

Abstract
Background
The contribution of glucocorticoids to sexual dimorphism in the heart is essentially unknown. Therefore, we sought to determine the sexually dimorphic actions of glucocorticoid signaling in cardiac function and gene expression. To accomplish this goal, we conducted studies on mice lacking glucocorticoid receptors (GR) in cardiomyocytes (cardioGRKO mouse model).
Methods and Results
Deletion of cardiomyocyte GR leads to an increase in mortality because of the development of spontaneous cardiac pathology in both male and female mice; however, females are more resistant to GR signaling inactivation in the heart. Male cardioGRKO mice had a median survival age of 6 months. In contrast, females had a median survival age of 10 months. Transthoracic echocardiography data showed phenotypic differences between male and female cardioGRKO hearts. By 3 months of age, male cardioGRKO mice exhibited left ventricular systolic dysfunction. Conversely, no significant functional deficits were observed in female cardioGRKO mice at the same time point. Functional sensitivity of male hearts to the loss of cardiomyocyte GR was reversed following gonadectomy. RNA‐Seq analysis showed that deleting GR in the male hearts leads to a more profound dysregulation in the expression of genes implicated in heart rate regulation (calcium handling). In agreement with these gene expression data, cardiomyocytes isolated from male cardioGRKO hearts displayed altered intracellular calcium responses. In contrast, female GR‐deficient cardiomyocytes presented a response comparable with controls.
Conclusions
These data suggest that GR regulates calcium responses in a sex‐biased manner, leading to sexually distinct responses to stress in male and female mice hearts, which may contribute to sex differences in heart disease, including the development of ventricular arrhythmias that contribute to heart failure and sudden death.
Keywords: cardiomyocyte, glucocorticoid, glucocorticoid receptor, heart failure, sex hormones
Subject Categories: Cardiomyopathy, Heart Failure, Basic Science Research
Clinical Perspective
What Is New?
Stress is an emerging risk factor for heart disease, yet the sex‐specific effects of stress on the heart are unknown. Using mice lacking the glucocorticoid receptor (GR) in cardiomyocytes, we investigate the contribution of GR signaling to maintain cardiac homeostasis in a sex‐specific manner.
Our findings showed that ablation of GR signaling in the heart leads to sex‐specific changes in cardiac gene expression and function.
We found that GR plays sex‐specific effects in preserving intracellular calcium handling in ventricular cardiomyocytes, and the mechanisms underlying these effects may result from cardiac GR signaling crosstalk with sex hormones signaling.
What Are the Clinical Implications?
Stress exerts direct effects on the heart, and these effects are sex‐specific.
Sex‐specific manipulation of the GR signaling in the heart may provide a therapeutic approach to improve cardiomyocyte intracellular calcium handling, which may improve myocardial contractility after a heart attack.
Introduction
Differences in gene expression profiles in the cardiovascular system are well documented in males and females.1 Although these distinct patterns of gene expression between sexes have been associated with sex chromosomes and sex hormones,2, 3 recent studies using genome‐wide expression profiling show that interactions between sex hormones and non‐sex hormone pathways, such as glucocorticoid receptor (GR, NR3C1) signaling, are significant contributors to the biological differences between males and females in health and disease.4, 5
Cardiovascular disease is a leading cause of mortality worldwide. Several risk factors contribute to cardiovascular disease and its complications. Exposure to chronic stress is increasingly recognized as an important risk factor for heart disease. Women appear to be more susceptible to the deleterious effects of stress as it relates to cardiac health.6 However, the molecular pathways underlying the direct and sex‐specific effects of stress on the heart are unknown.
Glucocorticoids are primary stress‐induced hormones that function as potent anti‐inflammatory and immune regulators.5, 7 In addition to the actions of these hormones on the immune system, glucocorticoids also have significant effects on the regulation of embryonic development, intermediate metabolism, central nervous system, growth, reproduction, and the cardiovascular system.8, 9 Glucocorticoids exert their physiological effects by binding the GR, a member of the nuclear receptor superfamily of ligand‐activated transcription factors.10, 11, 12 Studies have demonstrated that glucocorticoid signaling via GR leads to the regulation of intricate gene networks in a tissue‐specific manner.12 While sex differences in stress reactivity and gene expression have been found in the immune system and metabolism,2, 4, 5 sex‐specific effects of glucocorticoids on cardiovascular function have been largely unexplored. Recent studies have demonstrated for the first time that glucocorticoids have direct effects on the heart and that intact glucocorticoid signaling in cardiomyocytes is critical in normal physiology.13 Using a mouse model lacking GR in cardiomyocytes (cardiomyocyte‐specific GR knockout, cardioGRKO), Oakley et al showed that male cardioGRKO mice exhibit significantly compromised heart function characterized by left ventricular systolic dysfunction and dysregulation of genes involved in cardiovascular disease.13 To further expand on the role of GR signaling in cardiac health and to investigate the influence of sex on GR regulation of cardiac gene expression and function, we performed comparative studies on male and female cardioGRKO mice. Our studies revealed that female cardioGRKO mice are partially protected from the deleterious effects of GR ablation in cardiomyocytes. Female cardioGRKO mice present with a longer lifespan and more preserved cardiac function than their male counterparts. Consistent with the differences in phenotype, hearts from cardioGRKO males displayed a more pronounced dysregulation in the expression of genes implicated in heart rate regulation, calcium handling, and cardiomyocyte structure and viability, whereas female cardioGRKO hearts displayed no significant differences in the expression of these target genes at the same time point. The ryanodine receptor 2 (Ryr2) was among the dysregulated genes in male knockout hearts implicated in calcium handling. Normal Ryr2 expression and function are critical for cardiomyocyte calcium‐induced calcium release.14 Decreased Ryr2 in the male hearts resulted in a compromised intracellular calcium response in male GR‐deficient cardiomyocytes. The differential sensitivity of the male cardioGRKO hearts to pathology was reversed following gonadectomy. These data suggest that cardiac GR signaling crosstalk with hormones produced by the gonads might contribute to the differences in heart phenotype and gene expression, and may explain in part the mechanisms whereby stress hormones affect cardiac health in a sex‐specific manner.
Methods
Data Availability Disclosure Statement
The authors declare that all supporting data and method descriptions are available within the article or from the corresponding author upon reasonable request.
Animals
The cardioGRKO mice were generated by standard gene‐targeting procedures as previously described.13, 15 Briefly, the GR (Nr3c1) locus was modified by inserting loxP sites upstream of exon 3 and downstream of exon 4. Mice carrying the modified GR allele were then derived by blastocyst (albino B6) injection. Homozygous GR floxed (GRloxP,loxP) mice were then mated with mice expressing Cre recombinase under the direction of cardiomyocyte‐specific αMHCCre/+. The resulting offspring were GRloxP/loxPαMHCCre/+ mice (designated cardioGRKO) and Cre‐negative GRloxP/loxPαMHC+/+ (designated GR fl/fl or GR flox/flox) littermate mice that served as controls. Genotype was determined by polymerase chain reaction using DNA from tails as previously described.13 All mice were on a C57BL/6N/J background. Male and female cardioGRKO were born at the predicted Mendelian ratio. Castration and ovariectomy were performed in both GR floxed and cardioGRKO mice at 6 weeks of age. All experiments were approved and performed according to the guidelines of the Animal Care and Use Committee at the National Institute of Environmental Health Sciences, National Institutes of Health.
Echocardiographic Analysis
Transthoracic echocardiography was performed on conscious mice with a VisualSonics Vevo 770 ultrasound biomicroscopy system (VisualSonics, Inc) with a 30‐MHx 707B scan head as previously described.13, 16 Two‐dimensional guided M‐mode analysis of the left ventricle was performed on 3‐month old GR loxP/loxP (controls) and GRloxP/loxPα‐MHCCre/+ (cardioGRKO) male and female mice. Anterior wall thickness, posterior wall thickness, and left ventricular (LV) internal diameters (LV internal diameter at diastole [LVID;d] and LV internal diameter at systole [LVID;s]) were measured as previously described by Oakley et al.13 LV systolic function was assessed by fractional shortening (FS) and ejection fraction (EF), calculated from the equation FS%=(LVID;d−LVID;s)/LVID;d×100 and EF%=(LV vol;d−LV Vol;s)/LV Vol;d×100. M‐mode measurements represent 3 average consecutive cardiac cycles from each mouse.
Histological Analysis and Immunofluorescence
Hearts from control and cardioGRKO mice were perfused with PBS and fixed with 4% paraformaldehyde. Samples were processed, embedded in paraffin, cut in 5 μm sections, and stained with hematoxylin and eosin. Pictures were acquired using a Nikon Eclipse TE300 inverted microscope. For the immunofluorescence, Citrate buffer (Biocare Medical, CA) was used for antigen retrieval and blocked with 5% normal goat serum in 0.2% triton/PBS. After blocking, samples were incubated overnight with rabbit anti‐GR antibody (Cell Signaling, MA) and mouse anti‐troponin I antibody (Millipore, MA) followed by 2‐hour incubation with a goat anti‐rabbit IgG antibody, Alexa Fluor 594 (red) and a goat anti‐mouse IgG, Alexa Fluor 488 (green) from ThermoFisher Scientific. Nuclei was stained with DAPI. Images were obtained on a Leica TCS SP5 Spectral Confocal Microscope equipped with a ×40 (oil) objective.
Quantitative Real‐Time Polymerase Chain Reaction QTR‐PCR
Total RNA was isolated from the whole hearts of control and cardioGRKO mice with the RNeasy Mini kit and RNase‐Free DNase kit (Qiagen) according to the manufacturer's instructions. mRNA levels were measured with a CFX96 Real‐Time System C1000 Touch Thermal Cycler (Bio‐Rad). Predesigned primer/probe sets for Nr3c1, Acta1 (skeletal muscle α‐actin [Ska]), Myh7 (β‐myosin heavy chain [βMhc]), Nppb (brain natriuretic peptide [Bnp]), Atp2a2 (sarco(endo)plasmic reticulum calcium transport ATPase 2 [Serca2]), Ryr2 (ryanodine receptor 2), Slc25a4 (solute carrier family 25 member 4 [Ant‐1, Adenine Nucleotide Translocator]), Kcnq1 (potassium voltage‐gated channel subfamily Q member 1), and ppib (cyclophilin B, peptidylprolyl isomerase B) were obtained from ThermoFisher Scientific. Values measured for each primer/probe set were normalized to Ppib.
