

Abstract
The adherens junction (AJ) couples the actin cytoskeletons of neighboring cells to allow mechanical integration and tissue organization. The physiological demands of intercellular adhesion require that the AJ be responsive to dynamic changes in force while maintaining mechanical load. These demands are tested in the heart, where cardiomyocyte AJs must withstand repeated cycles of actomyosin-mediated contractile force. Here we show that force-responsive cardiomyocyte AJs recruit actin-binding ligands to selectively couple actin networks. We employed a panel of N-cadherin-αE-catenin fusion proteins to rebuild AJs with specific actin linkages in N-cadherin-null cardiomyocytes. In this system, vinculin recruitment was required to rescue myofibril integration at nascent contacts. In contrast, loss of vinculin from the AJ disrupted junction morphology and blocked myofibril integration at cell–cell contacts. Our results identify vinculin as a critical link to contractile actomyosin and offer insight to how actin integration at the AJ is regulated to provide stability under mechanical load.
INTRODUCTION
Adherens junctions (AJs) link the actin cytoskeletons of adjacent cells to provide the foundation for multicellular tissue organization. The dynamic demands of cell–cell adhesion require that the AJ be both responsive and resilient to mechanical force. This is especially true in the heart, where the AJ must transmit the mechanical forces of actomyosin contraction while maintaining adhesive homeostasis. How the AJ balances mechanical integration with contractile force to maintain tissue integrity is not clear.
Cardiomyocytes are linked through a specialized cell–cell contact called the intercalated disc (ICD). The ICD is the site of mechanical and electrical continuity between individual cardiomyocytes that allow the heart to function as a syncytium (Vite and Radice, 2014; Ehler, 2016; Vermij et al., 2017). Three junctional complexes form the ICD: the AJ, desmosome, and gap junction. The AJ and desmosome are responsible for mechanical integration by coupling the actin and intermediate filament cytoskeletons, respectively, of neighboring cells. Gap junctions permit electrical continuity through the free flow of ions. Importantly, the ICD AJ is the site of myofibril integration between cardiomyocytes and allows contractile force to be transduced across heart tissue (Luo and Radice, 2003).
The core of the AJ is the cadherin-catenin complex (Ratheesh and Yap, 2012; Mege and Ishiyama, 2017). N-cadherin, the sole classical cadherin expressed in cardiomyocytes (Kostetskii et al., 2005), is a single-pass transmembrane protein with an extracellular domain that mediates homotypic, calcium-dependent interactions (Shapiro and Weis, 2009; Harrison et al., 2011). The adhesive properties of classical cadherins are driven by the recruitment of cytosolic catenin proteins to the cadherin tail: p120-catenin binds to the juxtamembrane domain and β-catenin binds to the distal part of the tail. β-Catenin, in turn, recruits α-catenin to the cadherin-catenin complex. α-Catenin is an actin-binding protein and the primary link between the AJ and the actin cytoskeleton (Rimm et al., 1995; Drees et al., 2005; Yamada et al., 2005; Buckley et al., 2014; Pokutta et al., 2014; Ishiyama et al., 2018). Mammalian cardiomyocytes express two α-catenins: αE(epithelial)-catenin and αT(testes)-catenin (Janssens et al., 2001; Goossens et al., 2007; Li et al., 2012, 2015; Wickline et al., 2016).
AJ binding capabilities are modified by the forces of actomyosin contraction, largely through changes in α-catenin conformation (Hoffman and Yap, 2015; Charras and Yap, 2018). Force induces a conformational change in the middle(M)-domain of α-catenin to reveal binding sites for ligands, many of which bind F-actin (le Duc et al., 2010; Yonemura et al., 2010; Choi et al., 2012; Yao et al., 2014; Kim et al., 2015; Matsuzawa et al., 2018; Pang et al., 2019). The force required to unfurl αE-catenin and αT-catenin (5 pN; Yao et al., 2014; Pang et al., 2019) is well within the range of a myosin motor, demonstrating the physiological relevance for this model of regulation (Finer et al., 1994; Charras and Yap, 2018). The recruitment of actin-binding proteins in response to force is thought to help anchor F-actin to the AJ (Yap et al., 2018).
Cardiomyocytes have at least two distinct actin networks at cell–cell contacts—myofibrils and the cortical cytoskeleton (Li et al., 2019)—that must be integrated at AJs. Many actin-binding ligands interact with αE-catenin, including vinculin, afadin, ZO-1, and Eplin (Yonemura, 2017). In epithelia, vinculin is recruited to αE-catenin in a force-dependent manner and this interaction is thought to be important for reinforcing the association between αE-catenin and F-actin (le Duc et al., 2010; Yonemura et al., 2010; Huveneers et al., 2012; Kale et al., 2018). Likewise, epithelial afadin can also bind αE-catenin in a force-dependent manner (Matsuzawa et al., 2018), where it functions to strengthen the AJ under tension (Choi et al., 2016). Both vinculin and afadin localize to the ICD (Geiger et al., 1985; Borrmann et al., 2006) and are recruited to cardiomyocyte AJs (Li et al., 2019). Vinculin is required for proper heart development and functions in cardiomyocyte adhesion and contraction (Shiraishi et al., 1997; Xu et al., 1998). Afadin was recently identified as having a cardioprotective role at the ICD, as mice lacking afadin were shown to be more susceptible to stress-induced injury and myopathy (Zankov et al., 2017). How vinculin and afadin function in mechanical coupling at cardiomyocyte AJs is not well understood.
Here we sought to define the individual functions of αE-catenin, vinculin, and afadin in coupling actin to cardiomyocyte AJs. We demonstrate that cultured neonatal cardiomyocytes recruit vinculin and afadin to AJs in a force-dependent manner, similar to epithelia. We show that loss of N-cadherin in cardiomyocytes disrupts cell–cell adhesion and dissolves junctional complexes. These phenotypes defined in our in situ loss-of-N-cadherin system are strikingly similar to those shown for in vivo models (Kostetskii et al., 2005). We developed a series of N-cadherin:αE-catenin fusions to test how AJ ligand recruitment and actin-binding influences cell–cell contact architecture and myofibril coupling. We show for the first time that vinculin recruitment to the AJ is necessary to couple myofibrils to developing cell–cell contacts in cardiomyocytes. Our results offer new insight into actin linkage to the AJ and identify vinculin as a key link between contractile actin networks and the cardiomyocyte AJ.
RESULTS
Force regulates α-catenin ligand recruitment to cardiomyocyte AJs
Vinculin and afadin are recruited to epithelial AJs in a force-dependent manner (le Duc et al., 2010; Yonemura et al., 2010; Choi et al., 2016; Kale et al., 2018; Matsuzawa et al., 2018; Seddiki et al., 2018). Vinculin and afadin localize to the ICD in adult heart (Geiger et al., 1985; Mandai et al., 1997) and proximity proteomics revealed that both are enriched at the AJ in cultured neonatal cardiomyocytes (Li et al., 2019). We sought to determine whether vinculin and afadin recruitment to cardiomyocyte AJs is tension dependent. Cultured cardiomyocytes were treated with 100 µM blebbistatin to suppress myosin activity for up to 1 h and stained for vinculin or afadin (Figure 1, A, B, D, and E). Cardiomyocytes ceased contraction within 30 s of blebbistatin addition, whereas dimethyl sulfoxide (DMSO) did not affect contraction (unpublished observation). In blebbistatin-treated cells, both vinculin and afadin were significantly reduced at cell–cell contacts after 1 h, with significant loss of vinculin seen after 30 min (Figure 1, C and F). This is consistent with a requirement for tension at the AJ to recruit vinculin or afadin and indicates that nascent cardiomyocyte AJs retain the ability to respond to changes in mechanical force.
