

Abstract
Periodontitis is characterized by the progressive destruction of tooth-supporting alveolar bone, which is mainly caused by chronic inflammation in response to persistent bacterial insult. It has recently become clear that the pathogenesis of periodontitis is associated with a high ratio of proinflammatory M1 (classically activated) macrophages to anti-inflammatory M2 (alternatively activated). To decrease the inflammatory activity, we locally delivered the C-C motif chemokine ligand 2 (CCL2) using controlled-release microparticles (MPs). CCL2 is known to promote chemotaxis of M0 or M2 phenotype macrophages to the inflamed site and induce M2 phenotype polarization locally. Our in vitro data showed that CCL2 increased the number of M2 phenotype macrophages, decreased TNF-α secretion, and enhanced chemotaxis of RAW264.7 cells toward CCL2 MPs. Moreover, we induced periodontal disease in 2 animal models through inoculation of Porphyromonas gingivalis and ligature around the murine molar. Micro–computed tomography analysis showed significant reduction of alveolar bone loss in the CCL2 MP treatment group when compared with a blank MP group and a no-treatment periodontitis group in both models. Immunohistologic analysis showed a significant increase in the M2 phenotype subset and a decrease in the M1 phenotype subset in the CCL2 MP group of the P. gingivalis–induced model. Also, in both models, tartrate-resistant acidic phosphatase staining showed significantly fewer numbers of osteoclasts in the CCL2 MP group in alveolar bone area. Moreover, quantitative polymerase chain reaction results showed a significant increase in IL-1RA (interleukin 1 receptor antagonist) mRNA expression and a decrease in RANKL (receptor activator of nuclear factor kappa-Β ligand) mRNA expression in the CCL2 MP group in the ligature model. In summary, manipulation of endogenous M2 phenotype macrophages with CCL2 MPs decreased the M1 phenotype:M2 phenotype ratio and prevented alveolar bone loss in mouse periodontitis models. The delivery of CCL2 MPs provides a novel approach to treat periodontal disease.
Keywords: chemokines, controlled release, cytokines, host modulation therapy, PLGA, CCL2
Introduction
Periodontitis is characterized by the progressive destruction of tooth-supporting alveolar bone, which may eventually lead to tooth loss. Approximately 46% of adults aged ≥30 y in the United States suffer from periodontitis (Eke et al. 2015). Moreover, periodontal diseases may increase the risk of diabetes, cardiovascular disease, and preterm birth (Pihlstrom et al. 2005; Kuo et al. 2008). For treatment of periodontitis, eliminating pathogens and reestablishing normal oral flora has been the focus (Pihlstrom et al. 2005). Current standard treatment includes scaling and root planning, sometimes combined with adjunctive therapies such as systemic or local administration of antimicrobials (Smiley et al. 2015). However, considering the periodontitis recurrence rate, the nonresponding patient rate, and the growing concerns surrounding antibiotic resistance and overuse, we sought an alternative strategy for periodontitis treatment (Haffajee et al. 2004; Johnson et al. 2008; Varela et al. 2011).
In the pathogenic progression of periodontitis, bacterial infection initiates inflammation. However, the host immune-inflammatory response is what initially contributes to periodontium destruction (Kinane 2001; Pihlstrom et al. 2005; Garlet 2010). Specifically, cytokines such as TNF-α, IL-1β, and IL-6 trigger upregulation of the primary tissue destruction mediators. With the increased understanding of the determinate role of inflammation in periodontitis, considerable attention has been devoted to modulation of host immune response. Therapeutic blockade of proinflammatory cytokines TNF-α and IL-1 (Assuma et al. 1998; Delima et al. 2001), injection of anti-inflammatory cytokine IL-11 (Martuscelli et al. 2000), activation of inflammation downregulator PPAR-γ (proliferator-activated receptor–gamma; Di Paola et al. 2006; Hassumi et al. 2009), and recruitment of immune-modulatory regulatory T cells (Glowacki et al. 2013) were reported to reduce alveolar bone loss in diverse animal periodontitis models. All these findings support the concept that balancing immune response in periodontitis could serve as an alternative treatment strategy.
In this study, we hypothesized that modulation of the macrophage phenotype will effectively decrease the inflammatory response, since macrophages activated by sustained microbial challenge in the periodontitis microenvironment are one of the major sources for these destructive cytokines (Graves and Cochran 2003; Graves 2008). Macrophages are a group of phenotypic heterogenic cells, which are classified into 2 major subsets, depending on the stimuli: classically activated (M1) and alternatively activated (M2) (Gordon 2003; Stein et al. 1992; Martinez and Gordon 2014). M1 and M2 macrophages are the 2 extremes of the function continuum, with different mRNA expressions (Biswas et al. 2006) and proteome profiles (Zhang et al. 2015).
Specifically, M1 macrophages facilitate pathogen elimination by phagocytosis, stimulating the activation of polymorphonuclear neutrophils and T cells and producing proinflammatory cytokines such as IL-1β, IL-6, and TNF-α (Sima and Glogauer 2013). Also, M1 macrophages aid in osteoclast activation by secreting the cytokines that promote the Th1 response and stimulate preosteoclasts. This results in elevated receptor activator of nuclear factor kappa-Β ligand (RANKL) expression and bone destruction (Adamopoulos and Mellins 2015). M2 macrophages are involved in inflammation resolution and tissue regeneration by secretion of anti-inflammatory mediators such as interleukin 1 receptor antagonist (IL-1RA; Mantovani et al. 2004). In periodontitis, M1 macrophages are the predominant type, as shown in mouse and nonhuman primate experimental periodontitis models (Lam et al. 2014; Gonzalez et al. 2015) and human specimens (Yang et al. 2017). In addition, M1 was shown to play a role in periodontal disease (Yamaguchi et al. 2016). The activation of M1 macrophages is suggested to contribute to the initiation and maintenance of a proinflammatory state during chronic periodontitis (Lam et al. 2016).