Immunoblots
Heart tissue and cells were homogenized and lysed in Tris‐Glycine SDS sample buffer (Invitrogen, CA) supplemented with 2.5% β‐mercaptoethanol as previously described.17 Polyvinylidene difluoride (PVDF) membranes blotted with equal amounts of protein were incubated with primary antibodies the rabbit anti‐GR antibody (Cell Signaling, MA) and the rabbit anti‐Ryr2 anntibody (Abcam, MA) and developed using the ChemiDoc Imaging System (BioRad, CA). GR and Ryr2 protein levels were quantified by densitometry using National Institutes of Health ImageJ analysis software.
RNA‐Seq Analysis
Gene expression analyses were performed on RNA from the hearts of male and female control and cardioGRKO mice at 3 months of age. All RNA‐Seq libraries (non‐strand‐specific, paired end) were prepared with the TruSeq RNA Sample Prep kit (Illumina, San Diego, CA). The total RNA samples were subject to poly(A) enrichment as part of the TruSeq protocol. mRNA samples were sequenced using the paired‐end 75 bp protocol and the NextSeq 500 platform (Illumina) per the manufacturer's protocol. The RNA‐seq data will be available in the Gene Expression Omnibus repository at the National Center for Biotechnology Information (enter token slsjamwgzzathib into the box GSE120573: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120573). Raw reads (69–105 million reads per sample) were multiplexed to each sample according to their barcode information.
Trim_galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) was used to remove or trim adaptor‐containing reads and low‐quality reads, and the minimum length of trimmed reads was set at 20 bases. Remaining reads were aligned to the UCSC mm10 reference genome using TopHat2. The quantification results from htseq‐count were then analyzed with the Bioconductor package DESeq2, which fits a negative binomial distribution to estimate technical and biological variability. We made comparisons for knockout versus wild type across male and female samples. A gene was considered differentially expressed if the P‐value for differential expression was <0.01. The heatmaps were made using Partek Genomic Suite (Version 6.6).
Functional Analysis
The Pathway and Network analysis of the significant genes was conducted in the Ingenuity Pathway Analysis (IPA) tool (version 27821452, Ingenuity Systems, Redwood City, CA). To identify GR and mineralocorticoid receptor (MR) target genes, Nr3c1 and Nr3c2 (gene names for GR and MR, respectively) were entered in the search option of IPA, and gene networks were created using our lists of significant genes. Direct and indirect connections between genes and GR or MR were visualized using the pathway function of IPA. GR and MR target genes were further analyzed based on their reported role in cardiovascular disease by overlaying the GR and MR connected genes to the Diseases and Functions option. Venn Diagrams for all our analyses were generated using the Compare function of IPA.
Cardiomyocyte Isolation
Adult cardiomyocytes from control and cardioGRKO mice were isolated using a commercially available kit (Perfusion Adumyts Cardiomyocyte Isolation Kit, Cellutron, Baltimore, MD).
Calcium Measurements on Isolated Cardiomyocytes
Intracellular calcium measurements were performed as previously described by Oakley et al.13 Briefly, fresh isolated cardiomyocytes were incubated with a calcium sensitive indicator, fluo‐4 (Invitrogen). Following isolation, cardiomyocytes were allowed to attach to Laminin‐coated glass bottom wells in complete AW medium (Cellutron, Baltimore, MD) for at least 1 hour. Cells were incubated in serum‐free AW medium containing 1 μmol/L fluo‐4 for 15 minutes at room temperature in the dark. Cells were washed 3 times then incubated for 15 minutes at room temperature in a HEPES‐buffered Tyrode salt solution (135 mmol/L NaCl; 4 mmol/L KCl; 1.0 mmol/L MgCl2; 20 mmol/L HEPES; 1.0 mmol/L CaCl2 and 10 mmol/L glucose, with pH 7.4 adjusted by NaOH). Calcium measurements were performed on a Nikon Eclipse Ti‐E inverted confocal microscope equipped with 20× 0.75 NA objective. Fluo‐4 fluorescence was monitored by exciting the indicator at 488 nm, and collecting the emission wavelength at 500 to 630 nm. Data were collected in a Galvano frame mode at 2 frames per second. The change in fluo‐4 fluorescence emission intensity represents the change in intracellular calcium. Regions of interest were selected with cardiomyocytes displaying spontaneous intracellular calcium changes in the form of local calcium transients. Intracellular calcium changes were monitored at the single cell level. For each experiment 4 regions of interest were monitored with a total of at least 20 cells. A caffeine solution was diluted over the samples for stimulation of intracellular calcium signals. Cells were monitored at baseline and final concentrations of caffeine bathing the cells at 0.25 and 10 mmol/L.
Statistical Analysis
Statistics were performed with the GraphPad Prism software v6 (GraphPad Software, Inc). A Kaplan–Meier analysis was performed after birth to evaluate differences in survival for male and female control and cardioGRKO mice. Data are from control mice (27 males, 28 females) and cardioGRKO mice (24 males, 23 females). A Kruskal–Wallis test with Dunn's multiple comparisons analysis (males versus females and GR flox/flox versus CardioGRKO) was used to evaluate whether differences in echocardiogram measurements and gene expression data between groups were statistically significant. Differences were considered to be statistically significant when P<0.05. For all studies we used 3 to 8 mice per group.
Results
Inactivation of Cardiomyocyte GR Leads to an Increased Mortality Rate in Male Mice as Compared With Their Female Counterparts
Kaplan–Meier survival curve analysis shows that both male and female cardioGRKO mice have a reduced lifespan as compared with their littermate controls13; however, the effects of cardiomyocyte GR inactivation are more profound in males than in females (Figure 1). Male cardioGRKO had a median survival age of 6 months (Figure 1). In contrast, females had a median survival age of 10 months (Figure 1). These results suggest that inactivation of cardiomyocyte GR in vivo exhibits sexual dimorphism (Figure 1) and that deletion of cardiomyocyte GR leads to more profound effects on males than in females.
Figure 1.
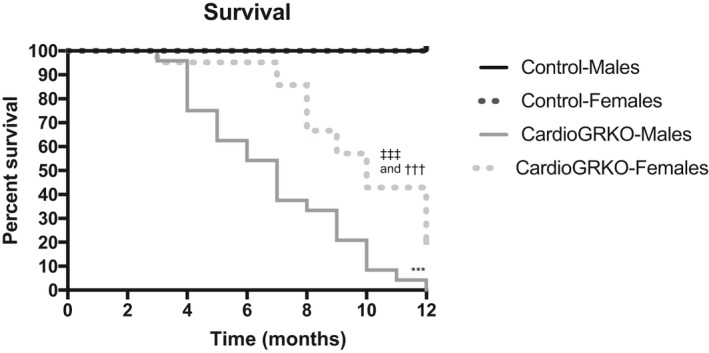
Ablation of the glucocorticoid receptor in cardiomyocytes leads to an increased mortality rate in male cardioGRKO mice as compared with their female counterparts. Kaplan–Meier analysis of survival after birth was performed for male and female control and cardioGRKO mice. Shown are survival male and female controls and cardioGRKO. Data are from control mice (27 males, 28 females) and cardioGRKO mice (24 males, 23 females). Male controls vs male cardioGRKO. cardioGRKO indicates cardiomyocyte‐specific glucocorticoid receptor knockout. ***P<0.001; female controls vs female cardioGRKO ††† P<0.001; male cardioGRKO vs female cardioGRKO ‡‡‡ P<0.001.
Male and Female cardioGRKO Mice Develop Left Ventricular Systolic Dysfunction That Progresses to Dilated Cardiomyopathy
Conscious transthoracic echocardiography was performed on male and female cardioGRKO at 3 and 6 months of age. Transthoracic echocardiography data showed a distinct cardiac phenotype between male and female cardioGRKO mice (Table 1). By 3 months of age, male knockout mice exhibited LV systolic dysfunction, as evidenced by significant decreases in ejection fraction (EF, 70.74±8.51%) and fractional shortening (FS, 37.44±5.90%) (Table 1). Although female cardioGRKO also presented decreases in EF (74.98±6.60%) and FS (42.93±8.84%), these changes were not statistically significant as compared with their littermate controls. Despite presenting a more preserved cardiac function at the 3‐month time point, cardioGRKO female mice present a similar degree of LV systolic dysfunction as their male counterparts at 6 months of age. Both male and female cardioGRKO mice have a significant decrease in EF (62.23±14.41 males versus 60.93±10.05 females) and FS (33.73±9.68 males versus 32.34±6.78 females). These functional data are in agreement with the pathology findings that show that cardioGRKO female mice, much like their male counterparts, have significantly enlarged hearts at 6 months of age (Figure 2A), which is consistent with the phenotypical characterization previously reported by Oakley et al.13 Histological evaluation of hematoxylin and eosin stained sections from 6 months old male and female cardioGRKO hearts showed a significant dilation of the left ventricle (consistent with dilated cardiomyopathy) and atrial thrombosis in some of the hearts which may be associated with atrial blood stasis (a risk factor for atrial fibrillation)18 (Figure 2B). Therefore, despite the preserved systolic function exhibited by female GR deficient hearts at 3 months of age, by 6 months of age female cardioGRKO mice exhibited a similar cardiac pathology as compared with their male counterparts (Table 2).
Table 1.
