

Abstract

Emerging chemicals of concern (ECCs), including phthalate plasticizers, flame retardants, and phenolic compounds, are likely present in electronic nicotine delivery system (ENDS) replacement solutions (e-liquids) which are often packaged, stored in, and/or can contact with, plastic, glass, and metal materials. Developing and validating an efficient analytical method for concurrent quantification of ECCs in e-liquids are thus needed to inform evidence-based safety evaluation of ENDS products. In this study, we developed and validated a “dilute-and-shoot” method using high-performance liquid chromatography coupled with tandem mass spectrometry to simultaneously measure organophosphate flame retardants (OPFRs), phthalate plasticizers, and tetrabromobisphenol A (TBBPA) in e-liquids. We analyzed samples in positive electrospray ionization mode (ESI+) for OPFRs and phthalates and negative ESI– for TBBPA. The method has a total runtime of 10 min. The optimized procedure was able to deliver broad dynamic linearity ranges with coefficients of determination (R2) above 0.995, limits of detection ranging from 0.020 to 10 ng/mL, average accuracy within ±15%, and imprecision ≤ 15.0% for all analytes. To our knowledge, this is the first multianalysis method for measuring ECCs in e-liquid samples, and the validation results show that it is sensitive, accurate, precise, and efficient.
Introduction
Electronic cigarettes are engineered to heat a liquid solution (hereafter called e-liquid) so that the generated aerosol can be inhaled by the user. e-Liquids are often packaged and/or stored in plastic, glass, and metal materials. This has raised concerns about potential contamination of e-liquids by many emerging chemicals of concern (ECCs),1 including phthalates, organophosphate flame retardants (OPFRs), and phenolic compounds (e.g., tetrabromobisphenol A, TBBPA), that have been widely used as plasticizers, flame retardants, lubricants, preservatives, antifoaming stabilizers, and surfactants in plastics, electronics, and packaging materials because many of these chemicals are not chemically bonded to the materials in which they are present, and they can leach or outgas with time and use. In addition, ingredients in e-liquids, for example, propylene glycol (PG) and/or vegetable glycerin (VG), flavoring compounds, and nicotine can be manufactured using materials that may contain these chemicals. Consequently, people may be at potential health risks of being exposed to ECCs when using electronic nicotine delivery system (ENDS) products. A number of studies have documented that ECC exposures may lead to adverse health outcomes, including carcinogenic activity, neurotoxicity, endocrine disruption, and reproductive and developmental abnormalities.2−11 As a matter of fact, ECCs are often overlooked in current debates with regard to the health impacts of e-cigarettes which are focused on the known toxicants present in cigarettes12 that are generally reduced in e-cigarettes, such as polycyclic aromatic hydrocarbons, nitrosamines, volatile organic compounds, nitrogen oxides (NOx), CO, and metals.13−17
For this, investigation of the types and concentrations of ECCs in e-liquids is needed to provide scientific data for a better evaluation of the safety of the ENDS products. Using gas chromatography (GC)–mass spectrometry (MS), Oh and Shin developed a method for measuring diethyl phthalate (DEP) and diethylhexyl phthalate (DEHP) in replacement liquid.18 Several other phthalates of interest were not included in their method. Besides, this method employed liquid–liquid extraction which is relatively time-consuming and required a volume of 0.5 mL for sample preparation to achieve the lowest limits of detection (LODs) that were 0.003 and 0.004 μg/mL for DEP and DEHP, respectively. As such, in many cases, the maximum volume of e-liquids contained in the ENDS products may be insufficient for repeating the analysis often required to ensure the reliability of the analytical data. Some of the limitations have been improved in a method based on high-performance liquid chromatography coupled with tandem MS (HPLC–MS/MS) reported by Moldoveanu and Yerabolu.19 For example, the sample amount was reduced to 100 mg, and six other phthalates including dibutyl (DBP), benzyl butyl (BBP), diphenyl, di-n-octyl, diisononyl, and diisodecyl phthalates were simultaneously measured along with DEP and DEHP. However, the LODs of the analytes in this method were around 20 ng/mL or higher, which are not as sensitive as those described in the method by Oh and Shin.18 Besides, this method also required a runtime of 20 min for these compounds, which is relatively inefficient for studies involving large sample sizes. Current liquid chromatography technology allows a short runtime of <10 min and is thus able to reduce the consumable costs in the long run. “Cost-effectiveness” is usually a factor that needs to be considered when developing analytical assays which have a high-frequency application potential.
In this study, we aimed to develop and validate the HPLC–MS/MS analytical method to simultaneously measure the concentrations of ECCs, including phthalates, OPFRs, and TBBPA, in e-liquid samples. To our knowledge, this is the first multianalysis method for measuring these chemicals in e-liquids with high sensitivity, accuracy, precision, efficiency, and robustness.
Results and Discussion
Selection of “Dilute–Shoot” and SPE Cleanup Sample Preparation
We first optimized SPE cleanup procedures aiming to obtain higher sensitivity. Although most of OPFRs and phthalates are lipophilic compounds, their polarity and solubility vary in broad ranges, resulting in a wide retention ability on SPEs. We found that a higher percentage of methanol in washing solution (e.g., 50%) provided cleaner samples but unfortunately also lower recoveries for some compounds (Figure 1), including triethyl phosphate (TEP) and dimethyl phthalate (DMP), compared to those washed using solvents consisting of a lower methanol percentage (15%). Choosing a solvent with a proper percentage of methanol in water relies on several considerations. One important consideration is the type of the sample matrix. Unlike many other types of samples, including human samples, e-liquid samples predominantly contain PG, VG, or both, which have minimal retention on C18. For this reason, washing solvents with a low methanol percentage in water can still be effective to elute PG/VG from SPE. Any PG/VG residuals in the treated samples can be separated and directed into a waste container by the liquid chromatography (LC) flow after injection.
Figure 1.
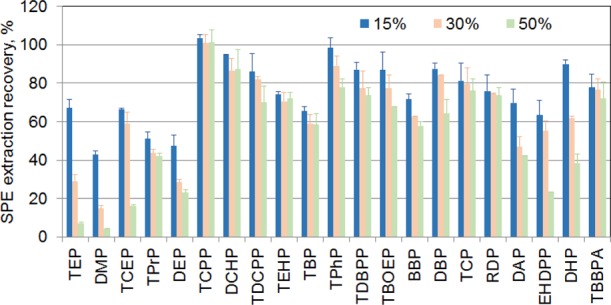
Examination of the effects of methanol fraction in the SPE washing solvent on the preparation recovery.
