

Abstract
Background
Heart fats (epicardial and paracardial adipose tissue [PAT]) are greater after menopause. Endogenous estrogen may regulate these fat depots. We evaluated the differential effects of hormone therapy formulations on heart fat accumulations and their associations with coronary artery calcification (CAC) progression in recently menopausal women from KEEPS (Kronos Early Estrogen Prevention Study).
Methods and Results
KEEPS was a multicenter, randomized, placebo‐controlled trial of the effects of 0.45 mg/d oral conjugated equine estrogens and 50 µg/d transdermal 17β‐estradiol, compared with placebo, on 48‐month progression of subclinical atherosclerosis among 727 early menopausal women. CAC progression was defined if baseline CAC score was 0 and 48‐month CAC score was >0 or if baseline CAC score was >0 and <100 and annualized change in CAC score was ≥10. Of 727 KEEPS participants, 474 (mean age: 52.7 [SD: 2.6]; 78.1% white) had computed tomography–based heart fat and CAC measures at both baseline and 48 months. Compared with women on placebo, women on oral conjugated equine estrogens were less likely to have any increase in epicardial adipose tissue (odds ratio for oral conjugated equine estrogens versus placebo: 0.62 [95% CI, 0.40–0.97]; P=0.03). PAT did not change in any group. Changes in epicardial adipose tissue and PAT did not differ by treatment group. CAC increased in 14% of participants. The assigned treatment modified the association between PAT changes and CAC progression (P=0.02) such that PAT increases were associated with CAC increases only in the transdermal 17β‐estradiol group.
Conclusions
In recently menopausal women, oral conjugated equine estrogens may slow epicardial adipose tissue accumulation, whereas transdermal 17β‐estradiol may increase progression of CAC associated with PAT accumulation.
Clinical Trial Registration
URL: http://www.clinicaltrials.gov. Unique identifier: NCT00154180.
Keywords: coronary artery disease, epicardial fat, estrogen, menopause
Subject Categories: Treatment, Women, Cardiovascular Disease, Aging, Obesity
Clinical Perspective
What Is New?
Oral conjugated equine estrogens may slow epicardial adipose tissue accumulation in recently menopausal women.
Transdermal 17β‐estradiol may increase risk associated with paracardial adipose tissue accumulation on progression of coronary artery calcification in recently menopausal women.
What Are the Clinical Implications?
Findings underscore the differential contributions of estrogen type and route of administration when assessing risk related to hormone therapy use.
Hormone therapy remains the best therapeutic option for the relief of debilitating menopausal symptoms.
Clinicians should continue to individualize hormone therapy prescription using the best available evidence to maximize benefits and minimize risks.
Introduction
Heart fat volumes are larger in postmenopausal than premenopausal women.1 Estrogen may have an important influence on heart fat buildup, as the accumulation of fat outside the pericardium is associated with lower levels and a steeper decline in endogenous estradiol (E2).1 Relative to the pericardium, 2 heart fat depots can be identified: (1) epicardial adipose tissue (EAT), which directly covers the heart and is located within the pericardial sac, and (2) paracardial adipose tissue (PAT), which is located anterior to EAT, outside the pericardial sac.2, 3
Heart fat in EAT and PAT depots may provide a readily detectable, noninvasive, novel risk marker for coronary arterial disease in postmenopausal women. Heart fat may contribute to the pathogenesis of coronary arterial disease4, 5, 6, 7, 8, 9, 10, 11 via both paracrine and endocrine release of anti‐ and proinflammatory adipokines into the coronary arteries and the myocardium.12 Interestingly, in midlife women, EAT was significantly associated with subclinical atherosclerosis, although this association was not modified by menopausal status or E2 levels. In contrast, a positive association between PAT and subclinical atherosclerosis was stronger in postmenopausal women with lower levels of E2.13 These findings are in line with the notion that EAT and PAT are distinct depots. EAT and PAT have different embryological origins, blood supplies, and metabolic activities,2, 3 supporting evaluation of heart fat depots separately.
The effect of exogenous estrogen use on cardiovascular disease (CVD) risk remains controversial.14, 15, 16, 17, 18 Current hormone therapy (HT) prescribing practices recommend HT to treat menopausal symptoms in early menopause but not to protect against CVD.19 Given the potential effects of endogenous estrogens on heart fat accumulations and the ongoing controversy about the CVD risks and benefits of HT in postmenopausal women, it is essential to assess the impact of HT on heart fat accumulations and the linkage of heart fat increases with development of atherosclerosis. Moreover, it is possible that HT may affect cardiometabolic risk differently based on formulation and route of administration.20 Thus, it is plausible to hypothesize that various formulations of HT will have differential impacts on heart fat accumulations and their association with atherosclerosis development. Furthermore, this impact may be more pronounced for the PAT depot, for which the association with subclinical atherosclerosis was modified by levels of E2 in midlife women.13
KEEPS (Kronos Early Estrogen Prevention Study) was a multicenter, randomized, placebo‐controlled clinical trial of the effects of oral and transdermal HT, compared with placebo, on 48‐month progression of subclinical atherosclerosis among recently menopausal women.21 The main results from KEEPS showed that 48 months of early use of HT did not affect progression of atherosclerosis despite improving some markers of CVD risk (lipids and insulin resistance).22 Whether early use of HT has an effect on heart fat accumulation is not known. KEEPS provides an ideal opportunity to examine possible differential effects of 2 routes of HT administration on 2 distinct depots of heart fat accumulation and their associations with the development of atherosclerosis, as measured by coronary artery calcification (CAC).
The main aims of this study were (1) to assess the effect of oral and transdermal HT formulations versus placebo on heart fat accumulations over 48 months of treatment and (2) to examine the extent to which HT formulations modify the longitudinal associations of heart fat depots with CAC progression in recently menopausal women.
Methods
The authors declare that all supporting results are available within the article and its online supplementary files.
Study Design and Participants
This study is a secondary analysis of data from KEEPS (ClinicalTrials.gov identifier: NCT00154180) and a completed ancillary study of KEEPS on ectopic fat. KEEPS participants were recruited between July 2005 and June 2008 and followed for 48 months. Visits were completed by March 2012. Full KEEPS inclusion and exclusion criteria have been published elsewhere.21 Briefly, women were eligible if they had an intact uterus, were aged 42 to 58 years, were between 6 and 36 months from their last menses, and had plasma follicle‐stimulating hormone levels ≥35 IU/L, E2 levels <147 pmol/L, or both. Women reporting a history of clinical CVD, current heavy smoking, body mass index (calculated as kg/m2) ≥35, LDL (low‐density lipoprotein) cholesterol >190 mg/dL or triglycerides >400 mg/dL, diabetes mellitus, uncontrolled hypertension, or moderate subclinical CVD, defined as a CAC score ≥50, were ineligible for randomization. Former or current HT users were screened only after having discontinued therapy for ≥90 days. A total of 727 women (77% white, 7% black, 3% Asian, 7% Hispanic, and 6% other) met inclusion criteria and were randomized to (1) oral‐conjugated equine estrogens (o‐CEE; 0.45 mg/d, n=230, 31.6%), (2) transdermal 17β‐estradiol (t‐E2; 50 μg/d, n=222, 30.5%), or (3) placebo (inactive pill and patch, n=275, 37.8%). Women receiving o‐CEE or t‐E2 also received oral micronized progesterone 200 mg/d for the first 12 days each month.