Echocardiogram Data on 3‐Month‐Old Male and Female GRflox and CardioGRKO
| Males | Females | |||
|---|---|---|---|---|
| Controls (n=5) | CardioGRKO (n=5) | Controls (n=5) | CardioGRKO (n=6) | |
| IVS;d, mm | 1.13±0.23 | 0.94±0.11 | 1.21±0.17 | 0.96±0.18 |
| LVID;d, mm | 2.73±0.32 | 2.89±0.51 | 2.56±0.57 | 2.88±0.29 |
| LVPW;d, mm | 1.13±0.22 | 1.18±0.28 | 1.12±0.45 | 0.95±0.19 |
| IVS;s, mm | 1.48±0.22 | 1.32±0.20 | 1.42±0.04 | 1.35±0.17 |
| LVID;s, mm | 1.42±0.18 | 2.14±0.42* | 1.40±0.43 | 1.73±0.40 |
| LVPW;s, mm | 1.47±0.28 | 1.27±0.12 | 1.47±0.41 | 1.24±0.19 |
| EF, % | 83.25±3.24 | 70.73±8.51* | 78.91±7.62 | 74.98±6.60 |
| FS, % | 48.66±3.10 | 37.44±5.90* | 46.33±6.37 | 42.93±8.84 |
| LV mass, mg | 89.63±25.75 | 99.96±19.50 | 82.73±8.86 | 82.26±11.17 |
| HW | 131.66±0.95 | 173.8±3.48†, ∥ | 137.44±1.80 | 144.66±9.09†, ¶ |
| BW, g | 31.84±0.74 | 32.70±0.72 | 31.05±1.11 | 28.38±1.33 |
| HW/BW | 4.13±0.10 | 5.29±0.81†, § | 4.43±0.15 | 5.10±0.33† |
| LV mass/BW | 2.82±0.85 | 3.05±0.65 | 2.66±0.29 | 2.89±0.34 |
| HR, bpm | 716.20±19.20 | 681.40±30.22 | 707.80±2.4 | 700.8±25.78 |
Echocardiographic measurements from transthoracic M‐mode tracings on conscious male and female. Data are mean±SD. Two‐way ANOVA with post hoc Tukey test multiple comparisons analysis (male controls vs female controls, male controls vs male cardioGRKO, male controls vs female cardioGRKO, female controls vs male cardioGRKO, female controls vs female cardioGRKO, and male cardioGRKO vs female cardioGRKO) were used to evaluate whether differences between groups were statistically significant. Values without asterisks were not significantly different. bpm indicates beats per min; HW, heart weight; BW, body weight; EF, ejection fraction; FS, fractional shortening; HR, heart rate; IVS;d, interventricular septal thickness in diastole; IVS;s, interventricular septal thickness in systole; LV, left ventricular; LVID;d, LV end‐diastolic dimension; LVID;s, LV end‐systolic dimension; LVPW;d, posterior wall thickness in diastole; LVPW;s, posterior wall thickness in systole.
*P<0.05 and † P<0.01 vs control males.
§ P<0.05, ∥ P<0.01 vs control females.
P<0.001 vs cardioGRKO males.
Figure 2.
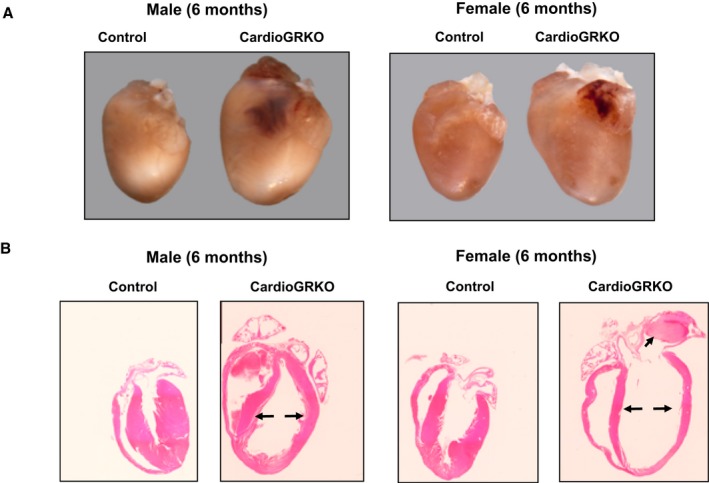
CardioGRKO male and female mice develop spontaneous left ventricular remodeling. A, Representative images of intact hearts from 6‐month‐old male and female control and cardioGRKO mice. B, Longitudinal hematoxylin & eosin‐stained heart sections from 6‐month‐old male and female control and cardioGRKO mice. Arrows (→) point out to increased left ventricle size and the presence of a thrombus in the left atria of the female cardioGRKO heart. The images are representative of 5 mice per group. CardioGRKO indicates cardiomyocyte‐specific glucocorticoid receptor knockout.
Table 2.
Echocardiogram Data on 6‐Month‐Old Male and Female GRflox/flox and CardioGRKO Mice
| Males | Females | |||
|---|---|---|---|---|
| Controls (n=5) | CardioGRKO (n=5) | Controls (n=3) | CardioGRKO (n=4) | |
| IVS;d, mm | 1.01±0.07 | 1.02±0.15 | 0.96±0.04 | 0.88±0.21* |
| LVID;d, mm | 3.06±0.27 | 3.87±0.74 | 3.03±0.05 | 3.77±0.79 |
| LVPW;d, mm | 1.11±0.06 | 0.98±0.06 | 1.04±0.07 | 0.87±0.17 |
| IVS;s, mm | 1.65±0.08 | 1.58±0.37 | 1.42±0.02 | 1.11±0.43* |
| LVID;s, mm | 1.44±0.10 | 2.61±0.86*, § | 1.28±0.09 | 2.59±0.78§ |
| LVPW;s, mm | 1.62±0.16 | 1.29±0.15 | 1.41±0.01 | 1.24±0.44 |
| EF, % | 84.91±2.29 | 62.23±14.41*, § | 88.33±1.54 | 60.93±10.05†, ∥ |
| FS, % | 52.71±2.90 | 33.73±9.68†, ∥ | 57.58±2.26 | 32.34±6.78†, ¶ |
| LV mass, mg | 94.47±14.36 | 122.38±23.69 | 85.30±4.46 | 94.69±9.62§ |
| BW, g | 34.4±1.28 | 37.44±2.66 | 33.23±0.74 | 36.32±2.32 |
| LV mass/BW | 2.75±0.48 | 3.29±0.75 | 2.56±0.13 | 2.61±0.33 |
| HR, bpm | 722.66±19.97 | 670.69±35.69 | 650.50±28.00 | 642.25±52.69 |
Echocardiographic measurements from transthoracic M‐mode tracings on conscious male and female. Data are mean±SD. Two‐way ANOVA with multiple comparisons analysis (male controls vs female controls, male controls vs male cardioGRKO, male controls vs female cardioGRKO, female controls vs male cardioGRKO, female controls vs female cardioGRKO, and male cardioGRKO vs female cardioGRKO) were used to evaluate whether differences between groups were statistically significant. Values without asterisks were not significantly different. bpm indicates beats per min; BW, body weight; EF, ejection fraction; FS, fractional shortening; HR, heart rate; IVS;d, interventricular septal thickness in diastole; IVS;s, interventricular septal thickness in systole; LV, left ventricular; LVID;d, LV end‐diastolic dimension; LVID;s, LV end‐systolic dimension; LVPW;d, posterior wall thickness in diastole; LVPW;s, posterior wall thickness in systole.
*P<0.05 vs male control mice.
† P<0.05 vs male cardioGRKO.
§ P<0.05, ∥ P<0.01, and ¶ P<0.001 vs control females.
Male But Not Female CardioGRKO Exhibit Increased Expression of Fetal Genes Associated With Pathological Cardiac Hypertrophy
Male and female cardioGRKO mice displayed a similar reduction (≈70%) in GR mRNA levels as compared with their male knockout counterparts (Figure 3A). In agreement with decreased GR mRNA levels, GR levels in the male and female knockouts are not significantly different (Figure 3B). The remaining GR is attributed to endothelial cells, cardiac fibroblasts, and smooth muscle cells. As shown in Figure 3C, GR was not detected in cardiomyocytes of 6 months old male and female cardioGRKO hearts. The figure also shows a decrease in the intensity of the staining for cardiac troponin I in the hearts of both male and female cardioGRKO. These changes in troponin I levels may be associated with the structural changes in the myocardium related to the progression of pathological cardiac hypertrophy in cardioGRKO mice. Decreased troponin I levels were found in all the cardioGRKO hearts (n=3 per sex, genotype).
Figure 3.
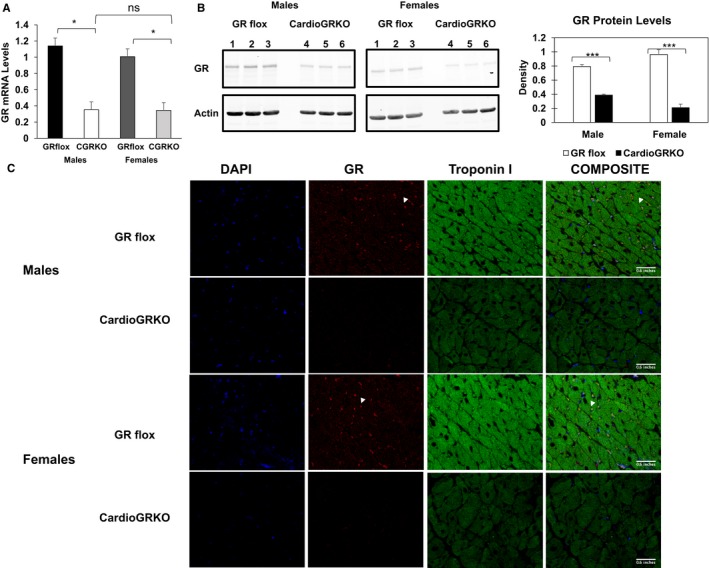
GR mRNA and protein are significantly reduced in the hearts of cardiomyocyte‐specific glucocorticoid receptor knockout (cardioGRKO) mice and no glucocorticoid receptor (GR) is detected in the cardiomyocytes of the knockout mice. A, RT‐polymerase chain reaction of GR mRNA from hearts of control and cardioGRKO mice. B, Representative immunoblot show GR levels in control (GR flox) and knockout hearts. Hearts from male cardioGRKO have reduction of ≈70% in GR total protein levels, whereas female hearts present a reduction of ≈80% in GR protein levels. Immunoblots were quantified by densitometry using National Institutes of Health ImageJ analysis software. Data are mean±SEM (n=5 mice per group). *P<0.05; ***P<0.0001. C, Representative immunofluorescence staining of heart sections from control and cardioGRKO mice with anti‐troponin I (green) and anti‐GR (red) antibodies. DAPI is shown in blue. Data represent n=3 mice per sex and genotype. All pictures were acquired on a Leica TCS SP5 Spectral Confocal Microscope equipped with a ×40 (oil) objective. CardioGRKO indicates cardiomyocyte‐specific glucocorticoid receptor knockout; CGRKO, cardioGRKO; GR, glucocorticoid receptor.
