

Abstract

The ligand, bis-β-diketone with an azobenzene bridge (4,4′-(4,4,4-trifluoro-1,3-butanedione)azobenzene, H2L), was prepared for the synthesis of a series of dinuclear lanthanide complexes with the formula [Ln2L3(DMSO)4] (Ln = Eu3+, Gd3+, Tb3+, and DMSO = dimethyl sulfoxide). X-ray crystallographic analysis reveals that the three complexes are triple-stranded dinuclear structures formed by three bis-β-diketonate ligands with two lanthanide ions (Ln3+). The trans-to-cis photoisomerization rates of the azobenzene group of the three [Ln2L3(DMSO)4] complexes in ethanol and acetonitrile solutions are similar to those of the pure H2L ligand and other azobenzene-containing mononuclear lanthanide complexes, but the trans-to-cis quantum yields (Φt→c = 10–3) are 1 order of magnitude smaller. The first-order rate constant for the cis-to-trans thermal isomerization at 50 °C of the H2L ligand is similar to those of azobenzene derivatives, while those for the [Ln2L3(DMSO)4] complexes (kiso = 10–4 s–1) are higher than those of the mononuclear azobenzene-containing lanthanide complexes. Furthermore, as the lanthanide ionic radius becomes smaller from Eu3+ to Gd3+ to Tb3+, the thermal isomerization rate constant decreases and the half-life increases. All these results are proposed to arise from the rigidity at both ends of the azo group by coordination to the dinuclear lanthanide ions and the different isomerization mechanisms. These are the first examples of bis-β-diketonate dinuclear lanthanide complexes with an azobenzene bridge and help illustrate the mechanism of azobenzene isomerization.
Introduction
It is well known that the azobenzene molecule can undergo trans-to-cis isomerization upon UV–visible light irradiation, which results in changes to the absorption spectrum, molecular configuration, dipole moment, geometric size, and surface free energy of the molecule, and also reverse cis-to-trans isomerization by visible light or heating (or in the dark).1−3 These special properties make it important for application in the fields of optoelectronic information storage materials,4,5 molecular switches,6,7 liquid crystal materials,8,9 solar energy storage materials,10−12 and in biology.13−15 Extensive studies have shown that azobenzene and its derivatives can exhibit an excellent reversible photoisomerization behavior. There are many factors that affect the azobenzene photoisomerization properties, which include the isomerization mechanism, rate, and pathway, such as the intensity of light, irradiation wavelength, temperature, pressure, solvent, and substituted groups on the azobenzene.16−26 Up to date, four mechanisms have been proposed to explain the possible mechanism for azobenzene photoisomerization—rotation, inversion, concerted inversion, and inversion-assisted rotation. These theories can be referred to in some previously published reviews.1,3,12,20
With the intense research on the isomerization properties of azobenzene, the modification of azobenzene provides an important theoretical basis to obtain more useful information on the photoresponsivity of azobenzene.7,10,12,18,26 Combining an azo moiety within coordination compounds is an alternative to modify pure organic azobenzene and can yield more versatile molecular properties such as redox, optical, and magnetic properties, accompanying with the photoisomerization reaction. Most of the studied azobenzene-modifed metal complexes were ferrocene–azobenzene, bis or tris(bipyridine), terpyridine metal complexes with azobenzene-conjugation, metalladithiolenes with azobenzene groups, and azobenzene-containing tris-cyclometalated complexes.27−35 The center metal ions of these complexes were all the d-block group VIII. They displayed reversible trans-to-cis photoisomerization. However, the efficiency of the photoisomerization was lower than those of the parent azobenzenes. To overcome this difficulty, we have focused on the f-block lanthanide ions [Ln(III)], which have unusual electronic properties caused by shielding of the 4f orbitals with the fully filled 5s25p6 subshells. Recently, we have studied the photoinduced trans–cis–trans isomerization properties of azobenzene-containing mononuclear and dinuclear lanthanide complexes in the solution and solid states.36−38 Our study showed that azobenzene-functionalized lanthanide complexes exhibit good reversible trans–cis–trans isomerization properties both in solvents and the solid state and can be recycled with good fatigue resistance. Owing to the coordination of lanthanide ions, the functional properties of the azobenzene group in the complexes are superior to those of pure azobenzene ligands. In addition to the good photoinduced trans–cis–trans isomerization properties, the complexes of Eu and Yb can also exhibit luminescence with the dual characteristics of photoinduced trans–cis–trans isomerism and luminescence, which can realize multiple practical applications. Therefore, these azobenzene functionalized lanthanide complexes deserve further exploration and development. Recently, studies on the photophysical properties of bis-β-diketonate lanthanide complexes with triple-stranded dinuclear helicate structures have received extensive interest.39−42 These triple-stranded dinuclear helicate bis-β-diketonate lanthanide complexes have the characteristic of rigidity, yet differ from macrocyclic azobenzene dimers whose effect of rigidity has been discussed and whose photochemical and thermal isomerizations have been reported.43−45 In this context, a new bis-β-diketone with an azobenzene bridged ligand, 4,4′-(4,4,4-trifluoro-1,3-butanedione)azobenzene (H2L), was prepared to synthesize three dinuclear lanthanide (Ln = Eu, Gd, Tb) complexes (Scheme 1). The photo and thermal isomerizations of the H2L ligand and its triple-stranded helicate dinuclear lanthanide complexes were investigated, and their properties were compared with those of nonrigid azobenzene-attached lanthanide complexes.
Scheme 1. Synthetic Procedures for Preparation of the Ligands and Complexes.

Results and Discussion
Synthesis and Characterization of the Complexes
Scheme 1 shows the synthetic method of the complexes [Ln2L3(DMSO)4] (Ln = Eu3+, Gd3+, Tb3+). The typical carbonyl stretching frequency of H2L (1600 cm–1) in the infrared (IR) spectra was red-shifted in the complexes, with values of 1614 cm–1 in [Eu2L3(DMSO)4]; 1615 cm–1 in [Gd2L3(DMSO)4]; and 1615 cm–1 in [Tb2L3(DMSO)4], which suggests the carbonyl oxygen was coordinated to the central Ln3+ ion. It is clear that no significant change was observed in the absorption peak position of the azo group (−N=N−) and trifluoromethyl group (−CF3), both in the free ligand and the complexes. At the same time, owing to the coordination of dimethyl sulfoxide (DMSO), the characteristic absorption peaks of the S=O double bond appeared at around 1308 and 1288 cm–1.
It is shown from the thermogravimetric analysis (TGA) data that complex [Eu2L3(DMSO)4] underwent a mass loss of 7.29% (calcd 7.87%) in the first step (155–247 °C), which corresponded to the loss of the two coordinated DMSO molecules, and then, it underwent a second step decomposition (247–312 °C) with a mass loss of 15.29% (calcd 15.74%), which corresponded to the loss of all four coordinated DMSO molecules (Figure S1). For the complexes [Gd2L3(DMSO)4] and [Tb2L3(DMSO)4], the two weight loss curves were almost overlapping. Complexes [Gd2L3(DMSO)4] and [Tb2L3(DMSO)4] were more stable than complex [Eu2L3(DMSO)4], and their first mass loss occurred between 207 and 268 °C, and the mass loss rate was 7.76 and 7.71%, respectively (calcd loss for two DMSO molecules was 7.83 and 7.81%, respectively); the second mass loss occurred at 268–312 °C, and the mass loss rate was 13.67 and 13.64%, which is also consistent with the calculated loss of four DMSO molecules (14.45%). All three complexes continued to lose mass and gradually decomposed as the temperature was increased after the complexes experienced the two mass loss steps. TGA showed that the three bis-β-diketonate lanthanide complexes exhibited high stability.