An ancillary study designed to measure ectopic fat before (baseline) and 48 months after randomization was conducted in KEEPS. The current analysis excluded all KEEPS participants who did not have heart fat and CAC measured at both baseline and 48 months, leaving 474 participants in the analytical sample (Figure 1. Excluded participants were more likely to have a longer time since menopause, to not be a college graduate, or to be a current smoker. Moreover, they had significantly lower baseline PAT volume than those who were included (Table S1).
Figure 1.
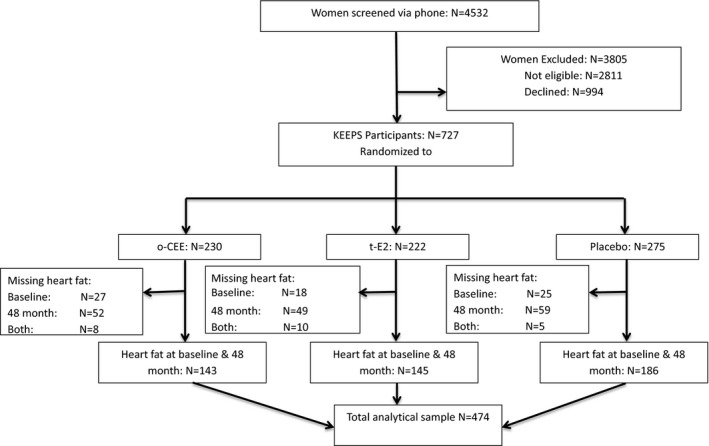
CONSORT flow diagram of the KEEPS (Kronos Early Estrogen Prevention Study) heart fat ancillary study. CONSORT indicates Consolidated Standards of Reporting Trials; o‐CEE, oral conjugated equine estrogens; t‐E2, transdermal β17‐estradiol.
The institutional review board at each participating site approved the trial, and all participants provided informed consent to participate in the trial.
Coronary Artery Calcification
CAC was a designated end point of the KEEPS main trial assessed at baseline and at 48 months.23, 24 Images of the coronary arteries were obtained using high‐speed axial tomography with rapid acquisition of 30 to 40 contiguous 3‐mm‐thick images during end diastole using ECG triggering during a single 30‐ to 35‐second breath hold.23 CAC score was quantified using the Agatston scoring method24 by an experienced reader blinded to study group. CAC was defined as a hyperattenuating lesion >130 Hounsfield units, with an area of at least 3 pixels. The total calcification score was the sum of the individual scores for the 4 major epicardial coronary arteries. A mean Agatston score was calculated from the results of 2 sequential scans among all participants for more precise estimates. CAC progression was defined using similar criteria applied in other studies of participants with low CVD risk.25 CAC was present when baseline CAC score was 0 and 48‐month CAC score was >0, when baseline CAC score was >0 and <100 and annualized change in CAC score was ≥10, or when baseline CAC score was ≥100 and annualized percentage change in CAC score was ≥10% (by design, all KEEPS participants had CAC scores <50 at baseline; therefore, none of the participants met this criterion).
Heart Fat Depots
Heart fat depot volumes were measured as part of a study ancillary to KEEPS (2009–2012) that utilized existing computed tomography scans before randomization (baseline) and 48 months after randomization. All images were read centrally by experienced readers who were blinded to study group, as described previously.26 Briefly, total heart fat volume (EAT plus PAT) was determined from 15 mm above to 30 mm below the superior extent of the left main coronary artery. This region of the heart was selected because it includes the epicardial fat surrounding the proximal coronary arteries. The anterior border of the heart fat volume was the chest wall, and the posterior borders were the aorta and the bronchus. Using volume analysis software, fat was distinguished from other heart tissue by a threshold of −190 to −30 Hounsfield units. EAT was measured by manually tracing out the pericardium every 2 to 3 slices below the start point and then using the software to automatically trace out the segments in between these selected slices. PAT was measured by subtracting EAT volume from total heart fat volume. Reproducibility measurements of EAT and PAT were performed on 20 randomly selected scans from another study that used a similar protocol. Both Spearman and intraclass correlation coefficients between readers (intrareader) were 0.99 each for EAT and 0.86 and 0.96, respectively, for PAT. Similarly, both Spearman and intraclass correlation coefficients between repeated readings (interreader) were 0.98 each for EAT and 0.96 and 0.90, respectively, for PAT.26
Covariates
At baseline and after 48 months of randomization, fasting levels of serum total cholesterol, HDL (high‐density lipoprotein) cholesterol, LDL cholesterol, and triglycerides were measured at the Kronos Science Laboratory (KSL) using standard methods. Glucose and insulin levels were also measured through studies ancillary to KEEPS. Total, HDL, and LDL cholesterol and triglycerides were assayed at KSL using Carolina Liquid Chemistries reagent on the Stanbio Sirrus chemistry analyzer. For total cholesterol, the intra‐assay coefficient of variation (CV) was 1.4% to 2.2% and the interassay CV was 4.3% to 5.0%. For HDL cholesterol, the intra‐assay CV was 2.7% to 3.1% and the interassay CV was 3.5% to 3.8%. For LDL cholesterol, the intra‐assay CV was 1.3% to 1.5% and the interassay CV was 5.3% to 7.1%. For triglycerides, both the intra‐assay and interassay CVs were 5.5% to 5.6%.
Glucose was measured at KSL on the Stanbio Sirrus chemistry analyzer using the Stanbio reagent standard, with an intra‐assay CV of 1.7% to 1.3%, and an interassay CV of 2.2% to 2.5%. Insulin assays were conducted at KSL on the Immulite 2000 by solid‐phase, chemiluminescent immunometric assay with a method detection limit of 2 μIU/mL, intra‐assay CV of 2.6% to 2.8%, and interassay CV of 2.8% to 3.3%. The homeostasis model assessment of insulin resistance was calculated as described by Matthews et al.27
Demographics, race/ethnicity, income, employment status, education level, history of smoking, medication use, alcohol consumption, and physical activity (metabolic equivalents, calculated as total energy expenditure from recreational physical activity in kcal/wk per kg)26 were all collected at screening or baseline visits.22 Physical measures were obtained at baseline and 48 months. Body mass index was calculated from measured weight in kilograms divided by height in square meters. Waist circumference (in cm) was measured at the smallest horizontal circumference using nonstretchable tape. Blood pressure was assessed in the right arm after at least 5 minutes of rest and averaged across 2 readings.
Statistical Analysis
Comparisons across the 3 treatment groups were performed using ANOVA for normally distributed variables or the Kruskal–Wallis test for skewed variables; the χ2 or Fisher exact test was used for categorical variables. Change in EAT and PAT at 48 months was calculated as the difference between baseline and 48‐month values. The distribution of EAT and PAT changes were highly skewed, and transformations did not improve the distribution; therefore, nonparametric tests were used to assess within‐ and between‐group differences in 48‐month heart fat changes. In addition, logistic regression was used to test whether any increase in each heart fat depot (48‐month change >0) differed by treatment group. For CAC progression, associations between 48‐month changes in each heart fat depot and CAC progression were evaluated using logistic regression. Covariates were selected a priori and based on univariate analysis showing an association with CAC progression at P<0.1. For variables that were highly correlated, models were assessed with each one separately, and the models with the best model‐fit diagnostics were chosen. Moderation effects of HT use were assessed, and treatment group–specific odds ratios (95% CI) per 1‐SD increase in change in heart fat depot were calculated.