Accordingly, we aim to develop a method for creating a local environment that induces M2 phenotype polarization to convert the impaired M1 phenotype:M2 phenotype ratio in the periodontitis microenvironment. C-C motif chemokine ligand 2 (CCL2) is a long-known macrophage chemoattractant (Deshmane et al. 2009) recently reported to be associated with macrophage polarization (Roca et al. 2009). In this study, we analyzed the effect of CCL2 on inducing macrophage polarization toward the M2 phenotype, as well as the effects of CCL2-releasing microparticles (MPs) on the prevention of alveolar bone resorption, in murine periodontitis models.
Materials and Methods
Cell Culture
Mouse macrophage cell line RAW264.7 (ATCC TIB-71) was cultured in DMEM (Life Technologies) containing 10% fetal bovine serum (Atlanta Biologicals), 1% penicillin and streptomycin (Life Technologies), and 1% L-glutamine (Life Technologies).
Mouse bone marrow macrophages were collected by flushing bone marrow cells out of the femur and tibia of CD1 female mice; then, red blood cells were removed with a lysing solution (eBioscience). Monocyte- and macrophage-lineage cells were sorted with CD11b magnetic beads (Miltenyi Biotec Inc). CD11b+ cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) and supplemented with 10 ng/mL of murine macrophage colony-stimulating factor (Peprotech), 10% fetal bovine serum, 1% penicillin and streptomycin, 1% HEPES (Life Technologies), and 2% minimum essential medium (MEM) nonessential amino acids (Life Technologies). The medium was changed every other day.
In Vitro Analyses of Biological Effects of CCL2 on Macrophages
M2 Phenotype Polarization
Mouse bone marrow–derived macrophages were stimulated with 10 ng/mL of CCL2-containing medium or normal culture medium for 48 h after 1 h of serum starvation. Cells were then stained with PE/Cy7 anti-mouse CD86 (BioLegend) and PE anti-mouse CD206 (BioLegend) or isotype control antibodies after Fc antigen blocking. Flow cytometry was performed on an LSR-II flow cytometer (BD Biosciences), and data analysis was done with FlowJo software. The experiment was repeated 3 times with different mice (biological triplicates). Each biological sample was assessed in triplicates (technical triplicates). In addition, TNF-α secretion from RAW264.7 cells after CCL2 stimulation was analyzed with an ELISA kit. RAW264.7 cells, in triplicates, were treated with 100 ng/mL of lipopolysaccharide for 24 h for M1 activation. Subsequently, cells were treated with 10 or 15 ng/mL of CCL2-containing serum-free medium for 24 or 48 h. The amount of TNF-α in the supernatant was determined by an ELISA kit (R&D Systems). This experiment was repeated twice.
Cell Migration Assay
RAW 264.7 cells were placed on the upper chambers of Transwell plates with a 5-µm pore (Corning Life Sciences) and serum-free DMEM containing 10 ng/mL of recombinant mouse CCL2 (rmCCL2) peptide (R&D System) or 10 ng/mL of CCL2 released from poly(lactic-co-glycolic acid) (PLGA) MPs. These were placed in the lower chambers. Serum-free DMEM medium and that containing the same amount of blank PLGA MPs were used as controls. After 3 h of incubation, cells that had migrated to the bottom side of the membrane were stained with DAPI. Images were taken under 200× final magnification with 6 random fields for each membrane (Eclipse TE2000-E; Nikon Instruments). The number of cells was determined with ImageJ (National Institute of Health).
CCL2 PLGA MP Fabrication and Characterization
rmCCL2-releasing PLGA MPs were fabricated with a water-oil-water double-emulsion procedure as described previously (detailed in Appendix 1; Glowacki et al. 2013). The aqueous phase contained 28 µg of rmCCL2 and 15mM NaCl in 200 µL of 1% (wt/vol) bovine serum albumin solution. The oil phase contained 200 mg of PLGA (719897-5G; Sigma-Aldrich) in 4 mL of dichloromethane. Scanning electron microscopy (JSM-6330F; JEOL) was used to determine the surface character of the MPs. A volume impedance test (Multisizer-3; Beckman Coulter) was conducted to determine the size distribution of the MPs. For CCL2 releasing assay, 10 mg of rmCCL2-containing PLGA MPs were dissolved in 1 mL of phosphate-buffered saline (PBS) and incubated at 37 °C on a rotator. The supernatant was taken out daily and replenished with an equal amount of fresh PBS. The amount of CCL2 in the supernatant was measured with an ELISA kit (R&D Systems).
Murine Periodontal Disease Models
All animal experiments were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh (protocols 14073096 and 15053781). All animals in this experiment were wild-type BALB/c mice aged 6 to 8 wk, purchased from Jackson Laboratory, and maintained in the Division of Laboratory Animal Resources animal facility during the whole experiment period.
Porphyromonas gingivalis–Induced Mouse Periodontitis Model
This Porphyromonas gingivalis (Pg)–induced murine periodontitis model was established as previously described (Glowacki et al. 2013). Briefly, mouse periodontitis was induced by oral inoculation of Pg (ATCC 33277) resuspended at 1 × 1011 colony-forming units in BBL brain heart infusion agar (BD Biosciences) containing 2% carboxymethylcellulose, 3 times at 48-h intervals (described in Appendix 2).
Ligature-Induced Periodontitis Model
BALB/c mice underwent anesthesia by intraperitoneal injection of a mixture containing 66.7 mg/kg of ketamine and 6.7 mg/kg of xylazine. Silk suture (6-0; Henry Schein) was then placed around the left maxillary second molar. The contralateral molar was left unligated and used as healthy control. The ligatures were kept in the same place throughout the duration of the experiment.