The powder XRD pattern (Figure S2) of the three complexes were recorded over the 2θ = 10°–80° range. They all exhibited a crystalline nature with sharp peaks. The powder X-ray diffraction pattern matched well with the simulated pattern and confirmed the phase purity of the complexes.
X-ray Crystallographic Analysis
The crystallographic data of the three complexes [Eu2L3(DMSO)4], [Gd2L3(DMSO)4], and [Tb2L3(DMSO)4] are listed in Table S1 in the Supporting Information. Selected bond lengths and angles are shown in Tables S2–S4. The single crystal structure data show that all the three complexes crystallized in the triclinic P1̅ space group with two molecules in the unit cell. The two central Ln(III) ions had the same coordination mode. Each lanthanide ion was coordinated with eight oxygen atoms to form a stable double-capped triangular prism structure, of which six oxygen atoms came from the three deprotonated H2L ligands and the other two came from two DMSOs (Figures 1 and S3–S4). The structure featured a triple-stranded dinuclear helicate with two crystallographically equivalent lanthanide(III) ions coordinated to three bis-β-diketonate ligands. The bond lengths of the Eu–O, Gd–O, and Tb–O bonds coordinated to the bis-β-diketonate in the complexes were 2.357–2.438, 2.347–2.425, and 2.327–2.416 Å, respectively, which are in accordance with the reported values in their diketonate complexes.46,47 The bond lengths of the Eu–O, Gd–O, and Tb–O bonds coordinated to the solvent DMSO molecules ranged from 2.367 to 2.403, 2.352 to 2.391, and 2.350 to 2.383 Å, respectively, which are also in the ranges of the reported values.36,37 The bond lengths of the N=N bond in the three molecule were 1.240, 1.247, and 1.249 Å, respectively, which are in the normal ranges compared with the bond lengths in azobenzene-attached lanthanide derivatives. It is evident that the bond length of Ln–O slightly decreased and the bond length of N=N increased with an increase of atomic number from Eu to Gd to Tb. In the three complexes, all the C–N=N–C dihedral angles were close to 180° and N=N–C angles were in the range 111.7°–115.9°, in good agreement with the values found in azobenzene derivatives.48
Figure 1.
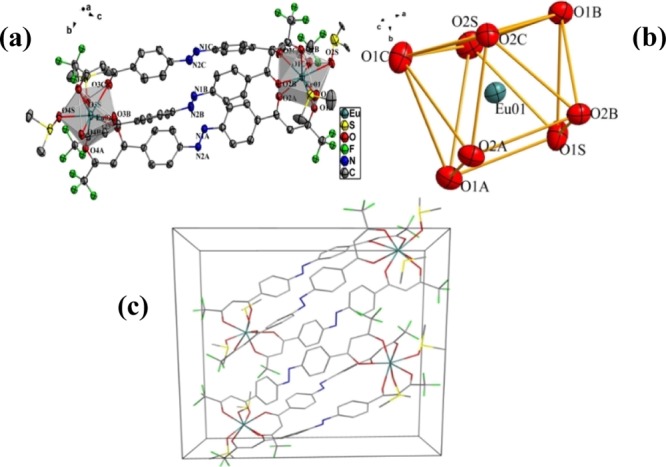
Crystal structure of [Eu2(L)3(DMSO)4] complex (all hydrogen atoms are omitted for clarity) (a), coordination polyhedron geometry of central Eu(III) (b) and the packing diagram in the unit cell (c).
Photoisomerization Behavior of Ligand H2L and Complexes [Ln2L3(DMSO)4] in Solutions
Figure 2 shows the UV–visible absorption spectral changes of the bis-β-diketone ligand H2L under 365 nm (UV) and 450 nm (visible) light irradiation with time in an ethanol solution. As shown in Figure 2a, the intensity of the π–π* transition absorption peak at 372 nm (ε372nm = 4.25 × 104 L mol–1 cm–1) gradually decreased as a function of time under UV light irradiation, and the π–π* transition absorption peak at 257 nm (ε257nm = 7.41 × 103 L mol–1 cm–1) increased, with an isosbestic point at 325 nm. The maximum wavelength of the absorption peak at 372 nm was gradually blue-shifted to 362 nm, which arose from the poor planarity of the cis configuration. The n−π* characteristic transition absorption peak of azobenzene in the range 400–500 nm was not observed. This may result from the close energy between the π–π* and n−π* transitions in ethanol solution and the two transition peaks overlapped, as is observed for aminoazobenzenes.1,37 This spectral change indicated that the bis-β-diketone ligand H2L gradually changed from the trans configuration to the cis configuration. When the solution was irradiated for 10 min, a photostable state was reached. Subsequently, when the solution was irradiated with visible light, the absorption band at 362 nm was gradually red-shifted and its intensity sharply increased in the first 8 s and remained unchanged after 12 s, which led to a 48% recovery of the absorption spectrum. The spectral changes of H2L in acetonitrile solution under UV light irradiation and its recoverable spectrum after irradiation with visible light were also examined. They were shown to be similar to those observed in ethanol solution (Figures S5–S6). The reversible isomerization reaction could be maintained for more than 10 cycles, which indicated good fatigue resistance and reversibility of the photoisomerization reaction. The first-order kinetics rate constants of the photoisomerization reaction of H2L in the ethanol and acetonitrile solutions were calculated to be 8.8 × 10–3 and 7.0 × 10–3 s–1, and the quantum yields were determined to be 0.041 and 0.023 (Table 1), respectively, which are slightly larger than those of azobenzene and its derivatives. The photoisomerization data showed a slightly higher isomerization efficiency in the higher polarity ethanol solvent.
Figure 2.

UV–vis spectral changes of H2L in ethanol solution (2 × 10–5 mol/L) upon irradiation at 365 nm and recoverable irradiation at 450 nm as a function of time.
Table 1. Quantum Yields (Φt-c) and Photoisomerization Rate Constants (kiso, s–1) of H2L and [Ln2L3(DMSO)4] Complexes in Different Solvents.