We conducted inverse probability weighting sensitivity analyses28 to account for missing data on heart fat depots and CAC at 48 months (n=160). With this method, completed cases are weighted by the inverse of their probability of being a completed case (Tables S1–S7). SAS v9.4 (SAS Institute) and STATA (release 15; StataCorp) were used for statistical analyses.
Results
Participants’ baseline variables and CAC progression are presented in Table 1. No significant differences were observed in baseline EAT and PAT volumes among the treatment groups (Table 2.
Table 1.
Baseline Variables and CAC Progression for Overall Analytical Sample and by Treatment Group
| Variable | Total N=474 | o‐CEE n=143 (30.2%) | t‐E2 n=145 (30.6%) | Placebo n=186 (39.2%) | P Value* |
|---|---|---|---|---|---|
| Age at baseline, y, mean (SD) | 52.7 (2.6) | 52. 8 (2.8) | 52.8 (2.6) | 52.5 (2.4) | 0.23 |
| Age at menopause, y, mean (SD) | 50.9 (2.6) | 51. 0 (2.9) | 50.9 (2.6) | 50.8 (2.3) | 0.36 |
| Time since menopause, y, mean (SD) | 1.8 (0.8) | 1.8 (0.8) | 1.8 (0.7) | 1.7 (0.8) | 0.33 |
| White race, n (%) | 370 (78.1) | 112 (78.3) | 111 (76.6) | 147 (79.0) | 0.86 |
| Education, n (%) | 0.06 | ||||
| Declined to answer | 5 (1.1) | 0 (0.0) | 2 (1.4) | 3 (1.6) | |
| High school graduate or less | 29 (6.1) | 11 (7.7) | 3 (2.1) | 15 (8.1) | |
| Some college | 80 (16.9) | 28 (19.6) | 20 (13.8) | 32 (17.2) | |
| College graduate | 360 (75.9) | 104 (72.7) | 120 (82.8) | 136 (73.1) | |
| Employed, n (%) | 391 (82.5) | 122 (85.3) | 120 (82.8) | 149 (80.1) | 0.47 |
| Income, n (%) | 0.56 | ||||
| <$60 000 | 84 (17.7) | 27 (18.9) | 29 (20.0) | 28 (15.1) | |
| $60 000 to <$100 000 | 65 (13.7) | 17 (11.9) | 19 (13.1) | 29 (15.6) | |
| >$100 000 | 85 (17.9) | 23 (16.1) | 22 (15.2) | 40 (21.5) | |
| Unknown | 240 (50.6) | 76 (53.1) | 75 (51.7) | 89 (47.8) | |
| Physical activity level, MET‐h/wk, median (IQR) | 16.7 (7.0, 28.6) | 17.5 (7.4, 32.7) | 14.6 (5.3, 24.6) | 17.2 (7.5, 28.0) | 0.16 |
| Alcohol consumption, n (%) | 361 (76.2) | 120 (83.9) | 103 (71.0) | 138 (74.2) | 0.03 |
| Smoking status, n (%) | 0.77 | ||||
| Never | 380 (80.2) | 119 (83.2) | 115 (79.3) | 146 (78.5) | |
| Past | 70 (14.8) | 19 (13.3) | 21 (14.5) | 30 (16.1) | |
| Current | 24 (5.1) | 5 (3.5) | 9 (6.2) | 10 (5.4) | |
| Ever use HT, n (%) | 100 (21.1) | 36 (25.2) | 29 (20.0) | 35 (18.8) | 0.35 |
| Antihypertensive medication use, n (%) | 0.50 | ||||
| Never | 392 (82.7) | 115 (80.4) | 119 (82.1) | 158 (84.9) | |
| Past | 26 (5.5) | 11 (7.7) | 9 (6.2) | 6 (3.2) | |
| Current | 56 (11.8) | 17 (11.9) | 17 (11.7) | 22 (11.8) | |
| BMI, kg/m2, mean (SD) | 26.0 (4.3) | 26.1 (4.2) | 25.7 (4.4) | 26.3 (4.3) | 0.55 |
| Waist circumference, cm, mean (SD) | 84.4 (11.7) | 84.3 (11.2) | 83.4 (11.9) | 85.1 (11.9) | 0.48 |
| Systolic blood pressure, mm Hg, mean (SD) | 118.4 (15.1) | 119.3 (14.7) | 116.6 (16.0) | 119.1 (14.7) | 0.99 |
| HDL cholesterol, mg/dL, mean (SD) | 72.2 (15.0) | 72.8 (15.1) | 74.5 (16.6) | 70.0 (13.4) | 0.06 |
| LDL cholesterol, mg/dL, mean (SD) | 111.4 (27.5) | 111.2 (26.7) | 111.1 (28.9) | 111.8 (27.0) | 0.84 |
| Triglycerides, mg/dL, median (IQR) | 69.5 (50.0, 105.5) | 69.0 (50.0, 105.0) | 66.0 (47.0, 101.0) | 73.5 (55.0, 110.0) | 0.16 |
| Fasting glucose, mg/dL, median (IQR) | 78.5 (74.0, 85.5) | 79.0 (74.0, 84.0) | 78.0 (74.0, 84.0) | 79.0 (73.0, 87.0) | 0.66 |
| Insulin, μIU/mL, median (IQR) | 4.2 (2.0, 7.4) | 3.9 (1.0, 6.9) | 3.7 (1.0, 6.7) | 4.7 (2.1, 8.3) | 0.28 |
| HOMA, median (IQR) | 0.8 (0.3, 1.5) | 0.8 (0.3, 1.3) | 0.8 (0.2, 1.4) | 0.9 (0.4, 1.7) | 0.24 |
| CAC Agatston score, median (IQR) | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | 0.35 |
| Any CAC (>0), n (%) | 55 (11.6) | 12 (8.4) | 20 (13.8) | 23 (12.4) | 0.33 |
| CAC progression, n (%) | 66 (13.9) | 20 (14.0) | 19 (13.1) | 27 (14.5) | 0.93 |
BMI indicates body mass index; CAC, coronary artery calcification; HDL, high‐density lipoprotein; HOMA, homeostasis model assessment of insulin resistance index; HT, hormone therapy; IQR, interquartile range; LDL, low‐density lipoprotein; MET, metabolic equivalents; o‐CEE, oral conjugated equine estrogen; t‐E2, transdermal 17β‐estradiol.
χ2 test was used for categorical variables, and ANOVA or the Kruskal–Wallis test was used for continuous variables, as appropriate.
Table 2.
