

Abstract
Hydrogels are commonly used as engineered extracellular matrix (ECM) mimics in applications ranging from tissue engineering to in vitro disease models. Ideal mechanisms used to crosslink ECM-mimicking hydrogels do not interfere with the biology of the system. However, most common hydrogel crosslinking chemistries exhibit some form of cross-reactivity. The field of bio-orthogonal chemistry has arisen to address the need for highly specific and robust reactions in biological contexts. Accordingly, bio-orthogonal crosslinking strategies have been incorporated into hydrogel design, allowing for gentle and efficient encapsulation of cells in various hydrogel materials. Furthermore, the selective nature of bio-orthogonal chemistries can permit dynamic modification of hydrogel materials in the presence of live cells and other biomolecules to alter matrix mechanical properties and biochemistry on demand. In this review, we provide an overview of bio-orthogonal strategies used to prepare cell-encapsulating hydrogels and highlight the potential applications of bio-orthogonal chemistries in the design of dynamic engineered ECMs.
Keywords: engineered extracellular matrix, bio-orthogonal chemistry, hydrogels, dynamic materials, biomaterials
Graphical Abstract
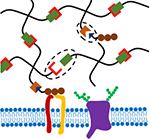
Bio-orthogonal strategies for hydrogel crosslinking and functionalization can facilitate efficient and gentle cell encapsulation and dynamic modification of hydrogel properties. This review discusses bio-orthogonal chemistries used to prepare cell encapsulating hydrogels and highlights the potential for bio-orthogonal reactions to dynamically tune the mechanics and biochemistry of engineered extracellular matrix mimics.
1. Introduction
The native extracellular matrix (ECM) serves not only as a scaffold to organize cells into tissues but also as a rich source of cues that guide cell fate decisions. Both the mechanics and biochemistry of the ECM modulate cellular behaviors such as migration, proliferation, and differentiation.[1] Accordingly, engineering synthetic ECMs with tunable properties is a promising approach to control cell phenotype for applications in tissue engineering, regenerative medicine, and in vitro disease models. The native ECM is a water-swollen network, predominantly consisting of proteins and polysaccharides. In order to mimic these properties of the ECM, hydrogels composed of natural polysaccharides, natural and engineered proteins, and synthetic polymers have been used to encapsulate cells for 3D culture and transplantation.[2]
To be a suitable platform for cell encapsulation, hydrogel crosslinking and functionalization chemistries must be compatible with living cells. Many approaches for 3D cell encapsulation utilize covalent crosslinking reactions, as covalent bonds provide greater stability than physical crosslinking and can thus better maintain matrix properties over time and can typically achieve a higher range of material stiffness. At present, most covalent hydrogel crosslinking chemistries make use of either photo-initiated radical polymerization or chemical crosslinkers that react with common functional groups such as amines and sulfhydryls. The potential off-target effects of these crosslinking methods must be taken into account when designing hydrogel ECM mimics for cell encapsulation. For example, photoinitiators used in radical polymerizations may be cytotoxic, and exposure to UV light commonly used in these polymerizations may induce DNA damage.[3, 4] Other chemical crosslinkers that react with amines and sulfhydryls can react with these same functional groups present in cell-surface proteins,[5] potentially altering the phenotype of the encapsulated cells. To limit the potential confounding effects of hydrogel crosslinking reactions on cellular phenotype, the ideal gelation chemistry would be bio-orthogonal (Figure 1). Here, we define bio-orthogonal reactions as those that make use of chemical reaction pairs that (1) do not naturally occur in biological systems, (2) do not cross-react with functional groups that are present in biology, and (3) do not require cytotoxic catalysts or produce cytotoxic byproducts.[6]
Figure 1.

Bio-orthogonal reactions make use of reactive groups that do not naturally occur in biological systems, react selectively to avoid cross-reactivity with various biological functional groups, and do not produce toxic byproducts or require toxic catalysis. In the context of hydrogels as engineered ECMs, bio-orthogonal chemistries can be used to crosslink the hydrogel and modulate the presentation of biochemical signals. Two example bio-orthogonal reactions, strain-promoted azide-alkyne cycloaddition (SPAAC) in blue and tetrazine-mediated inverse-electron demand Diels-Alder (IED-DA) reaction in green, are depicted to form a hydrogel (black network) adjacent to the cell surface (blue lipid bilayer). Common biological functional groups present on the cell surface that may be targets of cross-reactivity in non-bio-orthogonal reactions are also depicted in red.
The field of bio-orthogonal chemistry was pioneered by Bertozzi and colleagues with the development of the Staudinger ligation as a method for selectively labeling cell-surface glycans.[6, 7] The Staudinger ligation utilizes two reaction partners that are absent from biological systems, an azide and a functionalized triarylphosphine, to form a stable amide bond.[7] This reaction was demonstrated to be robust and highly specific in complex biological contexts, including labeling glycoproteins in cell lysates[8] and on cell surfaces within live mice.[9] Limitations including slow reaction kinetics and susceptibility of the triarylphosphines to oxidation led the Bertozzi lab to identify strain-promoted azide-alkyne cycloaddition (SPAAC) as a second bio-orthogonal ligation reaction.[6, 10] SPAAC makes use of azides and strained cyclooctynes to yield stable triazole linkages via 1,3-dipolar cycloaddition.[10] The initial cyclooctyne reaction partner exhibited similarly slow reaction kinetics to the Staudinger ligation, but further work by the Bertozzi,[11, 12] Boons,[13] van Delft,[14, 15] and Popik[16] groups generated variants with rate constants enhanced by up to two orders of magnitude. Similar to the Staudinger ligation, SPAAC ligation was demonstrated to be bio-orthogonal in vivo, permitting selective labeling of azide-bearing cell surface glycans in developing zebrafish embryos.[17]
The bio-orthogonal SPAAC reaction is sometimes termed “copper-free click chemistry” to contrast with the original “click” reaction, copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC), developed independently by the Sharpless and Meldal labs.[18] According to the criteria for bio-orthogonal reactions presented above, the original CuACC reaction would not be considered bio-orthogonal due to the use of a cytotoxic copper catalyst,[6] although copper-binding ligands have been developed to permit CuACC reactions on live cells.[19] This example serves to highlight an important distinction between click chemistry and bio-orthogonal chemistry, two often conflated terms. Sharpless and colleagues defined click chemistry as reactions that are “modular, wide in scope, give very high yields, generate only inoffensive byproducts that can be removed by nonchromatographic methods, and be stereospecific (but not necessarily enantioselective).”[20] Such reactions should require “simple reaction conditions (ideally, the process should be insensitive to oxygen and water), readily available starting materials and reagents, the use of no solvent or a solvent that is benign (such as water) or easily removed, and simple product isolation.”[20] Therefore, the term “click chemistry” encompasses a broader range of reactions that are robust, but not necessarily free of off-target effects, in biological contexts. Such click reactions that are commonly used in hydrogel synthesis would include 1,4-conjugate additions (i.e., thiol-vinyl sulfone and thiol-maleimide reactions), aldehyde-nucleophile reactions (hydrazone and oxime ligations), and photo-activated thiol-ene coupling. These reactions have the potential for substantial cross-reactivity with functional groups present in biological systems, including sulfhydryls, amines, and aldehydes, precluding these reactions from being truly bio-orthogonal. The distinctions between click chemistry and bio-orthogonal chemistry are summarized in Figure 2. This review focuses on bio-orthogonal techniques to crosslink and functionalize hydrogels as engineered ECMs. For detailed discussions of other click reactions used to prepare hydrogel matrices, the reader is referred to several excellent reviews.[5, 21]
Figure 2.

Various “click”-type reactions have been employed in hydrogel systems, but only a few examples fit a rigorous definition of bio-orthogonality.
In this review, we discuss bio-orthogonal approaches to prepare covalently-crosslinked hydrogels and identify supramolecular assembly as a candidate mechanism of bio-orthogonal physical crosslinking. Beyond crosslinking chemistries, the bio-orthogonal nature of the reactions discussed can facilitate dynamic modification of hydrogels in the presence of live cells. The native ECM is highly dynamic and is remodeled throughout development and in response to disease, changing its biochemical composition and mechanical properties.[22] We also explore how bio-orthogonal chemistries can be adapted to mimic this dynamism in engineered hydrogel matrices.
2. Bio-orthogonal chemistries for hydrogel crosslinking
The predominant application of bio-orthogonal chemistries in hydrogel-based cell matrices is in the design of cytocompatible and robust crosslinking reactions. Truly bio-orthogonal reactions can proceed under physiological conditions (pH 7.4 saline at 37°C) and in the presence of cell culture medium supplements and live cells. Such reactions do not produce cytotoxic byproducts and will not cross-react with functional groups present on the surface of cells. For these reasons, bio-orthogonal crosslinking chemistries are a very mild approach to produce cell-encapsulating hydrogels. An additional benefit of this selectivity is that the reactions are resilient against variations in biomolecular composition. For instance, cell-adhesive peptides or growth factors can be incorporated into bio-orthogonally-crosslinked hydrogel networks without off-target crosslinking during gelation. In contrast, covalent crosslinking strategies using amine or sulfhydryl reactive crosslinkers would have the potential to react with peptides and proteins, altering the bioactivity and/or release of these factors. This section provides an overview of bio-orthogonal ligation chemistries used to crosslink hydrogels and presents alternative chemistries that may be employed in future hydrogel design. Bio-orthogonal crosslinking chemistries used in hydrogel synthesis are summarized in Table 1.
Table 1.
Bio-orthogonal chemistries to crosslink hydrogels.
| Gelation Chemistry | Reactive Group #1 | Reactive Group #2 | Reaction Product | Notes | References |
|---|---|---|---|---|---|
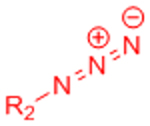
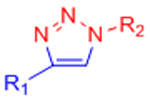
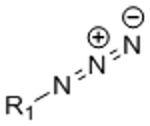
| Copper-catalyzed azide-alkyne cycloaddition (CuAAC) | 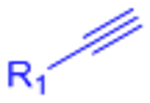 |
 |
 |
Potential Cu2+ toxicity may necessitate use of chelating ligands | [24] |
 |
 |
 |
X: H (Slow reaction kinetics) X: F (Improved kinetics) | [25, 27–30, 123, 124] | |
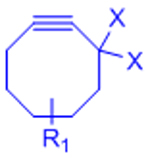
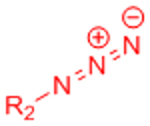
| Strain-promoted azide-alkyne cycloaddition (SPAAC) |  |
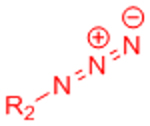 |
 |
[31–33] | |
 |
 |
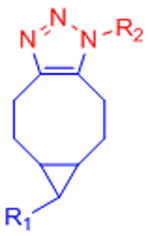 |
[34–37, 162] | ||
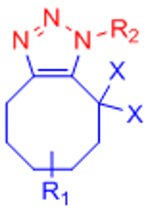
| Inverse electron demand Diels-Alder (IED-DA) | 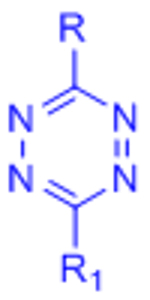 |
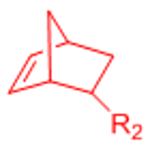 |
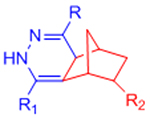 |
[51–54, 132] | |
 |
 |
 |
Extremely rapid gelation kinetics | [56] | |
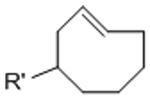
| Diels-Alder (DA) |  |
 |
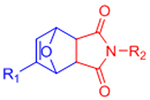 |
Maleimides can cross-react with thiols | [58–68, 70, 71] |
 |
 |
 |
Reversible under physiological conditions | [74] | |
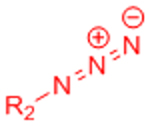
| Staudinger ligation | 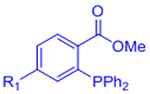 |
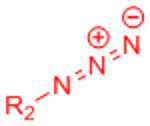 |
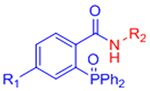 |
Slow reaction kinetics | [35, 75, 76] |
| Nitrile Oxide Cycloaddition | 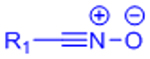 |
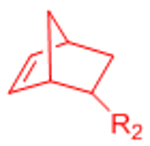 |
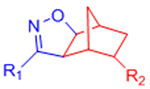 |
Highly reactive nitrile oxide must be generated in situ | [78, 79] |
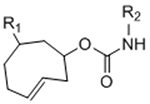
Two important considerations for choosing bio-orthogonal chemistries to prepare engineered ECMs are the kinetics of the crosslinking reaction and the complexity of the chemical synthesis for the hydrogel precursors functionalized with bio-orthogonal reaction pairs. Ideal gelation reactions for applications such as cell encapsulation would complete crosslinking within seconds to minutes to maintain homogeneous cell distribution in 3D. While numerous design factors collectively determine gelation time, including the reaction rate of the crosslinking moities, the concentration of the hydrogel precursors, and the degree of polymer functionalization, choosing a crosslinking chemsitry with an appropriate reaction rate is the first step in designing hydrogels suitable for cell encapsulation. Crosslinking time and number of synthetic steps are plotted in Figure 3 for the various bio-orthogonal crosslinking reactions discussed in this section.
Figure 3.

Crosslinking time and synthetic complexity are two important parameters to consider when selecting bio-orthogonal reactions to prepare hydrogel-based engineered ECMs. The time to complete crosslinking, as determined by the time to reach plateau storage modulus, is plotted on the x-axis, and the total number of synthetic steps to make the hydrogel precursors is plotted on the y-axis, for various previously published crosslinking chemistries. It is important to note that other factors, such as precursor concentration and degree of polymer functionalization, influence crosslinking time in addition to the chemical reaction used. The year in which each hydrogel system was reported is denoted next to each data point. Over time, as synthetic schemes have been optimized and bio-orthogonal reaction precursors have become commercially available, the number of synthetic steps required has in general decreased. Those data points with an asterix (*) next to the year could now be completed in 3 or fewer synthetic steps with commercially available reagents. These data were compiled from references 25, 28-35, 51-54, 56, 58, 59, 62, 64-66, 68, 70, 71, 74, 78, 132.