PLGA MP Administration
Mice were anesthetized by the ketamine-xylazine mixture. In the Pg-induced periodontitis model, 50 µL of solution containing 5 mg/mL of MPs in PBS with 2% carboxymethylcellulose was injected with a 28.5G insulin syringe into gingival tissue at the mesial side of the first molar, the distal side of the third molar, and the 2 interdental sites. The injection was made on the left and right sides of the maxillae. MPs were administrated on days −1, 20, and 40 relative to the first bacteria inoculation. The mice were sacrificed on day 60. In the ligature-induced periodontitis model, a 50-µL solution containing 5 mg/mL of MPs was injected into the gingival tissues surrounding the ligated tooth of the left maxillae on the same day of ligature placement, and mice were sacrificed on day 8. All MP injections in mice were conducted under stereomicroscope.
Micro–computed tomography
Mouse maxillary alveolar bone was fixed in 10% neutral buffered formalin for 24 h and then transferred into 70% ethanol for scanning with a micro–computed tomography system (vivaCT 40; SCANCO Medical) with a resolution of 10.5 µm and 55 kVp. All scans were reoriented with DataViewer (GE Healthcare) to the same position for bone loss evaluation. The images were reoriented with anatomic landmarks, and the distance between cementoenamel junction and alveolar bone crest was measured at a 52.5-µm interval (details in Appendix 3).
Quantitative Polymerase Chain Reaction
From the ligature-induced periodontitis model, the ligated sides of the mouse maxillary alveolar bones with gingival tissue attached were harvested and immediately frozen in liquid nitrogen. The RNA was extracted with the QIAshredder and RNeasy Mini Kit (Qiagen). Quantitative polymerase chain reaction was conducted with TaqMan One-Step RT-PCR Master Mix Reagents (Applied Biosystems) on a QuantStudio 6 Flex Real-Time PCR system (Thermo Fisher Scientific), and all primers and probes were purchased from TaqMan (Applied Biosystems). Data analysis was done with the delta-delta Ct method.
Histologic and Immunostaining Analysis
Maxillae samples from Pg- and ligature-induced periodontitis models were demineralized and embedded in paraffin; 4-µm-thick sagittal sections were made for histologic and immunostaining analysis. Osteoclasts were stained with the TRAP Kit (387A-1KTF; Sigma Chemical Co) following the manufacturer’s instructions with hematoxylin Gill 3 (Sigma-Aldrich) counterstaining. The number of osteoclasts (tartrate-resistant acidic phosphatase–positive cells) per square millimeter of alveolar bone surface was counted (details in Appendix 4). Samples from the Pg-induced periodontitis model were used to determine macrophage population changes under the influence of CCL2 MPs. Briefly, sagittal section slides were stained with goat-anti-mouse CD206 antibody (AF2535; R&D Systems), donkey anti-goat IgG Alexa Flour 594 (Invitrogen), F4/80 antibody (MCA497R; AbD Serotec; Austyn and Gordon 1981), donkey anti-rat IgG Alexa Flour 488 (Invitrogen), and DAPI. Microscopic images were taken with a fluorescent microscope (Eclipse TE2000-E; Nikon Instruments), and positively stained cells were counted with ImageJ software (described in Appendix 5).
Statistical Analysis
Statistical analysis was conducted with SPSS Statistics 23 (IBM). All data obey normal distribution, as determined by Levene’s test. One-way analysis of variance was followed by Tukey’s honestly significant difference post hoc test, which used to compare variances from multiple groups. Student’s t test was used to compare variances between the groups. For correlation analysis, Pearson product-moment correlation was conducted. All statistical significance was considered at P < 0.05.
Results
CCL2 Induces Polarization of Mouse Macrophages toward CD206+ M2 Phenotype
After 48 h of CCL2 (10 ng/mL) treatment, the CD206+/CD86– cell population increased by an average of 4.177% (SD, 1.26%; Fig. 1A). As for the CD206 mean fluorescent intensity marker, the ratio of CCL2-treated macrophages to that of unstimulated macrophages was 1.35 (P < 0.05; Fig. 1B). Also, after 24 or 48 h of 10- or 15-ng/mL CCL2 treatment, lipopolysaccharide-stimulated M1 phenotype RAW264.7 cells secreted a significantly smaller amount of TNF-α (Fig. 1C, D).
Figure 1.