Medians of EAT and PAT 48‐Month Changes Overall and by Treatment Group
| EAT Volume, cm3, Median (IQR) | PAT Volume, cm3, Median (IQR) | |||||||
|---|---|---|---|---|---|---|---|---|
| Baseline | 48 mo | Change | P Value* | Baseline | 48 mo | Change | P Value* | |
| All | 36.8 (25.6, 54.5) | 37.6 (26.3, 50.5) | 1.3 (−4.9, 5.7) | 0.004 | 14.5 (10.2, 21.3) | 14.1 (10.0, 20.8) | 0.5 (−3.5, 3.9) | 0.13 |
| o‐CEE | 40.6 (28.6, 59.0) | 39.9 (28.8, 52.9) | −0.1 (−5.9, 6.1) | >0.99 | 14.6 (11.1, 22.1) | 15.1 (10.2, 22.5) | 0.9 (−3.9, 4.5) | 0.18 |
| t‐E2 | 35.9 (25.0, 48.3) | 35.7 (25.3, 46.6) | 1.7 (−4.1, 4.9) | 0.07 | 13.3 (8.6, 20.8) | 13.3 (9.4, 18.9) | 0.4 (−3.2, 3.6) | 0.32 |
| Placebo | 36.1 (25.6, 51.7) | 37.9 (25.2, 51.9) | 2.1 (−4.6, 6.4) | 0.003 | 14.9 (10.5, 22.1) | 14.4 (10.5, 20.0) | 0.2 (−3.7, 4.0) | 0.83 |
| P value† | 0.07 | 0.07 | 0.40 | 0.12 | 0.13 | 0.89 | ||
EAT indicates epicardial adipose tissue; IQR, interquartile range; o‐CEE, oral conjugated equine estrogen; PAT, paracardial adipose tissue; t‐E2, transdermal 17β‐estradiol.
Sign test.
Kruskal–Wallis test was used to compare the medians.
PAT and EAT changes did not differ significantly across treatment groups. EAT did not increase in the o‐CEE group, increased marginally in the t‐E2 group, and increased significantly in the placebo group. PAT did not change in any group (Table 2. When analyzing any increase in EAT or PAT (change >0) as related to treatment groups, women on o‐CEE had significantly lower risk of developing any increase in EAT compared with placebo and also tended to have lower risk than the t‐E2 group (Table 3.
Table 3.
Any Increase in Heart Fat Depots Over 48 Months by Treatment Group
| Treatment | EAT* | PAT† | ||
|---|---|---|---|---|
| OR (95% CI) | P Value | OR (95% CI) | P Value | |
| 0.10 | 0.66 | |||
| o‐CEE | 0.62 (0.40, 0.97) | 0.03 | 1.22 (0.79, 1.88) | 0.38 |
| t‐E2 | 0.87 (0.56, 1.35) | 0.54 | 1.12 (0.72, 1.72) | 0.62 |
| Placebo | ··· | ··· | ··· | ··· |
EAT indicates epicardial adipose tissue; o‐CEE, oral conjugated equine estrogen; OR, odds ratio; PAT, paracardial adipose tissue; t‐E2: transdermal 17β‐estradiol.
EAT: o‐CEE vs t‐E2, OR: 0.72 (95% CI, 0.45, 1.14); P=0.05.
PAT: o‐CEE vs t‐E2, OR: 1.09 (95% CI, 0.69, 1.74); P=0.48.
CAC progressed in 14% of the study participants. Women who had CAC progression were more likely to be less educated, to have a longer time since menopause, to have higher triglycerides, insulin, and insulin resistance indexes compared with women who did not show CAC progression (Table S2).
Overall, changes in EAT and PAT were not significantly associated with CAC progression (Table S3). Assigned treatment significantly modified the association between PAT changes (Figure 2B), but not EAT changes (although a similar trend was observed; Figure 2A), and CAC progression in models that were both minimally adjusted (model 1) and fully adjusted (model 2; P≤0.02; Table S4). PAT changes were associated with greater CAC progression risk only in the t‐E2 group (Figure 2B). Further adjustment for time since menopause and index of homeostasis model assessment of insulin resistance provided similar results (data not shown). The results from the sensitivity analyses that accounted for missing data with inverse probability weighting were consistent with results from the main analyses, which excluded missing data (Tables S5–S7).
Figure 2.
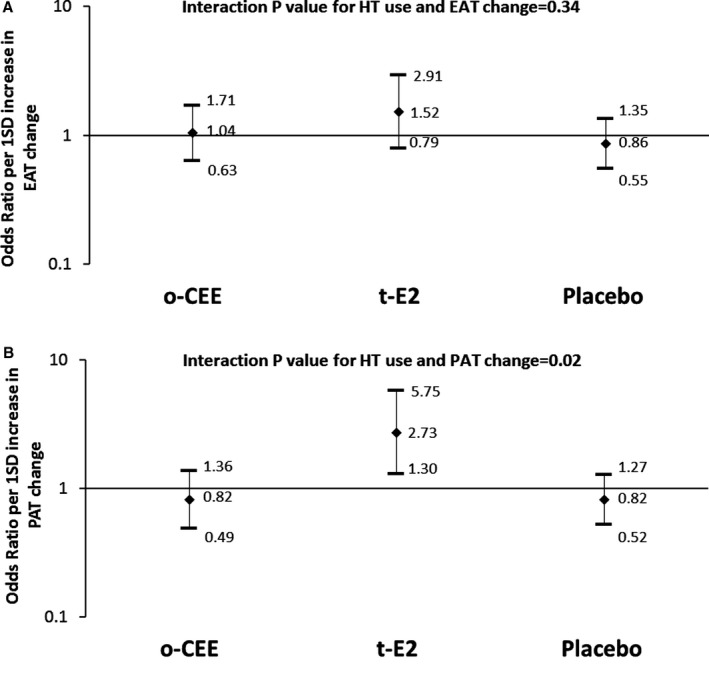
Effect modification of assigned HT use on the association between the change in EAT (A) or PAT (B) and CAC progression. Odds ratio (95% CI) represents the increase in the risk of coronary artery calcification progression per 1‐SD increase in the change of heart fat. Models adjusted for age, race and study site, education, smoking, physical activity, alcohol consumption, lipids, systolic blood pressure, waist circumference, antihypertensive medication and treatment, and baseline heart fat volume. EAT indicates epicardial adipose tissue; HT, hormone therapy; o‐CEE, oral conjugated equine estrogens; PAT, paracardial adipose tissue; t‐E2, transdermal β17‐estradiol.
Discussion
The current results provide the first evidence of potential differential impacts of exogenous estrogen preparations on heart fat accumulations and their associations with CAC development in recently menopausal women. We did not find a significant difference in PAT or EAT changes across treatment groups. However, early menopausal women on o‐CEE demonstrated no change in EAT volume over 48 months relative to women randomized to placebo, whose EAT increased over time. PAT did not change in any group. In addition, we found that the use of t‐E2, but not o‐CEE, augmented the positive association between increases in PAT and CAC progression.
