

Abstract
Background
In murine heart failure models and in humans with diabetic‐related heart hypertrophy, inhibition of phosphodiesterase 5 (PDE5) by sildenafil improves cardiac outcomes. However, the mechanism by which sildenafil improves cardiac function is unclear. We have observed a relationship between PDE5 and β2 adrenergic receptor (β2AR), which is characterized here as a novel mechanistic axis by which sildenafil improves symptoms of diabetic cardiomyopathy.
Methods and Results
Wild‐type and β2AR knockout mice fed a high fat diet (HFD) were treated with sildenafil, and echocardiogram analysis was performed. Cardiomyocytes were isolated for excitation‐contraction (E‐C) coupling, fluorescence resonant energy transfer, and proximity ligation assays; while heart tissues were implemented for biochemical and histological analyses. PDE5 selectively associates with β2AR, but not β1 adrenergic receptor, and inhibition of PDE5 with sildenafil restores the impaired response to adrenergic stimulation in HFD mice and isolated ventriculomyocytes. Sildenafil enhances β adrenergic receptor (βAR)‐stimulated cGMP and cAMP signals in HFD myocytes. Consequently, inhibition of PDE5 leads to protein kinase G–, and to a lesser extent, calcium/calmodulin‐dependent kinase II–dependent improvements in adrenergically stimulated E‐C coupling. Deletion of β2AR abolishes sildenafil's effect. Although the PDE5‐β2AR association is not altered in HFD, phosphodiesterase 3 displays an increased association with the β2AR‐PDE5 complex in HFD myocytes.
Conclusions
This study elucidates mechanisms by which the β2AR‐PDE5 axis can be targeted for treating diabetic cardiomyopathy. Inhibition of PDE5 enhances β2AR stimulation of cGMP and cAMP signals, as well as protein kinase G–dependent E‐C coupling in HFD myocytes.
Keywords: cardiac myocyte, cell signaling, diabetic cardiomyopathy, EC coupling, echocardiography, pathophysiology, pharmacology
Subject Categories: Basic Science Research, Cell Signalling/Signal Transduction, Contractile function, Myocardial Biology
Clinical perspective
What Is New?
Phosphodiesterase 5 is in a protein complex with β2 adrenergic receptor in cardiomyocytes. Inhibition of phosphodiesterase 5 promotes cGMP in a β2 adrenergic receptor–dependent fashion, leading to improvements in contractility in mice fed a high fat diet by enhancing cGMP‐protein kinase G signaling, as well as phosphodiesterase 3‐dependent cAMP signaling.
What Are the Clinical Implications?
The β2 adrenergic receptor–phosphodiesterase 5 axis represents a promising therapeutic target for treating diabetic‐related heart hypertrophy.
Introduction
Patients with diabetes mellitus in the United States are 2 to 4 times more likely to develop heart diseases than patients without diabetes mellitus, with diabetes mellitus placing a patient in a high‐risk category for the development of heart failure (HF).1 Studies suggest that differences in the cause of diabetic‐related HF2, 3 and many of the medications patients with diabetes mellitus take to manage their condition2, 4, 5, 6 can both contribute to worse HF outcomes. For instance, several publications have shown that patients with diabetes mellitus and HF with preserved ejection fraction (HFpEF) are 70% to 80% more likely to be hospitalized than HFpEF patients without diabetes mellitus.7, 8, 9 This highlights the need for development of more effective treatments for diabetic‐related HF, with an emphasis on individualized care.
Studies have shown that the phosphodiesterase 5 (PDE5) inhibitor, sildenafil, is effective at reversing symptoms in murine HF models10, 11, 12 and improving cardiac contractility in men with diabetic‐related cardiac hypertrophy.13, 14 However, the RELAX (Phosphodiesterase‐5 Inhibition to Improve Clinical Status and Exercise Capacity in Diastolic Heart Failure) trial showed that sildenafil has little effect in the general HF population,14, 15, 16 further begging a critical question: how does PDE5 recover cardiac contractile function in the heart? Earlier studies indicate that inhibition of PDE5 leads to pleiotropic beneficial downstream effects in different animal models of cardiac diseases, including reduced cardiac hypertrophy and altered protein expression,11, 17 improved cardiac contractile function via increased myofilament‐mediated relaxation,12 improved mitochondrial function and reduced oxidative stress,18, 19, 20, 21 and improved anti‐inflammatory and antiapoptotic responses during cardiac ischemia.19 In models using streptozotocin or a high fat diet (HFD) to induce diabetes mellitus or the related cardiomyopathy, the PDE5 inhibitor vardenafil improves heart function through nitric oxide synthase–cGMP‐PKG (protein kinase G) activation.22, 23 Meanwhile, PDE5 has been linked to the soluble guanylyl cyclase pathway, whereas PDE9 is linked to the particulate guanylyl cyclase pathway for differential regulation of local cGMP signaling induced by different neurohormonal stimuli.24 Still, it is unclear how PDE5‐dependent regulation of cGMP can be influenced by elevated neurohormonal stimulation in diabetic hearts, which may offer new strategies to effectively target a subpopulation of patients.
Previous work has shown that PDE5 inhibition leads to changes in adrenergic modulation in the heart,25 in which β3 adrenergic receptor (β3AR) has been linked to the effects of inhibition of PDE5, leading to a negative inotropic response through PKG phosphorylation of troponin I in mouse hearts.26 Recent studies indicate that PDE5 is also linked to cardiac β2 adrenergic signaling, which negatively modulates chronotropy in neonatal and atrial nodal cells.27 Meanwhile, we have recently shown that β2 adrenergic receptor (β2AR) plays an important role in E‐C coupling during diabetic‐related cardiomyopathy, with β2AR knockout (β2KO) being cardioprotective.23, 28 This is due to β2AR complexing with insulin receptor29 which leads to transactivation of β2AR.28, 30 Concurrently, diabetic cardiomyocytes have an increased expression in phosphodiesterase, which is correlated with a decrease in PKA (protein kinase A) activity.28 We speculate a potential connection between cardiac adrenergic signaling and PDE5 that may be relevant to diabetic cardiomyopathy, including a reduced cardiac inotropy. Moreover, the interaction between different phosphodiesterase isoforms is essential in regulation of cardiac adrenergic signaling to control both cAMP and cGMP concentrations in cardiomyocytes.31 For instance, the binding of cGMP to phosphodiesterase 3 (PDE3) can act as a competitive inhibitor for binding and breakdown of cAMP, whereas the binding of cGMP to PDE2 can act as a stimulator to increase cAMP hydrolysis. In this way, treatments such as sildenafil that increase cGMP concentrations may also indirectly modulate cAMP concentrations via cross‐talk with PDE2 and PDE3.31 Given the clinical research showing that PDE5 inhibition is effective in enhancing cardiac function in male diabetic cardiomyopathy, we hypothesize that there is a functional association between PDE5 and βARs, contributing to reduced cardiac contractility during development of diabetic cardiomyopathy; and inhibition of PDE5 with sildenafil can improve adrenergic stimulation of both cAMP and cGMP signals and enhance E‐C coupling of diabetic myocytes.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Reagents and Materials
All reagents and materials were obtained from Millipore‐Sigma unless otherwise specified.
Experimental Animals
UC Davis's Institutional Animal Care and Use Committee approved animal protocols in accordance with institutional and National Institutes of Health (NIH) guidelines and were approved by the Association for Assessment and Accreditation of Laboratory Animal Care. Male wild‐type (WT) C57BL/6J mice were purchased from Jackson Laboratories, and β2 adrenergic receptor knockout (B2KO) C57BL/6J mice were bred in‐house. Starting at 6 to 7 weeks old, mice received either a 10% fat diet (Research diets Inc., D12450J), deemed normal chow (NC), or a HFD containing 60% fat by calories (D12492J), deemed HFD. n≥10 mice per group. Mice that were gavaged received either vehicle (1% carboxymethyl cellulose) or 100 mg/kg per day of crushed sildenafil citrate tablets in vehicle at 20 μL/10 g body weight starting 3.5 months after the special diet was started. All gavaged NC mice were treated with vehicle only. Adult mice were euthanized at 8 weeks old (no special diet employed) or after 4.5 to 5 months of special diet.
Intraperitoneal Glucose Tolerance Test
Mice underwent fasting for 6 hours before intraperitoneal glucose tolerance testing. Blood glucose readings were taken from a tail nick with a Bayer Breeze2 standard glucometer at 0, 15, 30, 60, and 120 minutes after injection with 1 mg/g IP glucose solution. The area under the curve was calculated using GraphPad Prism 7.
Echocardiography
Short‐axis M‐mode and ventricular tissue Doppler were performed under isoflurane anesthesia (2–2.5% for induction and 1–1.5% for maintenance) in pure oxygen flow using a Vevo 2100 imaging system (VisualSonics) with a 22‐ to 55‐MHz MS550D transducer. Body temperature, ECG, and respiration rate were monitored during the test to maintain the health of the animal and reduce variation, as previously described.32, 33
Parasternal short‐axis M‐mode was also used to assess sympathetic reserve of the mice. Once each mouse's heart rate had equilibrated within the target heart rate between 400 and 500 beats per minute, the mouse was injected with (‐)‐isoproterenol hydrochloride (0.2 mg/kg IP), followed by sildenafil citrate (1.6 mg/kg IP, Millipore‐Sigma). Readings were taken once a minute for 20 minutes beginning immediately before injection with isoproterenol. All echocardiogram analysis was performed with group identifiers removed from the imaging.
Histology
Hearts were perfused with 10% formalin, dehydrated on an Autotechnicon Mono Tissue Processor (Technicon Corporation), embedded in paraffin with a Leica HistoEmbedder, and sectioned using a Leica RM2255 Microtome. Sections were stained with hematoxylin and eosin and Masson's trichrome according to the manufacturer's protocols. Apoptosis detection was performed using the Promega DeadEnd Fluorometric terminal deoxynucleotidyl transferase dUTP nick‐end labeling (TUNEL) system according to manufacturer's instructions. Hematoxylin and eosin and Masson's trichome images were collected on a Nikon eclipse with a Nikon DS‐Qi1Mc camera. TUNEL images were collected on a Zeiss LSM 700 confocal. Images were analyzed using Image J (NIH) by a group‐blinded researcher.
Adult Rabbit and Mouse Ventriculomyocytes
Thirteen‐ to 14‐week‐old male New Zealand white rabbits (no special diet implemented) were purchased from Harlan Laboratories (Indianapolis, IN) and euthanized under general anesthesia (induction with 2 mg/kg propofol and 2–5% isoflurane in 100% oxygen). After thoracotomy, the heart was excised and rinsed in Minimum Essential Medium without calcium. Proceeding steps were performed as previously described,34 with the heart being digested with 150 mL of recirculated perfusion buffer containing 150 mg collagenase II and 8 mg protease XIV until digestion was complete. The ventricles were cut into a dish containing perfusion buffer with BSA, cut into pieces, titurated for 30 seconds with a 3‐mL transfer pipette, filtered through a 250‐μm mesh, and centrifuged at 500 rpm for 1 minute. The cell pellet was resuspended and recovered in a gradient of calcium buffers to a concentration of 1 mmol/L.
