

Abstract
Mitochondria are unique organelles harboring two distinct membranes, the mitochondrial inner and outer membrane (MIM and MOM, respectively). Mitochondria comprise only a subset of metabolic pathways for the synthesis of membrane lipids; therefore most lipid species and their precursors have to be imported from other cellular compartments. One such import process is mediated by the ER mitochondria encounter structure (ERMES) complex. Both mitochondrial membranes surround the hydrophilic intermembrane space (IMS). Therefore, additional systems are required that shuttle lipids between the MIM and MOM. Recently, we identified the IMS protein Mcp2 as a high-copy suppressor for cells that lack a functional ERMES complex. To understand better how mitochondria facilitate transport and biogenesis of lipids, we searched for genetic interactions of this suppressor. We found that MCP2 has a negative genetic interaction with the gene TGL2 encoding a neutral lipid hydrolase. We show that this lipase is located in the intermembrane space of the mitochondrion and is imported via the Mia40 disulfide relay system. Furthermore, we show a positive genetic interaction of double deletion of MCP2 and PSD1, the gene encoding the enzyme that synthesizes the major amount of cellular phosphatidylethanolamine. Finally, we demonstrate that the nucleotide-binding motifs of the predicted atypical kinase Mcp2 are required for its proper function. Taken together, our data suggest that Mcp2 is involved in mitochondrial lipid metabolism and an increase of this involvement by overexpression suppresses loss of ERMES.
INTRODUCTION
Mitochondria are endosymbiotic organelles present in almost all eukaryotic cells. They are the main source of cellular ATP and play an important role in many cellular processes. Their involvement in many metabolic pathways is reflected by intimate connections to almost every other eukaryotic organelle at contact sites (CS)—areas of close apposition of organelle membranes. For a long time, the ER mitochondria encounter structure (ERMES) complex, which functions as a molecular tether, was the only known mitochondrial CS mediator in yeast (Kornmann et al., 2009). Therefore, this multiprotein complex was the focus of many studies (Lang et al., 2015a). More recently, contact sites between mitochondria and the vacuole, plasma membrane, lipid droplets, and peroxisomes, as well as the proteins mediating them, were reported and studied in some detail (Elbaz-Alon et al., 2014; Honscher et al., 2014; John Peter et al., 2017; Kakimoto et al., 2018; Shai et al., 2018). Several of these studies indicate that ER–mitochondria and vacuole–mitochondria contacts can functionally compensate for each other to some extent (Elbaz-Alon et al., 2014; Honscher et al., 2014; John Peter et al., 2017). The main function of the ERMES complex seems to be its role in lipid exchange and it was the first mitochondrial tethering complex for which direct binding of lipid molecules was shown (AhYoung et al., 2015; Kawano et al., 2018).
Because mitochondria contain two distinct membranes, it is obvious that mitochondrial biogenesis also depends on regulated lipid exchange between the mitochondrial outer and inner membrane (MOM and MIM, respectively). To ensure this exchange, lipid molecules and their lipophilic precursors have to traverse the hydrophilic intermembrane space (IMS). It has been shown that the major system regulating the inner boundary membrane, where the MOM and MIM are in close proximity, is the mitochondrial contact site and cristae organizing system (MICOS) complex (reviewed in Wollweber et al., 2017). Recently, it was reported that MICOS also contributes to the regulation of intramitochondrial lipid distribution (Aaltonen et al., 2016). Other factors that have been suggested to contribute to intramitochondrial phospholipid handling are the abundant MOM protein porin (Por1) and its interaction partner Mdm31 in the MIM (Miyata et al., 2018). Mdm31 was initially identified as a mitochondrial morphology factor (Dimmer et al., 2005). Although other sites of contact for MOM and MIM have been proposed, such as the interaction of the translocase of the outer membrane (TOM) and translocase of the inner membrane (TIM) machineries (Waegemann et al., 2015), their relevance to mitochondrial lipid trafficking is unknown. Recently, we demonstrated that another MOM protein, Mcp3, which functions as a weak suppressor of loss of ERMES function, interacts physically with the MIM protein Aim19 (Sinzel et al., 2018).
So far, the only system in Saccharomyces cerevisiae that has been identified as shuttling phospholipids across the IMS consists of the lipid-binding proteins Ups1 and Ups2 that act together with their mediator, Mdm35 (Potting et al., 2010; Connerth et al., 2012; Tamura et al., 2012a,b; Aaltonen et al., 2016). Ups1 is responsible for the transport of phosphatidic acid (PA) to the MIM, where cardiolipin (CL) is synthesized from this precursor. In contrast, Ups2 shuttles phosphatidylserine (PS) between the MOM and the MIM for the synthesis of phosphatidylethanolamine (PE) by Psd1. Both proteins are bound in the IMS to Mdm35, a protein initially identified to be essential for mitochondrial distribution and morphology in yeast (Dimmer et al., 2002). The mode by which other phospholipids (besides PS and PA) and their precursors travel between MOM and MIM is still unknown.
In this study, we present two novel IMS-exposed proteins in yeast that are involved in lipid metabolism. One of them is Tgl2, a lipase that is imported into the IMS via the Mia40 disulfide relay pathway. The other is Mcp2, an atypical kinase for which we show that its nucleotide binding motifs are required for proper function. The synthetic negative genetic interaction of both genes and the functional cross–talk of Mcp2 with the PS decarboxylase Psd1 strongly suggest a role of Mcp2 and Tgl2 in the handling of mitochondrial lipids and/or their precursors.
RESULTS
MCP2 displays negative synthetic genetic interaction with the mitochondrial lipase TGL2
We previously identified Mcp2 as a high-copy suppressor of loss of functional ERMES complex. However, the deletion of MCP2 did not result in any obvious growth phenotype (Tan et al., 2013). One possible reason for the absence of a growth phenotype upon deletion of a gene is the existence of parallel backup pathways to support viability. Hence, to better characterize the molecular function of Mcp2, we asked whether MCP2 shows synthetic genetic interactions with other genes. To this end, we crossed all ∼6000 yeast single mutants (either deletion for nonessential or down-regulation for essential genes) with an mcp2Δ strain and then monitored the growth of the double mutants. We reported previously, based on manual tetrad dissection analysis, that the four genes encoding the main subunits of the ERMES complex showed negative genetic interactions with MCP2 (Tan et al., 2013). In our current whole-genome synthetic genetic interaction analysis, we found that deletion of either MDM10, MDM34, or MDM12 in combination with deletion of MCP2 resulted in a smaller area of the growth zone (40–60% of the area of single deletions of ERMES components). The deletion strain of MMM1 was absent from our initial library and hence could not be assayed. Interestingly, mcp2Δgem1Δ cells also grew more slowly in the SGA analysis (only 44% of the area of gem1Δ single deletion). Gem1 is a MOM GTPase that was shown to be a peripheral subunit of the ERMES complex (Frederick et al., 2004; Kornmann et al., 2011; Stroud et al., 2011). Yet the most prominent genetic interaction with a gene encoding for a protein related to mitochondrial function was with TGL2. The double-deletion mutant mcp2Δtgl2Δ cells formed a much smaller growth area (5% in comparison with the tgl2Δ cells).
TGL2 encodes for a protein with a predicted lipase domain (Grillitsch and Daum, 2011). It was shown before that it cofractionates with the mitochondrial fraction in sucrose gradient centrifugation and that the enriched protein has lipolytic activity toward diacylglycerols (DAGs) and triacylglycerols (TAGs) (Ham et al., 2010). However, its role in mitochondria remains unclear.
For further analysis, we first confirmed the growth impairment of mcp2Δtgl2Δ cells at the single-gene level. We constructed the mcp2Δtgl2Δ strain by tetrad dissection of the heterozygous diploid double deletion in the W303 wild-type background. Whereas the single mutants tgl2Δ and mcp2Δ did not show growth impairment on standard media, the double-deletion mutant grew significantly slower (Figure 1A). The growth defect was most pronounced on the nonfermentable carbon source glycerol, consistent with a role of these proteins in mitochondrial functionality. The synthetic growth defect is in agreement with our observation with the double mutant in the BY4741 genetic background, which was used in the initial high-throughput synthetic interaction analysis. Growth of the double-deletion mutant could be restored by expression of plasmid-borne Mcp2 or Tgl2 (Figure 1B), demonstrating that the growth phenotype is directly linked to the absence of both proteins. Of note, this growth defect of the double-deletion strain was observed initially on all media for colonies retrieved by tetrad dissection. Yet after prolonged growth on fermentable carbon sources, the double-deleted cells adapted and then grew similarly to single-deletion mutants and the original control cells. The growth defect remained stable on nonfermentable carbon sources.