Male cardioGRKO hearts displayed increased expression of β‐myosin heavy chain (β‐Mhc) and skeletal muscle α‐actin (Ska) at 3 months of age that reached 2.5‐ and 5‐fold, respectively, over control mice (Figure 4A and 4B). The expression of brain natriuretic peptide (Nppb), another marker of hypertrophy, was also dysregulated in male GR‐deficient hearts (Figure 4C). In contrast, no significant changes in the expression of these hypertrophic markers were found in female cardioGRKO hearts at this time point (Figure 4A through 4C). Therefore, these data suggest that deletion of cardiomyocyte GR leads to reactivation of the fetal cardiac gene program and pathological cardiac hypertrophy in a sex‐specific manner.
Figure 4.
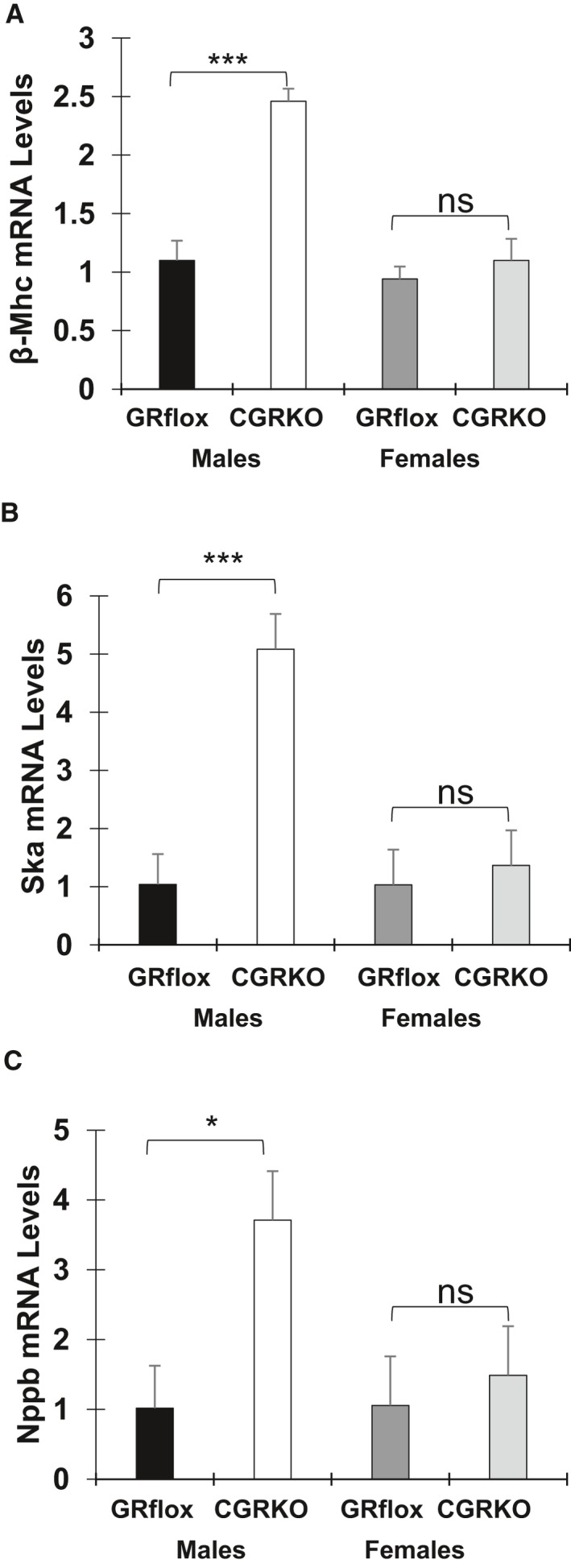
mRNA levels of genes associated with cardiac pathology and the glucocorticoid receptor (GR) levels in the heart. Total RNA was isolated from whole hearts of 3‐month‐old male and female control (GRflox) and cardiomyocyte‐specific glucocorticoid receptor knockout mice. Expression levels of β‐myosin heavy chain (A), skeletal muscle α‐actin (B), brain natriuretic peptide (C), were measured by QRT‐PCR. Data represent n=5 mice per sex and genotype. β‐Mhc indicates β‐myosin heavy chain; cCGRkO, cardiomyocyte‐specific glucocorticoid receptor knockout; GR, glucocorticoid receptor; Nppb, brain natriuretic peptide; Ska, skeletal muscle α‐actin. *P<0.05, and ***P<0.001.
GR Deletion in Cardiomyocytes Leads to Dysregulation of Different Signaling Pathways in the Male and Female Heart
Using a non‐biased genome‐wide profiling approach we sought to define the molecular signaling events underlying the cardiac pathology in male and female cardioGRKO mice. We performed RNA‐Seq on hearts from 3‐month‐old male and female control and cardioGRKO mice. Three biological replicates were evaluated for each treatment group. A heat map of sample replicates and hierarchical clustering analysis shows a distinct pattern of gene expression between male and female control hearts (Figure 5A). The expression of 6279 genes is significantly different in male and female control hearts (Figure 4B). In contrast, the expression of only 3879 genes is sexually dimorphic in male and female cardioGRKO hearts (Figure 5B). A comparison of the sexually dimorphic genes between control and GR knockout hearts revealed that 2299 genes are in common between controls and cardioGRKO hearts (Figure 5B). Although ablation of GR in cardiomyocytes resulted in the loss of 64% of the sexually dimorphic expressed genes in the heart (Figure 5B), GR deletion also led to sexual dimorphism in the heart, with 1580 genes becoming differentially expressed between male and female (Figure 5B). To further understand the effects of GR in sexual dimorphism in the heart we search for genes known to be GR targets. We found that of the 4262 genes differentially expressed in male cardioGRKO hearts 306 genes have been associated with GR signaling. In the female cardioGRKO hearts we identified 392 genes of the total of 4559 that have been associated with GR signaling (Figure 5C). Comparison of these GR‐associated genes by Venn diagrams shows that ≈45% of these genes are unique to the cardioGRKO male hearts, while 56% are unique to the female knockout hearts. These findings suggest that GR signaling in the heart has a prominent effect in sex‐specific gene expression.
Figure 5.
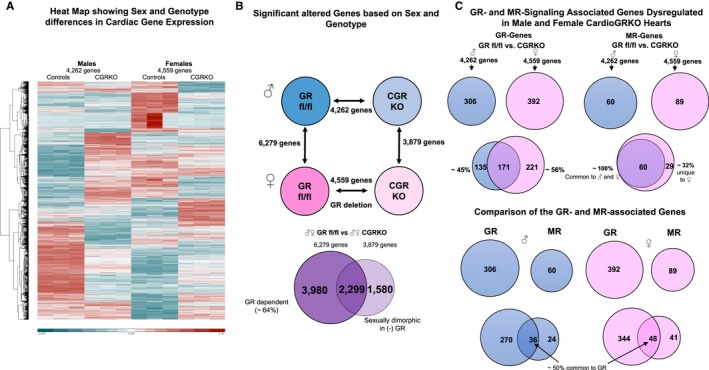
Pattern of gene expression between male and female hearts from control (glucocorticoid receptor [GR] fl/fl) and cardiomyocyte‐specific glucocorticoid receptor knockout mice. A, Heat map of RNA‐seq data with hierarchical clustering of genes that are differentially expressed between control and cardioGRKO male and female mice. Red indicates high‐expressing genes whereas blue indicates low‐expressing genes. B, Upper panel: Analysis of RNA‐seq results showing significantly altered genes in each comparison, male GRfl/fl to male cardioGRKO (4262 genes), female GRfl/fl to female cardioGRKO mice (4559 genes), male GRfl/fl to female GRfl/fl (6279 genes), and male cardioGRKO to female cardioGRKO (3879 genes). Lower panel: Venn diagram of the results overlaying male GRfl/fl vs female GRfl/fl (left circle) to male cardioGRKO vs female cardioGRKO mice (right circle). 2299 genes are in common between controls and cardioGRKO hearts (intersection). Cardiomyocyte GR regulates the expression 3980 sexually dimorphic genes in control hearts (64% of a total of 6279 genes are GR dependent). In the absence of GR, 1580 genes are differentially expressed between cardioGRKO male and female hearts. C, Upper right panel: Of the 4262 genes differentially expressed in male cardioGRKO hearts as compared with controls (GR fl/fl), 306 of these genes are directly or indirectly associated with GR. A similar number of GR associated genes, 392, were found among the 4559 genes with altered expression in the female cardioGRKO hearts. Comparison of these GR‐associated genes shows that ≈44% of these genes are unique to the cardioGRKO male hearts, while 56% are unique to the female knockout hearts. Upper left panel: Only 60 genes of the 4262 differentially expressed genes in the male cardioGRKO hearts have been related to mineralocorticoid receptor signaling, and only 89 genes of the total of 4559 female knockout dysregulated genes are associated to MR. Venn diagram of these genes overlaying male and female GRfl/fl vs cardioGRKO hearts shows that the majority (100% for males and ≈68% for females) of the reported MR‐associated genes are common between males and females. Lower panel: About 50% of the MR genes are commonly associated with GR. CardioGRKO indicates cardiomyocyte‐specific glucocorticoid receptor knockout; CGRkO, cardiomyocyte‐specific glucocorticoid receptor knockout; GR, glucocorticoid receptor.
In contrast to GR, only a few of the total number of genes dysregulated in the knockout hearts have been associated with the closely related mineralocorticoid receptor (MR) (Figure 5C), and the majority (100% for males and ≈68% for females) of these MR‐related genes are shared between male and female cardioGRKO hearts. We also found that about 50% of the MR‐related genes are commonly associated with GR (Figure 5C). Analysis of the GR and MR regulated genes revealed that 51 (males) and 68 (females) of the GR associated genes are related with “abnormalities of the heart ventricle” (according to the literature‐based Ingenuity Pathway Analysis [IPA] software) (Figure 6A). In contrast, only 17 and 19 of the MR‐associated genes dysregulated in cardioGRKO hearts are identified within this category (Figure 6A). Among the common GR and MR associated genes, we found atrial natriuretic peptide (Nppa), brain type natriuretic peptide (Nppb), and cyclooxygenase (Ptgs2). The vascular endothelial growth factor A (Vegfa) was found among the unique MR dependent genes in both male and female cardioGRKO hearts, and changes in its expression are associated with hypertrophy and angiogenesis. Angiotensinogen (Agt) was found among the unique GR‐regulated genes, and its expression appears to be compromised only in male cardioGRKO hearts. Genetic polymorphisms of the Agt and angiotensin‐converting enzyme genes are associated with increased risk of hypertension and left ventricular hypertrophy in humans.19 Therefore, alterations in the expression of this gene may explain in part the different sensitivity to alterations in GR signaling between male and female mice. Visualization of the genes connected to Agt showed more significant alterations in gene expression in the male cardioGRKO hearts as compared with their female counterparts (Figure 6B). Similarly, analysis of the Vegfa network showed more pronounced changes in the male knockout heart than in their female counterparts.