Another important consideration is the sensitivity needed for achieving specific research goals. Higher sensitivity is often desired for obtaining a higher detection rate. This is important because many of the compounds are potential endocrine-disrupting chemicals (EDCs) which can adversely affect human health even at low levels.20 For this reason, we chose to use 15% of methanol in water as the second washing solvent following the first washing step by pure water, and we observed that this procedure can provide an average extraction recovery of >60% for all analytes.
Using the SPE extraction method, we also observed that LODs for all analytes can reach ≤25 pg/mL. The enhanced sensitivity obtained using SPE cleanup procedures would provide a large capacity for obtaining high detection rates that are critical for accurately examining the adverse health effects attributable to e-cigarette use in future studies. However, we observed that SPE cleanup procedures can also enrich or lead to elevated background residual levels for most of the analytes, causing broad variations in analytical results, especially for those compounds whose concentrations were <5 ng/mL. For this reason, we chose to develop, optimize, and validate a direct “dilute–shoot” analytical assay for the analysis of e-liquid samples. As the “dilute–shoot” sample preparation is carried out in one single step, it is expected that it will be less time-consuming, and, most importantly, it can minimize or avoid undesirable errors and deviations that are often observed during the SPE cleanup procedures.
Matrix Ion Suppression Optimization
Because of the lipophilic property of most of the analytes, a high percentage of organic phase, averagely above 50%, is required to elute them from the HPLC C18 column. Compared to others, DEP has a relatively high polarity and low retention on the column with a retention time of 2.6 min observed in this method (Figure 2). One major concern about matrix effects is relevant to the PG and VG residuals resulting from sample preparation. We observed that this can be minimized by directing the LC flow during the first 2.2 min to a waste container. This is mainly because of the low retention of PG and VG on the C18 column because of their relative high polarity. This practice can also be friendly to the mass spectrometer as PG and VG are viscous liquids with boiling points of 188 and 290 °C, respectively. Without directing the LC flow, the PG and VG residuals can potentially deposit and build up onto the interior surface of the mass spectrometer detector over time, resulting in lower MS sensitivity. We also directed the LC flow in the last 3 min to a waste container, aiming to reduce the buildup of other potential interferences in the MS analysis. The calculated data show no significant matrix ion suppression occurred in the assay (Table 1), which may reflect the effectiveness of the LC gradient program used for minimizing the system contamination.
Figure 2.

(a) Representative chromatograms for a standard sample with a spiked concentration of 16 ng/mL for TEP, DMP, TCEP, TPrP, and DEP. (b) Representative chromatograms of a standard sample with a spiked concentration of 16 ng/mL for TCPP, DCHP, TDCPP, TEHP, and TnBP. (c) Representative chromatograms of a standard sample with a spiked concentration of 16 ng/mL for TPhP, TDBPP, TBOEP, BBP, and DBP. (d) Representative chromatograms of a standard sample with a spiked concentration of 16 ng/mL for TCP, RDP, DAP, EHDPP, and DHP. (e) Representative chromatograms of a standard sample with a spiked concentration of 16 ng/mL for TBBPA.
Table 1. Matrix Ion Suppression Evaluation (%)a.
| 100% PG | PG/VG (v/v: 50–50) | 100% VG | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | A | B | C | D | A | B | C | D | |
| TEP | 88 | 90 | 96 | 83 | 98 | 101 | 102 | 102 | 98 | 97 | 99 | 115 |
| DMP | 93 | 90 | 88 | 85 | 106 | 102 | 100 | 101 | 93 | 96 | 97 | 109 |
| TCEP | 102 | 96 | 83 | 109 | 97 | 102 | 104 | 109 | 109 | 85 | 109 | 106 |
| TPrP | 89 | 85 | 100 | 104 | 102 | 103 | 96 | 102 | 100 | 91 | 101 | 109 |
| DEP | 86 | 101 | 85 | 97 | 99 | 98 | 108 | 102 | 110 | 91 | 85 | 109 |
| TCPP | 92 | 105 | 93 | 108 | 104 | 109 | 103 | 103 | 91 | 94 | 102 | 90 |
| DCHP | 103 | 88 | 97 | 102 | 115 | 111 | 122 | 119 | 117 | 105 | 107 | 93 |
| TDCPP | 110 | 100 | 95 | 108 | 103 | 94 | 95 | 114 | 100 | 97 | 96 | 90 |
| TEHP | 90 | 110 | 96 | 96 | 105 | 107 | 111 | 114 | 94 | 109 | 100 | 86 |
| TnBP | 116 | 99 | 86 | 84 | 100 | 107 | 101 | 105 | 95 | 105 | 86 | 101 |
| TPhP | 86 | 95 | 105 | 87 | 110 | 109 | 99 | 102 | 104 | 107 | 107 | 87 |
| TDBPP | 118 | 81 | 112 | 99 | 104 | 103 | 106 | 98 | 105 | 94 | 101 | 109 |
| TBOEP | 102 | 82 | 88 | 110 | 106 | 104 | 104 | 120 | 106 | 107 | 86 | 92 |
| BBP | 115 | 83 | 82 | 93 | 105 | 102 | 103 | 101 | 92 | 102 | 111 | 92 |
| DBP | 108 | 89 | 107 | 88 | 113 | 117 | 117 | 109 | 101 | 92 | 97 | 88 |
| TCP | 114 | 96 | 97 | 109 | 102 | 103 | 102 | 106 | 114 | 85 | 112 | 108 |
| RDP | 110 | 94 | 85 | 90 | 99 | 109 | 105 | 110 | 96 | 88 | 92 | 90 |
| DAP | 112 | 110 | 103 | 107 | 105 | 107 | 100 | 106 | 112 | 110 | 89 | 86 |
| EHDPP | 94 | 95 | 88 | 111 | 109 | 105 | 84 | 88 | 99 | 105 | 110 | 94 |
| DHP | 117 | 108 | 87 | 91 | 104 | 103 | 103 | 108 | 110 | 98 | 108 | 89 |
| TBBPA | 114 | 100 | 93 | 92 | 94 | 110 | 81 | 106 | 92 | 85 | 93 | 96 |
Concentrations for the pools A, B, C, and D are 16, 40, 100, and 250 ng/mL, respectively.