FIGURE 1:

Synthetic negative genetic interaction of MCP2 and TGL2. (A) Growth defect of mcp2Δtgl2Δ cells. Wild-type (WT), mcp2∆, tgl2Δ, and mcp2Δtgl2Δ cells were grown to logarithmic phase and spotted on either SD or SG plates in a 1:5 dilution series. Plates were incubated for growth at 30°C. (B) Growth defect of mcp2Δtgl2Δ can be rescued by expression of Mcp2 or Tgl2. Wild-type (WT) or mcp2Δtgl2Δ cells were transformed with the empty plasmid pYX142 (∅). In addition, mcp2Δtgl2Δ cells were transformed with pYX142 encoding MCP2, MCP2-HA, or TGL2 under the control of the TPI promoter. Cells were grown to logarithmic phase and spotted on SD-Leu or SG-Leu plates in a 1:5 dilution series. Plates were incubated for growth at 30°C. (C) Deletion of MCP2 and TGL2 leads to changes in mitochondrial morphology. WT, mcp2∆, tgl2Δ, and mcp2Δtgl2Δ cells expressing mitochondrial-targeted GFP (mtGFP) were grown to midlogarithmic phase and then analyzed by fluorescence microscopy. Typical images of the four different strains are shown (scale bar = 5 µm). (D) Quantification of the strains depicted in C. The average percentages of three independent experiments with at least 100 cells per experiment are shown with SD bars. (E) Lipid droplet analysis of mcp2Δtgl2Δ cells by fluorescence microscopy. Cells were grown to logarithmic phase and stained with BODIPY 493/503. (F) Neutral lipid levels are increased in cells lacking Mcp2 and Tgl2. Cells were stained with BODIPY 493/503 as in E and overall fluorescence was analyzed by a fluorescence reader. The bar diagram shows the average percentages of three independent experiments with SD bars. Fluorescence of WT cells was set to 100%. (G) Cells lacking Mcp2 and Tgl2 show alterations in phospholipid composition. Lipids were extracted from yeast cells grown on YPD to logarithmic phase and then analyzed by mass spectrometric analysis. The level of each phospholipid species as a % of total PL is shown as a mean with SD bars (n = 6; * p < 0.05). (H) Increased TAG levels in mcp2Δtgl2Δ cells. Cells grown and analyzed as in G. The average ratio with SD bars is shown (n = 6; * p < 0.05).
Next, we analyzed the mitochondrial morphology of the deletion mutants. Loss of Mcp2 did not result in any apparent morphological aberrations. Similarly, the deletion of TGL2 did not affect mitochondrial morphology. Yet the loss of both proteins resulted in fragmented and shortened organelles in about half of the population (Figure 1, C and D).
Tgl2 is a homologue of the triacyl glycerol lipase (TGL) family, which includes in S. cerevisiae also Tgl1, Tgl3, Tgl4, and Tgl5. These other lipases are mainly found in lipid droplets (LD; Grillitsch and Daum, 2011). Whereas Tgl1 hydrolyzes ergosterol esters to release ergosterol from its storage variant in LDs, Tgl3, Tgl4, and Tgl5 cleave TAGs and have overlapping functions. To test a possible link of Tgl2 and/or Mcp2 to LDs, we visualized lipid droplets of tgl2Δ and mcp2Δ cells, as well as the double deletion, with the lipophilic dye BODIPY 493/503. Using fluorescence microscopy, we did not observe an eminent difference between the control cells and the mutated ones; on the average, we observed 3–10 lipid droplets per cell in all investigated populations (Figure 1E). Yet the overall fluorescence intensity, as evaluated by a fluorescence reader, was higher in the double-deletion cells, suggesting elevated neutral lipid levels in these cells (Figure 1F).
Because both mitochondrial morphology defects and increased lipophilic dye accumulation could be the result of an alteration of the lipid composition and distribution in cells, we performed lipidomic analysis of mcp2Δtgl2Δ cells in comparison with the corresponding wild type. We found a significant increase in phosphatidylcholine (PC) content and a reduction in phosphatidylethanolamine (PE) and cardiolipin (CL) amounts. Interestingly, the latter two PLs are synthesized mainly (PE) or exclusively (CL) in mitochondria (Figure 1G and Supplemental Table 1). This phospholipid (PL) imbalance could explain the observed morphology alterations (Figure 1, C and D). Yet we cannot exclude the possibility that the reduction in CL and PE levels is a result of an overall lowered mitochondrial mass in the double-deletion cells. Furthermore, we observed an increase of the TAG/PL ratio for mcp2Δtgl2Δ cells (Figure 1H and Supplemental Table 1), further supporting an increase of TAG in LDs. Of note, we did not detect a significantly increased DAG/PL ratio (Figure 1H).
Taken together, the combined loss of both Mcp2 and Tgl2 results in a change in the overall (membrane and neutral) lipid composition of yeast cells.
Tgl2 is a soluble IMS protein
We reported previously that the predicted kinase domain of Mcp2 is exposed to the mitochondrial IMS (Tan et al., 2013). The observed genetic interaction of MCP2 with TGL2 led us to study in detail the localization of Tgl2. The protein does not contain a predicted N-terminal mitochondrial targeting sequence, yet evidence that Tgl2 might be a mitochondrial protein was provided previously by cell fractionation experiments and immunofluorescence staining (Ham et al., 2010). To confirm these observations, we constructed C- or N-terminal HA-tagged versions of Tgl2. Next, we tested the functionality of these constructs by transforming them into the double deletion strain mcp2Δtgl2Δ followed by growth analysis. Interestingly, the C-terminal HA-tag resulted in a nonfunctional fusion protein, whereas the variant with the N-terminal tag rescued the growth defect to the same extent as the native protein (Figure 2A). To verify the mitochondrial localization of Tgl2, we used yeast cells expressing the N-terminally tagged Tgl2 (HA-Tgl2) and performed subcellular fractionation. Indeed, Tgl2 was found exclusively in the mitochondrial fraction (Figure 2B).
FIGURE 2:

Tgl2 is a soluble intermembrane space protein. (A) N-terminal HA-tagged Tgl2 is functional. WT or mcp2Δtgl2Δ cells were transformed with the empty plasmid pYX142 (∅). In addition, mcp2Δtgl2Δ cells were transformed with pYX142 encoding the indicated proteins under the control of the TPI promoter. Cells were grown to logarithmic phase and spotted on SD-Leu plates in a 1:5 dilution series. Plates were incubated for growth at 30°C. (B) Tgl2 is a mitochondrial protein. Whole cell lysate (WCL) and fractions corresponding to cytosol (Cyt.), light microsomal fraction (ER), and mitochondria (Mito.) of cells expressing HA-Tgl2 were analyzed by SDS–PAGE and immunodecoration with antibodies against the HA-tag, the mitochondrial protein Tom20, a marker protein for the cytosol (Bmh1), and an ER marker protein (Erv2). (C) Tgl2 is a soluble protein. Mitochondria isolated from HA-Tgl2–expressing cells (T, total) were subjected to alkaline extraction. The supernatant (S) and pellet (P) fractions were analyzed by SDS–PAGE and immunodecoration with antibodies against the indicated proteins. Tom20, an integral MOM protein; Hep1, a soluble matrix protein. (D) Tgl2 is an IMS protein. Mitochondria as in C were treated with proteinase K (PK) under different conditions. Mitochondria were kept intact, the MOM was ruptured by hypoosmolar swelling (SW), or mitochondria were lysed by the addition of the detergent Triton X-100 (TX). Samples were precipitated with TCA and analyzed by SDS–PAGE and immunodecoration with antibodies against the HA-tag or the indicated mitochondrial proteins. Tom20, a MOM protein exposed to the cytosol; Dld1, a MIM protein exposed to the IMS.
In silico analysis of the primary sequence of Tgl2 did not identify any transmembrane domain. Accordingly, we could confirm by carbonate extraction of mitochondria isolated from cells expressing HA-Tgl2 that the protein behaved as a soluble protein (Figure 2C). To analyze in which mitochondrial compartment Tgl2 resides, we incubated the HA-Tgl2–containing organelles with proteinase K (PK) under different conditions. Tgl2 was protected from the externally added protease as long as mitochondria were intact (Figure 2D, lane 2). Upon either rupture of the outer membrane (Figure 2D, lane 3) or detergent-mediated solubilization of mitochondria (Figure 2D, lane 4), Tgl2 became protease-accessible and was degraded by PK. Taken together, these results show that Tgl2 is a soluble protein of the IMS, where the putative kinase domain of Mcp2 is also localized.
The import of Tgl2 into the IMS relies on the Mia40 pathway
Next, we asked how Tgl2 reaches its destination in the IMS. To this end, we used an in vitro import assay. In this assay, radiolabeled Tgl2 was incubated with isolated mitochondria, followed by the addition of PK to degrade any nonimported protein. As shown in Figure 3A, Tgl2 was imported in a time-dependent manner and became partially protected from protease degradation, again supporting our finding that Tgl2 is a mitochondrial protein.