CCL2-induced macrophages polarizing toward M2 phenotype. Bone marrow–derived macrophages were cultured in vitro, unstimulated or stimulated with CCL2, stained with CD86 and CD206 (M1 and M2 phenotype–specific markers, respectively), then analyzed by flow cytometry. (a) CD206+ population was upregulated by CCL2 treatment. An additional 4.479% of the macrophage population (SD, 1.1%) was skewed toward the CD206-positive M2 phenotype versus the unstimulated M0 population. Representative flow cytometry zebra plots are shown in panel a. (b) CCL2 increased the CD206 median fluorescence intensity (MFI) by 1.35-fold. Relative CD206 MFI ratio of CCL2-treated macrophages to no cytokine–treated macrophages. From 3 repeated experiments; error bar indicates SD. *P < 0.05 by Student’s t test. The lipopolysaccharide (LPS)–stimulated, high TNF-α-secreting RAW264.7 macrophages were used as M1 phenotype macrophages in this experiment. RAW264.7 cells cultured in a LPS-free, CCL2-free environment were used as M0. After LPS stimulation, the M1 phenotype macrophages were treated with a medium containing 10 or 15 ng/mL of CCL2. Concentrations of TNF-α in supernatant were determined by ELISA. Either 1 d (c) or 2 d (d) after CCL2 treatment, TNF-α secretion from M1 phenotype macrophages was significantly reduced as compared with untreated M1 phenotype macrophages for both CCL2 concentrations tested. Each group is triplicated; error bar indicates SD. *P < 0.05 by 1-way ANOVA, post hoc Tukey HSD test. For release profile of the MPs, CCL2 MPs were dissolved in phosphate-buffered solution; the supernatant was collected on a daily basis; and the amount of CCL2 released was detected by ELISA. (e) The cumulative amount of CCL2 released from 1 mg of CCL2 PLGA MPs. Cell migration assay was used to test CCL2 MP bioviability. RAW264.7 cells were cultured in Transwell plates, and 10 ng/mL of CCL2 protein in serum-free medium was put into the lower chamber. CCL2 MPs were incubated in serum-free medium for 72 h at 37° in a 5% CO2 incubator, and the supernatant was used for the migration assay. The amount of CCL2 MPs was measured by ELISA assay to ensure an equal amount of CCL2 in the supernatant and positive control. Blank MPs resolved in serum-free medium (equal incubation time, condition, and MP weight as those of CCL2 MPs) and serum-free medium served as controls. (f) CCL2 released from MPs retained strong chemotaxis ability when compared with medium control and blank MP control, with no statistical difference as compared with an equal amount of CCL2. Each group is triplicated; error bar indicates SD. ***P < 0.001 by 1-way ANOVA, post hoc Tukey HSD test. ANOVA, analysis of variance; CCL2, C-C motif chemokine ligand 2; HSD, honestly significant difference; M1, classically activated macrophage; M2, alternatively activated macrophage; MP, microparticle.
Characterization of CCL2 PLGA MPs
As shown in the scanning electron microscopy images (Appendix Fig. 1A, B), the CCL2 MPs were spherical in shape and porous on the smooth surface and inside. Size distribution testing confirmed that the mean size of the MPs was 14.44 μm (SD, 5.22 μm; Appendix Fig. 1C) .
The release profile of CCL2 showed a burst release in the first 72 h, continued by a slow and steady release phase, then a lag phase of quick release up to 71 d when the particles were completely dissolved (Fig. 1E).
As shown in Figure 1F, the soluble CCL2 group and the CCL2 MP group had significantly higher numbers of cells migrated to the bottom side of the membrane as compared with controls (untreated and blank MPs).
CCL2-Releasing MPs Inhibit Alveolar Bone Resorption in a Murine Periodontitis Model
In the Pg-induced model, we observed that mice that received CCL2 MPs showed significantly reduced alveolar bone resorption when compared with the control groups (untreated or blank MPs) on the buccal side and interdental space (Fig. 2A–C).
Figure 2.

The sustained CCL2 release formulation significantly inhibited alveolar bone resorption in mouse periodontitis models. (a) Representative microCT 3-dimensional reconstruction pictures of mouse molars from Porphyromonas gingivalis (Pg)–induced periodontitis. (b, c) Quantification of periodontitis-induced alveolar bone loss, represented by linear measurement of CEJ-ABC distance on buccal and interdental root surfaces. Bone loss was indicated as the distance between the CEJ (white line) and ABC (yellow line) measured on microCT scans. The untreated group consisted of mice that were inoculated with Pg but did not receive an MP injection. The blank group consisted of mice inoculated with Pg that also received a blank MP injection. The CCL2 group consisted of mice that were inoculated with Pg and received a CCL2 MP injection. Mice in the no-Pg, no-PLGA group served as healthy controls. n = 5–8 mice, *P < 0.05, **P < 0.005 by 1-way ANOVA and a post hoc Tukey HSD test. (d) Representative microCT 3D reconstruction pictures of mouse molars from ligature model. (e) Linear measurements of CEJ-ABC distance were conducted around the second molars on transaxial and sagittal sections along the buccal, palatal, and interdental axes. Bone loss was determined by subtracting the ligated-side CEJ-ABC distance from the healthy contralateral side, based on measurements from microCT scans. n = 6 mice, *P < 0.05, 1-way ANOVA, followed by a post hoc Tukey HSD test. Error bars indicate SD. ANOVA, analysis of variance; CCL2, C-C motif chemokine ligand 2; CEJ-ABC, cementoenamel junction–alveolar bone crest; HSD, honestly significant difference; microCT, micro–computed tomography; MP, microparticle; Pg, Porphyromonas gingivalis.
In the ligature-induced mouse periodontitis model, administration of CCL2 MPs significantly reduced the alveolar bone loss around the second molar when compared with untreated or blank groups (Fig. 2D, E). Furthermore, as shown in Figure 3A and B, CCL2 MP injection indeed increased the M2 phenotype ratio. Intriguingly, there was no significant difference in pan-macrophages among groups (Fig. 3C).
Figure 3.