2.1. Azide-alkyne cycloaddition
Azide-alkyne cycloaddition is the most common bio-orthogonal technique used to prepare cell-encapsulating hydrogels. While the first hydrogel materials crosslinked via azide-alkyne cycloaddition made use of copper(I) as a catalyst,[23] the potential for copper cytotoxicity has prevented this approach from becoming widely used for cell encapsulation. Nevertheless, certain cell types and hydrogel formulations have been shown to tolerate exposure to copper during the crosslinking process,[24] and copper-binding ligands known to reduce cytotoxicity[19] could be employed during crosslinking in other systems.
To overcome limitations of copper cytotoxicity, DeForest et al. built upon work in the Bertozzi lab using difluorinated cyclooctynes (DIFO) for strain-promoted azide-alkyne cycloaddtion (SPAAC)[11] to prepare bio-orthogonally-crosslinked poly(ethylene glycol) (PEG) hydrogels for cell encapsulation.[25] Azide-functionalized 4-arm PEG was crosslinked with peptides functionalized with DIFO on the N- and C-terminal amino acids (Figure 4A).[25] The sequence of the peptide crosslinker was chosen to be degradable by cell-secreted matrix metalloproteinases (MMPs) to permit cell spreading and migration.[26] The onset of gelation occurred within minutes, and gelation was complete within one hour (Figure 4B).[25] As expected based on the bio-orthogonal nature of the crosslinking reaction, encapsulated fibroblasts remained highly viable 24 hours after gelation (Figure 4C).[25] Later studies demonstrated that altering the molecular weight of the PEG macromers or the molar ratio of azide to DIFO groups permitted control over the mechanical properties of the hydrogels.[27]
Figure 4.

(A) Schematic depicting hydrogel formation from azide-functionalized PEG and DIFO-functionalized peptides crosslinked via SPAAC. (B) The SPAAC-crosslinked materials gel within minutes and complete crosslinking in approximately 1 hour. (C) Fibroblast cells encapsulated within the gels remain viable 24 hours post-crosslinking, as observed by live/dead assay. Green: live, Red: dead. A-C reproduced with permission. Copyright 2009, Nature Publishing Group.[25] (D) Schematic depicting bio-orthogonal crosslinking of engineered elastin-like proteins via SPAAC or Staudinger ligation. (E) The SPAAC crosslinked samples complete crosslinking within minutes, while the Staudinger samples crosslink on the order of 1 hour. Both (F) human MSCs and (G) murine neural progenitor cells retain their appropriate phenotypes when cultured within the SPAAC-crosslinked ELP hydrogels, as observed by immunocytochemistry. Blue: nuclei (DAPI), Red: F-actin (phalloidin), Green: Nestin. D-G reproduced with permission.[35] Copyright 2016, John Wiley and Sons.
Following this initial work, other groups have used cyclooctyne-based SPAAC crosslinking to prepare injectable hydrogel formulations that gel in situ. Takahashi, et al., employed azide- and unsubstituted cyclooctyne-modified hyaluronic acid (HA) to prepare hydrogels for cell encapsulation and in vivo transplantation.[28] Gels injected into the murine peritoneum exhibited only mild inflammation with no detrimental tissue adhesions and were cleared within 3 weeks.[28] Similarly, PEG hydrogels prepared from azide- and monofluorinated cyclooctyne-functionalized polymers evoked a minimal immune response characterized by infiltration of immune cells and mild scar tissue formation when subcutaneously injected into mice.[29] Cyclooctyne-modified HA crosslinked with azide-modified PEG also served as cytocompatible hydrogel scaffolds, maintaining high viability and facilitating proliferation of encapsulated COS-7 cells.[30]
One limitation with the early cyclooctyne derivatives was slower than desired reaction kinetics,[6] which resulted in gelation times on the orders of tens of minutes to achieve complete crosslinking.[25, 28–30] In the context of improved kinetics for bio-orthogonal labeling, the Bertozzi, van Delft, and Popik groups independently reported up to an order of magnitude increase in reaction rates when using cyclooctynes fused to two benzyl rings, with an amide-bonded nitrogen inserted into the cyclooctyne ring.[12, 14, 16] Similar dibenzylcyclooctynes (DBCO) have been utilized to crosslink hydrogels for orthopedic tissue engineering applications. Hermann et al. used PEG macromers functionalized with azides and DBCO groups to form injectable hydrogels to deliver a bone morphogenetic protein inhibitor in a murine craniosynostosis model.[31] These DBCO gels exhibited more rapid gelation kinetics than earlier cyclooctyne variants, completing gelation in less than 2 minutes.[31] Wang et al. demonstrated that dextrans crosslinked by azides and DBCO groups facilitated matrix deposition by encapsulated chondrocytes,[32] and Zheng et al. used DBCO-modified PEG hydrogels as scaffolds for mesenchymal stem cell (MSC) culture.[33]
While the introduction of DBCO groups increased the rate of the crosslinking reaction, DBCO, like the previous cyclooctyne variants, required complicated and low-yielding syntheses.[15] To overcome this limitation, Dommerholt et al. fused a cyclopropane ring to cyclooctyne to generate a highly strained bicyclononyne (BCN) that reacted with comparable rates to DBCO while requiring fewer synthetic steps.[15] In a hydrogel context, DeForest and Tirrell later employed BCN-functionalized PEG macromers to prepare SPAAC-crosslinked gels.[34] BCN-based reactions have also been used to prepared hydrogels comprised of engineered recombinant elastin-like proteins (ELPs).[35–37] The modular design of ELPs permits independent tuning of matrix mechanics and biochemistry, including cell adhesive ligand presentation and susceptibility to proteolytic degradation.[38] However, traditional approaches to prepare hydrogels from these materials relied on crosslinking functional groups on the amino acid side chains, such as primary amines of lysines,[38, 39] resulting in possible undesired crosslinking of cell surface proteins. Functionalization of lysine residues with azide or BCN moieties,[35] or functionalizing tyrosine residues with an azide linker via an ene-type reaction,[36] permitted cell encapsulation in ELP hydrogels via bio-orthogonal crosslinking while retaining decoupled control of matrix stiffness and adhesive ligand density (Figure 4D). Crosslinking in these materials proceeds to completion on the order of 1–2 minutes, and the hydrogels support the 3D culture of multiple cells types, facilitating high viability and maintenance of the appropriate cellular phenotypes (Figure 4E-G).[35, 36]
An important caveat regarding the bio-orthogonality of SPAAC is the mild reactivity of cyclootynes toward free thiols, such as those found in cysteine side chains. Several groups have reported thiol-yne reactions using various cyclooctynes under physiological conditions.[12, 40, 41] Nevertheless, the low abundance of free thiols in most biological systems and the significantly higher reaction rates for SPAAC compared to thiol-yne reactions support the characterization of SPAAC as a nearly bio-orthogonal reaction. For example, the second order rate constant for the BCN-azide reaction (k2 ~ 10−1 M−1 s−1)[15] is approximately three orders of magnitude greater than the rate constant for the BCN-thiol reaction (k2 ~ 10−4 M−1 s−1).[41] For comparison, the rate constant for the thiol-maleimide reaction is over 700 M−1 s-1.[42] Thus, hydrogels crosslinked by traditional thiol-reactive chemistries have a much greater potential for undesired cell-surface protein crosslinking than those crosslinked by SPAAC.
In addition to SPAAC, uncatalyzed azide-alkyne cycloaddition can be achieved in water using terminal alkynes with adjacent electron-withdrawing substituents. Li et al. demonstrated that terminal alkynes connected to an ester carbonyl carbon readily reacted with azides in water at room temperature without the need for a copper catalyst.[43] Truong et al. employed this reaction to prepare cell-encapsulating hydrogels from azide-functionalized chitosan and alkyne-functionalized PEG.[44] The hydrogels completed crosslinking within an hour and supported the viability of encapsulated MSCs.[44]
2.2. Inverse-electron demand Diels-Alder (IED-DA) reaction
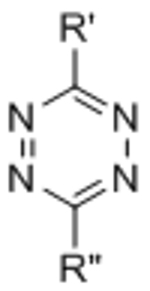
The inverse electron-demand Diels-Alder (IED-DA) reaction is a second bio-orthogonal cycloaddition that is gaining popularity as a crosslinking mechanism for cell-encapsulating hydrogels. IED-DA reactions typically involve tetrazine ligations with strained alkenes. Tetrazine-based IED-DA reactions were first reported as bio-orthogonal ligations by Fox and co-workers.[45] The reaction between a dipyridyl-functionalized tetrazine and trans-cyclooctene (TCO) proceeded at a rate 3 orders of magnitude greater than the optimized SPAAC reaction,[45] making tetrazine ligation the fastest bio-orthogonal conjugation chemistry reported to date.[46] Contemporaneously, the Hilderbrand lab and the Braun lab independently introduced tetrazine ligations with norbornenes[47] and a derivative of the tetracyclic Reppe anhydride,[48] respectively. While the reaction between tetrazine and norbornene is slower than between tetrazine and TCO, norbornene is significantly more stable than TCO in solution.[46] Cyclopropenes have also been introduced as alternative dienophiles to TCO and norbornene.[49]
The use of a tetrazine IED-DA reaction to prepare covalently crosslinked, DMSO-swollen gels was first reported by Zhou, et al.[50] Soon after, Alge, et al. published a study extending the use of bio-orthogonal IED-DA crosslinking to the synthesis of cell-encapsulating hydrogels.[51] Four-arm PEG macromers were functionalized with tetrazine and crosslinked with difunctionalized norbornene peptides. The peptide crosslinkers were designed to be MMP cleavable to permit proteolysis of the matrix, and norbornene functionalized RGD integrin-binding peptides were included to facilitate cell-matrix adhesion.[51] Gelation occurred within minutes, and crosslinking was completed within 20 minutes.[51] Encapsulated human MSCs exhibited excellent viability in the IED-DA crosslinked gels.[51]
Tetrazine-norbornene ligation has since been utilized in other hydrogel systems. Desai et al. functionalized alginate with norbornene and tetrazine groups to form stable, covalently-crosslinked hydrogels.[52] Compared to fibroblasts encapsulated in alginate gels crosslinked through ionic interactions with calcium ions, fibroblasts in the IED-DA alginate gels exhibited higher viability both directly following encapsulation and after 3 days in culture. When injected subcutaneously into mice, both ionically-crosslinked and IED-DA alginate gels elicted a minimal inflammatory response characterized by encapsulation within a thin layer of fibrous tissue. While the ionically-crosslinked gels exhibited significant degradation by one month post-implantation, the IED-DA gels remained largely intact for at least 2 months.[52] Koshy et al. later applied this same chemistry to covalently crosslinked gelatin hydrogels.[53] Gelatin hydrogels are commonly used in 3D cell culture, and photoinitiated radical polymerization of methacrylated gelatin (GelMA) is the most common method for preparing such hydrogels. The crosslinking scheme developed by Koshy et al. eliminated the need for UV light exposure and the use of potentially cytotoxic photoinitiators. Accordingly, higher viability of human MSCs was observed in tetrazine-norbornene crosslinked gels compared to GelMA.[53] Subcutaneous injection of the gelatin hydrogels was also well-tolerated by mice.[53] Truong et al. exploited the orthogonal nature of the tetrazine ligation to prepare mechanically tough double-network hydrogels.[54] One network was crosslinkined via tetrazine-norbornene ligation, and the second network was crosslinked via thiol-alkyne addition.[54] The resulting hydrogels were resilient to multiple rounds of mechanical loading, and human MSCs encapsulated within the hydrogels remained highly viable after 2 days in culture.[54]
TCO has been used as an alternative dienophile to norbornene to prepare hydrogels with extremely rapid gelation kinetics. Following the initial report of the tetrazine-TCO ligation, Fox and co-workers discovered that the additional strain imposed by fusing a cyclopropane ring to the cyclooctene resulted in an increase in the reaction rate by a further two orders of magnitude.[55] Using this modified TCO, the Jia and Fox labs synthesized tetrazine-modified hyaluronic acid and bifunctional TCO PEG crosslinkers to prepare hydrogels.[56] The rapid gelation rate permitted fabrication of multilayer hydrogel microspheres and hydrogel channels, and the resultant gels supported the formation of cancer cell spheroids from an initial single cell suspension, indicating significant cell proliferation. [56]
2.3. Diels-Alder (DA) reaction
Traditional Diels-Alder (DA) reactions have also found utility as hydrogel crosslinking chemistries. While DA reactions employed in organic synthesis often require high temperatures to proceed at a reasonable rate, DA reactions are accelerated in water due to hydrophobic stabilization of the transition state.[57] Thus, certain diene-dienophile reaction pairs make DA cycloadditions viable strategies for bio-orthogonal hydrogel crosslinking.