Background Residuals and Potential Carryover
We evaluated the carryover by measuring the residual levels of each compound in blank solvent samples analyzed following the samples with concentrations ranging from 250 to 500 ng/mL for all analytes. We compared the average peak area of each analyte in the first solvent blank sample with the average peak area for the corresponding analyte in the treated samples.21−24 To differentiate the potential carryover from the system background residual levels, multiple samples containing high-purity methanol and water were injected and analyzed prior to analyzing the samples with high concentration levels. In this assay, when the samples were diluted by 10 times, we did not identify the background residual levels for TEP, TPrP, tris(2-chloroethyl)phosphate (TCEP), dihexyl phthalate (DHP), tris(2,3-dibromopropyl)phosphate (TDBPP), DMP, diamyl phthalate (DAP), resorcinol bis(diphenyl phosphate) (RDP), TDBPP, and TBBPA. However, background residuals were identified for the rest of the compounds (Figure 3). On average, OPFRs had higher background residual levels compared to phthalate plasticizers. We observed that the background levels were generally decreased by increasing the dilution factor.
Figure 3.
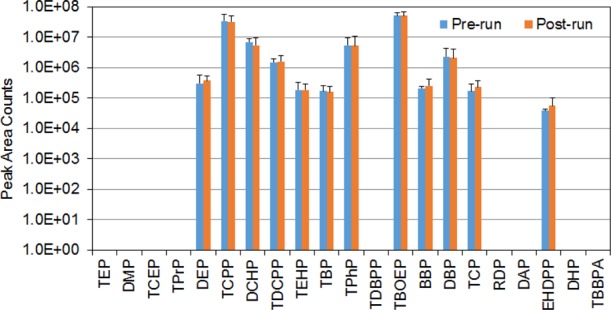
Examination of the residual levels in blank samples analyzed prior to and following the samples with a concentration level of 250 ng/mL.
To avoid carryover contamination, before each injection, the injection port was rinsed with 300 μL of the mixed solvents consisting of 60% acetonitrile, 45% isopropanol, and 5% water, and the measuring LC line was washed with 600 μL of 95% of acetonitrile in water (volume-based). Comparison analyses were then performed to evaluate the residual levels of each compound in the blank samples analyzed prior to and following the samples with high concentration levels (250–500 ng/mL). No significant difference was observed in the MS responses between the two sets of blank samples, suggesting that system background residuals were the main contamination sources. We observed that preparing mobile buffer solutions freshly on an as-needed basis could minimize the background levels.
The method was further validated by determining the LOD, limit of quantitation (LOQ), dynamic linear ranges, accuracy, and precision. The calculated LOD and LOQ concentrations are presented in Table 2 and the overall accuracy and imprecision are given in Table 3.
Table 2. LOD, LOQ, Dynamic Linearity Calibration Ranges and Retention Time.
| compounds | LODs (ng/mL) | LOQs (ng/mL) | dynamic linearity calibration range (ng/mL) | RT (min) |
|---|---|---|---|---|
| TEP | 0.050 | 0.167 | 0.010–500 | 2.46 |
| DMP | 0.150 | 0.500 | 0.100–500 | 3.36 |
| TCEP | 0.150 | 0.500 | 0.100–500 | 3.57 |
| TPrP | 0.025 | 0.083 | 0.010–250 | 4.19 |
| DEP | 0.150 | 0.500 | 0.100–500 | 4.38 |
| TCPP | 0.150 | 0.500 | 0.100–250 | 4.57 |
| DCHP | 10.0 | 33.3 | 2.50–500 | 4.60 |
| TDCPP | 0.150 | 0.500 | 0.100–500 | 5.28 |
| TEHP | 2.50 | 8.33 | 1.00–500 | 5.29 |
| TnBP | 0.026 | 0.087 | 0.010–250 | 5.36 |
| TPhP | 0.030 | 0.100 | 0.010–500 | 5.45 |
| TDBPP | 0.210 | 0.700 | 0.150–500 | 5.49 |
| TBOEP | 0.150 | 0.500 | 0.100–250 | 5.66 |
| BBP | 0.020 | 0.066 | 0.010–500 | 5.75 |
| DBP | 0.020 | 0.066 | 0.010–500 | 5.82 |
| TCP | 0.050 | 0.167 | 0.010–500 | 6.09 |
| RDP | 0.150 | 0.500 | 0.100–500 | 6.12 |
| DAP | 0.050 | 0.167 | 0.010–250 | 6.33 |
| EHDPP | 1.00 | 3.33 | 0.400–500 | 6.35 |
| DHP | 0.150 | 0.500 | 0.100–250 | 6.80 |
| TBBPA | 1.00 | 3.33 | 0.400–500 | 5.63 |
Table 3. Method Accuracy and Precisiona.