The method implemented for mouse cardiomyocyte isolation has been previously described.35 Adult mice were injected with 5000 units/kg heparin (Fresenius Kabi) and anesthetized in 2% to 5% isoflurane. The heart was predigested with 15 mL of perfusion buffer containing 2.5 mg of collagenase II (Worthington), 0.5 mg protease XIV, and 0.1% BSA. The heart was then digested with 20 mL of perfusion buffer containing 10 mg collagenase II, 2 mg protease XIV, 0.1% BSA, and 50 μmol/L CaCl2, which was recirculated through the system until the heart was digested. The heart was cut below the atria into a dish containing 5 mL of digestion buffer and dissociated using forceps. The supernatant was placed in 5 mL of stop buffer (12.5 μmol/L CaCl2 and 10% fetal bovine serum [FBS] in perfusion buffer) and was centrifuged at 500 rpm for 1 minute. Remaining particles of tissue were further digested with 5 mL of digestion buffer for 5 minutes at 37°C twice more to ensure maximal yield of cells. Cell pellets were resuspended and recovered in a gradient of calcium buffers to a concentration of 1 mmol/L.
Neonatal Mouse Cardiomyocytes
Neonatal pups that were less than 24 hours old were collected from WT, β1 adrenergic receptor (β1AR)‐knockout β1KO, β2KO, and β3AR knockout (B3KO) FVB mice bred in‐house. Hearts were minced and rotated in calcium‐ and bicarbonate‐free Hank's buffer with HEPES (136.9 mmol/L NaCl, 5.36 mmol/L KCl, 0.81 mmol/L MgSO4, 5.55 mmol/L glucose, 0.44 mmol/L KH2PO4, 0.34 Na2HPO4, 20 mmol/L HEPES) containing 1 mg/mL of collagenase type II (Worthington) for 10 minutes at 37°C. Tissue pieces settled for 1 minute before the supernatant was expunged to remove epithelial cells. Fresh collagenase type II–containing calcium‐ and bicarbonate‐free Hank's buffer with HEPES was then used in conjunction with a 10‐mL pipette to triturate the tissue and was followed by another 10‐minute incubation at 37°C. The tissue was then further triturated; the supernatant was collected and placed in 2 mL of horse serum to stop digestion and centrifuged at 800 rpm for 3 minutes to pellet cells. To ensure maximal yield of cells, remaining tissue pieces were further dissociated with another round of digestion, using the same procedure as outlined above. All cell pellets were combined through resuspension in Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS and plated on a 10‐cm Petri dish for 1 hour at 37°C. Cardiomyocytes were then gently washed from the Petri dish and collected, while fibroblasts that remained attached to the Petri dish were discarded. The collected cardiomyocytes were plated on laminin‐coated glass coverslips in DMEM with 10% FBS for 24 hours at 37°C before adenovirus infection with the cGi50036 fluorescence resonant energy transfer (FRET) biosensor in serum‐free DMEM for 48 hours.
Coimmunoprecipitation
Isolated rabbit adult ventriculomyocytes (AVMs) were incubated in DMEM + 10% FBS for 2 hours. The media was changed to serum‐free DMEM containing adenovirus coding for either Flag‐tagged mouse β2AR (Flag‐mβ2AR) or cyan fluorescent protein‐ and Flag‐tagged mouse β1AR (Flag‐mβ1AR) for 40 hours. Viruses were produced using the AdEasy system37 (Qbiogene). Cells were lysed with IP lysis buffer (25 mmol/L HEPES, 150 mmol/L NaCl, 5 mmol/L EDTA, 10% glycerol, 1% Triton‐X) containing inhibitors (100 mmol/L Na3F, 1 mmol/L Na2VO4, 1 mmol/L glycerol, 2.5 mmol/L NaP2O7, 10 μg/mL leupeptin, 1 mmol/L phenylmethane sulfonyl fluoride, 10 μg/mL aprotinin). Coimmunoprecipitation was performed for 15 hours at 4°C using Flag‐M2 beads. Western blot was used to detect PDE5 and Flag by a group‐blinded researcher.
Western Blotting
Left ventricular tissues were lysed as previously described29 using FastPrep‐24 lysing matrix D beads (MP Biomedicals) in lysis buffer (25 mmol/L Tris HCl pH=7.6, 150 mmol/L NaCl, 1% IGEPAL, 1% sodium deoxycholate, 0.1% SDS, 1 mmol/L EDTA) containing protease and phosphatase inhibitors (100 mmol/L NaF, 1 mmol/L Na2VO4, 1% glycerol, 2.5 mmol/L NaP2O7, 1 mmol/L phenylmethane sulfonyl fluoride, 10 μg/mL aprotinin, 10 μg/mL pepstatin A, and 20 μg/mL E‐64, 5 μg/mL bestatin). Protein concentrations were equilibrated after BCA assay. For most blots, 50 μg of each lysate was resolved by SDS‐PAGE using a 4% to 20% gradient gel and transferred to 0.45 μm polyvinylidene fluoride. For phospholamban blots, lysates were further treated with 10% β‐mercaptoethanol, sonicated, and heated at 37°C for 1 hour before resolution of 15 μg of protein per sample by SDS‐PAGE and transfer onto 0.2 μm of nitrocellulose membrane (Bio‐Rad). Membranes were incubated with primary antibody, followed by IRDye 680CW or 800CW secondary antibodies and scanned on an Odyssey scanner (Li‐Cor Biosciences). Primary antibodies implemented were phosphodiesterase 5A (PDE5A; sc‐398747, Santa Cruz), Flag (F7425, Millipore‐Sigma), phosphodiesterase 3A (NIH, PDE3A‐CT aa1098‐1115),38 phospho‐serine 282 and total cardiac‐specific myosin‐binding protein c (p282 and cMyBP‐C and total cMyBP‐C, generous gift from Dr Sakthivel Sadayappan,39 Loyola University), phospho‐serine 23/24 and total troponin I (p23/24 and total TnI; 4004 and 4002, Cell Signaling), sarcoplasmic‐endoplasmic reticulum calcium ATPase 2A (SERCA2A; 2A7‐A1, Thermo), phospho‐serine 16 and total phospholamban (p16 and total PLB; A010‐12 and A010‐14, Badrilla), and glyceraldehyde 3‐phosphate (GAPDH; 97166, Cell Signaling). Band intensities were quantified using ImageJ (NIH), divided by GAPDH intensities and normalized to the average of the first group.
Proximity Ligation Assay
Cells were fixed with buffered 4% paraformaldehyde for 10 minutes and stored in ddPBS at 4°C. Cells were permeabilized (0.3% Triton‐X, 2% goat serum in ddPBS) and incubated at 4°C overnight in primary antibodies. Primary antibodies instituted included PDE5A (2395, Cell Signaling) with β2AR (SC‐271322, Santa Cruz) for PDE5A:β2AR PLA, phosphodiesterase 3A (NIH, PDE3A‐CT aa1098‐1115)38 with β2AR (SC‐570, Santa Cruz) for PDE3A:β2AR PLA, and PDE5A (2395, Cell Signaling) with phosphodiesterase 3A (NIH, PDE3A‐CT aa1098‐1115)38 for PDE5:PDE3 PLA. The manufacturer's protocol was followed for proceeding steps. Images were collected on a Zeiss LSM 700 confocal and analyzed on ImageJ by a group‐blinded researcher.
Adult Cardiomyocyte Contractility and Calcium Transient
Mouse group identity was masked from the researcher performing the assay. Mouse AVMs were suspended in isolation buffer containing 1 mmol/L CaCl2 and placed on ice, and 10 mg/L Fluo‐4‐AM (Thermo‐Fisher) was added. Cells were incubated at room temperature for 12 minutes, and then the supernatant was replaced with fresh buffer. Cells were incubated at room temperature for another 15 minutes and then placed on ice for 15 minutes or more. As previously described,30 the cells were then transferred into beating buffer and were paced using platinum electrodes with 50 V at 1 Hz with a stimulator. Cells that were not sharply beating in pace and flashing green in pace were excluded from analysis.
A Zeiss Axio Observer A1 inverted fluorescence microscope with Hamamatsu Orca‐Flash 4.0 camera acquired phase contrast and fluorescein isothiocyanate images to assess contractility and calcium transient, respectively. The cells were treated with 100 nmol/L isoproterenol and/or 1 μmol/L sildenafil. 10 μmol/L Forskolin + 100 μmol/L IBMX were similarly added to act as positive control. Cells were pretreated with 100 nmol/L ICI‐118551, 100 nmol/L CGP‐20712, 1 μmol/L KN‐93, 1 μmol/L PKI 14‐22myr, or 100 nmol/L DT2. The percentage of myocyte contractility was defined as (maximal cell length−minimal cell length)/maximal cell length×100, assessed by MetaMorph software. Analysis of calcium transient dynamics was performed by a group‐blinded researcher using an in‐house routine written in interactive data language as previously described.40
Fluorescence Resonant Energy Transfer
Mouse group identity was masked from the researcher performing the assay. Mouse adult cardiomyocytes that were intended for FRET assays were plated, as previously described,41 on laminin‐coated glass coverslips with mouse FRET media (10.7 g/L M1018, 6.25 μmol/L blebbistatin, 10 mmol/L HEPES, 0.2% BSA, 4 mmol/L NaHCO3, 1× penicillin‐streptomycin‐glutamate [PSG]) containing 10% FBS and incubated at 37°C for 5 hours. The media was then changed to serum‐free mouse FRET media that contained adenovirus coding for an appropriate FRET biosensor, and cells were incubated at 37°C for 30 to 50 hours, based on biosensor expression. FRET biosensors implemented in this study were the A‐kinase activity reporter‐3 (AKAR3), Epac‐based sensor #H187 (Epac‐SH187), and cGMP indicator‐500 nmol/L (cGi500), which have all been previously described.36, 42, 43, 44, 45 Viruses were produced using the AdEasy system37 (Qbiogene).
For both neonatal and adult cardiomyocytes, the media was changed to serum‐free media without virus. Coverslips were transferred to glass bottom culture dishes (MatTek) containing ddPBS (137 mmol/L NaCl, 2.7 mmol/L KCl, 10 mmol/L Na2HPO4, 1.8 mmol/L KH2PO4, pH=7.4). For adult cells, ddPBS was supplemented with 1 μmol/L blebbistatin. A Zeiss Axio Observer A1 inverted fluorescence microscope with Hamamatsu Orca‐Flash 4.0 camera acquired FRET images. Images were recorded by excitation at 430 to 455 nm and emission on 2 filters (475DF40 for cyan and 535DF25 for yellow). Cells were pretreated with 100 nmol/L ICI‐118551 or 100 nmol/L CGP‐20712 as indicated. The change in FRET ratio was measured after 100 nmol/L isoproterenol and/or 1 μmol/L sildenafil citrate were added. 10 μmol/L Forskolin+100 μmol/L IBMX were similarly added to act as positive control for AKAR3 or Epac‐SH187, while 50 μmol/L sodium nitroprusside acted as positive control for cGi500. If no response was seen to initial drug addition, the appropriate positive control drug was added. Only cells that depicted a response were analyzed. Images were subjected to background subtraction and were acquired every 30 seconds with an exposure time of 200 ms. The donor/acceptor FRET ratio was normalized to the baseline.