| ethanol |
acetonitrile |
|||
|---|---|---|---|---|
| compounds | 103Φt→c | 104kiso | 103Φt→c | 104kiso |
| H2L | 41.1 ± 0.45 | 88.7 ± 5.7 | 22.7 ± 0.26 | 70.1 ± 5.3 |
| [Eu2(L)3(DMSO)4] | 7.07 ± 0.26 | 69.1 ± 2.3 | 6.82 ± 0.34 | 63.5 ± 3.1 |
| [Gd2(L)3(DMSO)4] | 5.80 ± 0.32 | 65.0 ± 3.6 | 4.30 ± 0.29 | 74.3 ± 4.1 |
| [Tb2(L)3(DMSO)4] | 6.37 ± 0.39 | 79.3 ± 4.2 | 6.16 ± 0.42 | 81.0 ± 5.1 |
The photoisomerization reactions of the three [Ln2L3(DMSO)4] complexes showed a similarity in the ethanol and acetonitrile solutions. The absorption spectra of the three complexes were almost identical, as shown by the UV–vis data in Table 2. The molar absorption coefficients at the maximum wavelengths were more than three times that of the free ligand, owing to the three ligands in the complexes. Taking the [Eu2L3(DMSO)4] complex as an example, Figure 3 shows the characteristic spectral changes of the [Eu2L3(DMSO)4] complex in ethanol solution under UV and visible light irradiation (the other spectral change data are provided in Figures S7–S9). The typical π–π* absorption band of the [Eu2L3(DMSO)4] complex in ethanol solution exhibited a peak at 366 nm with a shoulder peak at 300 nm. Upon irradiation of the solution with UV light, the spectral changes were similar to those of the pure H2L ligand but with no maximum wavelength shift. The [Eu2L3(DMSO)4] complex reached a photostable state after 20 min with irradiation at 365 nm, and 70% of the complex returned to the trans form after the UV irradiation was switched to visible light for 4 s. The recovery percentage of the trans form was higher than that of the pure H2L ligand. This spectral change indicated that the azo group in the [Eu2L3(DMSO)4] complex was in a more rigid environment and its trans form was more stable than that of the pure H2L ligand. Ten cycles of trans–cis–trans photoisomerization were performed by irradiating the [Eu2L3(DMSO)4] complex in ethanol solution with UV light for 30 min followed by visible light for 3 min. It was found that there were no signs of photodegradation. This indicated that the complex is stable under these conditions.
Table 2. UV–vis Absorption Data of H2L and [Ln2L3(DMSO)4] Complexes in Different Solvents (λmax[nm] and εmax[L mol–1 cm–1]).
| compounds | ethanol | acetonitrile |
|---|---|---|
| H2L | 364(39 860), 300(23 775) | 474(2170), 368(57 539) |
| [Eu2(L)3(DMSO)4] | 474(4380), 366(136 137), 303(55 095) | 474(4569), 366(143 910), 302(55 195) |
| [Gd2(L)3(DMSO)4] | 474(4660), 366(149 243), 303(58 691) | 474(4880), 366(152 261), 302(57 528) |
| [Tb2(L)3(DMSO)4] | 474(4659), 366(148 335), 303(58 967) | 474(4585), 366(145 229), 302(55 336) |
Figure 3.

UV–vis spectral changes of [Eu2L3(DMSO)4] complex in ethanol (1.0 × 10–5 mol/L) upon irradiation at 365 nm (a) and recoverable irradiation at 450 nm (b) as a function of time.
The first-order kinetics rate constants of the photoisomerization reactions of the three [Ln2L3(DMSO)4] complexes in ethanol and acetonitrile solutions are shown in Table 1, which were similar to those of the pure H2L ligand and our previously reported azobenzene-containing lanthanide complexes. This demonstrated that the photoisomerization reaction activation energies are similar in the azobenzene-containing lanthanide complexes. However, in these three complexes, the trans-to-cis quantum yields were 1 order of magnitude smaller than both the pure H2L ligand and other azobenzene-containing lanthanide complexes. This may be because of the rigidity at both ends of the azo group by coordination to the dinuclear lanthanide ions and the different photoisomerization pathways.
In addition, the photoisomerization properties of the [Ln2L3(DMSO)4] complexes doped in polymethyl methacrylate (PMMA) films were also investigated. The trans-to-cis isomerization in the PMMA film was not observed. Different from our previously studied lanthanide complexes doped in PMMA, the trans-to-cis photoisomerization was hampered in a rigid environment owing to the azo group being in a rigid environment of the complex in solid state.
Thermal cis-to-trans Isomerization of the [Ln2L3(DMSO)4] Complexes
Cis-to-trans isomerization of azobenzene and its derivatives can also spontaneously occur in the dark. In the ethanol and acetonitrile solutions, after solutions of the ligand H2L or the [Ln2L3(DMSO)4] complexes were irradiated by UV light for half an hour to reach the photostable state, the solutions were then placed in the dark and the UV–visible absorption spectrum was obtained as a function of time. It was observed that a spontaneous change from the cis form to the relatively more stable trans form was achieved in the dark. This showed that the ligand H2L in ethanol or acetonitrile solutions could be returned to its original trans-form after being placed in the dark for about 2 days. The complexes could also gradually change from cis to trans, albeit a little slower than the free ligand (about 4 days) because the degree of freedom of the ligand in the complex was relatively small and the azobenzene moiety in the complex was more rigid and restricted, owing to the dinuclear triple helix structure of the complex.
Thermal cis-to-trans isomerization of both the ligand H2L and the [Ln2L3(DMSO)4] complexes in ethanol or acetonitrile solutions was also investigated as a function of temperature (Figure S10). The results showed that the complexes were stable in solution and the ligands did not dissociate in the solvents as temperature increased. The rate constants of the [Ln2L3(DMSO)4] complexes were a little different from those of the pure ligands, whereas the order of magnitude of thermal isomerization rates was the same as those of the pure ligands. This indicated that the thermal isomerization pathway in the ligand and these complexes was simple, and also similar to that of the free azobenzene ligands and our previously reported azobenzene-attached lanthanide complexes.36,37 It should be pointed out that the conversion of cis-to-trans under heating was higher than that by light irradiation at room temperature.
Owing to the rates of cis-to-trans thermal isomerization of H2L and the [Ln2L3(DMSO)4] complexes at room temperature being very slow, the thermal cis-to-trans isomerization of H2L and the [Ln2L3(DMSO)4] complexes was investigated at 50 °C. The UV–visible spectral changes of thermal cis-to-trans isomerization of the [Eu2L3(DMSO)4] complex in ethanol solution are shown in Figure 4 and the others are shown in Figures S11 and S12 in the Supporting Information. The first-order rate constants for the cis-to-trans thermal isomerization at 50 °C of H2L were similar to those of the azobenzene derivatives, while the first-order rate constants for the [Ln2L3(DMSO)4] complexes were higher than those of the previously reported azobenzene-containing lanthanide complexes.38 This can be attributed to the restricted rigid structure of azobenzene in the complex. The first-order rate constants (k) and half-lives (τ1/2) at 50 °C are shown in Table 3. The thermal isomerization rate of H2L in ethanol solution was higher than that in acetonitrile solution as a result of the solvent effect, but the thermal isomerization rate and half-life of the complexes in ethanol and acetonitrile solutions were basically the same regardless of the solvent effect. This clearly proves that cis-to-trans thermal isomerization of H2L proceeds via a rotation mechanism while that of the [Ln2L3(DMSO)4] complexes proceeds via an inversion mechanism. These mechanism can be discriminated from the dependence of k on the solvent polarity.49,50 The rotation mechanism occurs through a transition state (TS) where one nitrogen atom is sp hybridized, whereas rotation proceeds by dipolar TS. In the rotation mechanism, the dipolar TS is stabilized in polar solvents and higher values for k should be obtained; for the inversion mechanism, very small variation of k with solvent should be observed. It can be also seen from the Table 3 that as the atomic number increased from Eu to Gd to Tb, the lanthanide ionic radius became smaller, the thermal isomerization rate constant decreased and the half-life increased, which is in agreement with the order of the complex stability.
Figure 4.

UV–vis spectral changes before and after 30 min irradiation with 365 nm and thermal cis-to-trans isomerization of [Eu2L3(DMSO)4] complex in ethanol solution (1.0 × 10–5 mol/L) at 50 °C.
