

Abstract
Background
Systemic iron status has been implicated in atherosclerosis and thrombosis. The aim of this study was to investigate the effect of genetically determined iron status on carotid intima‐media thickness, carotid plaque, and venous thromboembolism using Mendelian randomization.
Methods and Results
Genetic instrumental variables for iron status were selected from a genome‐wide meta‐analysis of 48 972 subjects. Genetic association estimates for carotid intima‐media thickness and carotid plaque were obtained using data from 71 128 and 48 434 participants, respectively, and estimates for venous thromboembolism were obtained using data from a study incorporating 7507 cases and 52 632 controls. Conventional 2‐sample summary data Mendelian randomization was performed for the main analysis. Higher genetically determined iron status was associated with increased risk of venous thromboembolism. Odds ratios per SD increase in biomarker levels were 1.37 (95% CI 1.14‐1.66) for serum iron, 1.25 (1.09‐1.43) for transferrin saturation, 1.92 (1.28‐2.88) for ferritin, and 0.76 (0.63‐0.92) for serum transferrin (with higher transferrin levels representing lower iron status). In contrast, higher iron status was associated with lower risk of carotid plaque. Corresponding odds ratios were 0.85 (0.73‐0.99) for serum iron and 0.89 (0.80‐1.00) for transferrin saturation, with concordant trends for serum transferrin and ferritin that did not reach statistical significance. There was no Mendelian randomization evidence of an effect of iron status on carotid intima‐media thickness.
Conclusions
These findings support previous work to suggest that higher genetically determined iron status is protective against some forms of atherosclerotic disease but increases the risk of thrombosis related to stasis of blood.
Keywords: atherosclerosis, Mendelian randomization, thrombosis
Subject Categories: Thrombosis; Genetic, Association Studies; Atherosclerosis
Clinical Perspective
What Is New?
Thrombotic disease is the leading cause of global mortality.
The Mendelian randomization technique uses randomly allocated genetic variants to instrument the effect of an exposure in investigating for a causal effect on a particular outcome and is less prone than traditional observational research to environmental confounding and reverse causation.
In this study Mendelian randomization analysis was performed to investigate for an effect of higher genetically determined iron status on venous thromboembolism, carotid plaque, and carotid artery intima‐media thickness.
What Are the Clinical Implications?
Higher iron status was found to increase the risk of venous thromboembolism, decrease the risk of carotid plaque, and have no significant effect on carotid artery intima‐media thickness.
These results are consistent with previous studies that suggest higher iron status has a protective role in atherosclerosis but increases the risk of thrombosis related to stasis of blood.
Introduction
Thrombosis is a common underlying mechanism for ischemic heart disease, ischemic stroke, and venous thromboembolism (VTE), and thrombotic disease processes together are the leading cause of global mortality and constitute the largest contributor to the global disease burden as measured by disability‐adjusted life years.1, 2, 3, 4 Iron has been implicated in multiple aspects of pathological thrombosis, including oxidative stress, thrombocytosis, and increased erythrocyte viscosity.5, 6 Previous observational studies have provided evidence of a nonlinear relationship between iron status and thrombotic disease, with both iron deficiency and iron overload shown to increase risk of VTE7, 8, 9 and carotid atherosclerosis.5, 10, 11, 12 However, the effect of iron‐status variation within the normal range is less well established.
Mendelian randomization (MR) is a technique that uses genetic variants as proxies for a modifiable exposure (genetic instruments) in order to investigate for a causal effect on risk of disease.13 If there is causal association between the exposure and disease of interest, the genetic variants instrumenting the exposure will relate to the disease, provided that the requisite assumptions of the model are met. Because these variants are randomly allocated at conception, their association with the disease outcome is less susceptible to the potential environmental confounding factors and reverse causation biases that can affect observational studies.13 MR can therefore provide more reliable estimates of causal relationships. We have previously used the MR approach to demonstrate a contrasting effect of higher genetically determined iron status on different thrombotic disease processes: increasing risk of cardioembolic stroke14 while conferring protection in coronary artery disease,15 consistent with observational analyses.16, 17, 18 Consequently, we have suggested that higher iron status may bestow a protective effect on atherosclerosis while, on the other hand, it increases the risk of thrombosis related to stasis of blood.14
Quantifiable biomarkers of iron status, including serum iron, ferritin, transferrin, and transferrin saturation, can be used as phenotypic proxies for overall iron status.19, 20 Genetic variants associated with these biomarkers in a pattern concordant with an overall relation to increased iron status (increased serum iron, ferritin and transferrin saturation, and decreased transferrin levels) therefore represent potential genetic instruments for iron status. In this study we used such instruments to perform an MR analysis to gain further insight into the role of iron status in thrombotic disease processes. Specifically, we investigated how iron status affects carotid artery intima‐media thickness (cIMT) and carotid plaque, 2 correlated but distinct phenotypes of vessel narrowing that may be used to facilitate mechanistic insight. Increasing evidence suggests that cIMT is associated with vessel hypertrophy and hyperplasia in response to shear stress associated with aging, whereas carotid plaque may represent the product of a dynamic inflammatory cascade in atherosclerosis.21, 22, 23 In addition, we investigated the association between iron status and VTE. These analyses were selected to offer further insight into the role of iron status in thrombotic disease, which, given the variations in iron status observed worldwide,24 could have significant potential clinical and public health implications.
