

Abstract
Background
Heart tube looping to form a 4‐chambered heart is a critical stage of embryonic heart development, but the gene drivers and their regulatory targets have not been extensively characterized at the cell‐type level.
Methods and Results
To study the interaction of signaling pathways, transcription factors (TFs), and genetic networks in the process, we constructed gene co‐expression networks and identified gene modules highly activated in individual cardiomyocytes at multiple anatomical regions and developmental stages using previously published single‐cell RNA‐seq data. Function analyses of the modules uncovered major pathways important for spatiotemporal cardiomyocyte differentiation. Interestingly, about half of the pathways were highly active in cardiomyocytes at the outflow tract (OFT) and atrioventricular canal, including well‐known pathways for cardiac development and many newly identified ones. We predicted that these OFT‐atrioventricular canal pathways were regulated by a large number of TFs actively expressed at the OFT–atrioventricular canal cardiomyocytes, with the prediction supported by motif enrichment analysis, including 10 TFs that have not been previously associated with cardiac development (eg, Etv5, Rbpms, and Baz2b). Furthermore, we found that TF targets in the OFT–atrioventricular canal modules were most significantly enriched with genes associated with mouse heart developmental abnormalities and human congenital heart defects, in comparison with TF targets in other modules, consistent with the critical developmental roles of OFT.
Conclusions
By analyzing gene co‐expression at single cardiomyocytes, our systematic study has uncovered many known and additional new important TFs and their regulated molecular signaling pathways that are spatiotemporally active during heart looping.
Keywords: bioinformatics, genetics, scRNA‐seq, transcription factors, transcriptome
Subject Categories: Functional Genomics, Gene Expression & Regulation, Genetics
Clinical Perspective
What Is New?
This study identifies gene modules that show distinct and enriched spatiotemporal expression in murine embryonic cardiomyocytes at the single‐cell level, and characterizes their relevance to functional pathway and congenital heart defects.
Using new bioinformatics approaches, this study investigates the transcription factor regulatory networks regulating these modules systematically.
What Are the Clinical Implications?
This work can help in better understanding the regulatory targets and functional roles of many well‐known cardiac transcription factors, but also predicts additional transcription factors likely playing similar roles in embryonic cardiomyocyte development.
Further studies are needed to confirm the molecular functions of the cardiac transcription factors newly implicated in embryonic heart developments and heart diseases.
Introduction
Prenatal heart development is controlled by evolutionarily conserved genetic networks1, 2 consisting of well‐recognized transcription factors (TFs) (eg, Gata4, Nkx2‐5, and Tbx5),3, 4, 5 signaling pathways (eg, WNT and BMP),6, 7 and dynamic epigenetic networks that are modulated by histone modifications or DNA methylation.8, 9, 10 These networks ensure the precise establishment of gene expression pattern and concurrent differentiation of cardiovascular cell types in an orderly spatial and temporal manner. However, transcriptional regulatory networks involved in cardiac looping have not been fully deciphered, partially because of the lack of appropriate experimental technology for resolving cellular and developmental heterogeneity.
The introduction of single‐cell RNA‐seq technology has started to overcome this limitation. It revolutionizes the studies of gene regulation in embryonic developments by providing a systematic and high‐throughput way to profile the expression of hundreds to thousands of cells simultaneously, resulting in the discovery of new cell types, cell state transitions and functions, as well as gene markers that are associated with unique cells populations and functions.11, 12, 13, 14, 15, 16, 17, 18 The single‐cell RNA‐seq (scRNA‐seq) data, however, have generally not been fully exploited for addressing the dynamic expression of transcription factors and their cooperative or competitive interactions, even though it is well appreciated that the expression and targets of TFs are fundamental for deciphering the cardiac genetic programs.
In this study, we took a systematic approach to analyze scRNA‐seq data to characterize gene co‐expression in differentiating cardiomyocytes at multiple anatomical locations of early cardiac developmental stages, and to investigate their regulation and maintenance by TFs. Our results indicate that the genetic programs for cardiac cell differentiation at the outflow tract–atrioventricular canal (OFT‐AVC) are extremely complex, involving many critical pathways that are regulated by a significantly large number of TFs. This finding suggests that mutations in the genes regulating OFT‐AVC development likely confer a high risk for congenital heart defects (CHD).
Methods
The authors declare that all supporting data are available within the article and its online supplemental files. The scRNA‐seq data sets were obtained from previous publications12, 14; data set 1 is accessible from the Gene Expression Omnibus (GEO; GSE76118), and data set 2 can be obtained from the GNomEx database (https://b2b.hci.utah.edu/gnomex/), with accession numbers 272R, 274R, 275R, 276R, 277R1, 279R1, 280R, 281R, and 439R.
Single Cell RNA‐Seq Data
The first single‐cell transcriptional data set of murine embryonic cardiac cells (data set 1) was obtained from GSE76118. The cells were captured from different anatomical locations at different time points and contained a total number of 2233 cells: cardiomyocytes (1252), epicardial cells (EPs) (40), endothelial cell (ECs) (191), mesenchymal cells (MCs) (281), and the cells with 2 or more cell‐type characters (360).14 The second data set (data set 2)12 was downloaded from the GNomEx database, with accession numbers 272R, 274R, 275R, 276R, 277R1, 279R1, 280R, 281R, and 439R. The 869 cells were mainly isolated from atrium or ventricle of murine heart cells from embryonic to postnatal days, with 484 cardiomyocytes, 146 ECs, 104 fibroblast‐enriched cells, and 135 other cells.
Data Processing and Quality Control
Raw counts of gene expression data in data set 1 were directly obtained from the GEO, with read aligned by STAR (2.4) and expression qualified by HTSeq (0.6.1), according to the original authors.14 For data set 2, only fastq files were available in the GNomEx database. Thus, we used the HISAT219 to align reads to the mouse genome (GRCm38.79) with its default “soft‐clip adapters,” followed by SAMtool 1.420 to generate alignment files and HTSeq (0.6.1)21 to compute read counts. To remove outlier cells (eg, doublets or abnormal cells), we qualified each cell by its total UMI and the number of expressed genes, and discarded cells not within standard normal distribution. After these quality controls, in data set 1, read counts of cells ranged from 0.55 to 3.3 million, and the numbers of expressed genes per cell were >4000. Similarly, cells in data set 2 were retained with counts from 0.3 to 2.1 million and >2500 expressed genes. Furthermore, genes with reads in <50% of cells were discarded, resulting in 12 792 genes for further analysis, including 1275 TF genes, 9694 protein‐coding genes, and 1823 noncoding genes.
Cell Clustering, Dimension Reduction, and Trajectory Construction
The raw gene count data were processed using packages tidyr (v0.8.0),22 dplyr (v0.7.4),23 and scater (v1.6.1) packages24 in R. Clustering cells was carried out mainly with principal component analysis, t‐distributed stochastic neighbor embedding by the package Rtsne (0.13),25 and k‐means clustering by the package SC3 (1.6)26 in R. Ordering cells in a trajectory corresponding to a biological process was also performed by the package monocle (v2.6.1).27 We carried out this analysis for atrial cardiomyocytes, using data from left atrium, right atrium, and AVC of E9.5 and 10.5, ventricular cardiomyocytes, using data from left ventricular septum, right ventricular septum, left ventricle, and right ventricle of E9.5 and 10.5, and OFT cardiomyocytes, using data from OFT, proximal outflow tract, and distal outflow tract. In each case, the top 1000 most dispersed genes were used in an unsupervised manner. Furthermore, plots in this study were generated by ggplot2 (v2.2.1)28 while heat‐map was built by pheatmap (v1.0.8)29 in R.
WGCNA Co‐Expression Network
Signed weighted gene co‐expression network was built from the expressed genes in data set 1, using the package weighted gene co‐expression network analysis (WGCNA) (v1.61)30 in R, with the scale‐free topology fit index over 0.8. More specifically, we estimated soft thresholding power and chose 10 to construct gene network using data set 1. In WGCNA, the adjacencies are used to represent gene connection strengths of gene network. In signed weighted network,
where Cor is correlation coefficient, and power is soft‐thresholding power.
From the intramodular connectivity analysis, we defined hub genes as the top connected genes (eigengene‐based connectivity, or called signed connectivity, between the gene and module eigengene [kME] ≥0.25).
Consensus Module Analysis
The preservation of the gene modules from data set 1 was supported by consensus module analysis using the package WGCNA.31 The analysis built consensus modules by combining data set 1 and data set 2 and then tested whether the modules generated with data set 1 alone were preserved in the consensus modules (ie, if the genes of a given module in data set 1 were significantly enriched in any consensus module from both data sets, with P<0.05). The Cytoscape32 and Gephi tools33 were used to visualize gene co‐expression networks of the modules; the node size represented connections and the length of an edge indicated the strength of the connection.
Differential Expression Analyses
The differentially expressed (DE) genes of different single‐cell clusters or subgroups were identified by the package edgeR (v3.20.1),34 with adjusted P<0.05 as significant. Batch effects were removed when detecting differentially expressed genes, including spatial factor, temporal factor, and the condition variable.
Microarray Data and Differentially Expressed Analysis
The microarray of cardiomyopathy patients was obtained from the GEO (GSE57338),35 and the data for Bmp2/4, Hey2, Nr2f2, Tbx3, and Tbx5 were also downloaded from the GEO, with accession numbers GSE34502, GSE6526, GSE63759, GSE73862, and GSE77576, respectively.36, 37, 38, 39, 40 All the microarray data were processed using the package oligo (v1.42.0)41 in R, and the DE genes were detected using the package limma (v3.34.5),42 with adjusted P<0.05.
Gene Sets, Phenotype, and Disease‐Associated Enrichment Analyses
Pathway (GO, KEGG and Reactome)43, 44, 45 and gene set enrichment analyses were performed using the ToppGene suite46 and gene set enrichment analysis (v3.0),47 respectively, with FDR <0.05 as a cutoff in David and FDR <0.1 in gene set enrichment analysis. Phenotype and disease‐associated enrichment analyses were also carried out using the ToppGene suite, with false discovery rate (FDR) <0.05. The enriched phenotype and disease terms (IDs) from the ToppGene were cross‐compared with the disease category information in the MGI (Mouse Genome Informatics),48 IMPC (International Mouse Phenotyping Consortium),49 and DisGeNET50 databases to retrieve terms related to cardiovascular systems, heart phenotypes, and cardiac diseases, resulting in 395 gene sets associated with abnormal heart phenotypes and 99 gene sets linked to cardiac diseases.
Cardiac, Literature‐Based, and Noncardiac TFs
To uncover potentially novel TFs involved in regulating cardiac development, we used heart phenotype‐ or disease‐associated gene sets from the MGI, IMPC, and DisGeNET databases, and literature search to separate TFs into 3 groups. “Cardiac TFs” were the TFs that have been associated (ie, annotated) within any of the heart‐phenotype gene sets from the 3 databases. The rest were then classified either as “literature‐based cardiac TFs,” if there was a literature support for a function in heart development, or otherwise as “noncardiac TFs,” to indicate their unknown roles in cardiac development or function.