Heart fat is associated with coronary arterial disease risk factors, CVD events, and all‐cause mortality.4, 5, 6, 7, 8, 9, 10, 11 Compared with premenopausal women, postmenopausal women have higher volumes of fat in heart fat depots.1 Greater PAT volume is significantly associated with atherosclerosis risk,13 and such associations are more evident at lower levels of E2,1, 13 suggesting a possible impact of estrogen on heart fat accumulation and raising the question of whether exogenous estrogen use could affect heart fat depots. Our study suggests that this is the case and that the specific exogenous estrogen preparations and/or routes of administration may contribute to or protect against heart fat deposition and CVD risk. The findings also underscore the likelihood that EAT and PAT are metabolically distinct adipose tissue depots that should be evaluated separately in assessing their contributions to CVD risk.2, 3
We are not aware of any other clinical trial of postmenopausal women that assessed the effect of exogenous estrogen on heart fat accumulations. However, several small scale clinical trials evaluated effects of different HT preparations on other fat depots and adiposity measures. Results of these studies were not consistent.29, 30, 31, 32, 33, 34, 35, 36, 37 The available data suggest that exogenous estrogen likely plays a complex role in adipose tissue accumulation that could vary by the type and route of administration of estrogen used, by the type of fat depot, and possibly by age, menopausal duration, or other population characteristics. Most recently, different HT preparations and routes of administration were found to be associated with different levels of risk for venous thromboembolism in 2 nested case–control studies that utilized large databases in the United Kingdom.38 This new evidence suggests that HT use produces different effects based on type, combination, dose, and route of administration of the preparations used.
In line with these results, we found differential effects of o‐CEE and t‐E2 on heart fat depots and their associations with CAC. Other exogenous estrogen preparations such as oral micronized 17β‐estradiol, ethinyl estradiol, and estrone might produce other effects. Interestingly, the transdermal estrogen used in this trial contains only 17β‐estradiol, whereas o‐CEE is a complex of at least 10 natural estrogens including estrone, 17β‐estradiol, and ring B unsaturated estrogens (equilin and equilenin) with almost 50% as estrone and ≤1% as 17β‐estradiol.39 Ring B unsaturated estrogens are believed to be stronger antioxidants than 17β‐estradiol and estrone, and some ring B unsaturated estrogens could act selectively in target tissues.39, 40 The stronger antioxidant property of ring B unsaturated estrogens in o‐CEE likely contributes to the observed favorable impact of o‐CEE on EAT accumulation in this study. Weak antioxidant mechanism together with overexpression of oxidative stress could contribute to adiposity‐related complications.41
Growing evidence supports vital roles of estrogen signaling in the development and function of adipose tissue.42, 43, 44, 45 Most of estrogen's effects are mediated by estrogen receptor α (ERα) and ERβ,41 both of which are expressed in human mature white adipocytes with a relative abundance of ERα.46, 47 ERα plays a critical role in maintaining adipose tissue function and preventing inflammatory damage.48 However, functionality of ERα depends on circulating estrogen levels. In the absence of adequate estrogen levels, adipocyte ERα may contribute to adipose tissue expansion and adipose tissue macrophage infiltration.48 When the ligand is lacking, ERα may bind to other estrogen response elements, adipocyte function‐specific factors, or other signaling factors that could produce detrimental metabolic effects or prompt a differential signaling program.48 It is possible that the use of t‐E2 in this study did not raise circulating estrogens to an adequate level to prevent the adverse metabolic activity of heart fat accumulation, explaining the negative impact of t‐E2 use on the association between CAC progression and PAT accumulation in our study. Interestingly, we observed a similar, but not significant, effect modification of t‐E2 on the association between EAT accumulation and CAC progression. In addition, we reported a marginal increase in EAT after 48 months of t‐E2 use.
Despite the neutral effect of HT use on CAC progression among recently menopausal women reported in the original KEEPS trial,22 we found that HT use significantly modifies the association between PAT increase and CAC progression in the same target population. The nonsignificant effect of HT use observed in the overall KEEPS trial was attributed to insufficient power to compare progression of CAC among treatment groups. In particular, KEEPS included only relatively healthy women with CAC scores <50 at baseline; therefore, it was challenging to detect modest progression in CAC with 48 months of follow‐up.22 This criterion may have limited our ability to detect main associations between heart fat depots and CAC progression, as in other studies.4, 5, 6, 7, 8, 9, 10, 11
This study has several inherent strengths and weaknesses. The ability to study a sample of well‐characterized, community‐dwelling women randomized to 2 different estrogen preparations is a paramount strength. However, some limitations are imposed by the design of KEEPS, as described earlier. Furthermore, the current findings are specific to the KEEPS population, and the durations, types, and doses of HT used cannot be readily generalized. Finally, participants excluded because of missing heart fat data at any of the 2 time points had lower PAT volumes at baseline than those included, and this may have reduced variability in PAT compared with EAT. However, sensitivity analyses accounting for missing data suggested similar findings. Regardless, this study is the first to assess the effects of 2 different routes of administration and estrogen preparations on heart fat accumulation, an evolving menopause‐specific risk factor.1, 13
Clinical Implications and Conclusions
Our results are in line with the recent North American Menopause Society position statement on HT, which underscores the differential contributions of estrogen type, dose, duration of use, route of administration, timing of initiation, and whether a progestogen is used when assessing risk related to HT use.49 We reported differential effects of HT use on heart fat depots in recently menopausal women. Use of o‐CEE may slow EAT accumulation, whereas t‐E2 appears to augment the association between PAT accumulation and CAC progression. HT remains the best therapeutic option for the relief of debilitating menopausal symptoms.49 Clinicians should continue to individualize HT prescription using best available evidence to maximize benefits and minimize risks.
Author Contributions
El Khoudary, Venugopal, and Black have full access to all of the analyzed data in the study and take responsibility for the integrity of the data. El Khoudary, Qian, and Venugopal take responsibility for the accuracy of the data analysis. Concept and design: El Khoudary, Brooks, Manson, Santoro, Black, Harman, and Budoff. Acquisition, analysis, or interpretation of data: all authors. Drafting of the article: El Khoudary, Critical revision of the article for important intellectual content: all authors, Statistical analysis: Zhao and Venugopal, Final approval of the version to be published: all authors. Heart fat ancillary study funding: El Khoudary.
Institutional Review Board Numbers for KEEPS Institutions
Central KEEPS (Kronos Early Estrogen Prevention Study) and Phoenix KEEPS (institutional review board protocol by the Western Institutional Review Board (WIRB)): study no. 1058663 and WIRB PRO no. 20040792; KEEPS (main study and cognitive substudy) no. 10‐02980 and Mammographic Density And Breast Health Ancillary Study no. 11‐05383.
Brigham and Women's Hospital (partners): 2004‐P‐002144 BWH.
Mayo Clinic: 2241‐04.
Columbia: AAAA‐8062.
Yale: 0409027022.
University of Utah: 13257.
Einstein/Montefiore: 04‐08‐213.
University of Wisconsin: H‐2005‐0059.
University of California, San Francisco: KEEPS (main study and cognitive substudy) no. 10‐02980.
University of Washington: 26702; Veterans Administration Puget Sound Health Care System: 01048.
KEEPS Investigators and Staff
Albert Einstein College of Medicine: Genevieve Neal‐Perry, Ruth Freeman, Hussein Amin (deceased), Barbara Isaac, Maureen Magnani, Rachel Wildman.