| pool A |
pool B |
pool C |
pool D |
||||||
|---|---|---|---|---|---|---|---|---|---|
| interday | intraday | interday | intraday | interday | intraday | interday | intraday | ||
| TPrP | measured | 16.5 | 17.1 | 41.4 | 42.3 | 102 | 110 | 246 | 244 |
| accuracy | 103 | 107 | 103 | 106 | 102 | 110 | 98.3 | 98 | |
| RSD % | 2.4 | 5 | 2.8 | 4 | 1.6 | 7.1 | 1.2 | 1.8 | |
| TPhP | measured | 18.4 | 17.5 | 40.4 | 43.5 | 111 | 116 | 264 | 262 |
| accuracy | 115 | 109 | 101 | 109 | 111 | 116 | 106 | 105 | |
| RSD % | 10.5 | 6.5 | 4.3 | 6.2 | 7.8 | 11 | 3.9 | 3.5 | |
| TnBP | measured | 15.3 | 16.9 | 39.9 | 41.6 | 102 | 103 | 246 | 238 |
| accuracy | 96 | 106 | 100 | 104 | 102 | 103 | 99 | 95 | |
| RSD % | 2.4 | 3.9 | 2.1 | 2.8 | 1.3 | 2.3 | 1 | 3.4 | |
| TEP | measured | 14.7 | 16.6 | 40.5 | 40.2 | 99 | 97 | 248 | 244 |
| accuracy | 92 | 104 | 101 | 101 | 99 | 97 | 99 | 98 | |
| RSD % | 3.1 | 2.7 | 0.6 | 0.3 | 0.7 | 2.1 | 0.7 | 1.6 | |
| TEHP | measured | 17.3 | 18.2 | 36.1 | 42.4 | 86 | 103 | 271 | 224 |
| accuracy | 108 | 114 | 90 | 106 | 86 | 103 | 108 | 90 | |
| RSD % | 8.7 | 9.8 | 11 | 4.2 | 9.7 | 6 | 5.8 | 7.4 | |
| TDCPP | measured | 18.1 | 13.7 | 44.5 | 46 | 88 | 86 | 239 | 240 |
| accuracy | 113 | 86 | 111 | 115 | 88 | 86 | 96 | 96 | |
| RSD % | 9.3 | 10 | 9.8 | 11 | 8.2 | 9.9 | 3.1 | 2.9 | |
| TDBPP | measured | 13.8 | 14.8 | 38 | 34.2 | 93 | 88 | 246 | 256 |
| accuracy | 87 | 93 | 95 | 86 | 93 | 88 | 98 | 102 | |
| RSD % | 9.5 | 5.2 | 6.8 | 10.2 | 4.8 | 8.6 | 1.1 | 1.6 | |
| TCPP | measured | 16.1 | 16.4 | 38.8 | 42.4 | 103 | 103 | 246 | 242 |
| accuracy | 100 | 102 | 97 | 106 | 103 | 103 | 99 | 97 | |
| RSD % | 0.2 | 1.6 | 3.8 | 4.2 | 2.2 | 1.8 | 1 | 2.4 | |
| TCP | measured | 16.6 | 16.5 | 42.8 | 43.5 | 103 | 100 | 254 | 241 |
| accuracy | 104 | 103 | 107 | 109 | 103 | 100 | 101 | 97 | |
| RSD % | 2.8 | 2.3 | 4.6 | 6.1 | 1.8 | 0.1 | 1 | 2.5 | |
| TCEP | measured | 15.7 | 16.6 | 40.6 | 41.5 | 102 | 97 | 250 | 232 |
| accuracy | 98.3 | 104 | 102 | 104 | 102 | 97 | 100 | 93 | |
| RSD % | 1.2 | 2.5 | 1.8 | 2.7 | 1.3 | 1.9 | 0.1 | 5.2 | |
| TBOEP | measured | 18.4 | 17.6 | 32.3 | 37.8 | 100 | 97 | 218 | 230 |
| accuracy | 115 | 110 | 80.7 | 94.4 | 100 | 97 | 87 | 92 | |
| RSD % | 10.4 | 6.9 | 9.1 | 4 | 0.3 | 2.2 | 9.2 | 5.8 | |
| RDP | measured | 13.8 | 14.6 | 32.4 | 34.7 | 90 | 88 | 246 | 225 |
| accuracy | 86 | 91 | 81 | 87 | 90 | 88 | 98 | 90 | |
| RSD % | 9.8 | 6.2 | 9.6 | 9.3 | 7.4 | 8.6 | 1.2 | 7.1 | |
| EHDPP | measured | 17.8 | 18.1 | 43.9 | 44.7 | 88 | 108 | 243 | 280 |
| accuracy | 111 | 113 | 110 | 112 | 88 | 108 | 97 | 112 | |
| RSD % | 12.2 | 9.4 | 6.3 | 8.3 | 8.5 | 5.3 | 2 | 8.5 | |
| DAP | measured | 17.2 | 17.2 | 40.8 | 42.6 | 105 | 108 | 241 | 231 |
| accuracy | 108 | 108 | 102 | 107 | 105 | 108 | 97 | 92 | |
| RSD % | 5.4 | 5.4 | 3.1 | 4.6 | 3.4 | 5.4 | 2.5 | 5.5 | |
| DMP | measured | 15.9 | 15.3 | 39.2 | 38.9 | 97 | 97 | 242 | 241 |
| accuracy | 99.5 | 95.7 | 97.9 | 97.1 | 97 | 97 | 97 | 96 | |
| RSD % | 0.3 | 3 | 1.5 | 2 | 1.8 | 1.8 | 2.3 | 2.6 | |
| DHP | measured | 17.2 | 17.5 | 40.3 | 40.4 | 102 | 102 | 246 | 229 |
| accuracy | 107 | 109 | 101 | 101 | 102 | 102 | 98 | 92 | |
| RSD % | 5.1 | 6.4 | 0.6 | 0.8 | 1.4 | 1.3 | 1.2 | 5.9 | |
| DEP | measured | 18.9 | 19.3 | 42.1 | 41.7 | 109 | 99 | 246 | 248 |
| accuracy | 118 | 121 | 105 | 104 | 109 | 99 | 98 | 99 | |
| RSD % | 12.6 | 14.7 | 2.8 | 3 | 6.2 | 0.5 | 1.2 | 0.5 | |
| DBP | measured | 17.7 | 14.1 | 31.9 | 44.7 | 118 | 92 | 261 | 241 |
| accuracy | 111 | 88.4 | 79.8 | 112 | 118 | 92 | 104 | 97 | |
| RSD % | 7.6 | 8.2 | 13.3 | 8.4 | 12.9 | 5.8 | 3.1 | 2.4 | |
| BBP | measured | 14.6 | 14.7 | 35 | 35.4 | 97 | 95 | 260 | 241 |
| accuracy | 90.9 | 91.9 | 87.4 | 88.4 | 97 | 95 | 104 | 96 | |
| RSD % | 6.4 | 5.7 | 7 | 8.2 | 1.9 | 3.3 | 2.8 | 2.5 | |
| TBBPA | measured | 14.3 | 15.4 | 36.4 | 40.5 | 90 | 110 | 238 | 227 |
| accuracy | 89.5 | 96.0 | 90.9 | 101 | 90 | 110 | 95 | 91 | |
| RSD % | 7.4 | 2.8 | 4.8 | 1 | 7 | 7.1 | 3.3 | 6.5 | |
Concentrations for the pools A, B, C, and D are 16, 40, 100, and 250 ng/mL, respectively.