TF‐Target Analysis
One hundred twenty‐three TFs considered as key drivers were selected by these criteria: (1) a member of the 5 selected modules (4, 5, 6, 7, 8), (2) high expression (average cpm >1 in the given module), and (3) a hub gene in the module. To predict the TF‐target relationship, we used the adjacency scores computed by WGCNA. Considering all the genes (n=12 792) in the signed weighted gene co‐expression network of data set 1, the top 20% adjacency values were ≥0.0017. Thus, for a TF, any gene with an adjacency ≥0.0017 was considered to be a candidate target of this TF. In TF‐target preservation analysis, this cutoff (adjacency ≥0.0017) was also applied to define TF‐targets in consensus gene co‐expression networks from both data sets 1 and 2. Because TF‐target analysis was based on the top adjacency, a TF and some of its targets might be in different modules, and our TF‐target consensus analysis included all the targets. On the other hand, to investigate the regulatory roles of TFs in the spatiotemporal modules, our enrichment analysis of a TF's targets discarded the targets that were in different module from the TF.
TF‐Driver Score
To investigate and predict the effects of a TF on a given pathway, GO term, or heart phenotype gene set, we imported an approach, termed “key driver analysis,”30, 51, 52, 53, 54 which estimates the functions of a key driver by enrichment analysis of the driver's targets. The targets of 123 TFs were qualified by this approach, and we scored the results by the following formula,
where Ts: TF targets in the pathway; Ttotal: total TF's targets; MEs: the over‐representing module genes in the pathway; MEtotal: total module genes
For each TF, we first investigated its module and the pathways, Gene Ontology (GO) terms and gene sets enriched in this module, then performed the enrichment analysis of this TF's targets, and finally selected the overlap between the 2 enrichment results. After determining the pathways, GO terms, and gene sets, we detected TF‐driver scores of individual TFs to explore their potential functions.
DNA Motif Enrichment Analysis
To obtain regulatory regions of mouse genes, we downloaded the murine heart E10.5 H3K27ac ChIP‐seq data from the ENCODE project55 (ENCSR582SPN). The peaks, corresponding to either promoters or enhances, from −50 kb to +50 kb of the transcription start site of a gene were used for motif analysis. The enrichment of TF binding motifs was performed using the AME (v4.12.0)56 of the MEME suite (FDR <0.05). Motif visualization and similarity analysis were carried out using package motifStack (v1.22.0)57 and MotIV (v1.34.0)58 in R.
Animals
Wild‐type CD1 mice were obtained from Charles River Laboratories. All mouse experiments were performed according to the guideline of the National Institute of Health and the protocol approved by the Institutional Animal Care and Use Committee of Albert Einstein College of Medicine. Noontime on the day of detecting vaginal plugs was designated as E0.5.
RNA Extraction and Quantitative Polymerase Chain Reaction
The hearts were isolated from E9.5 and E10.5 embryos and separated into 3 parts: AVC, OFT, and ventricle. Tissues from 4 hearts were pooled as 1 sample and subjected to total RNAs extraction using Trizol (Life Technology) according to the manufacturer's manual. First‐strand cDNA was synthesized using the Superscript IV Reverse Transcriptase Kit (Life Technology). Quantitative polymerase chain reaction was performed using the Power SYBR Green PCR Master Mix (Life Technology). Gene‐specific primers were used (Table S1). The relative expression level of genes was normalized to the expression level of Gapdh and calculated using the 2‐ΔΔCT method. Biological repeats were performed using 4 different samples. Student t test was used for comparison between groups and the P value <0.05 was considered significant.
Results
Extraction of CM scRNA‐seq Data
The general approaches of our study are composed of construction of gene coexpression networks from scRNA‐seq data, functional enrichment analysis, and prediction of genes and pathways regulated by transcription factors (Figure 1). Our study started from the scRNA‐seq data that were previously obtained from mouse embryonic hearts at embryonic day (E) 8.5 to postnatal day (P) 21.12, 14 In these 2 studies, ≈3000 single cells in total were captured for RNA‐seq from multiple dissected heart zones (eg, outflow tract and left atrium) at different developmental stages (Figure 2). To date, these scRNA‐seq data remain highly unique for their high gene coverage and anatomical spatial information, when compared with many subsequent scRNA‐seq data obtained by droplet‐based platforms,59, 60 and thus are especially suitable for our study. The data sets were composed of multiple cardiac cell types, including endothelial cells (EC), MC, and fibroblast‐enriched cells, and cardiomyocytes. To identify cardiomyocytes from these data, we used unsupervised clustering methods, similarly to the original reports. Principal component analysis showed that cells from each of the 2 studies (“data set 1” and “data set 2”) could be segregated into 2 parts, confirmed by k‐means clustering and t‐Distributed Stochastic Neighbor Embedding analysis (Figure S1A and S1D). Based on the expression of known cardiac markers, we were able to identify the cardiomyocytes (expressing Tnnc1 and Tnni3), EC (expressing Cdh5 and Tek), and MC (expressing Col1a1 and Ptn) clusters (Figure S1E). Although we were unable to directly compare our cell clustering result to the original reports because cell type information was not provided in the publicly released data,12, 14 the proportion of cells in these clusters and the expression profiles of known markers demonstrated that we had reproduced the original clustering sufficiently and separated cardiomyocytes from other cardiac cells successfully (Figure S1E).
Figure 1.
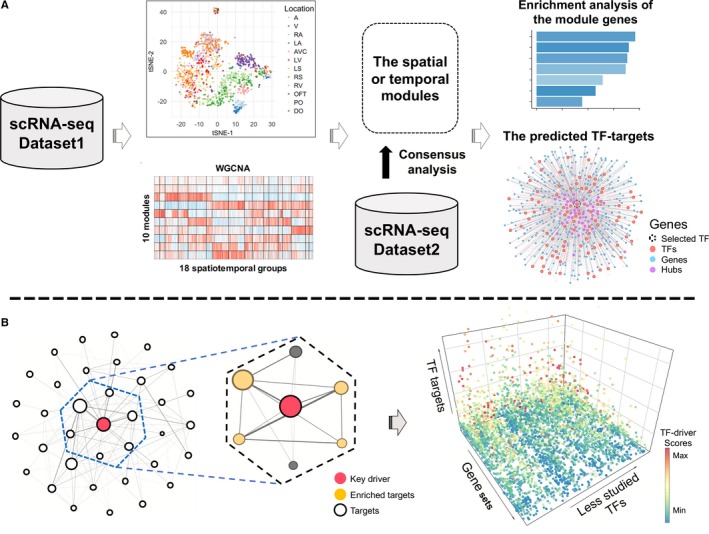
Workflow for inferring transcription factor (TF)‐target regulatory network. A, Overview of our approach for identifying and characterizing gene co‐expression patterns in cardiomyocytes. The t‐distributed stochastic neighbor embedding (tSNE) map indicates the gene expression similarity among cardiomyocytes, while the weighted gene co‐expression network analysis (WGCNA) heatmap shows co‐expression patterns of genes across cardiomyocytes in different spatial or temporal subgroups. The plot for the predicted TF targets display a TF (dash circle) and its targets that are in the same gene module, with colors for different types of module genes. B, Schematic illustration of function analysis for TF targets. Left panel shows that the potential functional roles of a TF could be predicted by enrichment analysis of its targets, with the dashed box highlighting targets of a TF (red). Right panel shows the enrichment results from the analysis of targets for less‐studied TFs, with colors for TF‐driver scores that quantify the potential regulatory effects of a TF on individual gene sets. See Methods for details. A indicates atrium; AVC, atrioventricular canal; DO, distal outflow tract; LA, left atrium; LS, left ventricular septum; LV, left ventricle; OFT, outflow tract; PO, proximal outflow tract; RA, right atrium; RS, right ventricular septum; RV, right ventricle.
Figure 2.
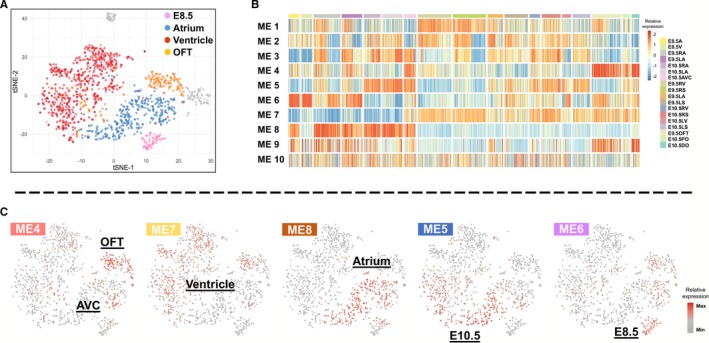
Identification of gene expression patterns for cardiomyocytes at different anatomical location or developmental stage. A, The t‐SNE map of the cardiomyocytes in data set 1. B, Ten gene modules generated by WGCNA (weighted gene co‐expression network analysis). Columns of the heatmap represent individual cardiomyocyte cells, grouped by their anatomical and developmental stage origins, while each row is a gene module, with heat‐map colors indicating the relative expression of its eigengene. C, Module eigengene patterns at t‐SNE map of cardiomyocyte cells, with colors for module eigengene expressions. AVC indicates atrioventricular canal; OFT, outflow tract; t‐SNE, t‐distributed stochastic neighbor embedding.
Spatiotemporal‐Specific Transcriptional Heterogeneity of Cardiomyocytes
As cardiomyocytes were captured at different developmental stages and anatomical locations, we first studied the contributions of these 2 factors (time and location) and additional technical factors to gene expression variation (see Methods). We found that the number of expressed genes in individual cardiomyocytes and anatomical origin explained the most expression variations, followed by developmental stage, but sequencing depth (“counts”) was not a main factor (Figure S2A). Thus, we considered that the gene expression heterogeneity across cardiomyocyte groups mostly reflected biological difference, consistent with the original reports.12, 14 We should point out that in this study we did not directly integrate the 2 data sets but analyzed them together, as they were generated with 2 quite different strategies and thus could have batch effects. Data set 1 served as the primary data source for our analyses and conclusions, while data set 2 was used mostly to confirm our findings of gene co‐expression among cardiomyocytes at different anatomical locations or developmental stages.