The first reported use of a DA reaction to prepare hydrogels employed a synthetic co-polymer containing furan moieties as dienes and bifunctional PEG maleimide crosslinkers as dienophiles.[58, 59] Despite the accelerated DA reaction rate in water, gelation of these materials occurred over the timescale of an hour.[58] Shoichet and co-workers later introduced furan-maleimide crosslinked DA hydrogels as potential scaffolds for tissue engineering, using furan-modified hyaluronic acid and bifunctional PEG maleimide crosslinkers.[60] These hydrogels were demonstrated to be cytocompatible using mammary epithelial cells,[60] and further optimizations permitted control over hydrogel stiffness and presentation of bioactive factors.[61] A later study confirmed that the DA hyaluronic acid hydrogels were well-tolerated in vivo, with no detectable immune cell activation or scar formation compared to untreated controls, and could deliver brain-derived neurotrophic factor to injured spinal cords in a rat model.[62]
Subsequently, the furan-maleimide DA reaction has been used to prepare hydrogels for various tissue engineering and drug delivery applications. To mimic native cartilage ECM, Yu et al. prepared interpenetrating network hydrogels composed of furan-functionalized gelatin and furan-functionalized hyaluronic acid, crosslinked by bifunctional PEG maleimide.[63] Yu et al. later used DA-crosslinked HA-PEG hydrogels as 3D cell culture scaffolds for chondrocytes.[64] DA-crosslinked hydrogels have also been used as passively releasing, drug delivery depots for small molecules,[65] growth factors,[66, 67] and therapeutic antibodies.[68] Because the DA reaction is reversible, furan-maleimide ligation can be used to covalently couple molecules to hydrogel networks and control their release.[69] Fan et al. exploited this reversibility to covalently immobilize furan-modified dexamethasone in DA-crosslinked hyaluronic acid hydrogels, controlling the release of dexamethasone over the course of two weeks.[70]
While the furan-maleimide DA reaction is the most commonly used DA reaction for hydrogel crosslinking, this reaction pair has significant limitations. Foremost in the context of this review, the reaction scheme is not truly bio-orthogonal. Maleimides are commonly used in bioconjugation of thiol-containing molecules via conjugate addition, and maleimides also exhibit some side reactivity with amines. Maleimides are additionally susceptible to hydrolysis, which was previously shown to play a role in the degradation of furan-maleimide crosslinked hydrogels.[71] Due to the reversible nature of the Diels-Alder reaction, an equilibrium exists between free maleimides and crosslinked maleimides. The free maleimides are susceptible to hydrolysis and once hydrolyzed do not form stable DA-based crosslinks. Thus, overtime, the number of potential crosslinks is decreased, and the gel degrades.[71] Adding a hydrophobic spacer between the PEG macromers and the maleimides can improve hydrogel stability by reducing the rate of maleimide hydrolysis.[68] Finally, the kinetics of the furan-maleimide reaction are slow relative to other potential crosslinking chemistries, with some reports of gelation requiring multiple hours under physiological conditions.[62, 64]
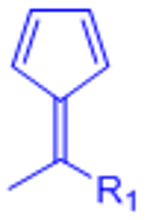
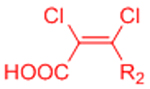
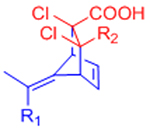
The application of other DA reaction pairs to DA reactions has been largely unexplored, with the exception of a reaction scheme using fulvenes as dienes. Lehn and co-workers introduced fulvene-based DA reactions as an example of room temperature dynamic chemistry.[72] Fulvenes undergo reversible DA cycloadditions with cyanoolefin dienophiles due to the strong electron-withdrawing character of the cyano groups.[72] This chemistry was initially employed to create self-healing polymeric materials.[73] Wei et al. later adapted this reaction scheme to prepare self-healing dextran hydrogels, replacing the cyanoolefin dienophiles with more stable dichloromaleic acid dieneophiles.[74] Fulvene-functionalized dextran was crosslinked via dichloromaleic acid-modified PEG to create hydrogels that exhibit reversible self-healing under physiological conditions.[74]
2.4. Staudinger ligation
Despite its place as the first demonstrated bio-orthogonal reaction, the Staudinger ligation has found limited utility as a hydrogel crosslinking chemistry, likely arising from its relatively slow reaction rate and limited stability of the triarylphosphine functional groups.[6] As an example, we recently reported that engineered elastin-like protein hydrogels crosslinked by Staudinger ligation completed gelation on the timescale of an hour, whereas gels crosslinked by BCN-mediated SPAAC completed gelation within a few minutes.[35] Nevertheless, Staudinger ligation has been effectively used as a means to stabilize alginate hydrogel microbeads.[75, 76] Initial crosslinking of the beads was accomplished by exposing the alginate solutions to divalent cations, and then azide-functionalized alginate reacted via Staudinger ligation with triarylphosphine-modifed PEG or alginate to produce dual-crosslinked networks.[75, 76] The Staudinger-crosslinked hydrogels remained stable, even after chelation of the divalent cations.[75, 76] Pancreatic endocrine cells cultured within the Staudinger-crosslinked gels remained viable and proliferated.[76]
2.5. Nitrile oxide-norbornene cycloaddition
Nitrile oxides and norbornenes undergo 1,3-dipolar cycloadditions similar to the azides and alkynes employed in SPAAC. Nitrile oxides are highly reactive, which initially made implementation of these cycloaddition reactions challenging in a biological context. However, the nitrile oxide can be stabilized by adjacent aromatic rings and generated in situ by hydrolysis of a hydroximoyl chloride precursor.[77] Truong et al. applied this chemistry to prepare cell-encapsulating PEG hydrogels.[78] PEG macromers were functionalized with norbornene and hydroximoyl chloride. Upon mixing under aqueous conditions, the hydroximoyl chloride is converted to a reactive nitrile oxide, which then undergoes 1,3-dipolar cycloaddition with the norbornene-functionalized PEG.[78] The mixture exhibits rapid gelation, with crosslinking completed in a few minutes.[78] Neural progenitor cells encapsulated within the resultant gels remained viable post-gelation.[78] Truong et al. later employed this chemistry to prepare bio-orthogonally crosslinked gelatin hydrogels.[79] Gelatin norbornene crosslinked with hydroximoyl chloride functionalized PEG served as suitable hydrogels for the 3D culture of fibroblasts, which maintained high viability and expression of phenotypic markers similar to Matrigel controls.[79]
2.6. Other bio-orthogonal reactions with potential applications in hydrogel crosslinking
Following the initial reports of the Staudinger ligation and SPAAC as bio-orthogonal reactions, significant effort has been expended to identify new bio-orthogonal chemistries with improved reaction kinetics, enhanced reactant stability, and facile synthesis.[46, 80] Of these more recently reported bio-orthogonal reactions, tetrazine ligation and nitrile oxide-norbornene cycloaddition have been employed to generate cell encapsulating hydrogels, as discussed in the preceding sections. This section briefly presents other bio-orthogonal ligation chemistries that have potential utility in the preparation of hydrogel biomaterials.
Dipolar cycloadditions between nitrones and cyclooctynes proceed more rapidly than the analogous SPAAC reaction between azides and cyclooctynes,[81] suggesting the strain-promoted alkyne–nitrone cycloaddition (SPANC) may find utility in preparing hydrogels with rapid crosslinking kinetics. The poor stability of some nitrones in water will likely dictate which reaction partners would be acceptable for aqueous crosslinking.[46] A second potential 1,3-dipolar cycloaddition for use in bio-orthogonal crosslinking is the reaction between nitrile imines and alkenes.[82, 83] The reaction proceeds relatively rapidly, with rate constants on the order of the SPANC and nitrile oxide-norbornene reactions.[46] The potential applicability for use in cell-encapsulating hydrogels may be limited by the requirement to generate the nitrile imine in situ by photolysis of a tetrazole precursor.[82, 83] The UV light employed in this scheme may prove mutagenic.[4] Furthermore, a recent study has reported potential off-target reactivity of nitrile imines with biological nucleophiles, raising questions about the bio-orthogonality of this approach.[84] Nevertheless, this chemistry has been employed to crosslink PEG hydrogels.[85] To achieve the greatest enhancement in gelation rates using 1,3-dipolar cylcoadditions, the reaction between azides and oxanorbornadienes could be implemented in a hydrogel context. Van Berkel et al. reported reaction rates approaching those of the tetrazine ligation.[86] Azides first react with oxanorbornadienes via 1,3-dipolar cycloaddition, followed by elimination of furan via a retro-Diels-Alder reaction to afford stable trizole linkages, similar to SPAAC and CuAAC.[86]
An additional class of bio-orthogonal reactions that may find applicability in hydrogel crosslinking is hetero-Diels-Alder reactions. Like traditional DA reactions, hetero-DA reactions proceed via a concerted pericyclic reaction mechanism that does not require external catalysis, generates no byproducts, and is accelerated in water due to hydrophobic interactions. In contrast to traditional DA reactions, hetero-DA reactions can possess significantly higher reaction rates. Glassner et al. employed hetero-DA cycloadditions between cyclopentadiene-functionalized PEG and dithioester-terminated poly(2-hydroxyethyl acrylate) or poly(glucopyranosyl acrylate) to form block co-polymers under aqueous conditions at ambient temperature.[87] The aqueous compatibility of this reaction is promising for hydrogel development, although the reaction timescales for this reaction pair are on the order of hours.[87] However, a recent report by Absil et al. demonstrated rapid crosslinking of microgels prepared from cyclopentadiene-functionalized polymers and a 4,4′-(4,4′-diphenylmethylene)-bis-(1,2,4-triazoline-3,5-dione) (bis-PTAD) crosslinker.[88] While the improved reaction rate is rapid enough for cell-encapsulating hydrogels, the reactivity of PTADs toward tyrosines and the potential for PTAD decomposition into amine-reactive isocyanates would limit the bio-orthogonality of this reaction pair.[88] An alterative hetero-DA pair is 2-napthoquinone-3-methide and a polarized olefin.[89] The reactive 2-napthoquinone-3-methide is generated in situ by photodehydration of a 3-(hydroxymethyl)-2-naphthol moiety.[89] The rapid reaction rates would facilitate efficient crosslinking, but the UV irradiation required to generate the reactive species may damage cells.[4] Shifting to visible light-mediated photodehydration or developing alternative chemistries to generate the reactive species would expand the potential utility of this reaction.
2.7. Supramolecular assemblies as bio-orthogonal approaches to physically-crosslinked hydrogels
The concept of bio-orthogonalilty has traditionally been applied to covalent ligation reactions, due to its first applications in cell surface glycan and metabolic labeling. In these applications, a permanent, covalent bond serves as a reliable reporter of specific chemical functionality. Many approaches to hydrogel crosslinking also rely on covalent bonds to produce mechanically robust gels. However, hydrogels crosslinked through non-covalent, physical interactions also comprise an important class of hydrogel-based materials with applications in engineered ECMs. The reversible nature of the crosslinks in physical hydrogels has been exploited to develop platforms for cell injection,[90, 91] bioprinting,[92, 93] and tuning of viscoelasticity to influence cellular phenotype.[94] The mechanisms for physical crosslinking can also be assessed on their bio-orthogonality, i.e. how likely the crosslinking pairs are to have off-target interactions with biological entities. Many of the common physical hydrogel networks are crosslinked by ionic interactions (such as with calcium ions that can interact with natural cellular signaling pathways or with transition metals that can be cytotoxic) or are comprised of protein or peptide components (which can be degraded by cell-secreted proteases). However, a subset of supramolecular materials that form physical hydrogels can be considered to crosslink via bio-orthogonal mechanisms. We briefly highlight two classes of supramolecular materials, guest-host hydrogels and polymer-nanoparticle hydrogels, and direct the reader to recent reviews on the topic for more details.[95, 96]
Guest-host hydrogels form due to non-covalent association of guest molecules within the cavities of host molecules.[96] Common host molecules are macrocycles such as cyclodextrins and cucurbit[n]urils.[96] While cyclodextrins occur in biology, cucurbit[n]urils are synthetic in origin, providing a potential bio-orthogonal handle for physical crosslinking. Due to the physical nature of the interactions between hosts and guests, any non-specific associations would be transient and thus unlikely to have lasting detrimental impacts on the biological system of interest. While some guest molecules previously implemented in guest-host gel systems, such as cholesterol[97] and the amino acids phenylalanine and tryptophan,[98] are present in biological systems, non-biologically occurring guests, notably adamantane,[99–102] have also been used. In one early example, Kretschmann et al. reported the preparation of physically-crosslinked hydrogels from adamantane-functionalized N-isopropylacrylamide co-polymers and bifunctional β-cyclodextrin.[99] This approach was later extended to a two component polymeric system, with separately prepared adamantane- and cyclodextrin-containing polymers forming hydrogel networks upon mixing.[100] Similar systems have since used adamantane- or n-butane-functionalized polyacrylamide guest components and β-cyclodextrin-functionalized polyacrylamide hosts[101] or adamantane- and β-cyclodextrin-functionalized hyaluronic acids (HA)[102] to prepare physical hydrogels. Guest-host HA hydrogels have proven to be a highly versatile system, with applications in cell transplantation[91] and bioprinting.[93] Cucurbit[n]urils have also been employed as physical crosslinking moieties in guest-host gels. Appel et al. reported the formation of self-healing and stimuli-responsive polymer networks crosslinked by a ternary complex of cucurbit[8]uril, methyl viologen-functionalized polymers (first guest) and naphthoxy-functionalized polymers (second guest).[103] HA hydrogels prepared from cucurbit[6]uril- and diaminohexane-functionalized polymers have shown promise as engineered ECMs (Figure 5)[104] to direct the chondrogenesis of human MSCs[105] and to support immunomodulation by transgenic MSCs transplanted in a murine cancer model.[106]
Figure 5.

(A) Schematic depicting supramolecular hydrogel formation from cucurbit[6]uril (CB[6])- and polyamine (PA)-modified hyaluronic acid (HA). The resulting hydrogels can be further modified with cucurbit[6]uril-tagged small molecules, such as RGD cell-adhesion peptides. (B,C) Increased proliferation of encapsulated fibroblasts is observed in gels to which RGD-cucurbit[6]uril was tethered via guest-host interactions compared to unmodified controls. Blue: DAPI (nuclei). Scale bars: 50 μm. Reproduced with permission.[104] Copyright 2012, American Chemical Society.
Polymer-nanoparticle gels comprise a second class of candidate supramolecular materials for bio-orthogonal, physically-crosslinked hydrogels. While various formulations of polymer-nanoparticle composites can have potential biomedical applications as engineered ECMs,[107, 108] here we focus specifically on supramolecular assemblies arising from physical adsorption of polymers onto nanoparticle surfaces. The physical gelation of poly(ethylene oxide) and Laponite clay nanoparticles has been previously studied for the unique rheological properties of the system.[107] PEO-Laponite composite gels have also been investigated as potential drug delivery systems[109] and cell culture substrates.[110] More recent approaches have used clay nanosheets crosslinked by dendrimeric PEGs[111] and polymeric nanoparticles crosslinked by biologically-derived polymers such as cellulose and HA.[112] Further studies are necessary to demonstrate utility of these generally biocompatible materials as 3D cell culture platforms.
