

Abstract
Periodontal diseases are initiated by bacteria that accumulate in a biofilm on the tooth surface and affect the adjacent periodontal tissue. Systemic diseases such as diabetes, rheumatoid arthritis (RA), and systemic lupus erythematosus (SLE) increase susceptibility to destructive periodontal diseases. In human studies and in animal models, these diseases have been shown to enhance inflammation in the periodontium and increase the risk or severity of periodontitis. All 3 systemic diseases are linked to a decrease in bacterial taxa associated with health and an increase in taxa associated with disease. Although there is controversy regarding the specific oral bacterial changes associated with each disease, it has been reported that diabetes increases the levels of Capnocytophaga, Porphyromonas, and Pseudomonas, while Prevotella and Selenomonas are increased in RA and Selenomonas, Leptotrichia, and Prevotella in SLE. In an animal model, diabetes increased the pathogenicity of the oral microbiome, as shown by increased inflammation, osteoclastogenesis, and periodontal bone loss when transferred to normal germ-free hosts. Moreover, in diabetic animals, the increased pathogenicity could be substantially reversed by inhibition of IL-17, indicating that host inflammation altered the microbial pathogenicity. Increased IL-17 has also been shown in SLE, RA, and leukocyte adhesion deficiency and may contribute to oral microbial changes in these diseases. Successful RA treatment with anti-inflammatory drugs partially reverses the oral microbial dysbiosis. Together, these data demonstrate that systemic diseases characterized by enhanced inflammation disturb the oral microbiota and point to IL-17 as key mediator in this process.
Keywords: bacteria, biofilm, dysbiosis, periodontitis, periodontium, inflammation
Oral Microbiome in Health
The microbiome has a significant impact on the host, as germ-free mice have increased immune diseases, such as asthma and inflammatory bowel disease, indicating a dynamic relationship between them (Olszak et al. 2012). The oral microbiome includes bacteria, fungi, archaea, viruses, and protozoa (Dewhirst et al. 2010). The bacterial component is the best understood and is the focus of this review. The formation of dental plaque is affected by the mode of delivery (vaginal or caesarean), breast or bottle-feeding, and proximity to siblings and pets (Dewhirst et al. 2010). Bacteria can be found on all oral tissues, and there is overlap in the bacteria found on each. The most abundant bacteria are Streptococcus oralis, Streptococcus mitis, and Streptococcus peroris. Bacteria associated with periodontal health include Streptococcus, Granulicatella, Neisseria, Haemophilus, Corynebacterium, Rothia, Actinomyces, Prevotella, and Capnocytophaga (Segata et al. 2012). A biofilm forms on the tooth surface, initiated by a pellicle that promotes bacterial adhesion, with Streptococcus and Actinomyces as early colonizers (Socransky and Haffajee 2005). The latter facilitate formation of a multispecies biofilm that is spatially organized and depends on coaggregation among bacterial taxa (Socransky and Haffajee 2005). The subgingival biofilm is typically more anaerobic than the supragingival biofilm (Socransky et al. 1998).
Changes in the Oral Microbiota Caused by Periodontal Disease
In a National Health and Nutrition Examination Study, 47% of US adults had evidence of periodontitis, and 10% to 15% had advanced periodontitis (Kinane et al. 2017). Periodontal diseases are thought to result from opportunistic infections. The specific factors leading to changes in bacteria that cause periodontal diseases are unknown, although it is recognized that nonideal restorations, genetic conditions that alter the host response, and systemic diseases, such as diabetes and rheumatoid arthritis (RA), predispose to disease (Kinane et al. 2017). The relationships between the biofilm and the host immune response are dynamic, and the ecologic interactions between them determine local homeostasis or transition to a state of disease (Dewhirst 2010; Griffen 2012). Inflammation occurs when bacteria or their products encounter leukocytes in the epithelium or underlying connective tissue. Bacterial challenge leads to the migration of dendritic cells (DCs) to lymph nodes and gingival epithelium. A loss of DCs in experimental periodontitis or a decrease in DC function through lineage-specific gene deletion increases periodontal inflammation, receptor activator of nuclear factor kappa-B ligand (RANKL) expression, and bone loss (Xiao et al. 2015). Interestingly, reduced DC activity diminishes activation of lymphocytes and formation of antibodies and is linked to increased periodontal disease susceptibility (Xiao et al. 2015).
Periodontitis is associated with a shift in the bacterial community structure and composition (Dewhirst 2010; Diaz 2012; Griffen et al. 2012). A noticeable change in this equilibrium is quantitative, with an increased bacterial biomass with up to a 3-log increase in subgingival bacterial load in periodontitis subjects (Diaz 2012). Qualitative changes also occur as a result of competitiveness among species (Diaz 2012; Griffen et al. 2012), leading to an increase in bacterial taxa that are pathogenic. Thus, the dynamic balance among various bacterial taxa is likely to be instrumental in determining periodontal disease activity. Socransky and colleagues (1998) identified combinations of Bacteroides forsythus, Porphyromonas gingivalis, and Treponema denticola as being highly associated with clinical measures of periodontal disease. Fine and coworkers (2013) reported that a combination of Aggregatibacter actinomycetemcomitans, Streptococcus parasanguinis, and Filifactor alocis was associated with bone loss in localized aggressive periodontitis (Fine et al. 2013).
In gingivitis and periodontitis, there is likely to be an alteration in microbial composition or bacterial pathogenicity which is currently referred to as microbial dysbiosis (Roberts and Darveau 2015). Unlike many infectious diseases, periodontitis is initiated by bacteria that are likely already present, rather than by the introduction of an exogenous taxa. Putative periodontal pathogens are often found at healthy sites with no evidence of periodontal breakdown (Dewhirst 2010; Diaz 2012; Griffen et al. 2012). The composition of the microbiota is governed by local factors, but systemic factors can also have a significant effect. One mechanism is that systemic inflammatory diseases may increase local inflammation, which alters the expression of inflammatory mediators locally and increases recruitment of leukocytes to the periodontium (see Fig. 1). An alternative hypothesis that was popular in the mid-1900s and has some recent support postulates that an overall increase in microbial biomass, rather than a specific change in composition, may promote periodontitis. This is supported by increased bacterial loads in humans with periodontitis and in mice as they age and develop periodontitis, with increased susceptibility to colonization by periodontal pathogens such as P. gingivalis (Wu et al. 2016).
