

Abstract
Background
Ischemia/reperfusion (I/R) injury is a critical issue in the development of treatment strategies for ischemic heart disease. MURC (muscle‐restricted coiled‐coil protein)/Cavin‐4 (caveolae‐associated protein 4), which is a component of caveolae, is involved in the pathophysiology of dilated cardiomyopathy and cardiac hypertrophy. However, the role of MURC in cardiac I/R injury remains unknown.
Methods and Results
The systems network genomic analysis based on PC‐corr network inference on microarray data between wild‐type and MURC knockout mouse hearts predicted a network of discriminating genes associated with reactive oxygen species. To demonstrate the prediction, we analyzed I/R‐injured mouse hearts. MURC deletion decreased infarct size and preserved heart contraction with reactive oxygen species–related molecule EGR1 (early growth response protein 1) and DDIT4 (DNA‐damage‐inducible transcript 4) suppression in I/R‐injured hearts. Because PC‐corr network inference integrated with a protein–protein interaction network prediction also showed that MURC is involved in the apoptotic pathway, we confirmed the upregulation of STAT3 (signal transducer and activator of transcription 3) and BCL2 (B‐cell lymphoma 2) and the inactivation of caspase 3 in I/R‐injured hearts of MURC knockout mice compared with those of wild‐type mice. STAT3 inhibitor canceled the cardioprotective effect of MURC deletion in I/R‐injured hearts. In cardiomyocytes exposed to hydrogen peroxide, MURC overexpression promoted apoptosis and MURC knockdown inhibited apoptosis. STAT3 inhibitor canceled the antiapoptotic effect of MURC knockdown in cardiomyocytes.
Conclusions
Our findings, obtained by prediction from systems network genomic analysis followed by experimental validation, suggested that MURC modulates cardiac I/R injury through the regulation of reactive oxygen species–induced cell death and STAT3‐meditated antiapoptosis. Functional inhibition of MURC may be effective in reducing cardiac I/R injury.
Keywords: apoptosis, caveolae, ischemia reperfusion injury, reactive oxygen species, systems biology
Subject Categories: Cell Signalling/Signal Transduction, Ischemia, Basic Science Research
Clinical Perspective
What Is New?
This study reveals the intracellular signaling network related to MURC (muscle‐restricted coiled‐coil protein)/Cavin‐4 (caveolae‐associated protein 4) in cardiac ischemia/reperfusion (I/R) injury.
MURC/Cavin‐4 deletion has a cardioprotective effect against I/R injury through the inhibition of reactive oxygen species–induced cell death and the promotion of STAT3 (signal transducer and activator of transcription 3)–meditated antiapoptosis.
The systems network genomic analysis, based on PC‐corr network inference integrated with a protein–protein interaction network prediction followed by experimental validation, uncovers a crucial role of MURC/Cavin‐4 in cardiac I/R injury.
What Are the Clinical Implications?
This study elucidates the novel and comprehensive pathophysiological function of MURC/Cavin‐4 in cardiac I/R injury.
Functional inhibition of MURC/Cavin‐4 may be effective in reducing cardiac I/R injury.
Introduction
Coronary heart disease, a leading cause of death worldwide, places tremendous burden on individuals and society given its high rates of mortality and morbidity.1 The severity of myocardial infarction depends in particular on delays in the initiation of treatment; therefore, early revascularization therapy is critical for survival and positive prognosis. Catheterization and thrombolytic therapy have improved the clinical scenario of myocardial infarction.2, 3 Although cardiac ischemia/reperfusion (I/R) is essential for cardiac‐cell survival, it also increases infarct size, deteriorates cardiac contraction, and induces heart failure.4, 5, 6 I/R injury produces oxidative damage, cell death, and aberrant immune response through the generation of mitochondrial reactive oxygen species (ROS).7, 8, 9 Neutrophil NADPH oxidase around the infarct area, xanthine oxidase, and uncoupled nitric oxide synthase also produce ROS.10 Several clinical trials have been launched to assess treatments that overcome I/R injury. However, few effective treatments exist to prevent myocardial I/R injury.5
In myocardial infarction, cell death by apoptosis and necrosis occurs because of hypoxia and I/R injury after revascularization therapy. STAT3 (signal transducer and activator of transcription 3) plays a central role in the JAK (Janus kinase)/STAT signaling pathway to transmit the signal from the membrane to the nucleus.11 JAK and STAT3 are essential components of some cytokine receptors. STAT3 regulates apoptosis by promoting the transcription of antiapoptotic molecules such as BCL2 (B‐cell lymphoma 2),12, 13 which inhibits ROS‐induced apoptosis.14 In cardiovascular diseases, STAT3 functions as a transcription factor and is involved in myocardial infarction, oxidative damage, myocarditis, hypertrophy, and remodeling.15 STAT3 activated in ischemic preconditioning has a cardioprotective effect for ischemic heart disease.16 Activation of STAT3 protects against cardiac I/R injury by reducing infarct size in cardiac‐specific transgenic mice expressing constitutively active STAT3.17
Caveolae are plasma membrane invaginations rich in cholesterol, glycosphingolipids, and lipid‐anchored proteins.18 Two distinct components, caveolins and cavins,19, 20 cooperate to form caveola structure and modulate its biogenesis and function.21, 22 Caveolin has 3 isoforms: Cav‐1 (caveolin‐1), Cav‐2, and Cav‐3.23, 24 Cavin has 4 isoforms: PTRF (polymerase 1 and transcript release factor)/Cavin‐1, SDPR (serum deprivation protein response)/Cavin‐2, SRBC (SDR‐related gene product that binds to C kinase)/Cavin‐3, and MURC (muscle‐restricted coiled‐coil protein)/Cavin‐4.25, 26, 27 MURC plays several roles in the pathophysiology of cardiovascular diseases28, 29, 30, 31, 32 and is involved in myofibrillar organization, cardiac dysfunction, conduction disturbance, and atrial arrhythmia through the Rho‐ROCK signaling pathway.27 Gene mutations are observed in patients with dilated cardiomyopathy.32 MURC deficiency attenuates α1‐adrenergic receptor‐induced ERK1/2 (extracellular signal‐regulated kinase 1/2) activation, inhibits cardiomyocyte hypertrophy, and promotes the decrease of hypertrophy‐related gene expression.31 Moreover, hypoxia induces the synthesis of the MURC protein in the week after myocardial infarction in rat heart,33 suggesting its important role in left ventricular (LV) remodeling after myocardial infarction. Nevertheless, its role in cardiac I/R injury remains unknown.
Omic science is developing rapidly to reveal the complex signaling network surrounding biosystems. An ever‐increasing amount of omic data precipitated the development of the network inference method.34 Principal component analysis (PCA) is widely used to explore differential patterns in omic data sets. PC‐corr is an algorithm for unsupervised and parameter‐free inference of a linear multivariate‐discriminative correlation network based on the PCA loadings.35 PC‐corr enables creation of a discriminative correlation network directly (between‐omics features), associated with the sample separation obtained by preliminary PCA analysis, and can be easily adapted for big data exploration in complex biosystems. In this study, separation is between wild‐type (WT) and MURC knockout mouse samples. The protein–protein interaction network (PPIN) is a systematic protein network compiled by protein–protein interaction for the understanding of biological processes and protein function in the cell. Network inference from genomic profiles can be followed by a PPIN prediction to understand cell‐signaling networks, and protein network–based prediction of novel candidate genes is used to suggest additional candidate genes.36
In this study, we elucidate the MURC‐related intracellular signaling network in cardiac I/R injury using an integrated omic analysis. The systems network genomic analysis, based on PC‐corr network inference integrated with a PPIN prediction followed by experimental validation, uncovers a crucial role of MURC in cardiac I/R injury and reveals that MURC deletion has a cardioprotective effect against I/R injury.
Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
Microarray Analysis
To examine the gene expression of mouse hearts perturbed by MURC deficiency, we collected LV tissue from 10‐ to 13‐week‐old WT and MURC knockout (MURC KO) male mouse hearts. After euthanizing the mice, the hearts were excised by cervical dislocation and stored at −80°C. Total RNA was extracted from the tissue using TRIzol reagent (Invitrogen/Thermo Fisher Scientific), according to the manufacturer's instructions. The GeneChip Gene 1.0 ST Array System for Mouse (Affymetrix/Thermo Fisher Scientific) was used for gene expression profiling. The microarray gene‐expression data set was normalized by log transformation (log[1+x], where x denotes the data matrix). The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession number GSE125308.
PCA, PC‐Corr Network Construction, and Enrichment Analysis
To disclose the similarity in multidimensional microarray compositions between WT and MURC KO mouse hearts, we analyzed the full microarray data set by PCA and subsequently applied PC‐corr to elucidate the network of genes that predominantly contribute to the sample segregation obtained by PCA. The processing of the PCA result and the construction of the PC‐corr network were performed as described previously.35 MATLAB R2018a (MathWorks) was used for computational analyses. The PC‐corr network was constructed with a cutoff of 0.6 and visually depicted in Cytoscape 3.6.1.37 Enrichment analysis was conducted using DAVID 6.8 (Leidos Biomedical Research, https://david.ncifcrf.gov/)38, 39 for the extracted gene data set. Results of the enrichment analysis were obtained using the Benjamini multiple test correction.
PPIN and Enrichment Analysis
PPIN was used to suggest additional candidate genes by means of first‐neighbor network interaction prediction. Ninety‐one genes extracted from PC‐corr at a cutoff of 0.6 were input into STRING 10.5 (https://string-db.org/),40 and then we described a PPIN. We selected their first neighbors (261 proteins) in the PPIN. DAVID 6.8 was used for the enrichment analysis of the 261 proteins. We reorganized the PPIN by inputting the 46 proteins present in the enriched pathways of our interest to STRING.