Materials and Methods
This work used summary data obtained from published studies that had each previously received appropriate ethics and institutional review board approvals, and further sanction was therefore not required. The data and statistical coding used in this work can be obtained from the corresponding author on reasonable request. All statistical analysis was performed using R version 3.4.2 (The R Foundation for Statistical Computing, Vienna, Austria) and the MendelianRandomization and MR‐PRESSO software packages.25, 26
Genetic Instrument Selection
Single‐nucleotide polymorphisms (SNPs) to proxy iron status were obtained from a genome‐wide association study (GWAS) meta‐analysis performed by the GIS (Genetics of Iron Status) consortium,27 combining data from 48 972 subjects of European descent. Genetic associations between SNPs and iron biomarkers were identified for each sex separately using standardized residuals after making study‐specific adjustments (Table S1).27
Increased systemic iron status is associated with increased serum iron, transferrin saturation, and ferritin and with decreased transferrin.19 These markers can therefore be used as proxies for systemic iron status—the independent (endogenous) variable under consideration in this study. Accordingly, SNPs shown to have significant directional association with these 4 biomarkers (increased serum iron, ferritin, transferrin saturation, and decreased transferrin levels) were considered as potential genetic instruments. The GWAS meta‐analysis performed by the GIS consortium identified 12 SNPs associated with the aforementioned biomarkers of iron status (Table S2). Three of these (rs1800562 and rs1799945 in the hemochromatosis [HFE] gene and rs855791 in the transmembrane protease [TMPRSS6] serine 6 gene) demonstrated an association with all 4 biomarkers that was concordant with an effect on systemic iron status at genome‐wide significance (P<5×10−8). These were therefore selected as genetic instruments. Linkage disequilibrium between the 2 loci within the HFE gene was low (r 2<0.01), consistent with their independence. The biological effects of the HFE and TMPRSS6 proteins on systemic iron status are detailed in Data S1.
Instrument strength was evaluated using the F statistic,28 derived from a measure of the exposure variance explained by each SNP. To limit potential weak instrument bias, only SNPs with an F statistic of >10 were used.28
Genetic Associations
Association estimates between the SNPs and risk of VTE were derived from a GWAS meta‐analysis performed by the International Network on Venous Thrombosis Consortium.29 Data from 12 studies were included (Table S3, with details of adjustments and exclusion criteria), incorporating 7507 cases of VTE and 52 632 controls. Subjects were of European ancestry and had a diagnosis of VTE (deep vein thrombosis or pulmonary embolism) made objectively by a physician following clinical evaluation.
A GWAS meta‐analysis performed by the Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium was used to derive association estimates between SNPs and cIMT and carotid plaque.30 The meta‐analysis included data from 31 studies for cIMT and 17 studies for carotid plaque trait (Table S4, with details of adjustments and exclusion criteria), incorporating 71 128 and 48 434 (21 540 cases and 26 894 controls) participants, respectively. Subjects were of European ancestry and were evaluated using high‐resolution B‐mode ultrasonography for carotid plaque and cIMT parameters.31 Carotid plaque was defined as atherosclerotic thickening of the carotid artery wall or luminal stenosis >25%. cIMT parameters were defined as the mean of maximal values from several common carotid artery measurements, measured in millimeters.
Participant overlap in the studies used to obtain genetic association estimates for the exposure and the outcome can introduce bias into MR analysis.32 Based on the cohorts included in the considered GWAS meta‐analyses (Tables S1, S3, and S4), the Erasmus Rucphen Family Study contributed participants for investigation of iron status, cIMT, and carotid plaque, whereas the Nikmegen Biomedial Study contributed participants for investigation of iron status and cIMT.27, 30 This therefore resulted in a potential overlap of 1420 participants in the investigation of cIMT and of 549 participants for the investigation of carotid plaque. No cohorts overlapped for the investigation of iron status and VTE.27, 29
Mendelian Randomization Analysis
The main MR effect estimates were derived using the Wald Estimator,33 with the Delta method used to calculate standard error.34 Individual MR estimates for each measure of iron status were then combined using fixed‐effect inverse‐variance–weighted (IVW) meta‐analysis, to establish their overall effect on VTE and carotid plaque risk (calculated as odds ratio [OR] per SD unit increase in iron‐status biomarker), and effect on carotid intimal artery thickness (calculated as millimeter variation in cIMT per SD change in iron‐status biomarker).28 A statistical significance threshold of P<0.05 was used for these main MR analyses. This threshold was not adjusted for multiple testing of the different iron‐status biomarkers, as they each represented a proxy for overall iron status, which was the clinically relevant trait under consideration. Furthermore, adjustment for multiple testing of distinct outcomes was also not required, as each trait was specifically investigated to follow up the findings of previous research that had already identified significant effects.14, 15
For the main IVW MR analyses, the minimum and maximum true causal effects required to achieve 80% statistical power were estimated to provide an indication of the potential for false‐negative findings.35
Pleiotropy
MR analysis is based on the assumption that SNP outcome effects are mediated solely through the exposure (iron status in this study). Violation of this assumption through horizontal pleiotropy, whereby there is an association between the instrument and disease independent of the exposure of interest, can introduce directional bias.36
Statistical sensitivity analyses more robust to the inclusion of potentially pleiotropic variants can be used to help establish the validity of causal inference from MR analysis. However, such analyses typically require more than 3 instruments. Therefore, to increase the number of genetic instruments and allow for such statistical sensitivity analyses, the instrument selection criteria were relaxed in the GIS GWAS meta‐analysis to also include other SNPs associated with at least 1 biomarker reflecting higher iron status (ie, increased serum iron, ferritin, and transferrin saturation and decreased transferrin levels) at genome‐wide significance, with concordant directions of association with the other biomarkers, even if they did not reach genome‐wide statistical significance.14 Three further SNPs were identified using these selection criteria: rs7385804 as part of the transferrin receptor 2 (TFR2) gene, rs9990333 from the transferrin receptor (TFRC) gene, and rs411988 in the testis‐expressed 14 intercellular bridge‐forming factor (TEX14) gene. IVW MR analysis was subsequently repeated using all 6 SNPs for risk of cIMT and carotid plaque and with 5 SNPs for VTE (association estimates were not available for the rs1799945 SNP and VTE, nor was a suitable proxy with linkage disequilibrium r 2>0.8).