Accounting for developmental stages and anatomical locations, the cardiomyocytes in data set 1 could be approximately classified into 4 main clusters (Figure 2A). Excluding outliers (cells in gray in Figure 2A), E8.5 cardiomyocytes appeared as 1 cluster, while the cardiomyocytes at E9.5 and E10.5 could be segregated into 3 clusters based on anatomical locations: “atrial cluster” (left atrium, right atrium, and AVC), “ventricular cluster” (LV, left ventricular septum, RV, right ventricular septum), and “outflow tract cluster” (E9.5 outflow tract and distal outflow tract) (Figure 2A and Figure S2B). We also noticed that E10.5 cardiomyocytes from proximal outflow tract showed higher gene expression similarity to RV cardiomyocytes than E9.5 OFT or E10.5 distal outflow tract cardiomyocytes. In summary, the E8.5 to E10.5 cardiomyocytes can be separated mainly by their temporal (developmental stages) and spatial (anatomical locations) origins that reflect their developmental trajectories.
We next carried out signed WGCNA (see Methods) to define co‐expressed gene modules, and studied their relationship to the 4 main cardiomyocytes clusters. The analysis resulted in 10 gene modules from the 12 792 genes expressed in both data sets (Figure 2B), with the module sizes ranging from 63 to 7524. Interestingly, the 10 modules contained different proportions of noncoding genes and TFs (Figure S2C and Table S2), suggesting potentially different degrees of regulation complexity. A comparison of the WGCNA result with the t‐distributed stochastic neighbor embedding cell clustering map and cell sample origins revealed that some of the module eigengenes, representing the first principal component of gene expression profiles in individual modules, displayed a strong correlation with specific cardiomyocytes clusters or groups (Figure 2B and 2C, Figure S2B). Therefore, our work unveiled cardiomyocytes from different temporal or spatial origins, for which we identified genes with highly similar expression patterns. To address the robustness of the gene modules, we performed consensus module analysis by incorporating the scRNA‐seq data of cardiomyocytes from E9.5 to 18.5 in data set 212 (see Methods). The results confirmed that all gene modules were reproducible (ie, “preserved,” Figure 3 and Figure S3), indicating that most modular genes have similar co‐expression correlation in the 2 data sets, and the gene expression pattern is most likely a result of gene programs regulating embryonic cardiomyocytes development.
Figure 3.
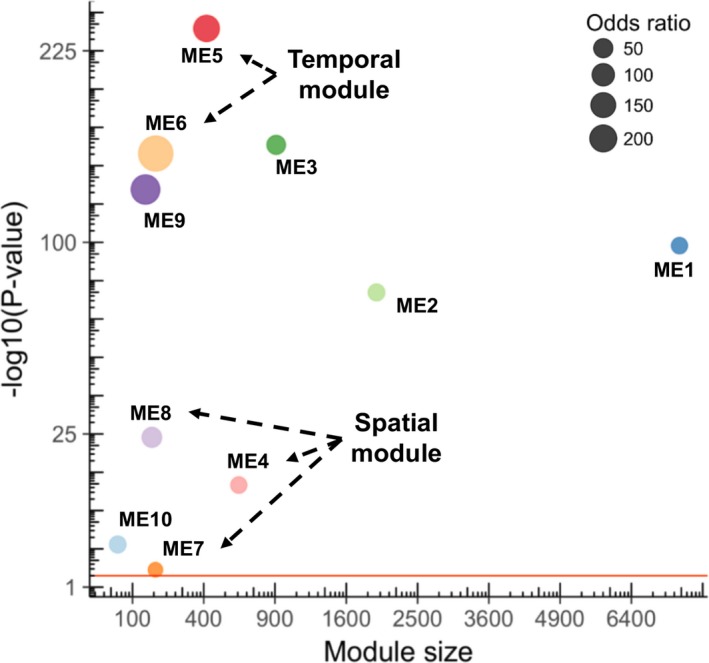
Preservation analysis of the weighted gene co‐expression network analysis modules. The dot size indicates the odds ratio and the y‐axis shows the significance. The red line indicates the threshold (P<0.05) for significant preservation.
Functional Analysis of the WGCNA Module Genes
We performed GO and pathway analyses to characterize the enriched functions of module genes (see Methods), focusing on the spatial and temporal modules (Figure 4A and Table S3). In the temporal modules, the genes of module 6 (E8.5 cardiomyocytes module) were enriched for function terms in regulation of cell differentiation, positive regulation of gene expression, and response to endogenous stimulus, including module genes Gsk3b, Notch2, and St13 (Figure 4B), indicating that this gene module is mainly involved in cell differentiation, concordant with the proliferation of early‐stage cardiomyocytes.61, 62 The module 5 (E10.5 cardiomyocytes module) genes were enriched in oxidative phosphorylation, electron transport chain, and cardiac muscle contraction signaling known to be highly active in late‐stage cardiomyocytes.63 Not only did this module include troponin and ryanodine receptor genes, but also it contained a number of important electron transport genes from mitochondrial genome, such as mt‐Co2, mt‐Co3, and mt‐Atp6 (Figure 4B), indicating distinct enhancement of mitochondrial ATP synthesis along with cardiomyocyte development (Table S3).
Figure 4.
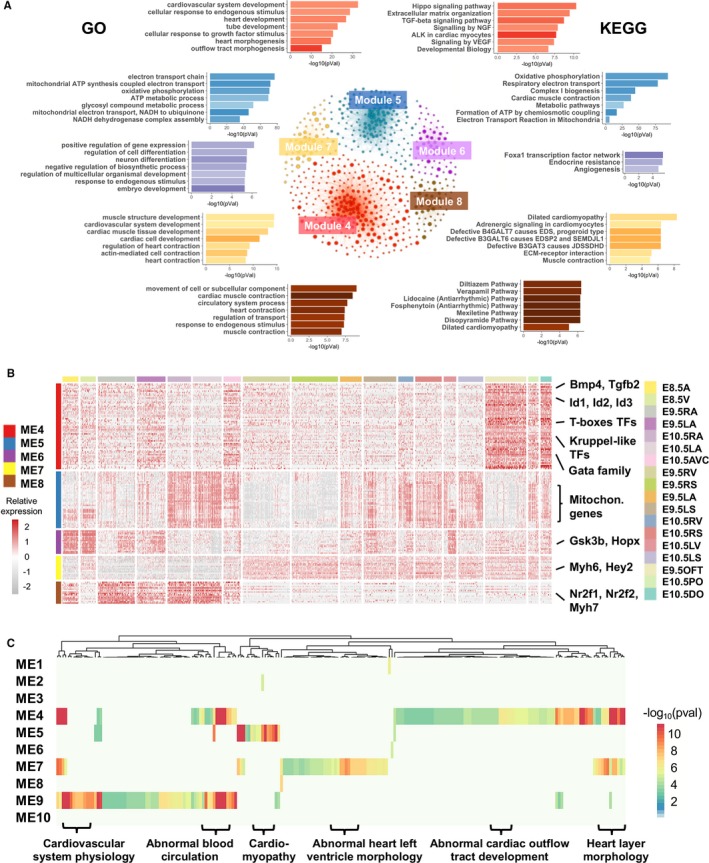
Enrichment analysis of genes in selected modules. A, The top 7 representative enriched Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways in the 5 selected modules. The networks in the middle illustrate the co‐expression networks of genes in these modules, with the nodes representing genes and node sizes proportion to the numbers of co‐expressed genes. Functional terms and pathways for individual modules are depicted by matching colors. B, Expression patterns of hub genes in 5 selected modules. C, Enriched phenotype‐ and disease‐associated gene sets in the modules. The gene sets (columns) associated with heart phenotypes and diseases were enriched in the gene modules. The colors in the heat‐map show statistical significance of enrichments, and some representative gene sets are labeled at the bottom. ALK indicates Anaplastic lymphoma kinase; EDS, Ehlers‐Danlos syndrome; EDSP, Ehlers‐Danlos syndrome progeroid; JDSSDHD, joint dislocations, short stature, craniofacial dysmorphism, and congenital heart defects; SEMDJL, Spondyloepimetaphyseal dysplasia with joint laxity; NGF, nerve growth factor.
In contrast, the OFT‐AVC module genes (from spatial module 4) were enriched for terms important in cardiac morphogenesis, tube development, cardiovascular system development, and cellular response to growth factor stimulus. These processes have been reported to activate OFT remodeling.62 Interestingly, transforming growth factor β (TGFβ), bone morphogenetic protein (BMP), and SMAD pathways, with their key regulators Bmp2/4, Tgfb2, Id1/2/3, and Smad 6/7 (Figure 4B), were enriched in module 4; they are known for roles in OFT and/or AVC development and morphogenesis.7, 62, 64 Interestingly, our analysis showed that genes in additional signaling pathways, such as nerve growth factor, vascular endothelial growth factor, SCF (stem cell factor)‐KIT, platelet‐derived growth factor, mitogen‐activated protein kinase, AKT, insulin‐like growth factor, interleukins, leptin, and insulin, were also significantly overrepresented in this module. These results indicate that OFT‐AVC formation and remodeling are tightly regulated by a complex network of signaling pathways in response to both external growth factors and endogenous stimuluses. Our finding is consistent with previous studies of protein and gene networks critical for heart development,65, 66 but our analysis also uncovered additional novel pathways that were not described before, such as DAP12 (also called KARAP) signaling (Figure 4A and Table S3). For module 7 (ventricular cardiomyocytes module), function analysis identified enriched terms in muscle structure development, cardiovascular system development, and regulation of cardiac contraction. The module 8 genes, actively expressed in atrial cardiomyocytes, exhibited significant enrichments in cell movement, response to endogenous stimulus, and ion signaling. Additionally, some well‐known cardiac genes involved in cardiac contraction were members of these 2 modules, such as cardiac myosin Myl2 and Myh7 in module 7, and Myh6 and tropomyosin Tpm2 in module 8 (Figure 4B and Table S2). All these results emphasize that our unbiased and systematic function analysis of spatiotemporal module genes uncovered both known and potentially novel signaling pathways in cardiomyocytes at different anatomical locations or heart looping stages, thus providing molecular profiles for the diverse developmental processes and cellular functions in embryonic cardiomyocytes.
Modular Enrichment of Cardiac Phenotype‐ and Disease‐Associated Genes
We next addressed how genes associated with different heart phenotypes or diseases were distributed in these gene modules, using the ToppGene and Fisher exact test (see Methods). Surprisingly, the results indicated that even though modules 1, 2, and 3 contained >80% of the expressed genes, phenotype or disease marker genes were most significantly enriched in modules 4, 5, 7, and 9 (adjusted P<0.05) (Figure 4C and Table S4). These results support the value of our demarcation of gene modules, as it separated correctly the developmental and disease‐relevant gene modules from others. In addition, this analysis uncovered the genetic relationship between developmental process and heart abnormalities, thus highlighting the developmental bases of some heart diseases.