Figure 1.
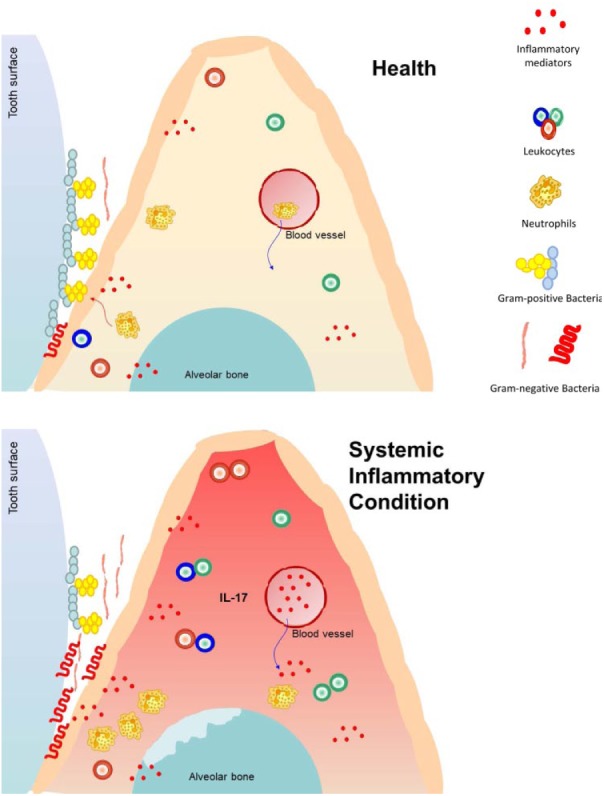
The impact of a systemic health condition on the periodontium and associated microbiota. In periodontal health there is little inflammation and gram-positive, aerobic species predominate. With a systemic inflammatory diseases associated with increased inflammation, there is a subsequent increase in periodontal inflammation, with recruitment of more inflammatory cells favoring colonization by facultative and anaerobic bacteria and gram-negative taxa. The change in bacteria may enhance inflammation.
Changes in the Oral Microbiota Caused by Diabetes
Systemic diseases with increased inflammation are frequently linked to increased risk of periodontal disease (Jepsen et al. 2018). Increased levels of periodontal disease, diabetes, systemic lupus erythematosus (SLE), and RA may reflect a common susceptibility, since each has inflammation as a common risk factor. Those individuals with a greater tendency toward inflammation have enhanced susceptibility and risk of periodontitis as well as increased levels of diabetes, SLE, and RA. The increased inflammation in each of these diseases provides a common framework for altering the oral microbiota.
Systemic diseases have a significant impact on periodontitis, with diabetes mellitus having one of the strongest linkages. Type 1 and type 2 diabetes mellitus (T1DM and T2DM) affect the periodontium in children and adults, with an increase in periodontal inflammation similar to the increased inflammation in other tissues affected by diabetes (Wu et al. 2015). Inoculation of the same bacterial load in the connective tissue of diabetic animals stimulates a greater inflammatory response compared with normoglycemic controls (Naguib et al. 2004). Diabetes potentially affects a number of factors that can enhance inflammation in periodontal tissues, including high glucose levels, increased formation of advanced glycation end products, and enhanced cytokine expression (e.g., tumor necrosis factor [TNF]; Wu et al. 2015). Neutrophils and monocytes/macrophages from diabetic patients express higher levels of cytokines in response to stimulation and are less effective in bacterial killing (Omori et al. 2008). Diabetes also affects mesenchymal cells in the host, including periodontal ligament cells, osteoblasts, and osteocytes that increase RANKL expression, and reduce coupled bone formation following an episode of periodontal bone loss (Wu et al. 2015). Cause-and-effect relationships have been established showing that blocking advanced glycation endproduct (AGE) signaling reduces inflammatory cytokine levels (including TNF), matrix metalloproteinase expression, and periodontal bone loss (Lalla et al. 2000).
T1DM and T2DM are associated with similar complications, particularly those linked to increased inflammation, such as cardiovascular disease, impaired wound healing, neuropathy, nephropathy, and periodontal disease (Chapple et al. 2013; Wu et al. 2015). T1DM and T2DM increase the inflammatory response to the same bacterial challenge (Naguib et al. 2004; Graves et al. 2005). The increased periodontal inflammation observed in T1DM and T2DM has been implicated in periodontal tissue damage triggered by bacteria that colonize the tooth surfaces. An important question is whether the increased periodontal destruction in diabetic subjects is strictly due to an altered host response, whether there is a change in bacterial pathogenicity that leads to greater inflammation and damage, or both.
A consensus report from the European Federation of Periodontology and the American Academy of Periodontology found no compelling evidence that diabetes has a significant impact on the oral microbiota (Chapple et al. 2013). Thus, human studies examining the impact of diabetes in the oral microbiome have not provided conclusive evidence for a significant change or no change. Similarly, there is no consensus that T1DM induces a specific bacterial change as compared with T2DM. However, some studies do report diabetes-induced alterations in the oral microbiome. Examples of these bacterial changes include 1) increased Capnocytophaga in patients with diabetes mellitus (Mashimo et al. 1983); 2) increased P. gingivalis and Tannerella forsythia (Campus et al. 2005; da Cruz et al. 2008); and 3) increased Capnocytophaga, Pseudomonas, Bergeyella, Sphingomonas, Corynebacterium, Propionibacterium, and Neisseria in hyperglycemic individuals (Ganesan et al. 2017). Note that these results are often not consistent, as it has been reported that diabetes reduces Porphyromonas, Filifactor, Eubacterium, Synergistetes, Tannerella, and Treponema genera (Casarin et al. 2012). Thus, previous human studies have not revealed consistent changes in microbial composition, in part because of the limited numbers of bacteria surveyed. It has also been proposed that there are differences in overall oral microbiome in normoglycemic and diabetic subjects rather than between healthy and diseased sites in the same individual (Ganesan et al. 2017). Recent high-throughput analysis suggests that there may be differences between diabetic and normal cohorts, but there is yet no consensus on the changes induced, as shown in Table 1 (Casarin et al. 2012; Aemaimanan et al. 2013; Zhou et al. 2013; Merchant et al. 2014; Sakalauskiene et al. 2014; Castrillon et al. 2015; Demmer et al. 2016). There may be a number of potential reasons for a lack of consensus—including statistical reasons based on large numbers of oral microorganisms yielding false positives; insufficient subject numbers, which may contribute to false negatives; presence of confounding factors, including the degree of hyperglycemia, duration of disease, and medications; technical limitations, such as the lack of unbiased approaches to identify oral bacteria; and a limited number of longitudinal studies.