FIGURE 1:
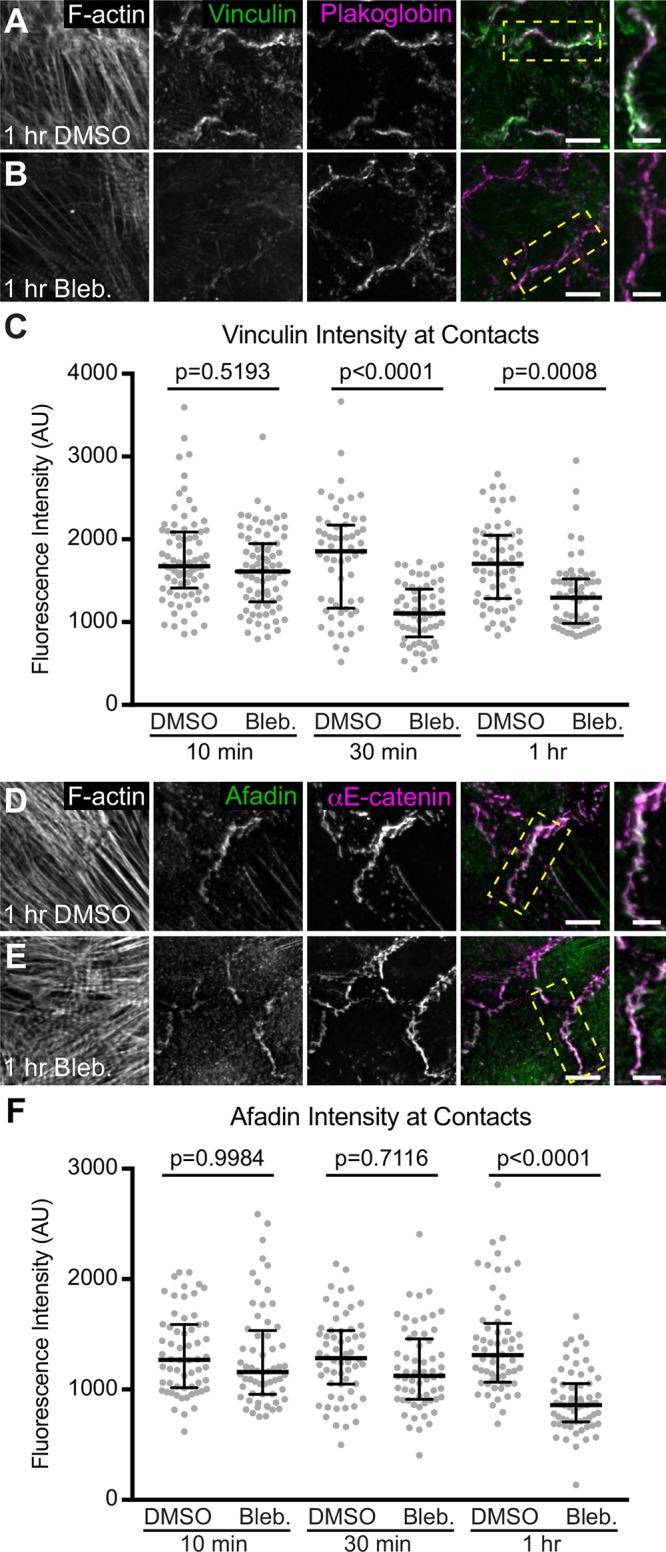
Vinculin and afadin recruitment to cardiomyocyte AJs is tension dependent. (A–E) Mouse neonatal cardiomyocytes were treated for 1 h with DMSO (A, D) or 100 μM blebbistatin (B, E) before fixation. Cells were stained for F-actin (A, B, D, and E), vinculin, and plakoglobin (A, B), or afadin and αE-catenin (D, E). Individual and merged vinculin (green) and plakoglobin (magenta) channels shown in A and B. Individual and merged afadin (green) and αE-catenin (magenta) channels shown in D and E. Far right column is a higher magnification of the boxed contact in merge. (C, F) Quantification of vinculin (C) or afadin (F) intensity at cell–cell contacts. Vinculin or afadin signal intensity was measured in cells treated with DMSO or blebbistatin for 10, 30, and 60 min before fixation. All data points are plotted. Middle horizontal bar is the median and error bars represent the quartile range. One-way ANOVA, n ≥ 60 images from at least three independent experiments. Images are maximum projections of 5 µm stacks. Scale bar is 10 μm in lower-magnification images, 5 μm in higher-magnification images.
Loss of N-cadherin disrupts cardiomyocyte cell–cell contacts
The force-responsive nature of cardiomyocyte AJs led us to question the roles of αE-catenin, vinculin, and afadin in linking the AJ to actin. To individually test these roles, we developed a system to selectively recruit actin-binding ligands and thus control the actin– interfaces at the cardiomyocyte AJ. We first needed to establish a cadherin-null system in which to rebuild AJs. In intact mouse heart tissue, conditional ablation of N-cadherin causes dissolution of all AJ components as well as loss of all desmosomal and gap junction proteins at the ICD (Kostetskii et al., 2005). We questioned whether loss of N-cadherin would disrupt ligand recruitment and junction organization (Figure 2A) in cultured neonatal cardiomyocytes. Cardiomyocytes from N-cadherin conditional knockout mice (Ncadfx/fx; Kostetskii et al., 2005) were isolated and infected with adenovirus expressing Cre-recombinase (hereby referred to as Cre).
FIGURE 2:
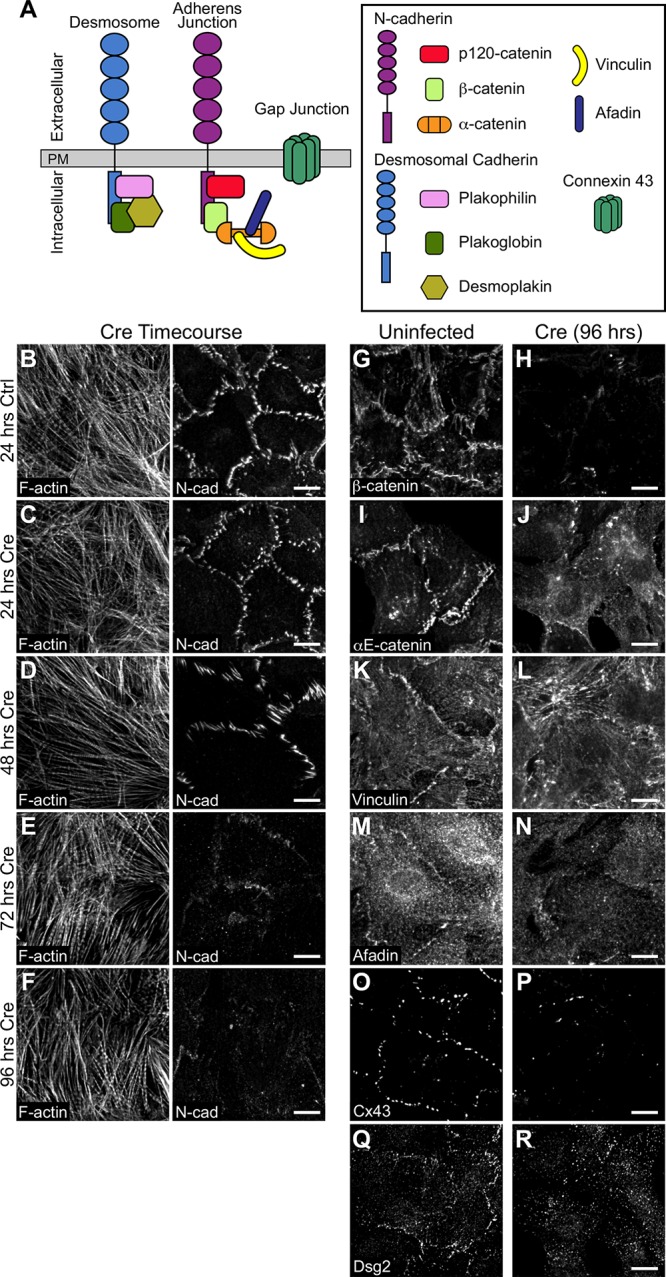
Loss of N-cadherin disrupts adhesion protein localization. (A) Cartoon schematic of desmosome, AJ, and gap junction proteins at cardiomyocyte cell–cell contacts. (B–F) Neonatal cardiomyocytes from Ncadfx/fx mice were uninfected (B) or infected with adenovirus expressing Cre recombinase (C–F) and fixed over 96 h to assess N-cadherin expression. Cells were stained for F-actin (left panel) and N-cadherin (N-cad, right panel). (G–R) Control and Cre-infected neonatal cardiomyocytes from Ncadfx/fx mice were fixed 96 h postinfection and stained for AJ proteins β-catenin and αE-catenin (G–J); AJ adapter proteins vinculin and afadin (K–N); connexin 43 (Cx43; O, P), and desmoglein 2 (Dsg2; Q, R). Images are maximum projections of 2–3 μm deconvolved stacks. Scale bar is 10 μm.
To determine the time required for N-cadherin depletion post–Cre-mediated recombination, we fixed Cre-infected cells at four different time points—24, 48, 72, and 96 h—postinfection and stained for N-cadherin (Figure 2, B–F). At 24 h postinfection, N-cadherin levels appeared similar to uninfected cells (Figure 2, B and C). At 48 h postinfection, N-cadherin levels remained high; however, cell–cell contacts began to appear jagged with N-cadherin clustering along more linear contacts (Figure 2D). We speculate that declining N-cadherin levels are compromising AJ strength and causing junctions to collapse and contort under contractile force. Notably, at 72 and 96 h postinfection, we observed a near complete loss of N-cadherin at cell–cell contacts (Figure 2, E and F) and ∼90% reduction in N-cadherin cellular levels (Supplemental Figure S1, A and B).
We next assessed cell–cell contact formation and protein recruitment at 96 h post Cre infection. In uninfected control cardiomyocytes, AJ markers N-cadherin, β-catenin, and αE-catenin were recruited to cell–cell contacts (Figure 2, G and I); likewise αE-catenin ligands vinculin and afadin (Figure 2, K and M), the gap junction protein connexin 43 (Cx43, Figure 2O), and desmosome markers desmoglein 2 (Dsg2, Figure 2Q), plakoglobin, and plakophilin 2 (Supplemental Figure S1, C and E) all localized to cell–cell contacts. In contrast, Cre expression dissolved cell–cell contacts and resulted in a loss of all AJ proteins, αE-catenin ligands, gap junctions, and desmosomes (Figure 2, H, J, L, N, P, and R; Supplemental Figure S1, D and F). As expected, N-cadherin is required for cell–cell adhesion in cultured cardiomyocytes and AJ formation is critical for the recruitment and organization of other junctional components.