In addition to guest-host and polymer-nanoparticle interactions, supramolecular assembly mechanisms inspired by biology, including protein- and DNA-based self-assembly, have also been employed to prepare hydrogels. As these systems make use of protein and DNA components that are inherently reactive with biomolecules such as proteases and nucleases, crosslinking of these hydrogels is less bio-orthogonal than guest-host and polymer-nanoparticle hydrogels. For a detailed discussion of DNA- and protein-based self-assembling systems, the reader is referred to recent review articles.[113]
3. Bio-orthogonal chemistries in the design of dynamic materials
The native ECM is highly dynamic, changing its biochemical and mechanical properties throughout development and aging and as a result of disease.[22] Recapitulating this dynamism in engineered ECM materials may facilitate advancements in regenerative medicine, for instance, by providing temporal control over cues for developmental processes such as stem cell differentiation and vasculogenesis. Dynamic ECM materials may also provide improved in vitro platforms to model disease progression, such as the stiffening of the ECM in tumor microenvironments.[114] Mechanisms to modify engineered ECMs should ideally be bio-orthogonal to ensure that only the altered matrix property (stiffness, cell-adhesive ligand density, etc.), and not any off-target effect of the modification chemistry, contributes to changes in cellular phenotype. This section describes strategies for two distinct modes of dynamic ECM modification: ligation reactions and bond cleavage reactions.
3.1. Adding functionality with bio-orthogonal ligations
Bio-orthogonal ligation reactions are promising tools to dynamically add functionality to engineered ECMs in the presence of live cells. Such reactions can facilitate the addition of cell-adhesive sites or increase the stiffness of the matrix.
3.1.1. Photo-mediated ligations
Photochemistry has proven to be a versatile tool for controlling the spatial and temporal presentation of biochemical and mechanical matrix properties. The first applications of photochemical regulation of ligation reactions in an engineered ECM context made use of photocaged reactive groups to selectively localize binding of bioactive factors to hydrogel scaffolds. Luo and Shoichet modified agarose hydrogels with S-2-nitrobenzyl-cysteine as a photocaged thiol.[115] Upon UV irradiation, the 2-nitrobenzyl photoactive group is released, leaving a free thiol. The resulting thiols were used to pattern maleimide-modified, cell-adhesive RGD peptides or full length proteins.[115, 116] The reliance on UV light and thiols limit the bio-orthogonality of this specific approach; however, advancements in photocaging groups has reduced the dependence on UV light. For instance, 6-bromo-7-hydroxy coumarin was developed as a two-photon-labile photocage,[117] permitting cleavage by a two-photon IR laser, substantially increasing the 3D resolution of the pattern while decreasing the potential for phototoxicity caused by UV irradiation. Wosnick and Shoichet replaced the 2-nitrobenzyl photocage with a 6-bromo-7-hydroxy coumarin photocage in the agarose gel system, which permitted selective 3D patterning of thiol-reactive molecules within hydrogels.[118] This system was later used to pattern vascular endothelial growth factor (VEGF) to direct stem cell differentiation[119] and to guide endothelial cell migration.[120] These systems still ultimately employed non-bio-orthogonal, thiol-reactive chemistries for ligation. To improve the selectivity of conjugation, recently 2-nitrobenzyl moieties have been used to photocage alkoxyamines.[34, 121, 122] Alkoxyamines react with aldehydes via oxime ligation to form stable covalent linkages under physiological conditions. The photomediated oxime ligation has been used to spatially and temporally control presentation of proteins[34, 122] and hydrogel stiffness.[122]
Instead of using photocaging moieties to mask and selectively reveal reactive groups, photocatalyzed ligation reactions have also been used to dynamically modify engineered ECM materials. DeForest et al. prepared SPAAC-crosslinked PEG hydrogels with pendant vinyl groups to permit photo-mediated thiol-ene reactions.[25] This system facilitated spatial control over cell spreading by localizing the covalent attachment of cell-adhesive RGD ligands to defined patterns.[25] Similar to photocaging approaches, the bio-orthogonality of this approach was limited by the use of UV light and thiols, in addition to the necessity for potentially cytotoxic photoinitiators. By changing photoinitiators, later studies were able to use visible light to mediate peptide conjugation.[123, 124] Photoinitiated reactions have also been employed to add crosslinks to hydrogel networks. Khetan et al. developed hyaluronic acid hydrogels with dual crosslinking to permit dynamic crosslinking in the presence of encapsulated cells.[125] Hydrogel networks were initially crosslinked with bifunctional, cell-degradable peptides to encapsulate cells, and then exposure to UV-light in the presence of a photoinitiator facilitated radical polymerization of pendant acrylate groups.[125] The additional crosslinking step was used to prevent degradation of the hydrogels by encapsulated cells, thereby blocking cell spreading in homogeneous[125] and patterned hydrogels.[126] The same approach was used to regulate MSC differentiation by blocking degradation-mediated generation of cellular traction.[127] Increasing crosslinking by photoinitiated radical polymerization in the acrylated-HA system also resulted in a stiffening of the material, which was used to probe the temporal effects of matrix mechanics on MSC differentiation.[128] Similar to other photo-mediated techniques, these crosslinking chemistries make use of UV light and photoinitiators. The radicals generated during polymerization can also have off-target reactions and negatively impact cellular function.
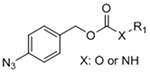
To overcome chemical cross-reactivity with biological functional groups, future approaches for photo-mediated ligation can utilize photo-activatable, bio-orthogonal reaction pairs (Table 2). Similar to photocaging strategies applied to reactive nucleophiles like thiols and alkoxyamines,[34, 115] one member of the bio-orthogonal reaction pair is initially masked and only released upon exposure to light. This approach has the potential to provide similar spatial and temporal control over biochemical and mechanical signals as those techniques discussed above. For instance, Poloukhtine et al. developed a photocaged strained cyclooctyne that can participate in SPAAC reactions after uncaging.[129] A cyclopropenone was used to mask the C-C triple bond in a DBCO molecule. UV irradiation leads to decarbonylation and generation of the strained alkyne that can react with azides via SPAAC.[129] Photocaged Staudinger ligation strategies have also been implemented by masking the reactive phosphene with a 2-nitrobenzyl group that can be removed by exposure to UV light.[130] In addition to unmasking relatively stable reaction partners, exposure to light can be used to generate unstable reactive groups in a spatially- and temporally-defined manner to participate in bio-orthogonal ligation reactions. UV irradiation of tetrazoles can yield nitrile imines for 1,3-dipolar cycloaddition reactions.[82] 2-Napthoquinone-3-methides generated by photodehydration of 3-(hydroxymethyl)-2-naphthol can participate in hetero-DA reactions with vinyl ethers to afford bio-orthogonally ligated products.[89] The use of potentially-cytotoxic UV-light is a significant drawback to all of these photocaged bio-orthogonal reactive groups. While the actual ligation reactions are bio-orthogonal, the UV irradiation used to uncage the reactive moieties may be mutagenic,[4] limiting the overall bio-orthogonality to these approaches. In a noteworthy improvement from these UV-mediated approaches, Zhang et al. recently reported a red-light-activatable tetrazine ligation.[131] Dihydrotetrazine is oxidized to tetrazine by exposure to 660 nm light in the presence of methylene blue as a photocatalyst.[131] The resulting tetrazines are then free to undergo IED-DA reactions with strained alkenes. Truong et al. employed this chemistry to prepare PEG hydrogels with temporal control over mechanical properties (Figure 6).[132] This crosslinking scheme permitted encapsulation of MSCs with high viability, indicating potential for dynamic modulation of the cellular microenvironment.[132] The visible light-activated tetrazine ligation can serve as a model for future development of fully bio-orthogonal, light-mediated ligation reactions.
Table 2.
Potential photocaged bio-orthogonal reactive groups for use in dynamic materials.
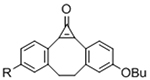
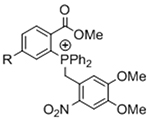
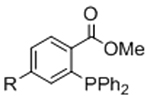
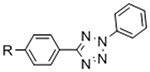
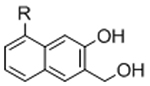
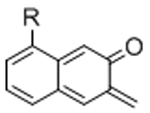
Figure 6.

(A) Schematic depicting the red light activation of a PEG-dihydrotetrazine precursor to PEG-tetrazine in the presence of a methylene blue photosensitizer to participate in bio-orthogonal IED-DA crosslinking with PEG-norbornene. (B) Upon irradiation with red light, the hydrogel crosslinks rapidly, reaching its plateau storage modulus within minutes. (C) Human MSCs encapsulated in photoactivated tetrazine hydrogels remain highly viable 24 hours post crosslinking, as assessed via a live/dead cytotoxicity assay. Green: live, Red: dead. Scale bar: 50 μm. Reproduced with permission.[132] Copyright 2017, American Chemical Society.
3.1.2. Enzyme-mediated ligation
Nature has evolved its own system of orthogonal reactivity through enzyme-substrate specificity. Careful choice of enzymes and substrates can permit selective modification of engineered ECMs without substantially altering the biology of the system being studied, providing a pseudo-bio-orthogonal approach to dynamically modulate matrix properties. The earliest work using enzyme-mediated coupling reactions to engineer hydrogel substrates employed transglutaminases to catalyze isopeptide bond formation between two peptide substrates.[133, 134] Optimization of the peptide substrates yielded robust gelation of hydrogels suitable for cell culture.[134, 135] Mosiewicz et al. combined transglutaminase-mediated ligation with photocaging strategies to facilitate dynamic pattern formation of biomolecules in PEG hydrogels.[136] A 2-nitrobenzyl photocage was used to mask the reactive lysine of a hydrogel-bound peptide substrate. Following UV irradiation, biomolecules with a partner glutamate-containing peptide reacted with the exposed lysine residue, catalyzed by transglutaminase.[136] This system was used to selectively pattern RGD cell-adhesive peptides to regulate MSC invasion in 3D gels.[136]
To achieve additional substrate specificity in the context of mammalian cell culture systems, others have turned to enzymes derived from bacteria. Zakeri et al. exploited the natural isopeptide bond formation within a bacterial adhesin to engineer the SpyTag-SpyCatcher system.[137] The short SpyTag peptide spontaneously associates with the SpyCatcher domain and forms a covalent bond between an aspartic acid residue on SpyTag and a lysine residue on SpyCatcher.[137] Sun et al. prepared recombinant elastin-like protein hydrogels crosslinked using genetically-encoded SpyTag and SpyCatcher reaction partners.[138] The resulting hydrogels supported the phenotypic maintenance of fibroblasts and embryonic stem cells in culture.[138] Following the introduction of the SpyTag/SpyCatcher reaction pair, additional protein pairs have been investigated for enzymatic ligation via isopeptide bond formation. Notably, Veggiani et al. identified a protein pair that reacts orthogonally to SpyTag/SpyCatcher, which the authors named SnoopTag/SnoopCatcher.[139] This orthogonality was exploited to conjugate two different antigens to opposite sides of nanoparticles for dual immunization vaccines.[140] Similar strategies could be employed in the future to control the temporal presentation of biaoctive signals from hydrogels by sequentially reacting SpyTag/SpyCatcher- and SnoopTag/SnoopCatcher-functionalized biomolecules.
In addition to isopeptide bond formation, bacterial enzymes have been employed for traditional peptide bond formation. Cambria et al. used Sortase A, another bacterial enzyme, to dynamically tether epidermal growth factor (EGF) to PEG hydrogels.[141] Sortase A mediates the ligation of a LPXTG peptide motif and a triglycine-containing polypetide.[142] PEG hydrogels with pedant LPRTG peptides were conjugated with EGF bearing an N-terminal triglycine sequence via sortase-mediated ligation.[141] EGF tethering enhanced the proliferation of epithelial cells cultured on the hydrogels.[141] The orthogonal chemical nature of sortase-mediated ligation has been demonstrated in vivo using an evolved Sortase A to dynamically-conjugate LPETG peptides with fluorescent probes onto the surface of a catheter in mice.[143]
3.1.3. Direct covalent ligation
The field of bio-orthogonal chemistry arose out of the desire to selectively label particular cellular products among the various chemical functionalities present in living systems, resulting in the development of chemical reactions that can occur even within live organisms.[144] Drawing inspiration from these early studies, bio-orthogonal ligation reactions can be directly applied in vitro or in vivo to dynamically alter the properties of hydrogel matrices without exposure to light or other external triggers. Utilizing excess azides present in SPAAC-crosslinked ELP hydrogels, we previously demonstrated selective hydrogel functionalization in the presence of live cells in serum-containing culture medium (Figure 7A,B).[35] As a model system, a BCN-modified fluorophore was selectively conjugated to an ELP hydrogel with encapsulated MSCs.[35] Brudno et al. used both SPAAC and tetrazine ligation to covalently modify alginate hydrogels in vivo.[145] Azide- or tetrazine-modified alginate gels were implanted in the hind limbs of mice, and the mice were then injected intravenously with DBCO- or TCO-modified fluorescent dye, respectively (Figure 7C,D).[145] Twenty-four hours post injection, the dyes localized to the hydrogels. Furthermore, the hydrogels could be sequentially loaded with dye over multiple injections, and SPAAC and tetrazine ligation gels could be independently targeted with different dyes within the same animal.[145] Oneto et al. later used a similar tetrazine-modified alginate hydrogel system to localize delivery of doxorubicin with a cleavable TCO moiety to soft tissue sarcomas in mice.[146] Animals treated with the localized drug exhibited improved reduction in tumor size and decreased systemic toxicity than animals treated with standard soluble doxorubicin.[146]
Figure 7.

(A) Schematic depicting the bio-orthogonal functionalization of SPAAC-crosslinked elastin-like protein hydrogels with BCN-modified fluorophores. (B) Hydrogels with live, encapsulated human MSCs were functionalized with a BCN-modified fluorescent dye (CF640R) in serum-containing medium, demonstrating selective incorporation of the BCN-bearing dye and not an amine-bearing control dye. A and B reproduced with permission.[35] Copyright 2016, John Wiley and Sons. (C,D) Alginate hydrogels functionalized with azide or tetrazine were implanted intramuscularly or into the mammary fat pads of mice, respectively. Intravenous injection of TCO-Cy5 and DBCO-Cy7 dyes resulted in localization to the tetrazine-alginate and azide-alginate hydrogels, respectively. C and D reproduced with permission.[145] Copyright 2015, John Wiley and Sons.