Table 3. Thermal Isomerization Rate Constants (kiso, min–1) and Half-Lives (τ1/2, min) of H2L and [Ln2L3(DMSO)4] Complexes in Different Solvents at 50 °C.
| ethanol |
acetonitrile |
|||
|---|---|---|---|---|
| compounds | 103kiso | τ1/2 | 103kiso | τ1/2 |
| H2L | 72.6 ± 5.3 | 9.54 ± 1.1 | 21.3 ± 1.9 | 32.3 ± 2.6 |
| [Eu2(L)3(DMSO)4] | 43.6 ± 3.4 | 15.7 ± 1.4 | 43.9 ± 3.9 | 15.8 ± 1.2 |
| [Gd2(L)3(DMSO)4] | 26.9 ± 2.3 | 25.7 ± 2.5 | 28.2 ± 2.9 | 24.5 ± 2.2 |
| [Tb2(L)3(DMSO)4] | 14.9 ± 1.7 | 46.3 ± 3.1 | 17.0 ± 2.3 | 40.5 ± 3.3 |
Luminescent Properties of the Complexes
It is a pity that no luminescence of the solution and solid state was observed for the three complexes at room temperature. This arose from the extremely weak fluorescence of the ligand itself and the inability to transfer its energy to the lanthanide ions.35 The singlet state energy levels of the ligand is estimated by referencing its absorbance edge, and they are equal of energy with 2.36 eV both in ethanol and acetonitrile solutions. From the phosphorescence spectrum of the [Gd2L3(DMSO)4] complex (Figure S13), the triplet energy level of ligand corresponding to its emission wavelength is calculated to be 1.98 eV. We also confirmed this experimental result by theoretically calculating the excited state energy levels of the ligand. Density functional theory (DFT) is a powerful and effective tool for solving many complicated problems in molecular electronic structures, and has successfully predicted the ground state and time-dependent excited state properties.51−53 The most commonly used DFT is the hybrid functional (Becke’s three-parameter correlation functional) and the Lee, Yang, and Parr exchange functional B3LYP method. We used the Gaussian 09a software to theoretically calculate the H2L structure at the 6-311++G(d,p) basis set with the B3LYP mixing functional using the DFT and time-dependent density functional theory (TD-DFT) method. The molecular geometry was optimized by frequency calculations until it no longer showed any imaginary frequency to ensure the optimized geometry was in the lowest energy and most stable state, and then the molecule excitation energies were calculated in the gas by TD-DFT. The polarization continuum model was also used to calculate the lowest singlet and triplet excitation energies of the ligand molecule in ethanol and acetonitrile solvents, as shown in Table 4.
Table 4. Excited States of Molecular H2La.
| excited state | multiplicity | excited energy/eV(gas) | oscillator strengths | excited energy/eV (ethanol) | oscillator strengths | excited energy/eV (acetonitrile) | oscillator strengths |
|---|---|---|---|---|---|---|---|
| 1 | T | 1.64 | 0.00 | 1.66 | 0.00 | 1.66 | 0.00 |
| 2 | T | 2.03 | 0.00 | 2.02 | 0.00 | 2.02 | 0.00 |
| 3 | T | 2.75 | 0.00 | 2.80 | 0.00 | 2.80 | 0.00 |
| 4 | S | 2.30 | 0.00 | 2.32 | 0.00 | 2.32 | 0.00 |
| 5 | S | 3.21 | 0.99 | 2.86 | 1.32 | 2.86 | 1.32 |
| 6 | S | 3.25 | 0.43 | 3.09 | 0.00 | 3.09 | 0.00 |
T = triplet state, S = singlet state.
The TD-DFT calculated energy of the first singlet excited state (2.32 eV) of the ligand in the solvent is very close to the experiment data, while the triplet excited state energy (1.66 eV) is a little smaller than the experimental data (1.98 eV). The deviation of the triplet excited state energy with 0.32 eV by TD-DFT calculation compared to the experimental result because the observed experimental phosphorescence emission energy as reference data may not exactly match the vertical excitation energy, and it is accepted that TD-DFT calculation underestimates the excitation energy in some case.54 However, both the experimental and the theoretical calculation results showed that the excitation energies of the ligand H2L in the lowest excited triplet state were all less than the first excited state of Eu3+, the energy level of the 5D0 was 2.1434 eV, and the 5D4 of Tb3+ 2.5416 eV and the 6P7/2 of Gd3+ was 3.9957 eV. According to the theory of Sato and Wada,55 the synthesized bis-β-diketone azobenzene ligand H2L is unable to efficiently transfer its energy to lanthanide ions. At the same time, the calculation results also showed that the oscillatory intensity of the first excited singlet state of azobenzene was zero, and the oscillator strength of the second excited singlet state was close to 1, which indicated that the ligand was mainly excited to the second excited singlet state; generally, relaxing to the first excited singlet state and then to the ground state also hampers the transfer of energy from the ligand to the lanthanide ions. The calculation results prove the reason why the luminescence of the three lanthanide complexes was not observed at room temperature. Therefore, it is necessary to determine the excitation energies of a ligand by theoretical calculation to assist in designing a reasonable ligand to obtain luminescent lanthanide complexes.
Conclusions
We synthesized three bis-β-diketonate lanthanide complexes with an azobenzene bridge and characterized their structures. The three complexes crystallize in the triclinic P1̅ space groups and the structure features a triple-stranded dinuclear helicate by the coordination of three bis-β-diketonate ligands to two crystallographically equivalent eight-coordinated lanthanide(III) ions. All three complexes show the photoisomerization reactions in solutions similar to their pure H2L ligand. The trans-to-cis quantum yields are 1 order of magnitude smaller than both the pure H2L ligand and the other azobenzene-containing lanthanide complexes. The photoisomerization reaction activation energy is similar in the azobenzene-containing lanthanide complexes. The rates of thermal isomerization of the [Ln2L3(DMSO)4] complexes at 50 °C are almost the same as those of the pure ligands and higher than our previously reported azobenzene-attached lanthanide complexes, and the conversion of cis-to-trans under heating is higher than that by light irradiation at room temperature. Furthermore, as the atomic number increases, the lanthanide ionic radius from Eu3+ to Gd3+ to Tb3+ becomes smaller, the thermal isomerization rate constant decreases and the half-life increases, which is in agreement with the order of the complex stability. All these characteristics can be supposed to arise from the rigidity at both ends of the azo group by coordination to the dinuclear lanthanide ions and the different photoisomerization pathway. These are the first examples of bis-β-diketonate lanthanide complexes with an azobenzene bridge and will assist to illustrate the azobenzene isomerization mechanism for theoretical chemists.
Methods
Materials and Methods
Ln(NO3)3·6H2O (Ln = Eu, Gd, Tb; Beijing Huawei Ruike Chemical Co., Ltd.), 4-aminoacetophenone, PMMA (Alfa Aesar), and ethyl trifluoroacetate (Adamas Reagent Co., Ltd.) were used as received. All other chemicals were of analytical grade and were purchased from Sinopharm Chemicals Group Co. Analytical-grade solvents were redistilled prior to use. The melting points were determined with X-4 micromelting point apparatus (Shanghai Precision & Scientific Instrument Co., Ltd) without correction. The elemental analyses were acquired with a Vario EL III elemental analyzer. The electrospray ionization mass spectrometry data were obtained with a Bruker ESQUIRE-3000 Plus LC–MS/MS spectrometer. The IR spectra (KBr pellets) were scanned with a Nicolet Avatar FT-IR 330 spectrometer. The solution 1H NMR spectra were obtained at 400 MHz with a Bruker ADVANCE 400 spectrometer at 25 °C. The chemical shifts (ppm) were determined using tetramethylsilane as the internal reference. TGA was carried out on a SDTQ 600 thermogravimeter under a nitrogen atmosphere with a heating rate of 10 °C/min. The UV–visible (vis) absorption spectra were scanned on a Shimadzu UV224012PC absorption spectrometer. A Shanghai Yihui ZF-3 UV analyzer was used to produce the 365 nm UV light and the 450 nm visible light was produced from a 300 W PLS-SXE300CUV Xe lamp and isolated by a sharp cut-off filter.