We then sought to determine whether cardiomyocyte cell–cell contacts could be restored with exogenous N-cadherin-GFP. Cardiomyocytes were sequentially infected with Cre and then N-cadherin-GFP adenovirus. Expression of N-cadherin-GFP restored cell–cell contacts and the localization of AJ, gap junction, and desmosome proteins (Figure 3; Supplemental Figure S1, G and H). The ability of N-cadherin-GFP to restore cell–cell contacts in an N-cadherin-null background demonstrated the pliable nature of this adhesion system and its tractability for probing cadherin and catenin function further.
FIGURE 3:

N-cadherin-GFP rescues cardiomyocyte junctional complexes. (A–F) Neonatal Ncadfx/fx cardiomyocytes infected sequentially with adenoviruses expressing Cre and N-cadherin-GFP, fixed, and stained for AJ-associated proteins (A–D), gap junctions (E), and desmosomes (F). Individual and merged N-cadherin-GFP (green) and ICD components (magenta) channels are shown. Far right columns are higher magnifications of the boxed contact in the merge. Individual and merged channels are shown. Images are maximum projections of 2–3 μm deconvolved stacks. Scale bar is 10 μm in lower-magnification images, 5 μm in higher-magnification images.
N-cadherin-αE-catenin fusions selectively recruit αE-catenin ligands
We designed a series of N-cadherin-αE-catenin fusion constructs to systematically delineate the individual and combined functions of αE-catenin, vinculin, and afadin in AJ-mediated cell–cell adhesion. Fusion constructs were created by taking the extracellular, transmembrane and p120 binding domains of N-cadherin and fusing them to GFP followed by the M-region and actin-binding domain (ABD) of αE-catenin (Figure 4A). The M-region of αE-catenin contains three separate domains: M1, M2, and M3 (Ishiyama et al., 2013; Rangarajan and Izard, 2013). The vinculin-binding site is found in M1 (Choi et al., 2012) and the afadin-binding site spans M2–M3 (Pokutta et al., 2002). The C-terminal tail of N-cadherin and the N-terminus of αE-catenin were removed to eliminate endogenous β-catenin and αE-catenin recruitment while allowing for proper N-cadherin trafficking (Davis et al., 2003; Wahl et al., 2003; Bianchini et al., 2015). Within the N-cadherin-GFP-αE-catenin fusion, we introduced various mutations or domain deletions to restrict ligand recruitment and actin-binding interfaces. Ncad-GFP-M1-ABD mimics the core cadherin-catenin complex as it possesses the αE-catenin ABD and contains both vinculin- and afadin-binding sites. Ncad-GFP-M1–M3 possesses the full M-region of αE-catenin but lacks the ABD and the ability to respond to tension. Ncad-GFP-M1–M2 has an open M-domain that can bind vinculin constitutively but lacks the αE-catenin ABD and afadin-binding domain (Choi et al., 2012). Two constructs were designed to selectively block vinculin recruitment while retaining afadin binding and actin binding through the αE-catenin ABD: Ncad-GFP-M1mutV-ABD, which contains five point mutations in M1 that ablate vinculin binding (Chen et al., 2015), and Ncad-GFP-M2-ABD, which lacks the entire M1 domain. Note that additional ligands may bind αE-catenin M2–M3: we focus on afadin recruitment and use it as proxy for ligand binding to M2-M3. Last, Ncad-GFP-ABD lacks M1–M3 but retains the αE-catenin ABD.
FIGURE 4:

N-cadherin-GFP-αE-catenin fusions selectively recruit ligands to cardiomyocyte cell–cell contacts. (A) Table of N-cadherin-GFP-αE-catenin fusion constructs used in this study. Nomenclature, domain schematic, and actin linkage cartoon are shown. (B) Quantification of N-cadherin-GFP-αE-catenin fusion cell–cell contact morphology. Mean contact length plotted against mean contact circularity. Error bars represent SEM. Data are from at least 180 individual cell–cell contacts from two independent infections with each fusion construct. (C–F) Cardiomyocytes infected with Cre and N-cadherin-GFP (C) or Cre and fusion adenoviruses (D–F), fixed, and stained for vinculin and afadin. Individual and merged GFP (green), vinculin (red), and afadin (blue) channels are shown. Images are a maximum projection of 2–3 μm deconvolved stacks. Bottom image is a higher magnification of boxed contact. (G–J) Quantification of vinculin and afadin intensities at cell–cell contacts. Signal intensity at contacts was divided by the average cytoplasmic intensity and a scatter plot of all data points is shown. The black horizontal line is the median and the error bars define the interquartile range. The shaded gray region in each plot defines the median (thick gray line) and interquartile range (thin gray lines) of vinculin or afadin recruitment observed with full-length N-cadherin-GFP (G) for comparison. One-way ANOVA, significance compared with recruitment with N-cadherin-GFP; n ≥ 50 images from at least two independent experiments. Scale bar is 10 μm in all images.
We tested the ability of Ncad-GFP-αEcat fusions to restore cell–cell contacts and selectively recruit vinculin and/or afadin in N-cadherin-null cells. Ncadfx/fx cardiomyocytes were sequentially infected with Cre plus individual adenoviral Ncad-GFP-αEcat fusions. We observed expression and proper localization of the fusion constructs by 24 h postinfection, which continued through 72 h postinfection, corresponding with the maximum loss of endogenous N-cadherin (Supplemental Figure S1, M-O). All Ncad-GFP-αEcat fusions localized to the membrane and reestablished cell–cell contacts (Figure 4, C–F; Supplemental Figure S2, A-C), though the gross morphology of these junctions differed markedly between constructs. We quantified and compared junction morphology, ligand recruitment, and the relationship between GFP expression and ligand binding for all fusion constructs (Figure 4, B–J; Supplemental Figure S2, A–M).
Ncad-GFP organized discrete, punctate junctions that recruited vinculin and afadin (Figure 4, B and C; Supplemental Figure S2G). Ncad-GFP vinculin and afadin recruitment levels (Figure 4G) were used as the standard for comparing all fusion constructs. Importantly, Ncad-GFP-M1-ABD formed cell–cell contacts that were morphologically similar to Ncad-GFP (Figure 4, B–D) and recruited afadin and enriched for vinculin (Figure 4, G and H; Supplemental Figure S2H). This indicates that the static Ncad-GFP-αEcat fusion can substitute for the cadherin-catenin complex to form cell–cell contacts in cardiomyocytes. Ncad-GFP-M1–M3, in contrast, which lacked the ABD and the ability to bind actin or respond to tension, formed long, more linear junctions (Figure 4B; Supplemental Figure S2A). Ncad-GFP-M1–M3 recruited a small amount of vinculin but no afadin (Supplemental Figure S2, A, D, and K). We speculate that the autoinhibited M1–M3 region is not capable of supporting strong vinculin binding and thus altering junction morphology. However, the constitutively active Ncad-GFP-M1–M2 enriched vinculin, but not afadin, and formed compact, discrete cell–cell contacts similar to Ncad-GFP-M1-ABD (Figure 4, B, E, and I; Supplemental Figure S2I). Ncad-GFP-M1–M2 was the only construct in which we observed a modest relationship between GFP expression and ligand recruitment (Supplemental Figure S2I), consistent with the ability of this construct to bind vinculin constitutively. Thus, the ability of a given fusion construct to restore junction formation was driven more by the functional properties of the construct rather than the expression level.
Ncad-GFP-M1mutV-ABD and Ncad-GFP-M2-ABD both recruited afadin, but not vinculin, and generated long, linear contacts that lacked the punctate morphology observed in Ncad-GFP-M1-ABD (Figure 4, B, F, and J; Supplemental Figure S2, B, E, J, and L). Last, Ncad-GFP-ABD formed poor contacts that failed to recruit vinculin and afadin (Supplemental Figure S2, C, F, and M). Thus, we were able to specifically recruit αE-catenin ligands to nascent cardiomyocyte contacts and observed morphological changes as a function of the selective association of AJ components. Notably, Ncad-GFP-M1–M2, which only recruits vinculin, formed cell–cell contacts similar to Ncad-GFP and Ncad-GFP-M1-ABD whereas Ncad-GFP-M1mutV-ABD and Ncad-GFP-M2-ABD, which do not recruit vinculin, organized long, linear contacts similar to Ncad-GFP-M1–M3 (Figure 4B).
To assess the effects of ligand recruitment on fusion stability along cell–cell contacts, we performed FRAP (fluorescence recovery after photobleaching) analysis of Ncad-GFP and three key Ncad-GFP-αEcat fusion constructs (Figure 5, A–D). Fluorescence recovery over 15 min was quantified, plotted, and fitted to a double exponential curve. The Ncad-GFP recovery profile was similar to previously published data from our group (Figure 5, E and I; Li et al., 2019), consistent with the ability of Ncad-GFP to restore AJs in an N-cadherin-null background. The mobile fraction of Ncad-GFP and all fusion constructs was ∼30%, similar to those observed for AJ components in cardiomyocytes and epithelial cells (Figure 5I; Yamada et al., 2005; Li et al., 2019). While F-actin binding is critical for AJ formation and extracellular and intracellular cadherin interactions cooperate to regulate AJ assembly (Hong et al., 2013), our results suggest that AJ plaque (immobile fraction) stability is regulated by cadherin extracellular interactions.