3.1.4. Supramolecular interactions
In addition to covalent ligation, physical interactions can also be used to control the presentation of bioactive factors from engineered ECMs. While traditional bio-orthogonal approaches have focused on the formation of stable covalent bonds, non-covalent interactions can achieve similarly selective material functionalization in the presence of live cells. As discussed in Section 2.7, supramolecular host-guest assemblies can be thought of as bio-orthogonal, physical binding interactions. The inherent reversibility of these supramolecular assemblies has been exploited to dynamically modulate the presentation of bioactive cues. Several studies have demonstrated the ability of host-guest complexes to localize peptides and proteins,[147] including cell-adhesive RGD peptides[148] and bone morphogenetic protein-6 (BMP-6).[149] In the context of cell-encapsulating hydrogels, Park et al. developed cucurbit[6]uril-diaminohexane crosslinked hyaluronic acid gels to which cucurbit[6]uril-conjugated RGD peptides could be dynamically linked via excess diaminohexane groups on the HA backbone (Figure 5).[104] Addition of RGD increased the spreading and proliferation of encapsulated fibroblasts.[104] Boekhoven et al. used alginate hydrogel surfaces with conjugated β-cyclodextrin as host molecules to dynamically regulate RGD presentation.[150] The authors took advantage of differences in the association constants for two different guest molecules, naphthalene and adamantane, to competitively remove bound RGD from the hydrogel surfaces. Cells were initially able to spread on surfaces presenting naphthyl-RGD, but treatment with non-adhesive, adamantane-RGE displaced the RGD and resulted in decreased cell spreading.[150]
3.2. Bio-orthogonal bond cleavage reactions
While the classic examples of bio-orthogonal reactions consist of ligation reactions, such as 1,3-dipolar cycloadditions and Diels-Alder reactions, recent interest has turned to developing bio-orthogonal bond cleavage reactions.[151] Such reactions have been applied to regulate prodrug uncaging and control protein activity in living systems.[151] These reactions may permit further advancement in the design of dynamic, engineered ECMs, complementing existing ligation reactions to facilitate removal of functionality, not just addition of functionality, on demand. Several elegant examples using photochemical and enzymatic techniques have demonstrated the power of bond cleavage reactions to regulate cellular behavior in hydrogel-based systems, and newer metal- and small molecule-catalyzed reactions have the potential to do the same.
3.2.1. Photo-mediated cleavage reactions
The first advancements toward bio-orthogonal bond cleavage reactions made use of photolabile groups. Various photocleavable moieties have been employed in many different applications. However, for the scope of this review, we will focus on the use of photocleavable groups to control the biochemical and mechanical properties of engineered ECM-mimicking hydrogels. For additional discussion of photocleavable groups, the reader is directed to a comprehensive review of the topic.[152]
Before discussing the application of photolabile groups to engineered ECMs, it is important to note that most of the photoreactive chemistries employed to date do not fit a strict definition of bio-orthogonality. 2-Nitrobenzyl groups are the most commonly used photocleavable groups in biomaterials applications. These groups often require the use of UV light to initiate cleavage, which can damage biological macromolecules such as DNA and harm living cells.[4, 151] Some variants of the 2-nitrobenzyl group are susceptible to 2-photon cleavage, which reduces concerns of light-induced cytotoxicity. However, nitrobenzyl groups still can be reduced by cellular nitroreductase enzymes, resulting in premature bond cleavage.[151, 153] Nevertheless, photolabile groups have proven to be an efficacious bio-selective strategy for spatially and temporally controlling the properties of engineered hydrogel ECMs.
The use of protected cell-adhesion ligands is one application of photocaging strategies to dynamically regulate matrix biochemistry. The del Campo and Tatsu labs independently developed two different photocaged RGD cell-adhesion peptides using the 2-nitrobenzyl group.[154, 155] Del Campo and colleagues used the 2-nitrobenzyl as a protecting group for the aspartic acid required for recognition of the peptide by cell surface integrins.[154] Tatsu and colleagues installed the 2-nitrobenzyl on the amide nitrogen between the arginine and glycine residues.[155] In both studies, UV irradiation of surfaces with conjugated, photocaged RGD peptide resulted in spatial and temporal control of cell adhesion and spreading.[154, 155] Lee et al. later applied photocaged RGD peptides to control the presentation of cell-adhesive ligands by implanted hydrogels in vivo. Uncaging the RGD ligand promoted cell adhesion and vascularization of the hydrogel constructs.[156] These photocaging systems have been applied to study various biological processes, including the effect of adhesive ligand presentation on myoblast differentiation,[157] cellular migration,[158] and the early stages of integrin-mediated adhesion to surfaces.[159]
Nitrobenzyl photolabile groups have also been employed for the converse purpose, that is, the selective removal of components from hydrogel constructs. Kloxin et al. inserted a nitrobenzyl group in the linker connecting an RGD peptide to a PEG hydrogel, such that UV irradiation would result in release of the peptide from the hydrogel.[160] The authors demonstrated that light-triggered release of RGD after 10 days of 3D culture enhanced the chondrogenic differentiation of MSCs.[160] DeForest and Anseth expanded this strategy to reversibly pattern RGD peptides in hydrogels.[124] Peptides were spatially patterned by photoactivated thiol-ene reactions using visible light and then selectively removed by UV irradiation to cleave the nitrobenzyl moiety, allowing dynamic patterning of cell adhesion.[124] DeForest and Tirrell generalized the approach to reversibly pattern proteins using photocaged alkoxyamines to dynamically control protein ligation and nitrobenzyl linkers to mediate protein release.[34]
In addition to regulating access to cell-adhesive ligands, photo-mediated bond cleavage reactions have been used to dynamically regulate hydrogel architecture and mechanics. Kloxin et al. also installed photolabile nitrobenzyl groups in the PEG backbone of their hydrogels.[160] Exposure to UV light resulted in cleavage of crosslinks and hydrogel degradation that could be spatially and temporally controlled by 2-photon patterning.[160] DeForest et al. later demonstrated that photo-mediated hydrogel degradation can direct cell spreading and migration in 3D hydrogels.[123] This technique has since been used to pattern neural progenitor cell outgrowth[161] and to create vascular networks within 3D hydrogel constructs.[162] Beyond bulk material degradation, photo-mediated cleavage of hydrogel crosslinks can temporally control the mechanical properties of the matrix. Yang et al. used PEG hydrogels that could be dynamically softened by exposure to light to elucidate the role of mechanical memory in biasing MSC differentiation.[163]
3.2.2. Enzyme-mediated cleavage reactions
Just as enzymes have been used to catalyze specific ligation reactions, enzymes have also been employed to selectively cleave bonds. In the context of engineered ECMs, crosslinks susceptible to degradation by cell-secreted proteases have become a common strategy to permit cell spreading, migration, and proliferation in 3D hydrogels.[164] More recent developments have seen the emergence of enzymatic strategies to enable user-directed matrix degradation through use of enzymes that are not typically present in the system being studied. Valdez et al. designed a PEG hydrogel that was crosslinked by modular peptides containing both a cell-secreted protease degradation site and a sortase recognition site.[165] Encapsulated endometrial cultures were able to remodel the matrix as necessary, and after the desired culture duration, the authors could fully degrade the gels by treatment with sortase and a glycine tripeptide to recover the cells and secreted proteins.[165] Due to the low frequency of the LPXTG sortase recognition sequence in the mammalian proteome, sortase-mediated cleavage was highly selective, leaving 26 of 27 tested cell-secreted proteins unmodified.[165] Sortase treatment has also been used to reversibly cleave EGF from hydrogel-based cell culture platforms.[141]
While enzymatic bond-cleavage reactions in a hydrogel context have been limited to the use of sortase, other fields have worked to identify or evolve specific enzyme-substrate pairs to achieve bio-orthogonal bond cleavage reactions. Tian et al. developed a family of fluorogenic esterase substrates to identify an ester structure that was not susceptible to hydrolysis by endogenous esterases in various animal cell types.[166] Once a target was identified, the authors screened exogenous enzymes and determined that a porcine liver esterase could selectively cleave their substrate.[166] Ritter et al. screened a library of cytochrome P450 mutants to identify enzymes that could selectively cleave propargyl and benzyl ether protecting groups.[167] Future work could seek to combine these enzyme-substrate pairs with synthetic hydrogels to develop novel, enzyme-responsive matrices.
3.2.3. Chemically-mediated cleavage reactions
Beyond photochemical uncaging and enzymatic catalysis, the use of more traditional chemical techniques for bond cleavage reactions is an area of active investigation.[151] While few examples have been applied to engineered ECMs, these techniques may be adapted to regulate matrix biochemistry and mechanics similar to the light- and enzyme-mediated techniques discussed above. The first developments toward bio-orthogonal, chemical triggering of bond cleavage made use of transition metal-catalyzed uncaging reactions.[151] Streu and Meggers reported the use of a ruthenium complex to catalyze the removal of an allylcarbamate protecting group, revealing a primary amine.[168] The reaction tolerated physiological conditions and could be performed in live cells.[168] Optimization of ligands in the ruthenium complex has increased the activity of the catalyst to permit efficient activation of prodrugs in mammalian cell culture.[169] Palladium-based strategies have also shown promise in bio-orthogonal bond cleavage reactions. Allylcarbamate deprotection can be achieved via heterogeneous catalysis using Pd0 microparticles within live cells.[170] Heterogeneous palladium catalysis can also be used to remove propargyl protecting groups on prodrugs in cellular environments.[171] Additionally, oxidized palladium species have been used to activate proteins within living cells[172] and remodel cell surface glycans.[173] In a recent study, gold nanoparticles embedded in a support resin were shown to catalyze cleavage of propargyl groups to activate prodrugs in cancer cell cultures.[174]
A potential concern with ruthenium- and palladium-mediated bond cleavage reactions is heavy metal-associated toxicity. Metallic palladium is known to be relatively inert,[175] so approaches employing heterogeneous catalysis with Pd0 are likely to have minimal cytotoxic effects. As soluble oxidized ruthenium and palladium species can exhibit significant toxicity,[175, 176] the ligands chosen for the metal catalysts in bond cleavage reactions should be selected to effectively bind the metal ions and limit off-target effects. Parallels can be drawn to the development of ruthenium complexes for targeted cancer chemotherapy.[177] Light- and pH-sensitive ligands can sequester the ruthenium ions, limiting systemic toxicity while allowing triggerable removal of the ligands in the tumor microenvironment to induce cytotoxicity.[178] The metal complexes used in the bond cleavage reactions discussed demonstrate minimal toxicity in typical cell culture environments,[151] suggesting these reactions may be sufficiently bio-orthogonal to enable dynamic modification of engineered ECMs in vitro. However, concerns about systemic toxicity and metabolized products must be addressed before using such strategies to modulate cellular microenvironments in vivo.
The use of organic small molecules in bio-orthogonal bond cleavage reactions is a young, but rapidly growing field. The first such cleavage reaction was reported by Versteegen et al. in which an IED-DA reaction ultimately results in scission of a carbamate linkage.[179] By positioning the carbamate linkage adjacent to the double bond of a TCO moiety, the dihydropyridazine product of tetrazine ligation can undergo rearrangement to form a pyridazine and expel carbon dioxide and an amine-terminated molecule.[179] This reaction has been applied to various systems,[180] including prodrug activation[179] and uncaging of proteins in live cells.[181] Using this bio-orthogonal bond cleavage reaction, Oneto et al. developed a hydrogel-based system for local activation of the anti-cancer drug doxorubicin (Figure 8).[146] The authors implanted tetrazine-modified alginate gels near tumors in a mouse model and systemically delivered a TCO-doxorubicin conjugate.[146] Doxorubicin was only released at the tumor site, increasing therapeutic efficacy and decreasing systemic toxicity.[146] A recent report has also demonstrated IED-DA-mediated cleavage of vinyl ethers to release alcohols, as opposed to the amines released from the TCO reaction scheme.[182]
Figure 8.

(A) Schematic depicting bio-orthogonal ligation of a drug to, followed by spontaneous release from, an alginate hydrogel functionalized with tetrazine. This system facilitated the localized activation of a doxorubicin prodrug to treat tumors in a mouse model. (B) Treatment with the hydrogel and doxorubicin prodrug resulted in significantly reduced tumor volume compared to systemic treatment with doxorubicin. (C) Localized release of doxorubicin via bio-orthogonal cleavage from the tetrazine-alginate hydrogel resulted in less non-specific toxicity than systemic doxorubicin treatment, as measured by weight change in the treated mice. Reproduced with permission.[146] Copyright 2016, American Chemical Society.
Other bio-orthogonal reactions have been developed more recently to mediate bond cleavage. Matikonda et al. reported a cleavage reaction initiated by the 1,3-dipolar cycloaddition of a p-aminobenzyloxycarbonyl azide and TCO.[183] The resulting triazole is unstable in aqueous environments and undergoes multiple rearrangements to ultimately eliminate an amine-bearing molecule.[183] Pawlak et al. used a traditional Staudinger reduction to unmask a lysine-containing antigen on the surface of dendritic cells to control T cell activation.[184] Finally, Kim and Bertozzi developed a novel, bio-orthogonal, bond cleavage system that uses N-oxides and boron-containing reagents.[185]
4. Conclusions and future directions
The chemical selectivity provided by bio-orthogonal reactions can enable careful manipulation of engineered ECMs to probe cell-matrix interactions and to provide signals that direct cell fate. Several different bio-orthogonal reactions have been employed to prepare covalently-crosslinked hydrogel platforms that support the culture of encapsulated cells. Advancements in bio-orthogonal reaction pairs have allowed for improved gelation kinetics in SPAAC- and IED-DA-crosslinked hydrogels, opening opportunities for applications in cell transplantation and bio-printing that require rapid gelation. Developments in supramolecular hydrogels represent potential bio-orthogonal, physically crosslinked systems with similar potential applications due to their shear-thinning properties.[186]
Translating bio-orthogonal gelation chemistries to these applications will require both optimized synthetic schemes for producing the hydrogel precursors at scale and additional in vivo studies to elucidate the longer tem effects of implanting materials crosslinked by bio-orthogonal reactions. As presented in Figure 3, progress has been made in minimizing the synthetic complexity of bio-orthogonal gel precursors. For instance, the development of BCN was motivated by a desire to decrease the number of synthetic steps and increase yields of strained cyclooctynes for SPAAC.[15] Additionally, the development of novel synthesis procedures, like continuous-flow UV-mediated isomerization in the synthesis of trans-cylcooctenes,[187] has resulted in improved yields. While bio-orthogonally crosslinked gels elicit minimal inflammation in vivo,[28, 29, 52, 53, 62] systemic effects of the degradation products of such materials should also be investigated. Previous reports indicate that phosphines used in Staudinger ligation[188] and alkynes used in 1,3-dipolar cycloadditions[189] may be metabolized by cytochrome P450 enzymes. The triazoles formed from 1,3-dipolar cycloadditions may also interact with cytochrome P450, as triazole derivatives have been reported as potential P450 inhibitors.[190] An additional concern when designing dynamic materials through bio-orthogonal chemistries for in vivo use is the stability of the reactive moities. For instance, many tetrazines exhibit poor serum stability,[191] but some methyl-terminated tetrazines have been reported with improved stability.[192] Highly-strained trans-cyclooctenes have been shown to undergo trans/cis isomerization in the presence of thiols,[55] suggesting these very reactive dienophiles may not be sufficiently stable for in vivo applications where thiols could result in inactivation of the functional group. To address such concerns, Taylor et al. recently developed a strained dioxolane-fused trans-cyclooctene with improved serum stability.[193] Continued optimization of bio-orthogonal reaction pairs may further expand the in vivo use of engineered ECMs utilizing these chemistries.