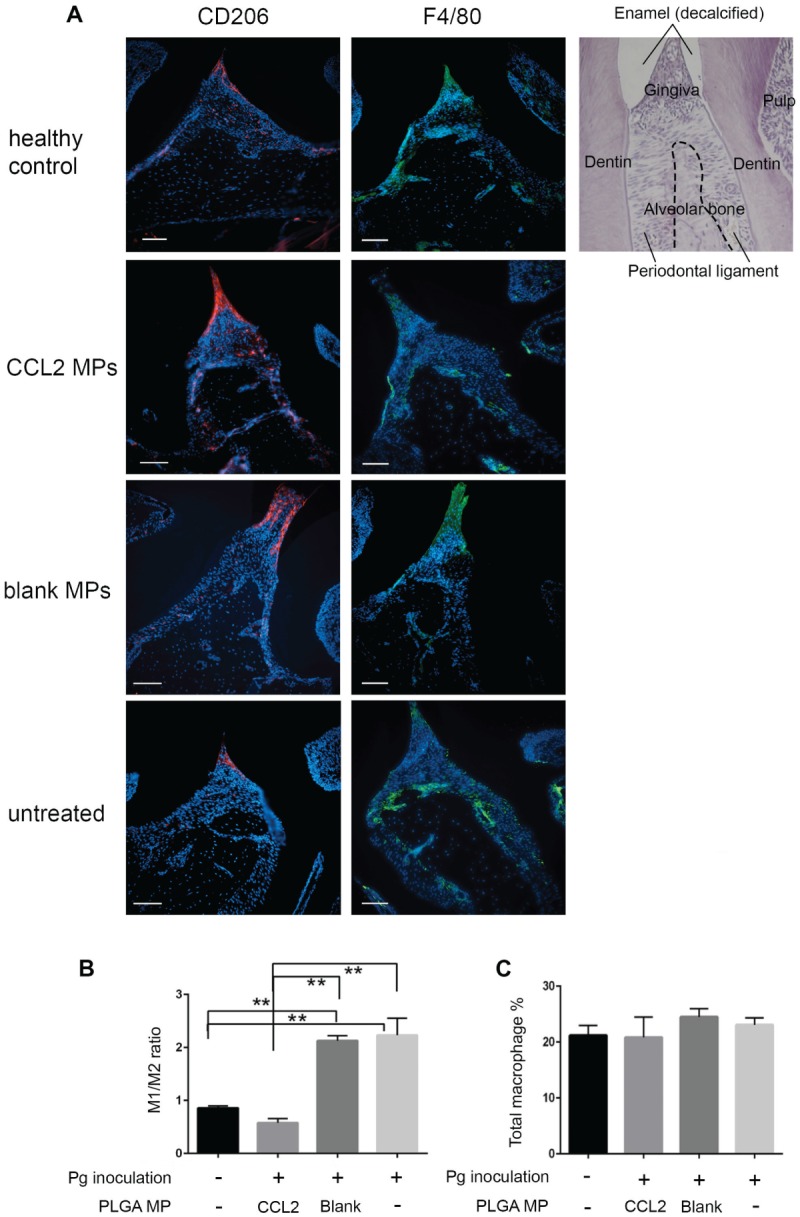
The sustained CCL2 release formulation significantly upregulates the M2 phenotype macrophage ratio in gingival papilla in the Pg-induced mouse periodontitis model. (a) Representative immunofluorescence staining of M2 phenotype macrophages (CD206+; red) and all mature macrophages (F4/80+ green) in gingival papillae in sagittal sections of maxillary molars from BALB/c mice infected with Pg 60 d after inoculation. DAPI counterstain presents location of all cells. Scale bar = 50 um. Final magnification 200×. AB, alveolar bone; DP, dental pulp; GP, gingival papillae; M, molar. Quantitative analysis of M2 phenotype macrophage percentage in gingival papillae per high-power field and M1 phenotype macrophage percentage (b), as well as M1 phenotype:M2 phenotype ratio (c). After CCL2 MP treatment, M2 phenotype percentage was increased. M1 phenotype percentage and M1 phenotype:M2 phenotype ratio both decreased, while no changes were made on the total load of macrophages when compared with untreated control or blank MP control. n = 3; *P < 0.05 and **P < 0.005 by 1-way analysis of variance and a post hoc Tukey honestly significant difference test. Error bars indicate SD. CCL2, C-C motif chemokine ligand 2; M1, classically activated macrophage; M2, alternatively activated macrophage; MP, microparticle; Pg, Porphyromonas gingivalis.
CCL2-Releasing MPs Reduce Numbers of Osteoclasts in the Alveolar Bone Region
With the Pg-induced periodontitis model, we observed that the number of osteoclasts was decreased significantly in the CCL2 MP group when compared with untreated or unloaded MP injection groups. In addition, our CCL2 MPs had the effect of reversing the osteoclast number in the periodontitis environment (Fig. 4A, B). Furthermore, there was a positive correlation between the number of osteoclasts and alveolar bone loss (r = 0.623, P = 0.018, n = 18; Fig. 4C). Overall, there was a moderate but significant correlation between periodontal osteoclast numbers and alveolar bone loss severity, with a correlation efficiency of 0.623. A significant decrease in osteoclast number in the CCL2 MP group was also observed in the ligature-induced model (Fig. 4D, E).
Figure 4.

The sustained CCL2 release formulation significantly downregulates osteoclast numbers in mouse periodontitis models. (a) Representative TRAP staining images from Pg mice model for osteoclasts. Row 1, left to right: healthy control, CCL2 MPs; row 2, left to right: blank, untreated. Each arrow or arrowhead indicates an osteoclast; final magnification, 200×. Scale bar = 100 um. (b) Comparison of osteoclasts number/mm2 of bone surface area among treatment groups; the healthy and CCL2 formulation–treated groups had statistically fewer osteoclasts than the untreated and blank groups. (c) Osteoclast numbers were significantly correlated with the severity of alveolar bone loss, measured as numbers of osteoclasts/mm2 of alveolar bone surface area to bone loss (the CEJ-ABC distance of the interdental planes), with a Pearson’s correlation coefficient of r = 0.623. n = 4 or 5/group. *P < 0.05, ***P < 0.001 by 1-way ANOVA, post hoc Tukey HSD test. (d) Representative TRAP staining pictures from ligature mice model. Row 1, left to right: healthy control, CCL2 MPs; row 2, left to right: blank MPs, untreated. Each arrow or arrowhead indicates an osteoclast; final magnification, 200×. All scale bars: 50 μm. (e) Comparison of osteoclasts numbers/mm2 of bone surface area among treatment groups; healthy and CCL2 formulation–treated groups had statistically fewer osteoclasts than either the untreated group or the blank PLGA MP group. n = 6/group. **P < 0.005 by 1-way ANOVA, post hoc Tukey HSD test. Error bars indicate SD. ANOVA, analysis of variance; CEJ-ABC, cementoenamel junction–alveolar bone crest; CCL2, C-C motif chemokine ligand 2; HSD, honestly significant difference; MP, microparticle; Pg, Porphyromonas gingivalis; TRAP, tartrate-resistant acidic phosphatase.