FIGURE 5:
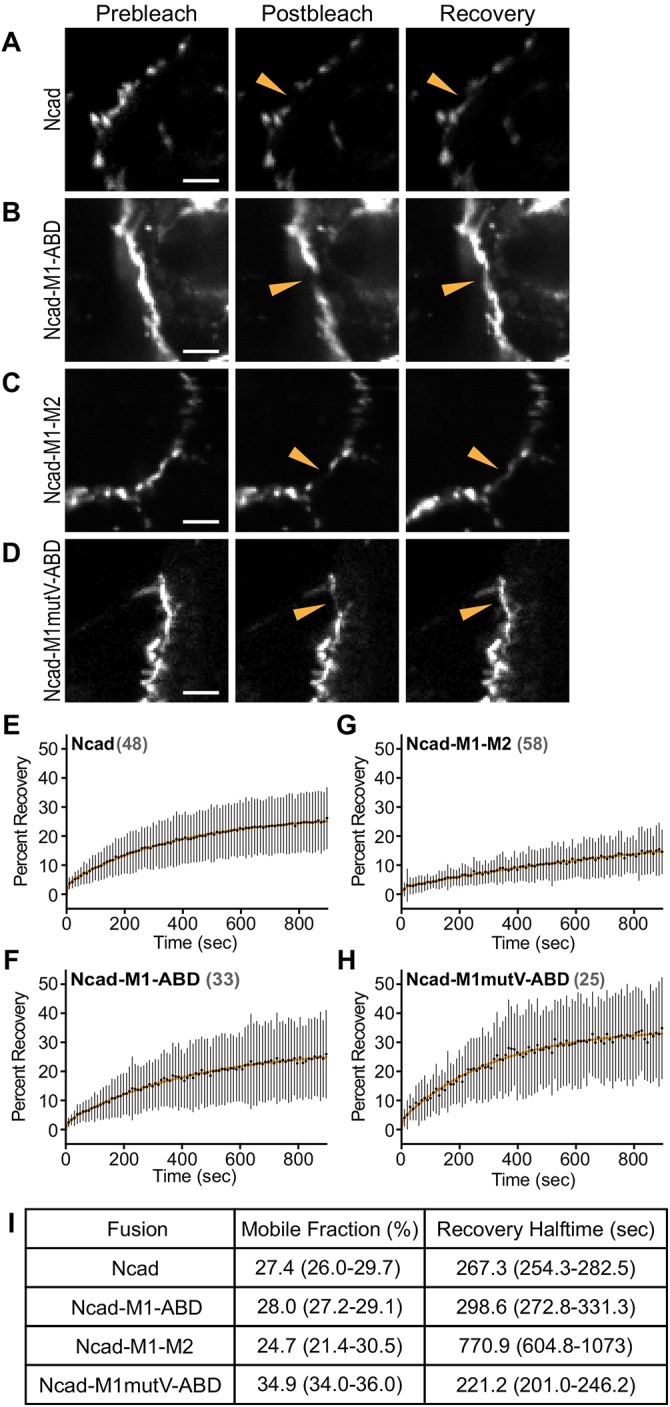
N-cadherin-GFP-αE-catenin fusion dynamics at cardiomyocyte cell–cell contacts. (A–D) Representative prebleach, postbleach, and recovery images from FRAP studies of Ncadfx/fx cardiomyocytes infected with Cre and N-cadherin-GFP (A) or Cre and N-cadherin-GFP-αE-catenin fusion adenoviruses (B–D). Orange arrowhead marks the FRAP region at a cell–cell contact. (E–H) Plots of mean ± SD FRAP recovery fraction over 15 min. The mean is represented by a black circle, and the SD is shown as a black line. The data were fitted to a double-exponential curve (orange line). The numbers of FRAP regions measured for each fusion construct are listed in gray. FRAP data were collected from at least two independent infections for each fusion. (I) Summary of the mobile fraction (percentage) and recovery halftime (seconds). Scale bar is 5 μm in A–D.
We then analyzed the recovery rates of the mobile fraction slow pools. Ncad-GFP and Ncad-GFP-M1-ABD had similar recovery rates (Figure 5, E, F, and I), consistent with the ability of Ncad-GFP-M1-ABD to reconstitute the AJ. Notably, Ncad-GFP-M1mutV-ABD had a recovery rate faster than Ncad-GFP, suggesting that vinculin regulates the dynamics of the AJ mobile pool (Figure 5, H and I). Consistent with this, Ncad-GFP-M1–M2, which binds vinculin constitutively, had a recovery rate that was nearly 3× slower than Ncad-GFP or Ncad-M1-ABD (Figure 5, G and I). We speculate that vinculin anchors the cadherin-catenin complex to actin to limit turnover of the mobile pool without affecting the immobile/mobile pool balance.
Vinculin links the AJ to contractile myofibrils
We then questioned whether the differences in cell–cell contact morphology and dynamics observed in cardiomyocytes expressing the Ncad-GFP-αEcat fusions could reflect fundamental changes in actin organization and/or linkage to the AJ. We used thin-section transmission electron microscopy (TEM) to assess the ultrastructural organization of the AJ-actin interface. In adult mouse cardiac tissue, the ICD is a contorted, electron-dense structure where myofibrils are coupled between adjacent cells at AJs (Bennett et al., 2006; Li et al., 2019). In wild-type cultured cardiomyocytes, we observed a similar junctional morphology with electron-dense AJs joining myofibrils across cells (Figure 6, A and B, contact highlighted in yellow). As expected, loss of N-cadherin dissolved cell junctions and prevented myofibril integration between neighboring cells (Figure 6, C and D). Importantly, AJ structure and myofibril pairing were rescued with N-cad-GFP expression in N-cadherin-null cells (Figure 6, E and F).
FIGURE 6:

Vinculin recruitment is required to couple myofibrils to the AJ. Ncadfx/fx cardiomyocytes uninfected (A, B), infected with Cre (C, D), infected with Cre and Ncad-GFP (E, F), or infected with Cre and N-cadherin-GFP-αE-catenin fusion adenoviruses (G–L) were fixed and processed for immunostaining or thin-section TEM. (A, C, E, G, I, K) Immunofluorescence lower-magnification (40×) images of control and infected cardiomyocytes. Uninfected control (A) and Cre-infected (C) cardiomyocytes were stained for N-cadherin and F-actin. Ncad-GFP (E) and N-cadherin-GFP-αE-catenin fusion adenovirus-infected (G, I, K) cardiomyocytes were stained for F-actin. Individual and merged N-cadherin/GFP (green) and F-actin (magenta) channels shown. Images are maximum projections of 5 μm stacks. (B, D, F, H, J, L) Representative TEM image of a cell–cell contact from >60 images from at least three independent experiments. Cell–cell junctions (J) are highlighted in yellow, Z-discs (Z) are colored orange, and a terminal myofibril sarcomere (S) is defined by a double arrow line. In L, the arrowhead marks electron density along the cell–cell junction and the asterisks mark poorly organized F-actin. Scale bar is 20 µm in A, C, E, G, I, and K and 1 μm in B, D, F, H, J, and L.
Next, we compared the ability of the Ncad-GFP-αE-cat fusions to organize actin along cardiomyocyte cell–cell contacts. Importantly, Ncad-GFP-M1-ABD restored myofibril coupling along thick, electron-dense junctions that were morphologically similar to controls (Figure 6, G and H compared to B and F). As expected, lack of strong F-actin binding in Ncad-M1–M3 prevented cytoskeletal integration at cell–cell contacts (Supplemental Figure S3A). Ncad-GFP-M1–M2, which connects N-cadherin to F-actin solely through vinculin, restored myofibril coupling, and generated electron-dense junctions morphologically similar to Ncad-GFP-M1-ABD and controls (Figure 6, I and J). In marked contrast, constructs that could bind actin but were incapable of recruiting vinculin failed to restore normal contact morphology or myofibril coupling (Figure 6, K and L; Supplemental Figure S3B). Ncad-GFP-M1mutV-ABD and Ncad-GFP-M2-ABD formed thin electron densities along elongated cell–cell contacts, but with little to no myofibril engagement. Likewise, Ncad-GFP-ABD organized poor junctions that lacked myofibril coupling (Supplemental Figure S3C). These results indicate that vinculin recruitment is required to link contractile actin to the cardiomyocyte AJ, as has been suggested in epithelia (Chen et al., 2015). Neither the αE-catenin ABD nor afadin recruitment was sufficient to restore myofibril coupling. The increase in electron density along contacts with afadin recruitment (Figure 6L) suggests that it may play a role in actin integration along contacts, but it is not sufficient to couple contractile actin in the absence of vinculin. Together, these results underscore the importance of vinculin in linking the AJ to actin under mechanical load and highlight how αE-catenin coordinates cytoskeletal integration to provide mechanical connections between cells.