Prediction of Targets for Spatiotemporal Transcription Factors
After studying the spatiotemporal expression of genes by co‐expression networks and functional enrichment analysis, we set out to address the primary goal of the current study: how the spatiotemporal expression and networks were regulated and maintained. In total, 186 TFs were present in the 5 selected modules (temporal‐ or spatial‐specific modules). One hundred three of them were highly expressed in module 4 (OFT‐AVC module), including T‐boxes TFs (Tbx2, Tbx3, and Tbx20), Kruppel‐like TFs (Klf3, Klf4, Klf6, and Klf7), Gata family (Gata2, Gata3, Gata5, and Gata6) and Id family (Id1, Id2 and Id3), as well as a large number of TFs whose functions in cardiac development remain unclear, like Bach2, Etv5, and Skil. We also found that 12 TFs were in the E10.5 module (module 5), 27 TFs were active expression in the E8.5 module (module 6), 20 and 24 TFs in ventricular module (module 7) or atrial module (module 8), respectively, including well‐studied cardiac genes Hey2, Irx4, Sox5, and Tbx5. The expression patterns of some representative TFs in these modules are shown in Figure 5A.
Figure 5.
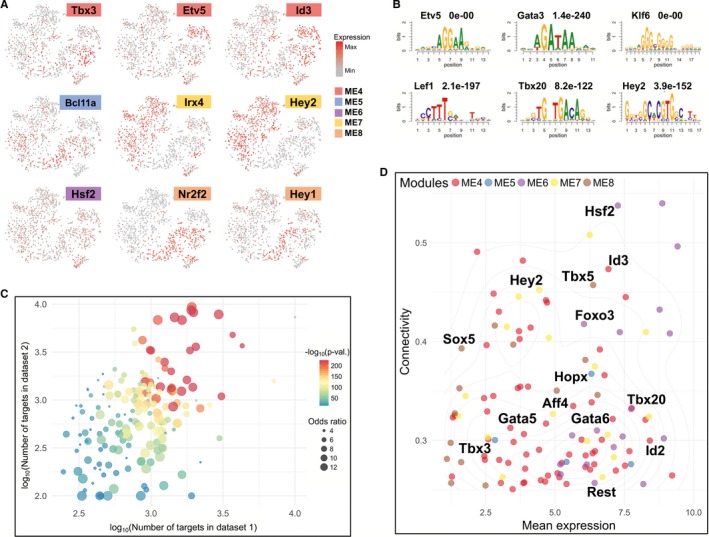
Spatiotemporally expressed TFs and their potential targeted genes. A, Expression patterns of 9 representative TFs (transcription factors) in the selected modules. B, The enriched motifs in the predicted targets of the representative TFs, with TF names and q‐values at the top. C, Preservation of TF‐targets. Bubbles in the plot represent different TFs, with colors for the significance of preservation and sizes for odds ratios of target overlap. D, Mean expression and eigengene‐based connectivity of the candidate TF drivers (n=123). In plot, x‐axis represents the correlations between TFs’ expression profiles and their corresponding module eigengenes, while y‐axis represents the average expression values for TFs. TFs indicates transcription factors.
To predict the targets for individual TFs, we used the adjacency scores computed by WGCNA for quantifying the co‐expression strengths between genes, whereas we considered a gene as a TF's target if their pairwise adjacency score was at the top 20% of all adjacency scores in the full co‐expression network (see Methods). As such, TFs in the same module could have a very different list of targets. To support our TF‐target prediction, we first performed motif analysis for 82 TFs that have known DNA binding motifs. The result showed that for ≈80% of them, the corresponding motifs (or the motifs for the same TF family) were significantly enriched in the regulatory regions (defined by enriched H3K27ac modification, see Methods) of their targets (Figure 5B and Table S5). Next, we re‐analyzed several published microarray gene expression data sets, in which the expression of Hey2, Nr2f2, Tbx3, and Tbx5 in hearts was disrupted.36, 37, 38, 39, 40 We asked whether the differentially expressed genes upon knockdown or knockout of these TFs were correlated with the targets predicted by our method, using the χ2 test. The results showed significant correlations, Hey2 (P<2.2e‐16), Nr2f2 (P=1.246e‐06), Tbx3 (P=0.00056), and Tbx5 (P=1.457e‐05). Lastly, we repeated the TF‐target prediction using the consensus gene network built by both scRNA‐seq data sets and found that for 184 of the 186 TFs the predicted targets were reproducible (P<0.05) (Figure 5C and Table S6, see Methods). To sum up, all these analyses and results support our TF‐target prediction.
To study TFs with potentially major roles in cardiac‐looping stage cardiomyocyte development, we then focused on TFs that are likely “key drivers,” central nodes in a co‐expression network,30, 51, 52, 53, 54 and studied what functional pathways they may regulate. Specifically, we selected TFs that were hub genes and actively expressed in individual modules, resulting in 123 TFs (Figure 5D and Table S6, see Methods).
Function Analysis of Well‐Known Transcription Factors Across Cardiac Looping Stages
From the 123 TFs, we started with 64 that were previously associated with cardiac phenotypes (referred to as “cardiac TFs,” see Methods), and asked in which pathways their targets were involved. Since we have already determined pathways, function terms, and heart phenotype‐associated gene sets enriched in each module, we only analyzed the enrichments of a TF's targets in these pathways or terms. The analysis identified key drivers by “TF‐driver scores,” which tested the association between a TF's targets and an enriched pathway or term in a gene module (see Methods). As an example, shown in Figure 6A, TGFβ signaling pathway was enriched in module 4 (Table S3), containing 17 module‐4 genes. Among them, 12 genes were predicted targets of Id3 and 7 for Pitx2, while both Id3 and Pitx2 were in this signaling, with TF‐driver score 1.28 and 2.84, respectively. This result suggested that these 2 TFs could be key drivers of this pathway (driver score >1, see Methods), and that Id3 was likely more important than Pitx2 to the TGFβ signaling because the former putatively regulated more targets in this pathway, consistent with a previous report that Id3 was directly activated by BMPs in TGFβ signaling.64 Extending this TF driver analysis to all the 64 cardiac TFs, we found that ≈80% (n=51) of cardiac TFs were likely key regulators of the signaling pathways enriched in our 5 selected modules, with most cardiac TFs controlling multiple pathways (Figure 6B, Figures S4 and S5 and Table S7), such as the well‐studied Gata family, T‐boxes TFs, Gli3, Hey1/2, Id1/3, Klf6, Loxl2, Mecom, Mef2c, and Nr2f2. On the other hand, ranking the pathways by the number of TF regulators, we found that regulations of the pathways in cardiomyocyte differentiation appeared extremely complex; the top‐ranked pathways, such as extracellular matrix organization, elastic fiber formation, TGFβ signaling, Hippo signaling, axon guidance, and Anaplastic lymphoma kinase (ALK) in cardiac myocytes, were predicted to be regulated by >30 cardiac TFs simultaneously.
Figure 6.
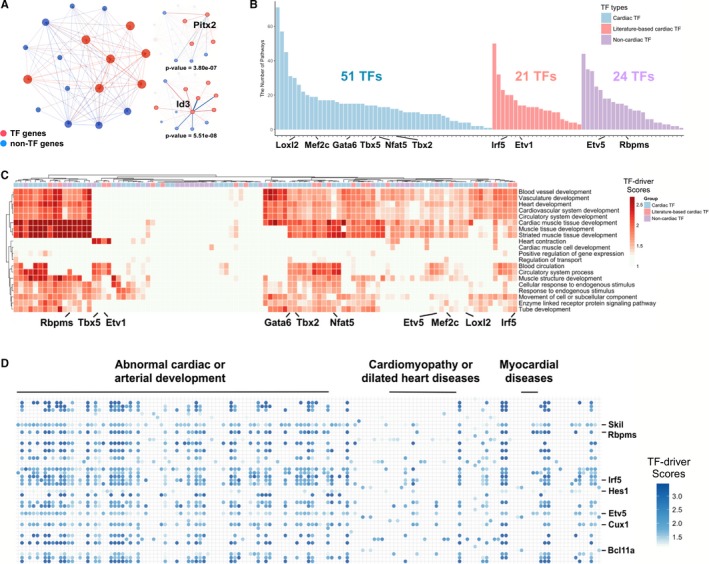
Function and phenotype analysis of cardiac and noncardiac TF targets. A, A network showing genes in the transforming growth factor β signaling. In left panel, 17 genes of module 4 were in this pathway, with overrepresentation P=1.91e‐09. Node and edge connections in right panel represent adjacencies between a TF and its targets, 12 genes targeted by Id3 and 7 genes for Pitx2. B, The number of enriched pathways (y‐axis) under the regulations of cardiac, literature‐based cardiac, and noncardiac TFs. C, Predicted regulations of cardiac, literature‐based cardiac, and noncardiac TFs in 20 representative heart terms enriched in the selected weighted gene co‐expression network analysis modules. The colors in the heat‐map show the TF‐driver scores in functional terms, representing the regulatory effects of individual TFs on the terms. D, Predicted functional roles of literature‐based cardiac and noncardiac TFs in different heart phenotype‐ or disease‐associated gene sets, with the colors for TF‐driver scores. TF indicates transcription factor.
We also extended this analysis to heart phenotype‐ or disease‐associated gene sets. The results suggested that ≈85% (n=54) of cardiac TFs were involved in the regulation of gene sets implicated in multiple heart abnormality or diseases (Table S7), concordant with these TF being defined as “cardiac TFs.” Interestingly, the cardiomyopathy‐associated genes were overrepresented in modules 4, 5, and 7 (Table S3), and mainly regulated by Gli3, Klf6, Loxl2, Hopx, Hey2, Irx4, and Myocd. In summary, our analysis, in an unsupervised manner, revealed the potentially functional roles of the spatiotemporally expressed cardiac TFs in differentiating cardiomyocytes at critical cardiac‐looping stages.
Analysis of Targets for Less‐Studied TFs in Heart Looping
We next studied the 59 less‐studied TFs (ie, associations with abnormal heart phenotypes have not been clearly established), as analyses of their targeted pathways could provide new insights to their potential functions. Compared with the above results for cardiac TFs, the targets of these less‐studied TFs, such as Cux1, Etv5, Id2, Irf5, Klf4, Nfil3, Myrf, and Skil, were significantly enriched in many of the same heart developmental terms or processes, supported by similarly high TF‐driver scores (Figure 6C and Figure S5). Furthermore, our analysis showed that 45 of the 59 TFs were likely to regulate multiple functional pathways that were enriched in the selected gene modules (Figure 6B, Figure S4 and Table S7), indicating their important functional roles. Interestingly, even though the mouse phenotype databases did not associate these TFs with an abnormal phenotype or disease, 21 of the 45 TFs have been reported to perform important functions in heart development: Hes1, Hbp1, Skil, Irf5, Id2, Cux1, Myrf, Kdm5b, Grhl1, Arhgap35, Gatad2b, Arid5b, Mga, Peg3, Nfil3, Nr2f1, Ptrf, Camta1, Etv1, Id4, and Nfib.67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89 As such, we named these TFs as “literature‐based cardiac TFs,” to distinguish them from the other “noncardiac TFs” that do not have known evidence for a cardiac function (Table S6). These findings support the biological relevance of our TF‐target prediction, and strongly indicate that the set of noncardiac TFs (n=24) also have high possibilities to regulate critical pathways in embryonic heart development.