Ligand Synthesis
4,4′-Acetylazobenzene56
p-Aminoacetophenone (0.068 g, 0.5 mmol) and sodium iodide (0.15 g, 1.0 mmol) were mixed in a 100 mL flask, then t-butyl hypochlorite (0.11 g, 1.0 mmol) was slowly added under a nitrogen atmosphere, and the reaction was kept at a temperature of −20 °C for 12 h. After the reaction was completed, sodium thiosulfate solution (10.0 mL, 1.0 mmol/L) was added to the reaction mixture solution, and the mixture was stirred for 2 min. The reaction solution was extracted with dichloromethane three times. The lower layer solution was collected and dried over anhydrous sodium sulfate. The filtrate was evaporated to dryness and washed several times with acetone, and the solid was collected to give a pure dark red solid L1 (0.074 g, yield 57%). m.p. 209–210 °C; ESI(+)-MS (m/z, methanol): 265.3 [M+]; 1H NMR (400 MHz, CDCl3): δ (ppm) 8.15 (d, J = 8.4 Hz, 4H), 8.04 (d, J = 8.4 Hz, 4H), 2.71 (s, 6H).
4,4′-(4,4,4-Trifluoro-1,3-butanedione)azobenzene42,57
Ethylene glycol dimethyl ether (5 mL) was added to a 100 mL flask containing 0.08 g (2 mmol) sodium hydride. The solution was stirred at room temperature for 10 min; then 0.284 g (2 mmol) of ethyl trifluoroacetate was added to the above solution, and the mixture was stirred at room temperature for another 30 min. After that, 0.18 g (0.68 mmol) of L1 was added to the above mixed solution and stirred for 24 h. When the reaction was completed, 10 mL of ice water was added to the mixtures. The pH of the solution was adjusted to 2–3 with 0.1 mol/L HCl, then the solution was filtered, and the precipitate was washed with 1:1 ethanol/water (V/V). The product was recrystallized from petroleum ether and chloroform mixed solvents to give a pure brown solid H2L (0.54 g, yield 62%). mp 166–168 °C; ESI(−)–MS (m/z, methanol): 489.8 [M + methanol]−; 1H NMR (400 MHz, CDCl3): δ (ppm) 15.00 (s, 2H), 8.15 (d, J = 8.6 Hz, 4H), 8.09 (d, J = 8.5 Hz, 4H), 6.72 (s, 2H); FT-IR (KBr, cm–1): 3433 (s, O–H), 3115, 3072 (w, C–H), 1600 (s, C=O), 1492 (m, N=N), 1266 (s, C–N), 1199, 1160 (s, C–O), 1104, 1064 (s, C–F), 1009 (m, C–C), 866, 807, 701 (m, Ph-H); elemental analysis calcd for C20H12O4N2F6 (%): C, 52.41; H, 2.63; N, 6.11. Found (%): C, 52.32; H, 2.60; N, 5.92.
Synthesis of Ln(III) Complexes
General Procedures
Ligand H2L (0.137 g, 0.3 mmol) was added to a round-bottom flask containing 10 mL of absolute ethanol and stirred at 60 °C, and then 0.6 mL of 1.0 mol/L sodium hydroxide solution was added dropwise to the solution. After the ligand L was completely dissolved, the solution was stirred continuously at room temperature. When the solution was cooled to room temperature, a solution of rare earth nitrate salt (0.2 mmol) in 2 mL of absolute ethanol was added slowly to the above reaction system, and the reaction was stirred at room temperature for 24 h. After the reaction was completed, 15 mL of deionized water was added to the solution and filtered. The precipitate was washed with 1:1 ethanol/water mixed solvents several times and dried in a vacuum drying oven. The solid powder was crystalized with mixed solvents of DMSO and absolute ethanol to obtain a final pure rare earth complex. Suitable crystals were obtained by volatilization of the solution in a test tube having a diameter of 0.5 cm.
[Eu2L3(DMSO)4]
It was obtained as an orange solid (0.13 g, yield 72%), 1H NMR (400 MHz, DMSO): δ (ppm) 8.37 (s, 4H), 7.83 (s, 4H), 6.98 (s, 2H), 2.46 (s, 8H). FT-IR (KBr, cm–1): 2917, 2845 (w, C–H), 1614 (s, C=O), 1572, 1528 (s, C=C), 1490, 1461 (m, N=N), 1308, 1288 (s, S=O), 1245 (m, C–N), 1193, 1139 (s, C–O), 1067 (s, C–F), 1009 (m, C–C), 863, 794, 696 (s, Ph-H); elemental analysis calcd for Eu2C68H54O16N6F18S4 (%): C, 41.13; H, 2.74; N, 4.23. Found (%): C, 40.64; H, 2.71; N, 4.13.
[Gd2L3(DMSO)4]
It was obtained as an orange solid (0.14 g, yield 70%), FT-IR (KBr, cm–1): 2912, 2849 (w, C–H), 1615 (s, C=O), 1572, 1528 (s, C=C), 1491, 1462 (m, N=N), 1309, 1289 (s, S=O), 1245 (m, C–N), 1193, 1139 (s, C–O), 1067 (s, C–F), 1009 (m, C–C), 863, 794, 696 (s, Ph-H); elemental analysis calcd for Gd2C68H54O16N6F18S4 (%): C, 40.92; H, 2.72; N, 4.21. Found (%): C, 40.82; H, 2.69; N, 4.21.
[Tb2L3(DMSO)4]
It was obtained as an orange solid (0.14 g, yield 72%), FT-IR (KBr, cm–1): 2914, 2853 (w, C–H), 1615 (s, C=O), 1572, 1528 (s, C=C), 1491, 1462 (m, N=N), 1309, 1289 (s, S=O), 1244 (m, C–N), 1193, 1139 (s, C–O), 1067 (s, C–F), 1009 (m, C–C), 862, 794, 696 (s, Ph-H); elemental analysis calcd for Tb2C68H54O16N6F18S4 (%): C, 40.85; H, 2.72; N, 4.20. Found (%) C, 40.06; H, 2.63; N, 3.92.
Structure Determination of[Ln2L3(DMSO)4]Complexes by X-ray Diffraction
The single-crystal data of [Eu2L3(DMSO)4], [Gd2L3(DMSO)4], and [Tb2L3(DMSO)4] were collected using an Oxford Diffraction Gemini R Ultra Ruby CCD diffractometer with MoKα (α = 0.71073 Å) radiation. Absorption correction was applied to the data using the multiscan CrysAlis Red program.58 The structures were solved by direct methods and refined by full-matrix least-squares calculations based on F2 using the SHELXTL-2014 software package.59 All of the non-hydrogen atoms were refined anisotropically. Data for the crystal structures of the three complexes have been deposited at the Cambridge Crystallographic Data Centre (CCDC 1546472, 1546473, 1546474 for complexes [Eu2L3(DMSO)4], [Gd2L3(DMSO)4] and [Tb2L3(DMSO)4], respectively); these data are obtainable free of charge via the internet at www.ccdc.cam.ac.uk/conts/retrieving.