CCL2-Releasing MPs Alter Gene Expression of Periodontium toward an Anti-inflammatory Profile
Quantitative polymerase chain reaction results showed that the group with CCL2 MP injection expressed a significantly higher anti-inflammatory marker, IL-1RA, when compared with untreated control and blank PLGA control groups (Fig. 5A). In the meantime, we also observed a significantly lower mRNA expression level of RANKL in the CCL2 MP group (Fig. 5B).
Figure 5.

The sustained CCL2 release formulation leads to an increased mRNA expression of anti-inflammatory marker IL-1RA and decreased mRNA expression of osteoclast activity marker RANKL. mRNA expression of (a) IL-1RA and (b) RANKL in half maxilla as measured by quantitative PCR. The mRNA expression levels were compared by the value of 2(−ΔΔCt), with the healthy control group normalized to 1 and GAPDH as endogenous control. n = 9 or 10 mice; *P < 0.05 by 1-way analysis of variance, followed by post hoc Tukey honestly significant difference test. Error bar indicates SD. (c) The CCL2 released from MPs creates a gradient of CCL2 in the gingival soft tissue and attracts M0 and M2 phenotype macrophages to the inflamed site. Also, the accumulated CCL2 induces phenotypical polarization of M0 and M1 phenotype macrophages to M2 phenotype macrophages. The increased population of M2 phenotype macrophages (proved by immunohistochemistry) in the periodontal tissue decreases osteoclast number and activity (proved by micro–computed tomography, tartrate-resistant acidic phosphatase staining, and quantitative PCR). The possible crosstalk between macrophages and osteoclasts needs further investigation. CCL2, C-C motif chemokine ligand 2; IL-1RA, interleukin 1 receptor antagonist; M1, classically activated macrophage; M2, alternatively activated macrophage; MP, microparticle; PCR, polymerase chain reaction.
Discussion
It was reported that the macrophage phenotype is plastic and that signals from the surrounding microenvironment have an influence on its activation and function (Sica and Mantovani 2012; Martinez and Gordon 2014). CCL2 has recently emerged as an M2 phenotype polarization inducer, besides being a potent chemoattractant for the monocyte-macrophage lineage. In our study, we hypothesized that the local controlled delivery of CCL2 would recruit M0 and M2 phenotype macrophages to the inflamed site, induce M2 phenotype differentiation, and prevent alveolar bone loss. As depicted in Figure 5C, we propose that homing of M0 and M2 phenotype macrophages and the increase of M2 phenotype macrophages would decrease the production of proinflammatory cytokines from the M1 phenotype macrophages and increase the production of anti-inflammatory cytokines from M2 phenotype macrophage. Therefore, this change of cytokine profile would shift the inflammatory profile toward protective and reduce osteoclastic activity and subsequent bone loss (Fig. 5C). To prove this hypothesis, we first demonstrated that CCL2 induces M2 phenotype characters of murine macrophages in vitro. Furthermore, the viability of the CCL2 protein released from the PLGA MPs was confirmed with macrophage chemotaxis assay, which turned out to be as effective as rmCCL2 of the same amount in the matter of macrophage recruitment.
To confirm that our CCL2-releasing MPs have the ability to induce M2 phenotype macrophage polarization and change in situ, we tested the CCL2 MP murine periodontitis models.
Indeed, CCL2 MPs could effectively inhibit inflammation-mediated alveolar bone loss in the Pg- and ligature-induced periodontitis models. Furthermore, we confirmed that local administration of CCL2-releasing MPs increased the number of M2 phenotype with the Pg-induced periodontitis model.
Immunofluorescent staining of M2 phenotype macrophage–specific marker CD206 and pan–mature macrophage marker F4/80 revealed the presence of different macrophage subsets in periodontium and the changes of their presence in the periodontitis condition with CCL2 MP treatment. The increase in the M2 phenotype population and the decrease in the M1 phenotype:M2 phenotype ratio suggested that CCL2 MP treatment changed the local macrophage population from M1 phenotype predominant to M2 phenotype predominant. In addition, we speculate that the stable number of total macrophages under CCL2 stimulation indicates redirecting local macrophage polarization toward the M2 phenotype rather than recruiting M2 phenotype macrophages.
Our goal in this article is to show that recruiting M2 macrophages could be a potential therapy for periodontal disease. Regulating the immune response is a fascinating field and is rapidly evolving; our understanding of the role of macrophages and the various subtypes (M2a, b, and c) is extremely important and interesting and will be addressed in future studies. The more that we understand these cells, the better our therapy can be targeted.
The ligature-induced mouse periodontitis model is known for its quick and aggressive alveolar bone loss after ligature placement (Abe and Hajishengallis 2013). With this ligature model, we observed that net bone loss was significantly reduced by CCL2 MP administration as compared with an untreated control group and the blank PLGA MP-injected group.