Brigham and Women's Hospital/Harvard Medical School: JoAnn Manson (PI), Maria Bueche, Marie Gerhard‐Herman, Kate Kalan, Jan Lieson, Kathryn M. Rexrode, Barbara Richmond, Frank Rybicki, Brian Walsh.
Columbia College of Physicians and Surgeons: Rogerio Lobo (PI), Luz Sanabria, Maria Soto, Michelle P. Warren, Ralf C. Zimmerman.
Kronos Longevity Research Institute: S. Mitchell Harman (PI), Mary Dunn, Panayiotis D. Tsitouras, Viola Zepeda.
Mayo Clinic: Virginia M. Mille (PI)r, Muthuvel Jayachandran, Philip A. Araoz, Rebecca Beck, Dalene Bott‐Kitslaar, Sharon L. Mulvagh, Lynne T. Shuster, Teresa G. Zais (deceased).
University of California, Los Angeles, CAC Reading Center: Matthew Budoff (PI), Chris Dailing, Yanlin Gao, Angel Solano.
University of California, San Francisco (UCSF) Medical Center: Marcelle I. Cedars (PI), Nancy Jancar, Jean Perry, Rebecca S. Wong, Robyn Pearl, Judy Yee, Brett Elicker, Gretchen A.W. Gooding; UCSF Statistical Center: Dennis Black, Eric Vittinghof, Lisa Palermo.
University of Southern California, Atherosclerosis Research Unit/Core Imaging and Reading Center: Howard N. Hodis (PI), Yanjie Li, Mingzhu Yan.
University of Utah School of Medicine: Eliot A. Brinton (PI), Paul N. Hopkins, M. Nazeem Nanjee, Kirtly Jones, Timothy Beals, Stacey Larrinaga‐Shum.
VA Puget Sound Health Care System and University of Washington School of Medicine: George R. Merriam (PI, deceased), Pamela Asberry, Sue Ann Brickle, Colleen Carney, Molly Carr, Monica Kletke, Lynna C. Smith.
Yale University, School of Medicine: Hugh Taylor (PI), Kathryn Czarkowski, Lubna Pal, Linda McDonald, Mary Jane Minkin, Diane Wall, Erin Wolff.
Others: Frederick Naftolin (co‐PI, New York University), Nanette Santoro (PI, formerly from Albert Einstein College of Medicine, currently University of Colorado).
Sources of Funding
KEEPS (Kronos Early Estrogen Prevention Study) was funded by grants from the Aurora Foundation to the Kronos Longevity Research Institute (S.M.H. and F.N., co‐PIs); from the National Institutes of Health (NIH) HL90639 to V.M.M. and R21 NS066147 to K.K.; and from the Mayo Clinic (Clinical and Translational Science Award [CTSA] UL1 RR024150), the Mayo Foundation, Brigham and Women's Hospital/Harvard Medical School CTSA UL1 RR024139, and University of California, San Francisco CTSA UL1 RR024131 from the National Center for Advancing Translational Sciences, a component of the NIH and NIH Roadmap for Medical Research. The Pfizer Company supported poststudy hormone measurements. Study medications were supplied in part by Bayer Health Care and by Abbott Pharmaceuticals. KEEPS Heart Fat ancillary study: NHLBI R21 HL140011 to El Khoudary, NIH HL094581 to Wildman. The article contents are solely the responsibility of the authors and do not necessarily represent the official view of the National Center for Advancing Translational Sciences or the National Institutes of Health.
Disclosures
El Khoudary reports grant support from NHLBI HL140011; Santoro reports consulting work for Menogenix (scientific advisory board, stock options) and Astellas/Ogeda (scientific advisory board); Miller reports grant support from NIH U54 AG44170 and the Mayo Foundation for Research and Education. The remaining authors have no disclosures to report.
Supporting information
Table S1. Baseline Variables and Coronary Artery Calcification Progression for Women Included and Excluded From the Analytical Sample
Table S2. Baseline Study Variables by 48‐Month Coronary Artery Calcification Progression
Table S3. Multivariable Logistic Regression Results of the Association Between the Change in Heart Fat Depots and Coronary Artery Calcification Progression
Table S4. Multivariable Logistic Regression Results of the Association Between the Change in Heart Fat Depots and Coronary Artery Calcification Progression by Treatment
Table S5. Inverse Probability Weighting Sensitivity Analysis for Odds Ratio (95% CI) of Any Increase in Heart Fat Depots Over 48 Months by Treatment Group
Table S6. Inverse Probability Weighting Sensitivity Analysis of the Association Between the Change in Heart Fat Depots and Coronary Artery Calcification Progression From Multivariable Logistic Regression
Table S7. Inverse Probability Weighting Sensitivity Analysis of the Association Between the Change in Heart Fat Depots and Coronary Artery Calcification Progression From Multivariable Logistic Regression by Treatment
Acknowledgments
Role of the Sponsors: The Aurora Foundation, Bayer HealthCare, Abbott Pharmaceuticals, and Pfizer Pharmaceuticals had no input into the design or conduct of the study or the writing, review, or approval of this article. We gratefully acknowledge the dedicated efforts of all the investigators and staff at the KEEPS (Kronos Early Estrogen Prevention Study) clinical centers, the KEEPS Data Coordinating Center at the Kronos Longevity Research Institute, and the National Institutes of Health institutes supporting ancillary studies. Above all, we recognize and thank the KEEPS participants for their dedication and commitment to the KEEPS research program.
(J Am Heart Assoc. 2019;8:e012763 DOI: 10.1161/JAHA.119.012763.)