FIGURE 3:
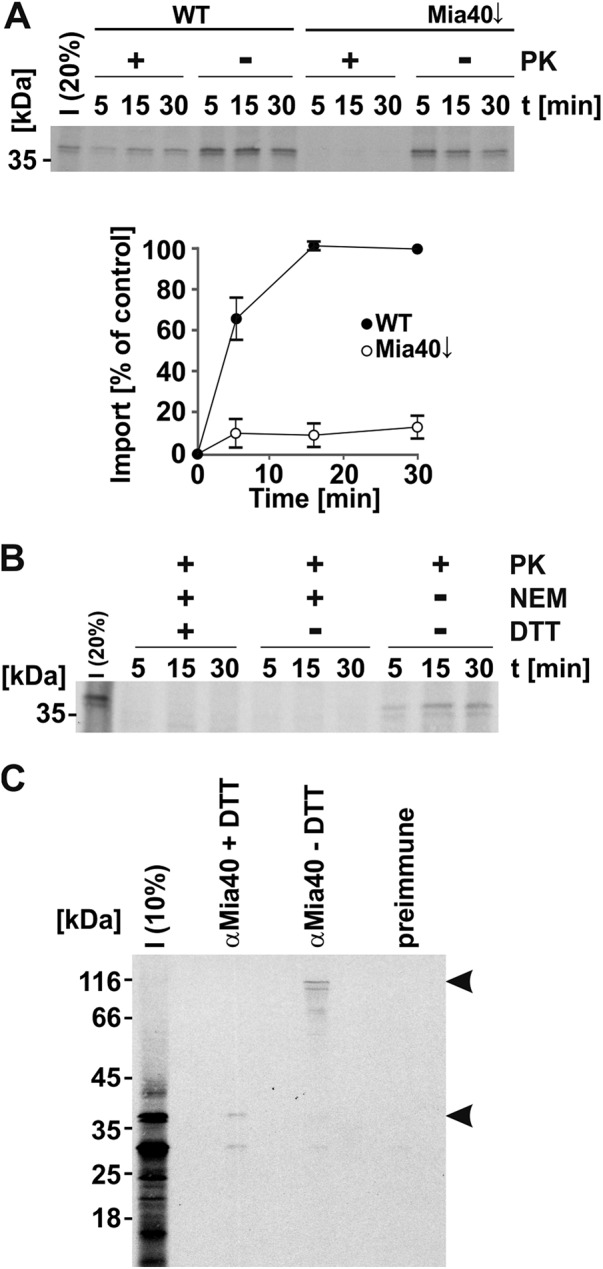
Tgl2 is imported into mitochondria via the Mia40 pathway. (A) Import of Tgl2 is dependent on Mia40. Mitochondria isolated from WT cells and cells depleted for Mia40 (Mia40↓) were incubated with radiolabeled Tgl2 for the indicated periods. After import, mitochondria were either treated with proteinase K (100 µg/ml) to digest nonimported Tgl2 (+PK) or left untreated (-PK). Next, mitochondria were reisolated and analyzed by SDS–PAGE and autoradiography. The autoradiography depicts a representative experiment; the quantification shows the mean of n = 3 independent experiments with SD bars. (B) Import of Tgl2 relies on redox-active sulfhydryl groups. Mitochondria isolated from WT cells were incubated with radiolabeled Tgl2 for the indicated periods. Tgl2 was preincubated with or without (+/−) NEM with or without (+/−) subsequent DTT treatment. All samples were treated with PK after import. (C) Tgl2 binds covalently to Mia40 as a mixed disulfide. After import of radioactively labeled Tgl2 for 10 min at 25°C, free thiols were blocked with NEM and mitochondria were lysed. Immunoprecipitation was performed with an antibody specific for Mia40 or preimmune serum. Bound proteins were eluted in either reducing (+DTT) or nonreducing (-DTT) buffer and analyzed by SDS–PAGE. The arrowheads depict Tgl2-containing protein bands. I, 10 or 20% of radiolabeled precursor protein used in each import reaction as indicated.
Several proteins of the IMS rely on the Mia40 pathway for import. This route is also known as the disulfide relay pathway, as the substrates of Mia40 usually contain several cysteine residues that can form intramolecular disulfide bonds in their mature form in the IMS. The formation of intramolecular disulfide bonds between cysteine residues is necessary for the stable accumulation of Mia40 substrates in the IMS (Peleh et al., 2016). Indeed, the sequence of Tgl2 contains eight different cysteine residues, none of these matching the Cys–X3–Cys or Cys–X9–Cys patterns that are typical for Mia40 IMS substrates (Longen et al., 2009). We observed that import into mitochondria with reduced levels of Mia40 was strongly reduced (Figure 3A). The compromised functionality of Mia40 was demonstrated by reduced import of the known Mia40 substrate Cmc1 (Supplemental Figure 1A). In contrast, the overall import capacity of these organelles was not impaired, as could be shown by the normal import of the Mia40-independent substrate Oxa1 (Supplemental Figure 1B). Furthermore, the covalent modification of the sulfhydryl groups of Tgl2 by the addition of the alkylating reagent N-ethyl-maleimide (NEM) inhibited its import into the IMS (Figure 3B).
To elucidate whether Tgl2 binds directly to Mia40, we imported radiolabeled Tgl2 into mitochondria for a short time, blocked free thiols by NEM to stabilize mixed disulfide intermediates, and isolated Mia40 and potential covalent intermediates by immunoprecipitation with an anti-Mia40 antibody. This procedure revealed a mixed Tgl2–Mia40 heterodimer that could be dissociated by reducing the disulfide bridges with DTT. Importantly, this adduct was not detected upon addition of the reducing agent DTT or by using preimmune serum (Figure 3C). Additionally, we analyzed a Tgl2 variant where all cysteine residues were replaced by serine (Tgl2(C-S); Supplemental Figure 1C). In contrast to the in vitro import of native Tgl2, the cysteineless variant gave rise to a number of smaller fragments, indicative of its proteolytic instability (Supplemental Figure 1D). Similar behavior was reported previously for the known Mia40 substrate Atp23, where Mia40 functions also as a chaperone mediating the folding in the IMS (Weckbecker et al., 2012). The appearance of proteolytic fragments of Tgl2(C-S) is not surprising, considering previous reports that misfolded proteins of the IMS are unstable and degraded by the i-AAA protease Yme1 (Baker et al., 2012; Schreiner et al., 2012). As expected, Tgl2 lacking cysteine residues did not form a mixed Tgl2–Mia40 dimer during in vitro import (Supplemental Figure 1E). The instability of the cysteineless variant was supported by the expression of this version in vivo, which did not result in a detectable protein at steady state levels (Supplemental Figure 1F) and which therefore could not rescue the growth defect of mcp2Δtgl2Δ cells (Supplemental Figure 1G).
In summary, we identified Tgl2 as a novel import substrate of Mia40 that differs from previously identified Mia40 substrates in the pattern of its cysteine residues.
Conserved residues of the kinase domain are crucial for the function of Mcp2
The IMS exposed protein Mcp2 has homologues in higher eukaryotes in the form of the five members of the aarF domain containing kinase (ADCK) family, ADCK1–ADCK5 (Lagier-Tourenne et al., 2008). ADCK1 shares the highest sequence similarity with Mcp2. To answer the question of whether the kinase domain is crucial for the function of Mcp2, we searched for residues that are conserved in the kinase domain of ADCK1 and Mcp2 (Lys210 and Asp223 of Mcp2) or among the members of the ADCK family and Mcp2 (Glu256 of Mcp2; Figure 4A). Next, we generated mutant Mcp2 versions, in which these three residues were individually replaced with alanine, and expressed them in a strain lacking the ERMES subunit Mdm10. Whereas expression of native Mcp2 partially rescued the growth defect, the mutated variants failed to do so (Figure 4B and Tan et al., 2013). Of note, native Mcp2 and its three variants were expressed to a similar extent (Figure 4C), excluding the possibility that differences in growth phenotype suppression resulted from variable protein levels. Taken together, mutations of conserved residues in the putative atypical kinase motifs of Mcp2 result in loss of function of Mcp2.
FIGURE 4:
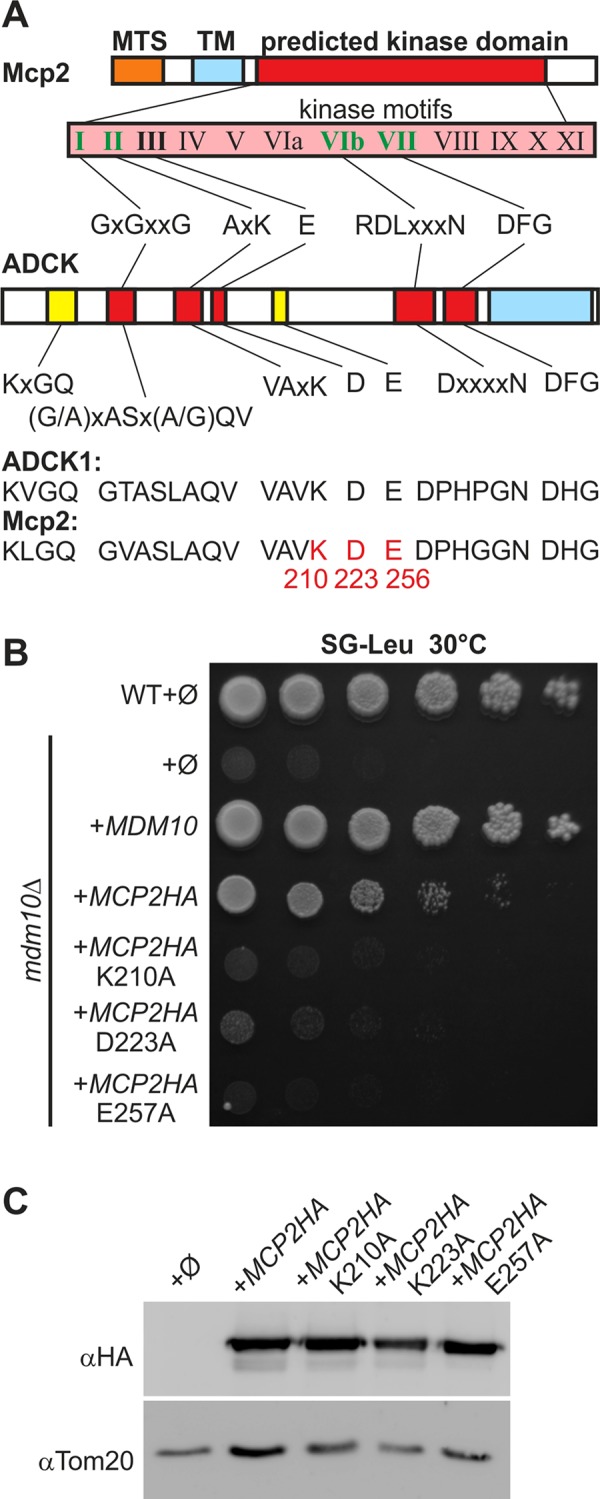
Conserved putative kinase residues of Mcp2 are important for function. (A) Mcp2 is a putative kinase with nucleotide-binding motifs and is homologous to the mammalian ADCK family members. Of the 12 hallmark motifs found in kinases (roman numerals), five can be identified in members of the ADCK family (partially adopted from Lagier-Tourenne et al., 2008) and Mcp2 (bold). All four nucleotide-binding motifs (green) are present in Mcp2. Yellow motifs are highly conserved among ADCK family members as well as Mcp2. MTS, mitochondrial targeting signal; TM, transmembrane domain; ADCK1, aarF domain containing kinase 1, the closest homologue to Mcp2. Highlighted in red are residues that were mutated in Mcp2 for further analysis. (B) Mutation of conserved kinase residues of Mcp2 leads to loss of function. WT or mdm10Δ cells were transformed with the empty plasmid pYX142 (∅). In addition, mdm10Δ cells were transformed with pYX142 encoding the indicated Mcp2 variants under control of the TPI promoter. Cells were grown to logarithmic phase and spotted on SG-Leu plates in a 1:5 dilution series. Plates were incubated for growth at 30°C. (C) Mutated Mcp2 variants are expressed to levels similar to those for native Mcp2. Cells described in B were grown to midlogarithmic phase and crude mitochondrial fractions were analyzed by SDS–PAGE and immunodecoration with antibodies against the HA-tag or Tom20 as loading control.