Mouse Model of Cardiac I/R Injury
A mouse model of cardiac I/R injury was created as described previously with slight modifications.41 The left anterior descending coronary arteries of 10‐week‐old C57BL/6 background male mice were ligated under 1.0% isoflurane anesthesia for 1 hour before reperfusion. Hearts were excised 30 minutes or 24 hours after reperfusion for analysis. Serum cardiac troponin I level was measured 24 hours after reperfusion by high‐sensitivity mouse cardiac troponin I ELISA (Life Diagnostics), according to the manufacturer's instruction. MURC KO mice were generated as described previously.31 Transgenic mice expressing MURC in the heart (Tg‐MURC) were generated as described previously.27 All animal use conformed with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH; Publication no. 85‐23, revised 1996) and was approved by the institutional animal care and use committee of the Kyoto Prefectural University of Medicine.
Echocardiography and Triphenyltetrazolium Chloride Staining
Echocardiography was performed using a Vevo 2100 system (VisualSonics) equipped with a 30‐MHz microprobe under isoflurane anesthesia 24 hours after I/R. Triphenyltetrazolium chloride staining was performed as described previously with slight modifications.42 To determine the area at risk, 10% Evans blue dye (0.5 g/kg) was injected into the retro‐orbital venous sinus after ligation of left anterior descending coronary arteries. Hearts were excised and stored at −80°C. Cross‐sections of the hearts (1 mm) were immersed in 1.0% triphenyltetrazolium chloride in 0.9% saline at 24°C for 2 to 3 minutes. The vial was continuously agitated in a water bath at 37°C for 15 minutes, and then cross‐sections were fixed in 10% neutral buffered formalin for 60 minutes. Sections were digitally photographed using a Leica MC120 HD microscope camera (Leica Microsystems). Infarct size and area at risk were quantified with ImageJ 1.49 software (NIH).
Dihydroethidium Staining
Dihydroethidium staining was performed as described previously with slight modifications.43 Cross‐sections through the left ventricles (1 mm) of freshly isolated mouse hearts were prepared and then equilibrated in Krebs buffer for 30 minutes at 37°C. Sections were then stained with 30 μmol/L dihydroethidium in fresh Krebs buffer and incubated in the dark for 30 minutes with gentle rotation. Dihydroethidium‐stained LV sections were imaged using a Carl Zeiss LSM 510 confocal microscope with a ×10 dry objective at 488 nm excitation. ZEN software (Carl Zeiss) was used to collect sequential Z‐stacked confocal line scans of each section and to assemble 2.5‐dimensional histogram plots of mean dihydroethidium intensity. Fluorescence intensity analysis of dihydroethidium was measured with ImageJ software.
Cell Culture
Neonatal rat cardiomyocytes (NRCMs) were prepared as described previously with slight modifications.27 Cardiomyocytes isolated from 1‐ to 3‐day‐old Wistar rats were cultured in serum‐containing medium (DMEM, 10% fetal bovine serum) for a total of 72 hours. Adult mouse cardiomyocytes were prepared as described previously with slight modifications.44 Cardiomyocytes isolated from 10‐week‐old WT and MURC KO male mouse hearts were cultured in the culture medium and exposed to hydrogen peroxide after 24‐hour incubation.
Gene Silencing and Transfer
Rat MURC and control small interfering RNA (siRNA) duplex oligonucleotides (Stealth RNAi siRNAs) were purchased from Invitrogen/Thermo Fisher Scientific. The siRNAs were transiently transfected into cardiomyocytes using Lipofectamine RNAiMAX reagent (Invitrogen/Thermo Fisher Scientific), according to the manufacturer's instructions. The medium was changed 24 hours after transfection. The siRNA sequences are provided in Table S1. Recombinant adenoviruses expressing FLAG‐tagged human MURC/Cavin‐4 (Ad‐MURC) and β‐galactosidase were described previously.27 Twenty‐four hours after seeding on a plate, the cardiomyocytes were infected with Ad‐MURC and Ad‐β‐galactosidase diluted in culture media at a multiplicity of infection of 30 and incubated at 37°C for 1 hour. The viral suspension was removed, and the cardiomyocytes were cultured with fresh media.
Hypoxia/Reoxygenation of NRCMs and ROS Detection Assay
We examined ROS activity of cardiomyocytes exposed to hypoxia/reoxygenation. The ROS activity was evaluated using a CellROX detection kit (Invitrogen/Thermo Fisher Scientific) after serum deprivation for 12 hours under hypoxic conditions (1% O2 and 5% CO2, 37°C) followed by 3 hours of reoxygenation (21% O2 and 5% CO2, 37°C). The CellROX staining was performed according to the manufacturer's instructions; the CellROX reagent was added directly to the hypoxia/reoxygenation–challenged cardiomyocytes, and the mixture was incubated for 1 hour. The ROS intensity was measured using ImageJ software.
TUNEL Assay and Stimulation of Hydrogen Peroxide
Cultured NRCMs were exposed to 200 μmol/L hydrogen peroxide (H2O2) for 2 hours. A TUNEL (TdT‐mediated dUTP nick‐end labeling) assay was performed to detect apoptosis 6 hours after H2O2 exposure using the In Situ Cell Death Detection Kit, TMR red (Roche), according to the manufacturer's instructions. Cell death number was assessed by the percentage of TUNEL‐ and DAPI (40,6‐diamidino‐2–7 phenylindole)‐positive cells.
STAT3 Cancellation Experiment
STAT3 inhibitor WP1066 (20 mg/kg; Santa Cruz Biotechnology) or an equal‐volume vehicle (5% dimethyl sulfoxide) was administered intraperitoneally to mice daily for 3 days. The WP1066 dose was determined based on published toxicity and efficacy data in mice.13, 45 WP1066 (1 μmol/L) or an equal‐volume vehicle 5% dimethyl sulfoxide was administered to NRCMs 1 hour before H2O2 exposure.
Western Blot Analysis
Cell lysates and tissue samples were extracted using a lysis buffer (50 mmol/L Tris‐HCl [pH 7.5], 150 mmol/L NaCl, 50 mmol/L EDTA, 1% Triton X‐100, and protease–phosphatase inhibitor mixture). Protein samples were subjected to SDS‐PAGE and then transferred to membranes that were subsequently incubated with primary antibodies against STAT3, phosphorylated STAT3, ERK1/2, phosphorylated ERK1/2, Akt, phosphorylated Akt, cleaved caspase 3, DDIT4, GAPDH, β‐actin, FLAG M2, EGR1, caveolin‐1, caveolin‐3, PTRF, SDPR, SRBC, BCL2, and MURC. Anti‐MURC antibody was originally produced as described previously.30 Horseradish peroxidase–conjugated antirabbit and antimouse IgGs were used as secondary antibodies.
Immunoprecipitation
NRCMs transfected with Ad‐MURC were washed with ice‐cold PBS and lysed with lysis buffer containing 50 mmol/L Tris‐HCl (pH 7.5), 150 mmol/L NaCl, 50 mmol/L EDTA, 1% Triton X‐100, and protease–phosphatase inhibitor mixture. Immunoprecipitation was carried out by incubating equal amounts of cell lysates with magnetic beads (Magnosphere MS300/Carboxyl; Cosmo Bio) coated with each antibody at 4°C overnight. Beads were washed with wash buffer (50 mmol/L Tris‐HCl [pH 7.5], 150 mmol/L NaCl, 50 mmol/L EDTA, 1% Triton X‐100, and protease–phosphatase inhibitor mixture) 5 times, and the precipitated proteins were separated by SDS‐PAGE, transferred to polyvinylidene difluoride membranes, and probed with each antibody.
Reverse Transcription–Mediated Quantitative Polymerase Chain Reaction
Reverse transcription–mediated quantitative polymerase chain reaction was conducted as described previously.27, 29, 31 Total RNA was extracted from cultured cells or tissues using TRIzol (Invitrogen/Thermo Fisher Scientific) and converted to cDNA using a High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems/Thermo Fisher Scientific). Synthesized cDNA was analyzed by kinetic real‐time polymerase chain reaction using Takara PCR Thermal Cycler Dice (Takara Bio) with Platinum SYBR Green qPCR Supermix (Invitrogen/Thermo Fisher Scientific). The primer sequences are provided in Table S2.
Statistical Analysis
Statistical analysis was done in R 3.3.2 (R Foundation for Statistical Computing).46 The Shapiro–Wilk test was used for normality testing. All 2‐group analysis used a 2‐tailed Student t test. Comparisons of multiple groups with normal distribution were done with 1‐way ANOVA followed by the Tukey post hoc test. The Kruskal–Wallis test was used as a nonparametric test, followed by the Dunn post hoc test. All data are displayed as mean±SEM. All in vitro experiments were performed at least 3 times. MATLAB R2018a was used for the computational analyses employing PC‐corr. The PC‐corr network was constructed with a cutoff of 0.6 (maximum is 1) because this value ensures that the connectivity links of the network have medium to high levels of correlation and discrimination simultaneously. Results of the enrichment analysis were obtained using Benjamini multiple test correction with the significance threshold of 0.05.
Results
Systems Network Genomic Analysis Based on PC‐Corr Network Inference Reveals MURC Deficiency Is Involved in Response to ROS and Regulation of the Force of Heart Contraction
We performed microarray analysis for WT and MURC KO mouse hearts to examine the gene expression of mouse hearts perturbed by MURC deficiency. Heat mapping showed clear differentiation of gene expression between WT and MURC KO mouse hearts (Figure S1). PCA also showed clear segregation of gene expression between WT and MURC KO mouse hearts (Figure 1A).
Figure 1.