Statistical Analysis
All values are presented as mean±SEM. Statistical tests implemented were calculated using GraphPad Prism 7. P≤0.05 was considered significant. Unpaired t tests were performed for comparisons of 2 groups. For comparisons between ≥3 groups with a group n=3, Kruskal–Wallis tests with Dunn's post‐tests were performed. For comparisons between ≥3 groups with n>3 per group, 1‐way ANOVAs were executed with Tukey post‐tests. For comparisons between groups over time, a repeated measures ANOVA with Sidak post‐test was implemented. Post hoc tests were only performed on ANOVAs, which had a significant F value. Area under the curve was calculated by determining the total peak area above y=0 for each mouse. For left ventricular wall thickness, fibrosis, and TUNEL measurements, average values from multiple readings for each mouse were determined before plotting data on the overall graph.
Results
Sildenafil Treatment Improves Cardiac Reserve in Response to Sympathetic Stimulation in Diabetic Hearts
High fat diet (HFD) mice displayed a reduction in glucose tolerance, as well as a reduced cardiac ejection fraction and fractional shortening associated with cardiac remodeling in the form of increased hypertrophy, fibrosis, and myocyte apoptosis (Figure 1A through 1K). Meanwhile, chronic phosphodiesterase 5 (PDE5) inhibition with sildenafil in diabetic mice improved basal heart function, which correlated with reduced hypertrophy, fibrosis, and myocyte apoptosis (Figure 1C through 1K), similarly to previous reports in diabetic humans and rats.22, 23 Moreover, while HFD mice displayed a reduced cardiac reserve in response to adrenergic stimulation with isoproterenol, acute treatment with sildenafil restored cardiac reserve in response to adrenergic stimulation (Figure 1L and 1M). Chronic PDE5 inhibition with sildenafil also restored cardiac reserve in response to adrenergic stimulation (Figure 1L and 1M). These results suggest that besides improved basal diabetic heart function, sildenafil can also improve cardiac reserve in response to sympathetic stimulation in diabetic cardiomyopathy.
Figure 1.
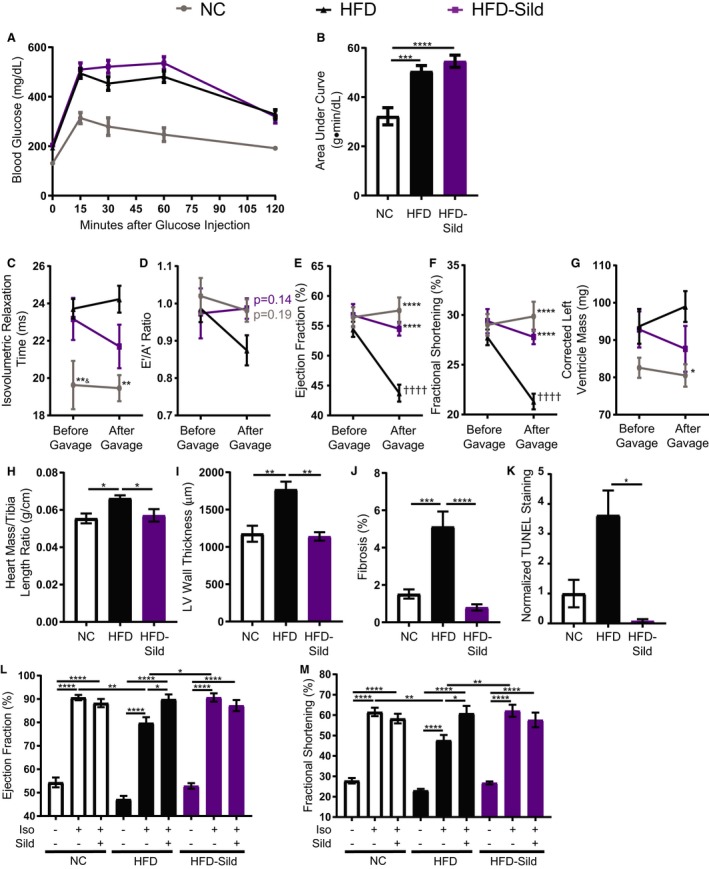
Sustained in vivo sildenafil treatment in mice fed a high fat diet (HFD) improves basal and adrenergic‐induced heart function and morphology. A, Intraperitoneal glucose tolerance testing (IPGTT) and (B) area under the curve of IPGTT from wild‐type (WT) mice on a special diet for 4.5 months and gavaged for 1 month with either vehicle or 100 mg/kg per day of sildenafil citrate. ***P≤0.001, ****P≤0.0001 by 1‐way ANOVA and Tukey post‐test. n≥9 mice per group. C, Isovolumetric relaxation time, (D) E’/A’ ratio, (E) ejection fraction, (F) fractional shortening, and (G) corrected left ventricular (LV) mass of WT mice after 3.5 and 4.5 months of normal chow or HFD, as measured by M‐mode and tissue Doppler echocardiogram. Between the 2 readings, all mice were gavaged daily with either vehicle only or 100 mg/kg sildenafil. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001 compared with HFD at the same time point; & P≤0.05 compared with HFD‐sildenafil at the same time point; †††† P≤0.0001 compared with the same group before gavage by a repeated measures ANOVA and Sidak post‐test. n≥9 mice per group. H, Heart mass to tibia length ratio of gavaged mice. n=5 mice per group. *P≤0.05 by 1‐way ANOVA and Tukey post‐test. (I) Quantification of LV wall thickness of gavaged mice measured from hematoxylin and eosin slices. n=5 mice per group. **P≤0.01 by 1‐way ANOVA and Tukey post‐test. J, Quantification of fibrosis percentage in gavaged mice, measured from 6 areas of each mouse left ventricle stained with Masson's trichome. n=5 mice per group. ***P≤0.001, ****P≤0.0001 by 1‐way ANOVA and Tukey post‐test. K, Quantification of terminal deoxynucleotidyl transferase dUTP nick‐end labeling (TUNEL)–positive cells in heart slices from gavaged mice. *P≤0.05 by Kruskal–Wallis test with Dunn's post‐test. n=3 mice per group. L, Ejection fraction and (M) fractional shortening of HFD mice gavaged with sildenafil (HFD‐sildenafil) or vehicle only (HFD) at baseline and after isoproterenol followed by sildenafil injections. n≥9 mice per group. *P≤0.05, **P≤0.01, ***P≤0.001, and ****P≤0.0001 by 1‐way ANOVA and Tukey post‐test.
Western blots were then performed to determine the changes in E‐C coupling proteins. First, NC and HFD heart tissue samples were compared. As we have previously observed,28 HFD mouse hearts have a reduction in expression of SERCA2A, which leads to an increase in the ratio of phospholamban‐to‐SERCA2A. A reduction in phosphorylation of p23/24 TnI and p16 PLB could also be similarly observed. Meanwhile, PDE5A and PDE3A showed no significant change between NC and HFD mouse hearts. A PKG substrate of p282 of cMyBP‐C promotes myocyte contractility upon phosphorylation.46 The expression of total cMyBP‐C, as well as the levels of p282 cMyBP‐C, did not show significant changes between NC and HFD mouse hearts (Figure 2A and 2B). When comparing sildenafil‐treated HFD animals with their HFD counterparts, the expression of PDE5A and PDE3A in hearts did not significantly change. Interestingly, chronic sildenafil treatment did increase the expression of total cMyBP‐C, as well as the levels of p282 cMyBP‐C (Figure 2C and 2D). Sildenafil also increased expression of TnI while decreasing PLB expression, but did not change the expression of SERCA2A in HFD hearts (Figure 2C and 2D). In addition, sildenafil led to an increase in p16 and PLB/total PLB (Figure 2C and 2D). These data suggest that sustained sildenafil treatment in vivo alters the expression and activities of proteins in a manner that favors improved E‐C coupling and heart function. These include increases in myofilament protein expression and myofilament calcium sensitivity, as well as increases in SERCA calcium reuptake activity.
Figure 2.
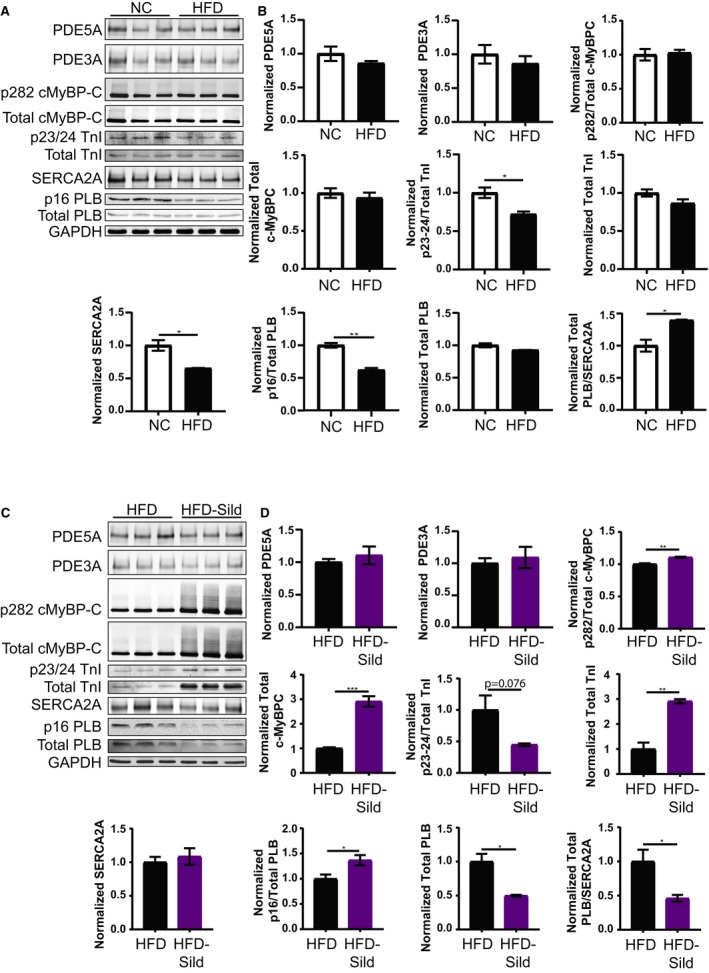
Sildenafil treatment improves E‐C coupling protein activation and expression in mice fed a high fat diet (HFD). Images and quantification of Western blots for phosphodiesterase 5A (PDE5A), phosphodiesterase 3A (PDE3A), phospho‐serine 282 (p282) and total cardiac myosin binding protein C (cMyBP‐C), phospho‐serine 23/24 (p23/24) and total troponin I (TnI), sarcoplasmic‐endoplasmic reticulum calcium ATPase 2A (SERCA2A), and phospho‐serine 16 (p16), and total phospholamban (PLB) for normal chow (NC) and HFD mice (A and B) followed by that for HFD and HFD‐sildenafil mice (C and D). Total protein normalized to glyceraldehyde 3‐phosphate (GAPDH). *P≤0.05, **P≤0.01, ***P≤0.001 by unpaired t test. n=3 mice per group.