Interestingly, the number of osteoclasts decreased significantly in CCL2 group in accordance with the amount of bone loss. Furthermore, 8 d after ligature placement, we observed that IL-1RA expression was significantly increased by CCL2 MPs as compared with untreated and blank MPs and healthy control. A recent study reported that IL-1RA could also promote macrophage polarization toward M2 phenotype (Luz-Crawford et al. 2016). In such a case, upregulation of IL-1RA in periodontal tissue could facilitate an additional polarization effect of local macrophages toward M2 phenotype. As for bone destruction markers, we found that the untreated group and blank PLGA group had an increased mRNA expression level of RANKL versus the healthy control group, which was consistent with previous reports and also explained the periodontitis-associated alveolar bone loss (Cochran 2008). Notably, the RANKL expression level was significantly decreased in the CCL2 MP group when compared with the untreated group and a blank MP group. Importantly, we observed a similar osteoclast number decrease and inhibition of inflammation-associated alveolar bone resorption in the Pg model and the ligature model, which indicates the efficacy of CCL2 MPs on both relatively rapid and slow bone resorption.
In conclusion, manipulation of endogenous M2 phenotype macrophages with CCL2 MPs decreased the M1 phenotype:M2 phenotype ratio and prevented alveolar bone loss in mouse periodontitis models. Thus, the delivery of CCL2 MPs provides a novel approach to treat periodontal disease.
Author Contributions
Z. Zhuang, S. Yoshizawa-Smith, contributed to conception, design, and data analysis, drafted the manuscript; A. Glowacki, K. Maltos, C. Pacheco, K. Verdelis, G.P. Garlet, S. Little, contributed to data analysis and interpretation, critically revised the manuscript; M. Shehabeldin, M. Mulkeen, N. Myers, R. Chong, contributed to data analysis, critically revised the manuscript; C. Sfeir, contributed to conception, design, and data analysis, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
Supplemental material, DS_10.1177_0022034518805984 for Induction of M2 Macrophages Prevents Bone Loss in Murine Periodontitis Models by Z. Zhuang, S. Yoshizawa-Smith, A. Glowacki, K. Maltos, C. Pacheco, M. Shehabeldin, M. Mulkeen, N. Myers, R. Chong, K. Verdelis, G.P. Garlet, S. Little and C. Sfeir in Journal of Dental Research
Acknowledgments
We also thank Dr. Albert Donnerberg for his flow data analysis assistance.
Footnotes
A supplemental appendix to this article is available online.
This work was supported by the National Institute of Dental and Craniofacial Research (grant 1R21DE025735-01). We thank Louis Riehl for graphic design and the SEM core facility and FACS facility from the University of Pittsburgh for their kind help and training.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
ORCID iD: G.P. Garlet  https://orcid.org/0000-0002-5071-8382
https://orcid.org/0000-0002-5071-8382
References
- Abe T, Hajishengallis G. 2013. Optimization of the ligature-induced periodontitis model in mice. J Immunol Methods. 394(1–2):49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamopoulos IE, Mellins ED. 2015. Alternative pathways of osteoclastogenesis in inflammatory arthritis. Nat Rev Rheumatol. 11(3):189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assuma R, Oates T, Cochran D, Amar S, Graves D. 1998. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. J Immunol. 160(1):403–409. [PubMed] [Google Scholar]
- Austyn JM, Gordon S. 1981. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol. 11(10):805–815. [DOI] [PubMed] [Google Scholar]
- Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, Bottazzi B, Doni A, Vincenzo B, Pasqualini F. 2006. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-κB and enhanced IRF-3/STAT1 activation). Blood. 107(5):2112–2122. [DOI] [PubMed] [Google Scholar]
- Cochran DL. 2008. Inflammation and bone loss in periodontal disease. J Periodontol. 79(8 Suppl):1569–1576. [DOI] [PubMed] [Google Scholar]
- Delima A, Oates T, Assuma R, Schwartz Z, Cochran D, Amar S, Graves D. 2001. Soluble antagonists to interleukin-1 (IL-1) and tumor necrosis factor (TNF) inhibits loss of tissue attachment in experimental periodontitis. J Clin Periodontol. 28(3):233–240. [DOI] [PubMed] [Google Scholar]
- Deshmane SL, Kremlev S, Amini S, Sawaya BE. 2009. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 29(6):313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paola R, Mazzon E, Maiere D, Zito D, Britti D, De Majo M, Genovese T, Cuzzocrea S. 2006. Rosiglitazone reduces the evolution of experimental periodontitis in the rat. J Dent Res. 85(2):156–161. [DOI] [PubMed] [Google Scholar]
- Eke PI, Dye BA, Wei L, Slade GD, Thornton-Evans GO, Borgnakke WS, Taylor GW, Page RC, Beck JD, Genco RJ. 2015. Update on prevalence of periodontitis in adults in the United States: NHANES 2009 to 2012. J Periodontol. 86(5):611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlet GP. 2010. Destructive and protective roles of cytokines in periodontitis: a re-appraisal from host defense and tissue destruction viewpoints. J Dent Res. 89(12):1349–1363. [DOI] [PubMed] [Google Scholar]
- Glowacki AJ, Yoshizawa S, Jhunjhunwala S, Vieira AE, Garlet GP, Sfeir C, Little SR. 2013. Prevention of inflammation-mediated bone loss in murine and canine periodontal disease via recruitment of regulatory lymphocytes. Proc Natl Acad Sci U S A. 110(46):18525–18530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez OA, Novak MJ, Kirakodu S, Stromberg A, Nagarajan R, Huang CB, Chen KC, Orraca L, Martinez-Gonzalez J, Ebersole JL. 2015. Differential gene expression profiles reflecting macrophage polarization in aging and periodontitis gingival tissues. Immunol Invest. 44(7):643–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. 2003. Alternative activation of macrophages. Nat Rev Immunol. 3(1):23–35. [DOI] [PubMed] [Google Scholar]