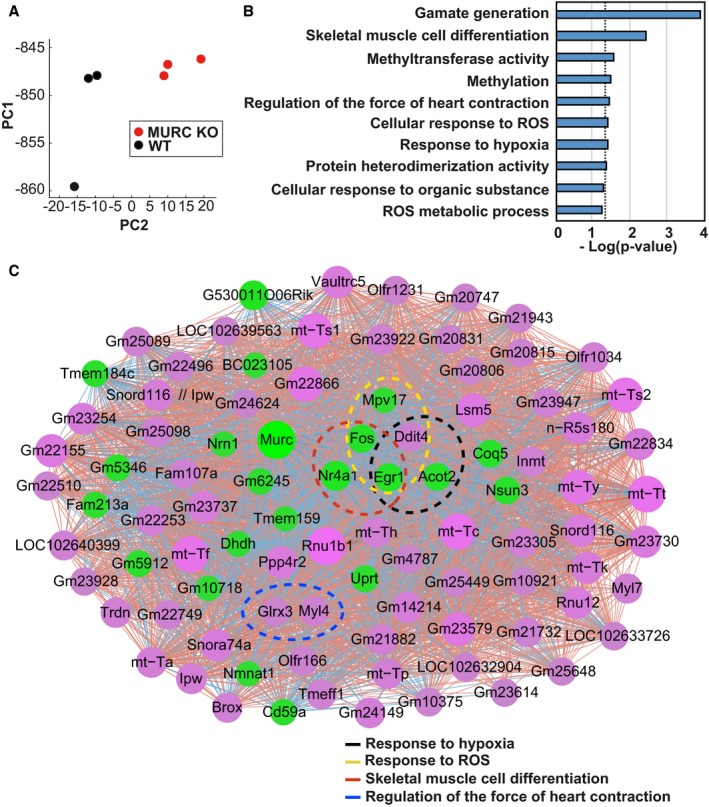
Microarray analysis of wild‐type (WT) and MURC KO (muscle‐restricted coiled‐coil protein knockout) mouse hearts shows MURC deficiency is involved in response to hypoxia and reactive oxygen species (ROS). A, Principal component analysis. Red nodes indicate MURC KO, black nodes indicate WT. PC1, first principal component; PC2, second principal component. B, Enrichment analysis conducted by DAVID. Top 10 GO pathways are ranked by Benjamini‐corrected P value and scaled according to the function −log10 (P value). The vertical dashed line indicates the threshold at 0.05. C, PC‐corr network was constructed according to the loading of PC2 at a cutoff of 0.6. Magenta nodes indicate genes with higher expression in MURC KO; green nodes indicate genes with higher expression in WT mouse hearts. Node size is proportional to node degree. The color of interaction denotes the direction of the Pearson correlation between the features: red for positive Pearson correlation and blue for negative case. Nodes circled with a dotted line highlight specific pathways as follows: black, response to hypoxia; yellow, response to ROS; red, skeletal muscle cell differentiation; blue, regulation of the force of heart contraction. The network was depicted by Cytoscape. n=3 per group.
PC‐corr was used to enlighten the discriminative correlation network according to the weights of the links at a cutoff of 0.6 (Table S3), as both groups were clearly separated along the second principal component PC2. The genes obtained in the PC‐corr network were input to DAVID for enrichment analysis (Figure 1B; Table S4). The genes involved in response to hypoxia, response to ROS, skeletal muscle cell differentiation, and regulation of the force of heart contraction and belonging to the top 10 pairs of enriched pathways were highlighted with the respective color circle in the PC‐corr network. This reorganized network enabled us to determine that expression levels for the following specific pathway‐related genes were markedly perturbed by MURC deficiency (Figure 1C): DDIT4, EGR1, and ACOT2 (acyl‐CoA thioesterase 2) in response to hypoxia; DDIT4, EGR1, FOS (Fos proto‐oncogene, AP‐1 transcription factor subunit), and MPV17 (mitochondrial inner membrane protein MPV17) in response to ROS; EGR1, FOS and NR4A1 (nuclear receptor subfamily 4 group A member 1) in skeletal muscle cell differentiation; and GLRX3 (glutaredoxin 3) and MYL4 (myosin light chain 4) in regulation of the force of heart contraction.
MURC Deficiency Preserves Heart Contraction With Infarct Size Reduction After I/R
Because I/R injury induces cardiac damage along with ROS production,7, 8, 9 we created a mouse model of I/R injury to demonstrate the relationship between ROS and MURC in the heart. By echocardiography, we evaluated cardiac function on WT and MURC KO mouse hearts after sham operation or 24 hours after I/R (Figure 2A and 2B). Fractional shortening and ejection fraction are representative indicators of LV systolic function. Although there was no difference in fractional shortening and ejection fraction between the WT and MURC KO groups of both sham‐operated control groups, fractional shortening and ejection fraction were markedly preserved in MURC KO compared with WT mouse hearts after I/R. Mouse heart rate was equivalent between groups during echocardiography (Figure S2). We then evaluated the infarct size by triphenyltetrazolium chloride and Evans blue dual staining (Figure 2C). The area at risk assessed as the proportion of left ventricle was equal between the 2 groups (ie, the procedure was performed equally). The infarct size assessed as the proportion of the area at risk was significantly lower in MURC KO compared with WT mouse hearts after I/R (Figure 2D). Cardiac troponin I level was significantly lower in MURC KO than WT mouse serum (Figure 2E). Similarly, we performed echocardiography and measurement of infarct size on the hearts of adult transgenic mice expressing cardiac‐specific MURC (Tg‐MURC) 24 hours after I/R; however, we found no difference in fractional shortening, ejection fraction, and infarct size between WT and Tg‐MURC mouse hearts (Figure S3A and S3B).
Figure 2.
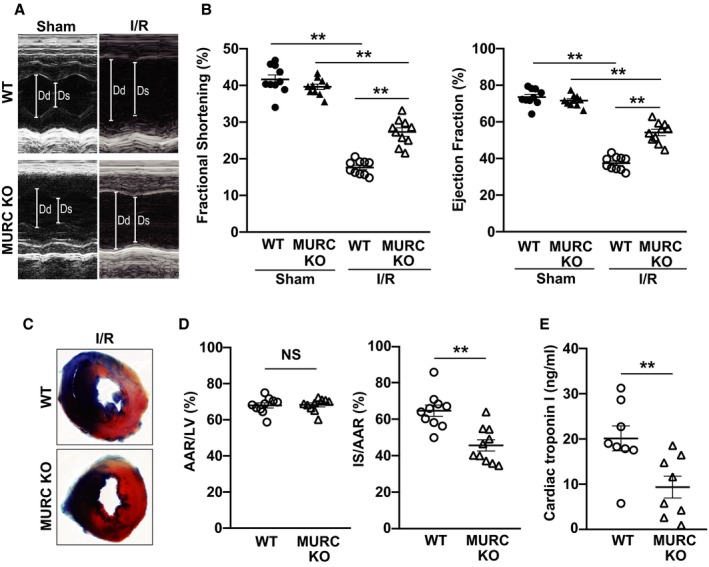
MURC (muscle‐restricted coiled‐coil protein) deficiency preserves left ventricular systolic function and infarct size in the heart after ischemia/reperfusion (I/R). Representative echocardiography (A) and quantification (B) of wild‐type (WT) and MURC knockout (MURC KO) mouse hearts after sham operation or 24 hours after I/R; n=10 per group. Representative triphenyltetrazolium chloride and Evans blue staining (C) and quantification (D) of WT and MURC KO mouse hearts 24 hours after I/R. In panel C, blue, white, and red regions represent the nonischemic area, the infarct area, and the noninfarct area at risk, respectively. In panel D, the area at risk (AAR) was assessed as a proportion of the left ventricle (LV; left), and the infarct size (IS) was assessed as a proportion of the AAR (right); n=10 per group. E, Cardiac troponin I level of WT and MURC KO mouse serum 24 hours after I/R; n=8 per group. Data are presented as mean±SEM. **P<0.01. Dd indicates left ventricular internal dimension in diastole; Ds, left ventricular internal dimension in systole; NS, not significant.
MURC Deficiency Ameliorates ROS Production in the Heart After I/R With Reduced EGR1 and DDIT4 mRNA Expression
We evaluated cardiac ROS production in WT and MURC KO mouse hearts 30 minutes after I/R by dihydroethidium of LV tissue from mouse hearts with confocal micrographs (Figure 3A). ROS production increased after I/R but was significantly lower in MURC KO compared with WT mouse hearts (Figure 3B). We also evaluated ROS production in NRCMs exposed to hypoxia/reoxygenation. ROS production was measured by CellROX staining in NRCMs transfected with control siRNA, MURC siRNA 1 or 2. MURC siRNAs had an induction of MURC knockdown in NRCMs (Figure S4A and S4B). ROS production increased after hypoxia/reoxygenation exposure but was significantly lower in NRCMs transfected with MURC siRNAs compared with control siRNA (Figure S5A and S5B).
Figure 3.