Lack of Psd1 renders yeast cells more sensitive to elevated levels of Mcp2
To investigate a potential role of Mcp2 in organellar lipid homeostasis, we focused on mitochondrial lipid biosynthesis pathways. In yeast, phosphatidylethanolamine (PE) is mainly synthesized in mitochondria from PS by the decarboxylase Psd1 and the majority of PE is then exported to the rest of the cell. To analyze whether Mcp2 plays a role in this process, we investigated its genetic interaction with PSD1. To that goal, we deleted PSD1 and overexpressed Mcp2, ERMES subunits, or Mcp1. Surprisingly, we found that overexpression of Mcp2 led to a growth defect in yeast cells lacking Psd1 (Figure 5A). This effect was specific, as it was not observed upon overexpression of the other genes tested. As a further control, overexpression of Mcp2 in combination with deletion of GEP4, which plays a central role in the biosynthesis pathway of CL, did not result in similar growth retardation (Figure 5B). Finally, to rule out the possibility that the growth defect results simply from an overload of proteins in the MIM, we also overexpressed Mdm31, Mdm32, or Mdm38 in psd1Δ cells, which were shown previously to be inner membrane proteins (Dimmer et al., 2005; Frazier et al., 2006). Overexpression of none of these proteins resulted in a growth defect of psd1Δ cells (Figure 5C). Additionally, we tested the overexpression of the third Mdm10-complementing protein, Mcp3, which was recently identified as an outer membrane component (Sinzel et al., 2016, 2018). Again, we did not observe an effect on the growth of cells lacking Psd1 (Figure 5C).
FIGURE 5:

Expression of Mcp2 is a burden for cells lacking Psd1. (A, B) Overexpression of Mcp2 is toxic in cells lacking Psd1. In A, WT or psd1Δ cells were transformed with the empty plasmid pYX142 (∅). In addition, psd1Δ cells were transformed with pYX142 encoding ERMES subunits, MCP1, or MCP2 under control of the TPI promoter. Cells were grown to logarithmic phase and spotted on SD-Leu plates in a 1:5 dilution series. Plates were incubated for growth at 30°C. In B as in A, for cells lacking Gep4. (C) WT or psd1Δ cells were transformed with the empty plasmid pYX142 (∅). In addition, psd1Δ cells were transformed with pYX142 encoding the indicated proteins under control of the TPI promoter. Cells were grown to logarithmic phase and spotted on either SD-Leu or YPG plates in a 1:5 dilution series. Plates were incubated for growth at 30°C. (D) Simultaneous deletion of MCP2 and PSD1 suppresses partially the growth defect of psd1Δ cells. WT, mcp2Δ, psd1Δ, or mcp2Δpsd1Δ cells were grown to logarithmic phase and spotted on full media containing fermentable (YPD) or nonfermentable (YPG) carbon sources in a 1:5 dilution series and analyzed for growth at either 30 or 37°C.
Of note, we did not identify PSD1 as a genetic interactor with MCP2 in our initial synthetic genetic interaction screen in the BY4741 background. To study the genetic interactions between the two genes in another context, we created a double-deletion mutant in the W303 background by tetrad dissection. It was reported previously that deletion of PSD1 leads to severe growth defects on synthetic media lacking ethanolamine, on media harboring nonfermentable carbon sources, or at elevated temperatures (Trotter et al., 1993). Interestingly, the defect at elevated temperatures could be partially restored by simultaneously deleting MCP2 (Figure 5D).
Collectively, our results suggest that elevated Mcp2 activity is a burden for yeast cells that lack Psd1-mediated PE synthesis, and accordingly, loss of Mcp2 facilitates growth of these psd1Δ cells.
MCP2 interacts genetically with genes encoding the Ups1/Ups2/Mdm35 system
Because Tgl2 and Mcp2 face the IMS, we asked whether they are functionally related to the intramitochondrial lipid transport system Ups1/Ups2/Mdm35. To address this issue, we analyzed genetic interactions of the respective genes in the W303a background. To this end, we created double-deletion strains by tetrad dissection of every possible combination of these genes and studied their growth in comparison with that of the respective single-deletion cells at either optimal or elevated temperature on rich or synthetic medium supplemented with either a fermentable or a nonfermentable carbon source.
Whereas the deletion of TGL2, together with the absence of UPS1, UPS2, or MDM35, did not result in any change of growth in comparison with that of the single-deletion cells (Supplemental Figure 2), the deletion of MCP2 seemed to be beneficial for cells lacking UPS1, UPS2, or MDM35 (Figure 6). Of note, the growth defects of ups2Δ and mdm35Δ cells are minor and only observable on synthetic medium at higher temperatures, whereas the growth defect of ups1Δ cells is more pronounced. Yet at elevated temperature on nonfermentable carbon sources, all the double-deletion cells mcp2Δups1Δ, mcp2Δups2Δ, and mcp2Δmdm35Δ grew better than the corresponding single-deletion cells ups1Δ, ups2Δ, or mdm35Δ. Because the observed growth defects are quite subtle, we confirmed the results with the same set of strains in the W303α background (Supplemental Figure 3).
FIGURE 6:

Growth analysis of double-deletion mutants lacking MCP2 and either UPS1, UPS2, or MDM35. Cells of the indicated genotype in the W303a WT background were grown to logarithmic phase on full medium containing the fermentable carbon source glucose (YPD) to logarithmic phase and spotted on full (YP) or synthetic (S) media containing either glucose (D) or the nonfermentable carbon source glycerol (G) in a 1:5 dilution series and analyzed for growth at either 30 or 37°C.
Taken together, loss of Mcp2 function is beneficial when the transport of PA or PS across the IMS is hampered.
Overexpression of Mcp2 is toxic for yeast cells
We showed before that moderate overexpression of Mcp2 in wild-type cells led to a slight distortion of mitochondrial morphology (Tan et al., 2013). To analyze the outcome of stronger overexpression of Mcp2, we placed Mcp2 or its HA-tagged variant under the control of the inducible galactose promoter. Strikingly, induction of the GAL1 promoter led to a strong growth defect on media containing galactose, whereas moderate overexpression under the control of the TPI promoter did not result in such growth retardation (Figure 7, A and B).
FIGURE 7:

Growth analysis of yeast cells devoid of or with strong overexpression of Mcp2. (A) Strong overexpression of Mcp2 is toxic for WT yeast cells. WT cells were transformed with the empty plasmids pYX113 and pYX142 (∅): the empty plasmid pYX113 together with the pYX142-Mcp2 construct (TPIprMCP2 or TPIprMCP2-HA) under control of the TPI promoter or the empty plasmid pYX142 together with the pYX113-Mcp2 construct (GAL1prMCP2 or GAL1prMCP2-HA) under control of the GAL1 promoter. Cells were grown to logarithmic phase and spotted on SD-Ura-Leu or SGal-Ura-Leu plates in a 1:5 dilution series. Plates were incubated for growth at 30°C. (B) Expression levels of HA-tagged Mcp2 under control of the TPI and GAL1 promoter. Cells as in A were grown to midlogarithmic phase and crude mitochondrial fractions were analyzed by SDS–PAGE and immunodecoration with antibodies against the HA-tag or Tom40, a MOM protein, as loading control. (C) The toxic effect of Mcp2 overexpression depends on the conserved kinase residues of the protein. WT cells were transformed with the empty plasmid pYX113 (∅) or with pYX113 encoding the indicated variants under control of the GAL1 promoter. Cells were grown to logarithmic phase and spotted on SD-Ura and SGal-Ura plates in a 1:5 dilution series. Plates were incubated for growth at 30°C. (D) Increased sensitivity to oleate of yeast cells lacking Mcp2 at high temperatures. WT cells of different mating type (W303a and α), as well as mcp2Δ cells with different genetic background, were grown to logarithmic phase and spotted on synthetic medium containing glucose (SD) as well as on oleate-containing medium containing 0.1% glucose (YNBGO) in a 1:5 dilution series. Plates were incubated for growth at 37°C.