References
- 1. El Khoudary SR, Shields KJ, Janssen I, Hanley C, Budoff MJ, Barinas‐Mitchell E, Everson‐Rose SA, Powell LH, Matthews KA. Cardiovascular fat, menopause, and sex hormones in women: the SWAN cardiovascular fat ancillary study. J Clin Endocrinol Metab. 2015;100:3304–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Iacobellis G, Willens HJ. Echocardiographic epicardial fat: a review of research and clinical applications. J Am Soc Echocardiogr. 2009; 22:1311–1319. quiz 1417–8. [DOI] [PubMed] [Google Scholar]
- 3. Kaushik M, Reddy YM. Distinction of “fat around the heart”. J Am Coll Cardiol. 2011;5:1640; author reply 1640‐1. [DOI] [PubMed] [Google Scholar]
- 4. Rosito GA, Massaro JM, Hoffmann U, Ruberg FL, Mahabadi AA, Vasan RS, O'Donnell CJ, Fox CS. Pericardial fat, visceral abdominal fat, cardiovascular disease risk factors, and vascular calcification in a community‐based sample: the Framingham Heart Study. Circulation. 2008;117:605–613. [DOI] [PubMed] [Google Scholar]
- 5. Mahabadi AA, Lehmann N, Kälsch H, Robens T, Bauer M, Dykun I, Budde T, Moebus S, Jöckel KH, Erbel R, Möhlenkamp S. Association of epicardial adipose tissue with progression of coronary artery calcification is more pronounced in the early phase of atherosclerosis: results from the Heinz Nixdorf Recall Study. JACC Cardiovasc Imaging. 2014;7:909–916. [DOI] [PubMed] [Google Scholar]
- 6. Mahabadi AA, Massaro JM, Rosito GA, Levy D, Murabito JM, Wolf PA, O'Donnell CJ, Fox CS, Hoffmann U. Association of pericardial fat, intrathoracic fat, and visceral abdominal fat with cardiovascular disease burden: the Framingham Heart Study. Eur Heart J. 2009;30:850–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ding J, Hsu FC, Harris TB, Liu Y, Kritchevsky SB, Szklo M, Ouyang P, Espeland MA, Lohman KK, Criqui MH, Allison M, Bluemke DA, Carr JJ. The association of pericardial fat with incident coronary heart disease: the Multi‐Ethnic Study of Atherosclerosis (MESA). Am J Clin Nutr. 2009;90:499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mahabadi AA, Berg MH, Lehmann N, Kälsch H, Bauer M, Kara K, Dragano N, Moebus S, Jöckel KH, Erbel R, Möhlenkamp S. Association of epicardial fat with cardiovascular risk factors and incident myocardial infarction in the general population: the Heinz Nixdorf Recall Study. J Am Coll Cardiol. 2013;61:1388–1395. [DOI] [PubMed] [Google Scholar]
- 9. Larsen BA, Laughlin GA, Saad SD, Barrett‐Connor E, Allison MA, Wassel CL. Pericardial fat is associated with all‐cause mortality but not incident CVD: the Rancho Bernardo Study. Atherosclerosis. 2015;239:470–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cetin M, Cakici M, Polat M, Suner A, Zencir C, Ardic I. Relation of epicardial fat thickness with carotid intima‐media thickness in patients with type 2 diabetes mellitus. Int J Endocrinol. 2013;2013:769175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abazid RM, Smettei OA, Kattea MO, Sayed S, Saqqah H, Widyan AM, Opolski MP. Relation between epicardial fat and subclinical atherosclerosis in asymptomatic individuals. J Thorac Imaging. 2017;32:378–382. [DOI] [PubMed] [Google Scholar]
- 12. Iacobellis G, Gao YJ, Sharma AM. Do cardiac and perivascular adipose tissue play a role in atherosclerosis? Curr Diab Rep. 2008;8:20–24. [DOI] [PubMed] [Google Scholar]
- 13. El Khoudary SR, Shields KJ, Janssen I, Budoff MJ, Everson‐Rose SA, Powell LH, Matthews KA. Postmenopausal women with greater paracardial fat have more coronary artery calcification than premenopausal women: the SWAN cardiovascular fat ancillary study. J Am Heart Assoc. 2017;6:e004545 DOI: 10.1161/JAHA.116.004545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nkonde‐Price C, Bender JR. Menopause and the heart. Endocrinol Metab Clin North Am. 2015;44:559–564. [DOI] [PubMed] [Google Scholar]
- 15. Hulley S, Grady D, Bush T, Furberg C, Herrington D, Riggs B, Vittinghoff E. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA. 1998;280:605–613. [DOI] [PubMed] [Google Scholar]
- 16. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J; Writing Group for the Women's Health Initiative Investigators . Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women's Health Initiative randomized controlled trial. JAMA. 2002;288:321–333. [DOI] [PubMed] [Google Scholar]
- 17. Naftolin F, Taylor HS, Karas R, Brinton E, Newman I, Clarkson TB, Mendelsohn M, Lobo RA, Judelson DR, Nachtigall LE, Heward CB, Hecht H, Jaff MR, Harman SM; Women's Health Initiative . The Women's Health Initiative could not have detected cardioprotective effects of starting hormone therapy during the menopausal transition. Fertil Steril. 2004;81:1498–1501. [DOI] [PubMed] [Google Scholar]
- 18. Manson JE, Chlebowski RT, Stefanick ML, Aragaki AK, Rossouw JE, Prentice RL, Anderson G, Howard BV, Thomson CA, LaCroix AZ, Wactawski‐Wende J, Jackson RD, Limacher M, Margolis KL, Wassertheil‐Smoller S, Beresford SA, Cauley JA, Eaton CB, Gass M, Hsia J, Johnson KC, Kooperberg C, Kuller LH, Lewis CE, Liu S, Martin LW, Ockene JK, O'Sullivan MJ, Powell LH, Simon MS, Van Horn L, Vitolins MZ, Wallace RB. Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the Women's Health Initiative randomized trials. JAMA. 2013;310:1353–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stuenkel CA, Gass ML, Manson JE, Lobo RA, Pal L, Rebar RW, Hall JE. A decade after the Women's Health Initiative—the experts do agree. J Clin Endocrinol Metab. 2012;97:2617–2618. [DOI] [PubMed] [Google Scholar]
- 20. Goodman MP. Are all estrogens created equal? A review of oral vs. transdermal therapy. J Womens Health (Larchmt). 2012;21:161–169. [DOI] [PubMed] [Google Scholar]
- 21. Harman SM, Brinton EA, Cedars M, Lobo R, Manson JE, Merriam GR, Miller VM, Naftolin F, Santoro N. KEEPS: the Kronos Early Estrogen Prevention Study. Climacteric. 2005;8:3–12. [DOI] [PubMed] [Google Scholar]
- 22. Harman SM, Black DM, Naftolin F, Brinton EA, Budoff MJ, Cedars MI, Hopkins PN, Lobo RA, Manson JE, Merriam GR, Miller VM, Neal‐Perry G, Santoro N, Taylor HS, Vittinghoff E, Yan M, Hodis HN. Arterial imaging outcomes and cardiovascular risk factors in recently menopausal women: a randomized trial. Ann Intern Med. 2014;161:249–260. [DOI] [PubMed] [Google Scholar]
- 23. Nasir K, Raggi P, Rumberger JA, Braunstein JB, Post WS, Budoff MJ, Blumenthal RS. Coronary artery calcium volume scores on electron beam tomography in 12,936 asymptomatic adults. Am J Cardiol. 2004;93:1146–1149. [DOI] [PubMed] [Google Scholar]
- 24. Agatston AS, Janowitz WR, Hildner FJ, Zusmer NR, Viamonte M Jr, Detrano R. Quantification of coronary artery calcium using ultrafast computed tomography. J Am Coll Cardiol. 1990;15:827–832. [DOI] [PubMed] [Google Scholar]