Table 1.
Major Changes in Bacterial Community Associated with Diabetes, RA, or SLE.
| Oral Bacterial Diversity |
Oral Bacterial Biomass |
Bacterial Changes Associated with
Disease |
Bacterial Changes Associated with
Health |
||||
|---|---|---|---|---|---|---|---|
| Human | Animals | Humans | Animals | Humans | Animals | Humans | Animals |
| Effect of diabetes | |||||||
| Reduceda; no change in alpha diversityb | Reducedc | Increasedd,e | No changec | Increased 1) facultative bacteria in subjects with periodontitis, 2) anaerobes in subjects with no periodontitis,a and 3) disease-associated bacteriaf | Increased disease- associated bacteriac | Reduced health-associated bacteriaa, f | Reduced health-associated bacteriac |
| Effect of SLE | |||||||
| Reducedg | No data | Increasedg | No data | Increased disease-associated bacteriag | No data | Reduced abundance of health-associated bacteriag | No data |
| Effect of RA | |||||||
| Increasedh | No data | Increasedh | Increasedi | Increased disease- associated bacteriah,j | Increased anaerobesk | Reduced abundance of health-associated bacteriah,j | Reduced abundance of health-associated bacteriak |
RA, rheumatoid arthritis; SLE, systemic lupus erythematosus.
Corrêa et al. (2018, unpublished results).
We carried out a longitudinal study in mice that spontaneously develop diabetes. The diabetes-prone and normoglycemic littermates initially had similar oral microbiomes. Development of hyperglycemia induced a shift in the bacterial composition (Xiao et al. 2017), increasing the levels of Proteobacteria (Enterobacteriaceae) and Firmicutes (Enterococcus, Staphylo-coccus, and Aerococcus) associated with pathologic changes (Demmer et al. 2016; Grice et al. 2010). Diabetes also reduced the diversity of oral microbiota as reported in other tissues where diabetes reduces bacterial diversity (Ussar et al. 2016). Interestingly, we have found that aging in mice also reduces oral bacterial diversity, which is accompanied by greater colonization of P. gingivalis when exposed to this human pathogen (Xiao et al. 2016).
Studies were undertaken to establish whether diabetes rendered the oral microbiota more pathogenic and to identify the potential mechanisms involved. To accomplish the former, the diabetic oral microbiota was transferred from diabetic hosts to normal germ-free mice and compared with the transfer of bacteria from normoglycemic donors to similar hosts. Transfer from hyperglycemic mice stimulated greater neutrophil infiltration and increased expression of bone-resorbing cytokines (IL-6 and RANKL), enhanced osteoclast numbers, and stimulated more periodontal bone loss than bacteria from normoglycemic controls. Inhibition of IL-17 altered the microbial composition in diabetic mice so that it was more similar to the normoglycemic oral microbiota and reduced the abundance of Enterococcus in particular. IL-17 inhibition decreased the capacity of bacteria from diabetic animals to stimulate inflammation and alveolar bone loss in normal germ-free recipients. We propose that chronic inflammation enhanced by diabetes causes microbial dysbiosis that increases bacterial pathogenicity (see Fig. 2). Inhibition of IL-17 could alter the microbiota by reducing inflammation, which in turn affects bacterial growth or colonization. This may occur as a result of reduced formation of substrates generated as a by-product of inflammation or, alternatively, by affecting antibacterial defenses (Curtis et al. 2011). Interestingly, high IL-17 levels are found in humans with periodontitis, and IL-17 induces RANKL (Abusleme and Moutsopoulos 2017). Leukocyte adhesion deficiency in humans results in greater IL-17 production and formation of dysbiotic bacterial communities, thereby identifying IL-17 as a cause of microbial dysbiosis (Abusleme and Moutsopoulos 2017). Similarly, patients with oral lichen planus have increased disease-associated bacterial species and increased IL-17 in saliva (Wang et al. 2014).
Figure 2.

Diabetes induces microbial changes in a murine model. Diabetes increases IL-17 expression, leading to increased periodontal inflammation and a change in bacterial composition that is more pathogenic when transferred to a germ-free host as compared with a transfer from normoglycemic animals. Inhibition of IL-17 reduces the pathogenicity of the diabetic microbiota (for details, see Xiao et al. 2017).
In skin wounds, diabetes alters the wound microbiota, which is linked to increased inflammation (Grice et al. 2010). Similarly, the gut microbiota changes with diabetes, which is subsequently altered when diabetes is reversed by bariatric surgery (Sweeney and Morton 2013). Moreover, when IL-17 was inhibited, the oral microbiota shifted in a direction of reduced pathogenicity and to a composition that was more similar to that of normoglycemic control animals (Xiao et al. 2017), suggesting that controlling inflammation increases the likelihood of having a “nonpathogenic” oral bacterial profile. Furthermore, there may be a bidirectional relationship between the gut microbiota and diabetes whereby 1) diabetes alters bacterial composition and 2) changes in bacterial profile enhance susceptibility to diabetes through a butyrate-mediated mechanism. Butyrate-producing bacteria are reduced in individuals with T2DM, and butyrate protects against the development of diabetes (Arora and Bäckhed 2016); thus, reduced production of butyrate may increase risk of diabetes. Oral bacteria may also have an impact on diabetes, although the evidence to support this link is limited. Oral administration of P. gingivalis coincides with the development of systemic inflammation and onset of insulin resistance in mice (Arimatsu et al. 2014). The increased systemic inflammation may be linked to a reduction in barrier function in the intestine caused by oral inoculation of P. gingivalis (Sato et al. 2017). Oral bacteria may contribute to a bidirectional relationship between periodontal disease and diabetes, whereby changes in the oral microbiota through increased systemic inflammation enhance insulin resistance to promote hyperglycemia (as discussed by Borgnakke et al. 2013).