We then asked whether the mere presence of high levels of Mcp2 molecules is the reason for the observed growth defect or whether functionality of Mcp2 is also required. To this end, we used the aforementioned mutated variants, which lack the capacity to rescue loss of ERMES function. Overexpression of these mutants led to reduced toxicity in comparison with overexpression of native Mcp2, suggesting that functionality contributes to toxicity (Figure 7C). Next, we wondered whether the massive overexpression of Mcp2 results in an alteration of the lipid composition of yeast cells. To this end, we analyzed the lipidome of yeast cells grown on galactose-containing medium to induce strong Mcp2 expression in comparison with the corresponding wild-type cells grown on the same medium. We could not find any statistically significant changes in the lipid composition of the cells with highly overexpressed Mcp2 (Supplemental Figure 4).
These findings suggest that high levels of functional Mcp2 result in an impairment of the growth of yeast cells.
Loss of Mcp2 leads to increased oleate sensitivity at elevated temperatures
Because MCP2 shows a negative synthetic genetic interaction with the lipase Tgl2, we tested the growth of mcp2Δ cells on a medium that contains the fatty acid (FA) oleate as the sole or an additional carbon source. We found that at elevated temperatures (37°C) on media containing oleate, cells lacking Mcp2 grew more slowly than control cells (Figure 7D). Because this growth impairment was rather subtle, we confirmed this behavior with yeast strains that were of different mating type and contained different cassettes for gene replacement of MCP2 (Figure 7D). These observations demonstrate that loss of the potential IMS kinase Mcp2 renders yeast cells more sensitive to high concentrations of oleate in the medium and suggests an involvement of Mcp2 in overall yeast lipid metabolism.
DISCUSSION
The processes by which mitochondria participate in lipid metabolism and trafficking are still ill-defined. In recent years, membrane contact sites have entered the stage as sites of lipid transfer. It seems that lipid-exchange proteins at regions where close proximity of membranes is mediated by tethering factors are playing a central role in these events.
In yeast cells, the most prominent platform for lipid exchange to and from mitochondria seems to be the ERMES complex. Subunits of this protein complex have been shown to bind phospholipids, especially PS and PE (AhYoung et al., 2015; Kawano et al., 2018). The whole problem of studying lipid trafficking of mitochondria is best illustrated by the fact that ERMES loss-of-function mutants adapt very quickly and partially lose their growth impairment (Sogo and Yaffe, 1994; Berger et al., 1997). The rescue of ERMES mutants can be the consequence of expression of high-copy suppressors (Tamura et al., 2012a; Tan et al., 2013; Kojima et al., 2016; Sinzel et al., 2016) or gain-of-function mutations in the protein Vps13 (Lang et al., 2015b). In addition, extended contacts of mitochondria with the vacuole can compensate for loss of ERMES function (Elbaz-Alon et al., 2014; Honscher et al., 2014). Indeed, yeast cells seem to harbor, in addition to their vesicular system, an extensive contact-site network providing a high degree of redundancy in lipid transport. Moreover, several lipids can be synthesized by different enzymes in various locations (e.g., PE can be synthesized by Psd1 in mitochondria and ER, and Psd2 in Golgi and vacuoles) or even by different metabolic pathways (PE by either decarboxylation of PS or the Kennedy pathway).
In the present work, we show that the lipase Tgl2 is localized to the IMS of mitochondria and that its import into this location depends on the Mia40 disulfide relay pathway. Most Mia40 substrates are small (8–12 kDa) helix–loop–helix proteins with two disulfide bonds connecting the helices (Banci et al., 2010). Tgl2 is certainly different from these proteins by being much larger in size (37.5 kDa) and, owing to its lipase domain, having a much more complex structure. It contains eight cysteine residues and in this property resembles Atp23, which also contains eight cysteine residues. These cysteine residues are essential for folding of Tgl2 into a stable protein; however, they are dispensable for its Mia40-dependent import, similar to what was previously reported for the Mia40 substrate Atp23 (Weckbecker et al., 2012).
Tgl2 is the only mitochondrial Tgl protein, as its homologues Tgl1, Tgl3, Tgl4, and Tgl5 function in lipid droplets (Grillitsch and Daum, 2011). Also, in its primary structure, Tgl2 considerably differs from the other Tgl proteins, pointing to a distinct role in lipid metabolism (Grillitsch and Daum, 2011). It was demonstrated that Tgl2 can hydrolyze TAGs and DAGs in vitro (Ham et al., 2010). Furthermore, it was shown that Escherichia coli cells expressing Tgl2 and disrupted for their DAG kinase DgkA are still growing under conditions where DAG accumulation becomes toxic (Van Heusden et al., 1998). This suggests that Tgl2 can also cleave DAGs within E. coli cells. Currently, the physiological substrates of Tgl2 in mitochondria are unknown.
Because, in S. cerevisiae, β-oxidation of FAs takes place solely in peroxisomes (Klug and Daum, 2014), one can speculate that the release of FAs for ATP production is not the purpose of Tgl2 function. A possible substrate in mitochondria therefore would be DAG, which derives from either phosphoglycerolipids or TAGs, which were never reported previously to play a role in mitochondria. Assuming that DAG is the physiological substrate of Tgl2 and a putative DAG-kinase activity of Mcp2 (as discussed below), the negative synthetic genetic interaction with MCP2 can be explained by the hypothesis that absence of both Tgl2 and Mcp2 leads to toxic elevated levels of DAG in mitochondria.
Although not all sequence motifs of a typical protein kinase are conserved in Mcp2 (Hanks et al., 1988; Lagier-Tourenne et al., 2008; summarized in Figure 4A), the motifs necessary for nucleotide binding (Leonard et al., 1998) are all present in Mcp2. We demonstrated that mutation of two conserved residues, which are predicted to be involved in nucleotide binding, are indeed necessary for the full rescue capacity for loss of ERMES. Moreover, the same mutations reduce toxicity of overexpression of Mcp2 in yeast cells.
These observations are the first evidence that nucleotide binding is important for Mcp2 function. It was suggested that the homologue ADCK family of kinases belongs to the so-called “atypical kinases” (Scheeff and Bourne, 2005; Lagier-Tourenne et al., 2008), which also comprises nonprotein kinases such as phosphoinositide and choline kinases. Therefore, it is tempting to assume that the substrate of Mcp2 might be a lipid metabolic intermediate or a lipid itself. Along this line, the negative genetic interaction of MCP2 with TGL2 might be explained by Mcp2 phosphorylating normally DAG to the CL precursor phosphatidic acid (PA). Of note, in mammalian cells acyl glycerol kinase (AGK/MuLK), a eukaryotic multisubstrate lipid kinase that can phosphorylate DAG (Waggoner et al., 2004), was recently reported to be present in the IMS as a subunit of the TIM22 translocase (Hung et al., 2014; Vukotic et al., 2017). According to this scenario, the absence of Mcp2 contributes further to the accumulation of DAG. In line with this hypothesis, an increase of mitochondrial DAG could result in an increase of whole cell DAG favoring TAG production for storage. This could explain the increased levels of lipid droplets (and presumably TAG) in the double-deletion mutant mcp2Δtgl2Δ (Figure 1, E and F).
Our lipidomic analysis of cells lacking both Mcp2 and Tgl2 showed neither a statistically significant increase of DAG levels nor a reduction of PA amounts. Yet the level of CL, which is synthesized from PA exclusively in mitochondria, is reduced (Figure 1, G and H). However, we cannot exclude the possibility that CL-reduction is the result of overall lowered mitochondrial mass in these cells. Another potential support for the hypothesis of elevated DAG levels in the double-deletion strain is the observed cellular TAG levels. Such an increase might be a consequence of enhanced synthesis from DAG and activated FAs. Another potential reason for the absence of detectable change is the fact that we analyzed lipids in whole-cell extract, while the relative changes in the mitochondrial levels of DAGs and PAs might not reflect on whole-cell lipid composition. Therefore, a goal for future studies is to analyze the lipid composition of mitochondria isolated from the double-deletion strain.
The genetic interaction of Mcp2 and Tgl2 led us to study a possible physical interaction between these proteins, which both function in the same compartment. We tried different pulldown and coimmunoprecipitation approaches with tagged proteins and different detergents for lysis of mitochondria but did not observe a stable interaction (unpublished data). These results suggest either that these proteins do not physically interact or that their association is very transient and might depend on the presence of a common substrate.
The genetic interaction between PSD1 and MCP2 further supports the involvement of the latter in lipid metabolism. The function of Mcp2 in the absence of Psd1 leads to a problematic outcome for yeast cells. Lack of Psd1 leads to reduced levels of PE, which like CL is a non-bilayer-forming phosphoglycerolipid (van den Brink-van der Laan et al., 2004; van Meer et al., 2008). PE and CL can compensate for each other, yet combined disruption of the Psd1 pathway and CL biosynthesis is lethal for yeast cells. PA, which is also a non-bilayer-forming lipid, is present in membranes only in trace amounts (van Meer et al., 2008). Thus, it is tempting to speculate that loss of Mcp2 could influence PA levels, especially in mitochondrial membranes, and therefore ameliorate the effects of Psd1 loss namely, lack of PE.