Performing enrichment analysis of TFs’ targets for the heart phenotype‐ or disease‐associated gene sets enriched in the 5 spatiotemporal modules, we found that the targets of the 21 literature‐based cardiac or the 24 noncardiac TFs were significantly overrepresented in multiple gene sets, such as abnormal cardiac OFT development, abnormal cardiovascular development, abnormal heart ventricle morphology, and abnormal cardiac muscle contractility (Figure 6D and Figures S6 through S8). Notably, >60 phenotype‐ and disease‐associated gene sets were found to be under the regulations of Hes1, Skil, Myrf, Etv5, Cux1, Irf5, and Id2. Most of them, except Etv5, have been reported to play a functional role in heart development, suggesting the potential heart genotype–phenotype associations of these TFs.
To confirm the high expression of the OFT‐AVC TFs, we selected 10 less‐studied TFs with the most predicted targets for quantitative polymerase chain reaction analysis. The results showed that almost all of these 10 TFs had significantly higher expressions at OFT‐AVC than ventricles at E10.5, with some also exhibiting higher expression in E9.5 OFT (Figure 7). Further analysis found that the more specific expression patterns they had, the more heart‐phenotype gene sets these TFs are involved in, with Spearman correlation >0.7 and P<0.05 at both E9.5 and 10.5 stages (Table S7).
Figure 7.
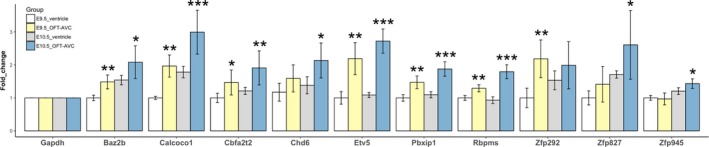
Quantitative polymerase chain reaction analysis of 10 TFs highly expressed in OFT‐AVC. The bar‐plots show expression fold‐changes (using Gapdh as reference). The statistical significances from comparisons of OFT‐AVC to ventricle at E9.5 and E10.5 separately are indicated by “*,” “**,” and “***” for P<0.05, 0.005, and 0.0005, respectively. OFT‐AVC indicates outflow tract–atrioventricular canal; TF, transcription factor.
Expression Changes and Functional Roles of TFs in Subgroups of Atrial or Ventricular Cardiomyocytes
The above analyses considered cardiomyocytes from the same spatial or temporal origin as a “homogeneous” group, but cardiomyocytes in the same group actually showed subtle transcriptional heterogeneity, because cardiomyocytes from the same anatomical location could be a mixture of cells at multiple developmental states. Therefore, we constructed putative developmental trajectories based on top variable genes (see Methods) in order to study potential cell state transitions of cardiomyocyte development at atrium, ventricle, or OFT (E9.5 OFT, E10.5 distal OFT and proximal OFT) (Figure 8A and 8B, Figure S9). The analysis revealed 3 distinct subgroups (“A”, “B,” and “C”) in each of the trajectories, largely reflecting the temporal and spatial relationship of cardiomyocytes at those locations (Figure S10).
Figure 8.
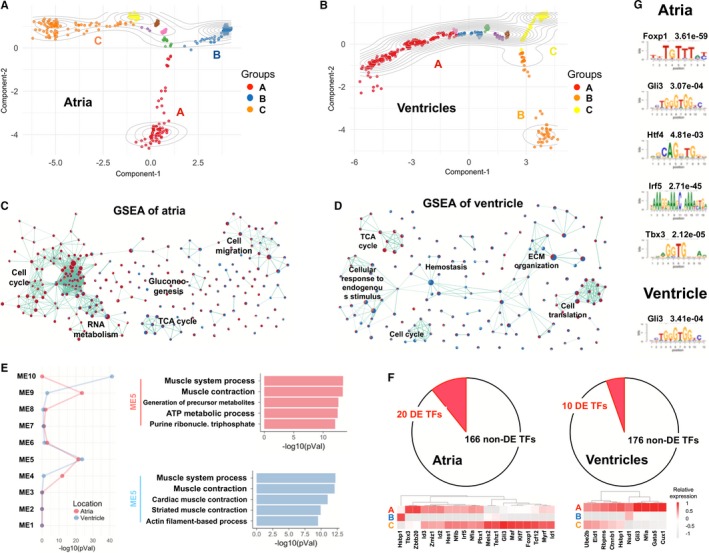
Differentially expressed genes and TFs in atrial or ventricular cardiomyocytes. A, A trajectory of atrial cardiomyocytes, indicating that the atrial cardiomyocytes consisted of 3 major subgroups (A through C). B, The trajectory of ventricular cardiomyocytes. C and D, Enriched gene sets for the DE genes among the 3 subgroups of atrial (C) and ventricular (D) cardiomyocytes. Here, DE genes from each of the pairwise comparisons were used for GSEA. The upregulated and downregulated genes are in red and blue nodes, respectively, while the edges of nodes represent the overlapping genes in 2 pathways. E, Module enrichments of atrial and ventricular DE genes. The enrichment significance of atrial or ventricular DE genes were represented by the red and blue lines, respectively, in left panel. The right panel shows the top 5 enriched pathways of atrial or ventricular DE genes that were also identified in module 5. F, Spatiotemporally DE TFs at atria and ventricles. G, The enriched motifs for atrial or ventricular DE TFs in (F). DE indicates differentially expressed; ECM, extracellular matrix; GSEA, gene set enrichment analysis; TCA, tricarboxylic acid cycle; TFs, transcription factors.
Gene set enrichment analysis indicated that DE genes from pairwise comparisons of the 3 atrial subgroups were enriched for developmental pathways, such as cell cycle, mRNA processing, protein metabolism, extracellular matrix organization, citric acid cycle, and gluconeogenesis (Figure 8C and Table S8). In comparison, the enrichment of ventricular DE genes among the 3 subgroups was mainly related to myogenesis, oxidative phosphorylation, angiogenesis, epithelial mesenchymal transition, apical junction, and Heme metabolism pathways (Figure 8D and Table S8). Interestingly, only a small number of DE genes (<60) were found among the subgroups of OFT cardiomyocytes, even though cardiomyocytes at this anatomical location were from 2 embryonic heart stages (E9.5 and E10.5). Mapping the DE genes to the above module genes showed that atrial DE genes were significantly enriched in modules 4, 5, and 9, while ventricular DE genes were enriched in modules 5 and 10 (Figure 8E). Pathway analysis of the overlapped DE genes suggested that both atrial and ventricular DE genes were dramatically overrepresented in module 5 and enriched in pathways related to muscle system process and muscle contraction (Figure 8E), indicating our trajectories could represent developmental states of cardiomyocytes at different anatomical heart regions.
To further support the importance of TFs in cardiomyocyte development, we examined TFs that were differentially expressed in atrial or ventricular subgroups, and contained in the 5 gene modules. This resulted in 20 DE TFs in the atrial and 10 in the ventricular subgroups (Figure 8F and Figure S11). Motif analyses showed that the regulatory regions of these atrial DE genes were enriched for 5 TF motifs, Foxp1 (q‐value 3.61e‐59), Gli3 (q‐value 3.07e‐04), Htf4 (q‐value 4.81e‐03), Irf5 (q‐value 2.71e‐45), and Tbx3 (q‐value 2.12e‐05) (Figure 8G). For the ventricular DE genes, Gli3 motif was found (q‐value 3.41e‐04) (Figure 8G). The findings strongly supported the regulations of cardiomyocytes differentiation by these TFs that are expressed in a highly spatiotemporal manner.
Discussion
Our analysis showed that across heart looping stages many signaling and pathways involved in cardiomyocyte differentiation were enriched in the OFT/AVC module (module 4) and they were predicted to be highly regulated by 66 TFs, and more so than genes in the other modules (Figure 9 and Figure S12). In addition, we found that the proportion of TFs in this module was much higher than those of other modules (Figure S2C). Integrating the results of function analysis of “cardiac,” “literature‐based cardiac,” and “noncardiac” TFs, our study strongly suggests that the genetic program in differentiating cardiomyocytes at OFT/AVC is not only regulated by well‐studied cardiac TFs, such as Tbx2/3, but also under the complex regulation of other 64 TFs (Figure 9 and Figure S12), such as Gli3, Mecom, Loxl2, and Meis2, including additional TFs whose cardiac functions are yet to be addressed. The high expression of the 10 noncardiac TFs in OFT/AVC was confirmed by quantitative polymerase chain reaction analysis (Figure 7). Interestingly, we also found a significant correlation between the expression patterns of the noncardiac TFs and the number of heart phenotypes they involved, with greater specific spatial expression correlated with more phenotypes. Considering all the results, we hypothesize that OFT/AVC formation and remodeling is likely the most tightly regulated embryonic heart developmental process. As such, mutations in the genes, especially TFs, important for OFT/AVC development, are likely to be the high‐risk factors for congenital heart defects. In fact, we analyzed 2 sets of manually curated CHD‐associated genes and found that they were significantly enriched in the OFT‐AVC module, with the odds ratios of 2.39 (P=0.001) and 2.35 (P=0.0003) for the 186 CHD genes in the article by Sifrim et al90 and the 253 CHD genes in the article by Jin et al,91 respectively. We also analyzed the genes with significantly higher expression in ECs and MCs of OFT‐AVC than the ECs/MCs of other locations. The results indicated that only the group of genes with higher expression in OFT‐AVC ECs was significantly enriched for the 253 CHD genes curated by Jin et al, suggesting cell‐type specificity of our finding.
Figure 9.
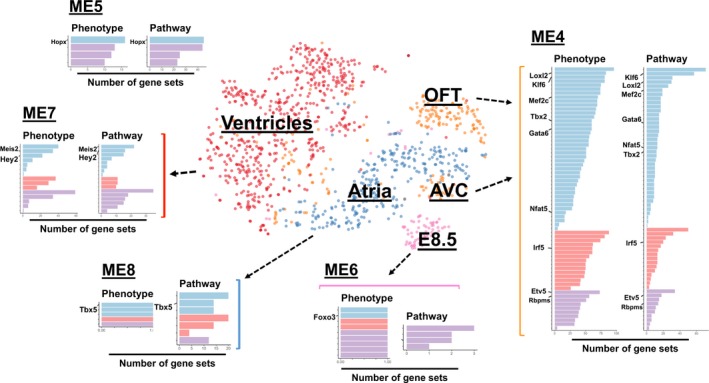
Summary of TFs and their regulatory functions in the spatiotemporal modules. The functional roles of the candidate TFs: cardiac, literature‐based cardiac, and noncardiac, in the 5 selected modules. Each bar‐plot panel of individual modules shows the number of the enriched functional pathways and heart phenotype‐ or disease‐associated gene sets under the regulations of the designated TFs. AVC indicates atrioventricular canal; OFTs, outflow tracts; TFs, transcription factors.