Changes in the Oral Microbiota Caused by Rheumatoid Arthritis
RA is a systemic autoimmune disease characterized by chronic inflammation (de Smit et al. 2015). Periodontal disease and RA share some pathogenic mechanisms, including enhanced inflammation and bone loss. Increased prevalence of periodontitis is observed in patients with RA (de Smit et al. 2015). There is also evidence that periodontitis can initiate RA by the production of enzymes that form malondialdehyde-acetaldehyde, citrullinated and carbamylated adducts that enhance self-antigenicity to initiate an autoimmune response (de Smit et al. 2015). We and others have shown that experimental arthritis in rodents promotes alveolar bone loss (Ramamurthy et al. 2005; Queiroz-Junior et al. 2011; Corrêa et al. 2016; Kim et al. 2017). Treatment with oral antiseptics that reduce the total oral bacterial load protects against RA-induced periodontal bone loss, indicating a role for oral microbiota (Queiroz-Junior et al. 2011). The data are consistent with a “2-hit” model, in which one hit represents the oral microbiota and the second hit the impact of the systemic disease on the local inflammation. RA may amplify the inflammatory response in the periodontium, which in turn modifies the microbiota (Golub et al. 2006). Chronic systemic inflammation in RA may affect levels of inflammatory cytokines in oral tissues (Mirrielees et al. 2010). RA in rodents increases the expression of inflammatory cytokines in the periodontium, such as TNF-α, IL-1, IL-6, and IL-17 (Queiroz-Junior et al. 2011). RA subjects also show increased salivary concentration of IL-17, TNF-α, and IL-33 (Corrêa et al. 2018, unpublished results) similar to observations in SLE (Corrêa et al. 2017). It is noteworthy that studies examining other diseases showed a link between IL-17 and disturbances in the microbiota, such as LAD-1 (leukocyte adhesion deficiency 1) and oral lichen planus (Moutsopoulos et al. 2014; Wang et al. 2014).
RA alters the oral microbiome qualitatively and quantitatively in animal studies. Mice with RA exhibit higher levels of Parvimonas micra, Selenomonas noxia, and Veionella parvula (Corrêa et al. 2016). In humans, the RA-associated microbiome is substantially different from that of healthy controls. Anaerobic species such as Lactobacillus salivarius, Atopobium, Leptotrichia, Prevotella, and Cryptobacterium curtum are enriched in the oral microbiota of RA patients, and health-associated Corynebacterium and Streptococcus genera are reduced (Scher et al. 2012; Zhang et al. 2015). We also found that RA increases the microbial biomass approximately 1-log and alters the composition with increased numbers of pathogenic bacteria (Corrêa et al. 2018, unpublished results). RA patients without periodontitis have an enrichment in periodontitis-associated bacteria, such as Prevotella (e.g., P. melaninogenica, P. denticola, P. histicola, P. nigrescens, P. oulorum, and P. maculosa), and other pathogenic species (S. noxia, S. sputigena, and Anaeroglobus geminatus) (Corrêa et al. 2018, unpublished data). In addition, RA subjects have a reduction of health-associated species (Streptococcus, Rothia aeria, Kingella oralis, Haemophilus, and Actinomyces) (Corrêa et al. 2018, unpublished data). These data are summarized in Table 2.
Table 2.
Specific Changes in the Bacterial Community Associated with Diabetes, RA, or SLE.
| Genera | ||
|---|---|---|
| Elevated in Human Diabetes | Elevated in Human SLE | Elevated in Human RA |
| Capnocytophaga, Pseudomonas, Bergeyella, Sphingomonas, Corynebacterium, Propionibacterium, and Neisseriaa | Prevotella oulorum, P. nigrescens, P. oris, Selenomonas noxia, Leptotrichia, and Lachnospiraceaeb | Lactobacillus salivarius, Atopobium, and Cryptobacterium curtumc |
| Streptococcus, Actinomyces, and Rothiad | Leptotrichia and Prevotellae | |
| Prevotella, Tannerella, and Pseudomonasf | Prevotella (e.g., P. melaninogenica, P. denticola, P. histicola, P. nigrescens, P. oulorum, P. maculosa), Selenomonas noxia, S. sputigena, and Anaeroglobus geminatusg | |
| TM7, Aggregatibacter, Neisseria, Gemella, Eikenella, Selenomonas, Actinomyces, Capnocytophaga, Fusobacterium, Veillonella, and Streptococcush | ||
| Reduced in Human Diabetes | Reduced in Human SLE | Reduced in Human RA |
| Actinomyces, Atopobium, Bifidobacterium, Scardovia, Atopobium, Corynebacterium, and Rothiai | Capnocytophaga, Rothia, Haemophilus parainfluenzae, and Streptococcusb | Corynebacterium, Streptococcus, and Haemophilusc |
| Porphyromonas, Filifactor, Eubacterium, Synergistetes, Tannerella, and Treponemah | Streptococcus, Rothia aeria, Kingella oralis, Haemophilus, and Actinomycesg | |
RA, rheumatoid arthritis; SLE, systemic lupus erythematosus.
Corrêa et al. (2018, unpublished results).
The microbial composition of the gut in early RA differs from controls, with a reduction of Bifidobacterium and Bacteroides and an increase in Prevotella (Horta-Baas et al. 2017). Similarly, Prevotella species are also enriched in the saliva (Zhang et al. 2015) and subgingival microbiota (Corrêa et al. 2018, unpublished) of RA subjects. Interestingly, Prevotella copri has a high capacity to induce Th17-related cytokines, and Prevotella sp. is associated with Th17-mediated mucosal inflammation (Larsen 2017).