- 25. Berry JD, Liu K, Folsom AR, Lewis CE, Carr JJ, Polak JF, Shea S, Sidney S, O'Leary DH, Chan C, Lloyd‐Jones DM. Prevalence and progression of subclinical atherosclerosis in younger adults with low short‐term but high lifetime estimated risk for cardiovascular disease: the coronary artery risk development in young adults study and Multi‐Ethnic Study of Atherosclerosis. Circulation. 2009;119:382–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang G, Wang D, Zeb I, Budoff MJ, Harman SM, Miller V, Brinton EA, El Khoudary SR, Manson JE, Sowers MR, Hodis HN, Merriam GR, Cedars MI, Taylor HS, Naftolin F, Lobo RA, Santoro N, Wildman RP. Intra‐thoracic fat, cardiometabolic risk factors, and subclinical cardiovascular disease in healthy, recently menopausal women screened for the Kronos Early Estrogen Prevention Study (KEEPS). Atherosclerosis. 2012;221:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. [DOI] [PubMed] [Google Scholar]
- 28. Seaman SR, White IR. Review of inverse probability weighting for dealing with missing data. Stat Methods Med Res. 2013;22:278–295. [DOI] [PubMed] [Google Scholar]
- 29. Kristensen K, Pedersen SB, Vestergaard P, Mosekilde L, Richelsen B. Hormone replacement therapy affects body composition and leptin differently in obese and non‐obese postmenopausal women. J Endocrinol. 1999;163:55–62. [DOI] [PubMed] [Google Scholar]
- 30. Gambacciani M, Ciaponi M, Cappagli B, Piaggesi L, De Simone L, Orlandi R, Genazzani ARJ. Body weight, body fat distribution, and hormonal replacement therapy in early postmenopausal women. J Clin Endocrinol Metab. 1997;82:414–417. [DOI] [PubMed] [Google Scholar]
- 31. Reubinoff BE, Wurtman J, Rojansky N, Adler D, Stein P, Schenker JG, Brzezinski A. Effects of hormone replacement therapy on weight, body composition, fat distribution, and food intake in early postmenopausal women: a prospective study. Fertil Steril. 1995;64:963–968. [DOI] [PubMed] [Google Scholar]
- 32. Mattiasson I, Rendell M, Törnquist C, Jeppsson S, Hulthén UL. Effects of estrogen replacement therapy on abdominal fat compartments as related to glucose and lipid metabolism in early postmenopausal women. Horm Metab Res. 2002;34:583–588. [DOI] [PubMed] [Google Scholar]
- 33. Sites CK, L'Hommedieu GD, Toth MJ, Brochu M, Cooper BC, Fairhurst PA. The effect of hormone replacement therapy on body composition, body fat distribution, and insulin sensitivity in menopausal women: a randomized, double‐blind, placebo‐controlled trial. J Clin Endocrinol Metab. 2005;90:2701–2707. [DOI] [PubMed] [Google Scholar]
- 34. Aloia JF, Vaswani A, Russo L, Sheehan M, Flaster E. The influence of menopause and hormonal replacement therapy on body cell mass and body fat mass. Am J Obstet Gynecol. 1995;172:896–900. [DOI] [PubMed] [Google Scholar]
- 35. Yuksel H. Effects of postmenopausal hormone replacement therapy on body fat composition. Gynecol Endocrinol. 2007;23:99–104. [DOI] [PubMed] [Google Scholar]
- 36. dos Reis CM, de Melo NR, Meirelles ES, Vezozzo DP, Halpern A. Body composition, visceral fat distribution and fat oxidation in postmenopausal women using oral or transdermal oestrogen. Maturitas. 2003;46:59–68. [DOI] [PubMed] [Google Scholar]
- 37. Hänggi W, Lippuner K, Jaeger P, Birkhäuser MH, Horber FF. Differential impact of conventional oral or transdermal hormone replacement therapy or tibolone on body composition in postmenopausal women. Clin Endocrinol (Oxf). 1998;48:691–699. [DOI] [PubMed] [Google Scholar]
- 38. Vinogradova Y, Coupland C, Hippisley‐Cox J. Use of hormone replacement therapy and risk of venous thromboembolism: nested case‐control studies using the QResearch and CPRD database. BMJ. 2019;364:k4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bhavnani BR, Stanczyk FZ. Pharmacology of conjugated equine estrogens: efficacy, safety and mechanism of action. J Steroid Biochem Mol Biol. 2014;142:16–29. [DOI] [PubMed] [Google Scholar]
- 40. Washburn SA, Honoré EK, Cline JM, Helman M, Wagner JD, Adelman SJ, Clarkson TB. Effects of 17 alpha‐dihydroequilenin sulfate on atherosclerotic male and female rhesus monkeys. Am J Obstet Gynecol. 1996;175:341–349; discussion 349–51. [DOI] [PubMed] [Google Scholar]
- 41. Marseglia L, Manti S, D'Angelo G, Nicotera A, Parisi E, Di Rosa G, Gitto E, Arrigo T. Oxidative stress in obesity: a critical component in human diseases. Int J Mol Sci. 2014;16:378–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gao H, Dahlman‐Wright K. Implications of estrogen receptor alpha and estrogen receptor beta for adipose tissue functions and cardiometabolic complications. Horm Mol Biol Clin Investig. 2013;15:81–90. [DOI] [PubMed] [Google Scholar]
- 43. Barros RP, Gustafsson JA. Estrogen receptors and the metabolic network. Cell Metab. 2011;14:289–299. [DOI] [PubMed] [Google Scholar]
- 44. Cooke PS, Naaz A. Role of estrogens in adipocyte development and function. Exp Biol Med (Maywood). 2004;229:1127–1135. [DOI] [PubMed] [Google Scholar]
- 45. Pallottini V, Bulzomi P, Galluzzo P, Martini C, Marino M. Estrogen regulation of adipose tissue functions: involvement of estrogen receptor isoforms. Infect Disord Drug Targets. 2008;8:52–60. [DOI] [PubMed] [Google Scholar]
- 46. Dieudonne MN, Leneveu MC, Giudicelli Y, Pecquery R. Evidence for functional estrogen receptors alpha and beta in human adipose cells: regional specificities and regulation by estrogens. Am J Physiol Cell Physiol. 2004;286:C655–C661. [DOI] [PubMed] [Google Scholar]
- 47. Rodriguez‐Cuenca S, Monjo M, Proenza AM, Roca P. Depot differences in steroid receptor expression in adipose tissue: possible role of the local steroid milieu. Am J Physiol Endocrinol Metab. 2005;288:E200–E207. [DOI] [PubMed] [Google Scholar]
- 48. Davis KE, Neinast MD, Sun K, Skiles WM, Bills JD, Zehr JA, Zeve D, Hahner LD, Cox DW, Gent LM, Xu Y, Wang ZV, Khan SA, Clegg DJ. The sexually dimorphic role of adipose and adipocyte estrogen receptors in modulating adipose tissue expansion, inflammation, and fibrosis. Mol Metab. 2013;2:227–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. The 2017 hormone therapy position statement of the North American Menopause Society. Menopause. 2018;25:1362–1387. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Baseline Variables and Coronary Artery Calcification Progression for Women Included and Excluded From the Analytical Sample
Table S2. Baseline Study Variables by 48‐Month Coronary Artery Calcification Progression
Table S3. Multivariable Logistic Regression Results of the Association Between the Change in Heart Fat Depots and Coronary Artery Calcification Progression
Table S4. Multivariable Logistic Regression Results of the Association Between the Change in Heart Fat Depots and Coronary Artery Calcification Progression by Treatment
Table S5. Inverse Probability Weighting Sensitivity Analysis for Odds Ratio (95% CI) of Any Increase in Heart Fat Depots Over 48 Months by Treatment Group
Table S6. Inverse Probability Weighting Sensitivity Analysis of the Association Between the Change in Heart Fat Depots and Coronary Artery Calcification Progression From Multivariable Logistic Regression
Table S7. Inverse Probability Weighting Sensitivity Analysis of the Association Between the Change in Heart Fat Depots and Coronary Artery Calcification Progression From Multivariable Logistic Regression by Treatment