CE. 2018. Discovery and structure-activity-relationship study of N-alkyl-5-hydroxypyrimidinone carboxamides as novel antitubercular agents targeting decaprenylphosphoryl-beta-d-ribose 2’-oxidase. J Med Chem 61:9952–9965. doi: 10.1021/acs.jmedchem.8b00883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alland D, Steyn AJ, Weisbrod T, Aldrich K, Jacobs WR Jr.. 2000. Characterization of the Mycobacterium tuberculosis iniBAC promoter, a promoter that responds to cell wall biosynthesis inhibition. J Bacteriol 182:1802–1811. doi: 10.1128/JB.182.7.1802-1811.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ioerger TR, Feng Y, Ganesula K, Chen X, Dobos KM, Fortune S, Jacobs WR Jr, Mizrahi V, Parish T, Rubin E, Sassetti C, Sacchettini JC. 2010. Variation among genome sequences of H37Rv strains of Mycobacterium tuberculosis from multiple laboratories. J Bacteriol 192:3645–3653. doi: 10.1128/JB.00166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park Y, Pacitto A, Bayliss T, Cleghorn LAT, Wang Z, Hartman T, Arora K, Ioerger TR, Sacchettini J, Rizzi M, Donini S, Blundell TL, Ascher DB, Rhee K, Breda A, Zhou N, Dartois V, Jonnala SR, Via LE, Mizrahi V, Epemolu O, Stojanovski L, Simeons F, Osuna-Cabello M, Ellis L, MacKenzie CJ, Smith ARC, Davis SH, Murugesan D, Buchanan KI, Turner PA, Huggett M, Zuccotto F, Rebollo-Lopez MJ, Lafuente-Monasterio MJ, Sanz O, Diaz GS, Lelièvre J, Ballell L, Selenski C, Axtman M, Ghidelli-Disse S, Pflaumer H, Bösche M, Drewes G, Freiberg GM, Kurnick MD, Srikumaran M, Kempf DJ, Green SR, Ray PC, Read K, Wyatt P, Barry CE, Boshoff HI. 2017. Essential but not vulnerable: indazole sulfonamides targeting inosine monophosphate dehydrogenase as potential leads against Mycobacterium tuberculosis. ACS Infect Dis 3:18–33. doi: 10.1021/acsinfecdis.6b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh R, Barry CE III, Boshoff HI. 2010. The three RelE homologs of Mycobacterium tuberculosis have individual, drug-specific effects on bacterial antibiotic tolerance. J Bacteriol 192:1279–1291. doi: 10.1128/JB.01285-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.