The increased levels of inflammatory mediators in the periodontal tissues of subjects with RA and other diseases may change the ecologic conditions to favor pathogenic bacterial species and promote periodontitis (Abusleme and Moutsopoulos 2017). Inflamed subgingival sites, which exhibit a low redox potential, support a microbial community with higher proportions of obligate anaerobic bacteria (Dewhirst 2010; Diaz 2012; Griffen et al. 2012). Local inflammation enhanced by systemic disease may change the microbial composition toward one that is adapted to an inflammatory environment and more capable of inducing inflammation. The increased inflammation caused by RA coupled with microbial changes may amplify periodontal inflammation and explain the greater susceptibility to periodontitis that we and others have observed in these patients (de Smit et al. 2015). Therefore, changes in systemic and local inflammation may disrupt microbial homeostasis and consequently increase bacterial pathogenicity and periodontal disease susceptibility (see Table 1). Conversely, RA treatment improves periodontal status and affects the oral microbiome (Zhang et al. 2015). Disease-modifying antirheumatic drugs reduce inflammation and the severity of RA, and alter the gut and oral microbiota (Zhang et al. 2015). For example, bacteria that are associated with oral health as Prevotella maculosa are increased after RA treatment. These results suggest that controlling inflammation can shift the microbiota to a “healthy status,” restoring systemic and oral/periodontal homeostasis.
Oral Microbiota and SLE
SLE is an autoimmune disease characterized by persistent inflammation that leads to tissue damage in multiple organs, including kidneys, lungs, joints, heart, and brain. The etiology of SLE includes genetic and environmental factors (López et al. 2016) and is linked to microbial dysbiosis (Hevia et al. 2014; López et al. 2016; Corrêa et al. 2017).
In the oral cavity, the manifestations of SLE include unspecified oral ulcers (Jensen et al. 1999), xerostomia, hyposalivation (Jensen et al. 1999), and increased periodontal disease (Mutlu et al. 1993; Kobayashi et al. 2003; Corrêa et al. 2017). In a meta-analysis, a 1.76-fold increased risk of periodontal disease in SLE subjects was reported (Rutter-Locher et al. 2017). The increased risk is associated with changes in local and systemic inflammatory pathways, as demonstrated by elevated levels of cytokines (e.g., IL-6, IL-17, and IL-33) in the saliva of SLE patients (Marques et al. 2016; Corrêa et al. 2017; Mendonça et al. 2018). The inflammatory changes have been linked to dysbiosis of the subgingival biofilm in SLE subjects, as described in Figure 1. These observations are based on human studies. However, functional studies linking specific microbiota disturbances and inflammatory pathways with periodontal damage in SLE have not been reported as they have been for diabetes (Xiao 2017).
It was demonstrated that SLE patients have a higher bacterial load as compared with healthy subjects (Jensen et al. 1999; Corrêa et al. 2017) with an altered bacterial composition. High counts of Lactobacilli and Candida albicans are found in the oral cavity of SLE patients versus controls (Jensen et al. 1999). SLE subjects have reduced microbial diversity with greater proportions of pathogenic bacteria (Corrêa et al. 2017). Bacteria associated with periodontal disease, including Prevotella oulorum, P. nigrescens, P. oris, S noxia, Leptotrichia, and Lachnospiraceae, are elevated in SLE subjects, even in periodontally healthy sites (Corrêa et al. 2017). In addition, bacteria commonly associated with periodontal health, such as Capnocytophaga, Rothia, Haemophilus parainfluenzae, and Streptococcus, are diminished in SLE patients with periodontitis. Changes in the microbiota in SLE patients are summarized in Tables 1 and 2. Moreover, the presence of pathogenic bacteria is positively correlated with the level of systemic inflammation, measured by serum C-reactive protein (Corrêa et al. 2017). Overall, a worsening periodontal condition is correlated with systemic inflammation (Corrêa et al. 2017). In line with these findings, periodontal treatment improves the response to conventional therapy in SLE patients with a reduction of disease activity (Fabbri et al. 2014). Increased inflammation may provide a source of nutrients in the form of tissue breakdown products and may alter the environment, favoring the growth of anaerobic bacteria (Hajishengallis 2014). In turn, changes in the microbiota might be important to amplify local inflammation and periodontal tissue damage, worsening the impact of the systemic disease on periodontal health. Taken together, these data highlight the connection between the microbiota and SLE and suggest that reduced inflammation will promote the formation of a less pathogenic oral microbial profile.
Changes in the gut microbiome in SLE patients have been reported and are reflected by increased diversity compared with healthy individuals (Hevia et al. 2014; López et al. 2016). Lupus-prone mice have depleted lactobacilli and an increase in Clostridial species (Lachnospiraceae) associated with an overall increase in bacterial diversity (Zhang et al. 2014; Mu et al. 2017). As a proof of concept, increasing Lactobacillales in the gut of mice by adding a mixture of 5 Lactobacillus strains led to an improvement of SLE severity and prolonged survival (Mu et al. 2017). In line with these results, the relative abundance of Lactobacillales seems to be restored in SLE patients without active disease (Hevia et al. 2014).
Concluding Remarks
Systemic diseases may share common risk factors that lead to an increased inflammatory response when there is an interruption in equilibrium. For example, the host response to oral bacteria may be altered by systemic diseases such as diabetes, RA, SLE, and LAD-1 so that the degree of inflammation induced is greater than it would be in normal conditions. The local periodontal inflammation may in turn alter the competition among bacterial species by affecting nutrient availability or redox potential and/or altering gene expression within a bacterial community. These changes may then enhance the pathogenicity of the microbiota to further activation of the inflammatory pathways. IL-17 and other cytokines may play a pivotal role in this process. Microbial changes observed in systemic diseases generally reduce microbial diversity and shift bacterial composition by increasing taxa that are generally considered to be “bad” and reducing those that are “good” (see Table 1). However, the assignment to “good” and “bad” categories is an oversimplification and needs to be experimentally justified, as recently reported for murine diabetic studies (Xiao et al. 2017). Furthermore, it is widely recognized that several systemic diseases cause increased periodontal inflammation with the assumption that the primary component was a straightforward upregulation of pro-osteoclastogenic factors (Xiao et al. 2016). However, this may be an oversimplification, as the inflammatory changes may induce an amplification loop in which the bacterial component becomes more pathogenic. We propose that a central mechanism for altering the microbiota is increased IL-17. We also recognize that IL-17 operates in concert with other inflammatory cytokines and that an overall dysregulation of these mediators may contribute to host alterations that lead to microbial dysbiosis. This concept and the downstream events that specifically alter the composition or behavior (gene expression) of bacteria need further investigation to render this hypothesis more precise.