Figure 6.
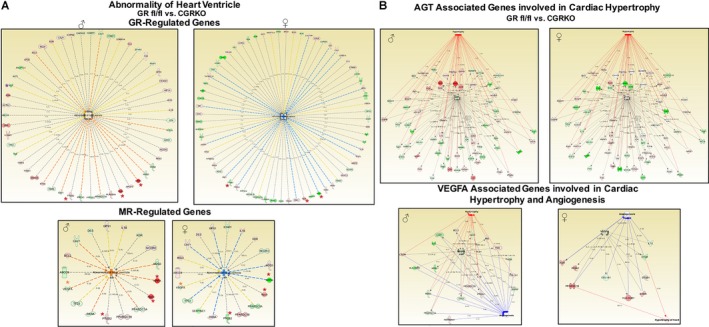
Analysis of the glucocorticoid receptor (GR) and the mineralocorticoid receptor (MR) dependent genes. A, Fifty‐one (male) and 68 (female) of the GR‐associated genes are related with abnormalities of the heart ventricle. Only 17 and 19 of the MR‐associated genes dysregulated in cardiomyocyte‐specific glucocorticoid receptor knockout (cardioGRKO hearts were identified within this category (lower panel). Atrial natriuretic peptide (Nppa), brain type natriuretic peptide (Nppb), and cyclooxygenase (Ptgs2) were identified among the common genes. Among the unique MR dependent genes was the vascular endothelial growth factor A (VegfA). Angiotensinogen (Agt) expression seems to be compromised only in male cardioGRKO hearts and its dysregulation is related to alterations in GR signaling. Dysregulation in Agt gene expression is associated with genes involved in cardiac hypertrophy. B, Visualization of the genes connected to Agt in male and female cardioGRKO (upper panel), and network of genes associated with VegfA. Alterations in VegA is associated with genes involved in hypertrophy and angiogenesis. Red depicts induced genes and green represents repressed genes. Orange lines (—) predict activation and blue lines predict inactivation. Yellow lines symbolize gene expression opposite to predicted effect, and gray lines represent that not prediction can be inferred based on the gene expression data. Nppa, Nppb, and Ptgs2 are marked with red asterisk (*). Agt is marked with a purple *, and VegA is marked with an orange *. CardioGRKO indicates cardiomyocyte‐specific glucocorticoid receptor knockout; CGRkO, cardiomyocyte‐specific glucocorticoid receptor knockout; GR, glucocorticoid receptor; MR, mineralocorticoid receptor.
To further evaluate the whole genome sexually dimorphic role of GR signaling in cardiac gene expression, analysis of the 1580 differentially expressed genes between cardioGRKO males versus females was performed by IPA software (Figure 7). This analysis identified “heart failure” and “hypertrophy” as the 2 more significantly activated (based on the z‐score) categories in male cardioGRKO hearts compared with the female cardioGRKO hearts (Figure 7A). One hundred and twenty‐six genes associated with “heart failure” were significantly altered in male hearts; in contrast, only 77 of these genes were dysregulated in the female hearts. Visualization of the genes involved in the heart failure category (Figure 7B) revealed that male cardioGRKO hearts presented more pronounced alterations in the expression of genes involved in calcium (Ca2+) handling (cardiomyocyte contraction), including ATPase sarcoplasmic/endoplasmic reticulum Ca2+ (Atp2a2, Serca), ryanodine receptor 2 (Ryr2), and genes implicated in maintaining the structure of cardiac sarcomeres and cardiomyocyte viability, such as desmin (Des), troponin C1 (Tnnc1), and the solute carrier family 25 member 4 (Slc25a4). Slc25a4 encodes the mitochondrial protein Ant‐1 (adenine nucleotide translocase type 1). Ant‐1 is involved in mitochondrial ADP/ATP transport. Alterations in the expression of Ant‐1 are associated with cardiomyopathy characterized by impaired mitochondrial ADP‐ATP exchange, decreased ADP‐stimulated tissue respiration, and increased mitochondrial reactive oxygen species production.20
Figure 7.
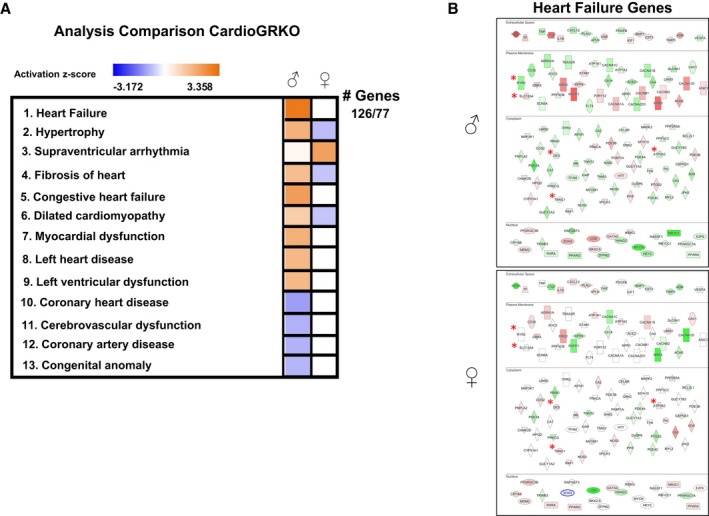
Effects of deleting cardiomyocyte glucocorticoid receptors on male and female heart gene expression. A, Differentially expressed genes between cardiomyocyte‐specific glucocorticoid receptor knockout (cardioGRKO) males vs females were analyzed by literature‐based Ingenuity Pathway Analysis (IPA) software. The top 13 diseases and biological functions were ranked and assigned an activation z‐score (orange indicates induced; blue indicates repressed). Heart failure was ranked as the most significant activated biological function. One hundred and twenty‐six genes in the heart failure function were significantly altered in male hearts; in contrast, only 77 of these genes were dysregulated in the female hearts. B, Visualization of the genes involved in the heart failure category in male and female cardioGRKO hearts. Genes involved in cardiomyocyte contraction, structure of cardiac sarcomeres, and cardiomyocyte viability are marked with an asterisk (*). ATPase sarcoplasmic/endoplasmic reticulum Ca2+ (Atp2a2, Serca), ryanodine receptor 2 (Ryr2), desmin (Des), troponin C1 (Tnnc1), and the solute carrier family 25 member 4 (Slc25a4). Red depicts induced genes and green represents repressed genes. CardioGRKO indicates cardiomyocyte‐specific glucocorticoid receptor knockout.
To validate the changes in these genes, we performed QRT‐PCR on hearts from additional cardioGRKO mice that were 3‐months‐old. In agreement with the RNA‐Seq data, the expression of all 5 of these genes was significantly dysregulated in the male cardioGRKO hearts (Figure 8). In contrast, only the levels of Tnnc1 were altered in the female cardioGRKO hearts (Figure 8B). In summary, our genome data indicate that deletion of cardiomyocyte GR leads to sexually dimorphic gene expression that predisposes males to an earlier and more profound deleterious cardiac phenotype compared with females. The functional impairments observed in the cardioGRKO hearts might be explained in part by the alterations in the expression of the validated target genes.
Figure 8.
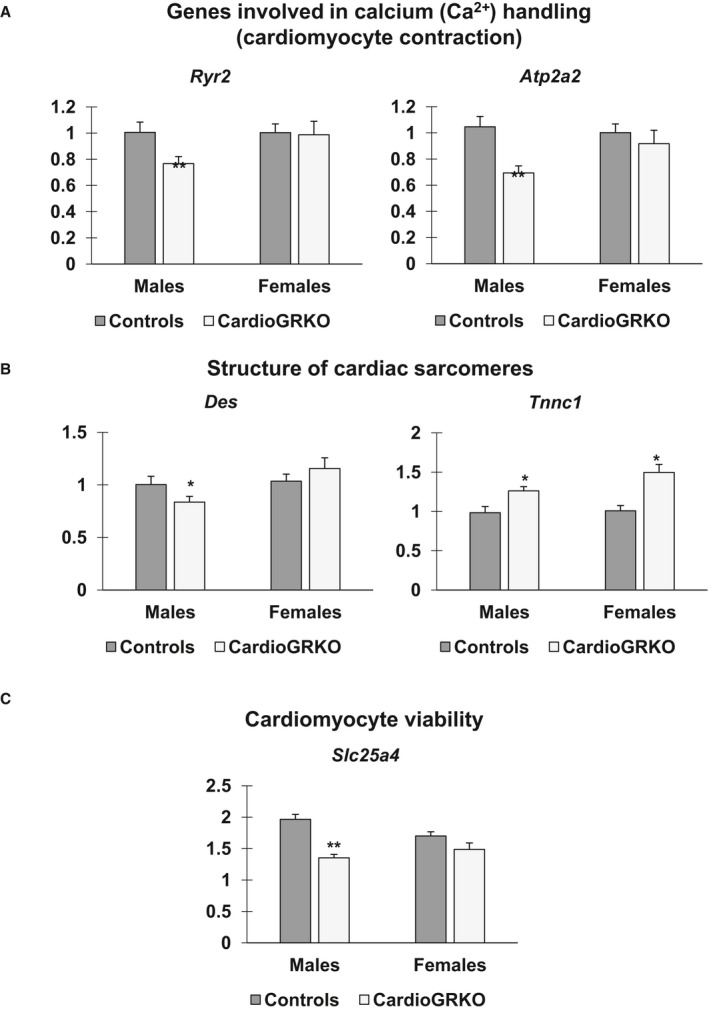
Genes associated with cardiomyocyte contraction, structure of cardiac sarcomeres, and cardiomyocyte viability are dysregulated in male cardiomyocyte‐specific glucocorticoid receptor knockout (cardioGRKO) hearts but not in their female counterparts. Total RNA was isolated from whole hearts of 3‐month‐old male and female control (glucocorticoid receptor flox/flox) and cardioGRKO mice. A, Expression levels of genes involved in calcium handling ryanodine receptor (Ryr2) and ATPase sarcoplasmic/endoplasmic reticulum Ca2+ (Atp2a2). B, Expression levels of genes involved in preserving the structure of cardiac sarcomeres desmin (Des) and troponin C1 (Tnnc1). C, Expression level of Slc25a4 (solute carrier family 25 member 4), a mitochondria carrier protein involved in cardiomyocyte viability. Data are mean±SEM (n=4–5 mice per group). *P<0.05, and **P<0.01. CardioGRKO indicates cardiomyocyte‐specific glucocorticoid receptor knockout.