Additional sensitivity analyses were performed using the MR‐Egger, weighted median and MR‐pleiotropy residual sum and outlier (PRESSO) methods.26, 37, 38 The MR‐Egger technique provides an estimate of horizontal pleiotropy from the intercept of a linear regression of SNP‐outcome and SNP‐exposure association estimates (deemed statistically significant based on P<0.05). In the absence of pleiotropic bias, either through the genetic instruments having no horizontal pleiotropy or directional pleiotropic effects canceling each other out, this intercept tends to 0. This method relies on the assumption that the SNP‐outcome association estimates are not correlated with the extent of pleiotropy arising from that instrument (instrument strength independent of direct effect assumption).39 In contrast, the weighted median MR sensitivity analysis does not rely on the instrument strength independent of direct effect assumption. This method calculates the median of an empirical distribution of MR association estimates weighted for their precision and provides consistent estimates when at least 50% of information for the analysis comes from valid instruments. Finally, MR‐PRESSO regresses the SNP‐outcome estimates on the SNP‐exposure estimates, with the gradient of the regression line representing the MR estimate.26 Furthermore, MR‐PRESSO is able to identify outlier variants based on their observed distance from the regression line, as compared with their expected distance based on the assumption of no horizontal pleiotropy.26
Given the lower statistical power of these sensitivity analyses,40 no formal significance threshold was set, and results were evaluated for consistency with the main analysis.
Results
Association estimates for SNP iron‐status biomarkers are shown in Table S5. The F statistics for genetic instruments were between 47 and 2127 across the 4 biomarkers of iron status. MR estimates, expressed as OR per SD unit increase in iron‐status biomarker for carotid plaque and VTE, and millimeter change in cIMT per SD unit increase in iron‐status biomarker for cIMT, are shown in Table S6. The minimum and maximum true causal effects required to achieve 80% statistical power for the main IVW MR analysis are detailed in Table S7.
The results demonstrate a detrimental effect on risk of VTE for serum iron (OR 1.37; 95% CI 1.14‐1.66; P=1×10−3), transferrin saturation (OR 1.25; 95% CI 1.09‐1.43; P=1×10−3) and (log‐transformed) ferritin (OR 1.92; 95% CI 1.28‐2.88; P=2×10−3) (Figure 1). Concordant with a detrimental effect of high iron status, transferrin levels (reflecting lower systemic iron) were associated with a decreased risk of VTE (OR 0.76; 95% CI 0.63‐0.92; P=0.01).
Figure 1.
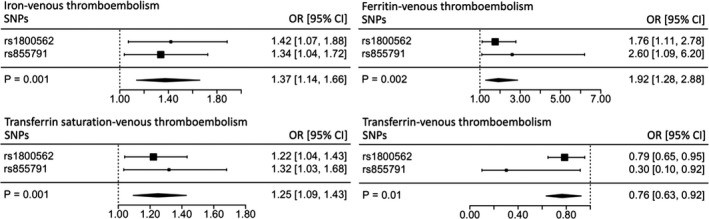
Individual SNP and pooled MR estimates for the effect of iron status on venous thromboembolism. Results for each biomarker are represented in a different forest plot. Each square represents an individual SNP MR estimate, with size proportional to the precision of the estimate, and horizontal lines representing 95% CIs. The diamonds underneath represent the pooled MR estimate, with corresponding widths representing 95% CIs. MR indicates Mendelian randomization; OR, odds ratio; SNP, single‐nucleotide polymorphism.
In contrast, the MR analysis demonstrated a protective effect on the risk of carotid plaque for serum iron (OR, 0.85; 95% CI, 0.73‐0.99; P=0.04) and transferrin saturation (OR, 0.89; 95% CI, 0.80‐1.00; P=0.05) (Figure 2). The other biomarkers reflected a protective role of higher iron status in carotid plaque, although their effect estimates did not reach significance ([log‐transformed] serum ferritin OR, 0.72; 95% CI, 0.51‐1.01; P=0.06; serum transferrin OR, 1.15; 95% CI, 0.97‐1.35; P=0.11).
Figure 2.
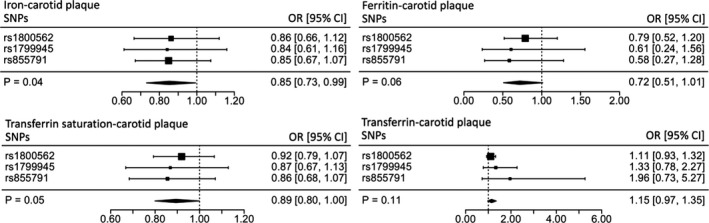
Individual SNP and pooled MR estimates for the effect of iron status on carotid plaque. Results for each biomarker are represented in a different forest plot. Each square represents an individual SNP MR estimate, with size proportional to the precision of the estimate, and horizontal lines representing 95% CIs. The diamonds underneath represent the pooled MR estimate, with corresponding widths representing 95% CIs. MR indicates Mendelian randomization; OR, odds ratio; SNP, single‐nucleotide polymorphism.
There was no significant association between iron status and cIMT (millimeter variation in cIMT per SD change in serum iron 0.00, 95% CI −0.01 to 0.01, P=0.90; transferrin saturation 0.00, 95% CI −0.01 to 0.01, P=0.75; [log‐transformed] serum ferritin 0.01, 95% CI −0.02 to 0.03, P=0.58; serum transferrin −0.01, 95% CI −0.01 to 0.01, P=0.32) (Figure 3).
Figure 3.