Author Contributions
D.T. Graves, contributed to conception, design, and data interpretation, drafted and critically revised the manuscript; J.D. Corrêa, T.A. Silva, contributed to design and data interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Footnotes
This study was supported by grants from the National Institute of Dental and Craniofacial Research (R01DE 017732 and R01DE 021921 for D.T.G.) and a research scholarship from the Brazilian National Council for Scientific and Technological Development (for T.A.S.).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Abusleme L, Moutsopoulos NM. 2017. IL-17: overview and role in oral immunity and microbiome. Oral Dis. 23(7):854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aemaimanan P, Amimanan P, Taweechaisupapong S. 2013. Quantification of key periodontal pathogens in insulin-dependent type 2 diabetic and non-diabetic patients with generalized chronic periodontitis. Anaerobe. 22:64–68. [DOI] [PubMed] [Google Scholar]
- Anbalagan R, Srikanth P, Mani M, Barani R, Seshadri KG, Janarthanan R. 2017. Next generation sequencing of oral microbiota in type 2 diabetes mellitus prior to and after neem stick usage and correlation with serum monocyte chemoattractant-1. Diabetes Res Clin Pract. 130:204–210. [DOI] [PubMed] [Google Scholar]
- Arimatsu K, Yamada H, Miyazawa H, Minagawa T, Nakajima M, Ryder MI, Gotoh K, Motooka D, Nakamura S, Iida T, et al. 2014. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci Rep. 4:4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora T, Bäckhed F. 2016. The gut microbiota and metabolic disease: current understanding and future perspectives. J Intern Med. 280(4):339–349. [DOI] [PubMed] [Google Scholar]
- Borgnakke WS, Ylöstalo PV, Taylor GW, Genco RJ. 2013. Effect of periodontal disease on diabetes: systematic review of epidemiologic observational evidence. J Periodontol. 84(4):S135–S152. [DOI] [PubMed] [Google Scholar]
- Campus G, Salem A, Uzzau S, Baldoni E, Tonolo G. 2005. Diabetes and periodontal disease: a case-control study. J Periodontol. 76(3):418–425. [DOI] [PubMed] [Google Scholar]
- Casarin R, Barbagallo A, Meulman B, Santos V, Sallum E, Nociti F, Duarte P, Casati M, Gonçalves R. 2012. Subgingival biodiversity in subjects with uncontrolled type-2 diabetes and chronic periodontitis. J Periodontal Res. 48(1):30–36. [DOI] [PubMed] [Google Scholar]
- Castrillon CA, Hincapie JP, Yepes FL, Roldan N, Moreno SM, Contreras A, Botero JE. 2015. Occurrence of red complex microorganisms and Aggregatibacter actinomycetemcomitans in patients with diabetes. J Investig Clin Dent. 6(1):25–31. [DOI] [PubMed] [Google Scholar]
- Chapple ILC, Genco R; Working Group 2 of Joint EFP/AAP Workshop. 2013. Diabetes and periodontal diseases: consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J Periodontol. 40 Suppl 14:S106–S112. [DOI] [PubMed] [Google Scholar]
- Coelho A, Paula A, Mota M, Laranjo M, Abrantes M, Carrilho F, Ferreira M, Silva M, Botelho F, Carrilho E. 2018. Dental caries and bacterial load in saliva and dental biofilm of type 1 diabetics on continuous subcutaneous insulin infusion. J Appl Oral Sci. 26:e20170500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrêa JD, Calderaro DCDC, Ferreira GA, Mendonça SMS, Fernandes GR, Xiao E, Teixeira ALAL, Leys EJ, Graves DT, Silva TATATA, et al. 2017. Subgingival microbiota dysbiosis in systemic lupus erythematosus: association with periodontal status. Microbiome. 5(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrêa JD, Saraiva AM, Queiroz-Junior CM, Madeira MF, Duarte PM, Teixeira MM, Souza DG, da Silva TA. 2016. Arthritis-induced alveolar bone loss is associated with changes in the composition of oral microbiota. Anaerobe. 39:91–96. [DOI] [PubMed] [Google Scholar]
- Curtis MA, Zenobia C, Darveau RP. 2011. The relationship of the oral microbiotia to periodontal health and disease. Cell Host Microbe. 10(4):302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cruz GA, de Toledo S, Sallum EA, Sallum AW, Ambrosano GM, de Cássia Orlandi Sardi J, da Cruz SE, Gonçalves RB. 2008. Clinical and laboratory evaluations of non-surgical periodontal treatment in subjects with diabetes mellitus. J Periodontol. 79(7):1150–1157. [DOI] [PubMed] [Google Scholar]
- de Groot PF, Belzer C, Aydin Ö, Levin E, Levels JH, Aalvink S, Boot F, Holleman F, van Raalte DH, Scheithauer TP, et al. 2017. Distinct fecal and oral microbiota composition in human type 1 diabetes, an observational study. PLoS One. 12:e0188475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demmer RT, Breskin A, Rosenbaum M, Zuk A, LeDuc C, Leibel R, Paster B, Desvarieux M, Jacobs DR, Jr, Papapanou PN. 2016. The subgingival microbiome, systemic inflammation and insulin resistance: the Oral Infections, Glucose Intolerance and Insulin Resistance Study. J Clin Periodontol. 44(3):255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Smit MJ, Westra J, Brouwer E, Janssen KM, Vissink A, van Winkelhoff AJ. 2015. Periodontitis and rheumatoid arthritis: what do we know? J Periodontol. 86(9):1013–1019. [DOI] [PubMed] [Google Scholar]
- Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, Lakshmanan A, Wade WG. 2010. The human oral microbiome. J Bacteriol. 192(19):5002–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz PI. 2012. Microbial diversity and interactions in subgingival biofilm communities. Front Oral Biol. 15:17–40. [DOI] [PubMed] [Google Scholar]
- Fabbri C, Fuller R, Bonfá E, Guedes LK, D’Alleva PS, Borba EF. 2014. Periodontitis treatment improves systemic lupus erythematosus response to immunosuppressive therapy. Clin Rheumatol. 33(4):505–509. [DOI] [PubMed] [Google Scholar]