Mcp2 closest homologue among the ADCK family is ADCK1 (Tan et al., 2013). The role of most ADCKs is unknown. Only for ADCK3 is it known that it plays a role in coenzyme Q biosynthesis (Lagier-Tourenne et al., 2008). ADCK3, in turn, has a homologue in yeast called Coq8 (Xie et al., 2011). We did not find any functional connection of Mcp2 and Coq8 in different growth assays, such as expression of Coq8 in ERMES mutants or expression of Mcp2 in a coq8Δ strain (unpublished data). For quite some time, it was suggested that Coq8 was a protein kinase that regulates coenzyme Q biosynthesis in yeast by phosphorylation of the biosynthetic enzymes in the inner membrane (Xie et al., 2011). However, recent observations suggest another role for Coq8. In this model, Coq8, after import to the mitochondrial matrix, is recruited to the MIM by binding to CL. In its activated membrane-bound state, Coq8 can extract hydrophobic phenolic coenzyme Q intermediates from the MIM upon ATP hydrolysis and/or couple ATP hydrolysis to complex Q (consisting of the different enzymatic subunits for coenzyme Q biosynthesis) formation at the MIM (Reidenbach et al., 2018). Because Mcp2 also belongs to the same family of “atypical kinases,” a similar mode of function by extraction of lipids or lipid intermediates from mitochondrial membranes can be envisaged. In this context, the positive genetic interactions of lack of MCP2 with the absence of UPS1, UPS2, or MDM35 become notable, since it is tempting to assume that possible reduced Mcp2-mediated extraction of lipids from membranes that are altered due to the absence of Ups1, Ups2, or Mdm35 has a stabilizing effect. Currently, this hypothesis is highly speculative and awaits experimental data to support it. Regardless of the actual molecular mechanism, these genetic interactions fit nicely into a scenario where Mcp2 contributes to lipid homeostasis of the two mitochondrial membranes by a so far unknown mechanism.
Finally, the increased sensitivity of mcp2Δ cells at high temperatures to high concentrations of external oleate also suggests an overall role of Mcp2 in yeast lipid homeostasis. A possible scenario could be that in the absence of Mcp2, salvage mechanisms from lipotoxic effects of free FAs are disturbed. Facing high FA concentrations, yeast cells store excess FA by increasing the TAG levels, which in turn leads to increased size and number of lipid droplets. This process could be disturbed by an overall imbalance of lipid metabolism, especially under extreme conditions such as elevated temperature, where even backup systems cannot cope with alterations in metabolite levels.
Taking these results together, the current study suggests that two IMS enzymes, a lipase (Tgl2) and an atypical kinase (Mcp2), are involved in yeast lipid metabolism. The next crucial step will be to identify the physiological substrates of these enzymes.
MATERIALS AND METHODS
Yeast strains and growth conditions
Yeast strains (all isogenic to W303a/α) were grown in and on standard rich or synthetic liquid and solid media, respectively (Green and Moehle, 2001). For drop dilution assays, cells were cultured to an OD600 of 1.0 and diluted in fivefold increments, followed by spotting 5 µl of each dilution on the corresponding medium. To delete complete ORFs by homologous recombination, gene-specific primers were used to amplify either the HIS3MX6 cassette from the plasmid pFA6a-HIS3MX6 (Wach et al., 1997) or the KanMX4 cassette from the plasmid pFA6a-KanMX4 (Wach et al., 1994). Double-deletion strains were retrieved by mating and tetrad dissection of the corresponding single-deletion strains. All deletion strains were confirmed by PCR with gene-specific primers. Transformation of yeast cells was performed by the lithium acetate method. Yeast strains used in this study are summarized in Table 1; primer sequences are listed in Table 2.
TABLE 1:
S. cerevisiae strains used in this study.
| Name | Genotype | Reference |
|---|---|---|
| W303a | MAT a; ade2-1; can1-100; his3-11; leu2-3,112; trp1∆2; ura3-52 | Thomas and Rothstein, 1989 |
| W303α | MAT α; ade2-1; can1-100; his3-11; leu2-3,112; trp1∆2; ura3-52 | Thomas and Rothstein, 1989 |
| YKD432 | W303a; mcp2∆::HIS3MX6 | Tan et al., 2013 |
| YKD898 | W303a; tgl2∆:: KanMX4 | This study |
| YKD1009 | W303a; mcp2∆:: HIS3MX6; tgl2∆::KanMX4 | This study |
| YKD291 | W303a; mdm10∆::HIS3MX6 | Tan et al., 2013 |
| YKD472 | W303a; psd1∆:: KanMX4 | This study |
| YKD492 | W303a; gep4∆:: KanMX4 | This study |
| YKD702 | W303a; mcp2∆:: HIS3MX6; psd1∆::KanMX4 | This study |
| YKD433 | W303α; mcp2∆::HIS3MX6 | Tan et al., 2013 |
| YKD450 | W303a; mcp2∆::KanMX4 | Tan et al., 2013 |
| YKD451 | W303α; mcp2∆::KanMX4 | Tan et al., 2013 |
| YMS721 | MATα, his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 can1Δ::STE2pr-spHIS5 lyp1Δ::STE3pr-LEU2 | Breslow et al., 2008 |
| YKD876 | YMS721; mcp2∆::NatMX4 | This study |
| CW143 | W303a; ups1Δ::NatMX4 | Potting et al., 2010 |
| CW130 | W303a; ups2Δ::NatMX4 | Potting et al., 2010 |
| YKD805 | W303a; mdm35Δ::HIS3MX6 | This study |
| YKD1043 | W303α; ups1Δ::NatMX4 | This study |
| YKD1045 | W303α; ups2Δ::NatMX4 | This study |
| YKD1046 | W303α; mdm35Δ::HIS3MX6 | This study |
| YKD1051 | W303a; mcp2∆:: HIS3MX6; ups1∆::NatMX4 | This study |
| YKD1052 | W303α; mcp2∆:: HIS3MX6; ups1∆::NatMX4 | This study |
| YKD1053 | W303a; mcp2∆:: HIS3MX6; ups2∆::NatMX4 | This study |
| YKD1054 | W303α; mcp2∆:: HIS3MX6; ups2∆::NatMX4 | This study |
| YKD1047 | W303a; mcp2∆:: KanMX4; mdm35∆::HIS3MX6 | This study |
| YKD1048 | W303α; mcp2∆:: KanMX4; mdm35∆::HIS3MX6 | This study |
| YKD1055 | W303a; tgl2∆:: HIS3MX6; ups1∆::NatMX4 | This study |
| YKD1056 | W303α; tgl2∆:: HIS3MX6; ups1∆::NatMX4 | This study |
| YKD1057 | W303a; tgl2∆:: HIS3MX6; ups2∆::NatMX4 | This study |
| YKD1058 | W303α; tgl2∆:: HIS3MX6; ups2∆::NatMX4 | This study |
| YKD1049 | W303a; tgl2∆:: KanMX4; mdm35∆::HIS3MX6 | This study |
| YKD1050 | W303α; tgl2∆:: KanMX4; mdm35∆::HIS3MX6 | This study |
TABLE 2:
Primers used in this study.
| Name | Sequence | Remarks |
|---|---|---|
| Primers for gene-targeting | ||
| KSD082 | 5′ TTC TTG GTC GTT ATT TTT TGA AGA AGA AGG AAA AGCAAA GCC AGC CGT ACG CTG CAG GTC GAC | Deletion of PSD1 ORF |
| KSD083 | 5′ TAC TAT ATA CAG CAA AAT AAA TGC TAA CTT TAC ATA TGA TTG CTT ATC GAT GAA TTC GAG CTC | Deletion of PSD1 ORF |
| KSD080 | 5′ AAC TGA AAG GCG GCA GTT ACA TTA CAT CGT CTC CTC TAC CTA GTC CGT ACG CTG CAG GTC GAC | Deletion of GEP4 ORF |
| KSD081 | 5′ TAT ATA AAA AAT TAA AAT GTT TTA CTT TTT ATT AAA GTT GCC TAA ATC GAT GAA TTC GAG CTC | Deletion of GEP4 ORF |
| KSD_Tgl2fwd | 5′ AGA GAA TAA AGC GTC TTG TTT TAA AGG AGG AAA AGC ATA AAA AAA CGT ACG CTG CAG GTC GAC | Deletion of TGL2 ORF |
| KSD_Tgl2rev | 5′ AAA AAA AGA GTA TTC TAT AAA CAG TTC TTA CGA ATA GAA CAA CTC ATC GAT GAA TTC GAG CTC | Deletion of TGL2 ORF |
| Primers for cloning | ||
| KSD_Tgl2s | 5′ GGG GAA TTC ATG AAA AAT GAT AAT AAA GCT AAT G | TGL2 ORF in pYX plasmids |
| KSD_Tgl2a1 | 5′ GGG AAG CTT TTA AAA TCC TTT TCT TGC CAA GT | TGL2 ORF in pYX plasmids |
| KSD_Tgl2a2 | 5′ GGG AAG CTT AAA TCC TTT TCT TGC CAA GTC AT | TGL2 ORF without Stop codon in pYX plasmids (C-terminal HA-tag) |
| KSD_Tgl2sHA | 5′ GGG GAA TTC ATG TAT CCG TAT GAT GTG CCT GAC TAC GCA ATG AAA AAT GAT AAT AAA GCT AAT G | TGL2 ORF with N-terminal HA-tag |
| K210A-1 | 5′ CTA GTG TAG CTG TCG CAT GTC AGC ATC CAT G | K210A site-directed mutagenesis |
| K210A-2 | 5′ GAT GGA TGC TGA VAT GCG ACA GCT ACA CTA G | K210A site-directed mutagenesis |
| D223A-1 | 5′ GTT TAT ACC ATT AGC TGT TAT GCT GAC AAG | D223A site-directed mutagenesis |
| D223A-2 | 5′ CTT GTC AGC ATA ACA GCT AAT GGT ATA AAC | D223A site-directed mutagenesis |
| E257A-1 | 5′ CTT CAA TCT ACG TGG CAC TGA ATT TTA CCA A | E257A site-directed mutagenesis |
| E257A-2 | 5′ TTG GTA AAA TTC AGT GCC ACG TAG ATT GAA G | E257A site-directed mutagenesis |
SGA screen for genetic interactions of MCP2
To identify genetic interactions of MCP2, an SGA technique–based growth phenotype screen was performed as described before (Cohen and Schuldiner, 2011). In brief, the complete open-reading frame of MCP2 was replaced by a NatMX4 module in the BY4741 wild-type background by homologous recombination. This query strain was crossed using a RoToR benchtop colony arrayer (Singer Instruments, United Kingdom) to manipulate libraries in high-density formats (1536 strains per plate) with the yeast knockout library of nonessential genes (Giaever et al., 2002) or the yeast DAmP library for essential genes (Breslow et al., 2008), both harboring KanMX4 modules. To this end, haploid strains from opposing mating types, each harboring a different genomic alteration, were mated on complete medium. Diploid cells were selected on plates containing CloNat and G418. Sporulation was then induced by transferring cells to nitrogen-starvation plates. Haploid cells containing all desired mutations were selected for by transferring cells to plates containing all selection markers alongside the toxic amino acid derivatives canavanine and thialysine to select against remaining diploids. The new yeast libraries, in which each colony harbored the mcp2Δ::NatMX4 deletion on the genetic background of a single mutation, were spotted on YPD medium. Growth of the single strains was quantified using the Balony free software for the analysis of images of plates containing arrays of yeast (the software package is maintained by Barry Young at the University of British Columbia, Vancouver, Canada). The SGA was performed twice with the query strain and twice with a reference BY4741 wild-type strain harboring the NatMX4 module in the URA3 locus, resulting in single-deletion strains for size comparison.