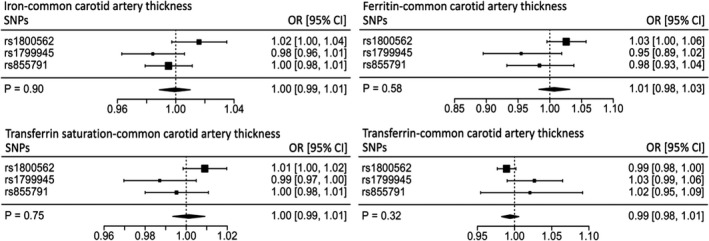
Individual SNP and pooled MR estimates for the effect of iron status on carotid intima‐media thickness. Results for each biomarker are represented in a different forest plot. Each square represents an individual SNP MR estimate, with size proportional to the precision of the estimate, and horizontal lines representing 95% CIs. The diamonds underneath represent the pooled MR estimate, with corresponding widths representing 95% CIs. MR indicates Mendelian randomization; OR, odds ratio; SNP, single‐nucleotide polymorphism.
Consistent directional effects for all analyses were observed in the IVW MR, MR‐Egger, weighted median, and MR‐PRESSO sensitivity analyses (incorporating the aforementioned genetic instruments selected from the GWAS search for loci with association with at least 1 biomarker of iron status) (Table S6). The MR‐Egger intercepts did not provide evidence of directional pleiotropy in any analysis, and neither did MR‐PRESSO identify outliers (Table S6).
Discussion
Contextual Findings and Mechanistic Insight
This study provides MR evidence of a contrasting role of higher genetically determined iron status on different thrombotic disease processes—increasing VTE risk, reducing risk of carotid plaque, and having no significant effect on cIMT.
Several observational studies have investigated the association between iron status and carotid atherosclerotic disease, with inconsistent results. Three studies found a sex‐specific positive association between serum ferritin and carotid plaque41, 42 or cIMT,43 2 studies provided evidence for a positive association with carotid plaque in both sexes combined,10, 44 and 2 others did not find any association between serum ferritin and carotid atherosclerosis.45, 46 In contrast, 2 further case‐control studies have reported a negative association between serum ferritin and cIMT.47, 48 These discrepancies may in part be due to unmeasured confounding such as that related to inflammation. Furthermore, they may represent a contrasting role of iron in different atherosclerotic phenotypes, with cIMT representing arterial hyperplasia (in response to hypertension) and carotid plaque representing fatty atherosclerotic lesions.49 The mechanisms by which iron may affect these processes remain unclear, although higher iron status has been implicated in carotid plaque development through oxidative modification of circulating lipids.50, 51 Within the wider context of atherosclerotic disease, there is evidence of a protective role of higher iron status in coronary heart disease in both observational18 and genetic studies.15
In contrast to atherosclerosis there have been relatively few studies investigating the association between iron status and VTE. Consistent with our results, a nested case‐control study found evidence of an increased risk of VTE in patients with higher hepcidin, a biomarker positively associated with iron levels.52 The study, which included 390 patients with confirmed VTE along with 802 age‐ and sex‐matched controls, identified a dose‐dependent relationship between hepcidin and risk of VTE (independent of C‐reactive protein, a marker of inflammation). However, the authors noted that their results were limited by potential confounding from other unmeasured mediators of iron metabolism (eg, underlying comorbidities, medications/supplements) as well as by the delay between sampling and recorded VTE events. Our current MR study, which utilized genetic instruments associated with 4 different biomarkers of iron status, provides further evidence of a detrimental role of higher iron status in VTE, which is more robust to the confounding suffered in traditional observational studies.
Taken together, these findings provide evidence of a protective role of higher iron status in some atherosclerotic processes, although it increased the risk of thromboembolic phenomena related to stasis of blood. This is consistent with previous MR analyses, which demonstrated a positive association between higher iron status and cardioembolic stroke,14 despite a reduced risk of coronary artery disease.15 The underlying mechanisms for this dichotomous relationship are unclear but may in part be due to the oxidizing properties of unliganded iron.53 Indeed, iron‐induced oxidative stress has been implicated in endothelial dysfunction, platelet activation, fibrin formation, and impaired plasminogen activation, which may in turn potentiate thromboembolic disease.54, 55, 56 Consistent with our results, a systematic analysis of iron status and coronary heart disease concluded that serum iron is associated with lower risk of coronary heart disease, for which atherosclerosis is a major mediator.18 A possible explanation for this protective effect is due to a reduction in circulating low‐density lipoprotein cholesterol levels attributable to a higher iron status.57 This may explain why our results demonstrate a protective role of higher iron status in carotid plaque only, since this is a marker of dyslipidemia and fatty plaque formation, whereas cIMT reflects vessel hyperplasia in response to hypertension.49 This is consistent with 2 observational analyses that demonstrated a positive association between serum ferritin and carotid plaque but not cIMT.41, 42 Alternatively, higher iron status may demonstrate a protective effect by acting as a surrogate marker for normal hemoglobin levels, which may be protective in atherosclerosis.18 Indeed, lower iron status is associated with iron‐deficiency anemia, which is in itself an established risk factor for coronary heart disease.18
Strengths and Limitations
A key strength of this MR analysis is its ability to overcome the environmental confounding encountered in traditional observational studies by using genetic variants to instrument the exposure. Indeed, biomarkers of iron status are implicated in other pathologies, including inflammation, liver disease, renal failure, and malignancy, all of which could affect observational associations with thrombotic disease.58, 59 Furthermore, our study offers insight into how iron status affects distinct thrombotic disease processes and supports evidence from 2 previous MR analyses investigating related pathophysiological mechanisms.14, 15 The minimum and maximum true causal estimates required to achieve 80% statistical power for the main IVW MR analysis (Table S7) also indicate that this study had adequate statistical power to detect clinically relevant effects.
Although these results have potentially significant clinical implications, it is important they be interpreted in context. Iron status exhibits a nonlinear relationship with thrombotic disease, with both iron deficiency and overload potential risk factors for atherosclerosis and thromboembolic processes.5, 7, 8, 9, 10, 11, 12, 16 Because MR analysis assumes a linear relationship between the instrumental variable and disease process,60 these findings should not be extrapolated beyond the normal range of iron status. Furthermore, by using genetic variants as proxies for iron status, we consider the lifetime effect of genetically determined iron status on thrombotic disease; hence, association estimates are likely to be greater than seen in comparable observational analyses.