Application to e-Liquid Samples
We measured the concentrations of ECCs for 20 refill e-liquid samples purchased from online commercial sources and presented the blank-corrected concentration (ng/mL) for each target analyte in Table 4. Among all analytes, DMP was detected in 80% of the samples, followed by DBP, DEP, and BBP. TEP, 2-ethylhexyl diphenyl phosphate (EHDPP), tris(1,3-dichloro-2-propyl)phosphate (TDCPP), dicyclohexyl phthalate (DCHP), tris(2-butoxyethyl)phosphate (TBOEP), tris(2-ethylhexyl)phosphate (TEHP), diphenyl phosphate, and TCEP were detected in 10–25% of the samples. TPrP, TBP, triphenyl phosphate (TPhP), and tris(1-chloro-2-propyl)phosphate (TCPP) were identified in 5% of the samples. TDBPP, RDP, DEHP, and TBBPA were not identified in the 20 e-liquid samples. We also observed that at least one of the analytes can be detected among these e-liquid samples and that the concentrations for certain analytes can be extremely high in some e-liquid samples. A highest concentration of 1776 ng/mL was observed for DBP (average: 176 ng/mL), followed by DCHP (451 ng/mL), DMP (153 ng/mL), and TEHP (194 ng/mL). Overall, the application results suggest that this method can be useful to quantify the emerging chemicals of health concern in e-liquid samples in future studies, thus to address the information gap pertinent to the safety of e-cigarette products.
Table 4. Blank-Corrected Concentrations of Phthalates and OPFRs in 20 e-Cigarette Refill Liquid Samples (ng/mL).
| refill e-liquid sample | EHDPP | TCP | TEP | TBOEP | TCEP | TDCPP | TEHP | DMP | DEP | BBP | DBP | DCHP | DAP |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID_01 | 0.142 | 3.04 | 3.71 | 4.18 | 451 | ||||||||
| ID_02 | 0.224 | 0.409 | 2.68 | 5.27 | |||||||||
| ID_03 | 0.249 | 2.69 | 8.84 | ||||||||||
| ID_04 | 0.056 | 4.89 | 0.144 | 0.253 | 4.69 | ||||||||
| ID_05 | 1.45 | 438 | 0.260 | ||||||||||
| ID_06 | 0.146 | ||||||||||||
| ID_07 | 2.43 | 0.314 | 0.256 | 0.498 | 2.61 | ||||||||
| ID_08 | 1.16 | 0.123 | |||||||||||
| ID_09 | 26.9 | 45.1 | |||||||||||
| ID_10 | 0.104 | ||||||||||||
| ID_11 | 0.15 | 0.194 | 1.47 | ||||||||||
| ID_12 | 6.07 | 0.294 | 1.41 | ||||||||||
| ID_13 | 0.076 | 117 | 0.152 | ||||||||||
| ID_14 | 0.378 | 0.465 | 8.82 | 0.188 | |||||||||
| ID_15 | 18.2 | 4.23 | |||||||||||
| ID_16 | 0.046 | 42.5 | 34.9 | 74.8 | 2.57 | 26.8 | |||||||
| ID_17 | 11.7 | 9.68 | 17.4 | 75.1 | 232 | ||||||||
| ID_18 | 5.82 | 7.71 | 7.78 | 0.256 | 194 | 152 | 82.4 | 51.7 | 1776 | 2.48 | 0.090 | ||
| ID_19 | 0.079 | 4.20 | 0.966 | 95.3 | |||||||||
| ID_20 | 4.29 | 12.07 | 1.68 | 1.02 | 7.01 |
Materials and Methods
Reagents and Standards
LC–MS-grade solvents (e.g., acetonitrile, water, and methanol), ammonium formate, formic acid, and USP-grade PG and VG were bought from Fisher Scientific (Fairlawn, NJ, USA). Native standards, including DBP, DEP, DEHP, BBP, DHP, DCHP, DAP, DMP, TBOEP, TCEP, TCPP, tricresyl phosphate (TCP), TPhP, TDCPP, tributyl phosphate (TnBP), TEHP, TDBPP, TEP, EHDPP, RDP, bis(1,3-dichloro-2-propyl)phosphate, TBBPA, and isotope-labeled standards, including DHP-d4, DEP-d4, DBP-d4, DMP-d4, and DCHP-d4, were purchased from AccuStandard (New Haven, CT, USA). Isotopically labeled compounds, including TPhP-d15, TCPP-d18, TBOEP-d27, TDCPP-d15, TEP-d15, TDBPP-d15, TBP-d27, TCEP-d12, and TPrP-d12, were purchased from Toronto Research Chemicals (North York, ON, Canada). All chemicals were directly used for preparing standard solutions, and no further purification was performed. TruView LCMS Certified Glass inject vials and SPE (C18, 100 mg) columns were bought from Waters (Milford, MA, USA) and Biotage (Charlotte, NC, USA), respectively.
Standard Preparation
An internal standard (ISTD) spiking solution, including DEP-d4, DBP-d4, DMP-d4, DHP-d4, TBP-d27, TEP-d15, TCEP-d12, TPrP-d21, TDCPP-d15, TDBPP-d15, TPhP-d15, TCPP-d18, TDCPP-d15, and TBOEP-d27, was prepared by mixing isotope-labeled stock solutions and diluting them with the mixed solvents consisting of 60% methanol and 40% water in volume, which yielded a spiking solution containing concentrations of 25 ng/mL for each isotope-labeled compound. Primary stock solutions and 12 working solutions, containing all target analytes with their concentrations ranging from 0.01 to 5000 ng/mL, were prepared by diluting the native standards with the mixed solvents consisting of 60% of methanol and 40% of water in volume. Subsequently, 12 calibrator solutions, with the concentrations of each native analyte ranging from 0.001 to 500 ng/mL, were prepared by adding 50 μL of each working solution and 50 μL of the ISTD spiking solution to 400 μL of blank PG and VG (v/v: 50:50) during sample preparation. Unknown e-liquid samples with targeted concentrations > dynamic linearity ranges were diluted with appropriate dilution factors, and reprepared and analyzed, to avoid mass spectrometer detector saturation. We used similar procedures for preparing high (250 ng/mL), mid (50 ng/mL), and low (16 ng/mL) quality control (QC) samples. Calibration, QC, and ISTD spiking solutions were kept in amber glass vials and stored at −20 °C.