Recombinant DNA techniques
The ORF of the TGL2 gene was amplified by PCR from yeast genomic DNA with specific primers, either with the stop codon or without introducing a C-terminal HA-tag, and cloned into the plasmid pYX142 with the restriction enzymes EcoRI and HindIII. To obtain the N-terminal HA-tag, a special primer encoding the HA encoding sequence was used. For in vitro transcription and translation, pGEM4-Tgl2 was constructed by subcloning the TGL2 ORF from pYX142-Tgl2 with the restriction enzymes EcoRI and HindIII. The synthetic gene for the ORF of Tgl2 where all Cys residues are replaced by Ser in the plasmid pEX-A128 was purchased from Eurofins Genomics, Ebersberg. The ORF was flanked by EcoRI and HindIII restriction sites at the 5′ and 3′ end, respectively, used for subcloning in pGEM4. To obtain the N-terminal HA-tag, the same primer set described above was used for PCR cloning into pYX142. Site-directed mutagenesis by PCR was performed to obtain the K210A, D223A, and E256A amino acid exchange mutants of Mcp2. Primer sequences are listed in Table 2. Plasmids for expression of Mcp1, Mcp2, Mcp2-HA, Mcp3, Mdm10, Mdm12, Mdm34, and Mmm1 were described previously (Tan et al., 2013; Sinzel et al., 2016). All expression plasmids are of the pYX series. Most experiments were performed with pYX142 constructs harboring the TPI promoter for overexpression. Mcp2 variants in the plasmid pYX113 were constructed by subcloning of the Mcp2 insert from pYX142 constructs. The pYX113 constructs are under the control of the GAL1 promoter. All constructs were confirmed by DNA sequencing.
Biochemical methods
Subcellular fractionation was performed according to published procedures (Walther et al., 2009). Mitochondria were isolated by differential centrifugation as described previously (Daum et al., 1982). For hypoosmolar rupture of the outer membrane, mitochondria were incubated for 30 min in hypotonic buffer (20 mM HEPES, pH 7.2) on ice. Soluble proteins were precipitated by trichloroacetic acid (TCA). Protein samples were analyzed by SDS–PAGE and immunoblotting using the ECL system. Carbonate extraction to precipitate integral membrane protein was performed as described previously (Tan et al., 2013). Proteinase K accessibility of proteins was analyzed by incubation of (pretreated) mitochondria with PK (20–100 μg/ml) for 30 min on ice, inhibiting the protease by 2 mM PMSF, and finally TCA precipitation of the samples. For preparation of mitochondria by mechanical rupture of cells with glass beads, cells were grown in minimal medium containing glucose to mid-logarithmic phase at 30°C. Cells were disrupted by repeated cycles of vortexing with glass beads at 4°C. Mitochondria were enriched by differential centrifugation.
In vitro import assays
Mitochondria for import assays were isolated from yeast cells as described above. The yeast strain with down-regulated levels of Mia40 and its corresponding wild type have been described previously (Mesecke et al., 2005). Cells were grown at 30°C in lactate medium containing 0.1% glucose in order to repress the GAL-10 promoter. Radiolabeled precursor proteins were synthesized in the presence of [35S]-methionine in rabbit reticulocyte lysate. The import reaction was performed by incubating the radiolabeled protein with isolated mitochondria in import buffer (1 M sorbitol, 160 mM KCl, 100 mM HEPES, 20 mM Mg(Ac)2, 4 mM KH2PO4, 2 mM ATP, and 2 mM NADH, pH 7.2). In some cases, the import reactions were treated with PK as described above. To block free thiols, radiolabeled Tgl2 was preincubated with 10 mM NEM (10 min at 25°C) with or without later addition of 50 mM DTT (2 min at 25°C). The immunoprecipitation was performed as described previously (Peleh et al., 2016). In short, after in vitro import of Tgl2 free thiols was blocked with NEM, mitochondria were reisolated and lysed in Triton X-100–containing buffer. Cleared lysate was immunoprecipitated with Mia40 antibody coupled to Sepharose-A. Bound proteins were analyzed by SDS–PAGE and autoradiography.
Fluorescence microscopy
For visualization of mitochondria, yeast cells were transformed with a vector harboring the mitochondrial presequence of subunit 9 of Fo-ATPase of Neurospora crassa fused to GFP (Westermann and Neupert, 2000). For visualization of lipid droplets, yeast cells were grown to midlogarithmic phase and incubated with the fluorescent dye BODIPY 493/503 (1 µg/ml final concentration) for 15 min. Cells were washed once with PBS and directly embedded in low–melting point agarose for analysis by microscopy. Microscopy images were acquired with an Axioskop20 fluorescence microscope equipped with an Axiocam MRm camera using the 43 Cy3 filter set and the AxioVision software (Carl Zeiss).
Neutral lipid quantification by BODIPY staining
Cells were grown to midlogarithmic phase. Cells corresponding to 1 ml of a culture with OD600 = 0.5 were harvested by centrifugation washed once with PBS, resuspended in PBS, and incubated for 30 min with 1 µg/ml BODIPY 493/503. Cells were transferred on ice and harvested by centrifugation at 4°C. After being washed with cold PBS, cells were resuspended in 1 ml PBS and analyzed by a fluorescence plate reader (Tecan Trading AG, Switzerland).
Mass spectrometric lipid analysis
Quantitative analysis of lipids was performed by standard nanoelectrospray ionization mass spectrometry (Ozbalci et al., 2013; Tatsuta, 2017). Lipids were extracted from 0.5 OD units of yeast cells in the presence of internal standards for the major phospholipid classes and neutral lipids (PC 17:0-14:1, PE 17:0-14:1, PI 17:0-14:1, PS 17:0-14:1, PG 17:0-14:1, 15:0-18:1-d7-PA; Cardiolipin mix I; d5-TG ISTD Mix I; d5-DG ISTD Mix I; d5-DG ISTD Mix II, all from Avanti Polar Lipids). Extraction was performed according to Bligh and Dyer with some optimization for whole yeast cells (Miliara et al., 2015). Lipids were dissolved in 10 mM ammonium acetate in methanol and analyzed in a QTRAP 6500 triple quadrupole mass spectrometer (SCIEX) equipped with a nanoinfusion spray device (TriVersa NanoMate, Advion). Mass spectra were processed by LipidView Software Version 1.2 (SCIEX) for identification and quantification of lipids. Lipid amounts (pmol) were corrected for response differences between internal standards and endogenous lipids.
Supplementary Material
Acknowledgments
We thank E. Kracker for technical assistance and the students Johannes Heimgärtner and Sabrina Eschbach for their help during their internships. This research was funded by the German Research Foundation (DFG) through GRK2364 and DI1386/2-1 to K.S.D. K.S.D. was additionally supported by the PROFILplus program of the Faculty of Medicine of the University of Tübingen. Collaborative work in the D.R., M.S., T.L., and J.M.H. labs was supported by a DFG DIP program (mitobalance 117516). D.R. and M.S. are supported by a German–Israeli Foundation (GIF) grant (Grant I-1458-412.13/2018). M.S. is an incumbent of the Dr. Gilbert Omenn and Martha Darling Professorial Chair in Molecular Genetics.