It is worth noting that among the less‐studied TFs, Etv5 and Rbpms are especially strong candidates for future studies. First of all, our analysis showed that Etv5 and Rbpms were associated with hundreds of gene sets for various cardiac GO terms or heart phenotypes (Figure 6C and 6D, Table S7). Then, our quantitative polymerase chain reaction results confirmed the high expression of Etv5 and Rbpms in OFT‐AVC at E9.5 and E10.5. Furthermore, these 2 TFs have been implicated in development or diseases. Rbpms, as a coactivator for the enhancement of TGFB1/Smad‐mediated transactivation, has been shown to play important roles in transcriptional regulation of human retinas,92 while Etv5 was demonstrated to enhance angiogenesis in colorectal cancer.93 More interestingly, a recent study suggested that Etv5 is a candidate gene‐regulatory‐network driver in mouse embryonic stem cells,94 consistent with our finding. Lastly, Etv1 and Etv2 play key roles in regulating cardiomyocyte95 or vascular endothelial cell development.96 As a member of the same ETS family, Etv5 may work with Etv1/2 cooperatively or independently during heart development, an interesting area to explore in the future.
Although our study has analyzed more than 1000 cardiac cardiomyocytes from multiple developmental stages and anatomical locations, the number of total cells is relatively small, considering that new scRNA‐seq technology allows expression profiling of tens of thousands or even up to millions of cells.97 The inclusion of more cells from more time points can certainly improve our results, especially in terms of finding key TFs regulating cell fate commitment and lineage specification. In theory, with all cardiac cells from the same heart being sampled simultaneously, one can also study the TF‐regulatory networks and signaling cross‐talks among different cardiac cell types, an important matter not addressed in the current study. Finally, it will be necessary to validate our TF‐target predictions using scRNA‐seq data from wild‐type hearts and mutant hearts with the expression of selected TFs compromised, such as previous studies of Nkx2‐5 function.12, 14 Nevertheless, the current study has identified key TFs, including not fully characterized ones, that can play important regulatory roles in embryonic heart development.
Sources of Funding
This study is supported by National Institutes of Health (NIH) of USA (Grants HL133120, HD092944, and HD070454).
Disclosures
None.
Supporting information
Table S2. Gene Expression Matrix of Individual Modules (see Excel file)
Table S3. Enriched GO Terms and Reactome and KEGG Pathways in Individual Modules (see Excel file)
Table S4. Enriched Cardiac Phenotype‐ and Disease‐Associated Gene Sets in Individual Modules (see Excel file)
Table S5. Motif Enrichment of 186 TFs (see Excel file)
Table S6. 186 TFs in the 5 Selected Modules and Their Predicted Targets (see Excel file)
Table S7. Enrichments of TF Targets in GO Terms, Pathways and Cardiac Phenotype‐ and Disease‐Associated Gene Sets (see Excel file)
Table S8. DE Genes of Atrial, Ventricular, or OFT Subgroups (see Excel file)
Table S1. The Primers of qPCRs
Figure S1. Clustering analysis of cardiac cell types in both datasets.
Figure S2. t‐SNE map and module preservation of cardiomyocytes.
Figure S3. Module preservation.
Figure S4. Numbers of pathways regulated by candidate TFs.
Figure S5. TF‐targets in enriched heart developmental pathways.
Figure S6. TF‐targets in enriched cardiac disease‐associated gene sets. Heat‐map shows the detailed version of Figure 6D, and displays the predicted roles of all the literature‐based cardiac and noncardiac TFs (transcription factors), measured as their TF‐driver scores (colors) for different heart phenotype‐ or disease‐associated gene sets.
Figure S7. Enrichment analysis of TF‐targets in cardiac disease–associated gene sets.
Figure S8. The number of TF‐targets in cardiac disease–associated gene sets.
Figure S9. A trajectory of CMs at outflow tract.
Figure S10. Trajectories of atrial and ventricular CMs (cardiomyocytes).
Figure S11. Expression patterns of representative atrial and ventricular TFs.
Figure S12. Summary of the full list of the spatiotemporal TFs.
Acknowledgments
We would like to thank the groups of Sean M. Wu and Christine E. Seidman for sharing the scRNA‐seq data, and Sean M. Wu provided the information of their single‐cell types.
(J Am Heart Assoc. 2019;8:e012941 DOI: 10.1161/JAHA.119.012941.)
References
- 1. Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Waardenberg AJ, Ramialison M, Bouveret R, Harvey RP. Genetic networks governing heart development. Cold Spring Harb Perspect Med. 2014;4:a013839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Takeuchi JK, Ohgi M, Koshiba‐Takeuchi K, Shiratori H, Sakaki I, Ogura K, Saijoh Y, Ogura T. Tbx5 specifies the left/right ventricles and ventricular septum position during cardiogenesis. Development. 2003;130:5953. [DOI] [PubMed] [Google Scholar]
- 4. He A, Gu F, Hu Y, Ma Q, Ye LY, Akiyama JA, Visel A, Pennacchio LA, Pu WT. Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat Commun. 2014;5:4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang L, Nomura‐Kitabayashi A, Sultana N, Cai W, Cai X, Moon AM, Cai CL. Mesodermal Nkx2.5 is necessary and sufficient for early second heart field development. Dev Biol. 2014;390:68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Afouda BA, Martin J, Liu F, Ciau‐Uitz A, Patient R, Hoppler S. GATA transcription factors integrate Wnt signalling during heart development. Development. 2008;135:3185–3190. [DOI] [PubMed] [Google Scholar]
- 7. Wang RN, Green J, Wang Z, Deng Y, Qiao M, Peabody M, Zhang Q, Ye J, Yan Z, Denduluri S, Idowu O, Li M, Shen C, Hu A, Haydon RC, Kang R, Mok J, Lee MJ, Luu HL, Shi LL. Bone morphogenetic protein (BMP) signaling in development and human diseases. Genes Dis. 2014;1:87–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chamberlain AA, Lin MY, Lister RL, Maslov AA, Wang YD, Suzuki M, Wu BR, Greally JM, Zheng DY, Zhou B. DNA methylation is developmentally regulated for genes essential for cardiogenesis. J Am Heart Assoc. 2014;3:e000976 DOI: 10.1161/JAHA.114.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang QJ, Liu ZP. Histone methylations in heart development, congenital and adult heart diseases. Epigenomics. 2015;7:321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang D, Wu B, Wang P, Wang Y, Lu P, Nechiporuk T, Floss T, Greally JM, Zheng D, Zhou B. Non‐CpG methylation by DNMT3B facilitates REST binding and gene silencing in developing mouse hearts. Nucleic Acids Res. 2017;45:3102–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen X, Chakravarty T, Zhang Y, Li X, Zhong JF, Wang C. Single‐cell transcriptome and epigenomic reprogramming of cardiomyocyte‐derived cardiac progenitor cells. Sci Data. 2016;3:160079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, Hinson JT, Homsy J, Gray J, Pu W, Bruneau BG, Seidman JG, Seidman CE. Single‐cell resolution of temporal gene expression during heart development. Dev Cell. 2016;39:480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gladka MM, Molenaar B, De Ruiter H, Versteeg D, Tsui H, Lacraz GPA, Van Der Elst S, Huibers MMH, Van Oudenaarden A, Van Rooij E. Single‐cell sequencing of the healthy and diseased heart reveals Ckap4 as a new modulator of fibroblasts activation. Cardiovasc Res. 2018;138:166–180. [DOI] [PubMed] [Google Scholar]
- 14. Li G, Xu A, Sim S, Priest JR, Tian X, Khan T, Quertermous T, Zhou B, Tsao PS, Quake SR, Wu SM. Transcriptomic profiling maps anatomically patterned subpopulations among single embryonic cardiac cells. Dev Cell. 2016;39:491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Z, Wang L, Welch JD, Ma H, Zhou Y, Vaseghi HR, Yu S, Wall JB, Alimohamadi S, Zheng M, Yin C, Shen W, Prins JF, Liu J, Qian L. Single‐cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nature. 2017;551:100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. See K, Tan WLW, Lim EH, Tiang Z, Lee LT, Li PYQ, Luu TDA, Ackers‐Johnson M, Foo RS. Single cardiomyocyte nuclear transcriptomes reveal a lincRNA‐regulated de‐differentiation and cell cycle stress‐response in vivo. Nat Commun. 2017;8:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sereti K‐I, Nguyen NB, Kamran P, Zhao P, Ranjbarvaziri S, Park S, Sabri S, Engel JL, Sung K, Kulkarni RP, Ding Y, Hsiai TK, Plath K, Ernst J, Sahoo D, Mikkola HKA, Iruela‐Arispe ML, Ardehali R. Analysis of cardiomyocyte clonal expansion during mouse heart development and injury. Nat Commun. 2018;9:754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Skelly DA, Squiers GT, McLellan MA, Bolisetty MT, Robson P, Rosenthal NA, Pinto AR. Single‐cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart. Cell Rep. 2018;22:600–610. [DOI] [PubMed] [Google Scholar]
- 19. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; Genome Project Data Processing S . The sequence alignment/map format and samtools. Bioinformatics. 2009;25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high‐throughput sequencing data. Bioinformatics. 2015;31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wickham H, Henry L. Tidyr: easily tidy data with ‘spread()’ and ‘gather()’ functions. 2018.
- 23. Wickham H, Francois R, Henry L, Muller K. Dplyr: a grammar of data manipulation. 2017.
- 24. McCarthy DJ, Campbell KR, Lun AT, Wills QF. Scater: pre‐processing, quality control, normalization and visualization of single‐cell RNA‐seq data in R. Bioinformatics. 2017;33:1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van der Maaten L, Hinton GE. Visualizing high‐dimensional data using t‐SNE. J Mach Learn Res. 2008;15:3221–3245. [Google Scholar]
- 26. Kiselev VY, Kirschner K, Schaub MT, Andrews T, Yiu A, Chandra T, Natarajan KN, Reik W, Barahona M, Green AR, Hemberg M. SC3: consensus clustering of single‐cell RNA‐seq data. Nat Methods. 2017;14:483–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wickham H. Ggplot2: Elegant Graphics for Data Analysis. New York: Springer‐Verlag; 2016. [Google Scholar]
- 29. Kolde R. Pheatmap: pretty heatmaps. 2013.
- 30. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol. 2011;7:e1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks. 2009.