Sample Preparation
For SPE cleanup assay, first, 50 μL of the ISTD solution was added to each glass tube precleaned with methanol. Then, 100 μL of each sample (e.g., e-liquid samples, QC solutions, calibration standards, and batch blanks) and 850 μL of methanol and water (v/v: 15:85) were transferred to the same tube. After gently mixing, the samples were transferred onto the C18 cartridges, which were precleaned and equilibrated with LC–MS-grade methanol (1.0 mL), followed by acetonitrile (1.0 mL) and water (1.0 mL), in order. After 10 min, the mixtures were pushed through the SPE column (approximately 1.0 psi positive pressure). Subsequently, the SPE column was washed with 1.0 mL of water and 1.0 mL of mixed solvents consisting of 15% methanol and 85% water (volume-based). High-purity nitrogen (25 psi) was then applied to dry the SPE column. After 15 min, 1.0 mL of LC–MS-grade methanol was loaded onto the SPE column, and the eluted samples were collected in 1.5 mL LC injection vials (“total recovery”, Waters, Milford, MA, USA) and evaporated to dryness under high-purity nitrogen at room temperature. The evaporated residuals were reconstituted with 100 μL of 1:1 (v/v) methanol/water mixture. The LC injection volume for each sample in this study was 10 μL.
For the direct “dilute-and-shoot” assay, 50 μL of the ISTD spiking solution was first added into each LC injection vial. Then, 50 μL of each sample (e.g., e-liquid samples, QC samples, calibration standards, and batch control blanks) and 400 μL of the mixed solvents consisting of 60% methanol in water (volume-based) were transferred to the same vial. After gently vortexing, 10 μL of each sample was injected into the LC column for analysis.
Instrumentation
The HPLC system used in this study consisted of a CTO-20AC column oven, a DGU-20A5R degasser, two LC-20ADXR pumps, and a SIL-30AC autosampler (Shimadzu Corp). Chromatographic separation was performed using a Kinetex C18 column (particle size, 2.6 μm; length, 10 cm; diameter, 2.1 mm, Phenomenex) at 40 °C. Two tandem in-line filters (2 and 0.5 μm) were connected prior to the LC column.
The mobile phase “A” consisted of 5.0 mM of ammonium formate and 0.1% formic acid prepared in LC–MS-grade water, and the organic phase “B” was 100% acetonitrile. An LC flow rate of 0.40 mL/min was used during the entire analysis. Detailed gradient elution conditions are given in Table 5. After sample injection, the LC flow occurring only during the period between 2.2 and 7.0 min was directed to the mass spectrometer by the switching valve, whereas the flow during the first 2.2 min and the last 3 min was directed to a waste container.
Table 5. HPLC Gradient Elution.
| time | module | event | percentage of buffer B (%) |
|---|---|---|---|
| 0.01 | system controller | start | |
| 1.00 | pumps | % B | 20 |
| 7.00 | pumps | % B | 100 |
| 8.50 | pumps | % B | 100 |
| 8.51 | pumps | % B | 20 |
| 10.0 | system controller | stop |
A Sciex triple quadrupole 6500+ mass spectrometer with a TurboIonSpray source (Foster City, CA, USA) was used for method development and sample analysis. Electrospray positive mode (ESI+) was used to acquire scheduled multipole reaction monitoring (MRM) precursor/product transition data for OPFRs and phthalates, and negative mode (ESI–) was used for TBBPA. The MS source parameter settings are provided in Table 6. Compound-dependent MS parameter settings were optimized manually. MRM transitions for native analytes and ISTD are given in Table 7. Figure 2a–e shows the representative chromatograms of all analytes included in the current method.
Table 6. MS Source Settings.
| parameter | optimized value |
|---|---|
| ion source | turbo spray |
| polarity | positive/negative |
| SC type | scheduled MRM |
| MRM detection window | 30 s |
| target scan time | 0.041 s |
| curtain gas (CUR) | 35 |
| collision gas (CAD) | 7 |
| ion spray voltage (IS) | 5500/–4500 V |
| temperature (TEM) | 400 °C |
| ion source gas 1 (GS1) | 60 |
| ion source gas 2 (GS2) | 70 |
Table 7. MS Settings of Native and Isotope-Labeled Compoundsa.
| MS settings, V |
||||||
|---|---|---|---|---|---|---|
| compound | ESI mode | precursor/(product ions) | DP | CE | EP | CXP |
| TPrP | + | 225.1/(99, 183.2, 141) | 50 | (27, 13, 15) | 10 | 11 |
| TnBP | + | 267.2/(99, 155.1, 211.3) | 50 | (25, 16, 12) | 10 | 10 |
| TPhP | + | 326.8/(152.1, 215, 153) | 65 | (47, 36, 35) | 10 | 11 |
| EHDPP | + | 363.2/(251.1, 363, 159.1) | 60 | (30, 10, 26) | 10 | 11 |
| TCP | + | 368.9/(166.2, 243, 165) | 50 | (38, 39, 45) | 10 | 11 |
| TEP | + | 183/(99.1, 127) | 60 | (25, 15) | 10 | 11 |
| TCEP | + | 285.2/(223, 161.2, 99) | 40 | (18, 23, 28) | 8 | 11 |
| TCPP | + | 327/(99, 329/99) | 35 | (45, 45) | 10 | 10 |
| TBOEP | + | 399.1/(299.1, 199.1) | 50 | (19, 22) | 10 | 10 |
| TDCPP | + | 431/(99, 209) | 45 | (32, 24) | 6 | 10 |
| TEHP | + | 435.3/(99.1, 211.1) | 40 | (25, 12) | 10 | 11 |
| TDBPP | + | 698.6/(99, 698.5) | 70 | (35, 10) | 10 | 11 |
| RDP | + | 575/(481, 419) | 70 | (50, 49) | 10 | 11 |
| DMP | + | 195.1/(163.1, 133.1) | 60 | (15, 32) | 10 | 11 |
| DEP | + | 223.1/(149.1, 177) | 50 | (23, 18) | 10 | 11 |
| BBP | + | 313.3/(205.1, 149) | 60 | (11, 18) | 10 | 11 |
| DBP | + | 279.1/(205, 149.1) | 80 | (11, 20) | 10 | 11 |
| DHP | + | 335.1/(149.1, 335.2) | 60 | (25, 10) | 10 | 11 |
| DCHP | + | 331.1/(99, 331.1) | 60 | (55, 10) | 10 | 10 |
| DAP | + | 307.2/(149.1, 219) | 50 | (27, 22) | 10 | 10 |
| TBBPA | – | 542.5/(417.8, 445.8, 419.6) | –80 | (−55, −44, −53) | 10 | 10 |
| TBP-d27 | + | 294.2/102 | 50 | 25 | 10 | 10 |
| TEP-d15 | + | 198/102 | 60 | 25 | 10 | 10 |
| TCEP-d12 | + | 297/232 | 40 | 18 | 8 | 11 |
| TPrP-d21 | + | 246.2/(102, 150.1) | 50 | (27, 15) | 10 | 11 |
| TDCPP-d15 | + | 446/(102, 216) | 45 | (32, 24) | 6 | 10 |
| TDBPP-d15 | + | 713.7/102 | 70 | 35 | 10 | 11 |
| TPhP-d15 | + | 342.1/160.2 | 65 | 47 | 10 | 11 |
| TCPP-d18 | + | 347/102 | 35 | 45 | 10 | 11 |
| TDCPP-d15 | + | 446/102 | 45 | 32 | 10 | 11 |
| TBOEP-d27 | + | 426.3/208 | 50 | 22 | 10 | 10 |
| DEP-d4 | + | 227.2/(153.1, 181) | 50 | (25, 18) | 10 | 10 |
| DBP-d4 | + | 283.2/(153.2, 209.2) | 60 | (22, 11) | 10 | 10 |
| DMP-d4 | + | 199.1/(167.1, 137.1) | 60 | (15, 32, 55) | 10 | 10 |
| DHP-d4 | + | 339.1/153.1 | 60 | 25 | 10 | 10 |
Abbreviations: ESI—electrospray ionization; DP—declustering potential; CE—collision energy; CXP—collision cell exit potential; EP—entrance potential.