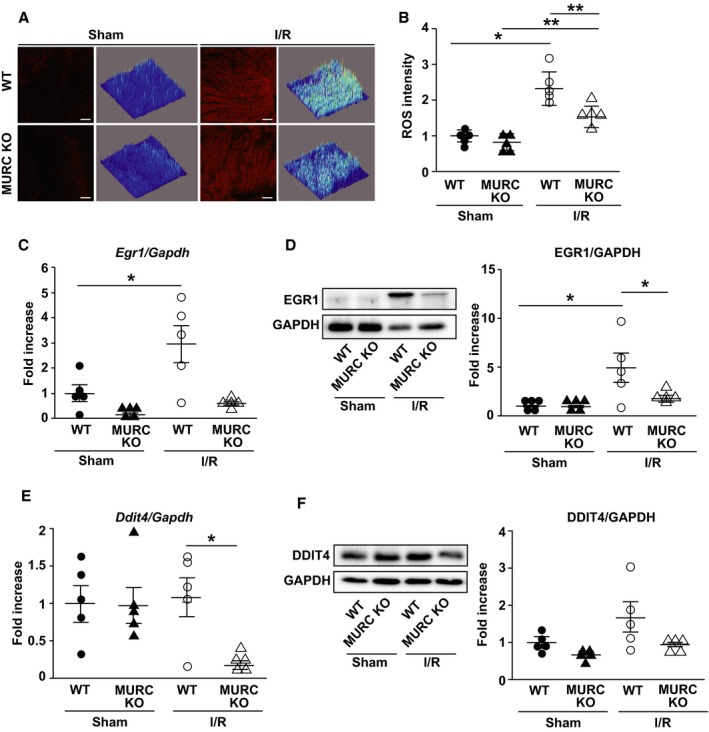
MURC (muscle‐restricted coiled‐coil protein) deficiency ameliorates reactive oxygen species (ROS) production in the heart after ischemia/reperfusion (I/R). A and B, Cardiac ROS production as measured by dihydroethidium staining of left ventricle tissue from mouse hearts with confocal micrographs and Z‐stack–generated 2.5‐dimensional reconstructions. Representative dihydroethidium staining (A) and quantification (B) of wild‐type (WT) and MURC knockout (MURC KO) mouse hearts after sham operation or 30 minutes after I/R. Scale bar=100 μm. C through F, Measurement of mRNA expressions (C and E) and the protein levels (D and F) of EGR1 (early growth response protein 1) and DDIT4 (DNA‐damage‐inducible transcript 4) in WT and MURC KO mouse hearts after sham operation or 24 hours after I/R; n=5 per group. *P<0.05, **P<0.01. Data are presented as mean±SEM.
Because ROS‐related gene expression was perturbed in microarray analysis of mouse hearts (Figure 1C), we examined mRNA expression and protein level of EGR1 and DDIT4. EGR1 mRNA expression and protein level were elevated 24 hours after I/R but significantly lower in MURC KO compared with WT mouse hearts (Figure 3C and 3D). DDIT4 mRNA expression was also significantly lower in MURC KO compared with WT mouse hearts 24 hours after I/R, whereas protein level was not significantly different (Figure 3E and 3F). Although Nox (NADPH oxidase) family genes are involved in the pathophysiology of cardiac I/R injury,47, 48 Nox4 mRNA expression was reduced only in MURC KO compared with WT mouse hearts 24 hours after I/R (Figure S6).
Protein Network–Based Prediction of Novel Candidate Genes Reveals a Pivotal Role of Antiapoptotic Signaling With STAT3 Activation and Increased BCL2 Expression in MURC KO Mouse Hearts
Protein network–based prediction of novel candidate genes was conducted to explore additional pathways involved in MURC deficiency. PPIN was used to suggest additional candidate genes by means of first‐neighbor network interaction prediction, as previously described.36 Ninety‐one genes extracted from PC‐corr at a cutoff of 0.6 were input into the STRING database to obtain their first neighbors (261 proteins) selected in the PPIN; we applied a biclustering algorithm to the matrix of 46 proteins present in the significant pathways of interest (Figure 4A). Then we reorganized the PPIN by inputting the 46 proteins into STRING. The pathways of regulation of apoptosis, response to ROS, response to hypoxia, and regulation of the force of heart contraction were interacted through several proteins in the MURC‐deficient heart (Figure 4B). DAVID was used for enrichment analysis (Figure 4C; Table S5).
Figure 4.
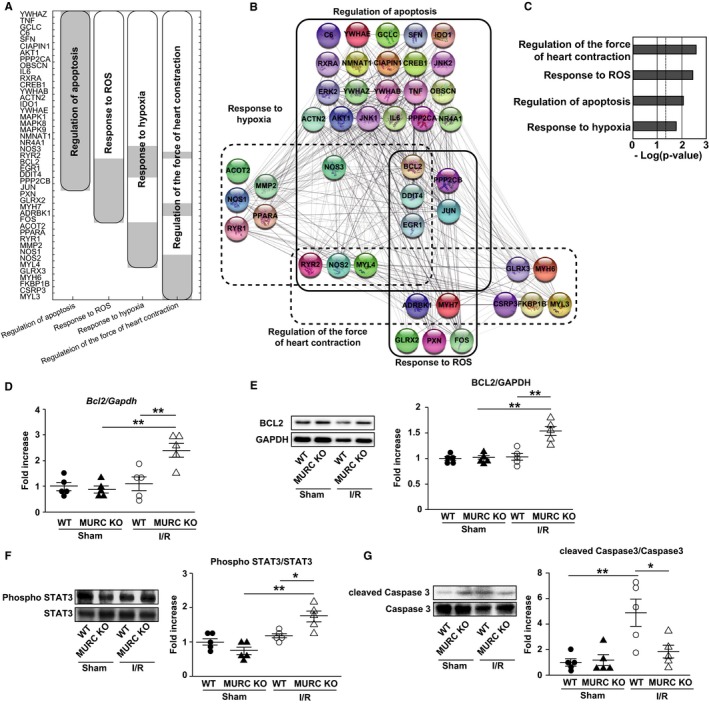
MURC (muscle‐restricted coiled‐coil protein) deficiency promotes antiapoptotic signaling with STAT3 (signal transducer and activator of transcription 3) activation in the heart after ischemia/reperfusion (I/R). A and B, Protein‐network–based prediction of novel candidate genes. The protein–protein interaction network (PPIN) was used to suggest additional candidate genes by means of first‐neighbor network interaction prediction. Gene data set extracted from PC‐corr at a cutoff of 0.6 was input into STRING. A, Biclustering of the matrix of all proteins obtained by PPIN present in the significant pathways of interest. The matrix consists of 46 proteins (rows) and 4 enriched pathways (columns). The black lines indicate the modules corresponding to the clusters of interacting proteins in the respective pathways. B, PPIN reorganized by STRING. Full and dashed lines delimit the diverse overlapped protein pathway modules. C, Enrichment analysis conducted by DAVID for PPIN. Pathways of interest are ranked by Benjamini‐corrected P value and scaled according to the function −log10 (P value). The vertical dashed line indicates the threshold at 0.05. Measurement of BCL2 (B‐cell lymphoma 2) expression in wild‐type (WT) and MURC knockout (MURC KO) mouse hearts after sham operation or 24 hours after I/R: mRNA expression (D) and representative Western blots (left) and quantification (right) of BCL2 (E). Representative Western blots (left) and quantification (right) of STAT3 phosphorylation (F) and cleaved caspase 3 (G) in WT and MURC KO mouse hearts after sham operation or 24 hours after I/R; n=5 per group. Data are presented as mean±SEM. *P<0.05, **P<0.01. Phospho indicates phosphorylated.
The PPIN reorganized by STRING implied the involvement of MURC in the apoptotic pathway, which includes BCL2, ROS‐responsive molecule EGR1, and ROS‐related molecule DDIT4. BCL2 and DDIT4 are involved in STAT3‐mediated antiapoptosis. In I/R‐injured hearts, mRNA expression and protein level of BCL2, which is a key molecule of STAT3‐mediated antiapoptotic signaling, were significantly higher in MURC KO compared with WT mice (Figure 4D and 4E). STAT3 plays an important role in antiapoptotic signaling through inhibiting DDIT4 transcription.49 DDIT4 mRNA expression was suppressed in I/R‐injured hearts of MURC KO mice (Figure 3E). Consequently, we evaluated STAT3 activation in the hearts after I/R. The phosphorylation of STAT3 was significantly higher in MURC KO compared with WT mouse hearts 24 hours after I/R (Figure 4F). The protein level of cleaved caspase 3 was significantly elevated 24 hours after I/R but significantly lower in MURC KO compared with WT mouse hearts (Figure 4G). Activation of other apoptosis‐related proteins such as ERK, p38, and Akt were not different between WT and MURC KO mouse hearts 24 hours after I/R (Figure S7).
MURC Modulates Apoptosis in Cardiomyocytes and Cardiac Function in Mouse Hearts Through STAT3 Activation
To determine whether MURC modulates the apoptosis of cardiomyocytes, including STAT3‐related antiapoptosis, we first performed TUNEL staining for NRCMs exposed to H2O2. Apoptosis induced by H2O2 was significantly inhibited in cardiomyocytes transfected with MURC siRNAs compared with control siRNA (Figure 5A and 5B). Similarly, H2O2‐induced apoptosis was significantly inhibited in adult mouse cardiomyocytes of MURC KO mice compared with those of WT mice (Figure S8). In contrast, TUNEL staining for NRCMs infected with Ad‐β‐galactosidase or Ad‐MURC showed that MURC overexpression significantly accelerated H2O2‐induced cardiomyocyte apoptosis (Figure 5C and 5D). Next we evaluated the relationship between MURC and STAT3 in cardiac I/R injury and apoptotic signaling, after confirming WP1066, a STAT3 inhibitor, inhibited the phosphorylation of STAT3 in mouse hearts (Figure S9). WP1066 treatment abolished the effect of the preservation of LV systolic function and the reduction of infarct size in MURC KO mouse hearts 24 hours after I/R (Figure 6A and 6B). TUNEL staining for H2O2‐exposed NRCMs also showed that WP1066 treatment abolished the MURC knockdown–induced antiapoptotic effect (Figure 6C). Consequently, STAT3 inhibition canceled the cardioprotective effect of MURC deficiency in mouse hearts after I/R. To investigate the direct association between MURC and STAT3, we performed immunoprecipitation of MURC and STAT3 in NRCMs transduced with β‐galactosidase or FLAG‐tagged human MURC. Unexpectedly, immunoprecipitation showed no direct protein interaction between MURC and STAT3 (Figure S10A and S10B).