Pleiotropy, whereby genetic instruments affect the disease outcome through pathways independent of the instrumented exposure, can also introduce bias into MR analysis.13 Indeed, previous MR work using the same instrument SNPs has identified potential pleiotropic associations with low‐density lipoprotein cholesterol levels and systolic blood pressure.15 In this study we relaxed the criteria for instrument selection to include additional SNPs associated with at least 1 biomarker of iron status at genome‐wide significance. Although this increased the risk of including invalid instruments, it did allow for statistical sensitivity analyses that are more robust to the inclusion of pleiotropic variants.40 MR‐Egger, weighted median, and MR‐PRESSO analyses with these instruments demonstrated consistent casual effects of iron status on each thrombotic disease, supporting the validity of our results. Furthermore, MR‐Egger did not provide evidence of directional pleiotropy, and MR‐PRESSO did not identify any outliers. However, MR‐Egger often suffers particularly low statistical power,40, 61 in keeping with the generally wider CIs and weaker P‐values of our results with this method as compared with the other approaches, and the findings from this should therefore be interpreted cautiously.
The analyses performed in this study were undertaken entirely in individuals of European ancestry. Further work will therefore be required to investigate whether similar findings are found in studying populations of different ethnicities. Finally, although there was likely a small degree of participant overlap in the studies used to obtain genetic association estimates for the iron‐status biomarkers and carotid traits,27, 30 the overlapping cohorts make up <3% of the overall population considered in any given GWAS and are therefore unlikely to have introduced significant bias.32
Conclusion
In this study we used MR analysis to investigate the association between iron status and different thrombotic disease processes. We found that higher iron status is associated with increased risk of VTE and reduced risk of carotid plaque disease but has no relation with carotid thickness. These results provide further evidence for a protective role of higher iron status in some forms of atherosclerotic disease along with increasing risk of a thromboembolic phenomenon related to stasis of blood. Given the scale of variation in iron status worldwide and the burden of thrombotic disease, these results have potentially significant clinical and public health implications. Further investigation is required to determine the precise mechanism of the suggested effects.
Author Contributions
Gill designed the study. Monori and Gill performed the analysis. Brewer and Gill drafted this article. All authors interpreted results. All authors critically revised the manuscript for intellectual content and approved the submitted version of the manuscript. All authors are accountable for the accuracy and integrity of the work.
Sources of Funding
Gill was funded by the Wellcome 4i Clinical PhD Programme at Imperial College London. Trégouët was financially supported by the EPIDEMIOM‐VTE Senior Chair from the Initiative of Excellence of the University of Bordeaux.
Disclosures
None.
Supporting information
Appendix S1. INVENT Consortium Members and Their Affiliations
Data S1. Biological Effects of the HFE and TMPRSS6 Proteins on Systemic Iron Status
Table S1. Cohort Demographics and Covariates for the Genetics of Iron Status Consortium GWAS Meta‐Analysis Adapted From Benyamin et al 20146
Table S2. Association Estimates for SNPs Associated With Biomarkers of Iron Status at Genome‐Wide Significance Identified From the Genetics of Iron Status Consortium GWAS Meta‐Analysis6
Table S3. Cohort Demographics and Covariates for the International Network Against Thrombosis Collaboration GWAS Meta‐Analysis7
Table S4. Cohort Demographics and Covariates for the Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium GWAS Meta‐Analysis8
Table S5. SNP‐Iron Association Estimates Obtained From the Genetics of Iron Status Consortium GWAS Meta‐Analysis6
Table S6. Mendelian Randomization Estimates and Statistical Sensitivity Analyses
Table S7. The Minimum and Maximum True Causal Effects Required to Achieve 80% Statistical Power for the Main Inverse‐Variance–Weighted Mendelian Randomization Analysis
Acknowledgments
The authors would like to acknowledge and thank the Genetics of Iron Status consortium, the International Network on Venous Thrombosis Consortium, and the Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium for making the data used in this project available.
(J Am Heart Assoc. 2019;8:e012994 DOI: 10.1161/JAHA.119.012994.)
References
- 1. Mathers C, Stevens GA, Mahanani WR, Fat DM, Hogan D. WHO methods and data sources for country‐level causes of death 2000–2016. 2018. Available at: http://terrance.who.int/mediacentre/data/ghe/GlobalCOD_method_2000_2016.pdf. Accessed March 9, 2019.