To determine whether the increases in whole‐heart sympathetic responses produced by sildenafil in WT HFD mice were caused by improvements in E‐C coupling, AVMs were treated ex vivo with isoproterenol and/or sildenafil and their contractility, calcium transient amplitude, and calcium halftime recovery (tau) were recorded. HFD myocytes displayed an impaired excitation‐contraction response to isoproterenol stimulation while acute treatment with sildenafil fully rescued isoproterenol‐induced contractility and calcium transient dynamics in HFD AVMs (Figure 3A through 3C). Meanwhile, chronic therapy with sildenafil also restored isoproterenol‐induced contractility and calcium transient dynamics in HFD AVMs. Additional treatment with sildenafil on myocytes isolated from sildenafil‐treated mice did not further enhance isoproterenol‐induced contractility or calcium transient. These data suggest that either acute in vitro or sustained in vivo sildenafil treatment is sufficient to rescue adrenergic stimulation of contractility in diabetic cardiomyocytes with improvements of calcium amplitude and reuptake. This opposes previous reports in which sildenafil attenuates isoproterenol‐induced contractility response in myocytes from 6‐ to 8‐week‐old mice.25, 26 As control, E‐C coupling was measured in myocytes from 8‐week‐old mice. The addition of sildenafil reduced contractility and calcium transient dynamics induced by isoproterenol in young AVMs (Figure 3D through 3F). These data suggest that a remodeling of PDE5 in modulation of adrenergic stimulation for E‐C coupling during development of diabetic cardiomyopathy occurs.
Figure 3.
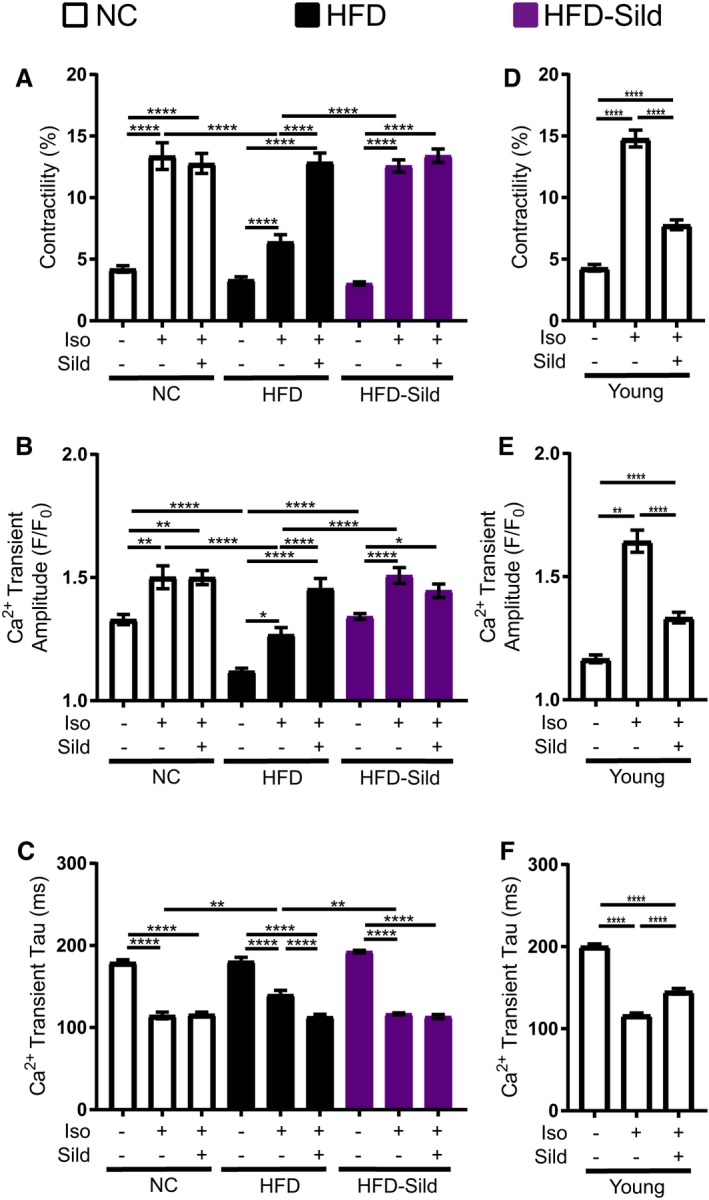
Sustained In vivo or acute in vitro sildenafil treatment is sufficient to rescue adrenergic response in diabetic adult ventriculomyocytes (AVMs). A, Contractility, (B) calcium transient amplitude, and (C) calcium transient halftime recovery (tau) of AVMs from wild‐type mice fed normal chow (NC) or high fat diet (HFD) for 4.5 months and gavaged with either sildenafil or vehicle only for 1 month. n≥20 cells per group. (D) Contractility, (E) calcium transient amplitude, and (F) calcium transient halftime recovery in isolated adult cardiomyocytes from young 8‐week‐old mice. n≥15 cells per group. For all graphs, *P≤0.05, **P≤0.01, ***P≤0.001, and ****P≤0.0001 by 1‐way ANOVA and Tukey post‐test.
Sildenafil Bolsters Adrenergically Stimulated Cyclic Nucleotide Concentration in HFD AVMs
FRET‐based analysis of cGMP with the cGi500 biosensor in AVMs was then applied to understand the mechanism underlying sildenafil's effect on E‐C coupling (Figure 4A). Sildenafil alone did not affect the baseline levels of cGMP in AVMs isolated from any group. Isoproterenol induced robust increases in cGMP in NC AVMs, which were significantly impaired in HFD AVMs (Figure 4A). Acute addition of sildenafil enhanced isoproterenol‐induced cGMP in HFD AVMs (Figure 4A). Chronic treatment with sildenafil in vivo also recovered isoproterenol‐induced increases in cAMP in HFD‐sildenafil AVMs, while acute addition of sildenafil to AVMs in vitro did not further enhance HFD‐sildenafil AVM responses induced by isoproterenol (Figure 4A). Eight‐week‐old mouse ventriculomyocytes responded in a similar fashion to the 6‐month‐old NC cells (Figure 4B). These data together suggest an increased coupling between PDE5 and adrenergic regulation in diabetic cardiomyopathy.
Figure 4.
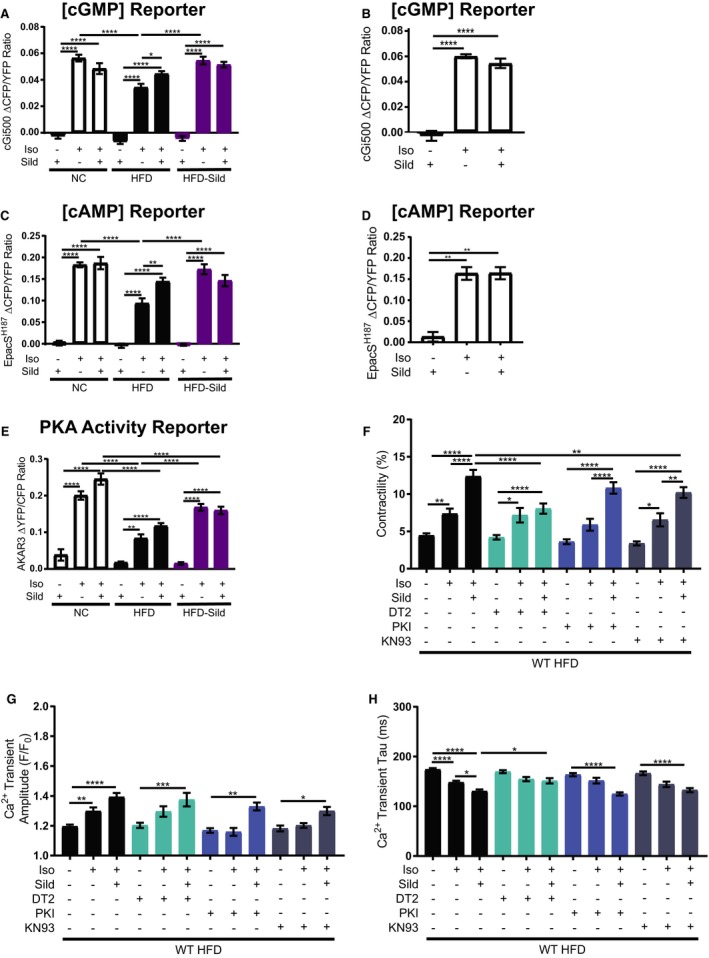
Sildenafil rescues cyclic nucleotide concentration, increases PKA (protein kinase A) activity, and improves adrenergic E‐C coupling response in a PKG (protein kinase G)– and calcium/calmodulin kinase II–dependent manner. Average maximal fluorescence resonant energy transfer (FRET) response of the cGMP probe, cGi500 in adult ventriculomyocytes (AVMs) from gavaged mice (A) and from young 8‐week‐old mice (B). Average maximal FRET response of the cAMP probe, EpacSH187 in AVMs from gavaged mice (C) and from young 8‐week‐old mice (D). Average maximal FRET response of the PKA activity probe, A‐kinase activity reporter‐3 in AVMs from gavaged mice (E). In all FRET response graphs, n≥15 cells per group; *P≤0.05, **P≤0.01, ***P≤0.001, and ****P≤0.0001 by 1‐way ANOVA with Tukey post‐test. Contractility (F), calcium transient amplitude (G), and calcium transient tau (H) of AVMs from wild‐type mice fed an HFD for 4.5 months. Cells were pretreated with and without DT2 peptide, myrisoylated PKI peptide (PKI), or KN93 before stimulation with isoproterenol and sildenafil. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001 by 1‐way ANOVA and Tukey post‐test. n≥25 cells per group.
Adrenergically induced cAMP signal and PKA activity in NC, HFD, and HFD‐sildenafil AVMs were also assessed with FRET‐based biosensors EpacSH187 and AKAR3, respectively. Sildenafil alone did not affect cAMP signal nor PKA activity (Figure 4C throguh 4E). Stimulation with isoproterenol alone induced robust increases in cAMP signal and PKA activity in NC AVMs. Similar increases in cAMP as seen in 6‐month‐old NC AVMs were also seen upon stimulation of 8‐week‐old mouse AVMs (Figure 4D). When compared with the increases seen in healthy controls, the HFD AVM response was significantly impaired (Figure 4C through 4E). Acute addition of sildenafil rescued isoproterenol‐induced cAMP signal and partially enhanced PKA activity in HFD AVMs (Figure 4C through 4E). Chronic treatment with sildenafil in vivo also rescued isoproterenol‐induced increases in cAMP signal and partially recovered PKA activity in HFD‐sildenafil AVMs; additional sildenafil in vitro did not further enhance the response induced by isoproterenol in AVMs from sildenafil‐treated animals (Figure 4C through 4E). These data suggest that both acute and sustained inhibition of PDE5 not only change cGMP signal but also cAMP signal and PKA activity in diabetic AVMs in response to adrenergic stimulation in HFD myocytes. In comparison, in both age‐matched NC and young mouse control AVMs, sildenafil did not further enhance cGMP nor cAMP concentrations induced by isoproterenol (Figure 4A through 4E).
Sildenafil Improves E‐C Coupling in Diabetic Myocytes in a PKG‐Dependent Manner
Based on the results from FRET assays, the function of downstream kinases of cGMP and cAMP that are important for E‐C coupling were evaluated. WT HFD AVMs were pretreated with the PKG inhibitor, DT2, the PKA inhibitor, PKI, or the calcium/calmodulin kinase II (CaMKII) inhibitor, KN93, and then their contractility and calcium transient dynamics were measured in response to isoproterenol and/or sildenafil. Inhibition of PKG caused a dramatic reduction in contractility that was accompanied by a slowing of tau but not a significant change in calcium transient amplitude (Figure 4F throguh 4H). To a lesser extent, inhibition of CaMKII also significantly attenuated contractility response to isoproterenol+sildenafil. CaMKII inhibition did not affect calcium transient dynamics to isoproterenol+sildenafil (Figure 4F through 4H). In contrast, inhibition of PKA did not affect contractility nor calcium cycling response to isoproterenol+sildenafil (Figure 4F through 4H). Together, these data indicate that in vitro sildenafil improves E‐C coupling primarily through PKG in HFD AVMs.