Data acquisition was performed using the Analyst software (version 1.7.0), and the subsequent chromatogram integration and concentration quantitation were performed using MultiQuant (version 3.0.3). Peak area ratios of analytes to the corresponding ISTD for each batch with a 1/x weighting factor were used to construct linear least-squares regression calibration curves.
Matrix Ion Suppression Evaluation
To evaluate the potential matrix ion suppression, we first prepared a set of four spiking standard solutions with concentrations of 160, 400, 1000, and 2500 ng/mL, and then we prepared four sample pools (A–D) by adding 50 μL of each spiking solution to 450 μL of blank PG or/and VG solvents, yielding concentrations of 16, 40, 100, and 250 ng/mL for the four pools A, B, C, and D, respectively. We prepared three sets of samples in PG/VG solution with their volume ratios of 1:0, 1:1, and 0:1, respectively. The fourth set of sample was prepared in mixed solvents consisting of 60% methanol and 40% water (volume-based). Three replicated analyses for each pool were performed. Following the same sample preparation procedures, matrix ion suppression was calculated by comparing the average peak area of each analyte in the samples prepared in PG/VG (first to third sets) with the average peak area of the analyte prepared in the mixed solvents: methanol/water (v/v: 60/40) (fourth set).
LODs and LOQs
We calculated the method LODs and the LOQs for all analytes as 3 times and 10 times the standard deviation (SD0) of the samples with zero analytic concentration,22,25 respectively. To determine SD0, we prepared and analyzed 10 pools (0.010, 0.026, 0.066, 0.164, 0.40, 1.0, 2.56, 6.40, 16, and 40 ng/mL) with low levels of targeted native analytes. For the analytes with detectable background residual levels, we used isotope-labeled standards to prepare the pools. We plotted the SD of each pool (Y axis) against the concentration (X axis) and defined the Y-intercept as SD0.
Accuracy and Precision
To assess the accuracy and precision of this assay, we spiked 50 μL of each working standard solution (160, 400, 1000, and 2500 ng/mL), which were prepared and used for matrix ion suppression evaluation, into 450 μL of blank PG and VG (v/v: 50:50), yielding a set of four pools with concentrations of 16, 40, 100, and 250 ng/mL for each analyte, respectively. We evaluated the intra- and interday accuracies, which were calculated as a percent of the target concentration, and imprecision, which was calculated as the relative SD (% RSD), for each analyte with the four different pools, following the “dilute-and-shoot” procedures described in the “Sample Preparation” section.
QC Measures
To ensure the reliability of the analytical data, several QC measures were applied. All samples, including unknown e-liquid samples, calibration standards, QCs, and laboratory control blanks, were prepared following the same sample preparation and analysis procedures. Calibration curves were constructed using 12 calibration standard solutions prepared in PG/VG (v/v: 50:50) to account for the potential matrix effects. A laboratory control blank for every 10 samples was prepared and analyzed in each analytical batch. All reportable results are blank-corrected concentrations. Calibration curves were regularly evaluated using certified standards purchased from a second commercial source or lot. Instruments were also regularly maintained to ensure high sensitivity.
Conclusions
In this study, we first evaluated the preparation efficiency using the SPE method and compared its performance with a simple “dilute-and-shoot” approach for preparing e-liquid samples. As the SPE method could cause increased background residual levels, the “dilute-and-shoot” method was chosen for sample preparation. We then optimized and validated HPLC–MS/MS assay to concurrently measure OPFRs, phthalate plasticizers, and TBBPA in the prepared e-liquid samples. The validation results indicated that this method was sensitive (LOD: 0.02–10 ng/mL), accurate (average inter-/intraday bias, <15%), precise (average inter-/intraday imprecision, <15%), and efficient (10 min runtime). To our knowledge, this is the first multianalysis method for measuring ECCs in e-liquid samples.
Acknowledgments
Research reported in this publication was supported by FDA Center for Tobacco Products and National Institute of Environmental Health Sciences (grant #: R21ES030028, PI: Wei). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the Food and Drug Administration.
The authors declare no competing financial interest.