Figure 5.
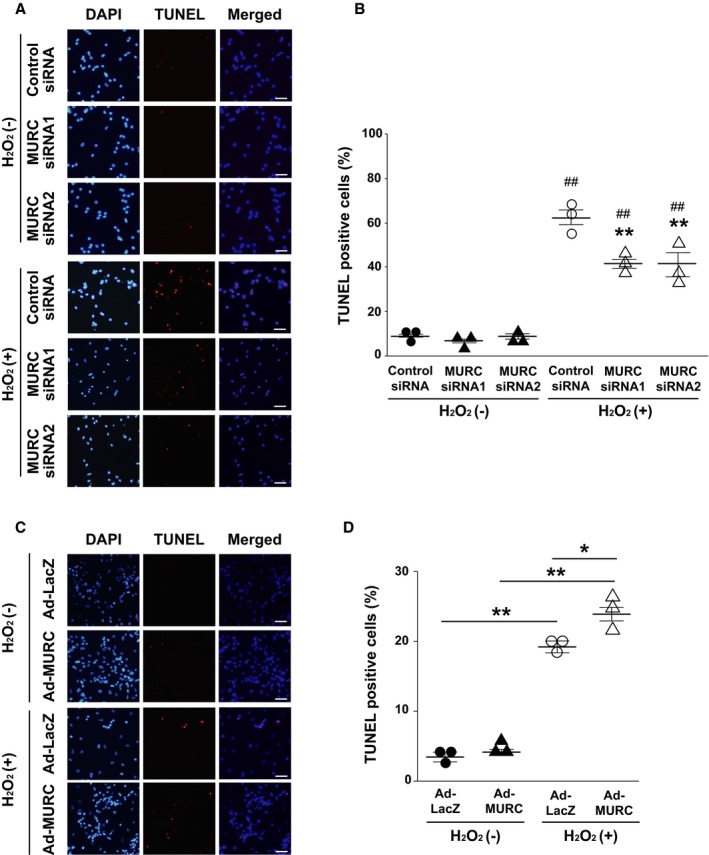
MURC (muscle‐restricted coiled‐coil protein) modulates apoptosis in neonatal rat cardiomyocytes (NRCMs). Representative TUNEL (TdT‐mediated dUTP nick‐end labeling) staining (A) and quantification (B) of NRCMs transfected with control small interfering RNA (siRNA), MURC siRNA 1, or MURC siRNA 2 before H2O2 exposure. ## P<0.01 vs corresponding H2O2(−) group; **P<0.01 vs control siRNA in H2O2(+) group. Representative TUNEL staining (C) and quantification (D) of NRCMs transduced with adenovirus‐mediated β‐galactosidase (Ad‐LacZ) or MURC (Ad‐MURC) before H2O2 exposure. *P<0.05, **P<0.01. Cell death number was assessed by the percentage of TUNEL‐ and DAPI (4′,6‐diamidino‐2‐phenylindole)–positive cells; n=3 per group. Data are presented as mean±SEM. Scale bar=50 μm.
Figure 6.
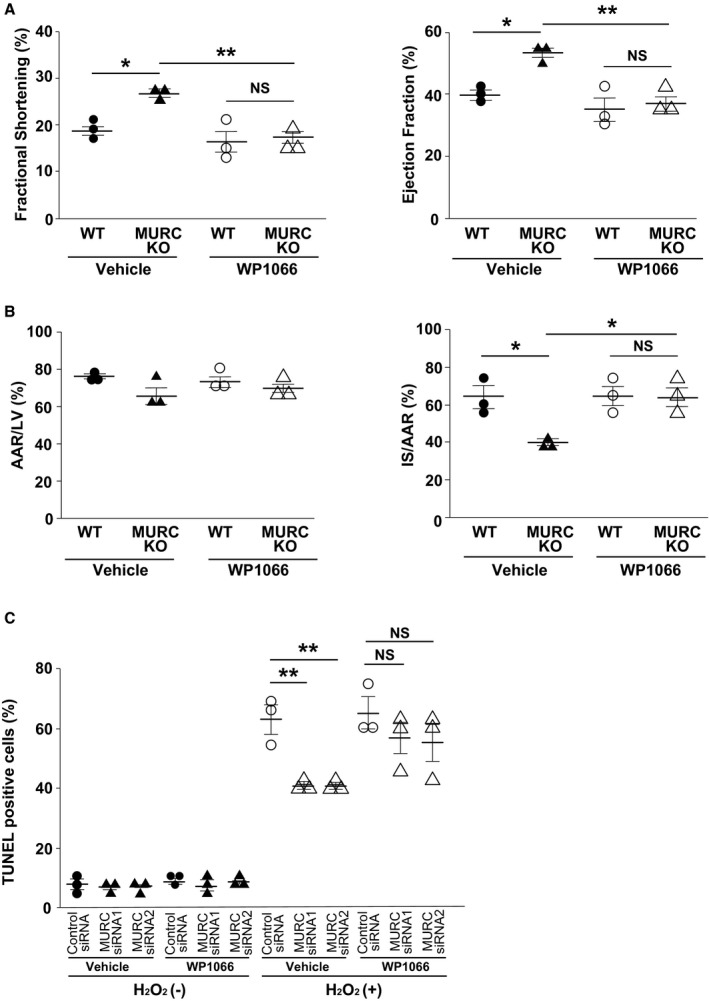
STAT3 (signal transducer and activator of transcription 3) inhibitor cancels the cardioprotective effect of MURC (muscle‐restricted coiled‐coil protein) deficiency. A, Left ventricle (LV) systolic function as measured by echocardiography of wild‐type (WT) and MURC knockout (MURC KO) mouse hearts 24 hours after I/R. Vehicle (5% dimethyl sulfoxide) or WP1066 was injected in mice intraperitoneally for 3 consecutive days; n=3 per group. B, Infarct size as measured by triphenyltetrazolium chloride and Evans blue staining of LV tissue from mouse hearts 24 hours after ischemia/reperfusion. The area at risk (AAR) was assessed as a proportion of LV (left), and the infarct size (IS) was assessed as a proportion of the AAR (right); n=3 per group. C, Evaluation of TUNEL (TdT‐mediated dUTP nick‐end labeling) staining for NRCMs transfected with control small interfering RNA (siRNA), MURC siRNA 1, or MURC siRNA 2 after H2O2 exposure. Vehicle or WP1066 (a STAT3 inhibitor) was administered to cardiomyocytes; n=3 per group. *P<0.05, **P<0.01. NS indicates not significant.
Discussion
Systems network genomic analysis is a useful tool for predicting complex intracellular‐signaling networks. In this study, we applied the systems network genomic analysis based on PC‐corr network inference integrated with a PPIN prediction to assess the functional role of MURC in cardiac I/R injury. MURC is the muscle‐restricted caveolar component expressed exclusively in myocytes.27 It has been reported that MURC is involved in dilated cardiomyopathy,32 cardiac hypertrophy,31 hypoxia,33 and skeletal muscle cell differentiation.50 Caveolae are considered platforms for some receptors and signal components. Caveolins and cavins cooperatively form the caveola structure and modulate its biogenesis and function.21, 22 However, because caveolins and cavins including MURC are trafficking or adaptor proteins,28 not enzymes, it is difficult to assess the function of these caveolae‐related proteins by conventional kinase assays. The systems network genomic analysis of MURC KO mouse hearts predicted a comprehensive role for MURC in cardiac I/R injury.
MURC deletion reduced infarct size after cardiac I/R and preserved cardiac contraction with a decrease in ROS production and expression of EGR1 and DDIT4. PC‐corr network inference on microarray data sets from WT and MURC KO mouse hearts predicted that MURC‐related pathways included response to hypoxia and ROS, skeletal muscle cell differentiation, and regulation of the force of heart contraction. We confirmed the prediction with experimental validation using a mouse model of cardiac I/R injury. EGR1 acts as a ROS‐sensitive transcription factor and promotes apoptosis and inflammation.51, 52, 53 DDIT4 also promotes apoptosis and inflammation in a STAT3‐dependent manner in response to various cellular stresses including ROS.49, 54, 55 Although the findings from MURC KO mouse hearts suggested that MURC overexpression aggravated cardiac I/R injury, cardiac‐specific Tg‐MURC mice did not exhibit aggravated contraction and infarct size in I/R hearts. We reported that the hearts of Tg‐MURC mice show cardiac hypertrophy at a young age and subsequently show cardiac enlargement and dysfunction.27 The phenotype possessed by Tg‐MURC mice may have shown a counteracting effect in cardiac I/R injury. PC‐corr network inference contributed greatly in elucidating the comprehensive role of MURC in cardiac I/R injury. However, the results of EGR1 and DDIT4 expression in PC‐corr network inference and the validation study were inconsistent. PC‐corr network inference predicted that EGR1 would be poorly expressed and DDIT4 highly expressed in MURC KO mouse hearts; however, the expression of EGR1 or DDIT4 was not different between the sham‐operated hearts of WT and MURC KO mice and decreased in I/R‐injured hearts of MURC KO mice compared with those of WT mice. These discrepancies may be caused by the small sample size, which should be regarded as a limitation of this study.