Acknowledgments
This work is supported by the National Natural Science Foundation of China (grant no. 21271150). We thank Liwen Bianji, Edanz Group China, for editing the English text of the draft of this manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01817.
Crystallographic data; selected bond lengths; thermogravimetric analysis curves; observed and calculated X-ray powder diffraction patterns; crystal structures; UV-vis spectral changes; phosphorescence spectra; 1H NMR spectra; IR spectra for H2L and all complexes, and NMR spectra of H2L and complex [Eu2L3(DMSO)4] (PDF)
Crystallographic data for C68 H54 F18 N6 O16 S4 Eu2 (CIF)
Crystallographic data for C68 H54 F18 N6 O16 S4 Gd2 (CIF)
Crystallographic data for C68 H54 F18 N6 O16 S4 Tb2 (CIF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bandara H. M. D.; Burdette S. C. Photoisomerization in Different Classes of Azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. 10.1039/c1cs15179g. [DOI] [PubMed] [Google Scholar]
- Tamai N.; Miyasaka H. Ultrafast Dynamics of Photochromic Systems. Chem. Rev. 2000, 100, 1875–1890. 10.1021/cr9800816. [DOI] [PubMed] [Google Scholar]
- Kumar G. S.; Neckers D. C. Photochemistry of Azobenzene-Containing Polymers. Chem. Rev. 1989, 89, 1915–1925. 10.1021/cr00098a012. [DOI] [Google Scholar]
- Rasmussen P. H.; Ramanujam P. S.; Hvilsted S.; Berg R. H. A Remarkably Efficient Azobenzene Peptide for Holographic Information Storage. J. Am. Chem. Soc. 1999, 121, 4738–4743. 10.1021/ja981402y. [DOI] [Google Scholar]
- Kumar A. S.; Ye T.; Takami T.; Yu B.-C.; Flatt A. K.; Tour J. M.; Weiss P. S. Reversible Photo-Switching of Single Azobenzene Molecules in Controlled Nanoscale Environments. Nano Lett. 2008, 8, 1644–1648. 10.1021/nl080323+. [DOI] [PubMed] [Google Scholar]
- Fan C. B.; Liu Z. Q.; Gong L. L.; Zheng A. M.; Zhang L.; Yan C. S.; Wu H. Q.; Feng X. F.; Luo F. Photoswitching Adsorption Selectivity in a Diarylethene-Azobenzene Mof. Chem. Commun. 2017, 53, 763–766. 10.1039/c6cc08982h. [DOI] [PubMed] [Google Scholar]
- Zhou H. W.; Xue C. G.; Weis P.; Suzuki Y.; Huang S. L.; Koynov K.; Auernhammer G. K.; Berger R.; Butt H. J.; Wu S. Photoswitching of Glass Transition Temperatures of Azobenzene-Containing Polymers Induces Reversible Solid-to-Liquid Transitions. Nat. Chem. 2017, 9, 145–151. 10.1038/nchem.2625. [DOI] [PubMed] [Google Scholar]
- Blinov L. M.; Kozlovsky M. V.; Cipparrone G. Photochromism and Holographic Grating Recording on a Chiral Side-Chain Liquid Crystalline Copolymer Containing Azobenzene Chromophores. Chem. Phys. 1999, 245, 473–485. 10.1016/s0301-0104(99)00104-4. [DOI] [Google Scholar]
- Piosik E.; Korbecka I.; Galewski Z.; Martynski T. Molecular Organization and Optical Switching of Liquid-Crystalline Azobenzenes in Monomolecular Films. Opt. Mater. 2018, 86, 475–483. 10.1016/j.optmat.2018.10.050. [DOI] [Google Scholar]
- Kucharski T. J.; Ferralis N.; Kolpak A. M.; Zheng J. O.; Nocera D. G.; Grossman J. C. Templated Assembly of Photoswitches Significantly Increases the Energy-Storage Capacity of Solar Thermal Fuels. Nat. Chem. 2014, 6, 441–447. 10.1038/nchem.1918. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Liu H. P.; Luo W.; Liu E. Z.; Zhao N. Q.; Yoshino K.; Feng W. Covalent Functionalization of Graphene by Azobenzene with Molecular Hydrogen Bonds for Long-Term Solar Thermal Storage. Sci. Rep. 2013, 3, 3260–3267. 10.1038/srep03260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L.; Feng Y.; Wang L.; Feng W. Azobenzene-Based Solar Thermal Fuels: Design, Properties, and Applications. Chem. Soc. Rev. 2018, 47, 7339–7368. 10.1039/c8cs00470f. [DOI] [PubMed] [Google Scholar]
- Deiana M.; Pokladek Z.; Olesiak-Banska J.; Młynarz P.; Samoc M.; Matczyszyn K. Photochromic Switching of the DNA Helicity Induced by Azobenzene Derivatives. Sci. Rep. 2016, 6, 28605–28614. 10.1038/srep28605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panja A.; Matsuo T.; Nagao S.; Hirota S. DNA Cleavage by the Photocontrolled Cooperation of Zn(II) Centers in an Azobenzene-Linked Dizinc Complex. Inorg. Chem. 2011, 50, 11437–11445. 10.1021/ic201244y. [DOI] [PubMed] [Google Scholar]
- Chen E.; Kumita J. R.; Woolley G. A.; Kliger D. S. The Kinetics of Helix Unfolding of an Azobenzene Cross-Linked Peptide Probed by Nanosecond Time-Resolved Optical Rotatory Dispersion. J. Am. Chem. Soc. 2003, 125, 12443–12449. 10.1021/ja030277+. [DOI] [PubMed] [Google Scholar]
- Samai S.; Bradley D. J.; Choi T. L. Y.; Yan Y.; Ginger D. S. Temperature-Dependent Photoisomerization Quantum Yields for Azobenzene-Modified DNA. J. Phys. Chem. C 2017, 121, 6997–7004. 10.1021/acs.jpcc.6b12241. [DOI] [Google Scholar]
- Moldt T.; Przyrembel D.; Schulze M.; Bronsch W.; Boie L.; Brete D.; Gahl C.; Klajn R.; Tegeder P.; Weinelt M. Differing Isomerization Kinetics of Azobenzene-Functionalized Self-Assembled Monolayers in Ambient Air and in Vacuum. Langmuir 2016, 32, 10795–10801. 10.1021/acs.langmuir.6b01690. [DOI] [PubMed] [Google Scholar]
- Castellanos S.; Goulet-Hanssens A.; Zhao F.; Dikhtiarenko A.; Pustovarenko A.; Hecht S.; Gascon J.; Kapteijn F.; Bléger D. Structural Effects in Visible-Light-Responsive Metal-Organic Frameworks Incorporating Ortho-Fluoroazobenzenes. Chem.—Eur. J. 2016, 22, 746–752. 10.1002/chem.201503503. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Heinke L.; Jelic J.; Cakici M.; Dommaschk M.; Maurer R. J.; Oberhofer H.; Grosjean S.; Herges R.; Brase S.; et al. Photoswitching in Nanoporous, Crystalline Solids: An Experimental and Theoretical Study for Azobenzene Linkers Incorporated in Mofs. Phys. Chem. Chem. Phys. 2015, 17, 14582–14587. 10.1039/c5cp01372k. [DOI] [PubMed] [Google Scholar]