Vinculin ligand recruitment to AJs does not correlate with myofibril coupling
Myofibrils are a highly specialized, contractile actin network, distinct from actin cables or stress fibers found in epithelial cells. However, the importance of vinculin in linking this unique network to the cardiomyocyte AJ is reminiscent of contractile actin linkages in epithelial cells. Vinculin anchors F-actin to the AJ (le Duc et al., 2010; Yonemura et al., 2010; Kale et al., 2018) and can also recruit Ena/VASP proteins to promote actin assembly at junctions under tension (Leerberg et al., 2014).To determine whether cardiomyocytes use a similar linkage mechanism to epithelial cells, we probed for the Ena/VASP protein Mena (Krause et al., 2003). Mena is recruited to epithelial contacts under tension (Leerberg et al., 2014), is localized to the ICD in heart tissue (Aguilar et al., 2011), and was identified in a proximity proteomics screen for N-cadherin-associated proteins in cardiomyocytes (Li et al., 2019). Immunostaining revealed limited recruitment of Mena to cell–cell contacts in uninfected control cardiomyocytes (Supplemental Figure S4, A and E). Relative to the control, we observed no measurable difference in Mena recruitment among Ncad-GFP-M1-ABD, Ncad-GFP-M1–M2, and Ncad-GFP-M1mutV-ABD (Supplemental Figure S4, B–D and F–H) despite significant differences in vinculin recruitment between these constructs (Figure 4). Thus, while Mena may function in linking contractile F-actin to nascent cardiomyocyte AJs, its recruitment is limited and not correlated with vinculin levels, suggesting a more peripheral role in regulating cardiomyocyte junctional actin.
α-Actinin binds vinculin (Kroemker et al., 1994) and αE-catenin (Knudsen et al., 1995; Nieset et al., 1997). α-Actinin cross-links actin filaments at myofibril Z-discs and is critical for cardiomyocyte organization (Frank and Frey, 2011). Immunostaining revealed limited α-actinin recruitment to cell–cell contacts in control cardiomyocytes (Supplemental Figure S5, A and E). Similar levels of recruitment were observed in N-cadherin-null cardiomyocytes rescued with Ncad-GFP-M1-ABD and Ncad-GFP-M1mutV-ABD (Supplemental Figure S5, B, D, F, and H). However, in Ncad-GFP-M1–M2, we observed a significant recruitment of α-actinin to contacts (Supplemental Figure S5, C and G). We were not able to determine whether α-actinin was recruited through vinculin and/or αE-catenin M1–M2. While increased α-actinin recruitment could impact Ncad-GFP-M1–M2 dynamics (Figure 5, G and I), α-actinin enrichment does not correlate with AJ coupling to F-actin.
DISCUSSION
The physiological demands of cell–cell adhesion require the AJ to both sense and resist mechanical force. In the heart, these properties are challenged by the demands of myocyte contraction. Cardiomyocytes are connected through the ICD, a structure specific to the heart that functions to mechanically couple the myofibrils of neighboring cells through the AJ. The cardiomyocyte thus offers a unique system to investigate how the AJ functions under mechanical load. In addition, the organization of the cardiomyocyte cytoskeleton at intercellular junctions—well-defined, contractile actomyosin bundles terminating at AJs under tension—permits direct analysis of how changes in AJ composition impact protein dynamics, cytoskeletal organization, and mechanical coupling.
In epithelia, mechanical force induces a conformational change in the M-domain of α-catenin to reveal binding sites for ligands such as vinculin (le Duc et al., 2010; Yonemura et al., 2010; Choi et al., 2012; Yao et al., 2014; Kim et al., 2015; Matsuzawa et al., 2018; Pang et al., 2019). When we inhibited myosin function with blebbistatin, we observed a significant reduction in vinculin and afadin from cardiomyocyte cell–cell junctions (Figure 1), indicating a loss of tension-based recruitment to α-catenin. The mechanical properties of α-catenin are retained in cardiomyocytes, at least at nascent junctions. Note that cardiomyocytes express two α-catenins—αE-catenin and αT-catenin—and both have been shown to bind vinculin in a force-dependent manner (Yao et al., 2014; Pang et al., 2019). While our study focused on the better defined αE-catenin, it is likely that both α-catenins cooperate to regulate AJ organization through tension-mediated ligand recruitment (Vite et al., 2015).
It is not clear whether the source of the activating tension is from the cortical or myofibril actin networks. While our results indicate that sustained myofibril coupling to AJs requires vinculin, α-catenin may bind and sense tension through myofibril actin filaments to recruit vinculin and organize a strong, load-bearing AJ. Once vinculin is recruited, α-catenin could remain bound to the myofibril end. Of note, the ability of the Ncad-GFP-M1–M2 construct, which is linked to actin only through vinculin, to couple myofibrils suggests that vinculin alone is sufficient to bind myofibrils in the absence of α-catenin. In contrast, the inability of the Ncad-GFP-M1mutV-ABD and Ncad-GFP-M2-ABD fusions to couple myofibrils at junctions suggests that afadin enrichment is not sufficient to link the AJ to F-actin under load. In epithelial, afadin recruitment strengthens AJs under tension (Choi et al., 2016; Matsuzawa et al., 2018). Afadin may function similarly in cardiomyocytes, but only in the presence of vinculin. Future work will provide more details on the actin architecture at cardiomyocyte AJs and the individual roles of actin-binding proteins in organizing AJ actin networks.
Our results indicate that vinculin is essential for linking the AJ to actin under sustained mechanical load. Whether vinculin does so by creating an additional linkage between F-actin and/or ligand recruitment is not clear. Vinculin can recruit ligands such as Mena to promote actin assembly at junctions under tension (Leerberg et al., 2014). While Mena localizes to cardiomyocyte cell–cell contacts, we did not observe a concomitant enrichment of Mena with constitutive vinculin recruitment to cardiomyocyte AJs (Supplemental Figure S5). We did not detect VASP at cardiomyocyte AJs (data not shown). Thus, our results suggest that vinculin-mediated ligand recruitment may not be the primary driver of increased stability and myofibril integration at the AJ. Instead, the ABD of vinculin itself may play a critical role in coupling the AJ to contractile actin.
Myofibrils arrange their actin filaments so that the myosin motors exert force as they move toward the barbed end. Recent biophysical data have shown that the vinculin–actin interaction is asymmetrical, where the bond is strengthened when an actin filament is under pointed (-) end-directed load (Huang et al., 2017). Additional work has demonstrated that differential recruitment of vinculin to sites of high tension in epithelial cells is used to balance tensile and shear forces across cell contacts (Kale et al., 2018). We propose a model in which tension activates αE-catenin at nascent cardiomyocyte contacts to promote ligand binding. Vinculin recruitment, in turn, promotes myofibril binding to strengthen the AJ under load and provide mechanical integration (Figure 7). Vinculin functions as both a mechanical linchpin and critical organizer of actomyosin and AJ architecture to regulate cardiomyocyte adhesion. We suggest that this is a general mechanism cells use to link the AJ to contractile actin networks across various cells types. For example, vinculin is enriched at tricellular junctions in epithelial sheets where contractile actin terminates perpendicularly to AJs (Higashi and Miller, 2017), similar to cardiomyocytes. Linking to specific actin networks through selective ligand recruitment would allow the AJ to control mechanical load and respond to changes in cellular tension.
FIGURE 7:
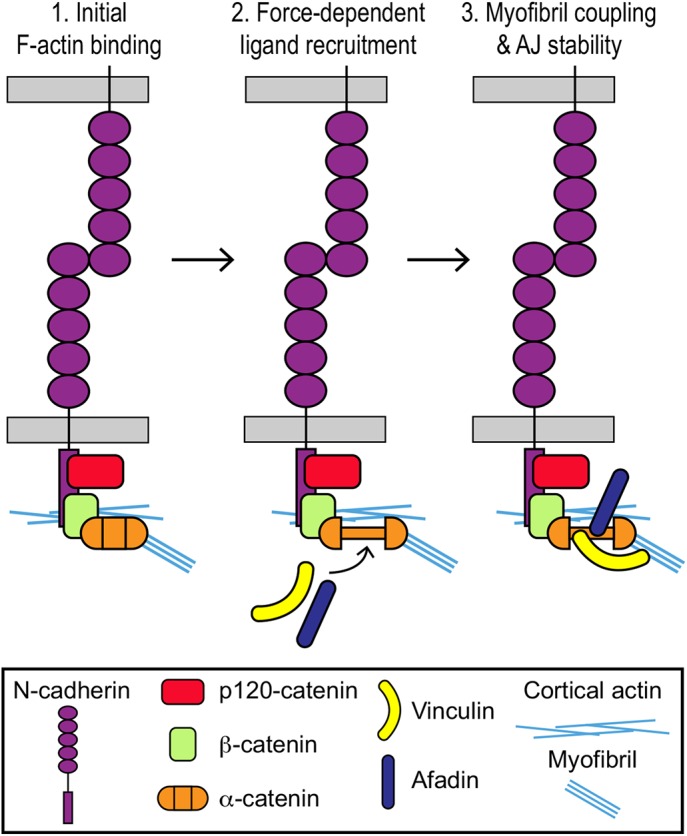
Model for linking contractile actin to cardiomyocyte AJs. (1) The cadherin-catenin complex makes initial contact with F-actin through α-catenin. Mechanical force is required to promote strong binding of the αE-catenin ABD to F-actin. (2) Increased tension induces conformational changes in the central M-region of α-catenin to promote recruitment of vinculin and afadin. (3) Vinculin recruitment stabilizes the AJ-actin interface under mechanical load to promote myofibril coupling and mechanical continuity between cardiomyocytes. The role of afadin at cardiomyocyte AJs is not clear.