PC‐corr network inference integrated with a PPIN prediction also indicated the involvement of antiapoptotic signaling in cardioprotection by MURC deletion. The protein network–based prediction of novel candidate genes indicated that several pathways such as regulation of apoptosis, response to ROS, and response to hypoxia included not only EGR1 and DDIT4 but also BCL2. ROS modulates several apoptosis‐related signaling pathways, thereby controlling cell death in cardiac I/R injury.8 In the validation study, BCL2 was significantly increased in the hearts of MURC KO mice compared with those of WT mice after I/R injury. BCL2 is a key regulator of antiapoptotic signaling and inhibits several apoptoses including ROS‐induced apoptosis.14 EGR1 and DDIT4 are also involved in ROS‐related apoptosis.
MURC deletion promoted the activation of STAT3, which regulates apoptosis by facilitating BCL2 transcription,12, 13 and preserved cardiac function in cardiac I/R injury. MURC knockdown in cardiomyocytes promoted the antiapoptotic pathway via STAT3 activation. STAT3 is a pivotal regulator in various cardiovascular diseases; it acts as a transcription factor and is involved in myocardial infarction, oxidative damage, myocarditis, hypertrophy, and remodeling.15 STAT3 activation in ischemic preconditioning has a cardioprotective effect for ischemic heart disease.16 Constitutive activation of STAT3 also protects cardiac I/R injury.17 Although PC‐corr network inference integrated with a PPIN prediction did not directly suggest the involvement of STAT3, STAT3 is a key molecule of BCL2‐mediated antiapoptosis; therefore, we examined the STAT3‐mediated antiapoptotic signal pathway. The phosphorylation of STAT3 was significantly higher in MURC KO compared with WT mouse hearts after I/R. A STAT3 inhibitor canceled a cardioprotective effect due to MURC deletion.
Immunoprecipitation analysis showed no direct association between MURC and STAT3 in cardiomyocytes. Our results suggests that MURC modulates apoptosis in cardiomyocytes and cardiac function in mouse hearts through STAT3 activation with indirect protein interaction. A recent study demonstrated that the phosphorylation of STAT3 is regulated by the interaction of Cavin‐1 with SOCS3 (suppressor of cytokine signaling 3) in a shared signal‐transducing receptor for a family of cytokines, gp130‐mediated cytokine signaling.56 Cavin‐1 localizes SOCS3 in the caveolae at the plasma membrane and suppresses gp130/JAK/STAT3 signaling. MURC forms large complexes with other cavins and caveolins in caveolae and has a direct association with Cavin‐1.21 The mechanism by which MURC regulates STAT3 activation remains ambiguous in our study; it is unclear whether MURC inhibits JAK/STAT3 signaling in coordination with Cavin‐1 or whether MURC has the same function as Cavin‐1 in cardiomyocytes. MURC deletion may facilitate the phosphorylation of JAK in caveolae at the plasma membrane and subsequently induce the STAT3 transition from the cell membrane to the nucleus in coordination with activated JAK.
Protein network–based prediction of novel candidate genes indicates that MURC is highly relevant to ERK in mouse hearts; however, there was no difference in ERK phosphorylation between WT and MURC KO mouse hearts during I/R injury. MURC facilitates recruitment of ERK to caveolae and concentric cardiac hypertrophy.31 Hypoxia induces MURC expression in cardiomyocytes during hypertrophy with ERK activation.33 ERK is an apoptosis‐related molecule in I/R injury. Its activity has been implicated in neurodegenerative diseases and brain injury following I/R in rodents.57, 58 Recently, Yu et al reported that the MAPK/ERK‐CREB pathway promotes I/R‐induced cardiomyocyte apoptosis by inhibiting FUNDC1 (FUN14 domain containing 1)–related mitophagy.59 Accordingly, we tried to identify the relationship between MURC and ERK activities in I/R‐induced cardiomyocyte apoptosis. MURC, however, showed no association with ERK phosphorylation 24 hours after I/R.
MURC interacts with caveolin‐3 at the plasma membrane.29, 60 Cardiac‐specific caveolin‐3 overexpression increases the formation of caveolae and induces cardiac protection against I/R injury.61 In contrast, caveolin‐3 knockout mice show no isoflurane‐induced cardiac protection against I/R injury.62 In the present study, there was no significant change in caveolin‐1 and other caveolae‐related protein levels except for MURC/Cavin‐4 between WT and MURC KO mouse hearts (Figure S11A–S11F). Mouse left anterior descending coronary arteries were ligated for 60 minutes during ischemia according to the previous report.63 The long period of ischemia might have affected the cardioprotective effect of isoflurane in MURC KO mouse hearts. We previously reported that there was no reduction or deformation of caveolae in MURC/Cavin‐4 KO mouse hearts.31 Therefore, the cardioprotective effect of MURC/Cavin‐4 deletion is not derived from the caveolae formation.
In our study, we mimic a cardiac I/R model by hydrogen peroxide exposure or a hypoxia/reoxygenation model in vitro. The in vitro assay reflects only ROS or hypoxia, not the changes in glucose availability and pH. The discrepancy between in vivo conditions and the in vitro model should be considered a study limitation.
In conclusion, our study applied systems network genomic analysis based on PC‐corr network inference integrated with a PPIN prediction to identify a previously undescribed function of MURC in the underlying pathogenetic mechanism of cardiac I/R injury (Figure 7). MURC deletion reduced infarct size and preserved heart contraction through the inhibition of ROS‐induced cell death and the promotion of STAT3‐mediated antiapoptotic signaling in cardiac I/R injury. Functional inhibition of MURC may be effective for reducing cardiac I/R injury.
Figure 7.
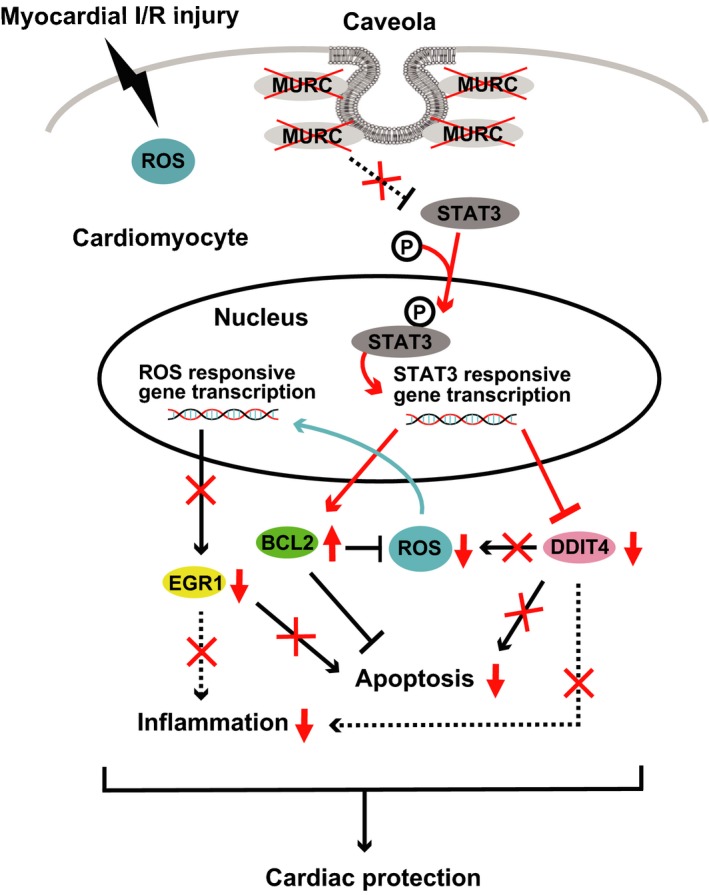
Schematic illustration of the cardioprotective effect of MURC (muscle‐restricted coiled‐coil protein) deficiency in myocardial ischemia/reperfusion (I/R) injury. Intracellular signaling network including reactive oxygen species (ROS) reduction and activated antiapoptotic signaling with STAT3 (signal transducer and activator of transcription 3) phosphorylation leads to the cardioprotective effect in cardiomyocytes of MURC deficiency after I/R. Intracellular signaling modified by MURC deletion is highlighted (red). BCL2 indicates B‐cell lymphoma 2; EGR1, early growth response protein 1; DDIT4, DNA‐damage‐inducible transcript 4.
Sources of Funding
This work was supported in part by Japan Society for the Promotion of Science Grants‐in‐Aid for Scientific Research (JSPS KAKENHI; grant nos. JP18K07046, JP18K08111) and the Takeda Science Foundation.
Disclosures
None.
Supporting information
Table S1. Small Interfering RNA Sequences
Table S2. Reverse Transcription–Mediated Quantitative Polymerase Chain Reaction Primers
Table S3. PC‐Corr Network Nodes and Links
Table S4. Benjamini‐Corrected P Values for the Enrichment Pathways in Figure 1B
Table S5. Benjamini‐Corrected P Values for the Enrichment Pathways in Figure 4C
Figure S1. Microarray analysis of the hearts of wild‐type and MURC (muscle‐restricted coiled‐coil protein) knockout mice.
Figure S2. Heart rate of wild‐type and MURC (muscle‐restricted coiled‐coil protein) knockout mouse hearts during echocardiography after sham operation or 24 hours after ischemia/reperfusion; n=10 per group.
Figure S3. Effect of cardiospecific MURC (muscle‐restricted coiled‐coil protein) overexpression in ischemia/reperfusion injury.
Figure S4. Gene silencing of MURC (muscle‐restricted coiled‐coil protein) through small interfering RNA in neonatal rat cardiomyocytes.
Figure S5. MURC (muscle‐restricted coiled‐coil protein) knockdown ameliorates reactive oxygen species production in cardiomyocytes after hypoxia/reoxygenation.
Figure S6. mRNA expression of Nox (NADPH oxidase) family genes in wild‐type and MURC (muscle‐restricted coiled‐coil protein) knockout mouse hearts 24 hours after ischemia/reperfusion.