PDE5 Interacts With β2 Adrenergic Receptor (B2AR) and Its Signaling Cascade
To determine whether sildenafil would affect adrenergically stimulated cGMP concentration in a βAR‐subtype–specific manner, FRET analysis of cGMP concentration was performed in WT, β1KO, β2AR knockout, and β3KO neonatal cardiomyocytes after treatment with sildenafil and/or isoproterenol. In WT cardiomyocytes, sildenafil significantly enhanced cGMP concentrations induced by isoproterenol. Deletion of β2AR abolished the effect of sildenafil, whereas deletion of either β1AR or β3AR did not do so (Figure 5A through 5D). These data suggest that sildenafil enhances cGMP induced by β2AR in neonatal cardiomyocytes. To determine whether this interaction also occurs in diabetic cardiomyopathy, AVMs from HFD mice were treated with the β1AR inhibitor, CGP‐20712, or the β2AR inhibitor, ICI‐118551, and adrenergically stimulated cGMP levels were measured by FRET (Figure 5E). While inhibition of β1AR led to a significant reduction in response to isoproterenol, the addition of sildenafil could still cause a significant rescue of signal. Meanwhile, HFD cells treated with the β2AR inhibitor had no additional response to the addition of sildenafil. In contrast, the addition of sildenafil to isoproterenol in NC AVMs inhibited with CGP‐20712 or ICI‐118551 showed no significant difference to the response to isoproterenol alone (Figure 5E). This suggests that PDE5 has an important functional interaction with β2AR in HFD AVMs.
Figure 5.
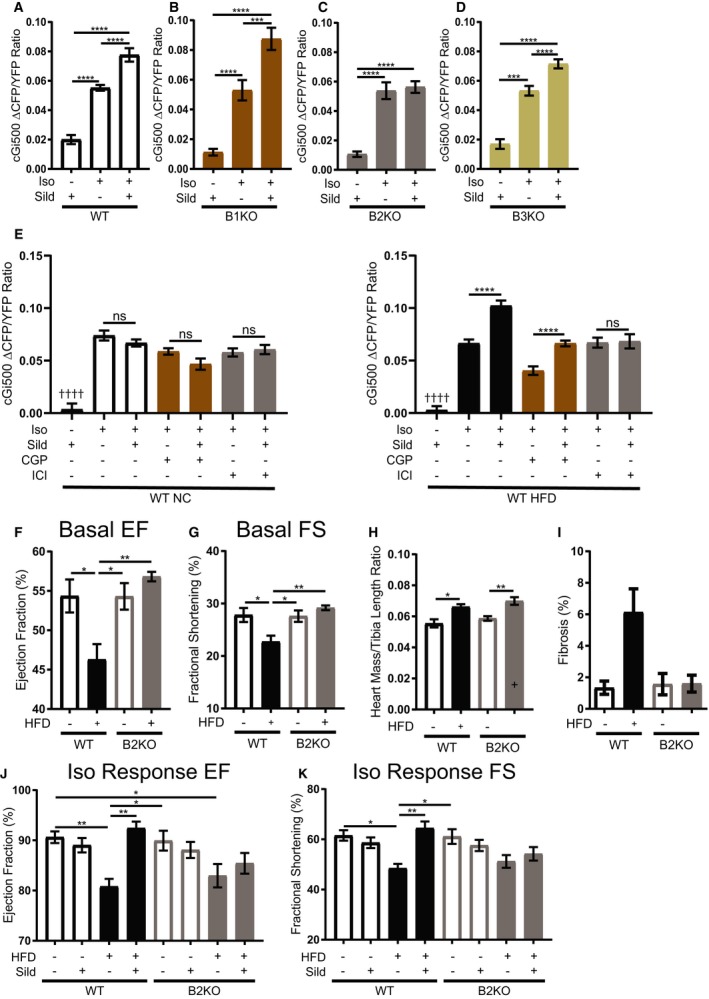
Phosphodiesterase 5 preferentially interacts with β2 adrenergic receptor (β2AR) and its signaling cascade. A through D, Average maximal fluorescence resonant energy transfer (FRET) response of the cGMP probe to isoproterenol, cGi500 in isolated neonatal cardiomyocytes from wild‐type (WT), β1 adrenergic receptor (β1AR) knockout (β1KO), β2AR knockout (β2KO), and β3 adrenergic receptor knockout (β3KO) mice. n≥30 cells per group. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001 by 1‐way ANOVA with Tukey post‐test. E, Average maximal FRET response of the cGMP probe to isoproterenol, cGi500 in isolated NC and HFD ACMs with pretreatment of the β1AR inhibitor, CGP‐20712 (CGP), or the β2AR inhibitor, ICI‐118551 (ICI). n≥20 cells per group. †††† P≤0.0001 compared with all other groups, *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001 by 1‐way ANOVA with Tukey post‐test. F, Ejection fraction and (G) fractional shortening echocardiogram measurements of WT and β2KO mice fed normal chow (NC) or high fat diet (HFD) after 4.5 months of special diet. H, Heart mass/tibia length ratio of WT and β2KO mice fed NC or HFD for 4.5 months. n≥7 mice per group. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001 by 1‐way ANOVA with Tukey post‐test. I, Interstitial cardiac fibrosis percentage in WT and β2KO mice fed NC or HFD for 4.5 months. n=3 mice per group. Kruskal–Wallis test with Dunn's post‐test performed. J, Ejection fraction and (K) fractional shortening echocardiogram measurements of WT and β2KO mice fed NC and HFD for 4.5 months after isoproterenol injection followed by sildenafil injection. n≥8 mice per group. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001 by 1‐way ANOVA with Tukey post‐test.
In vivo echocardiogram studies were then performed to test whether β2AR is necessary for sildenafil‐mediated modulation of adrenergic stimulation. As previously observed,28 β2KO mice fed a HFD are protected from decreases in basal heart function and worsening cardiac fibrosis and hypertrophy that is seen in WT HFD mice (Figure 5F through 5I). In contrast, both WT and β2KO mice fed HFD had significant reductions in response to isoproterenol when compared with age‐matched WT NC controls (Figure 5J and 5K). Additional administration of sildenafil enhanced the increase in ejection fraction and fractional shortening induced by isoproterenol in WT HFD mice, but not in WT NC controls. In contrast, sildenafil did not boost isoproterenol‐induced ejection fraction and fractional shortening in β2KO HFD mice, which suggests that sildenafil improves diabetic heart stress response in a β2AR fashion (Figure 5J and 5K).
Consistent with the in vivo observations, HFD feeding led to impaired E‐C coupling after isoproterenol stimulation, which is rescued by sildenafil treatment (Figure 6A through 6C). Deletion of β2AR normalized the isoproterenol‐induced E‐C coupling in HFD AVMs. Treatment with sildenafil did not cause an additional response in the E‐C coupling parameters, again supporting that sildenafil improves E‐C coupling in diabetic myocytes in a β2AR‐dependent manner. To ensure that the results collected from β2KO HFD myocytes were not compensatory from genetic deletion, AVMs from HFD WT mice were pretreated with the β1AR inhibitor, CGP‐20712, or the β2AR inhibitor, ICI‐118551. While CGP‐20712 abolished isoproterenol response completely, ICI‐118551 did not significantly affect isoproterenol response (Figure 6D through 6F), consistent with the primary regulatory role of β1AR in sympathetic E‐C coupling response.47 However, when sildenafil is added to isoproterenol in ICI‐118551‐treated AVMs, there is not a significant enhancement in the isoproterenol response like that in WT HFD cells without pretreatment. Addition of sildenafil to isoproterenol in ICI‐118551–pretreated AVMs is associated with a slowing of calcium tau, but not a significant decrease in calcium amplitude (Figure 6D through 6F). These data reiterate that sildenafil improves sympathetically induced E‐C coupling dynamics in a β2AR‐dependent fashion. Moreover, these results underscore the importance of β1AR in sympathetically induced contractility, where modulation of the β2AR signal only further affects downstream signaling.
Figure 6.
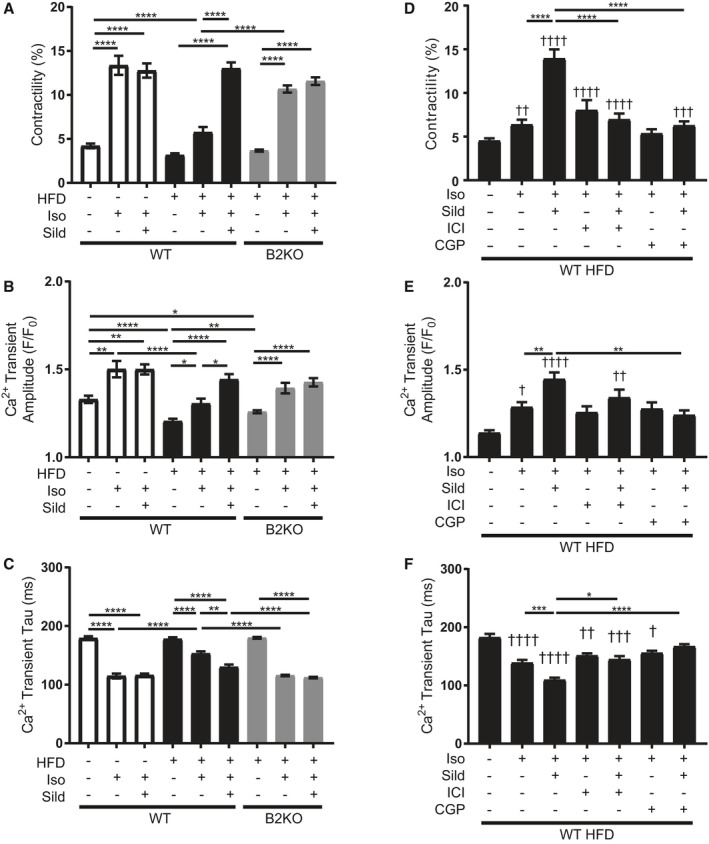
Sildenafil acts through β2 adrenergic receptor (β2AR) to improve adrenergic excitation‐contraction coupling response in high fat diet (HFD) adult ventriculomyocytes (AVMs). A, Contractility, (B) calcium transient amplitude, and (C) calcium transient tau of AVMs from wildtype (WT) and β2AR knockout (β2KO) mice fed NC or HFD for 4.5 months and treated ex vivo with drugs, as indicated. n≥20 cells per group. D, Contractility, (E) calcium transient amplitude, and (F) calcium transient tau of AVMs from WT mice fed HFD for 4.5 months. Cells were pretreated with and without ICI‐118551 (ICI) or CGP‐20712 (CGP) before stimulation with isoproterenol and/or sildenafil. n≥25 cells per group. For all graphs, † P≤0.05, †† P≤0.01, ††† P≤0.001, †††† P≤0.0001 compared with basal; and *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001 by 1‐way ANOVA and Tukey post‐test.