- 34. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Y, Morley M, Brandimarto J, Hannenhalli S, Hu Y, Ashley EA, Tang WH, Moravec CS, Margulies KB, Cappola TP, Li M; Consortium MA . RNA‐Seq identifies novel myocardial gene expression signatures of heart failure. Genomics. 2015;105:83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bonilla‐Claudio M, Wang J, Bai Y, Klysik E, Selever J, Martin JF. BMP signaling regulates a dose‐dependent transcriptional program to control facial skeletal development. Development. 2012;139:709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Luna‐Zurita L, Stirnimann CU, Glatt S, Kaynak BL, Thomas S, Baudin F, Samee MA, He D, Small EM, Mileikovsky M, Nagy A, Holloway AK, Pollard KS, Muller CW, Bruneau BG. Complex interdependence regulates heterotypic transcription factor distribution and coordinates cardiogenesis. Cell. 2016;164:999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Russell R, Ilg M, Lin Q, Wu G, Lechel A, Bergmann W, Eiseler T, Linta L, Kumar PP, Klingenstein M, Adachi K, Hohwieler M, Sakk O, Raab S, Moon A, Zenke M, Seufferlein T, Scholer HR, Illing A, Liebau S, Kleger A. A dynamic role of TBX3 in the pluripotency circuitry. Stem Cell Reports. 2015;5:1155–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shirvani SM, Mookanamparambil L, Ramoni MF, Chin MT. Transcription factor CHF1/Hey2 regulates the global transcriptional response to platelet‐derived growth factor in vascular smooth muscle cells. Physiol Genomics. 2007;30:61–68. [DOI] [PubMed] [Google Scholar]
- 40. Wu SP, Kao CY, Wang L, Creighton CJ, Yang J, Donti TR, Harmancey R, Vasquez HG, Graham BH, Bellen HJ, Taegtmeyer H, Chang CP, Tsai MJ, Tsai SY. Increased COUP‐TFII expression in adult hearts induces mitochondrial dysfunction resulting in heart failure. Nat Commun. 2015;6:8245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010;26:2363–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. The Gene Ontology C . Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res. 2017;45:D331–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, Milacic M, Roca CD, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Viteri G, Weiser J, Wu G, Stein L, Hermjakob H, D'Eustachio P. The reactome pathway knowledgebase. Nucleic Acids Res. 2018;46:D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–W311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, Meehan TF, Weninger WJ, Westerberg H, Adissu H, Baker CN, Bower L, Brown JM, Caddle LB, Chiani F, Clary D, Cleak J, Daly MJ, Denegre JM, Doe B, Dolan ME, Edie SM, Fuchs H, Gailus‐Durner V, Galli A, Gambadoro A, Gallegos J, Guo S, Horner NR, Hsu CW, Johnson SJ, Kalaga S, Keith LC, Lanoue L, Lawson TN, Lek M, Mark M, Marschall S, Mason J, McElwee ML, Newbigging S, Nutter LM, Peterson KA, Ramirez‐Solis R, Rowland DJ, Ryder E, Samocha KE, Seavitt JR, Selloum M, Szoke‐Kovacs Z, Tamura M, Trainor AG, Tudose I, Wakana S, Warren J, Wendling O, West DB, Wong L, Yoshiki A; International Mouse Phenotyping Consortium, Jackson L, Infrastructure Nationale Phenomin, Institut Clinique de la Souris (ICS), Charles River Laboratories, Harwell MRC, Toronto Centre for Phenogenomics, Wellcome Trust Sanger I, Center RB , MacArthur DG, Tocchini‐Valentini GP, Gao X, Flicek P, Bradley A, Skarnes WC, Justice MJ, Parkinson HE, Moore M, Wells S, Braun RE, Svenson KL, de Angelis MH, Herault Y, Mohun T, Mallon AM, Henkelman RM, Brown SD, Adams DJ, Lloyd KC, McKerlie C, Beaudet AL, Bucan M, Murray SA. High‐throughput discovery of novel developmental phenotypes. Nature. 2016;537:508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Collins FS, Finnell RH, Rossant J, Wurst W. A new partner for the International Knockout Mouse Consortium. Cell. 2007;129:235. [DOI] [PubMed] [Google Scholar]
- 50. Pinero J, Bravo A, Queralt‐Rosinach N, Gutierrez‐Sacristan A, Deu‐Pons J, Centeno E, Garcia‐Garcia J, Sanz F, Furlong LI. DisGeNET: a comprehensive platform integrating information on human disease‐associated genes and variants. Nucleic Acids Res. 2017;45:D833–D839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Carcamo‐Orive I, Hoffman GE, Cundiff P, Beckmann ND, D'Souza SL, Knowles JW, Patel A, Papatsenko D, Abbasi F, Reaven GM, Whalen S, Lee P, Shahbazi M, Henrion MYR, Zhu K, Wang S, Roussos P, Schadt EE, Pandey G, Chang R, Quertermous T, Lemischka I. Analysis of transcriptional variability in a large human ipsc library reveals genetic and non‐genetic determinants of heterogeneity. Cell Stem Cell. 2017;20:518–532.e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Makinen VP, Civelek M, Meng Q, Zhang B, Zhu J, Levian C, Huan T, Segre AV, Ghosh S, Vivar J, Nikpay M, Stewart AF, Nelson CP, Willenborg C, Erdmann J, Blakenberg S, O'Donnell CJ, Marz W, Laaksonen R, Epstein SE, Kathiresan S, Shah SH, Hazen SL, Reilly MP, Coronary ADG‐WR, Meta‐Analysis C, Lusis AJ, Samani NJ, Schunkert H, Quertermous T, McPherson R, Yang X, Assimes TL. Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS Genet. 2014;10:e1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shu L, Zhao Y, Kurt Z, Byars SG, Tukiainen T, Kettunen J, Orozco LD, Pellegrini M, Lusis AJ, Ripatti S, Zhang B, Inouye M, Makinen VP, Yang X. Mergeomics: multidimensional data integration to identify pathogenic perturbations to biological systems. BMC Genomics. 2016;17:874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, Lamb JR, Bennett DA, Molony C, Stone DJ, Gudnason V, Myers AJ, Schadt EE, Neumann H, Zhu J, Emilsson V. Integrated systems approach identifies genetic nodes and networks in late‐onset Alzheimer's disease. Cell. 2013;153:707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis C, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McLeay RC, Bailey TL. Motif enrichment analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics. 2010;11:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ou J, Wolfe SA, Brodsky MH, Zhu LJ. motifStack for the analysis of transcription factor binding site evolution. Nat Methods. 2018;15:8–9. [DOI] [PubMed] [Google Scholar]
- 58. Mercier E, Gottardo R. Motiv: motif identification and validation. 2014.
- 59. Ackers‐Johnson M, Tan WLW, Foo RS. Following hearts, one cell at a time: recent applications of single‐cell RNA sequencing to the understanding of heart disease. Nat Commun. 2018;9:4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Molenaar B, van Rooij E. Single‐cell sequencing of the mammalian heart. Circ Res. 2018;123:1033–1035. [DOI] [PubMed] [Google Scholar]
- 61. Naito AT, Akazawa H, Takano H, Minamino T, Nagai T, Aburatani H, Komuro I. Phosphatidylinositol 3‐kinase‐Akt pathway plays a critical role in early cardiomyogenesis by regulating canonical Wnt signaling. Circ Res. 2005;97:144–151. [DOI] [PubMed] [Google Scholar]
- 62. de la Pompa JL, Epstein JA. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev Cell. 2012;22:244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chung S, Dzeja PP, Faustino RS, Perez‐Terzic C, Behfar A, Terzic A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat Clin Pract Cardiovasc Med. 2007;4(suppl 1):S60–S67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hollnagel A, Oehlmann V, Heymer J, Ruther U, Nordheim A. Id genes are direct targets of bone morphogenetic protein induction in embryonic stem cells. J Biol Chem. 1999;274:19838–19845. [DOI] [PubMed] [Google Scholar]
- 65. Lage K, Mollgard K, Greenway S, Wakimoto H, Gorham JM, Workman CT, Bendsen E, Hansen NT, Rigina O, Roque FS, Wiese C, Christoffels VM, Roberts AE, Smoot LB, Pu WT, Donahoe PK, Tommerup N, Brunak S, Seidman CE, Seidman JG, Larsen LA. Dissecting spatio‐temporal protein networks driving human heart development and related disorders. Mol Syst Biol. 2010;6:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lage K, Greenway SC, Rosenfeld JA, Wakimoto H, Gorham JM, Segre AV, Roberts AE, Smoot LB, Pu WT, Pereira AC, Mesquita SM, Tommerup N, Brunak S, Ballif BC, Shaffer LG, Donahoe PK, Daly MJ, Seidman JG, Seidman CE, Larsen LA. Genetic and environmental risk factors in congenital heart disease functionally converge in protein networks driving heart development. Proc Natl Acad Sci USA. 2012;109:14035–14040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ahuja S, Dogra D, Stainier DYR, Reischauer S. Id4 functions downstream of BMP signaling to restrict TCF function in endocardial cells during atrioventricular valve development. Dev Biol. 2016;412:71–82. [DOI] [PubMed] [Google Scholar]
- 68. Albert M, Schmitz SU, Kooistra SM, Malatesta M, Morales Torres C, Rekling JC, Johansen JV, Abarrategui I, Helin K. The histone demethylase Jarid1b ensures faithful mouse development by protecting developmental genes from aberrant H3K4me3. PLoS Genet. 2013;9:e1003461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen X, Qin L, Liu Z, Liao L, Martin JF, Xu J. Knockout of SRC‐1 and SRC‐3 in mice decreases cardiomyocyte proliferation and causes a noncompaction cardiomyopathy phenotype. Int J Biol Sci. 2015;11:1056–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cunnington RH, Nazari M, Dixon IM. c‐Ski, Smurf2, and Arkadia as regulators of TGF‐beta signaling: new targets for managing myofibroblast function and cardiac fibrosis. Can J Physiol Pharmacol. 2009;87:764–772. [DOI] [PubMed] [Google Scholar]
- 71. da Rosa MS, Seminotti B, Ribeiro CA, Parmeggiani B, Grings M, Wajner M, Leipnitz G. 3‐Hydroxy‐3‐methylglutaric and 3‐methylglutaric acids impair redox status and energy production and transfer in rat heart: relevance for the pathophysiology of cardiac dysfunction in 3‐hydroxy‐3‐methylglutaryl‐coenzyme A lyase deficiency. Free Radic Res. 2016;50:997–1010. [DOI] [PubMed] [Google Scholar]