Figure S7. A through C, Representative Western blots (top) and quantification (bottom) of the activation of apoptosis‐related proteins in wild‐type and MURC (muscle‐restricted coiled‐coil protein) knockout mouse hearts 24 hours after ischemia/reperfusion.
Figure S8. A, Cardiomyocytes isolated from adult mouse heart after incubation for 24 hours. Rod‐shaped cells are alive cardiomyocytes. B and C, Representative TUNEL (TdT‐mediated dUTP nick‐end labeling) staining (B) and quantification (C) of wild‐type and MURC (muscle‐restricted coiled‐coil protein) knockout adult mouse cardiomyocytes after H2O2 exposure.
Figure S9. STAT3 inhibitor cancels the cardioprotective effect of MURC (muscle‐restricted coiled‐coil protein) deficiency.
Figure S10. Immunoprecipitation of MURC (muscle‐restricted coiled‐coil protein) and STAT3 (signal transducer and activator of transcription 3) in neonatal rat cardiomyocytes transduced with β‐galactosidase (LacZ) or FLAG‐tagged human MURC.
Figure S11. A through F, Representative Western blots (top) and quantification (bottom) of the caveola‐related proteins in wild‐type and MURC (muscle‐restricted coiled‐coil protein) knockout mouse hearts.
Acknowledgments
The authors thank Dr Yuji Naito for assistance with microarray analysis.
(J Am Heart Assoc. 2019;8:e012047 DOI: 10.1161/JAHA.119.012047.)
References
- 1. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P; American Heart Association Statistics C, Stroke Statistics S . Heart disease and stroke statistics—2017 update: a report from the American Heart Association. Circulation. 2017;135:e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. De Luca G, Suryapranata H, Stone GW, Antoniucci D, Tcheng JE, Neumann F‐J, Van de Werf F, Antman EM, Topol EJ. Abciximab as adjunctive therapy to reperfusion in acute ST‐segment elevation myocardial infarction. JAMA. 2005;293:1759–1765. [DOI] [PubMed] [Google Scholar]
- 3. Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet. 2003;361:13–20. [DOI] [PubMed] [Google Scholar]
- 4. Braunwald E, Kloner RA. Myocardial reperfusion: a double‐edged sword? J Clin Invest. 1985;76:1713–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hausenloy DJ, Yellon DM. Myocardial ischemia‐reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. [DOI] [PubMed] [Google Scholar]
- 7. Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu C‐H, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb‐Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eltzschig HK, Eckle T. Ischemia and reperfusion—from mechanism to translation. Nat Med. 2011;17:1391–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia‐reperfusion injury. Physiol Rev. 2008;88:581–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 2015;6:524–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. [DOI] [PubMed] [Google Scholar]
- 12. Bhattacharya S, Ray RM, Johnson LR. STAT3‐mediated transcription of Bcl‐2, Mcl‐1 and c‐IAP2 prevents apoptosis in polyamine‐depleted cells. Biochem J. 2005;392:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McGaffin KR, Zou B, McTiernan CF, O'Donnell CP. Leptin attenuates cardiac apoptosis after chronic ischaemic injury. Cardiovasc Res. 2009;83:313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hockenbery DM, Oltvai ZN, Yin X‐M, Milliman CL, Korsmeyer SJ. Bcl‐2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. [DOI] [PubMed] [Google Scholar]
- 15. Barry SP, Townsend PA, Latchman DS, Stephanou A. Role of the JAK‐STAT pathway in myocardial injury. Trends Mol Med. 2007;13:82–89. [DOI] [PubMed] [Google Scholar]
- 16. Xuan YT, Guo Y, Han H, Zhu Y, Bolli R. An essential role of the JAK‐STAT pathway in ischemic preconditioning. Proc Natl Acad Sci USA. 2001;98:9050–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oshima Y, Fujio Y, Nakanishi T, Itoh N, Yamamoto Y, Negoro S, Tanaka K, Kishimoto T, Kawase I, Azuma J. STAT3 mediates cardioprotection against ischemia/reperfusion injury through metallothionein induction in the heart. Cardiovasc Res. 2005;65:428–435. [DOI] [PubMed] [Google Scholar]
- 18. Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007;8:185–194. [DOI] [PubMed] [Google Scholar]
- 19. Briand N, Dugail I, Le Lay S. Cavin proteins: new players in the caveolae field. Biochimie. 2011;93:71–77. [DOI] [PubMed] [Google Scholar]
- 20. Hansen CG, Nichols BJ. Exploring the caves: cavins, caveolins and caveolae. Trends Cell Biol. 2010;20:177–186. [DOI] [PubMed] [Google Scholar]
- 21. Bastiani M, Parton RG. Caveolae at a glance. J Cell Sci. 2010;123:3831–3836. [DOI] [PubMed] [Google Scholar]
- 22. Hayashi YK, Matsuda C, Ogawa M, Goto K, Tominaga K, Mitsuhashi S, Park YE, Nonaka I, Hino‐Fukuyo N, Haginoya K, Sugano H, Nishino I. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest. 2009;119:2623–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gratton JP, Bernatchez P, Sessa WC. Caveolae and caveolins in the cardiovascular system. Circ Res. 2004;94:1408–1417. [DOI] [PubMed] [Google Scholar]
- 24. Williams TM, Lisanti MP. The caveolin genes: from cell biology to medicine. Ann Med. 2004;36:584–595. [DOI] [PubMed] [Google Scholar]
- 25. Bastiani M, Liu L, Hill MM, Jedrychowski MP, Nixon SJ, Lo HP, Abankwa D, Luetterforst R, Fernandez‐Rojo M, Breen MR, Gygi SP, Vinten J, Walser PJ, North KN, Hancock JF, Pilch PF, Parton RG. MURC/Cavin‐4 and cavin family members form tissue‐specific caveolar complexes. J Cell Biol. 2009;185:1259–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McMahon K‐A, Zajicek H, Li W‐P, Peyton MJ, Minna JD, Hernandez VJ, Luby‐Phelps K, Anderson RGW. SRBC/Cavin‐3 is a caveolin adapter protein that regulates caveolae function. EMBO J. 2009;28:1001–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ogata T, Ueyama T, Isodono K, Tagawa M, Takehara N, Kawashima T, Harada K, Takahashi T, Shioi T, Matsubara H, Oh H. MURC, a muscle‐restricted coiled‐coil protein that modulates the Rho/ROCK pathway, induces cardiac dysfunction and conduction disturbance. Mol Cell Biol. 2008;28:3424–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miyagawa K, Ogata T, Ueyama T, Kasahara T, Nakanishi N, Naito D, Taniguchi T, Hamaoka T, Maruyama N, Nishi M, Kimura T, Yamada H, Aoki H, Matoba S. Loss of MURC/Cavin‐4 induces JNK and MMP‐9 activity enhancement in vascular smooth muscle cells and exacerbates abdominal aortic aneurysm. Biochem Biophys Res Commun. 2017;487:587–593. [DOI] [PubMed] [Google Scholar]
- 29. Naito D, Ogata T, Hamaoka T, Nakanishi N, Miyagawa K, Maruyama N, Kasahara T, Taniguchi T, Nishi M, Matoba S, Ueyama T. The coiled‐coil domain of MURC/Cavin‐4 is involved in membrane trafficking of caveolin‐3 in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2015;309:H2127–H2136. [DOI] [PubMed] [Google Scholar]
- 30. Nakanishi N, Ogata T, Naito D, Miyagawa K, Taniguchi T, Hamaoka T, Maruyama N, Kasahara T, Nishi M, Matoba S, Ueyama T. MURC deficiency in smooth muscle attenuates pulmonary hypertension. Nat Commun. 2016;7:12417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ogata T, Naito D, Nakanishi N, Hayashi YK, Taniguchi T, Miyagawa K, Hamaoka T, Maruyama N, Matoba S, Ikeda K, Yamada H, Oh H, Ueyama T. MURC/Cavin‐4 facilitates recruitment of ERK to caveolae and concentric cardiac hypertrophy induced by alpha1‐adrenergic receptors. Proc Natl Acad Sci USA. 2014;111:3811–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rodriguez G, Ueyama T, Ogata T, Czernuszewicz G, Tan Y, Dorn GW II, Bogaev R, Amano K, Oh H, Matsubara H, Willerson JT, Marian AJ. Molecular genetic and functional characterization implicate muscle‐restricted coiled‐coil gene (MURC) as a causal gene for familial dilated cardiomyopathy. Circ Cardiovasc Genet. 2011;4:349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shyu KG, Cheng WP, Wang BW, Chang H. Hypoxia activates muscle‐restricted coiled‐coil protein (MURC) expression via transforming growth factor‐beta in cardiac myocytes. Clin Sci (Lond). 2014;126:367–375. [DOI] [PubMed] [Google Scholar]