To ascertain whether PDE5 physically interacts with β2AR, coimmunoprecipitation was performed on rabbit AVMs expressing either Flag‐mβ2AR or Flag‐mβ1AR, where Flag was pulled down. PDE5A was pulled down with Flag‐β2AR but not with Flag‐mβ1AR (Figure 7A and 7B), depicting that PDE5A specifically interacts in a protein complex with β2AR. Proximity ligation assay was then performed between PDE5A and β2AR in AVMs from NC and HFD mice to determine how this interaction may change. There is no significant change in colocalization of PDE5A and β2AR between these groups (Figure 7C), but there is a significant increase in the interaction of both PDE5A and β2AR with PDE3A in HFD cardiomyocytes (Figure 7D and 7E). This suggests that during disease development there is a change in the interaction of the β2AR:PDE5A complex with the dual cAMP/cGMP phosphodiesterase PDE3.
Figure 7.
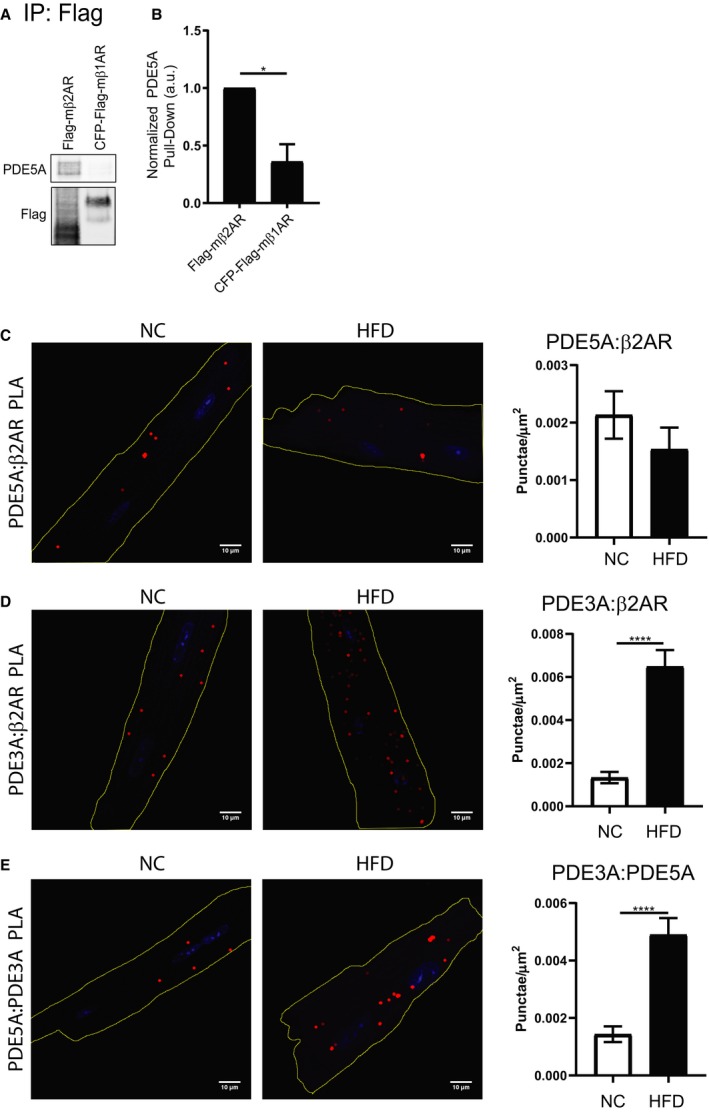
Phosphodiesterase 3 (PDE3) displays an increased association with the phosphodiesterase 5 (PDE5)–β2 adrenergic receptor (β2AR) protein complex in high fat diet (HFD) cardiomyocytes. A, Representative images and (B) quantification of coimmunoprecipitation of Flag‐tagged βARs from rabbit adult ventriculomyocytes (AVMs) expressing Flag‐mβ2AR or CFP (cyan fluorescent protein) – and Flag‐tagged mouse β1 adrenergic receptor (β1AR), with detection of PDE5A. n=3 rabbits. *P≤0.05 by unpaired t test. Proximity ligation assay (PLA) representative images and quantification in normal chow (NC) and HFD AVMs for the interaction between PDE5A and β2AR (C), between PDE3A and β2AR (D), and between PDE3A and PDE5A (E). Scale bar=10 μm. Red=PLA signal. Blue=DAPI. n=18 cells per group. ****P≤0.0001 by unpaired t test.
Discussion
Phosphodiesterase 5 (PDE5) has been linked to cardiac adrenergic signaling regulation in both physiological and pathological conditions. The important finding in this study is that PDE5A exists in a protein complex with β2 adrenergic receptor (β2AR) and mediates the impacts of sildenafil on adrenergic response in diabetic cardiomyopathy in a β2AR‐specific manner. The inhibition of PDE5 by sildenafil not only rescues adrenergically induced cGMP but also cAMP in diabetic cardiomyocytes through an increased interaction with PDE3. Sildenafil boosts E‐C coupling dynamics of diabetic AVMs through activation of PKG and, to a lesser extent, CaMKII (Figure 8A). Furthermore, chronic therapy with sildenafil in vivo improves cardiac function at baseline and after adrenergic stimulation. Deletion of β2AR abolishes the effects of sildenafil on adrenergic modulation. These data reveal a novel molecular mechanism of a functional PDE5‐β2AR complex underlying the effects of sildenafil in healthy and diseased hearts.
Figure 8.
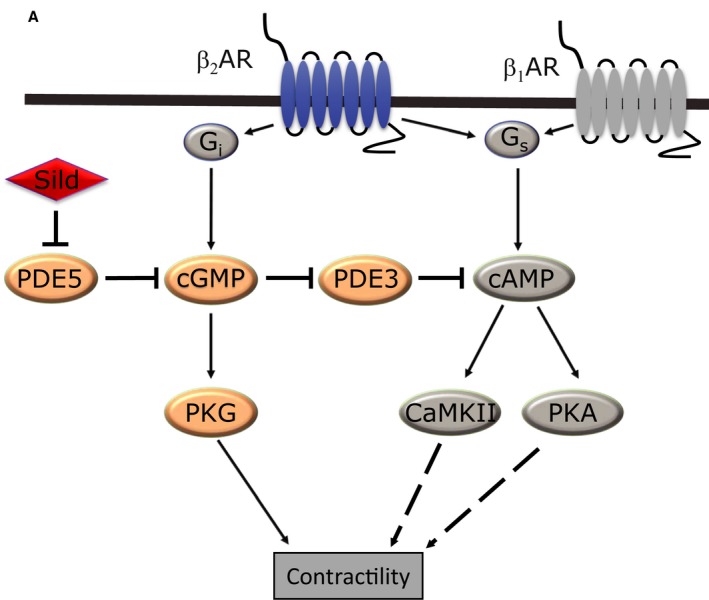
Inhibition of phosphodiesterase 5 (PDE5) by sildenafil improves contractility in a β2 adrenergic receptor (β2 AR)–dependent mechanism (A) depiction of the pathway sildenafil uses in mice fed a high fat diet to improve function. CaMKII indicates calcium/calmodulin kinase II; Gi, inhibitory G protein; Gs, stimulatory G protein; PKA, protein kinase A; PKG, protein kinase G; PDE3, phosphodiesterase 3; β1AR, β1 adrenergic receptor.
Biochemical, proximity ligation, and FRET‐based live cell imaging assays show that PDE5 selectively associates with cardiac β2AR but not β1AR and specifically enhances β2AR‐induced cGMP signaling in myocytes to improve E‐C coupling in a model of diabetic cardiomyopathy. These data, for the first time, place PDE5 in a receptor complex to facilitate the hydrolysis of cGMP, which exerts local control over a pool of cGMP after adrenergic stimulation in both neonatal and HFD myocytes. A previous report also linked a pool of cGMP promoted by PDE5 inhibition to β3AR in young adult cardiomyocytes, which interestingly leads to PKG‐dependent reduction of isoproterenol‐induced contractility via phosphorylation of TnI.26 However, a specific interaction between PDE5 and β3AR remains to be determined. Our observation also parallels the well‐documented association of cAMP‐specific PDE4 with both β1AR and β2AR in AVMs,48 suggesting that both cyclic nucleotides are subjected to tight regulation in space and time. Future use of PDE5 inhibitors in the study of developmental‐ and disease‐related roles that PDEs have in the heart will be a valuable tool to disentangle this important signaling network.
Our data show that inhibition of PDE5, a cGMP‐specific PDE, also enhances adrenergic stimulation of cAMP levels in diabetic cardiomyocytes. This could have been caused by cross‐talk between cAMP and cGMP through the dual substrate‐specific PDEs, PDE3, and/or PDE2. We have previously shown that inhibition of PDE5 leads to a decrease in cAMP in neonatal cardiomyocytes through an activation of PDE2.27 In contrast, PDE3, which is well expressed in cardiomyocytes,49 breaks down both cAMP and cGMP through competitive inhibition.50 While proteins important for cAMP signal management are scaffolded by A‐kinase anchoring proteins, proteins used in cGMP signal management attach to the leucine‐zipper motif in PKG.51 Moreover, in compartments that contain PKA regulatory subunit I, cGMP increases cAMP concentration, which suggests that PDE3 is the predominant phosphodiesterase. In compartments that contain PKA regulatory subunit II, cGMP decreases cAMP, which suggests that PDE2 is the predominant phosphodiesterase in this compartment.52 Therefore, the deliciate association of different PDEs allows for a fine‐tuned interplay between cAMP and cGMP.53, 54
Our data indicate an increase in association between PDE3 and PDE5‐β2AR complex in diabetic cardiomyocytes, supporting that the increase in cGMP caused by the inhibition of PDE5 by sildenafil can inhibit the breakdown of cAMP by PDE3 and leads to an enhancement of isoproterenol‐induced cAMP concentration by sildenafil. However, in cardiomyocytes from mice subjected to transaortic constriction, there are switches of PDE2/3 between β1AR and β2AR. The cAMP from β2AR is sensitive to PDE2 in transaortic constriction myocytes when compared with the sham controls.53 This suggests that β2AR interacts more with PDE2 in a model of cardiac hypertrophy after increased afterload, which is contrary to the results in the current model of diet‐induced hypertrophy. These data indicate again that, dependent on cellular context, PDE5 can switch coupling to different signaling cascades to modulate levels of specific pools of cyclic nucleotides in a given cardiomyocyte/heart. This difference may underlie the reasons for why sildenafil is effective at reducing symptoms of HF in men with diabetes mellitus13, 14 but not in the wider population of patients with general HF.14, 15, 16 Future work to determine how the localization of βARs and PDEs in diabetic cardiomyocytes affects subcellular cyclic nucleotide signaling will be highly informative to understanding diabetic‐related heart disease. In healthy individuals, β2AR is found mostly in the T‐tubule structures of the cardiomyocytes.55 During HF and to a lesser extent during aging, T‐tubules shrink in size,56, 57 with the structural proteins important for T‐tubule maintenance, junctophilin and caveolin, being reduced in expression.57, 58 As this structural change occurs, β2ARs move from the shrinking T‐tubules to the crest of the cell, which can interact with different proteins, leading to changes in signal propagation.55 Meanwhile, β1AR but not β2AR expression is reduced during HF; the ratio of β2AR: β1AR increases. While β2AR signaling does not trigger changes in E‐C coupling in healthy cardiomyocytes, it plays a significant role in modulating E‐C coupling dynamics and arrhythmogenesis in HF.59 Future studies exploring the development‐ and age‐related changes in βAR/phosphodiesterase coupling will be informative in developing more effective treatment strategies, especially for pediatric patients with HF, where there is growing evidence that PDE3 inhibition has varying outcomes based on age.60, 61
In the current study, sildenafil significantly enhanced cGMP signal induced by isoproterenol stimulation in both neonatal and HFD cardiomyocytes. However, sildenafil did not affect cGMP signal induced by isoproterenol stimulation in young adult 8‐week‐old and 6‐month‐old NC cardiomyocytes despite the presence of the β2AR/PDE5 complex. This is juxtaposed to data depicting that addition of sildenafil to isoproterenol causes a decrease in adrenergically stimulated E‐C coupling in 8‐week‐old myocytes while it causes no change in 6‐month‐old NC myocytes. The discrepancy between signaling and contractile function at different ages is currently not understood. Nevertheless, these data underlie the complex and dynamic changes of βAR complex during aging and disease development and indicate that PDE5 may switch association to different PDEs such as PDE3 to modulate specific pools of cyclic nucleotides to affect E‐C coupling in cardiomyocytes. Future study into where these pools are located, what PDEs control them, and how they change with age will be able to identify possible new targets for pharmacological intervention in pediatric HF care.