Beyond uses in hydrogel crosslinking, the highly specific nature of bio-orthogonal reactions makes them ideal mechanisms to dynamically modify engineered ECMs. Studies using light- and enzyme-induced changes in matrix biochemistry and mechanics have demonstrated that cells sense and respond to changes in their microenvironment. The development of bio-orthogonally tunable dynamic materials can permit enhanced selectivity in modulating matrix properties without concerns for potential off-target effects on the cells being manipulated. While a few studies have demonstrated the potential for traditional bio-orthogonal ligation reactions to dynamically alter hydrogel matrices in vitro and in vivo,[35, 145, 146] the possibilities to adapt bio-orthogonal ligation and bond cleavage reactions to alter matrix biochemistry and mechanics remain underexplored. Ultimately, engineered ECMs exploiting bio-orthogonal interactions may permit more sensitive studies of cell-matrix interactions and more robust therapeutic platforms for drug delivery and tissue engineering applications.
Table 3.
Potential bio-orthogonal bond cleavage reactions for dynamic materials.
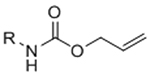
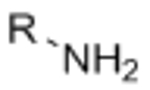
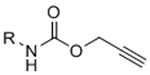
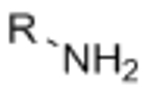
Acknowledgements
The authors acknowledge funding from the National Science Foundation (DMR 1508006), the California Institute for Regenerative Medicine (RT3–07948), and the National Institutes of Health (R21 EB020235, R21 HL138042, and U19 AI116484). C.M.M. was supported by an NIH NRSA pre-doctoral fellowship (F31 EB020502) and a Siebel Scholars fellowship.
Biographies

Christopher M. Madl obtained a B.A. and a M.S. in Biomedical Engineering from Harvard University in 2012 and a Ph.D. in Bioengineering from Stanford University in 2017. He is now a postdoctoral fellow in the Baxter Laboratory for Stem Cell Biology at Stanford Medical School. His research interests focus on the design of dynamic hydrogel platforms to study and regulate stem cell behavior.

Sarah C. Heilshorn is the Lee Otterson Faculty Scholar and Associate Professor in the Department of Materials Science & Engineering at Stanford University. She holds courtesy faculty appointments in the Departments of Chemical Engineering and Bioengineering and is a Bass University Fellow in Undergraduate Education. Her laboratory integrates concepts from materials engineering and protein science to design new, bioinspired materials for applications in disease modeling and regenerative medicine.
Contributor Information
Dr. Christopher M. Madl, Department of Bioengineering, Stanford University, Stanford, CA 94305, USA
Prof. Sarah C. Heilshorn, Department of Materials Science and Engineering, Stanford University, Stanford, CA 94305, USA, heilshorn@stanford.edu
References:
- [1].a) Guilak F, Cohen DM, Estes BT, Gimble JM, Liedtke W, Chen CS, Cell Stem Cell 2009, 5, 17; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang NF, Li S, Ann. Biomed. Eng 2011, 39, 1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Lee KY, Mooney DJ, Chem. Rev 2001, 101, 1869; [DOI] [PubMed] [Google Scholar]; b) Ratner BD, Bryant SJ, Annu. Rev. Biomed. Eng 2004, 6, 41. [DOI] [PubMed] [Google Scholar]
- [3].a) Sabnis A, Rahimi M, Chapman C, Nguyen KT, J. Biomed. Mater. Res., Part A 2009, 91A, 52; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lin H, Zhang D, Alexander PG, Yang G, Tan J, Cheng AW-M, Tuan RS, Biomaterials 2013, 34, 331; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dubbin K, Tabet A, Heilshorn SC, Biofabrication 2017, 9, 044102; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Grzelak A, Rychlik B, Bartosz G, Free Radical Biol. Med 2001, 30, 1418. [DOI] [PubMed] [Google Scholar]
- [4].Pattison DI, Davies MJ, in Cancer: Cell Structures, Carcinogens and Genomic Instability, Birkhäuser; Basel, Switzerland: 2006, 131. [Google Scholar]
- [5].Medina SH, Schneider JP, in Chemoselective and Bioorthogonal Ligation Reactions: Concepts and Applications, (Eds: Algar WR, Dawson PE, Medintz IL), Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany: 2017. [Google Scholar]
- [6].Sletten EM, Bertozzi CR, Acc. Chem. Res 2011, 44, 666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Saxon E, Bertozzi CR, Science 2000, 287, 2007. [DOI] [PubMed] [Google Scholar]
- [8].Kiick KL, Saxon E, Tirrell DA, Bertozzi CR, Proc. Natl. Acad. Sci. USA 2002, 99, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Prescher JA, Dube DH, Bertozzi CR, Nature 2004, 430, 873. [DOI] [PubMed] [Google Scholar]
- [10].Agard NJ, Prescher JA, Bertozzi CR, J. Am. Chem. Soc 2004, 126, 15046. [DOI] [PubMed] [Google Scholar]
- [11].Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR, Proc. Natl. Acad. Sci. USA 2007, 104, 16793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jewett JC, Sletten EM, Bertozzi CR, J. Am. Chem. Soc 2010, 132, 3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ning X, Guo J, Wolfert MA, Boons G-J, Angew. Chem. Int. Ed 2008, 120, 2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Debets MF, van Berkel SS, Schoffelen S, Rutjes FPJT, van Hest JCM, van Delft FL, Chem. Commun 2010, 46, 97. [DOI] [PubMed] [Google Scholar]
- [15].Dommerholt J, Schmidt S, Temming R, Hendriks LJA, Rutjes FPJT, van Hest JCM, Lefeber DJ, Friedl P, van Delft FL, Angew. Chem. Int. Ed 2010, 49, 9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kuzmin A, Poloukhtine A, Wolfert MA, Popik VV, Bioconjugate Chem. 2010, 21, 2076. [DOI] [PubMed] [Google Scholar]
- [17].Laughlin ST, Baskin JM, Amacher SL, Bertozzi CR, Science 2008, 320, 664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB, Angew. Chem. Int. Ed 2002, 41, 2596; [DOI] [PubMed] [Google Scholar]; b) Tornøe CW, Christensen C, Meldal M, J. Org. Chem 2002, 67, 3057. [DOI] [PubMed] [Google Scholar]
- [19].a) Hong V, Steinmetz NF, Manchester M, Finn MG, Bioconjugate Chem. 2010, 21, 1912; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) del Amo DS, Wang W, Jiang H, Besanceney C, Yan AC, Levy M, Liu Y, Marlow FL, Wu P, J. Am. Chem. Soc 2010, 132, 16893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kolb HC, Finn MG, Sharpless KB, Angew. Chem. Int. Ed 2001, 40, 2004. [DOI] [PubMed] [Google Scholar]
- [21].a) Nimmo CM, Shoichet MS, Bioconjugate Chem. 2011, 22, 2199; [DOI] [PubMed] [Google Scholar]; b) Azagarsamy MA, Anseth KS, ACS Macro Lett. 2013, 2, 5; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jiang Y, Chen J, Deng C, Suuronen EJ, Zhong Z, Biomaterials 2014, 35, 4969. [DOI] [PubMed] [Google Scholar]
- [22].Lu P, Takai K, Weaver VM, Werb Z, Cold Spring Harbor Perspect. Biol 2011, 3, a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a) Ossipov DA, Hilborn J, Macromolecules 2006, 39, 1709; [Google Scholar]; b) Malkoch M, Vestberg R, Gupta N, Mespouille L, Dubois P, Mason AF, Hedrick JL, Liao Q, Frank CW, Kingsbury K, Hawker CJ, Chem. Commun 2006, 14, 2774. [DOI] [PubMed] [Google Scholar]
- [24].a) Testera AM, Girotti A, de Torre IG, Quintanilla L, Santos M, Alonsso M, Rodrguez-Cabello JC, J. Mater. Sci.: Mater. Med 2015, 26, 105; [DOI] [PubMed] [Google Scholar]; b) Crescenzi V, Cornelio L, Di Meo C, Nardecchia S, Lamanna R, Biomacromolecules 2007, 8, 1844; [DOI] [PubMed] [Google Scholar]; c) Huerta-Angeles G, Němcová M, Příkopová E, Šmejkalová D, Pravda M, Kučera L, Velebný V, Carbohydr. Polym 2012, 90, 1704. [DOI] [PubMed] [Google Scholar]
- [25].DeForest CA, Polizzotti BD, Anseth KS, Nat. Mater 2009, 8, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA, Proc. Natl. Acad. Sci. USA 2003, 100, 5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].DeForest CA, Sims EA, Anseth KS, Chem. Mater 2010, 22, 4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Takahashi A, Suzuki Y, Suhara T, Omichi K, Shimizu A, Hasegawa K, Kokudo N, Ohta S, Ito T, Biomacromolecules 2013, 14, 3581. [DOI] [PubMed] [Google Scholar]
- [29].Jiang H, Qin S, Dong H, Lei Q, Su X, Zhuo R, Zhong Z, Soft Matter 2015, 11. [DOI] [PubMed] [Google Scholar]
- [30].Fu S, Dong H, Deng X, Zhuo R, Zhong Z, Carbohydr. Polym 2017, 169, 332. [DOI] [PubMed] [Google Scholar]
- [31].Hermann CD, Wilson DS, Lawrence KA, Ning X, Olivares-Navarrete R, Williams JK, Guldberg RE, Murthy N, Schwartz Z, Boyan BD, Biomaterials 2014, 35, 9698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang X, Li Z, TingShi P. Zhao, An K, Lin C, Liu H, Mater. Sci. Eng., C 2017, 73, 21. [DOI] [PubMed] [Google Scholar]
- [33].Zheng J, Callahan LAS, Hao J, Guo K, Wesdemiotis C, Weiss RA, Becker ML, ACS Macro Lett. 2012, 1, 1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].DeForest CA, Tirrell DA, Nat. Mater 2015, 14, 523. [DOI] [PubMed] [Google Scholar]
- [35].Madl CM, Katz LM, Heilshorn SC, Adv. Funct. Mater 2016, 26, 3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Madl CM, Heilshorn SC, Bioconjugate Chem. 2017, 28, 724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].a) de Torre IG, Santos M, Quintanilla L, Testera A, Alonso M, Rodríguez-Cabello JC, Acta Biomater. 2014, 10, 2495; [DOI] [PubMed] [Google Scholar]; b) de Torre IG, Wolf F, Santos M, Rongen L, Alonso M, Jockenhoevel S, Rodríguez-Cabello JC, Mela P, Acta Biomater. 2015, 12, 146. [DOI] [PubMed] [Google Scholar]
- [38].Straley KS, Heilshorn SC, Soft Matter 2009, 5, 114. [Google Scholar]
- [39].Chung C, Lampe KJ, Heilshorn SC, Biomacromolecules 2012, 13, 3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].van Geel R, Pruijn GJM, van Delft FL, Boelens WC, Bioconjugate Chem. 2012, 23, 392. [DOI] [PubMed] [Google Scholar]
- [41].Tian H, Sakmar TP, Huber T, Chem. Commun 2016, 52, 5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Saito F, Noda H, Bode JW, ACS Chem. Biol 2015, 10, 1026. [DOI] [PubMed] [Google Scholar]
- [43].Li Z, Seo TS, Ju J, Tetrahedron Lett. 2004, 45, 3143. [Google Scholar]
- [44].Truong VX, Ablett MP, Gilbert HTJ, Bowen J, Richardson SM, Hoyland JA, Dove AP, Biomater. Sci 2014, 2, 167. [DOI] [PubMed] [Google Scholar]
- [45].Blackman ML, Royzen M, Fox JM, J. Am. Chem. Soc 2008, 130, 13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Patterson DM, Nazarova LA, Prescher JA, ACS Chem. Biol 2014, 9, 592. [DOI] [PubMed] [Google Scholar]
- [47].Devaraj NK, Weissleder R, Hilderbrand SA, Bioconjugate Chem. 2008, 19, 2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pipkorn R, Waldeck W, Didinger B, Koch M, Mueller G, Wiessler M, Braun K, J. Pept. Sci 2009, 15, 235. [DOI] [PubMed] [Google Scholar]
- [49].a) Yang J, Šečkutė J, Cole CM, Devaraj NK, Angew. Chem. Int. Ed 2012, 51, 7476; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Patterson DM, Nazarova LA, Xie B, Kamber DN, Prescher JA, J. Am. Chem. Soc 2012, 134, 18638. [DOI] [PubMed] [Google Scholar]