In summary, our results provide novel insights into the AJ-myofibril linkage in cardiomyocytes. We show that vinculin recruitment through α-catenin is required to couple the cardiomyocyte AJ to contractile actin. Our findings underscore the importance of vinculin at the AJ and the role it plays in stabilizing the AJ-actin interface under mechanical load.
MATERIALS AND METHODS
Plasmids
To build the N-cadherin-GFP-αE-catenin fusions, αE-catenin fragments encoding amino acids 273–510 (M1–M2), 273–651 (M1–M3), 273–906 (M1-ABD), 396-906 (M2-ABD), and 671–906 (ABD) were cloned into pEGFP-C1 by PCR. Next, Gibson Assembly (NEBuilder HiFi DNA Assembly Kit; New England Biolabs) was used to clone the N-cadherin fragment amino acids 1–839 into pcDNA3.1 (Thermo Fisher). Gibson assembly was then used to insert the EGFP-αE-catenin fragments into the pcDNA3.1 N-cadherin amino acids 1–839 backbone, downstream and in-frame with N-cadherin. During construction, a 12-amino-acid glycine and alanine linker was inserted between N-cadherin and αE-catenin to increase flexibility.
The point mutations R329A, R330A, L347A, L348A, and Y351A were introduced by site-directed mutagenesis (QuikChange Lightening Kit; Agilent) in the αE-catenin M-region of M1-ABD to inhibit vinculin binding and create the M1mutV-ABD construct (Chen et al., 2015).
Cardiomyocyte isolation and culture
All animal work was approved by the University of Pittsburgh Division of Laboratory Animal Resources. Outbred Swiss Webster mice were used to generate wild-type cardiomyocytes for blebbistatin experiments. N-cadfx'fx conditional knockout mice (Jackson Labs, stock #007611; Kostetskii et al., 2005) were used to generate N-cadherin-null cardiomyocytes.
Tissue culture dishes or MatTek dishes (35-mm dish with 10-mm microwell) were coated with rat tail Type I collagen (Millipore) diluted to 0.5 μg/μl in phosphate-buffered saline (PBS) for 30 min at room temperature. Dishes were dried and treated with UV radiation for 1 h, after which they were washed with PBS, dried, and stored at room temperature in the dark.
Neonatal mouse cardiomyocytes were isolated as described (Ehler et al., 2013). Briefly, mouse pups were killed at P1–P3, and the hearts were removed, cleaned, minced, and digested overnight at 4°C in 20 mM BDM (2,3-butanedione monoxime) and 0.0125% trypsin in Hank's balanced salt solution. The following day, heart tissue was digested further in 15 mg/ml Collagenase/Dispase (Roche) in Leibovitz media with 20 mM BDM to create a single-cell suspension. Cells were preplated for 1.5–2 h in plating media (65% high glucose DMEM, 19% M-199, 10% horse serum, 5% fetal bovine serum, and 1% penicillin-streptomycin) to remove fibroblasts and endothelial cells. Cardiomyocytes were plated on MatTek dishes (1.5 × 105) or 12-well dishes (4.5 × 105) in plating media. Sixteen hours postplating, the plating media were exchanged for maintenance media (78% high glucose DMEM, 17% M-199, 4% horse serum, 1% penicillin-streptomyocin, 1 μM AraC, and 1 μM Isoproternol).
Adenovirus production and infection
N-cadherin-GFP-αE-catenin fusions were expressed as adenoviruses using the AdEasy System as described (Luo et al., 2007; Li et al., 2019). Briefly, N-cadherin-GFP-αE-catenin fusions were moved in to pShuttle-CMV using NEBBuilder HiFi DNA Assembly Master Mix (New England Biolabs). Positive clones were transformed into AdEasier Escherichia coli cells to generate recombinant adenovirus DNA. Adenoviral plasmids were then transfected into HEK293 cells for virus production. Virus was amplified and purified using AdenoPACK 20 Adenovirus (Ad5) purification and concentration kit (Sartorius). Virus titer was determined by qPCR using Adeno-X qPCR Titration Kit (Clontech) on an Applied Biosystems 7900HT.
Adenovirus expressing Cre (Ad(RGD)-CMV-iCre) was purchased from Vector Biolabs.
Cdh2fx/fx cardiomyocytes were infected with adenovirus Cre at MOI (multiplicity of infection) 75 on the day of plating to achieve 100% infection. The media were replaced with maintenance media 16 h after virus addition, and 6–8 h later (22–24 h after Cre infection), cardiomyocytes were infected with the N-cadherin-GFP-αE-catenin fusion adenovirus at MOI 10–15 to achieve >50% infection rate. Cardiomyocytes were fixed 96 h after the initial Cre infection for analysis.
Immunofluorescence
Cells were processed for immunofluorescence as follows: cells were fixed in warmed (37°C) 4% EM grade paraformaldehyde in PHM buffer (60 mM PIPES, pH 7.0, 25 mM HEPES, pH 7.0, 2 mM MgCl2, and 0.12 M sucrose) for 10 min and washed twice with PBS. Cells were permeabilized with 0.2% Triton X-100 in PBS for 4 min and washed twice with PBS. Cells were blocked in 10% bovine serum albumin (BSA; Sigma) in PBS for 1 h at room temperature. Samples were incubated with primary antibodies in PBS + 1% BSA for 1 h at room temperature, washed 2× in PBS, incubated with secondary antibodies in PBS + 1% for 1 h at room temperature, washed 2× in PBS, and then mounted in Prolong Diamond (Thermo Fisher Scientific). All samples were cured at least 24 h before imaging.
For blebbistatin experiments, cardiomyocytes (96 h postplating) were treated with 100 μM blebbistatin in DMSO or DMSO alone for 10 min to 1 h. Cells were incubated at 37°C during treatment. After incubation, cells were first prepermeabilized in 0.2% Triton X-100 in PBS for 2 min, then fixed and labeled as described.
Antibodies
Primary antibodies used for immunostaining were: anti-αE-catenin (1:100; Enzo Life Sciences ALX-804-101-C100), anti-β-catenin (1:250; BD Transduction Laboratories 610154), anti-plakoglobin (1:100; Cell Signaling 2309), anti-vinculin (1:800; Sigma Aldrich V9131), anti-N-cadherin (1:250; Invitrogen 99-3900), anti-l-afadin (1:500; Sigma Aldrich A0349), anti-connexin-43 (1:100; ProteinTech 15386-1-AP), anti-plakophilin 2 (1:10; Progen 651101), anti-desmoglein 2 (1:250; Abcam EPR6768), anti–α-actinin (1:250; Sigma A7811), anti-Mena (1:300, mouse monoclonal, a kind gift from Frank Gertler, Massachusetts Institute of Technology), and anti-Cre recombinase (1:300; Cell Signaling 12830). Secondary antibodies used were goat anti-mouse or anti-rabbit immunoglobulin G conjugated to Alexa Fluor-488, 568, or 647 (1:250; Invitrogen). F-actin was visualized using an Alexa Fluor dye conjugated to phalloidin (1:100; Thermo Fisher Scientific).
Whole cell lysis and immunoblotting
Cardiomyocytes were cultured on collagen-coated 12-well dishes. 96 h after plating, cardiomyocytes were lysed with RIPA buffer supplemented with 1× protease inhibitors (Millipore). Lysate protein concentration was determined by BCA Assay (Bio-Rad). Lysate (15 μg) was loaded per well and resolved by 10% SDS–PAGE and then transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked in 5% BSA in 1× TBST + 0.02% NaN3 for 1 h at room temperature. Primary antibodies (N-cadherin 1:2500, GAPDH 1:750; Abcam ab9485) diluted in 5% BSA in 1× TBST + 0.02% NaN3 were added to the membrane and incubated overnight at 4°C. The membrane was washed in 1× TBST three times and then LI-COR secondary antibodies (goat anti-mouse 680, 1:15,000; goat anti-rabbit 800, 1:15,000) diluted in 1× TBST + 0.02% NaN3 were added. The membrane was incubated for 1 h at room temperature, washed three times in 1× TBST and once in PBS before imaging on a LI-COR Odyssey imaging system. Band intensities were quantified in ImageJ and plotted in Prism (GraphPad).
Confocal microscopy
Cells were imaged with a 100 × 1.49 NA objective or a 40 × 1.30 objective on a Nikon Eclipse Ti inverted microscope outfitted with a Prairie swept field confocal system, Agilent monolithic laser launch and Andor iXon3 camera using NIS-Elements (Nikon) imaging software. Image stacks were deconvolved (3D Deconvolution) in NIS-Elements and then maximum projections of the image stacks were created for presentation. Expression and staining levels were adjusted for presentation purposes in Photoshop (Adobe). All levels were corrected the same across each figure except Figure 6 where the phalloidin labeling of F-actin was modified individually to account for differences in staining. Note that Ncad-GFP-ABD levels were adjusted individually in Supplemental Figure S2 as this construct localized to cell–cell contacts less efficiently than the other fusions.