Decreases in Ryr2 Gene Expression and Protein Levels Lead to Abnormal Calcium Handling in Cardiomyocytes Isolated From Male CardioGRKO
Consistent with a reduction in Ryr2 gene expression, Ryr2 protein levels were significantly reduced in 3 months old cardioGRKO male hearts. Female cardioGRKO hearts displayed normal Ryr2 protein levels as compared with control hearts (Figure 9A). To test if decreased Ryr2 levels translated to abnormal calcium handling, we isolated cardiomyocytes from male and female adult control and cardioGRKO hearts and evaluated intracellular calcium release following treatment with the classic Ryr2 activator caffeine (Figure 9B). The data show that cardiomyocyte stimulation with low concentration caffeine (0.25 mmol/L) led to a pronounced increase in calcium release in cardiomyocytes isolated from controls and female cardioGRKO hearts (19/20 cells, 95%) (Figure 9B). In contrast, cardiomyocytes isolated from male cardioGRKO hearts showed a significantly decreased response (10/20, 50%) in response to caffeine stimulation (Figure 9B). The isolated cardiomyocytes were responsive to high caffeine concentrations (10 mmol/L) regardless of genotype or sex. The results obtained using male GR‐deficient cardiomyocytes are consistent with our previously published data.13 These findings indicate that reduced mRNA and protein levels of Ryr2 in male cardioGRKO hearts translated into functional abnormalities in caffeine‐triggered intracellular calcium release in cardiomyocytes, which may be associated with the earlier cardiac hypertrophy and systolic dysfunction found in male cardioGRKO mice as compared with their female counterparts.
Figure 9.
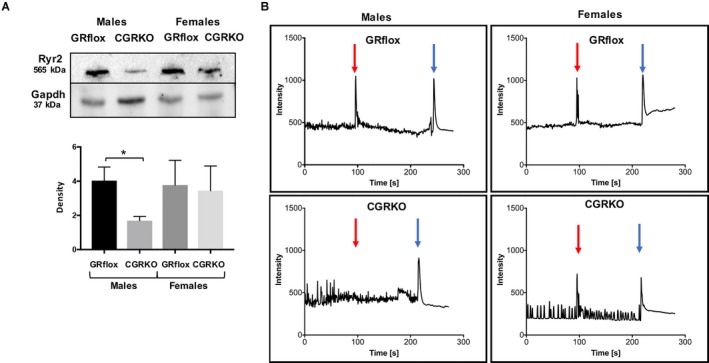
Expression and quantification of RyR2 (ryanodine receptor 2) protein with altered calcium responses in male cardiomyocyte‐specific glucocorticoid receptor knockout (cardioGRKO) mice. A, Representative immunoblot show Ryr2 protein levels in control (glucocorticoid receptor flox) and knockout hearts. Hearts from male cardioGRKO have reduction of ≈50% in Ryr2 total protein levels, whereas female hearts do not present a significant reduction in Ryr2 protein levels. Immunoblots were quantified by densitometry using National Institutes of Health ImageJ analysis software. Data are mean±SEM (n=3 mice per group). *P<0.05. B, Intracellular calcium changes were monitored using a calcium sensitive indicator, fluo‐4 in adult cardiomyocytes isolated from control and cardioGRKO male and female mice. Changes in intracellular calcium were monitored before and after application of a low concentration of caffeine (0.2 mmol/L, red arrow) and high concentration of caffeine (10 mmol/L, blue arrow) and are represented as the change in fluo‐4 fluorescence emission intensity. Upper panel graphs are representative data from different cardiomyocytes isolated from control mice. Lower panel graphs are representative data from different cardiomyocytes isolated from cardioGRKO mice. Cardiomyocytes displaying spontaneous intracellular calcium changes in the form of local calcium transients were selected for study. Intracellular calcium changes were monitored at the single cell level with data collected from a region of interest representing an individual cell. Typically, 4 regions of interest were monitored per experiment. CGRKO indicates cardiomyocyte‐specific glucocorticoid receptor knockout; GR, glucocorticoid receptor; Ryr2, ryanodine receptor 2.
Castration Protects Male CardioGRKO Mice From Developing LV Systolic Dysfunction
Conscious transthoracic echocardiography was performed on 3‐month‐old gonadectomized male and female cardioGRKO mice and their littermate controls. Castration protected male cardioGRKO mice from developing cardiac dysfunction. Castrated knockout mice presented with a preserved EF (81.54±10.36%) and FS (50.76±7.13%) as compared with their littermate controls (EF, 83.57±7.89% and FS, 52.08±9.99) (Table 3), and no significant abnormalities were found in LV end‐systolic dimension (LVID;s, 1.29±0.22 knockouts versus 1.44±0.37 controls), LV Vol; s (4.39±2.02 knockouts versus 6.04±2.02 controls), or heart weight (132.22±2.99 knockouts versus 128.30±2.99 controls). Ovariectomized female cardioGRKO did not show any significant functional changes as compared with their littermate controls (Table 3). Despite a slight reductions in EF (72.94±6.42% knockouts versus 81.69±3.52% controls) and FS (41.06±5.47% knockouts versus 49.24±3.86% controls) (Table 3), and an increase in LVID;s (1.65±0.26% knockouts versus 1.42±0.16% controls), ovariectomy did not exacerbate the susceptibility to the deleterious effects of GR inactivation in the female heart.
Table 3.
Echocardiogram Data on 3‐Month‐Old Castrated Males and Ovariectomized Female GRflox/flox and CardioGRKO
| Males | Females | |||
|---|---|---|---|---|
| Controls (n=3) | CardioGRKO (n=8) | Controls (n=3) | CardioGRKO (n=6) | |
| IVS;d, mm | 0.9±0.05 | 0.877±0.11 | 0.90±0.08 | 0.88±0.21 |
| LVID;d, mm | 3.01±0.14 | 2.75±0.31 | 2.84±0.10 | 2.80±0.33 |
| LVPW;d, mm | 1.04±0.23 | 0.99±0.20 | 0.91±0.13 | 0.92±0.26 |
| IVS;s, mm | 1.37±0.07 | 1.33±0.15 | 1.44±0.08 | 1.32±0.19 |
| LVID;s, mm | 1.44±0.37 | 1.29±0.22 | 1.42±0.16 | 1.65±0.26 |
| LVPW;s, mm | 1.56±0.40 | 1.33±0.24 | 1.31±0.08 | 1.14±0.25 |
| EF, % | 83.57±7.89 | 81.54±10.36 | 81.69±3.52 | 72.94±6.42 |
| FS, % | 52.08±9.99 | 50.76±7.13 | 49.24±3.86 | 41.06±5.47 |
| LV mass, mg | 81.33±6.95 | 84.94±10.72 | 83.16±12.44 | 84.66±10.12 |
| HW | 128.30±2.53 | 132.22±2.99 | 134.00±1.52 | 140.02±6.14* , † |
| BW, g | 33.56±0.51 | 32.47±2.60 | 34.06±0.75 | 35.98±0.88† |
| HW/BW | 3.82±0.12 | 4.09±0.28 | 3.93±0.12 | 3.89±0.23 |
| LV mass/BW | 2.42±0.24 | 2.01±0.39 | 2.44±0.41 | 2.35±0.28 |
| HR, bpm | 718±10.81 | 704.87±21.72 | 707±7.09 | 696.83±17.31 |
Echocardiographic measurements from transthoracic M‐mode tracings on conscious male and female. Data are mean±SD. Two‐way ANOVA with multiple comparisons analysis (male controls vs female controls, male controls vs male cardioGRKO, male controls vs female cardioGRKO, female controls vs male cardioGRKO, female controls vs female cardioGRKO, and male cardioGRKO vs female cardioGRKO) were used to evaluate whether differences between groups were statistically significant. Values without asterisks were not significantly different. bpm indicates beats per min; HW, heart weight; BW, body weight; EF, ejection fraction; FS, fractional shortening; HR, heart rate; IVS;d, interventricular septal thickness in diastole; IVS;s, interventricular septal thickness in systole; LV, left ventricular; LVID;d, LV end‐diastolic dimension; LVID;s, LV end‐systolic dimension; LVPW;d, posterior wall thickness in diastole; LVPW;s, posterior wall thickness in systole.
P<0.05 vs male control mice.
P<0.05 vs male cardioGRKO.
Discussion
Heart disease is a significant and growing public health problem in the United States and worldwide. There are significant disparities in heart disease prevention, clinical presentation, and treatment based on sex. The risk of heart disease in females has been underestimated because of the misperception that women are protected by estrogen and do not develop cardiovascular disease until several years post‐menopause.21 Because of this misconception, women have been underrepresented in clinical trials, and therefore most of the available treatments are tailored to males but not to females. Moreover, there is a gap in knowledge on the sex‐specific effects of cardiovascular risk factors, including obesity, diabetes mellitus, hypertension, and stress.22
Regarding the effects of stress, studies show that women have a unique vulnerability compared with men. For example, women aged <50 years have a significantly increased rate (3‐fold higher) of mental stress‐induced myocardial infarction than do similarly aged men.6, 23 Although sex hormones (estrogen and testosterone) are considered critical regulators of the physiological differences between men and women, the effects of sex in heart disease are more complex and are unlikely because of sex hormones alone.