- 2. Wendelboe AM, Raskob GE. Global burden of thrombosis. Circ Res. 2016;118:1340–1347. [DOI] [PubMed] [Google Scholar]
- 3. Katan M, Luft A. Global burden of stroke. Semin Neurol. 2018;38:208–211. [DOI] [PubMed] [Google Scholar]
- 4. Raskob GE, Angchaisuksiri P, Blanco AN, Buller H, Gallus A, Hunt BJ, Hylek EM, Kakkar A, Konstantinides SV, McCumber M, Ozaki Y, Wendelboe A, Weitz JI; ISTH Steering Committee for World Thrombosis Day . Thrombosis. Arterioscler Thromb Vasc Biol. 2014;34:2363–2371. [DOI] [PubMed] [Google Scholar]
- 5. Franchini M, Targher G, Montagnana M, Lippi G. Iron and thrombosis. Ann Hematol. 2008;87:167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Basuli D, Stevens RG, Torti FM, Torti SV. Epidemiological associations between iron and cardiovascular disease and diabetes. Front Pharmacol. 2014;5:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Keung Y‐K, Owen J. Iron deficiency and thrombosis: literature review. Clin Appl Thromb Hemost. 2004;10:387–391. [DOI] [PubMed] [Google Scholar]
- 8. Hung S‐H, Lin H‐C, Chung S‐D. Association between venous thromboembolism and iron‐deficiency anemia. Blood Coagul Fibrinolysis. 2015;26:368–372. [DOI] [PubMed] [Google Scholar]
- 9. Xie YG, Lillicrap DP, Taylor SA. An association between the common hereditary hemochromatosis mutation and the factor V Leiden allele in a population with thrombosis. Blood. 1998;92:1461–1462. [PubMed] [Google Scholar]
- 10. Ahluwalia N, Genoux A, Ferrieres J, Perret B, Carayol M, Drouet L, Ruidavets J‐B. Iron status is associated with carotid atherosclerotic plaques in middle‐aged adults. J Nutr. 2010;140:812–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kraml P. The role of iron in the pathogenesis of atherosclerosis. Physiol Res. 2017;66:S55–S67. [DOI] [PubMed] [Google Scholar]
- 12. Grammer TB, Kleber ME, Silbernagel G, Pilz S, Scharnagl H, Tomaschitz A, König W, März W. Hemoglobin, iron metabolism and angiographic coronary artery disease (The Ludwigshafen Risk and Cardiovascular Health Study). Atherosclerosis. 2014;236:292–300. [DOI] [PubMed] [Google Scholar]
- 13. Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gill D, Monori G, Tzoulaki I, Dehghan A. Iron status and risk of stroke. Stroke. 2018;49:2815–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gill D, Del Greco MF, Walker AP, Srai SKS, Laffan MA, Minelli C. The effect of iron status on risk of coronary artery disease. Arterioscler Thromb Vasc Biol. 2017;37:1788–1792. [DOI] [PubMed] [Google Scholar]
- 16. Gillum RF, Sempos CT, Makuc DM, Looker AC, Chien CY, Ingram DD. Serum transferrin saturation, stroke incidence, and mortality in women and men. The NHANES I Epidemiologic Followup Study. National Health and Nutrition Examination Survey. Am J Epidemiol. 1996;144:59–68. [DOI] [PubMed] [Google Scholar]
- 17. van der A DL, Grobbee DE, Roest M, Marx JJM, Voorbij HA, van der Schouw YT. Serum ferritin is a risk factor for stroke in postmenopausal women. Stroke. 2005;36:1637–1641. [DOI] [PubMed] [Google Scholar]
- 18. Das De S, Krishna S, Jethwa A. Iron status and its association with coronary heart disease: systematic review and meta‐analysis of prospective studies. Atherosclerosis. 2015;238:296–303. [DOI] [PubMed] [Google Scholar]
- 19. Wish JB. Assessing iron status: beyond serum ferritin and transferrin saturation. Clin J Am Soc Nephrol. 2006;1:S4–S8. [DOI] [PubMed] [Google Scholar]
- 20. Lopez A, Cacoub P, Macdougall IC, Peyrin‐Biroulet L. Iron deficiency anaemia. Lancet. 2016;387:907–916. [DOI] [PubMed] [Google Scholar]
- 21. Plichart M, Celermajer DS, Zureik M, Helmer C, Jouven X, Ritchie K, Tzourio C, Ducimetière P, Empana J‐P. Carotid intima‐media thickness in plaque‐free site, carotid plaques and coronary heart disease risk prediction in older adults. The Three‐City Study. Atherosclerosis. 2011;219:917–924. [DOI] [PubMed] [Google Scholar]
- 22. Spence JD. Measurement of intima‐media thickness vs. carotid plaque: uses in patient care, genetic research and evaluation of new therapies. Int J Stroke. 2006;1:216–221. [DOI] [PubMed] [Google Scholar]
- 23. Rundek T, Gardener H, Della‐Morte D, Dong C, Cabral D, Tiozzo E, Roberts E, Crisby M, Cheung K, Demmer R, Elkind MS, Sacco RL, Desvarieux M. The relationship between carotid intima‐media thickness and carotid plaque in the Northern Manhattan Study. Atherosclerosis. 2015;241:364–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. von Haehling S, Jankowska EA, van Veldhuisen DJ, Ponikowski P, Anker SD. Iron deficiency and cardiovascular disease. Nat Rev Cardiol. 2015;12:659–669. [DOI] [PubMed] [Google Scholar]