Of note, despite the increases in cAMP signal in diabetic cardiomyocytes, both CaMKII and PKA are only marginally involved in sildenafil's effect on isoproterenol‐induced cardiomyocyte contractility. Future study into the roles that PKA and CaMKII activity play in diabetic cardiomyopathy may provide insight into changes in E‐C coupling protein expression profile.62 In contrast, PKG is the major downstream kinase involved in sildenafil‐induced enhancement of E‐C coupling in HFD AVMs after isoproterenol stimulation. Our data indicate that the increased PKG activity for phosphorylation of myofilament proteins may contribute to the improvement of E‐C coupling in HFD myocytes, consistent with previous studies in various HF models.63
Conclusions
Our data are consistent with previous studies showing that the benefits of the PDE5 inhibitor are dependent on activation of NO‐cGC‐cGMP‐PKG signaling cascades in diabetic‐related cardiac diseases.22, 23 In those studies, inhibition of PDE5 leads to potentiation of NO‐cGMP signaling cascades and subsequent pleiotropic effects in the myocardium ranging from reduced hypertrophy, altered cytoskeletal gene expression, enhanced myofilament relaxation, improved mitochondrial function, and reduced oxidative stresses, as well as anti‐inflammatory and apoptotic responses during ischemic stress.18, 19, 20, 21 Here, we reveal an additional benefit of PDE5 inhibition in increasing cardiac E‐C coupling. Moreover, we have revealed a novel network linking PDE5 to PDE3, as well as cardiac β2AR, indicating that some of the previously reported beneficial effects of PDE5 inhibition may be the result of modulation by adrenergic stimulation and also dependent on increases in cAMP signal in diabetic hearts. Future study into the link among β2AR, PDE5, and PDE3 may shed light on how cyclic nucleotide levels affect development of diabetic cardiomyopathy. Together, our data provide a novel PDE5‐β2AR axis as a promising treatment target for improving heart function in diabetes mellitus.
Author Contributions
West, Barbagallo, Isidori, and Xiang conceived the experimental design. West, Deng, and Zhang isolated mouse AVMs, bred the needed mice, and cared for mice within the study. West, Wang and, Reddy performed myocyte contraction, calcium cycling, and FRET assays. Xiang, Xu, and West performed coimmunoprecipitations. West performed Western blots, stained histology samples, and performed echocardiogram assays. Chen analyzed contractility and calcium cycling data. Phan performed histological analysis. Xiang provided overall supervision of the study. West and Xiang procured funding for the study and wrote the original article. West and Xiang had full access to all of the data in the study and take responsibility for its integrity and the data analysis.
Sources of Funding
This study was supported by NIH grants R01‐HL127764 and R01‐HL112413 (Xiang), and the US Veteran's Administration Merit grant 01BX002900 (Xiang). West is a recipient of the American Heart Association predoctoral fellowship. Xiang is an established American Heart Association investigator.
Disclosures
None.
Acknowledgments
We would like to thank Mark Jaradeh in Dr Donald M. Bers's laboratory at the University of California, Davis, for isolating rabbit AVMs for our use. We thank Dr Sakthivel Sadayappan from the University of Cincinnati for the generous gift of cMyBP‐C antibodies and Dr Vincent Manganiello (deceased) from NIH for the generous gift of PDE3A antibody.
(J Am Heart Assoc. 2019;8:e012273 DOI: 10.1161/JAHA.119.012273.)
References
- 1. Benjamin EJ, Virani SS, Callaway CW, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O'Flaherty M, Palaniappan LP, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P. Heart Disease and Stroke Statistics‐2018 Update: A Report From the American Heart Association. Circulation. 2018;137:e67–e492. [DOI] [PubMed] [Google Scholar]
- 2. Nassif M, Kosiborod M. Effect of glucose‐lowering therapies on heart failure. Nat Rev Cardiol. 2018;15:282. [DOI] [PubMed] [Google Scholar]
- 3. Lindman BR, Dávila‐Román VG, Mann DL, McNulty S, Semigran MJ, Lewis GD, de las Fuentes L, Joseph SM, Vader J, Hernandez AF, Redfield MM. Cardiovascular phenotype in HFpEF patients with or without diabetes: a RELAX trial ancillary study. 2014;64:541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, Ohman P, Frederich R, Wiviott SD, Hoffman EB, Cavender MA, Udell JA, Desai NR, Mosenzon O, McGuire DK, Ray KK, Leiter LA, Raz I. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317–1326. [DOI] [PubMed] [Google Scholar]
- 5. Monami M, Dicembrini I, Mannucci E. Dipeptidyl peptidase‐4 inhibitors and heart failure: a meta‐analysis of randomized clinical trials. Nutr Metab Cardiovasc Dis. 2014;24:689–697. [DOI] [PubMed] [Google Scholar]
- 6. Simpson SH, Majumdar SR, Tsuyuki RT, Eurich DT, Johnson JA. Dose–response relation between sulfonylurea drugs and mortality in type 2 diabetes mellitus: a population‐based cohort study. Can Med Assoc J. 2006;174:169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aguilar D, Deswal A, Ramasubbu K, Mann DL, Bozkurt B. Comparison of patients with heart failure and preserved left ventricular ejection fraction among those with versus without diabetes mellitus. Am J Cardiol. 2010;105:373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. MacDonald MR, Petrie MC, Varyani F, Östergren J, Michelson EL, Young JB, Solomon SD, Granger CB, Swedberg K, Yusuf S, Pfeffer MA, McMurray JJV; and for the CI . Impact of diabetes on outcomes in patients with low and preserved ejection fraction heart failure: an analysis of the Candesartan in Heart failure: assessment of Reduction in Mortality and morbidity (CHARM) programme. Eur Heart J. 2008;29:1377–1385. [DOI] [PubMed] [Google Scholar]
- 9. Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355:251–259. [DOI] [PubMed] [Google Scholar]
- 10. Eskesen K, Olsen NT, Dimaano VL, Fritz‐Hansen T, Sogaard P, Chakir K, Steenbergen C, Kass D, Abraham TP. Sildenafil treatment attenuates ventricular remodeling in an experimental model of aortic regurgitation. SpringerPlus. 2015;4:592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nagayama T, Hsu S, Zhang M, Koitabashi N, Bedja D, Gabrielson KL, Takimoto E, Kass DA. Sildenafil stops progressive chamber, cellular, and molecular remodeling and improves calcium handling and function in hearts with pre‐existing advanced hypertrophy caused by pressure overload. J Am Coll Cardiol. 2009;53:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Prysyazhna O, Burgoyne J, Scotcher J, Grover S, Kass D, Eaton P. Phosphodiesterase 5 inhibition limits doxorubicin‐induced heart failure by attenuating protein kinase G Iα oxidation. J Biol Chem. 2016;291:17427–17436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Giannetta E, Isidori AM, Galea N, Carbone I, Mandosi E, Vizza CD, Naro F, Morano S, Fedele F, Lenzi A. Chronic Inhibition of cGMP phosphodiesterase 5A improves diabetic cardiomyopathy: a randomized, controlled clinical trial using magnetic resonance imaging with myocardial tagging. Circulation. 2012;125:2323–2333. [DOI] [PubMed] [Google Scholar]
- 14. Santi D, Giannetta E, Isidori AM, Vitale C, Aversa A, Simoni M. Therapy of endocrine disease. Effects of chronic use of phosphodiesterase inhibitors on endothelial markers in type 2 diabetes mellitus: a meta‐analysis. Eur J Endocrinol. 2015;172:R103–R114. [DOI] [PubMed] [Google Scholar]
- 15. Wang H, Anstrom K, Ilkayeva O, Muehlbauer MJ, Bain JR, McNulty S, Newgard CB, Kraus WE, Hernandez A, Felker GM, Redfield M, Shah SH. Sildenafil treatment in heart failure with preserved ejection fraction: targeted metabolomic profiling in the RELAX trial. JAMA Cardiol. 2017;2:896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O'Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E. Effect of phosphodiesterase‐5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309:1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koitabashi N, Aiba T, Hesketh GG, Rowell J, Zhang M, Takimoto E, Tomaselli GF, Kass DA. Cyclic GMP/PKG‐dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation: novel mechanism of cardiac stress modulation by PDE5 inhibition. J Mol Cell Cardiol. 2010;48:713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koka S, Aluri HS, Xi L, Lesnefsky EJ, Kukreja RC. Chronic inhibition of phosphodiesterase 5 with tadalafil attenuates mitochondrial dysfunction in type 2 diabetic hearts: potential role of NO/SIRT1/PGC‐1α signaling. Am J Physiol Heart Circ Physiol. 2014;306:H1558–H1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koka S, Das A, Salloum FN, Kukreja RC. Phosphodiesterase‐5 inhibitor tadalafil attenuates oxidative stress and protects against myocardial ischemia/reperfusion injury in type 2 diabetic mice. Free Radic Biol Med. 2013;60:80–88. [DOI] [PubMed] [Google Scholar]
- 20. Koka S, Xi L, Kukreja RC. Chronic treatment with long acting phosphodiesterase‐5 inhibitor tadalafil alters proteomic changes associated with cytoskeletal rearrangement and redox regulation in Type 2 diabetic hearts. Basic Res Cardiol. 2012;107:249. [DOI] [PubMed] [Google Scholar]
- 21. Varma A, Das A, Hoke NN, Durrant DE, Salloum FN, Kukreja RC. Anti‐inflammatory and cardioprotective effects of tadalafil in diabetic mice. PLoS One. 2012;7:e45243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mátyás C, Németh BT, Oláh A, Török M, Ruppert M, Kellermayer D, Barta BA, Szabó G, Kökény G, Horváth EM, Bódi B, Papp Z, Merkely B, Radovits T. Prevention of the development of heart failure with preserved ejection fraction by the phosphodiesterase‐5A inhibitor vardenafil in rats with type 2 diabetes. Eur J Heart Fail. 2017;19:326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Radovits T, Bömicke T, Kökény G, Arif R, Loganathan S, Kécsán K, Korkmaz S, Barnucz E, Sandner P, Karck M, Szabó G. The phosphodiesterase‐5 inhibitor vardenafil improves cardiovascular dysfunction in experimental diabetes mellitus. Br J Pharmacol. 2009;156:909–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, Jo SH, Danner T, Zhang M, Rainer PP, Bedja D, Kirk JA, Ranek MJ, Dostmann WR, Kwon C, Margulies KB, Van Eyk JE, Paulus WJ, Takimoto E, Kass DA. Phosphodiesterase 9A controls nitric‐oxide‐independent cGMP and hypertrophic heart disease. Nature. 2015;519:472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takimoto E, Belardi D, Tocchetti CG, Vahebi S, Cormaci G, Ketner EA, Moens AL, Champion HC, Kass DA. Compartmentalization of cardiac β‐adrenergic inotropy modulation by phosphodiesterase type 5. Circulation. 2007;115:2159–2167. [DOI] [PubMed] [Google Scholar]