FRAP experiments
FRAP experiments were conducted on a Nikon swept field confocal microscope (describe above) outfitted with a Tokai Hit cell incubator and Bruker miniscanner. Actively contracting cells were maintained at 37°C in a humidified, 5% CO2 environment. User-defined regions along cell–cell contacts were bleached with a 488 laser and recovery images collected every 10 s for 15 min. FRAP data were quantified in ImageJ (National Institutes of Health [NIH]) and average recovery plots were measured in Excel (Microsoft). All recovery plots represent data from two independent transfections of unique cell preps. The data were fitted to a double-exponential curve to determine the mobile fraction and half time of recovery in Prism (GraphPad). Only recovery rates of the slow pool are reported as this was the dominant mobile pool (87–91%) for all constructs.
Electron microscopy
Cardiomyocytes were grown on collagen-coated MatTek dishes and fixed as described above. After fixation and washing, cells were incubated with 1% OsO4 for 1 h. After several PBS washes, dishes were dehydrated through a graded series of 30%–100% ethanol and then infiltrated for 1 h in Polybed 812 epoxy resin (Polysciences, Warrington, PA). After several changes of 100% resin over 24 h, cells were embedded in inverted Beem capsules, cured at 37°C overnight, and then hardened for 2 d at 65°C. Blocks were removed from the glass dish via freeze/thaw method by alternating liquid nitrogen and 100°C water. Ultrathin (60-nm) sections were collected on to 200-mesh copper grids and stained with 2% uranyl acetate in 50% methanol for 10 min and 1% lead citrate for 7 min. Samples were photographed with a JEOL JEM 1400 PLUS Transmission Electron Microscope (Peabody, MA) at 80 kV with a Hamamatsu ORCA-HR bottom mount camera.
Image analysis
To measure vinculin and afadin recruitment to cell–cell contacts in blebbistatin treatment experiments, a single plane was selected from the z-stack where the cell–cell contact was most in focus and used for analysis in ImageJ (NIH). IsoJ Dark thresholding was then used to create a mask of either the plakoglobin staining channel (vinculin measurements) or the αE-catenin staining channel (afadin measurements) to define the region of analysis (cell–cell contacts). The average vinculin or afadin signal intensity was measured in the masked region and plotted in Prism software (Graphpad). A one-way analysis of variance (ANOVA) with multiple comparisons was performed; p < 0.05.
To quantify AJ ligands vinculin, afadin, Mena, and α-actinin recruitment to N-cadherin-GFP-αE-catenin fusions, a single plane was selected from the z-stack and IsoJ Dark thresholding was used to create a mask of the GFP channel to define the region of analysis in ImageJ. Ligand (vinculin, afadin, Mena, or α-actinin) signal intensity was then measured within the masked region. Next, three random intensity measurements of the ligand were taken in the cell cytoplasm and these values were averaged. Finally, the ratio of ligand intensity within the mask was divided by the cytoplasmic signal to normalize between samples and calculate the contact/cytoplasmic ratio. Colocalization data were plotted with Prism software (GraphPad). A one-way ANOVA with multiple comparisons was performed to determine significance; p < 0.05.
To examine the relationship between N-cadherin-GFP or N-cadherin-GFP-αE-catenin fusion expression levels and ligand recruitment, the vinculin or afadin contact/cytoplasmic ratio was plotted against the average GFP intensity of each fusion construct within the masked cell contact region (described above). Linear regression analysis was performed to calculate the slope, 95% confidence intervals, R square value and p value using Prism software (GraphPad).
Contact morphology was measured in Nikon Elements. Similar to above, a single plane was selected from the z-stack where the cell–cell contact was in focus. Thresholding was used to create a binary mask of the GFP channel that was then manually pruned to remove noncontact signal and better define cell–cell contacts for analysis. The length and circularity of all regions >1 μm2 in area was measured. Length was based on the rod model and calculated using the formula
Circularity was calculated from the area and perimeter of a contact using the formula
A circle has a circularity value of 1; all other shapes have a circularity value <1. Measurements were calculated from at least 190 contacts for each construct from 20 images collected from two independent experiments (10 images per experiment). Length and circularity mean and SEM was calculated in Excel (Microsoft) and plotted in Prism (Graphpad).
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Kwiatkowski lab for their helpful comments on the manuscript. This work was supported by National Institutes of Health F31 HL136069 (C.D.M.) and R01 HL127711 (A.V.K.).
Abbreviations used:
- ABD
actin-binding domain
- AJ
adherens junctions
- BDM
2,3-butanedione monoxime
- BSA
bovine serum albumin
- Cre
Cre-recombinase
- FRAP
fluorescence recovery after photobleaching
- ICD
intercalated disc
- MOI
multiplicity of infection
- PBS
phosphate-buffered saline
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-04-0216) on September 4, 2019.
REFERENCES
- Aguilar F, Belmonte SL, Ram R, Noujaim SF, Dunaevsky O, Protack TL, Jalife J, Todd Massey H, Gertler FB, Blaxall BC. (2011). Mammalian enabled (Mena) is a critical regulator of cardiac function. Am J Physiol Heart Circ Physiol , H1841–H1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett PM, Maggs AM, Baines AJ, Pinder JC. (2006). The transitional junction: a new functional subcellular domain at the intercalated disc. Mol Biol Cell , 2091–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchini JM, Kitt KN, Gloerich M, Pokutta S, Weis WI, Nelson WJ. (2015). Reevaluating alphaE-catenin monomer and homodimer functions by characterizing E-cadherin/alphaE-catenin chimeras. J Cell Biol , 1065–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrmann CM, Grund C, Kuhn C, Hofmann I, Pieperhoff S, Franke WW. (2006). The area composita of adhering junctions connecting heart muscle cells of vertebrates. II. Colocalizations of desmosomal and fascia adhaerens molecules in the intercalated disk. Eur J Cell Biol , 469–485. [DOI] [PubMed] [Google Scholar]
- Buckley CD, Tan J, Anderson KL, Hanein D, Volkmann N, Weis WI, Nelson WJ, Dunn AR. (2014). Cell adhesion. The minimal cadherin-catenin complex binds to actin filaments under force. Science , 1254211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras G, Yap AS. (2018). Tensile forces and mechanotransduction at cell-cell junctions. Curr Biol , R445–R457. [DOI] [PubMed] [Google Scholar]
- Chen CS, Hong S, Indra I, Sergeeva AP, Troyanovsky RB, Shapiro L, Honig B, Troyanovsky SM. (2015). alpha-Catenin-mediated cadherin clustering couples cadherin and actin dynamics. J Cell Biol , 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HJ, Pokutta S, Cadwell GW, Bobkov AA, Bankston LA, Liddington RC, Weis WI. (2012). alphaE-catenin is an autoinhibited molecule that coactivates vinculin. Proc Natl Acad Sci USA , 8576–8581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W, Acharya BR, Peyret G, Fardin MA, Mege RM, Ladoux B, Yap AS, Fanning AS, Peifer M. (2016). Remodeling the zonula adherens in response to tension and the role of afadin in this response. J Cell Biol , 243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MA, Ireton RC, Reynolds AB. (2003). A core function for p120-catenin in cadherin turnover. J Cell Biol , 525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI. (2005). Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell , 903–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehler E. (2016). Cardiac cytoarchitecture—why the “hardware” is important for heart function! Biochim Biophys Acta , 1857–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehler E, Moore-Morris T, Lange S. (2013). Isolation and culture of neonatal mouse cardiomyocytes. J Vis Exp , E50154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finer JT, Simmons RM, Spudich JA. (1994). Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature , 113–119. [DOI] [PubMed] [Google Scholar]
- Frank D, Frey N. (2011). Cardiac Z-disc signaling network. J Biol Chem , 9897–9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Volk T, Volberg T. (1985). Molecular heterogeneity of adherens junctions. J Cell Biol , 1523–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goossens S, Janssens B, Bonne S, De Rycke R, Braet F, van Hengel J, van Roy F. (2007). A unique and specific interaction between alphaT-catenin and plakophilin-2 in the area composita, the mixed-type junctional structure of cardiac intercalated discs. J Cell Sci , 2126–2136. [DOI] [PubMed] [Google Scholar]
- Harrison OJ, Jin X, Hong S, Bahna F, Ahlsen G, Brasch J, Wu Y, Vendome J, Felsovalyi K, Hampton CM, et al. (2011). The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure , 244–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi T, Miller AL. (2017). Tricellular junctions: how to build junctions at the TRICkiest points of epithelial cells. Mol Biol Cell , 2023–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman BD, Yap AS. (2015). Towards a dynamic understanding of cadherin-based mechanobiology. Trends Cell Biol , 803–814. [DOI] [PubMed] [Google Scholar]