- 25. Yavorska OO, Burgess S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol. 2017;46:1734–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benyamin B, Esko T, Ried JS, Radhakrishnan A, Vermeulen SH, Traglia M, Gögele M, Anderson D, Broer L, Podmore C, Luan Ja, Kutalik Z, Sanna S, van der Meer P, Tanaka T, Wang F, Westra H‐J, Franke L, Mihailov E, Milani L, Hälldin J, Winkelmann J, Meitinger T, Thiery J, Peters A, Waldenberger M, Rendon A, Jolley J, Sambrook J, Kiemeney LA, Sweep FC, Sala CF, Schwienbacher C, Pichler I, Hui J, Demirkan A, Isaacs A, Amin N, Steri M, Waeber G, Verweij N, Powell JE, Nyholt DR, Heath AC, Madden PAF, Visscher PM, Wright MJ, Montgomery GW, Martin NG, Hernandez D, Bandinelli S, van der Harst P, Uda M, Vollenweider P, Scott RA, Langenberg C, Wareham NJ, van Duijn C, Beilby J, Pramstaller PP, Hicks AA, Ouwehand WH, Oexle K, Gieger C, Metspalu A, Camaschella C, Toniolo D, Swinkels DW, Whitfield JB. Novel loci affecting iron homeostasis and their effects in individuals at risk for hemochromatosis. Nat Commun. 2014;5:4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Palmer TM, Lawlor DA, Harbord RM, Sheehan NA, Tobias JH, Timpson NJ, Smith GD, Sterne JAC. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res. 2012;21:223–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Germain M, Chasman DI, de Haan H, Tang W, Lindström S, Weng L‐C, de Andrade M, de Visser MCH, Wiggins KL, Suchon P, Saut N, Smadja DM, Le Gal G, van Hylckama Vlieg A, Di Narzo A, Hao K, Nelson CP, Rocanin‐Arjo A, Folkersen L, Monajemi R, Rose LM, Brody JA, Slagboom E, Aïssi D, Gagnon F, Deleuze J‐F, Deloukas P, Tzourio C, Dartigues J‐F, Berr C, Taylor KD, Civelek M, Eriksson P; Cardiogenics Consortium C , Psaty BM, Houwing‐Duitermaat J, Goodall AH, Cambien F, Kraft P, Amouyel P, Samani NJ, Basu S, Ridker PM, Rosendaal FR, Kabrhel C, Folsom AR, Heit J, Reitsma PH, Trégouët D‐A, Smith NL, Morange P‐E. Meta‐analysis of 65,734 individuals identifies TSPAN15 and SLC44A2 as two susceptibility loci for venous thromboembolism. Am J Hum Genet. 2015;96:532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Franceschini N, Giambartolomei C, de Vries PS, Finan C, Bis JC, Huntley RP, Lovering RC, Tajuddin SM, Winkler TW, Graff M, Kavousi M, Dale C, Smith AV, Hofer E, van Leeuwen EM, Nolte IM, Lu L, Scholz M, Sargurupremraj M, Pitkänen N, Franzén O, Joshi PK, Noordam R, Marioni RE, Hwang S‐J, Musani SK, Schminke U, Palmas W, Isaacs A, Correa A, Zonderman AB, Hofman A, Teumer A, Cox AJ, Uitterlinden AG, Wong A, Smit AJ, Newman AB, Britton A, Ruusalepp A, Sennblad B, Hedblad B, Pasaniuc B, Penninx BW, Langefeld CD, Wassel CL, Tzourio C, Fava C, Baldassarre D, O'Leary DH, Teupser D, Kuh D, Tremoli E, Mannarino E, Grossi E, Boerwinkle E, Schadt EE, Ingelsson E, Veglia F, Rivadeneira F, Beutner F, Chauhan G, Heiss G, Snieder H, Campbell H, Völzke H, Markus HS, Deary IJ, Jukema JW, de Graaf J, Price J, Pott J, Hopewell JC, Liang J, Thiery J, Engmann J, Gertow K, Rice K, Taylor KD, Dhana K, Kiemeney LALM, Lind L, Raffield LM, Launer LJ, Holdt LM, Dörr M, Dichgans M, Traylor M, Sitzer M, Kumari M, Kivimaki M, Nalls MA, Melander O, Raitakari O, Franco OH, Rueda‐Ochoa OL, Roussos P, Whincup PH, Amouyel P, Giral P, Anugu P, Wong Q, Malik R, Rauramaa R, Burkhardt R, Hardy R, Schmidt R, de Mutsert R, Morris RW, Strawbridge RJ, Wannamethee SG, Hägg S, Shah S, McLachlan S, Trompet S, Seshadri S, Kurl S, Heckbert SR, Ring S, Harris TB, Lehtimäki T, Galesloot TE, Shah T, de Faire U, Plagnol V, Rosamond WD, Post W, Zhu X, Zhang X, Guo X, Saba Y, Dehghan A, Seldenrijk A, Morrison AC, Hamsten A, Psaty BM, van Duijn CM, Lawlor DA, Mook‐Kanamori DO, Bowden DW, Schmidt H, Wilson JF, Wilson JG, Rotter JI, Wardlaw JM, Deanfield J, Halcox J, Lyytikäinen L‐P, Loeffler M, Evans MK, Debette S, Humphries SE, Völker U, Gudnason V, Hingorani AD, Björkegren JLM, Casas JP, O'Donnell CJ. GWAS and colocalization analyses implicate carotid intima‐media thickness and carotid plaque loci in cardiovascular outcomes. Nat Commun. 2018;9:5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després J‐P, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jiménez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB; American Heart Association Statistics Committee and Stroke Statistics Subcommittee . Heart disease and stroke statistics—2016 update. Circulation. 2016;133:e38–e360. [DOI] [PubMed] [Google Scholar]
- 32. Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two‐sample Mendelian randomization. Genet Epidemiol. 2016;40:597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lawlor DA, Harbord RM, Sterne JAC, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133–1163. [DOI] [PubMed] [Google Scholar]
- 34. Thomas DC, Lawlor DA, Thompson JR. Re: estimation of bias in nongenetic observational studies using “Mendelian triangulation” by Bautista et al. Ann Epidemiol. 2007;17:511–513. [DOI] [PubMed] [Google Scholar]
- 35. Brion MJ, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol. 2013;42:1497–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sheehan NA, Didelez V, Burton PR, Tobin MD. Mendelian randomisation and causal inference in observational epidemiology. PLoS Med. 2008;5:e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR‐Egger method. Eur J Epidemiol. 2017;32:377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology. 2017;28:30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wolff B, Völzke H, Lüdemann J, Robinson D, Vogelgesang D, Staudt A, Kessler C, Dahm JB, John U, Felix SB. Association between high serum ferritin levels and carotid atherosclerosis in the study of health in Pomerania (SHIP). Stroke. 2004;35:453–457. [DOI] [PubMed] [Google Scholar]
- 42. Rossi E, McQuillan BM, Hung J, Thompson PL, Kuek C, Beilby JP. Serum ferritin and C282Y mutation of the hemochromatosis gene as predictors of asymptomatic carotid atherosclerosis in a community population. Stroke. 2000;31:3015–3020. [DOI] [PubMed] [Google Scholar]