Glucocorticoids are steroid hormones secreted by the adrenal gland in response to stress. As previously reported by our group24 and others25, 26 supraphysiological increases in glucocorticoid levels induce hypertrophy in cardiomyocytes, which may contribute to cardiac dysfunction and progression to heart failure.27 However, under normal physiology, sharp increases in glucocorticoid levels in response to stressors (eg, physical and emotional stress, disturbances in daily rhythms of waking and sleep, etc) benefit the heart by promoting the expression of genes involved in preserving cardiomyocyte contractility and survival.13 Therefore, glucocorticoids can exert both positive and negative effects on cardiomyocytes depending on the physiological context. As previously reported GR expression in cardiomyocytes is essential for cardiac homeostasis.13 The present study shows that glucocorticoids signaling via GR plays a sex‐specific role in preserving intracellular calcium in cardiomyocytes. These findings are important because they suggest that activation of GR signaling in the heart may provide a sex‐specific therapeutic approach to improve the contractile performance of the heart in disease, for example after a heart attack.
Both male and female mice lacking GR in cardiomyocytes present with increased mortality rates as compared with their littermate counterparts. However, female cardioGRKO mice have a better survival rate than males. While the median survival rate for male cardioGRKO mice is ≈6 months of age, female mice have a median survival rate of ≈10 months. Of the 24 male cardioGRKO that were used for our survival study, 20 males were found dead in their cage, and 4 were euthanized because of signs of severe pain/distress, including labored breathing, slow to move or non‐responsive when coaxed, severe dehydration, and hunched. Of the 23 cardioGRKO female used for this study, 18 were found dead in their cage and 5 were euthanized after showing signs of distress similar to those presented by their male counterparts. Regardless of sex, about 90% of the necropsied cardoGRKO mice presented enlarged hearts and have excess fluid in the thoracic cavity.13 As shown in Figure 2, heart sections from 6‐month‐old male and female cardioGRKO mice euthanized because of severe respiratory distress showed enlarged atrium and significant dilation of the left ventricle. These findings are consistent with atrial fibrillation and dilated cardiomyopathy, common causes of heart failure and sudden death. Therefore, based on our previous13 and present findings, we concluded that the cardioGRKO mice are dying from heart failure.
In agreement with delayed mortality observed in cardioGRKO female mice, echocardiogram data show that female cardioGRKO mice present with less profound changes in LV function as compared with their male counterparts. These findings indicate that females are more resilient to cardiomyocyte GR inactivation than males, perhaps because of protective actions of ovarian hormones on the heart.28 However, ovariectomy only had a minor effect on LV function in cardioGRKO mice. Castration had a significant effect on LV function in male cardioGRKO mice. Castrated cardioGRKO mice presented with a preserved cardiac function, suggesting that male hormones, perhaps testosterone, exacerbate the detrimental effects of GR‐inactivation in the heart. Studies have demonstrated that male hearts are more susceptible to hypertrophy relative to female hearts.29 Clinical data also show that men are a higher risk for pathological cardiac hypertrophy than aged‐matched women.29 However, little is known about the underlying molecular mechanism that leads to males’ increased susceptibility to cardiac hypertrophy. Exposure to androgens has been associated with differences in hypertrophic response between males and females.30 Androgens increase the proliferation of vascular smooth muscle cells.31 Several studies have shown that castration and pharmacological suppression of androgen production protect the heart from hypertrophy and heart failure in animal models of hypertension‐induced hypertrophy.29, 32, 33 Most of the hypertrophic effects of androgens have been attributed to testosterone signaling via the androgen receptors, which are expressed at high levels in the myocardium.30, 34 However, because of conflicting data showing that androgen‐deprivation increased the risk of heart failure in prostate cancer patients, the role and physiological actions of androgens are still controversial.33 In the context of the present study, the reduced survival rate observed in male cardioGRKO mice might result from the detrimental effects of androgens on cardiomyocytes. Data from prostate cancer studies have shown that GR can bind androgen receptor responsive elements on target genes regulated by androgens.35 Therefore, a possible explanation for the increased susceptibility to the deleterious effects of deleting GR in cardiomyocytes in males is that lack of GR increases the availability of androgen receptor responsive elements for the androgen receptors in the heart, resulting in an increased androgen signaling in cardiomyocytes. Studies by Pugach et al36 have shown that long‐term Cre recombinase expression in cardiomyocytes can lead to cardiac dysfunction and cardiotoxicity characterized by mild inflammation and fibrosis.36 Also, sexual dimorphic phenotypes have been observed in response to Cre expression, and female mice appear to be more resistant to the effects of early Cre expression in the heart.36 However, our previously published echocardiography data13 echocardiography data showed that expression of Cre alone had little to no effect on heart function in mice up to 6 months of age, indicating that the cardiac phenotype in 1‐ to 6‐month‐old cardioGRKO mice is because of the GR inactivation, and perhaps to GR interactions with sex hormones signaling. Future studies on the effects of estrogen and testosterone replacement or treatment with an androgen receptor antagonist need to be performed to test this hypothesis. Future studies on the effects of estrogen and testosterone replacement or treatment with an androgen receptor antagonist need to be performed to test this hypothesis.
Consistent with the exacerbated phenotype in male cardioGRKO mice, differences between males and females were found at the gene expression level. While the gene expression of traditional cardiac hypertrophy markers (β‐myosin heavy chain [β‐Mhc] and skeletal muscle α‐actin [Ska]) is altered in male cardioGRKO hearts, no significant changes in the levels of these genes were observed in female hearts at the same time point. These data suggest that molecular changes underlying the phenotype of cardioGRKO males and females might correlate with distinct patterns of gene expression. Our results correlate with recent studies by Richardson et al37 that showed that male mice lacking GR in cardiomyocytes and vascular smooth muscle cells developed pathological cardiomyocyte hypertrophy, associated with increased myosin heavy chain‐β expression. Interestingly, in the Richardson et al study, it was found that MR mRNA levels were elevated in the hearts of their knockout mouse model. As previously reported by our group, no differences in the expression of MR were found in the cardioGRKO hearts.13 Analysis of RNA‐Seq data showed that while some of genes associated with LV dysfunction and hypertrophy are regulated by both GR and MR in both male and female hearts, GR seems to play a more significant role in cardiac sexual dimorphism, and its deletion in cardiomyocytes leads to MR‐independent changes in cardiac gene expression.
A global analysis comparison between male versus female cardioGRKO hearts revealed that gene networks involved in heart failure, hypertrophy, and dilated cardiomyopathy were activated in male cardioGRKO hearts; in contrast, these gene networks were not significantly altered in the hearts of female cardioGRKO mice at the same time point. Visualization of the genes involved in heart failure revealed dysregulation in the expression of genes involved in cardiomyocyte contractility, mitochondrial ADP/ATP transport, and sarcomere structure. Among the genes involved in cardiac contractility, we found Ryr2 and Atp2a2 (Serca2). The expression of these 2 genes was significantly dysregulated in male cardioGRKO hearts; in contrast, their levels were normal in female hearts. Cardiac Ryr2 is an intracellular receptor that modulates the release of Ca2+ from the sarcoplasmic reticulum leading to cardiomyocyte contraction.38 Atp2a2 encodes the Serca2 pump which is involved in the transport of calcium ions from the cytoplasm into the sarcoplasmic reticulum. Reductions in Ryr2 and Serca2 levels lead to perturbations in intracellular Ca2+ cycling.39 Elegant studies by Rog‐Zielinska et al40 showed that GR regulates the expression of Ryr2 and Serca2 in cardiomyocytes. Therefore, defects in the expression of these 2 genes may explain in part the early deterioration of cardiac function observed in cardioGRKO males as compared with their female counterparts. Functional data showed that reductions in Ryr2 expression in male cardioGRKO hearts correlate with abnormal calcium handling. Male cardiomyocytes isolated from cardioGRKO hearts exhibited a decreased sensitivity to caffeine‐induced intracellular calcium responses as compared with their female counterparts. Studies have shown that estrogen can protect the female heart from pathological cardiac hypertrophy by inducing rapid intracellular Ca2+ mobilization through the regulation of L‐type voltage‐operated Ca2+ channels and Atp2a2 and via indirect interactions with the Ryr2s.41, 42 Our data show that cardioGRKO female hearts express normal levels of Ryr2 and Serca2, perhaps resulting from ovarian hormones (in particular, estrogen) signaling compensating for the lack of GR in cardiomyocytes. Also, female cardioGRKO hearts present with a preserved expression of Slc25a4, which is a mitochondria carrier protein integral to mitochondrial ADP/ATP transport. Alterations in Slc25a4 expression are associated with abnormal myocardial repolarization and contractile mechanics, impaired left ventricular functions, and concentric cardiac hypertrophy, characterized by cardiomyocyte degeneration and structurally abnormal mitochondria.20 Therefore, the decrease in Slc25a4 expression in the heart might contribute to the development of pathological cardiac hypertrophy. The fact that female cardioGRKO mice do not display changes in the functionality of Ryr2 also could be related to the lack of androgens, as increased signaling through androgen receptors because of the lack of GR in cardiomyocytes may influence Ryr2 gene expression and function. Further studies are needed to investigate if the underlying mechanisms behind the differences in the onset of the phenotype between male and female cardioGRKO mice result from the detrimental effects of androgens, and whether these differences play a role in male and female differential sensitivity to the effects of stress as it relates to heart disease, in particular in myocardial infarction.
Sources of Funding
This research was supported by the Intramural Research Program of the National Institutes of Health. National Institute of Environmental Health Sciences (Cruz‐Topete, Oakley, He, and Cidlowski), Louisiana State University Health Sciences Center‐Shreveport (Cruz‐Topete, Carroll, Dominic, Watts, Trosclair, and Glasscock), and National Institutes of Health: R01 NS‐100954 and R01 NS‐099188 (Glasscock).
Disclosures
None.
Acknowledgments
We thank members of the Pathology Support Group (National Institute of Environmental Health Sciences) and LSU Health Sciences Center Shreveport Microscopy Core Facility for their assistance with tissue collection, histology, and image acquisition.
(J Am Heart Assoc. 2019;8:e011012 DOI: 10.1161/JAHA.118.011012.)
Contributor Information
Diana Cruz‐Topete, Email: dcruz2@lsuhsc.edu.
John A. Cidlowski, Email: cidlows1@niehs.nih.gov.