- 34. Marbach D, Prill RJ, Schaffter T, Mattiussi C, Floreano D, Stolovitzky G. Revealing strengths and weaknesses of methods for gene network inference. Proc Natl Acad Sci USA. 2010;107:6286–6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ciucci S, Ge Y, Duran C, Palladini A, Jimenez‐Jimenez V, Martinez‐Sanchez LM, Wang Y, Sales S, Shevchenko A, Poser SW, Herbig M, Otto O, Androutsellis‐Theotokis A, Guck J, Gerl MJ, Cannistraci CV. Enlightening discriminative network functional modules behind principal component analysis separation in differential‐omic science studies. Sci Rep. 2017;7:43946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cannistraci CV, Ogorevc J, Zorc M, Ravasi T, Dovc P, Kunej T. Pivotal role of the muscle‐contraction pathway in cryptorchidism and evidence for genomic connections with cardiomyopathy pathways in RASopathies. BMC Med Genomics. 2013;6:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shannon P. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 39. da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, Jensen LJ, von Mering C. The STRING database in 2017: quality‐controlled protein‐protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim SC, Boehm O, Meyer R, Hoeft A, Knufermann P, Baumgarten G. A murine closed‐chest model of myocardial ischemia and reperfusion. J Vis Exp. 2012;e3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bohl S, Medway DJ, Schulz‐Menger J, Schneider JE, Neubauer S, Lygate CA. Refined approach for quantification of in vivo ischemia‐reperfusion injury in the mouse heart. Am J Physiol Heart Circ Physiol. 2009;297:H2054–H2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vagnozzi RJ, Gatto GJ Jr, Kallander LS, Hoffman NE, Mallilankaraman K, Ballard VL, Lawhorn BG, Stoy P, Philp J, Graves AP, Naito Y, Lepore JJ, Gao E, Madesh M, Force T. Inhibition of the cardiomyocyte‐specific kinase TNNI3K limits oxidative stress, injury, and adverse remodeling in the ischemic heart. Sci Transl Med. 2013;5:207ra141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li D, Wu J, Bai Y, Zhao X, Liu L. Isolation and culture of adult mouse cardiomyocytes for cell signaling and in vitro cardiac hypertrophy. J Vis Exp. 2014;87:51357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kong LY, Abou‐Ghazal MK, Wei J, Chakraborty A, Sun W, Qiao W, Fuller GN, Fokt I, Grimm EA, Schmittling RJ, Archer GE Jr, Sampson JH, Priebe W, Heimberger AB. A novel inhibitor of signal transducers and activators of transcription 3 activation is efficacious against established central nervous system melanoma and inhibits regulatory T cells. Clin Cancer Res. 2008;14:5759–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. R Core Team 2016. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- 47. Braunersreuther V, Montecucco F, Asrih M, Pelli G, Galan K, Frias M, Burger F, Quindere AL, Montessuit C, Krause KH, Mach F, Jaquet V. Role of NADPH oxidase isoforms NOX1, NOX2 and NOX4 in myocardial ischemia/reperfusion injury. J Mol Cell Cardiol. 2013;64:99–107. [DOI] [PubMed] [Google Scholar]
- 48. Hoffmeyer MR, Jones SP, Ross CR, Sharp B, Grisham MB, Laroux FS, Stalker TJ, Scalia R, Lefer DJ. Myocardial ischemia/reperfusion injury in NADPH oxidase‐deficient mice. Circ Res. 2000;87:812–817. [DOI] [PubMed] [Google Scholar]
- 49. Kabat AM, Pearce EJ. Inflammation by way of macrophage metabolism. Science. 2017;356:488–489. [DOI] [PubMed] [Google Scholar]
- 50. Tagawa M, Ueyama T, Ogata T, Takehara N, Nakajima N, Isodono K, Asada S, Takahashi T, Matsubara H, Oh H. MURC, a muscle‐restricted coiled‐coil protein, is involved in the regulation of skeletal myogenesis. Am J Physiol Cell Physiol. 2008;295:C490–C498. [DOI] [PubMed] [Google Scholar]
- 51. Arora S, Wang Y, Jia Z, Vardar‐Sengul S, Munawar A, Doctor KS, Birrer M, McClelland M, Adamson E, Mercola D. Egr1 regulates the coordinated expression of numerous EGF receptor target genes as identified by ChIP‐on‐chip. Genome Biol. 2008;9:R166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roy S, Clark CJ, Mohebali K, Bhatt U, Wallace WA, Nahman NS, Ellison EC, Melvin WS, Sen CK. Reactive oxygen species and EGR‐1 gene expression in surgical postoperative peritoneal adhesions. World J Surg. 2004;28:316–320. [DOI] [PubMed] [Google Scholar]
- 53. Zhang Y, Shi G, Zheng J, Tang Z, Gao P, Lv Y, Guo F, Jia Q. The protective effects of N‐n‐butyl haloperidol iodide on myocardial ischemia‐reperfusion injury in rats by inhibiting Egr‐1 overexpression. Cell Physiol Biochem. 2007;20:639–648. [DOI] [PubMed] [Google Scholar]
- 54. Horak P, Crawford AR, Vadysirisack DD, Nash ZM, DeYoung MP, Sgroi D, Ellisen LW. Negative feedback control of HIF‐1 through REDD1‐regulated ROS suppresses tumorigenesis. Proc Natl Acad Sci USA. 2010;107:4675–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wolff NC, McKay RM, Brugarolas J. REDD1/DDIT4‐independent mTORC1 inhibition and apoptosis by glucocorticoids in thymocytes. Mol Cancer Res. 2014;12:867–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Williams JJL, Alotaiq N, Mullen W, Burchmore R, Liu L, Baillie GS, Schaper F, Pilch PF, Palmer TM. Interaction of suppressor of cytokine signalling 3 with cavin‐1 links SOCS3 function and cavin‐1 stability. Nat Commun. 2018;9:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chu CT, Levinthal DJ, Kulich SM, Chalovich EM, DeFranco DB. Oxidative neuronal injury. The dark side of ERK1/2. Eur J Biochem. 2004;271:2060–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Subramaniam S, Unsicker K. ERK and cell death: ERK1/2 in neuronal death. FEBS J. 2010;277:22–29. [DOI] [PubMed] [Google Scholar]
- 59. Yu W, Xu M, Zhang T, Zhang Q, Zou C. Mst1 promotes cardiac ischemia‐reperfusion injury by inhibiting the ERK‐CREB pathway and repressing FUNDC1‐mediated mitophagy. J Physiol Sci. 2019;69:113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rota R, Faggi F, Codenotti S, Poliani PL, Cominelli M, Chiarelli N, Colombi M, Vezzoli M, Monti E, Bono F, Tulipano G, Fiorentini C, Zanola A, Lo HP, Parton RG, Keller C, Fanzani A. MURC/cavin‐4 is co‐expressed with caveolin‐3 in rhabdomyosarcoma tumors and its silencing prevents myogenic differentiation in the human embryonal RD cell line. PLoS One. 2015;10:e0130287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tsutsumi YM, Horikawa YT, Jennings MM, Kidd MW, Niesman IR, Yokoyama U, Head BP, Hagiwara Y, Ishikawa Y, Miyanohara A, Patel PM, Insel PA, Patel HH, Roth DM. Cardiac‐specific overexpression of caveolin‐3 induces endogenous cardiac protection by mimicking ischemic preconditioning. Circulation. 2008;118:1979–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Horikawa YT, Patel HH, Tsutsumi YM, Jennings MM, Kidd MW, Hagiwara Y, Ishikawa Y, Insel PA, Roth DM. Caveolin‐3 expression and caveolae are required for isoflurane‐induced cardiac protection from hypoxia and ischemia/reperfusion injury. J Mol Cell Cardiol. 2008;44:123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kaiser RA, Bueno OF, Lips DJ, Doevendans PA, Jones F, Kimball TF, Molkentin JD. Targeted inhibition of p38 mitogen‐activated protein kinase antagonizes cardiac injury and cell death following ischemia‐reperfusion in vivo. J Biol Chem. 2004;279:15524–15530. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Small Interfering RNA Sequences
Table S2. Reverse Transcription–Mediated Quantitative Polymerase Chain Reaction Primers
Table S3. PC‐Corr Network Nodes and Links
Table S4. Benjamini‐Corrected P Values for the Enrichment Pathways in Figure 1B
Table S5. Benjamini‐Corrected P Values for the Enrichment Pathways in Figure 4C
Figure S1. Microarray analysis of the hearts of wild‐type and MURC (muscle‐restricted coiled‐coil protein) knockout mice.
Figure S2. Heart rate of wild‐type and MURC (muscle‐restricted coiled‐coil protein) knockout mouse hearts during echocardiography after sham operation or 24 hours after ischemia/reperfusion; n=10 per group.
Figure S3. Effect of cardiospecific MURC (muscle‐restricted coiled‐coil protein) overexpression in ischemia/reperfusion injury.
Figure S4. Gene silencing of MURC (muscle‐restricted coiled‐coil protein) through small interfering RNA in neonatal rat cardiomyocytes.
Figure S5. MURC (muscle‐restricted coiled‐coil protein) knockdown ameliorates reactive oxygen species production in cardiomyocytes after hypoxia/reoxygenation.
Figure S6. mRNA expression of Nox (NADPH oxidase) family genes in wild‐type and MURC (muscle‐restricted coiled‐coil protein) knockout mouse hearts 24 hours after ischemia/reperfusion.
Figure S7. A through C, Representative Western blots (top) and quantification (bottom) of the activation of apoptosis‐related proteins in wild‐type and MURC (muscle‐restricted coiled‐coil protein) knockout mouse hearts 24 hours after ischemia/reperfusion.
Figure S8. A, Cardiomyocytes isolated from adult mouse heart after incubation for 24 hours. Rod‐shaped cells are alive cardiomyocytes. B and C, Representative TUNEL (TdT‐mediated dUTP nick‐end labeling) staining (B) and quantification (C) of wild‐type and MURC (muscle‐restricted coiled‐coil protein) knockout adult mouse cardiomyocytes after H2O2 exposure.
Figure S9. STAT3 inhibitor cancels the cardioprotective effect of MURC (muscle‐restricted coiled‐coil protein) deficiency.
Figure S10. Immunoprecipitation of MURC (muscle‐restricted coiled‐coil protein) and STAT3 (signal transducer and activator of transcription 3) in neonatal rat cardiomyocytes transduced with β‐galactosidase (LacZ) or FLAG‐tagged human MURC.
Figure S11. A through F, Representative Western blots (top) and quantification (bottom) of the caveola‐related proteins in wild‐type and MURC (muscle‐restricted coiled‐coil protein) knockout mouse hearts.