- Fine DH, Markowitz K, Fairlie K, Tischio-Bereski D, Ferrendiz J, Furgang D, Paster BJ, Dewhirst FE. 2013. A consortium of Aggregatibacter actinomycetemcomitans, Streptococcus parasanguinis, and Filifactor alocis is present in sites prior to bone loss in a longitudinal study of localized aggressive periodontitis. J Clin Microbiol. 51(9):2850–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan SM, Joshi V, Fellows M, Dabdoub SM, Nagaraja HN, O’Donnell B, Deshpande NR, Kumar PS. 2017. A tale of two risks: smoking, diabetes and the subgingival microbiome. ISME J. 11(9):2075–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub LM, Payne JB, Reinhardt RA, Nieman G. 2006. Can systemic diseases co-induce (not just exacerbate) periodontitis? A hypothetical “two-hit” model. J Dent Res. 85(2):102–105. [DOI] [PubMed] [Google Scholar]
- Graves DT, Naguib G, Lu H, Leone C, Hsue H, Krall E. 2005. Inflammation is more persistent in type 1 diabetic mice. J Dent Res. 84(4):324–328. [DOI] [PubMed] [Google Scholar]
- Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, Podar M, Leys EJ. 2012. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 6(6):1176–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Snitkin ES, Yockey LJ, Bermudez DM; NISC Comparative Sequencing Program, Liechty KW, Segre JA. 2010. Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proc Natl Acad Sci U S A. 107(33):14799–14804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G. 2014. The inflammophilic character of the periodontitis-associated microbiota. Mol Oral Microbiol. 29(6):248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevia A, Milani C, López P, Cuervo A, Arboleya S, Duranti S, Turroni F, González S, Suárez A, Gueimonde M, et al. 2014. Intestinal dysbiosis associated with systemic lupus erythematosus. MBio. 5(5):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horta-Baas G, Romero-Figueroa MDS, Montiel-Jarquín AJ, Pizano-Zárate ML, García-Mena J, Ramírez-Durán N. 2017. Intestinal dysbiosis and rheumatoid arthritis: a link between gut microbiota and the pathogenesis of rheumatoid arthritis. J Immunol Res. 2017:4835189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JL, Bergem HO, Gilboe IM, Husby G, Axéll T. 1999. Oral and ocular sicca symptoms and findings are prevalent in systemic lupus erythematosus. J Oral Pathol Med. 28(7):317–322. [DOI] [PubMed] [Google Scholar]
- Jepsen S, Caton JG, Albandar JM, Bissada NF, Bouchard P, Cortellini P, Demirel K, de Sanctis M, Ercoli C, Fan J, et al. 2018. Periodontal manifestations of systemic diseases and developmental and acquired conditions: consensus report of workgroup 3 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J Clin Periodontol. 45 Suppl 20:S219–S229. [DOI] [PubMed] [Google Scholar]
- Kim D, Lee G, Huh YH, Lee SY, Park KH, Kim S, Kim J, Koh J, Ryu J. 2017. NAMPT is an essential regulator of RA-mediated periodontal inflammation. J Dent Res. 96(6):703–711. [DOI] [PubMed] [Google Scholar]
- Kinane DF, Stathopoulou PG, Papapanou PN. 2017. Periodontal diseases. Nat Rev Dis Prim. 3:17038. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Ito S, Yamamoto K, Hasegawa H, Sugita N, Kuroda T, Kaneko S, Narita I, Yasuda K, Nakano M, et al. 2003. Risk of periodontitis in systemic lupus erythematosus is associated with Fcgamma receptor polymorphisms. J Periodontol. 74(3):378–384. [DOI] [PubMed] [Google Scholar]
- Lalla E, Lamster IB, Feit M, Huang L, Spessot A, Qu W, Kislinger T, Lu Y, Stern DM, Schmidt AM. 2000. Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J Clin Invest. 105(8):1117–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen JM. 2017. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology. 151(4):363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J, Cai Q, Steinwandel M, Hargreaves MK, Bordenstein SR, Blot WJ, Zheng W, Shu XO. 2017. Association of oral microbiome with type 2 diabetes risk. J Periodontal Res. 52:636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López P, de Paz B, Rodríguez-Carrio J, Hevia A, Sánchez B, Margolles A, Suárez A. 2016. Th17 responses and natural IgM antibodies are related to gut microbiota composition in systemic lupus erythematosus patients. Sci Rep. 6:24072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques CP, Victor EC, Franco MM, Fernandes JM, Maor Y, de Andrade MS, Rodrigues VP, Benatti BB. 2016. Salivary levels of inflammatory cytokines and their association to periodontal disease in systemic lupus erythematosus patients: a case-control study. Cytokine. 85:165–170. [DOI] [PubMed] [Google Scholar]
- Mashimo PA, Yamamoto Y, Slots J, Park BH, Genco RJ. 1983. The periodontal microflora of juvenile diabetics: culture, immunofluorescence, and serum antibody studies. J Periodontol. 54(7):420–430. [DOI] [PubMed] [Google Scholar]
- Mendonça SMS, Corrêa JD, Souza AF, Travassos DV, Calderaro DC, Rocha NP, Vieira ÉLM, Teixeira AL, Ferreira GA, Silva TA. 2018. Immunological signatures in saliva of systemic lupus erythematosus patients: influence of periodontal condition. Clin Exp Rheumatol [epub ahead of print 19 Jul 2018] in press. [PubMed] [Google Scholar]
- Merchant AT, Shrestha D, Chaisson C, Choi YH, Hazlett LJ, Zhang J. 2014. Association between serum antibodies to oral microorganisms and hyperglycemia in adults. J Dent Res. 93(8):752–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirrielees J, Crofford LJ, Lin Y, Kryscio RJ, Dawson DR, 3rd, Ebersole JL, Miller CS. 2010. Rheumatoid arthritis and salivary biomarkers of periodontal disease. J Clin Periodontol. 37(12):1068–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, Abusleme L, Zenobia C, Hosur KB, Abe T, et al. 2014. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med. 6(229):229ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Q, Zhang H, Liao X, Lin K, Liu H, Edwards MR, Ahmed SA, Yuan R, Li L, Cecere TE, et al. 2017. Control of lupus nephritis by changes of gut microbiota. Microbiome. 5(1):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutlu S, Richards A, Maddison P, Scully C. 1993. Gingival and periodontal health in systemic lupus erythematosus. Community Dent Oral Epidemiol. 21(3):158–161. [DOI] [PubMed] [Google Scholar]