References
- 1. Isensee J, Witt H, Pregla R, Hetzer R, Regitz‐Zagrosek V, Noppinger PR. Sexually dimorphic gene expression in the heart of mice and men. J Mol Med (Berl). 2008;86:61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Quinn M, Ramamoorthy S, Cidlowski JA. Sexually dimorphic actions of glucocorticoids: beyond chromosomes and sex hormones. Ann N Y Acad Sci. 2014;1317:1–6. [DOI] [PubMed] [Google Scholar]
- 3. Dupont C, Gribnau J. Different flavors of X‐chromosome inactivation in mammals. Curr Opin Cell Biol. 2013;25:314–321. [DOI] [PubMed] [Google Scholar]
- 4. Quinn MA, Cidlowski JA. Endogenous hepatic glucocorticoid receptor signaling coordinates sex‐biased inflammatory gene expression. FASEB J. 2016;30:971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Duma D, Collins JB, Chou JW, Cidlowski JA. Sexually dimorphic actions of glucocorticoids provide a link to inflammatory diseases with gender differences in prevalence. Sci Signal. 2010;3:ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vaccarino V, Shah AJ, Rooks C, Ibeanu I, Nye JA, Pimple P, Salerno A, D'Marco L, Karohl C, Bremner JD, Raggi P. Sex differences in mental stress‐induced myocardial ischemia in young survivors of an acute myocardial infarction. Psychosom Med. 2014;76:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N Engl J Med. 2005;353:1711–1723. [DOI] [PubMed] [Google Scholar]
- 8. Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. 2013;34:518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burford NG, Webster NA, Cruz‐Topete D. Hypothalamic‐pituitary‐adrenal axis modulation of glucocorticoids in the cardiovascular system. Int J Mol Sci. 2017;18:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamamoto KR. Steroid receptor regulated transcription of specific genes and gene networks. Annu Rev Genet. 1985;19:209–252. [DOI] [PubMed] [Google Scholar]
- 11. Beato M, Herrlich P, Schutz G. Steroid hormone receptors: many actors in search of a plot. Cell. 1995;83:851–857. [DOI] [PubMed] [Google Scholar]
- 12. Phuc Le P, Friedman JR, Schug J, Brestelli JE, Parker JB, Bochkis IM, Kaestner KH. Glucocorticoid receptor‐dependent gene regulatory networks. PLoS Genet. 2005;1:e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oakley RH, Ren R, Cruz‐Topete D, Bird GS, Myers PH, Boyle MC, Schneider MD, Willis MS, Cidlowski JA. Essential role of stress hormone signaling in cardiomyocytes for the prevention of heart disease. Proc Natl Acad Sci USA. 2013;110:17035–17040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol. 2010;2:a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oakley RH, Cruz‐Topete D, He B, Foley JF, Myers PH, Xu X, Gomez‐Sanchez CE, Chambon P, Willis MS, Cidlowski JA. Cardiomyocyte glucocorticoid and mineralocorticoid receptors directly and antagonistically regulate heart disease in mice. Sci Signal. 2019;12:eaau9685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cruz‐Topete D, Myers PH, Foley JF, Willis MS, Cidlowski JA. Corticosteroids are essential for maintaining cardiovascular function in male mice. Endocrinology. 2016;157:2759–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cruz‐Topete D, He B, Xu X, Cidlowski JA. Kruppel‐like factor 13 is a major mediator of glucocorticoid receptor signaling in cardiomyocytes and protects these cells from DNA damage and death. J Biol Chem. 2016;291:19374–19386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ohara K, Hirai T, Fukuda N, Sakurai K, Nakagawa K, Nozawa T, Inoue H. Relation of left atrial blood stasis to clinical risk factors in atrial fibrillation. Int J Cardiol. 2009;132:210–215. [DOI] [PubMed] [Google Scholar]
- 19. Wang AY, Chan JC, Wang M, Poon E, Lui SF, Li PK, Sanderson J. Cardiac hypertrophy and remodeling in relation to ACE and angiotensinogen genes genotypes in Chinese dialysis patients. Kidney Int. 2003;63:1899–1907. [DOI] [PubMed] [Google Scholar]
- 20. Strauss KA, DuBiner L, Simon M, Zaragoza M, Sengupta PP, Li P, Narula N, Dreike S, Platt J, Procaccio V, Ortiz‐Gonzalez XR, Puffenberger EG, Kelley RI, Morton DH, Narula J, Wallace DC. Severity of cardiomyopathy associated with adenine nucleotide translocator‐1 deficiency correlates with mtDNA haplogroup. Proc Natl Acad Sci USA. 2013;110:3453–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maas AH, Appelman YE. Gender differences in coronary heart disease. Neth Heart J. 2010;18:598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wokhlu A, Pepine CJ. Mental stress and myocardial ischemia: young women at risk. J Am Heart Assoc. 2016;5:e004196 DOI: 10.1161/JAHA.116.004196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vaccarino V, Goldberg J, Magruder KM, Forsberg CW, Friedman MJ, Litz BT, Heagerty PJ, Huang GD, Gleason TC, Smith NL. Posttraumatic stress disorder and incidence of type‐2 diabetes: a prospective twin study. J Psychiatr Res. 2014;56:158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ren R, Oakley RH, Cruz‐Topete D, Cidlowski JA. Dual role for glucocorticoids in cardiomyocyte hypertrophy and apoptosis. Endocrinology. 2012;153:5346–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ohtani T, Mano T, Hikoso S, Sakata Y, Nishio M, Takeda Y, Otsu K, Miwa T, Masuyama T, Hori M, Yamamoto K. Cardiac steroidogenesis and glucocorticoid in the development of cardiac hypertrophy during the progression to heart failure. J Hypertens. 2009;27:1074–1083. [DOI] [PubMed] [Google Scholar]
- 26. Kuster DW, Merkus D, Kremer A, van Ijcken WF, de Beer VJ, Verhoeven AJ, Duncker DJ. Left ventricular remodeling in swine after myocardial infarction: a transcriptional genomics approach. Basic Res Cardiol. 2011;106:1269–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Souverein PC, Berard A, Van Staa TP, Cooper C, Egberts AC, Leufkens HG, Walker BR. Use of oral glucocorticoids and risk of cardiovascular and cerebrovascular disease in a population based case‐control study. Heart. 2004;90:859–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lagranha CJ, Silva TLA, Silva SCA, Braz GRF, da Silva AI, Fernandes MP, Sellitti DF. Protective effects of estrogen against cardiovascular disease mediated via oxidative stress in the brain. Life Sci. 2018;192:190–198. [DOI] [PubMed] [Google Scholar]
- 29. Huang CK, Lee SO, Chang E, Pang H, Chang C. Androgen receptor (AR) in cardiovascular diseases. J Endocrinol. 2016;229:R1–R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marsh JD, Lehmann MH, Ritchie RH, Gwathmey JK, Green GE, Schiebinger RJ. Androgen receptors mediate hypertrophy in cardiac myocytes. Circulation. 1998;98:256–261. [DOI] [PubMed] [Google Scholar]
- 31. Cabral AM, Vasquez EC, Moyses MR, Antonio A. Sex hormone modulation of ventricular hypertrophy in sinoaortic denervated rats. Hypertension. 1988;11:I93–I97. [DOI] [PubMed] [Google Scholar]
- 32. Li Y, Kishimoto I, Saito Y, Harada M, Kuwahara K, Izumi T, Hamanaka I, Takahashi N, Kawakami R, Tanimoto K, Nakagawa Y, Nakanishi M, Adachi Y, Garbers DL, Fukamizu A, Nakao K. Androgen contributes to gender‐related cardiac hypertrophy and fibrosis in mice lacking the gene encoding guanylyl cyclase‐A. Endocrinology. 2004;145:951–958. [DOI] [PubMed] [Google Scholar]
- 33. Ikeda Y, Aihara K, Yoshida S, Akaike M, Matsumoto T. Effects of androgens on cardiovascular remodeling. J Endocrinol. 2012;214:1–10. [DOI] [PubMed] [Google Scholar]
- 34. Shen JS, Meng XL, Wight‐Carter M, Day TS, Goetsch SC, Forni S, Schneider JW, Liu ZP, Schiffmann R. Blocking hyperactive androgen receptor signaling ameliorates cardiac and renal hypertrophy in Fabry mice. Hum Mol Genet. 2015;24:3181–3191. [DOI] [PubMed] [Google Scholar]
- 35. Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, Zheng D, Sawyers CL. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pugach EK, Richmond PA, Azofeifa JG, Dowell RD, Leinwand LA. Prolonged cre expression driven by the alpha‐myosin heavy chain promoter can be cardiotoxic. J Mol Cell Cardiol. 2015;86:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Richardson RV, Batchen EJ, Thomson AJ, Darroch R, Pan X, Rog‐Zielinska EA, Wyrzykowska W, Scullion K, Al‐Dujaili EA, Diaz ME, Moran CM, Kenyon CJ, Gray GA, Chapman KE. Glucocorticoid receptor alters isovolumetric contraction and restrains cardiac fibrosis. J Endocrinol. 2017;232:437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SR. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store‐overload‐induced Ca2+ release (SOICR). Proc Natl Acad Sci USA. 2004;101:13062–13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. George CH. Sarcoplasmic reticulum Ca2+ leak in heart failure: mere observation or functional relevance? Cardiovasc Res. 2008;77:302–314. [DOI] [PubMed] [Google Scholar]
- 40. Rog‐Zielinska EA, Craig MA, Manning JR, Richardson RV, Gowans GJ, Dunbar DR, Gharbi K, Kenyon CJ, Holmes MC, Hardie DG, Smith GL, Chapman KE. Glucocorticoids promote structural and functional maturation of foetal cardiomyocytes: a role for PGC‐1alpha. Cell Death Differ. 2015;22:1106–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rybalchenko V, Grillo MA, Gastinger MJ, Rybalchenko N, Payne AJ, Koulen P. The unliganded long isoform of estrogen receptor beta stimulates brain ryanodine receptor single channel activity alongside with cytosolic Ca2+ . J Recept Signal Transduct Res. 2009;29:326–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xin HB, Senbonmatsu T, Cheng DS, Wang YX, Copello JA, Ji GJ, Collier ML, Deng KY, Jeyakumar LH, Magnuson MA, Inagami T, Kotlikoff MI, Fleischer S. Oestrogen protects FKBP12.6 null mice from cardiac hypertrophy. Nature. 2002;416:334–338. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors declare that all supporting data and method descriptions are available within the article or from the corresponding author upon reasonable request.