- [50].Zhou H, Woo J, Cok AM, Wang M, Olsen BD, Johnson JA, Proc. Natl. Acad. Sci. USA 2012, 109, 19119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Alge DL, Azagarsamy MA, Donohue DF, Anseth KS, Biomacromolecules 2013, 14, 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Desai RM, Koshy ST, Hilderbrand SA, Mooney DJ, Joshi NS, Biomaterials 2015, 50, 30. [DOI] [PubMed] [Google Scholar]
- [53].Koshy ST, Desai RM, Joly P, Li J, Bagrodia RK, Lewin SA, Joshi NS, Mooney DJ, Adv. Healthcare Mater 2016, 5, 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Truong VX, Ablett MP, Richardson SM, Hoyland JA, Dove AP, J. Am. Chem. Soc 2015, 137, 1618. [DOI] [PubMed] [Google Scholar]
- [55].Taylor MT, Blackman ML, Dmitrenko O, Fox JM, J. Am. Chem. Soc 2011, 133, 9646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang H, Dicker KT, Xu X, Jia X, Fox JM, ACS Macro Lett. 2014, 3, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Rideout DC, Breslow R, J. Am. Chem. Soc 1980, 102, 7816. [Google Scholar]
- [58].Wei H-L, Yang Z, Zheng L-M, Shen Y-M, Polymer 2009, 50. [Google Scholar]
- [59].Wei H-L, Yang Z, Chen Y, Chu H-J, Zhu J, Li Z-C, Eur. Polym. J 2010, 46, 1032. [Google Scholar]
- [60].Nimmo CM, Owen SC, Shoichet MS, Biomacromolecules 2011, 12, 824. [DOI] [PubMed] [Google Scholar]
- [61].Owen SC, Fisher SA, Tam RY, Nimmo CM, Shoichet MS, Langmuir 2013, 29, 7393–7400. [DOI] [PubMed] [Google Scholar]
- [62].Führmann T, Obermeyer J, Tator CH, Shoichet MS, Methods 2015, 84, 60. [DOI] [PubMed] [Google Scholar]
- [63].Yu F, Cao X, Zeng L, Zhang Q, Chen X, Carbohydr. Polym 2013, 97, 188. [DOI] [PubMed] [Google Scholar]
- [64].Yu F, Cao X, Li Y, Zeng L, Zhu J, Wang G, Chen X, Polym. Chem 2014, 5, 5116. [Google Scholar]
- [65].Montiel-Herrera M, Gandini A, Goycool FM, Jacobsen NE, Lizardi-Mendoza J, Recillas-Mota M, Argüelles-Monal WM, Carbohydr. Polym 2015, 128, 220. [DOI] [PubMed] [Google Scholar]
- [66].Tan H, Rubin JP, Marra KG, Macromol. Rapid Commun 2011, 32, 905. [DOI] [PubMed] [Google Scholar]
- [67].Bai X, Lü S, Cao Z, Ni B, Wang X, Ning P, Ma D, Wei H, Liu M, Carbohydr. Polym 2017, 166, 123. [DOI] [PubMed] [Google Scholar]
- [68].Kirchhof S, Gregoritza M, Messmann V, Hammer N, Goepferich AM, Brandl FP, Eur. J. Pharm. Biopharm 2015, 96, 217. [DOI] [PubMed] [Google Scholar]
- [69].a) Koehler KC, Anseth KS, Bowman CN, Biomacromolecules 2013, 14, 538; [DOI] [PubMed] [Google Scholar]; b) Koehler KC, Alge DL, Anseth KS, Bowman CN, Biomaterials 2013, 34, 4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Fan M, Ma Y, Zhang Z, Mao J, Tan H, Hu X, Mater. Sci. Eng., C 2015, 56, 311. [DOI] [PubMed] [Google Scholar]
- [71].Kirchhof S, Brandl FP, Hammer N, Goepferich AM, J. Mater. Chem. B 2013, 1, 4855. [DOI] [PubMed] [Google Scholar]
- [72].Boul PJ, Reutenauer P, Lehn J-M, Org. Lett 2005, 7, 15. [DOI] [PubMed] [Google Scholar]
- [73].Reutenauer P, Buhler E, Boul PJ, Candau SJ, Lehn J-M, Chem. - Eur. J 2009, 15, 1893. [DOI] [PubMed] [Google Scholar]
- [74].Wei Z, Yang JH, Du XJ, Xu F, Zrinyi M, Osada Y, Li F, Chen YM, Macromol. Rapid Commun 2013, 34, 1464. [DOI] [PubMed] [Google Scholar]
- [75].Gattas-Asfura KM, Stabler CL, Biomacromolecules 2009, 10, 3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Gattás-Asfura KM, Fraker CA, Stabler CL, J. Biomed. Mater. Res., Part A 2011, 99A, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Gutsmiedl K, Wirges CT, Ehmke V, Carell T, Org. Lett 2009, 11, 2405. [DOI] [PubMed] [Google Scholar]
- [78].Truong VX, Zhou K, Simon GP, Forsythe JS, Macromol. Rapid Commun 2015, 36, 1729. [DOI] [PubMed] [Google Scholar]
- [79].Truong VX, Hun ML, Li F, Chidgey AP, Forsythe JS, Biomater. Sci 2016, 4, 1123. [DOI] [PubMed] [Google Scholar]
- [80].Ramil CP, Lin Q, Chem. Commun 2013, 49, 11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].a) McKay CS, Moran J, Pezacki JP, Chem. Commun 2010, 46, 931; [DOI] [PubMed] [Google Scholar]; b) Ning X, Temming RP, Dommerholt J, Guo J, Ania DB, Debets MF, Wolfert MA, Boons G-J, van Delft FL, Angew. Chem. Int. Ed 2010, 49, 3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Song W, Wang Y, Qu J, Madden MM, Lin Q, Angew. Chem. Int. Ed 2008, 47, 2832. [DOI] [PubMed] [Google Scholar]
- [83].Yu Z, Pan Y, Wang Z, Wang J, Lin Q, Angew. Chem. Int. Ed 2012, 51, 10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Li Z, Qian L, Li L, Bernhammer JC, Huynh HV, Lee J-S, Yao SQ, Angew. Chem. Int. Ed 2016, 55, 2002. [DOI] [PubMed] [Google Scholar]
- [85].Fan Y, Deng C, Cheng R, Meng F, Zhong Z, Biomacromolecules 2013, 14, 2814. [DOI] [PubMed] [Google Scholar]
- [86].van Berkel SS, Dirks ATJ, Debets MF, van Delft FL, Cornelissen JJLM, Nolte RJM, Rutjes FPJT, ChemBioChem 2007, 8, 1504. [DOI] [PubMed] [Google Scholar]
- [87].Glassner M, Delaittre G, Kaupp M, Blinco JP, Barner-Kowollik C, J. Am. Chem. Soc 2012, 134, 7274. [DOI] [PubMed] [Google Scholar]
- [88].a) Ban H, Gavrilyuk J, Barbas CF III, J. Am. Chem. Soc 2010, 132, 1523; [DOI] [PubMed] [Google Scholar]; b) Ban H, Nagano M, Gavrilyuk J, Hakamata W, Inokuma T, Barbas CF III, Bioconjugate Chem. 2013, 24, 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Arumugam S, Popik VV, J. Am. Chem. Soc 2011, 133, 5573. [DOI] [PubMed] [Google Scholar]
- [90].a) Aguado BA, Mulyasasmita W, Su J, Lampe KJ, Heilshorn SC, Tissue Eng Part A 2012, 18, 806; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mulyasasmita W, Cai L, Dewi RE, Jha A, Ullmann SD, Luong RH, Huang NF, Heilshorn SC, J. Controlled Release 2014, 191, 71; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yan C, Mackay ME, Czymmek K, Nagarkar RP, Schneider JP, Pochan DJ, Langmuir 2012, 28, 6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Gaffey AC, Chen MH, Venkataraman CM, Trubelja A, Rodell CB, Dinh PV, Hung G, MacArthur JW, Soopan RV, Burdick JA, Atluri P, J. Thorac. Cardiovasc. Surg 2015, 150, 1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].a) Jia W, Gungor-Ozkerim PS, Zhang YS, Yue K, Zhu K, Liu W, Pi Q, Byambaa B, Dokmeci MR, Shin SR, Khademhosseini A, Biomaterials 2016, 106, 58; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dubbin K, Hori Y, Lewis KK, Heilshorn SC, Adv. Healthcare Mater 2016, 5, 2488. [DOI] [PubMed] [Google Scholar]
- [93].Highley CB, Rodell CB, Burdick JA, Adv. Mater 2015, 27, 5075. [DOI] [PubMed] [Google Scholar]
- [94].a) Chaudhuri O, Gu L, Darnell M, Klumpers D, Bencherif SA, Weaver JC, Huebsch N, Mooney DJ, Nat. Commun 2015, 6, 6365; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chaudhuri O, Gu L, Klumpers D, Darnell M, Bencherif SA, Weaver JC, Huebsch N, Lee H.-p., Lippens E, Duda GN, Mooney DJ, Nat. Mater 2016, 15, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].a) Webber MJ, Appel EA, Meijer EW, Langer R, Nat. Mater 2016, 15, 13; [DOI] [PubMed] [Google Scholar]; b) Ma X, Zhao Y, Chem. Rev 2015, 115, 7794. [DOI] [PubMed] [Google Scholar]
- [96].Rodell CB, Mealy JE, Burdick JA, Bioconjugate Chem. 2015, 26, 2279. [DOI] [PubMed] [Google Scholar]
- [97].van de Manakker F, van der Pot M, Vermonden T, van Nostrum CF, Hennink WE, Macromolecules 2008, 41, 1766. [Google Scholar]
- [98].Rowland MJ, Appel EA, Coulston RJ, Scherman OA, J. Mater. Chem. B 2013, 1, 2904. [DOI] [PubMed] [Google Scholar]
- [99].Kretschmann O, Choi SW, Miyauchi M, Tomatsu I, Harada A, Ritter H, Angew. Chem. Int. Ed 2006, 45, 4361. [DOI] [PubMed] [Google Scholar]
- [100].Koopmans C, Ritter H, Macromolecules 2008, 41, 7418. [Google Scholar]
- [101].Kakuta T, Takashima Y, Nakahata M, Otsubo M, Yamaguchi H, Harada A, Adv. Mater 2013, 25, 2849. [DOI] [PubMed] [Google Scholar]
- [102].Rodell CB, Kaminski AL, Burdick JA, Biomacromolecules 2013, 14, 4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].a) Appel EA, Biedermann F, Rauwald U, Jones ST, Zayed JM, Scherman OA, J. Am. Chem. Soc 2010, 132, 14251; [DOI] [PubMed] [Google Scholar]; b) Appel EA, Loh XJ, Jones ST, Biedermann F, Dreiss CA, Scherman OA, J. Am. Chem. Soc 2012, 134, 11767. [DOI] [PubMed] [Google Scholar]
- [104].Park KM, Yang J-A, Jung H, Yeom J, Park JS, Park K-H, Hoffman AS, Hahn SK, Kim K, ACS Nano 2012, 6, 2960. [DOI] [PubMed] [Google Scholar]
- [105].Jung H, Park JS, Yeom J, Selvapalam N, Park KM, Oh K, Yang J-A, Park KH, Hahn SK, Kim K, Biomacromolecules 2014, 15, 707. [DOI] [PubMed] [Google Scholar]
- [106].Yeom J, Kim SJ, Jung H, Namkoong H, Yang J, Hwang BW, Oh K, Kim K, Sung YC, Hahn SK, Adv. Healthcare Mater 2015, 4, 237. [DOI] [PubMed] [Google Scholar]
- [107].Schexnailder P, Schmidt G, Colloid Polym. Sci 2009, 287, 1. [Google Scholar]
- [108].Wu C-J, Gaharwar AK, Schexnailder PJ, Schmidt G, Materials 2010, 3, 2986. [DOI] [PubMed] [Google Scholar]
- [109].Takahashi T, Yamada Y, Kataoka K, Nagasaki Y, J. Controlled Release 2005, 107, 408. [DOI] [PubMed] [Google Scholar]
- [110].Jin Q, Schexnailder P, Gaharwar AK, Schmidt G, Macromol. Biosci 2009, 9, 1028. [DOI] [PubMed] [Google Scholar]
- [111].a) Wang Q, Mynar JL, Yoshida M, Lee E, Lee M, Okuro K, Kinbara K, Aida T, Nature 2010, 463, 339; [DOI] [PubMed] [Google Scholar]; b) Tamesue S, Ohtani M, Yamada K, Ishida Y, Spruell JM, Lynd NA, Hawker CJ, Aida T, J. Am. Chem. Soc 2013, 135, 15650. [DOI] [PubMed] [Google Scholar]
- [112].a) Appel EA, Tibbitt MW, Webber MJ, Mattix BA, Veiseh O, Langer R, Nat. Commun 2015, 6, 6295; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Appel EA, Tibbitt MW, Greer JM, Fenton OS, Kreuels K, Anderson DG, Langer R, ACS Macro Lett. 2015, 4, 848. [DOI] [PubMed] [Google Scholar]
- [113].a) Shao Y, Jia H, Cao T, Liu D, Acc. Chem. Res 2017, 50, 659; [DOI] [PubMed] [Google Scholar]; b) Kopeček J, Yang J, Angew. Chem. Int. Ed 2012, 51, 7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SFT, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM, Cell 2009, 139, 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Luo Y, Shoichet MS, Nat. Mater 2004, 3, 249. [DOI] [PubMed] [Google Scholar]
- [116].Luo Y, Shoichet MS, Biomacromolecules 2004, 5, 2315. [DOI] [PubMed] [Google Scholar]
- [117].Furuta T, Wang SS-H, Dantzker JL, Dore TM, Bybee WJ, Callaway EM, Denk W, Tsien RY, Proc. Natl. Acad. Sci. USA 1999, 96, 1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Wosnick JH, Shoichet MS, Chem. Mater 2008, 20, 55. [Google Scholar]
- [119].Rahman N, Purpura KA, Wylie RG, Zandstra PW, Shoichet MS, Biomaterials 2010, 31, 8262. [DOI] [PubMed] [Google Scholar]
- [120].Aizawa Y, Wylie R, Shoichet M, Adv. Mater 2010, 22, 4831. [DOI] [PubMed] [Google Scholar]
- [121].Dendane N, Hoang A, Guillard L, Defrancq E, Vinet F, Dumy P, Bioconjugate Chem. 2007, 18, 671. [DOI] [PubMed] [Google Scholar]
- [122].Farahani PE, Adelmund SM, Shadish JA, DeForest CA, J. Mater. Chem. B 2017, 5, 4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].DeForest CA, Anseth KS, Nat. Chem 2011, 3, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].DeForest CA, Anseth KS, Angew. Chem. Int. Ed 2012, 51, 1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Khetan S, Katz JS, Burdick JA, Soft Matter 2009, 5, 1601. [Google Scholar]
- [126].Khetan S, Burdick JA, Biomaterials 2010, 31, 8228. [DOI] [PubMed] [Google Scholar]