- 72. Fletcher A, Raza F, Buursma J, Livingston S, Kearn D, Herndon B, Vanden Heauvel G, Baybutt R, Molteni A. Pathological changes in the heart of the strain CUX1 transgenic mice. FASEB J. 2015;29:LB432. [Google Scholar]
- 73. Hirose‐Yotsuya L, Okamoto F, Yamakawa T, Whitson RH, Fujita‐Yamaguchi Y, Itakura K. Knockdown of AT‐rich interaction domain (ARID) 5B gene expression induced AMPKalpha2 activation in cardiac myocytes. Biosci Trends. 2015;9:377–385. [DOI] [PubMed] [Google Scholar]
- 74. Jongbloed MR, Vicente‐Steijn R, Douglas YL, Wisse LJ, Mori K, Yokota Y, Bartelings MM, Schalij MJ, Mahtab EA, Poelmann RE, Gittenberger‐De Groot AC. Expression of Id2 in the second heart field and cardiac defects in Id2 knock‐out mice. Dev Dyn. 2011;240:2561–2577. [DOI] [PubMed] [Google Scholar]
- 75. Loirand G, Pacaud P. Involvement of Rho GTPases and their regulators in the pathogenesis of hypertension. Small GTPases. 2014;5:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Monnier V, Iche‐Torres M, Rera M, Contremoulins V, Guichard C, Lalevee N, Tricoire H, Perrin L. dJun and Vri/dNFIL3 are major regulators of cardiac aging in Drosophila. PLoS Genet. 2012;8:e1003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Muller‐Borer B, Esch G, Aldina R, Woon W, Fox R, Bursac N, Hiller S, Maeda N, Shepherd N, Jin JP, Hutson M, Anderson P, Kirby ML, Malouf NN. Calcium dependent CAMTA1 in adult stem cell commitment to a myocardial lineage. PLoS One. 2012;7:e38454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nassiri M, Liu J, Kulak S, Uwiera RR, Aird WC, Ballermann BJ, Jahroudi N. Repressors NFI and NFY participate in organ‐specific regulation of von Willebrand factor promoter activity in transgenic mice. Arterioscler Thromb Vasc Biol. 2010;30:1423–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Quaranta R, Fell J, Ruhle F, Rao J, Piccini I, Arauzo‐Bravo MJ, Verkerk AO, Stoll M, Greber B. Revised roles of ISL1 in a hES cell‐based model of human heart chamber specification. Elife. 2018;7:e31706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Rochais F, Dandonneau M, Mesbah K, Jarry T, Mattei MG, Kelly RG. Hes1 is expressed in the second heart field and is required for outflow tract development. PLoS One. 2009;4:e6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Seneviratne AN, Edsfeldt A, Cole JE, Kassiteridi C, Swart M, Park I, Green P, Khoyratty T, Saliba D, Goddard ME, Sansom SN, Goncalves I, Krams R, Udalova IA, Monaco C. Interferon regulatory factor 5 controls necrotic core formation in atherosclerotic lesions by impairing efferocytosis. Circulation. 2017;136:1140–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shekhar A, Lin X, Liu FY, Zhang J, Mo H, Bastarache L, Denny JC, Cox NJ, Delmar M, Roden DM, Fishman GI, Park DS. Transcription factor ETV1 is essential for rapid conduction in the heart. J Clin Invest. 2016;126:4444–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Song X, Gao X, Lu J, Liang H, Su P, Li Q, Pang Y. High mobility group box transcription factor 1 (HBP1) from Lampetra japonica affects cell cycle regulation. Dev Growth Differ. 2018;60:146–157. [DOI] [PubMed] [Google Scholar]
- 84. Taniguchi T, Maruyama N, Ogata T, Kasahara T, Nakanishi N, Miyagawa K, Naito D, Hamaoka T, Nishi M, Matoba S, Ueyama T. PTRF/cavin‐1 deficiency causes cardiac dysfunction accompanied by cardiomyocyte hypertrophy and cardiac fibrosis. PLoS One. 2016;11:e0162513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Theis JL, Sharpe KM, Matsumoto ME, Chai HS, Nair AA, Theis JD, de Andrade M, Wieben ED, Michels VV, Olson TM. Homozygosity mapping and exome sequencing reveal GATAD1 mutation in autosomal recessive dilated cardiomyopathy. Circ Cardiovasc Genet. 2011;4:585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Waldron L, Steimle JD, Greco TM, Gomez NC, Dorr KM, Kweon J, Temple B, Yang XH, Wilczewski CM, Davis IJ, Cristea IM, Moskowitz IP, Conlon FL. The cardiac TBX5 interactome reveals a chromatin remodeling network essential for cardiac septation. Dev Cell. 2016;36:262–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Walkowska A, Pawlak M, Jane S, Kompanowska‐Jezierska E, Wilanowski T. Effects of high and low sodium diet on blood pressure and heart rate in mice lacking the functional grainyhead‐like 1 gene. Physiol Res. 2017;66:163–165. [DOI] [PubMed] [Google Scholar]
- 88. Yaniz‐Galende E, Roux M, Nadaud S, Mougenot N, Bouvet M, Claude O, Lebreton G, Blanc C, Pinet F, Atassi F, Perret C, Dierick F, Dussaud S, Leprince P, Trégouët D‐A, Marazzi G, Sassoon D, Hulot J‐S. Fibrogenic potential of PW1/Peg3 expressing cardiac stem cells. J Am Coll Cardiol. 2017;70:728–741. [DOI] [PubMed] [Google Scholar]
- 89. Qi H, Yu L, Zhou X, Wynn J, Zhao H, Guo Y, Zhu N, Kitaygorodsky A, Hernan R, Aspelund G, Lim FY, Crombleholme T, Cusick R, Azarow K, Danko ME, Chung D, Warner BW, Mychaliska GB, Potoka D, Wagner AJ, ElFiky M, Wilson JM, Nickerson D, Bamshad M, High FA, Longoni M, Donahoe PK, Chung WK, Shen Y. De novo variants in congenital diaphragmatic hernia identify MYRF as a new syndrome and reveal genetic overlaps with other developmental disorders. PLoS Genet. 2018;14:e1007822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sifrim A, Hitz M‐P, Wilsdon A, Breckpot J, Turki SHA, Thienpont B, McRae J, Fitzgerald TW, Singh T, Swaminathan GJ, Prigmore E, Rajan D, Abdul‐Khaliq H, Banka S, Bauer UMM, Bentham J, Berger F, Bhattacharya S, Bu'Lock F, Canham N, Colgiu I‐G, Cosgrove C, Cox H, Daehnert I, Daly A, Danesh J, Fryer A, Gewillig M, Hobson E, Hoff K, Homfray T; the IS , Kahlert A‐K, Ketley A, Kramer H‐H, Lachlan K, Lampe AK, Louw JJ, Manickara AK, Manase D, McCarthy KP, Metcalfe K, Moore C, Newbury‐Ecob R, Omer SO, Ouwehand WH, Park S‐M, Parker MJ, Pickardt T, Pollard MO, Robert L, Roberts DJ, Sambrook J, Setchfield K, Stiller B, Thornborough C, Toka O, Watkins H, Williams D, Wright M, Mital S, Daubeney PEF, Keavney B, Goodship J; the UKKC , Abu‐Sulaiman RM, Klaassen S, Wright CF, Firth HV, Barrett JC, Devriendt K, FitzPatrick DR, Brook JD; the Deciphering Developmental Disorders S , Hurles ME. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet. 2016;48:1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, Hung WC, Haider S, Zhang J, Knight J, Bjornson RD, Castaldi C, Tikhonoa IR, Bilguvar K, Mane SM, Sanders SJ, Mital S, Russell MW, Gaynor JW, Deanfield J, Giardini A, Porter GA Jr, Srivastava D, Lo CW, Shen Y, Watkins WS, Yandell M, Yost HJ, Tristani‐Firouzi M, Newburger JW, Roberts AE, Kim R, Zhao H, Kaltman JR, Goldmuntz E, Chung WK, Seidman JG, Gelb BD, Seidman CE, Lifton RP, Brueckner M. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ye L, Gu L, Caprioli J, Piri N. RNA‐binding protein Rbpms is represented in human retinas by isoforms A and C and its transcriptional regulation involves Sp1‐binding site. Mol Genet Genomics. 2018;293:819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Cheng X, Jin Z, Ji X, Shen X, Feng H, Morgenlander W, Ou B, Wu H, Gao H, Ye F, Zhang Y, Peng Y, Liang J, Jiang Y, Zhang T, Qiu W, Lu X, Zhao R. ETS variant 5 promotes colorectal cancer angiogenesis by targeting platelet‐derived growth factor BB. Int J Cancer. 2019;145:179–191. [DOI] [PubMed] [Google Scholar]
- 94. Kalkan T, Bornelov S, Mulas C, Diamanti E, Lohoff T, Ralser M, Middelkamp S, Lombard P, Nichols J, Smith A. Complementary activity of ETV5, RBPJ, and TCF3 drives formative transition from naive pluripotency. Cell Stem Cell. 2019;24:785–801.e787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Shekhar A, Lin X, Lin B, Liu FY, Zhang J, Khodadadi‐Jamayran A, Tsirigos A, Bu L, Fishman GI, Park DS. ETV1 activates a rapid conduction transcriptional program in rodent and human cardiomyocytes. Sci Rep. 2018;8:9944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Se‐Yeong O, Ju Young K, Changwon P. The ETS factor, ETV2: a master regulator for vascular endothelial cell development. Mol Cells. 2015;38:1029–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall‐Levin M, Wyatt PW, Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, Bielas JH. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Gene Expression Matrix of Individual Modules (see Excel file)
Table S3. Enriched GO Terms and Reactome and KEGG Pathways in Individual Modules (see Excel file)
Table S4. Enriched Cardiac Phenotype‐ and Disease‐Associated Gene Sets in Individual Modules (see Excel file)
Table S5. Motif Enrichment of 186 TFs (see Excel file)
Table S6. 186 TFs in the 5 Selected Modules and Their Predicted Targets (see Excel file)
Table S7. Enrichments of TF Targets in GO Terms, Pathways and Cardiac Phenotype‐ and Disease‐Associated Gene Sets (see Excel file)
Table S8. DE Genes of Atrial, Ventricular, or OFT Subgroups (see Excel file)
Table S1. The Primers of qPCRs
Figure S1. Clustering analysis of cardiac cell types in both datasets.
Figure S2. t‐SNE map and module preservation of cardiomyocytes.
Figure S3. Module preservation.
Figure S4. Numbers of pathways regulated by candidate TFs.
Figure S5. TF‐targets in enriched heart developmental pathways.
Figure S6. TF‐targets in enriched cardiac disease‐associated gene sets. Heat‐map shows the detailed version of Figure 6D, and displays the predicted roles of all the literature‐based cardiac and noncardiac TFs (transcription factors), measured as their TF‐driver scores (colors) for different heart phenotype‐ or disease‐associated gene sets.
Figure S7. Enrichment analysis of TF‐targets in cardiac disease–associated gene sets.
Figure S8. The number of TF‐targets in cardiac disease–associated gene sets.
Figure S9. A trajectory of CMs at outflow tract.
Figure S10. Trajectories of atrial and ventricular CMs (cardiomyocytes).
Figure S11. Expression patterns of representative atrial and ventricular TFs.
Figure S12. Summary of the full list of the spatiotemporal TFs.