- 43. Xu H, Song Y, Xu J, Gu Y, Zhang Q, Liu L, Meng G, Wu H, Xia Y, Bao X, Shi H, Su Q, Fang L, Yu F, Yang H, Sun S, Wang X, Zhou M, Jia Q, Wang G, Song K, Wu Y, Sun Z, Niu K. Increased serum ferritin levels are independently associated with carotid atherosclerosis in women. Br J Nutr. 2017;117:1623–1630. [DOI] [PubMed] [Google Scholar]
- 44. Kiechl S, Aichner F, Gerstenbrand F, Egger G, Mair A, Rungger G, Spögler F, Jarosch E, Oberhollenzer F, Willeit J. Body iron stores and presence of carotid atherosclerosis. Results from the Bruneck Study. Arterioscler Thromb. 1994;14:1625–1630. [DOI] [PubMed] [Google Scholar]
- 45. Vergnaud AC, Bertrais S, Zureik M, Galan P, Blacher J, Hercberg S, Czernichow S. Dietary iron intake and serum ferritin in relation to 7.5 years structure and function of large arteries in the SUVIMAX cohort. Diabetes Metab. 2007;33:366–371. [DOI] [PubMed] [Google Scholar]
- 46. Yunker LM, Parboosingh JS, Conradson HE, Faris P, Bridge PJ, Buithieu J, Title LM, Charbonneau F, Verma S, Lonn EM, Anderson TJ. The effect of iron status on vascular health. Vasc Med. 2006;11:85–91. [DOI] [PubMed] [Google Scholar]
- 47. Moore M, Folsom AR, Barnes RW, Eckfeldt J. No association between serum ferritin and asymptomatic carotid atherosclerosis. Am J Epidemiol. 1995;141:719–723. [DOI] [PubMed] [Google Scholar]
- 48. Raumaraa R, Vaisanen S, Mecuri M, Raniken T, Penttila I, Bond MG. Association of risk factors and body iron status to carotid atherosclerosis in middle‐aged eastern Finnish men. Eur Heart J. 1994;15:1020–1027. [DOI] [PubMed] [Google Scholar]
- 49. Touboul PJ, Hennerici MG, Meairs S, Adams H, Amarenco P, Bornstein N, Csiba L, Desvarieux M, Ebrahim S, Hernandez Hernandez R, Jaff M, Kownator S, Naqvi T, Prati P, Rundek T, Sitzer M, Schminke U, Tardif JC, Taylor A, Vicaut E, Woo KS. Mannheim carotid intima‐media thickness and plaque consensus (2004–2006–2011). Cerebrovasc Dis. 2012;34:290–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fuhrman B, Oiknine J, Aviram M. Iron induces lipid peroxidation in cultured macrophages, increases their ability to oxidatively modify LDL, and affects their secretory properties. Atherosclerosis. 1994;111:65–78. [DOI] [PubMed] [Google Scholar]
- 51. Yuan XM, Brunk UT, Olsson AG. Effects of iron‐ and hemoglobin‐loaded human monocyte‐derived macrophages on oxidation and uptake of LDL. Arterioscler Thromb Vasc Biol. 1995;15:1345–1351. [DOI] [PubMed] [Google Scholar]
- 52. Ellingsen TS, Lappegård J, Ueland T, Aukrust P, Brækkan SK, Hansen J‐B. Plasma hepcidin is associated with future risk of venous thromboembolism. Blood Adv. 2018;2:1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kell DB. Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med Genomics. 2009;2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Feng YH, Hart G. In vitro oxidative damage to tissue‐type plasminogen activator: a selective modification of the biological functions. Cardiovasc Res. 1995;30:255–261. [PubMed] [Google Scholar]
- 55. Cooke JP. Does ADMA cause endothelial dysfunction? Arterioscler Thromb Vasc Biol. 2000;20:2032–2037. [DOI] [PubMed] [Google Scholar]
- 56. Upchurch GR Jr, Ramdev N, Walsh MT, Loscalzo J. Prothrombotic consequences of the oxidation of fibrinogen and their inhibition by aspirin. J Thromb Thrombolysis. 1998;5:9–14. [DOI] [PubMed] [Google Scholar]
- 57. Ozdemir A, Sevinc C, Selamet U, Turkmen F. The relationship between iron deficiency anemia and lipid metabolism in premenopausal women. Am J Med Sci. 2007;334:331–333. [DOI] [PubMed] [Google Scholar]
- 58. Dignass A, Farrag K, Stein J. Limitations of serum ferritin in diagnosing iron deficiency in inflammatory conditions. Int J Chronic Dis. 2018;2018:9394060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pfeiffer CM, Looker AC. Laboratory methodologies for indicators of iron status: strengths, limitations, and analytical challenges. Am J Clin Nutr. 2017;106:1606S–1614S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Smith GD, Ebrahim S. What can Mendelian randomisation tell us about modifiable behavioural and environmental exposures? BMJ. 2005;330:1076–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Slob EAW, Burgess S. A comparison of robust Mendelian randomization methods using summary data. bioRxiv. 2019. Available at: 10.1101/577940. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. INVENT Consortium Members and Their Affiliations
Data S1. Biological Effects of the HFE and TMPRSS6 Proteins on Systemic Iron Status
Table S1. Cohort Demographics and Covariates for the Genetics of Iron Status Consortium GWAS Meta‐Analysis Adapted From Benyamin et al 20146
Table S2. Association Estimates for SNPs Associated With Biomarkers of Iron Status at Genome‐Wide Significance Identified From the Genetics of Iron Status Consortium GWAS Meta‐Analysis6
Table S3. Cohort Demographics and Covariates for the International Network Against Thrombosis Collaboration GWAS Meta‐Analysis7
Table S4. Cohort Demographics and Covariates for the Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium GWAS Meta‐Analysis8
Table S5. SNP‐Iron Association Estimates Obtained From the Genetics of Iron Status Consortium GWAS Meta‐Analysis6
Table S6. Mendelian Randomization Estimates and Statistical Sensitivity Analyses
Table S7. The Minimum and Maximum True Causal Effects Required to Achieve 80% Statistical Power for the Main Inverse‐Variance–Weighted Mendelian Randomization Analysis