- [127].Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick JA, Nat. Mater 2013, 12, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Guvendiren M, Burdick JA, Nat. Commun 2012, 3, 792. [DOI] [PubMed] [Google Scholar]
- [129].Poloukhtine AA, Mbua NE, Wolfert MA, Boons G-J, Popik VV, J. Am. Chem. Soc 2009, 131, 15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Shah L, Laughlin ST, Carrico IS, J. Am. Chem. Soc 2016, 138, 5186–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Zhang H, Trout WS, Liu S, Andrade GA, Hudson DA, Scinto SL, Dicker KT, Li Y, Lazouski N, Rosenthal J, Thorpe C, Jia X, Fox JM, J. Am. Chem. Soc 2016, 138, 5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Truong VX, Tsang KM, Ercole F, Forsythe JS, Chem. Mater 2017, 29, 3678. [Google Scholar]
- [133].Sperinde JJ, Griffith LG, Macromolecules 1997, 30, 5255. [Google Scholar]
- [134].Hu B-H, Messersmith PB, J. Am. Chem. Soc 2003, 125, 14298. [DOI] [PubMed] [Google Scholar]
- [135].Ehrbar M, Rizzi SC, Hlushchuk R, Djonov V, Zisch AH, Hubbell JA, Weber FE, Lutolf MP, Biomaterials 2007, 28, 3856. [DOI] [PubMed] [Google Scholar]
- [136].Mosiewicz KA, Kolb L, Vlies A. J. v. d., Martino MM, Lienemann PS, Hubbell JA, Ehrbar M, Lutolf MP, Nat. Mater 2013, 12, 1072. [DOI] [PubMed] [Google Scholar]
- [137].Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, Howarth M, Proc. Natl. Acad. Sci. USA 2012, 109, E690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Sun F, Zhang W-B, Mahdavi A, Arnold FH, Tirrell DA, Proc. Natl. Acad. Sci. USA 2014, 111, 11269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Veggiani G, Nakamura T, Brenner MD, Gayet RV, Yan J, Robinson CV, Howarth M, Proc. Natl. Acad. Sci. USA 2016, 113, 1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Brune KD, Buldun CM, Li Y, Taylor IJ, Brod F, Biswas S, Howarth M, Bioconjugate Chem. 2017, 28, 1544. [DOI] [PubMed] [Google Scholar]
- [141].Cambria E, Renggli K, Ahrens CC, Cook CD, Kroll C, Krueger AT, Imperiali B, Griffith LG, Biomacromolecules 2015, 16, 2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Ton-That H, Mazmanian SK, Faull KF, Schneewind O, J. Biol. Chem 2000, 275, 9876. [DOI] [PubMed] [Google Scholar]
- [143].Ham HO, Qu Z, Haller CA, Dorr BM, Dai E, Kim W, Liu DR, Chaikof EL, Nat. Commun 2016, 7, 11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Prescher JA, Bertozzi CR, Nat. Chem. Biol 2005, 1, 13. [DOI] [PubMed] [Google Scholar]
- [145].Brudno Y, Desai RM, Kwee BJ, Joshi NS, Aizenberg M, Mooney DJ, ChemMedChem 2015, 10, 617. [DOI] [PubMed] [Google Scholar]
- [146].Oneto JMM, Khan I, Seebald L, Royzen M, ACS Cent. Sci 2016, 2, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].a) Tian F, Cziferszky M, Jiao D, Wahlström K, Geng J, Scherman OA, Langmuir 2011, 27, 1387; [DOI] [PubMed] [Google Scholar]; b) González-Campo A, Brasch M, Uhlenheuer DA, Gómez-Casado A, Yang L, Brunsveld L, Huskens J, Jonkheijm P, Langmuir 2012, 28, 16364; [DOI] [PubMed] [Google Scholar]; c) Yang L, Gomez-Casado A, Young JF, Nguyen HD, Cabanas-Danés J, Huskens J, Brunsveld L, Jonkheijm P, J. Am. Chem. Soc 2012, 134, 19199. [DOI] [PubMed] [Google Scholar]
- [148].a) An Q, Brinkmann J, Huskens J, Krabbenborg S, de Boer J, Jonkheijm P, Angew. Chem. Int. Ed 2012, 51, 12233; [DOI] [PubMed] [Google Scholar]; b) Neirynck P, Brinkmann J, An Q, van der Schaft DWJ, Milroy L-G, Jonkheijm P, Brunsveld L, Chem. Commun 2013, 49, 3679. [DOI] [PubMed] [Google Scholar]
- [149].Cabanas-Danés J, Rodrigues ED, Landman E, Weerd J. v., Blitterswijk C. v., Verrips T, Huskens J, Karperien M, Jonkheijm P, J. Am. Chem. Soc 2014, 136, 12675. [DOI] [PubMed] [Google Scholar]
- [150].Boekhoven J, Pérez CMR, Sur S, Worthy A, Stupp SI, Angew. Chem. Int. Ed 2013, 52, 12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Li J, Chen PR, Nat. Chem. Biol 2016, 12, 129. [DOI] [PubMed] [Google Scholar]
- [152].Klán P, Šolomek T, Bochet CG, Blanc A, Givens R, Rubina M, Popik V, Kostikov A, Wirz J, Chem. Rev 2013, 113, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Bae J, McNamara LE, Nael MA, Mahdi F, Doerksen RJ, Bidwell GL, Hammer NI, Jo S, Chem. Commun 2015, 51, 12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Petersen S, Alonso JM, Specht A, Duodu P, Goeldner M, del Campo A, Angew. Chem. Int. Ed 2008, 47, 3192. [DOI] [PubMed] [Google Scholar]
- [155].Ohmuro-Matsuyama Y, Tatsu Y, Angew. Chem. Int. Ed 2008, 47, 7527. [DOI] [PubMed] [Google Scholar]
- [156].Lee TT, García JR, Paez JI, Singh A, Phelps EA, Weis S, Shafiq Z, Shekaran A, del Campo A, García AJ, Nat. Mater 2015, 14, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Weis S, Lee TT, del Campo A, García AJ, Acta Biomater. 2013, 9, 8059. [DOI] [PubMed] [Google Scholar]
- [158].a) Salierno MJ, García AJ, del Campo A, Adv. Funct. Mater 2013, 23, 5974; [Google Scholar]; b) Salierno MJ, García-Fernandez L, Carabelos N, Kiefer K, García AJ, del Campo A, Biomaterials 2016, 82, 113. [DOI] [PubMed] [Google Scholar]
- [159].Iturri J, García-Fernández L, Reuning U, García AJ, del Campo A, Salierno MJ, Sci. Rep 2015, 5, 9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Kloxin AM, Kasko AM, Salinas CN, Anseth KS, Science 2009, 324, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Tsurkan MV, Wetzel R, Pérez-Hernández HR, Chwalek K, Kozlova A, Freudenberg U, Kempermann G, Zhang Y, Lasagni AF, Werner C, Adv. Healthcare Mater 2015, 4, 516. [DOI] [PubMed] [Google Scholar]
- [162].Arakawa CK, Badeau BA, Zheng Y, DeForest CA, Adv. Mater 2017, 29, 1703156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Yang C, Tibbitt MW, Basta L, Anseth KS, Nat. Mater 2014, 13, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].a) Lutolf MP, Raeber GP, Zisch AH, Tirelli N, Hubbell JA, Adv. Mater 2003, 15, 888; [Google Scholar]; b) Burdick JA, Murphy WL, Nat. Commun 2012, 3, 1269. [DOI] [PubMed] [Google Scholar]
- [165].Valdez J, Cook CD, Ahrens CC, Wang AJ, Brown A, Kumar M, Stockdale L, Rothenberg D, Renggli K, Gordon E, Lauffenburger D, White F, Griffith L, Biomaterials 2017, 130, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Tian L, Yang Y, Wysocki LM, Arnold AC, Hu A, Ravichandran B, Sternson SM, Looger LL, Lavis LD, Proc. Natl. Acad. Sci. USA 2012, 109, 4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Ritter C, Nett N, Acevedo-Rocha CG, Lonsdale R, Kräling K, Dempwolff F, Hoebenreich S, Graumann PL, Reetz MT, Meggers E, Angew. Chem. Int. Ed 2015, 54, 13440. [DOI] [PubMed] [Google Scholar]
- [168].Streu C, Meggers E, Angew. Chem. Int. Ed 2006, 45, 5645. [DOI] [PubMed] [Google Scholar]
- [169].a) Völker T, Dempwolff F, Graumann PL, Meggers E, Angew. Chem. Int. Ed 2014, 53, 10536; [DOI] [PubMed] [Google Scholar]; b) Völker T, Meggers E, ChemBioChem 2017, 18, 1083. [DOI] [PubMed] [Google Scholar]
- [170].Yusop RM, Unciti-Broceta A, Johansson EMV, Sánchez-Martín RM, Bradley M, Nat. Chem 2011, 3, 239. [DOI] [PubMed] [Google Scholar]
- [171].a) Weiss JT, Dawson JC, Macleod KG, Rybski W, Fraser C, Torres-Sánchez C, Patton EE, Bradley M, Carragher NO, Unciti-Broceta A, Nat. Commun 2014, 5, 3277; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Weiss JT, Rybski JCDCFW, Torres-Sánchez C, Bradley M, Patton EE, Carragher NO, Unciti-Broceta A, J. Med. Chem 2014, 57, 5395; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rubio-Ruiz B, Weiss JT, Unciti-Broceta A, J. Med. Chem 2016, 59, 9974. [DOI] [PubMed] [Google Scholar]
- [172].Li J, Yu J, Zhao J, Wang J, Zheng S, Lin S, Chen L, Yang M, Jia S, Zhang X, Chen PR, Nat. Chem 2014, 6, 352. [DOI] [PubMed] [Google Scholar]
- [173].Wang J, Cheng B, Li J, Zhang Z, Hong W, Chen X, Chen PR, Angew. Chem. Int. Ed 2015, 54, 5364. [DOI] [PubMed] [Google Scholar]
- [174].Pérez-López AM, Rubio-Ruiz B, Sebastián V, Hamilton L, Adam C, Bray TL, Irusta S, Brennan PM, Lloyd-Jones GC, Sieger D, Santamaría J, Unciti-Broceta A, Angew. Chem. Int. Ed 2017, 56, 12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].a) Wataha J, Hanks C, J. Oral Rehabil 1996, 23, 309; [DOI] [PubMed] [Google Scholar]; b) Lebeau A, in Hamilton & Hardy’s Industrial Toxicology, (Eds: Harbison RD, Bourgeois MM, Johnson GT), John Wiley & Sons, Inc., Hoboken, New Jersey: 2015, 187. [Google Scholar]
- [176].Liu TZ, Lee SD, Bhatnagar RS, Toxicol. Lett 1979, 4, 469. [Google Scholar]
- [177].Antonarakis ES, Emadi A, Cancer Chemother. Pharmacol 2010, 66, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Qu F, Park S, Martinez K, Gray JL, Thowfeik FS, Lundeen JA, Kuhn AE, Charboneau DJ, Gerlach DL, Lockart MM, Law JA, Jernigan KL, Chambers N, Zeller M, Pirog NA, Kassel WS, Schmehl RH, Paul JJ, Merino EJ, Kim Y, Papish ET, Inorg. Chem 2017, 56, 7519. [DOI] [PubMed] [Google Scholar]
- [179].Versteegen RM, Rossin R, Hoeve W. t., Janssen HM, Robillard MS, Angew. Chem. Int. Ed 2013, 52, 14112. [DOI] [PubMed] [Google Scholar]
- [180].Oliveira BL, Guo Z, Bernardes GJL, Chem. Soc. Rev 2017, 46, 4895. [DOI] [PubMed] [Google Scholar]
- [181].Li J, Chen SJPR, Nat. Chem. Biol 2014, 10, 1003. [DOI] [PubMed] [Google Scholar]
- [182].Jiménez-Moreno E, Guo Z, Oliveira BL, Albuquerque IS, Kitowski A, Guerreiro A, Boutureira O, Rodrigues T, Jiménez-Osés G, Bernardes GJL, Angew. Chem. Int. Ed 2017, 56, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Matikonda SS, Orsi DL, Staudacher V, Jenkins IA, Fiedler F, Chena J, Gamble AB, Chem. Sci 2015, 6, 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Pawlak JB, Gential GPP, Ruckwardt TJ, Bremmers JS, Meeuwenoord NJ, Ossendorp FA, Overkleeft HS, Filippov DV, van Kasteren SI, Angew. Chem. Int. Ed 2015, 54, 5628. [DOI] [PubMed] [Google Scholar]
- [185].Kim J, Bertozzi CR, Angew. Chem. Int. Ed 2015, 54, 15777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Marquardt LM, Heilshorn SC, Curr. Stem Cell Rep 2016, 2, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Royzen M, Yap GPA, Fox JM, J. Am. Chem. Soc 2008, 130, 3760. [DOI] [PubMed] [Google Scholar]
- [188].Wiley RA, Sternson LA, Sasame HA, Gillette JR, Biochem. Pharmacol 1972, 21, 3235. [DOI] [PubMed] [Google Scholar]
- [189].Ortiz de Montellano PR, Kunze KL, J. Biol. Chem 1980, 255, 5578. [PubMed] [Google Scholar]
- [190].a) McNulty J, Nair JJ, Vurgun N, DiFrancesco BR, Brown CE, Tsoi B, Crankshaw DJ, Holloway AC, Bioorg. Med. Chem. Lett 2012, 22, 718; [DOI] [PubMed] [Google Scholar]; b) Conner KP, Vennam P, Woods CM, Krzyaniak MD, Bowman MK, Atkins WM, Biochemistry 2012, 51, 6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Šečkutė J, Devaraj NK, Curr. Opin. Chem. Biol 2013, 17, 761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Karver MR, Weissleder R, Hilderbrand SA, Bioconjugate Chem. 2011, 22, 2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Taylor MT, Blackman ML, Dmitrenko O, Fox JM, J. Am. Chem. Soc 2011, 133, 9646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Darko A, Wallace S, Dmitrenko O, Machovina MM, Mehl RA, Chin JW, Fox JM, Chem. Sci 2014, 5, 3770. [DOI] [PMC free article] [PubMed] [Google Scholar]