References
- Wei B.; Goniewicz M. L.; O’Connor R. J.; Travers M. J.; Hyland A. J. Urinary Metabolite Levels of Flame Retardants in Electronic Cigarette Users: A Study Using the Data from NHANES 2013–2014. Int. J. Environ. Res. Public Health 2018, 15, 201. 10.3390/ijerph15020201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornehag C.-G.; Sundell J.; Weschler C. J.; Sigsgaard T.; Lundgren B.; Hasselgren M.; Hägerhed-Engman L. The Association between Asthma and Allergic Symptoms in Children and Phthalates in House Dust: A Nested Case-Control Study. Environ. Health Persp. 2004, 112, 1393. 10.1289/ehp.7187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya S. V.; Kulkarni H. Association of Urinary Bisphenol A Concentration with Allergic Asthma: Results from the National Health and Nutrition Examination Survey 2005-2006. J. Asthma 2012, 49, 800–806. 10.3109/02770903.2012.721041. [DOI] [PubMed] [Google Scholar]
- Araki A.; Saito I.; Kanazawa A.; Morimoto K.; Nakayama K.; Shibata E.; Tanaka M.; Takigawa T.; Yoshimura T.; Chikara H.; Saijo Y.; Kishi R. Phosphorus flame retardants in indoor dust and their relation to asthma and allergies of inhabitants. Indoor Air 2014, 24, 3–15. 10.1111/ina.12054. [DOI] [PubMed] [Google Scholar]
- Meeker J. D.; Stapleton H. M. House dust concentrations of organophosphate flame retardants in relation to hormone levels and semen quality parameters. Environ. Health Persp. 2010, 118, 318. 10.1289/ehp.0901332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippat C.; Mortamais M.; Chevrier C.; Petit C.; Calafat A. M.; Ye X.; Silva M. J.; Brambilla C.; Pin I.; Charles M.-A.; Cordier S.; Slama R. Exposure to phthalates and phenols during pregnancy and offspring size at birth. Environ. Health Persp. 2012, 120, 464. 10.1289/ehp.1103634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loganathan S. N.; Kannan K. Occurrence of bisphenol A in indoor dust from two locations in the eastern United States and implications for human exposures. Archiv. Environ. Contam. Toxicol. 2011, 61, 68–73. 10.1007/s00244-010-9634-y. [DOI] [PubMed] [Google Scholar]
- US-DHHS Toxicology and carcinogenesis: a studies of tris(2-chloroethyl)phosphate. https://ntp.niehs.nih.gov/ntp/htdocs/lt_rpts/tr391.pdf (accessed on Jan 11, 2018).
- OEHHA . Current Proposition 65 List; California Office of Environmental Health Hazard Assessment, 2016.
- Ehrlich S.; Williams P. L.; Missmer S. A.; Flaws J. A.; Ye X.; Calafat A. M.; Petrozza J. C.; Wright D.; Hauser R. Urinary bisphenol A concentrations and early reproductive health outcomes among women undergoing IVF. Hum. Reprod. 2012, 27, 3583–3592. 10.1093/humrep/des328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO, World Health Organization . Environmental Health Criteria 209, Flame Retardants: 420 Tris(chloropropyl)phosphate and Tris(2-chloroethyl)phosphate; World Health Organization: 421 Geneva, Switzerland, 1998.
- US-FDA(e) Harmful and Potentially Harmful Constituents in Tobacco Products and Tobacco Smoke: Established List. https://www.fda.gov/TobaccoProducts/Labeling/RulesRegulationsGuidance/ucm297786.htm (accessed on Feb 1, 2018).
- Hess C. A.; Olmedo P.; Navas-Acien A.; Goessler W.; Cohen J. E.; Rule A. M. E-cigarettes as a source of toxic and potentially carcinogenic metals. Environ. Res. 2017, 152, 221–225. 10.1016/j.envres.2016.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal V.; Kim K.-H. Review on quantitation methods for hazardous pollutants released by e-cigarette (EC) smoking. TrAC-Trend Anal. Chem. 2016, 78, 120–133. 10.1016/j.trac.2016.02.015. [DOI] [Google Scholar]
- Kavvalakis M. P.; Stivaktakis P. D.; Tzatzarakis M. N.; Kouretas D.; Liesivuori J.; Alegakis A. K.; Vynias D.; Tsatsakis A. M. Multicomponent analysis of replacement liquids of electronic cigarettes using chromatographic techniques. J. Anal. Toxicol. 2015, 39, 262–269. 10.1093/jat/bkv002. [DOI] [PubMed] [Google Scholar]
- Hahn J.; Monakhova Y. B.; Hengen J.; Kohl-Himmelseher M.; Schüssler J.; Hahn H.; Kuballa T.; Lachenmeier D. W. Electronic cigarettes: overview of chemical composition and exposure estimation. Tob. Induc. Dis. 2014, 12, 23. 10.1186/s12971-014-0023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.-J.; Shin H.-S. Determination of tobacco-specific nitrosamines in replacement liquids of electronic cigarettes by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2013, 1291, 48–55. 10.1016/j.chroma.2013.03.035. [DOI] [PubMed] [Google Scholar]
- Oh J.-A.; Shin H.-S. Identification and Quantification of Several Contaminated Compounds in Replacement Liquids of Electronic Cigarettes by Gas Chromatography-Mass Spectrometry. J. Chromatogr. Sci. 2014, 53, 841–848. 10.1093/chromsci/bmu146. [DOI] [PubMed] [Google Scholar]
- Moldoveanu S. C.; Yerabolu R. Critical evaluation of several techniques for the analysis of phthalates and terephthalates: Application to liquids used in electronic cigarettes. J. Chromatogr. A 2018, 1540, 77–86. 10.1016/j.chroma.2018.02.001. [DOI] [PubMed] [Google Scholar]
- NIEHS . Endocrine Disruptors; National Institute of Environmental Health Sciences, National Institutes of Health, U.S. Department of Health and Human Services, 2010. [Google Scholar]
- Wei B.; Feng J.; Rehmani I. J.; Miller S.; McGuffey J. E.; Blount B. C.; Wang L. A high-throughput robotic sample preparation system and HPLC-MS/MS for measuring urinary anatabine, anabasine, nicotine and major nicotine metabolites. Clin. Chim. Acta 2014, 436, 290–297. 10.1016/j.cca.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei B.; Wang L.; Blount B. C. Analysis of cannabinoids and their metabolites in human urine. Anal. Chem. 2015, 87, 10183–10187. 10.1021/acs.analchem.5b02603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei B.; McGuffey J. E.; Blount B. C.; Wang L. Sensitive Quantification of Cannabinoids in Milk by Alkaline Saponification-Solid Phase Extraction Combined with Isotope Dilution UPLC-MS/MS. ACS Omega 2016, 1, 1307–1313. 10.1021/acsomega.6b00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffey J. E.; Wei B.; Bernert J. T.; Morrow J. C.; Xia B.; Wang L.; Blount B. C. Validation of a LC-MS/MS Method for Quantifying Urinary Nicotine, Six Nicotine Metabolites and the Minor Tobacco Alkaloids-Anatabine and Anabasine-in Smokers’ Urine. PLoS One 2014, 9, e101816 10.1371/journal.pone.0101816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J. K.Quality Assurance of Chemical Measurements; CRC Press, 1987. [Google Scholar]