- Angelini G.; Canilho N.; Emo M.; Kingsley M.; Gasbarri C. Role of Solvent and Effect of Substituent on Azobenzene Isomerization by Using Room-Temperature Ionic Liquids as Reaction Media. J. Org. Chem. 2015, 80, 7430–7434. 10.1021/acs.joc.5b00898. [DOI] [PubMed] [Google Scholar]
- Neukirch A. J.; Shamberger L. C.; Abad E.; Haycock B. J.; Wang H.; Ortega J.; Prezhdo O. V.; Lewis J. P. Nonadiabatic Ensemble Simulations of Cis-Stilbene and Cis-Azobenzene Photoisomerization. J. Chem. Theory Comput. 2014, 10, 14–23. 10.1021/ct4009816. [DOI] [PubMed] [Google Scholar]
- Duarte L.; Fausto R.; Reva I. Structural and Spectroscopic Characterization of E- and Z-Isomers of Azobenzene. Phys. Chem. Chem. Phys. 2014, 16, 16919–16930. 10.1039/c4cp00240g. [DOI] [PubMed] [Google Scholar]
- Bao J.; Weber P. M. Electronic Effects on Photochemistry: The Diverse Reaction Dynamics of Highly Excited Stilbenes and Azobenzene. J. Am. Chem. Soc. 2011, 133, 4164–4167. 10.1021/ja108598w. [DOI] [PubMed] [Google Scholar]
- Dokić J.; Gothe M.; Wirth J.; Peters M. V.; Schwarz J.; Hecht S.; Saalfrank P. Quantum Chemical Investigation of Thermal Cis-to-Trans Isomerization of Azobenzene Derivatives: Substituent Effects, Solvent Effects, and Comparison to Experimental Data. J. Phys. Chem. A 2009, 113, 6763–6773. 10.1021/jp9021344. [DOI] [PubMed] [Google Scholar]
- Yamamura M.; Okazaki Y.; Nabeshima T. Photoisomerization Locking of Azobenzene by Formation of a Self-Assembled Macrocycle. Chem. Commun. 2012, 48, 5724–5726. 10.1039/c2cc18014f. [DOI] [PubMed] [Google Scholar]
- Bléger D.; Schwarz J.; Brouwer A. M.; Hecht S. O-Fluoroazobenzenes as Readily Synthesized Photoswitches Offering Nearly Quantitative Two-Way Isomerization with Visible Light. J. Am. Chem. Soc. 2012, 134, 20597–20600. 10.1021/ja310323y. [DOI] [PubMed] [Google Scholar]
- Yan J.-F.; Lin D.-Q.; Wang X.-G.; Wu K.-Q.; Xie L.-L.; Yuan Y.-F. Ferrocenylethynyl-Terminated Azobenzenes: Synthesis, Electrochemical, and Photoisomerization studies. Chem.—Asian J. 2015, 10, 614–621. 10.1002/asia.201403327. [DOI] [PubMed] [Google Scholar]
- Kume S.; Kurihara M.; Nishihara H. Reversible Trans-Cis Photoisomerization of Azobenzene-Attached Bipyridine Ligands Coordinated to Cobalt Using a Single UV Light Source and the Co(III)/Co(II) Redox Change. Chem. Commun. 2001, 1656–1657. 10.1039/b104381c. [DOI] [PubMed] [Google Scholar]
- Yamamura M.; Yamakawa K.; Okazaki Y.; Nabeshima T. Coordination-Driven Macrocyclization for Locking of Photo- and Thermalcis→transIsomerization of Azobenzene. Chem.—Eur. J. 2014, 20, 16258–16265. 10.1002/chem.201404620. [DOI] [PubMed] [Google Scholar]
- Guezguez I.; Ayadi A.; Ordon K.; Iliopoulos K.; Branzea D. G.; Migalska-Zalas A.; Makowska-Janusik M.; El-Ghayoury A.; Sahraoui B. Zinc Induced a Dramatic Enhancement of the Nonlinear Optical Properties of an Azo-Based Iminopyridine Ligand. J. Phys. Chem. C 2014, 118, 7545–7553. 10.1021/jp412204f. [DOI] [Google Scholar]
- Pérez-Miqueo J.; Telleria A.; Muñoz-Olasagasti M.; Altube A.; García-Lecina E.; de Cózar A.; Freixa Z. Azobenzene-Functionalized Iridium(III) Triscyclometalated Complexes. Dalton Trans. 2015, 44, 2075–2091. 10.1039/c4dt02018a. [DOI] [PubMed] [Google Scholar]
- Sierocki P.; Maas H.; Dragut P.; Richardt G.; Vögtle F.; De Cola L.; Brouwer F.; Zink J. I. Photoisomerization of Azobenzene Derivatives in Nanostructured Silica. J. Phys. Chem. B 2006, 110, 24390–24398. 10.1021/jp0641334. [DOI] [PubMed] [Google Scholar]
- Yutaka T.; Mori I.; Kurihara M.; Mizutani J.; Kubo K.; Furusho S.; Matsumura K.; Tamai N.; Nishihara H. Synthesis, Characterization, and Photochemical Properties of Azobenzene-Conjugated Ru(II) and Rh(III) Bis(Terpyridine) Complexes. Inorg. Chem. 2001, 40, 4986–4995. 10.1021/ic010351u. [DOI] [PubMed] [Google Scholar]
- Yutaka T.; Mori I.; Kurihara M.; Mizutani J.; Tamai N.; Kawai T.; Irie M.; Nishihara H. Photoluminescence Switching of Azobenzene-conjugated Pt(II) Terpyridine Complexes by Trans-Cis Photoisomerization. Inorg. Chem. 2002, 41, 7143–7150. 10.1021/ic0260116. [DOI] [PubMed] [Google Scholar]
- Telleria A.; Pérez-Miqueo J.; Altube A.; García-Lecina E.; de Cózar A.; Freixa Z. Azobenzene-Appended Bis-Cyclometalated Iridium(III) Bipyridyl Complexes. Organometallics 2015, 34, 5513–5529. 10.1021/acs.organomet.5b00838. [DOI] [Google Scholar]
- Lin L.-R.; Wang X.; Wei G.-N.; Tang H.-H.; Zhang H.; Ma L.-H. Azobenzene-Derived Tris-Beta-Diketonate Lanthanide Complexes: Reversible Trans-to-Cis Photoisomerization in Solution and Solid State. Dalton Trans. 2016, 45, 14954–14964. 10.1039/c6dt01310d. [DOI] [PubMed] [Google Scholar]
- Lin L.-R.; Tang H.-H.; Wang Y.-G.; Wang X.; Fang X.-M.; Ma L.-H. Functionalized Lanthanide(III) Complexes Constructed from Azobenzene Derivative and Beta-Diketone Ligands: Luminescent, Magnetic, and Reversible Trans-to-Cis Photoisomerization Properties. Inorg. Chem. 2017, 56, 3889–3900. 10.1021/acs.inorgchem.6b02819. [DOI] [PubMed] [Google Scholar]
- Wang Y.-G.; Li Y.-Q.; Tang H.-H.; Lin L.-R.; Ma L.-H. Near-Infrared Photoluminescence and Reversible Trans-to-Cis Photoisomerization of Mononuclear and Binuclear Ytterbium(III) Complexes Functionalized by Azobenzene Groups. ACS Omega 2018, 3, 5480–5490. 10.1021/acsomega.8b00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y.; Shi J.; Chu W.; Sun Z. Synthesis, Crystal Structure, and near-Ir Luminescent Properties of Lanthanide Bis(-Diketonate) Complexes. Eur. J. Inorg. Chem. 2013, 3063–3069. 10.1002/ejic.201300013. [DOI] [PubMed] [Google Scholar]