- 26. Lee DI, Vahebi S, Tocchetti CG, Barouch LA, Solaro RJ, Takimoto E, Kass DA. PDE5A suppression of acute β‐adrenergic activation requires modulation of myocyte beta‐3 signaling coupled to PKG‐mediated troponin I phosphorylation. Basic Res Cardiol. 2010;105:337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Isidori AM, Cornacchione M, Barbagallo F, Di Grazia A, Barrios F, Fassina L, Monaco L, Giannetta E, Gianfrilli D, Garofalo S, Zhang X, Chen X, Xiang YK, Lenzi A, Pellegrini M, Naro F. Inhibition of type 5 phosphodiesterase counteracts β2‐adrenergic signalling in beating cardiomyocytes. Cardiovasc Res. 2015;106:408–420. [DOI] [PubMed] [Google Scholar]
- 28. Wang Q, Liu Y, Fu Q, Xu B, Zhang Y, Kim S, Tan R, Barbagallo F, West T, Anderson E, Wei W, Abel ED, Xiang YK. Inhibiting insulin‐mediated beta2‐adrenergic receptor activation prevents diabetes‐associated cardiac dysfunction. Circulation. 2017;135:73–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fu Q, Xu B, Liu Y, Parikh D, Li J, Li Y, Zhang Y, Riehle C, Zhu Y, Rawlings T, Shi Q, Clark RB, Chen X, Abel ED, Xiang YK. Insulin inhibits cardiac contractility by inducing a G(i)‐biased β(2)‐adrenergic signaling in hearts. Diabetes. 2014;63:2676–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fu Q, Xu B, Parikh D, Cervantes D, Xiang YK. Insulin induces IRS2‐dependent and GRK2‐mediated β2AR internalization to attenuate βAR signaling in cardiomyocytes. Cell Signal. 2015;27:707–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao CY, Greenstein JL, Winslow RL. Interaction between phosphodiesterases in the regulation of the cardiac β‐adrenergic pathway. J Mol Cell Cardiol. 2015;88:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qi Y, Xu Z, Zhu Q, Thomas C, Kumar R, Feng H, Dostal DE, White MF, Baker KM, Guo S. Myocardial loss of IRS1 and IRS2 causes heart failure and is controlled by p38α MAPK during insulin resistance. Diabetes. 2013;62:3887–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, Makarewich C, Ai X, Li Y, Tang A, Wang J, Gao H, Wang F, Ge XJ, Kunapuli SP, Zhou L, Zeng C, Xiang KY, Chen X. Cardiotoxic and cardioprotective features of chronic β‐adrenergic signaling. Circ Res. 2013;112:498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pogwizd SM. Nonreentrant mechanisms underlying spontaneous ventricular arrhythmias in a model of nonischemic heart failure in rabbits. Circulation. 1995;92:1034–1048. [DOI] [PubMed] [Google Scholar]
- 35. Zhou YY, Wang SQ, Zhu WZ, Chruscinski A, Kobilka BK, Ziman B, Wang S, Lakatta EG, Cheng H, Xiao RP. Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology. Am J Physiol Heart Circ Physiol. 2000;279:H429–H436. [DOI] [PubMed] [Google Scholar]
- 36. Russwurm M, Mullershausen F, Friebe A, Jäger R, Russwurm C, Koesling D. Design of fluorescence resonance energy transfer (FRET)‐based cGMP indicators: a systematic approach. Biochem J. 2007;407:69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Luo J, Deng ZL, Luo X, Tang N, Song WX, Chen J, Sharff KA, Luu HH, Haydon RC, Kinzler KW, Vogelstein B, He TC. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc. 2007;2:1236. [DOI] [PubMed] [Google Scholar]
- 38. Beca S, Ahmad F, Shen W, Liu J, Makary S, Polidovitch N, Sun J, Hockman S, Chung YW, Movsesian M, Murphy E, Manganiello V, Backx PH. Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a signaling complexes in mouse heart. Circ Res. 2013;112:289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sadayappan S, Gulick J, Osinska H, Barefield D, Cuello F, Avkiran M, Lasko VM, Lorenz JN, Maillet M, Martin JL, Brown JH, Bers DM, Molkentin JD, James J, Robbins J. A critical function for Ser‐282 in cardiac myosin binding protein‐C phosphorylation and cardiac functionnovelty and significance. Circ Res. 2011;109:141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pereira L, Cheng H, Lao DH, Na L, van Oort RJ, Brown JH, Wehrens XH, Chen J, Bers DM. Epac2 mediates cardiac β1‐adrenergic dependent SR Ca(2+) leak and arrhythmia. Circulation. 2013;127:913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reddy GR, West TM, Jian Z, Jaradeh M, Shi Q, Wang Y, Chen‐Izu Y, Xiang YK. Illuminating cell signaling with genetically encoded FRET biosensors in adult mouse cardiomyocytes. J Gen Physiol. 2018;150:1567–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Allen MD, Zhang J. Subcellular dynamics of protein kinase A activity visualized by FRET‐based reporters. Biochem Biophys Res Commun. 2006;348:716–721. [DOI] [PubMed] [Google Scholar]
- 43. Klarenbeek J, Goedhart J, van Batenburg A, Groenewald D, Jalink K. Fourth‐generation Epac‐based FRET sensors for cAMP feature exceptional brightness, photostability and dynamic range: characterization of dedicated sensors for FLIM, for ratiometry and with high affinity. PLoS One. 2015;10:e0122513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takao K, Okamoto K‐I, Nakagawa T, Neve RL, Nagai T, Miyawaki A, Hashikawa T, Kobayashi S, Hayashi Y. Visualization of synaptic Ca2+/calmodulin‐dependent protein kinase II activity in living neurons. J Neurosci. 2005;25:3107–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. DiPilato LM, Zhang J. The role of membrane microdomains in shaping [small beta]2‐adrenergic receptor‐mediated cAMP dynamics. Mol BioSyst. 2009;5:832–837. [DOI] [PubMed] [Google Scholar]
- 46. Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Seidman CE, Seidman JG, Robbins J. Cardiac myosin binding protein c phosphorylation is cardioprotective. Proc Natl Acad Sci USA. 2006;103:16918–16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fu Q, Shi Q, West TM, Xiang YK. Cross‐talk between insulin signaling and G protein–coupled receptors. J Cardiovasc Pharmacol. 2017;70:74–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Richter W, Day P, Agrawal R, Bruss MD, Granier S, Wang YL, Rasmussen SGF, Horner K, Wang P, Lei T, Patterson AJ, Kobilka B, Conti M. Signaling from β1‐ and β2‐adrenergic receptors is defined by differential interactions with PDE4. EMBO J. 2008;27:384–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ding B, Abe JI, Wei H, Huang Q, Walsh RA, Molina CA, Zhao A, Sadoshima J, Blaxall BC, Berk BC, Yan C. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: implication in heart failure. Circulation. 2005;111:2469–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Degerman E, Belfrage P, Manganiello VC. Structure, localization, and regulation of cGMP‐inhibited phosphodiesterase (PDE3). J Biol Chem. 1997;272:6823–6826. [DOI] [PubMed] [Google Scholar]
- 51. Kokkonen K, Kass DA. Nanodomain regulation of cardiac cyclic nucleotide signaling by phosphodiesterases. Annu Rev Pharmacol Toxicol. 2017;57:455–479. [DOI] [PubMed] [Google Scholar]
- 52. Stangherlin A, Gesellchen F, Zoccarato A, Terrin A, Fields LA, Berrera M, Surdo NC, Craig MA, Smith G, Hamilton G, Zaccolo M. cGMP signals modulate cAMP levels in a compartment‐specific manner to regulate catecholamine‐dependent signaling in cardiac myocytes. Circ Res. 2011;108:929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Perera RK, Sprenger JU, Steinbrecher JH, Hubscher D, Lehnart SE, Abesser M, Schuh K, El‐Armouche A, Nikolaev VO. Microdomain switch of cGMP‐regulated phosphodiesterases leads to ANP‐induced augmentation of beta‐adrenoceptor‐stimulated contractility in early cardiac hypertrophy. Circ Res. 2015;116:1304–1311. [DOI] [PubMed] [Google Scholar]
- 54. Barbagallo F, Xu B, Reddy GR, West T, Wang Q, Fu Q, Li M, Shi Q, Ginsburg KS, Ferrier W, Isidori AM, Naro F, Patel HH, Bossuyt J, Bers D, Xiang YK. Genetically encoded biosensors reveal PKA hyperphosphorylation on the myofilaments in rabbit heart failure. Circ Res. 2016;119:931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, Lohse MJ, Korchev YE, Harding SE, Gorelik J. β2‐adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 2010;327:1653–1657. [DOI] [PubMed] [Google Scholar]
- 56. Guo A, Zhang C, Wei S, Chen B, Song LS. Emerging mechanisms of T‐tubule remodelling in heart failure. Cardiovasc Res. 2013;98:204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kong CHT, Bryant SM, Watson JJ, Gadeberg HC, Roth DM, Patel HH, Cannell MB, Orchard CH, James AF. The effects of aging on the regulation of T‐tubular ICa by caveolin in mouse ventricular myocytes. J Gerontol A Biol Sci Med Sci. 2018;73:711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Minamisawa S, Oshikawa J, Takeshima H, Hoshijima M, Wang Y, Chien KR, Ishikawa Y, Matsuoka R. Junctophilin type 2 is associated with caveolin‐3 and is down‐regulated in the hypertrophic and dilated cardiomyopathies. Biochem Biophys Res Commun. 2004;325:852–856. [DOI] [PubMed] [Google Scholar]
- 59. DeSantiago J, Ai X, Islam M, Acuna G, Ziolo MT, Bers DM, Pogwizd SM. Arrhythmogenic effects of β(2)‐adrenergic stimulation in the failing heart are due to enhanced SR Ca load. Circ Res. 2008;102:1389–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Price JF, Towbin JA, Dreyer WJ, Moffett BS, Kertesz NJ, Clunie SK, Denfield SW. Outpatient continuous parenteral inotropic therapy as bridge to transplantation in children with advanced heart failure. J Card Fail. 2006;12:139–143. [DOI] [PubMed] [Google Scholar]
- 61. Berg AM, Snell L, Mahle WT. Home inotropic therapy in children. J Heart Lung Transplant. 2007;26:453–457. [DOI] [PubMed] [Google Scholar]
- 62. Drews O, Tsukamoto O, Liem D, Streicher J, Wang Y, Ping P. Differential regulation of proteasome function in isoproterenol‐induced cardiac hypertrophy. Circ Res. 2010;107:1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rainer PP, Kass DA. Old dog, new tricks: novel cardiac targets and stress regulation by protein kinase G. Cardiovasc Res. 2016;111:154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]