- Graves D. 2008. Cytokines that promote periodontal tissue destruction. J Periodontol. 79(8 Suppl):1585–1591. [DOI] [PubMed] [Google Scholar]
- Graves D, Cochran D. 2003. The contribution of interleukin-1 and tumor necrosis factor to periodontal tissue destruction. J Periodontol. 74(3):391–401. [DOI] [PubMed] [Google Scholar]
- Haffajee AD, Uzel NG, Arguello EI, Torresyap G, Guerrero DM, Socransky SS. 2004. Clinical and microbiological changes associated with the use of combined antimicrobial therapies to treat “refractory” periodontitis. J Clin Periodontol. 31(10):869–877. [DOI] [PubMed] [Google Scholar]
- Hassumi MY, Silva-Filho VJ, Campos-Junior JC, Vieira SM, Cunha FQ, Alves PM, Alves JB, Kawai T, Goncalves RB, Napimoga MH. 2009. PPAR-gamma agonist rosiglitazone prevents inflammatory periodontal bone loss by inhibiting osteoclastogenesis. Int Immunopharmacol. 9(10):1150–1158. [DOI] [PubMed] [Google Scholar]
- Johnson JD, Chen R, Lenton PA, Zhang G, Hinrichs JE, Rudney JD. 2008. Persistence of extracrevicular bacterial reservoirs after treatment of aggressive periodontitis. J Periodontol. 79(12):2305–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinane DF. 2001. Causation and pathogenesis of periodontal disease. Periodontology 2000. 25(1):8–20. [DOI] [PubMed] [Google Scholar]
- Kuo LC, Polson AM, Kang T. 2008. Associations between periodontal diseases and systemic diseases: a review of the inter-relationships and interactions with diabetes, respiratory diseases, cardiovascular diseases and osteoporosis. Public Health. 122(4):417–433. [DOI] [PubMed] [Google Scholar]
- Lam RS, O’Brien-Simpson NM, Holden JA, Lenzo JC, Fong SB, Reynolds EC. 2016. Unprimed, M1 and M2 macrophages differentially interact with Porphyromonas gingivalis. PLoS One. 11(7):e0158629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam RS, O’Brien-Simpson NM, Lenzo JC, Holden JA, Brammar GC, Walsh KA, McNaughtan JE, Rowler DK, Van Rooijen N, Reynolds EC. 2014. Macrophage depletion abates Porphyromonas gingivalis–induced alveolar bone resorption in mice. J Immunol. 193(5):2349–2362. [DOI] [PubMed] [Google Scholar]
- Luz-Crawford P, Djouad F, Toupet K, Bony C, Franquesa M, Hoogduijn MJ, Jorgensen C, Noel D. 2016. Mesenchymal stem cell–derived interleukin 1 receptor antagonist promotes macrophage polarization and inhibits B cell differentiation. Stem Cells. 34(2):483–492. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. 2004. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25(12):677–686. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Gordon S. 2014. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martuscelli G, Fiorellini JP, Crohin CC, Howard Howell T. 2000. The effect of interleukin-11 on the progression of ligature-induced periodontal disease in the beagle dog. J Periodontol. 71(4):573–578. [DOI] [PubMed] [Google Scholar]
- Pihlstrom BL, Michalowicz BS, Johnson NW. 2005. Periodontal diseases. Lancet. 366(9499):1809–1820. [DOI] [PubMed] [Google Scholar]
- Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. 2009. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J Biol Chem. 284(49):34342–34354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Mantovani A. 2012. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 122(3):787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sima C, Glogauer M. 2013. Macrophage subsets and osteoimmunology: tuning of the immunological recognition and effector systems that maintain alveolar bone. Periodontology 2000. 63(1):80–101. [DOI] [PubMed] [Google Scholar]
- Smiley CJ, Tracy SL, Abt E, Michalowicz BS, John MT, Gunsolley J, Cobb CM, Rossmann J, Harrel SK, Forrest JL, et al. 2015. Systematic review and meta-analysis on the nonsurgical treatment of chronic periodontitis by means of scaling and root planing with or without adjuncts. J Am Dent Assoc. 146(7):508–524e5. [DOI] [PubMed] [Google Scholar]
- Stein M, Keshav S, Harris N, Gordon S. 1992. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 176(1):287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela VM, Heller D, Silva-Senem MX, Torres MC, Colombo AP, Feres-Filho EJ. 2011. Systemic antimicrobials adjunctive to a repeated mechanical and antiseptic therapy for aggressive periodontitis: a 6-month randomized controlled trial. J Periodontol. 82(8):1121–1130. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Movila A, Kataoka S, Wisitrasameewong W, Ruiz Torruella M, Murakoshi M, Murakami S, Kawai T. 2016. Proinflammatory M1 macrophages inhibit RANKL-induced osteoclastogenesis. Infect Immun. 84(10):2802–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zhu Y, Duan D, Wang P, Xin Y, Bai L, Liu Y, Xu Y. 2017. Enhanced activity of macrophage M1/M2 phenotypes in periodontitis. Arch Oral Biol [epub ahead of print 11 Mar 2017]. doi: 10.1016/j.archoralbio.2017.03.006 [DOI] [PubMed] [Google Scholar]
- Zhang F, Liu H, Jiang G, Wang H, Wang X, Wang H, Fang R, Cai S, Du J. 2015. Changes in the proteomic profile during the differential polarization status of the human monocyte-derived macrophage THP-1 cell line. Proteomics. 15(4):773–786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, DS_10.1177_0022034518805984 for Induction of M2 Macrophages Prevents Bone Loss in Murine Periodontitis Models by Z. Zhuang, S. Yoshizawa-Smith, A. Glowacki, K. Maltos, C. Pacheco, M. Shehabeldin, M. Mulkeen, N. Myers, R. Chong, K. Verdelis, G.P. Garlet, S. Little and C. Sfeir in Journal of Dental Research