- Bai J.; Gu H.; Hou Y.; Wang S. Luminescence Properties and Molecular Mechanics Calculation of Bis-Beta-Diketonate Eu3+ Complex/Polymer Hybrid Fibers. Opt. Mater. 2018, 79, 310–316. 10.1016/j.optmat.2018.03.029. [DOI] [Google Scholar]
- Chen P.; Li H.; Sun W.; Tang J.; Zhang L.; Yan P. Crystallization of Triple- and Quadruple-Stranded Dinuclear Bis-Beta-Diketonate-Dy(III) Helicates: Single Molecule Magnetic Behavior. Cryst. Eng. Comm. 2015, 17, 7227–7232. 10.1039/c5ce01067e. [DOI] [Google Scholar]
- Li H.-F.; Yan P.-F.; Chen P.; Wang Y.; Xu H.; Li G.-M. Highly Luminescent Bis-Diketone Lanthanide Complexes with Triple-Stranded Dinuclear Structure. Dalton Trans. 2012, 41, 900–907. 10.1039/c1dt11496d. [DOI] [PubMed] [Google Scholar]
- Norikane Y.; Tamaoki N. Photochemical and Thermal Cis/Trans Isomerization of Cyclic and Noncyclic Azobenzene Dimers: Effect of a Cyclic Structure on Isomerization. Eur. J. Org. Chem. 2006, 1296–1302. 10.1002/ejoc.200500682. [DOI] [Google Scholar]
- Norikane Y.; Kitamoto K.; Tamaoki N. Novel Crystal Structure, Cis-Trans Isomerization, and Host Property of Meta-Substituted Macrocyclic Azobenzenes with the Shortest Linkers. J. Org. Chem. 2003, 68, 8291–8304. 10.1021/jo0341160. [DOI] [PubMed] [Google Scholar]
- Nagamani S. A.; Norikane Y.; Tamaoki N. Photoinduced Hinge-Like Molecular Motion: Studies on Xanthene-Based Cyclic Azobenzene Dimers. J. Org. Chem. 2005, 70, 9304–9313. 10.1021/jo0513616. [DOI] [PubMed] [Google Scholar]
- Batista H. J.; de Andrade A. V. M.; Longo R. L.; Simas A. M.; de Sá G. F.; Thompson L. C.; Thompson L. C. Synthesis, X-Ray Structure, Spectroscopic Characterization, and Theoretical Prediction of the Structure and Electronic Spectrum of Eu(Btfa)3Bipy and an Assessment of the Effect of Fluorine as a Beta-Diketone Substituent on the Ligand-Metal Energy Transfer Process. Inorg. Chem. 1998, 37, 3542–3547. 10.1021/ic971602v. [DOI] [PubMed] [Google Scholar]
- Pettinari C.; Marchetti F.; Pettinari R.; Drozdov A.; Troyanov S.; Voloshin A. I.; Shavaleev N. M. Synthesis, Structure and Luminescence Properties of New Rare Earth Metal Complexes with 1-Phenyl-3-Methyl-4-Acylpyrazol-5-Ones. J. Chem. Soc., Dalton Trans. 2002, 1409–1415. 10.1039/b108058j. [DOI] [Google Scholar]
- Gajda K.; Zarychta B.; Daszkiewicz Z.; Domański A. A.; Ejsmont K. Substituent Effects in Trans-P, P ′-Disubstituted Azobenzenes: X-Ray Structures at 100 K and DFT-Calculated Structures. Acta Crystallogr., Sect. C: Struct. Chem. 2014, 70, 575–579. 10.1107/s2053229614009942. [DOI] [PubMed] [Google Scholar]
- Asano T.; Okada T. Further Kinetic Evidence for the Competitive Rotational and Inversional Z-E Isomerization of Substituted Azobenzenes. J. Org. Chem. 1986, 51, 4454–4458. 10.1021/jo00373a021. [DOI] [Google Scholar]
- Jerca F. A.; Jerca V. V.; Anghel D. F.; Stinga G.; Marton G.; Vasilescu D. S.; Vuluga D. M. Novel Aspects Regarding the Photochemistry of Azo-Derivatives Substituted with Strong Acceptor Groups. J. Phys. Chem. C 2015, 119, 10538–10549. 10.1021/jp511925n. [DOI] [Google Scholar]
- Grotjahn R.; Maier T. M.; Michl J.; Kaupp M. Development of a Tddft-Based Protocol with Local Hybrid Functionals for the Screening of Potential Singlet Fission Chromophores. J. Chem. Theory Comput. 2017, 13, 4984–4996. 10.1021/acs.jctc.7b00699. [DOI] [PubMed] [Google Scholar]
- Rubešová M.; Muchova E.; Slavicek P. Optimal Tuning of Range-Separated Hybrids for Solvated Molecules with Time-Dependent Density Functional Theory. J. Chem. Theory Comput. 2017, 13, 4972–4983. 10.1021/acs.jctc.7b00675. [DOI] [PubMed] [Google Scholar]
- Jacquemin D.; Moore B.; Planchat A.; Adamo C.; Autschbach J. Performance of an Optimally Tuned Range-Separated Hybrid Functional for 0-0 Electronic Excitation Energies. J. Chem. Theory Comput. 2014, 10, 1677–1685. 10.1021/ct5000617. [DOI] [PubMed] [Google Scholar]
- Silva-Junior M. R.; Schreiber M.; Sauer S. P. A.; Thiel W. Benchmarks for Electronically Excited States: Time-dependent Density Functional Theory and Density Functional Theory Based Multireference Configuration Interaction. J. Chem. Phys. 2008, 129, 104103–104114. 10.1063/1.2973541. [DOI] [PubMed] [Google Scholar]
- Sato S.; Wada M. Relations between Intramolecular Energy Transfer Efficiencies and Triplet State Energies in Rare Earth B-Diketone Chelates. Bull. Chem. Soc. Jpn. 1970, 43, 1955–1962. 10.1246/bcsj.43.1955. [DOI] [Google Scholar]
- Takeda Y.; Okumura S.; Minakata S. Oxidative Dimerization of Aromatic Amines Using Tbuoi: Entry to Unsymmetric Aromatic Azo Compounds. Angew. Chem., Int. Ed. 2012, 51, 7804–7808. 10.1002/anie.201202786. [DOI] [PubMed] [Google Scholar]
- Liu S.; He P.; Wang H.; Shi J.; Gong M. A Luminescent Quadruple Stranded Dinuclear Eu(Iii) Complex Based on 2,8-Bis(4 ′,4 ′,4 ′-Trifluoro-1 ′,3 ′-Dioxobutyl)-Dibenzothiophene for Light-Emitting Diodes. Inorg. Chem. Commun. 2009, 12, 506–508. 10.1016/j.inoche.2009.04.004. [DOI] [Google Scholar]
- Sheldrick G. M. Shelxt - Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. 10.1107/s2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement with Shelxl. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/s2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.