- Naguib G, Al-Mashat H, Desta T, Graves DT. 2004. Diabetes prolongs the inflammatory response to a bacterial stimulus through cytokine dysregulation. J Invest Dermatol. 123(1):87–92. [DOI] [PubMed] [Google Scholar]
- Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, Glickman JN, Siebert R, Baron RM, Kasper DL, et al. 2012. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 336(6080):489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori K, Ohira T, Uchida Y, Ayilavarapu S, Batista EL, Yagi M, Iwata T, Liu H, Hasturk H, Kantarci A, et al. 2008. Priming of neutrophil oxidative burst in diabetes requires preassembly of the NADPH oxidase. J Leukoc Biol. 84(1):292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queiroz-Junior CM, Madeira MF, Coelho FM, Costa VV, Bessoni RL, Sousa LF, Garlet GP, Souza Dda G, Teixeira MM, Silva TA. 2011. Experimental arthritis triggers periodontal disease in mice: involvement of TNF-α and the oral microbiota. J Immunol. 187(7):3821–3830. [DOI] [PubMed] [Google Scholar]
- Ramamurthy NS, Greenwald RA, Celiker MY, Shi EY. 2005. Experimental arthritis in rats induces biomarkers of periodontitis which are ameliorated by gene therapy with tissue inhibitor of matrix metalloproteinases. J Periodontol. 76(2):229–233. [DOI] [PubMed] [Google Scholar]
- Roberts FA, Darveau RP. 2015. Microbial protection and virulence in periodontal tissue as a function of polymicrobial communities: symbiosis and dysbiosis. Periodontol 2000. 69(1):18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter-Locher Z, Smith TO, Giles I, Sofat N. 2017. Association between systemic lupus erythematosus and periodontitis: a systematic review and meta-analysis. Front Immunol. 8:1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakalauskiene V, Kubilius R, Gleiznys A, Vitkauskiene A, Ivanauskiene E, Šaferis V. 2014. Relationship of clinical and microbiological variables in patients with type 1 diabetes mellitus and periodontitis. Med Sci Monit. 20:1871–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Takahashi N, Kato T, Matsuda Y, Yokoji M, Yamada M, Nakajima T, Kondo N, Endo N, Yamamoto R, et al. 2017. Aggravation of collagen-induced arthritis by orally administered Porphyromonas gingivalis through modulation of the gut microbiota and gut immune system. Sci Rep. 7(1):6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher JU, Ubeda C, Equinda M, Khanin R, Buischi Y, Viale A, Lipuma L, Attur M, Pillinger M, Weissmann G, et al. 2012. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arthritis Rheum. 64(10):3083–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Haake SK, Mannon P, Lemon KP, Waldron L, Gevers D, Huttenhower C, Izard J. 2012. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 13(6):R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socransky SS, Haffajee AD. 2005. Periodontal microbial ecology. Periodontol 2000. 38:135–187. [DOI] [PubMed] [Google Scholar]
- Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr 1998. Microbial complexes in subgingival plaque. J Clin Periodontol. 25(2):134–144. [DOI] [PubMed] [Google Scholar]
- Sweeney TE, Morton JM. 2013. The human gut microbiome: a review of the effect of obesity and surgically induced weight loss. JAMA Surg. 148(6):563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussar S, Fujisaka S, Kahn CR. 2016. Interactions between host genetics and gut microbiome in diabetes and metabolic syndrome. Mol Metab. 5(9):795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Han Q, Luo Z, Xu C, Liu J, Dan H, Xu Y, Zeng X, Chen Q. 2014. Oral lichen planus may enhance the expression of Th17-associated cytokines in local lesions of chronic periodontitis. Clin Oral Investig. 18(6):1647–1654. [DOI] [PubMed] [Google Scholar]
- Wu Y, Dong G, Xiao W, Xiao E, Miao F, Syverson A, Missaghian N, Vafa R, Cabrera-Ortega AA, Rossa C, Jr, et al. 2016. Effect of aging on periodontal inflammation, microbial colonization, and disease susceptibility. J Dent Res. 95(4):460–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YY, Xiao E, Graves DT. 2015. Diabetes mellitus related bone metabolism and periodontal disease. Int J Oral Sci. 7(2):63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao E, Mattos M, Vieira GHA, Chen S, Corrêa JD, Wu Y, Albiero ML, Bittinger K, Graves DT. 2017. Diabetes enhances IL-17 expression and alters the oral microbiome to increase its pathogenicity. Cell Host Microbe. 22(1):120–128.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Dong G, Pacios S, Alnammary M, Barger LA, Wang Y, Wu Y, Graves DT. 2015. FOXO1 deletion reduces dendritic cell function and enhances susceptibility to periodontitis. Am J Pathol. 185(4):1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Wang Y, Pacios S, Li S, Graves DT. 2016. Cellular and molecular aspects of bone remodeling. Front Oral Biol. 18:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liao X, Sparks JB, Luo XM. 2014. Dynamics of gut microbiota in autoimmune lupus. Appl Environ Microbiol. 80(24):7551–7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, Wu X, Li J, Tang L, Li Y, et al. 2015. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med. 21(8):895–905. [DOI] [PubMed] [Google Scholar]
- Zhou M, Rong R, Munro D, Zhu C, Gao X, Zhang Q, Dong Q. 2013. Investigation of the effect of type 2 diabetes mellitus on subgingival plaque microbiota by high-throughput 16S rDNA pyrosequencing. PLoS One. 8(4):e61516. [DOI] [PMC free article] [PubMed] [Google Scholar]