- Hong S, Troyanovsky RB, Troyanovsky SM. (2013). Binding to F-actin guides cadherin cluster assembly, stability, and movement. J Cell Biol , 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DL, Bax NA, Buckley CD, Weis WI, Dunn AR. (2017). Vinculin forms a directionally asymmetric catch bond with F-actin. Science , 703–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huveneers S, Oldenburg J, Spanjaard E, van der Krogt G, Grigoriev I, Akhmanova A, Rehmann H, de Rooij J. (2012). Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J Cell Biol , 641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama N, Sarpal R, Wood MN, Barrick SK, Nishikawa T, Hayashi H, Kobb AB, Flozak AS, Yemelyanov A, Fernandez-Gonzalez R, et al. (2018). Force-dependent allostery of the alpha-catenin actin-binding domain controls adherens junction dynamics and functions. Nat Commun , 5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama N, Tanaka N, Abe K, Yang YJ, Abbas YM, Umitsu M, Nagar B, Bueler SA, Rubinstein JL, Takeichi M, Ikura M. (2013). An autoinhibited structure of alpha-catenin and its implications for vinculin recruitment to adherens junctions. J Biol Chem , 15913–15925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens B, Goossens S, Staes K, Gilbert B, van Hengel J, Colpaert C, Bruyneel E, Mareel M, van Roy F. (2001). alphaT-catenin: a novel tissue-specific beta-catenin-binding protein mediating strong cell-cell adhesion. J Cell Sci , 3177–3188. [DOI] [PubMed] [Google Scholar]
- Kale GR, Yang X, Philippe JM, Mani M, Lenne PF, Lecuit T. (2018). Distinct contributions of tensile and shear stress on E-cadherin levels during morphogenesis. Nat Commun , 5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TJ, Zheng S, Sun J, Muhamed I, Wu J, Lei L, Kong X, Leckband DE, Wang Y. (2015). Dynamic visualization of alpha-catenin reveals rapid, reversible conformation switching between tension states. Curr Biol , 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen KA, Soler AP, Johnson KR, Wheelock MJ. (1995). Interaction of alpha-actinin with the cadherin/catenin cell-cell adhesion complex via alpha-catenin. J Cell Biol , 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostetskii I, Li J, Xiong Y, Zhou R, Ferrari VA, Patel VV, Molkentin JD, Radice GL. (2005). Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ Res , 346–354. [DOI] [PubMed] [Google Scholar]
- Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. (2003). Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol , 541–564. [DOI] [PubMed] [Google Scholar]
- Kroemker M, Rudiger AH, Jockusch BM, Rudiger M. (1994). Intramolecular interactions in vinculin control alpha-actinin binding to the vinculin head. FEBS Lett , 259–262. [DOI] [PubMed] [Google Scholar]
- le Duc Q, Shi Q, Blonk I, Sonnenberg A, Wang N, Leckband D, de Rooij J. (2010). Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J Cell Biol , 1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leerberg JM, Gomez GA, Verma S, Moussa EJ, Wu SK, Priya R, Hoffman BD, Grashoff C, Schwartz MA, Yap AS. (2014). Tension-sensitive actin assembly supports contractility at the epithelial zonula adherens. Curr Biol , 1689–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Gao E, Vite A, Yi R, Gomez L, Goossens S, van Roy F, Radice GL. (2015). Alpha-catenins control cardiomyocyte proliferation by regulating Yap activity. Circ Res , 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Goossens S, van Hengel J, Gao E, Cheng L, Tyberghein K, Shang X, De Rycke R, van Roy F, Radice GL. (2012). Loss of alphaT-catenin alters the hybrid adhering junctions in the heart and leads to dilated cardiomyopathy and ventricular arrhythmia following acute ischemia. J Cell Sci , 1058–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Merkel CD, Zeng X, Heier JA, Cantrell PS, Sun M, Stolz DB, Watkins SC, Yates NA, Kwiatkowski AV. (2019). The N-cadherin interactome in primary cardiomyocytes as defined by quantitative proximity proteomics. J Cell Sci , jcs221606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Deng ZL, Luo X, Tang N, Song WX, Chen J, Sharff KA, Luu HH, Haydon RC, Kinzler KW, et al. (2007). A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc , 1236–1247. [DOI] [PubMed] [Google Scholar]
- Luo Y, Radice GL. (2003). Cadherin-mediated adhesion is essential for myofibril continuity across the plasma membrane but not for assembly of the contractile apparatus. J Cell Sci , 1471–1479. [DOI] [PubMed] [Google Scholar]
- Mandai K, Nakanishi H, Satoh A, Obaishi H, Wada M, Nishioka H, Itoh M, Mizoguchi A, Aoki T, Fujimoto T, et al. (1997). Afadin: a novel actin filament-binding protein with one PDZ domain localized at cadherin-based cell-to-cell adherens junction. J Cell Biol , 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa K, Himoto T, Mochizuki Y, Ikenouchi J. (2018). alpha-Catenin controls the anisotropy of force distribution at cell-cell junctions during collective cell migration. Cell Rep , 3447–3456. [DOI] [PubMed] [Google Scholar]
- Mege RM, Ishiyama N. (2017). Integration of cadherin adhesion and cytoskeleton at adherens junctions. Cold Spring Harb Perspect Biol , a028738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieset JE, Redfield AR, Jin F, Knudsen KA, Johnson KR, Wheelock MJ. (1997). Characterization of the interactions of alpha-catenin with alpha-actinin and beta-catenin/plakoglobin. J Cell Sci (Pt 8), 1013–1022. [DOI] [PubMed] [Google Scholar]
- Pang SM, Le S, Kwiatkowski AV, Yan J. (2019). Mechanical stability of alphaT-catenin and its activation by force for vinculin binding. Mol Biol Cell , 1930–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokutta S, Choi HJ, Ahlsen G, Hansen SD, Weis WI. (2014). Structural and thermodynamic characterization of cadherin.beta-catenin.alpha-catenin complex formation. J Biol Chem , 13589–13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokutta S, Drees F, Takai Y, Nelson WJ, Weis WI. (2002). Biochemical and structural definition of the l-afadin- and actin-binding sites of alpha-catenin. J Biol Chem , 18868–18874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan ES, Izard T. (2013). Dimer asymmetry defines alpha-catenin interactions. Nat Struct Mol Biol , 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratheesh A, Yap AS. (2012). A bigger picture: classical cadherins and the dynamic actin cytoskeleton. Nat Rev Mol Cell Biol , 673–679. [DOI] [PubMed] [Google Scholar]
- Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS. (1995). Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci USA , 8813–8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddiki R, Narayana G, Strale PO, Balcioglu HE, Peyret G, Yao M, Le AP, Teck Lim C, Yan J, Ladoux B, Mege RM. (2018). Force-dependent binding of vinculin to alpha-catenin regulates cell-cell contact stability and collective cell behavior. Mol Biol Cell , 380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro L, Weis WI. (2009). Structure and biochemistry of cadherins and catenins. Cold Spring Harb Perspect Biol , a003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi I, Simpson DG, Carver W, Price R, Hirozane T, Terracio L, Borg TK. (1997). Vinculin is an essential component for normal myofibrillar arrangement in fetal mouse cardiac myocytes. J Mol Cell Cardiol , 2041–2052. [DOI] [PubMed] [Google Scholar]
- Vermij SH, Abriel H, van Veen TA. (2017). Refining the molecular organization of the cardiac intercalated disc. Cardiovasc Res , 259–275. [DOI] [PubMed] [Google Scholar]
- Vite A, Li J, Radice GL. (2015). New functions for alpha-catenins in health and disease: from cancer to heart regeneration. Cell Tissue Res , 773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vite A, Radice GL. (2014). N-cadherin/catenin complex as a master regulator of intercalated disc function. Cell Commun Adhes , 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl JK, 3rd, Kim YJ, Cullen JM, Johnson KR, Wheelock MJ. (2003). N-cadherin-catenin complexes form prior to cleavage of the proregion and transport to the plasma membrane. J Biol Chem , 17269–17276. [DOI] [PubMed] [Google Scholar]
- Wickline ED, Dale IW, Merkel CD, Heier JA, Stolz DB, Kwiatkowski AV. (2016). alphaT-Catenin is a constitutive actin-binding alpha-catenin that directly couples the cadherin.catenin complex to actin filaments. J Biol Chem , 15687–15699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Baribault H, Adamson ED. (1998). Vinculin knockout results in heart and brain defects during embryonic development. Development , 327–337. [DOI] [PubMed] [Google Scholar]
- Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ. (2005). Deconstructing the cadherin-catenin-actin complex. Cell , 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao M, Qiu W, Liu R, Efremov AK, Cong P, Seddiki R, Payre M, Lim CT, Ladoux B, Mege RM, Yan J. (2014). Force-dependent conformational switch of alpha-catenin controls vinculin binding. Nat Commun , 4525. [DOI] [PubMed] [Google Scholar]
- Yap AS, Duszyc K, Viasnoff V. (2018). Mechanosensing and mechanotransduction at cell-cell junctions. Cold Spring Harb Perspect Biol , a028761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura S. (2017). Actin filament association at adherens junctions. J Med Invest , 14–19. [DOI] [PubMed] [Google Scholar]
- Yonemura S, Wada Y, Watanabe T, Nagafuchi A, Shibata M. (2010). alpha-Catenin as a tension transducer that induces adherens junction development. Nat Cell Biol , 533–542. [DOI] [PubMed] [Google Scholar]
- Zankov DP, Sato A, Shimizu A, Ogita H. (2017). Differential effects of myocardial afadin on pressure overload-induced compensated cardiac hypertrophy. Circ J , 1862–1870. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.