Abbreviations used:
- ADCK
aarF domain containing kinase
- CL
cardiolipin
- CS
contact site
- DAG
diacylglycerol
- ERMES
ER mitochondria encounter structure
- FA
fatty acid
- IMS
intermembrane space
- LD
lipid droplet
- MICOS
mitochondrial contact site and cristae organizing system
- MIM
mitochondrial inner membrane
- MOM
mitochondrial outer membrane
- MTS
mitochondrial targeting signal
- PA
phosphatidic acid
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PI
phosphatidylinositol
- PL
phospholipid
- PS
phosphatidylserine
- SGA
synthetic genetic array
- TAG
triacylglycerol
- TIM
translocase of the inner membrane
- TM
transmembrane domain
- TOM
translocase of the outer membrane
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-03-0166) on September 4, 2019.
REFERENCES
- Aaltonen MJ, Friedman JR, Osman C, Salin B, di Rago JP, Nunnari J, Langer T, Tatsuta T. (2016). MICOS and phospholipid transfer by Ups2–Mdm35 organize membrane lipid synthesis in mitochondria. J Cell Biol , 525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AhYoung AP, Jiang J, Zhang J, Khoi Dang X, Loo JA, Zhou ZH, Egea PF. (2015). Conserved SMP domains of the ERMES complex bind phospholipids and mediate tether assembly. Proc Natl Acad Sci USA , E3179–E3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MJ, Mooga VP, Guiard B, Langer T, Ryan MT, Stojanovski D. (2012). Impaired folding of the mitochondrial small TIM chaperones induces clearance by the i-AAA protease. J Mol Biol , 227–239. [DOI] [PubMed] [Google Scholar]
- Banci L, Bertini I, Cefaro C, Cenacchi L, Ciofi-Baffoni S, Felli IC, Gallo A, Gonnelli L, Luchinat E, Sideris D, Tokatlidis K. (2010). Molecular chaperone function of Mia40 triggers consecutive induced folding steps of the substrate in mitochondrial protein import. Proc Natl Acad Sci USA , 20190–20195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger KH, Sogo LF, Yaffe MP. (1997). Mdm12p, a component required for mitochondrial inheritance that is conserved between budding and fission yeast. J Cell Biol , 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, Cameron DM, Collins SR, Schuldiner M, Stewart-Ornstein J, Newman HW, Braun S, Madhani HD, Krogan NJ, Weissman JS. (2008). A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat Methods , 711–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen Y, Schuldiner M. (2011). Advanced methods for high-throughput microscopy screening of genetically modified yeast libraries. Methods Mol Biol , 127–159. [DOI] [PubMed] [Google Scholar]
- Connerth M, Tatsuta T, Haag M, Klecker T, Westermann B, Langer T. (2012). Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science , 815–818. [DOI] [PubMed] [Google Scholar]
- Daum G, Bohni PC, Schatz G. (1982). Import of proteins into mitochondria. Cytochrome b2 and cytochrome c peroxidase are located in the intermembrane space of yeast mitochondria. J Biol Chem , 13028–13033. [PubMed] [Google Scholar]
- Dimmer KS, Fritz S, Fuchs F, Messerschmitt M, Weinbach N, Neupert W, Westermann B. (2002). Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiae. Mol Biol Cell , 847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer KS, Jakobs S, Vogel F, Altmann K, Westermann B. (2005). Mdm31 and Mdm32 are inner membrane proteins required for maintenance of mitochondrial shape and stability of mitochondrial DNA nucleoids in yeast. J Cell Biol , 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz-Alon Y, Rosenfeld-Gur E, Shinder V, Futerman AH, Geiger T, Schuldiner M. (2014). A dynamic interface between vacuoles and mitochondria in yeast. Dev Cell , 95–102. [DOI] [PubMed] [Google Scholar]
- Frazier AE, Taylor RD, Mick DU, Warscheid B, Stoepel N, Meyer HE, Ryan MT, Guiard B, Rehling P. (2006). Mdm38 interacts with ribosomes and is a component of the mitochondrial protein export machinery. J Cell Biol , 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick RL, McCaffery JM, Cunningham KW, Okamoto K, Shaw JM. (2004). Yeast Miro GTPase, Gem1p, regulates mitochondrial morphology via a novel pathway. J Cell Biol , 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, et al (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature , 387–391. [DOI] [PubMed] [Google Scholar]
- Green SR, Moehle CM. (2001). Media and culture of yeast. Curr Protoc Cell Biol Chapter 1:Unit 1.6. [DOI] [PubMed] [Google Scholar]
- Grillitsch K, Daum G. (2011). Triacylglycerol lipases of the yeast. Front Biol , 219–230. [Google Scholar]
- Ham HJ, Rho HJ, Shin SK, Yoon HJ. (2010). The TGL2 gene of Saccharomyces cerevisiae encodes an active acylglycerol lipase located in the mitochondria. J Biol Chem , 3005–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Quinn AM, Hunter T. (1988). The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science , 42–52. [DOI] [PubMed] [Google Scholar]
- Honscher C, Mari M, Auffarth K, Bohnert M, Griffith J, Geerts W, van der Laan M, Cabrera M, Reggiori F, Ungermann C. (2014). Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev Cell , 86–94. [DOI] [PubMed] [Google Scholar]
- Hung V, Zou P, Rhee HW, Udeshi ND, Cracan V, Svinkina T, Carr SA, Mootha VK, Ting AY. (2014). Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol Cell , 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John Peter AT, Herrmann B, Antunes D, Rapaport D, Dimmer KS, Kornmann B. (2017). Vps13–Mcp1 interact at vacuole-mitochondria interfaces and bypass ER-mitochondria contact sites. J Cell Biol , 3219–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakimoto Y, Tashiro S, Kojima R, Morozumi Y, Endo T, Tamura Y. (2018). Visualizing multiple inter-organelle contact sites using the organelle-targeted split-GFP system. Sci Rep , 6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano S, Tamura Y, Kojima R, Bala S, Asai E, Michel AH, Kornmann B, Riezman I, Riezman H, Sakae Y, et al (2018). Structure–function insights into direct lipid transfer between membranes by Mmm1–Mdm12 of ERMES. J Cell Biol , 959–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug L, Daum G. (2014). Yeast lipid metabolism at a glance. FEMS Yeast Res , 369–388. [DOI] [PubMed] [Google Scholar]
- Kojima R, Kajiura S, Sesaki H, Endo T, Tamura Y. (2016). Identification of multi-copy suppressors for endoplasmic reticulum–mitochondria tethering proteins in Saccharomyces cerevisiae. FEBS Lett , 3061–3070. [DOI] [PubMed] [Google Scholar]
- Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P. (2009). An ER–mitochondria tethering complex revealed by a synthetic biology screen. Science , 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B, Osman C, Walter P. (2011). The conserved GTPase Gem1 regulates endoplasmic reticulum–mitochondria connections. Proc Natl Acad Sci USA , 14151–14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Tazir M, Lopez LC, Quinzii CM, Assoum M, Drouot N, Busso C, Makri S, Ali-Pacha L, Benhassine T, et al (2008). ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet , 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang A, John Peter AT, Kornmann B. (2015a). ER-mitochondria contact sites in yeast: beyond the myths of ERMES. Cur Opin Cell Biol , 7–12. [DOI] [PubMed] [Google Scholar]
- Lang AB, John Peter AT, Walter P, Kornmann B. (2015b). ER–mitochondrial junctions can be bypassed by dominant mutations in the endosomal protein Vps13. J Cell Biol , 883–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard CJ, Aravind L, Koonin EV. (1998). Novel families of putative protein kinases in bacteria and archaea: evolution of the “eukaryotic” protein kinase superfamily. Genome Res , 1038–1047. [DOI] [PubMed] [Google Scholar]
- Longen S, Bien M, Bihlmaier K, Kloeppel C, Kauff F, Hammermeister M, Westermann B, Herrmann JM, Riemer J. (2009). Systematic analysis of the twin cx(9)c protein family. J Mol Biol , 356–368. [DOI] [PubMed] [Google Scholar]
- Mesecke N, Terziyska N, Kozany C, Baumann F, Neupert W, Hell K, Herrmann JM. (2005). A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell , 1059–1069. [DOI] [PubMed] [Google Scholar]
- Miliara X, Garnett JA, Tatsuta T, Abid Ali F, Baldie H, Perez-Dorado I, Simpson P, Yague E, Langer T, Matthews S. (2015). Structural insight into the TRIAP1/PRELI-like domain family of mitochondrial phospholipid transfer complexes. EMBO Rep , 824–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata N, Fujii S, Kuge O. (2018). Porin proteins have critical functions in mitochondrial phospholipid metabolism in yeast. J Biol Chem , 17593–17605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbalci C, Sachsenheimer T, Brugger B. (2013). Quantitative analysis of cellular lipids by nano-electrospray ionization mass spectrometry. Methods Mol Biol , 3–20. [DOI] [PubMed] [Google Scholar]
- Peleh V, Cordat E, Herrmann JM. (2016). Mia40 is a trans-site receptor that drives protein import into the mitochondrial intermembrane space by hydrophobic substrate binding. eLife , e16177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potting C, Wilmes C, Engmann T, Osman C, Langer T. (2010). Regulation of mitochondrial phospholipids by Ups1/PRELI-like proteins depends on proteolysis and Mdm35. EMBO J , 2888–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidenbach AG, Kemmerer ZA, Aydin D, Jochem A, McDevitt MT, Hutchins PD, Stark JL, Stefely JA, Reddy T, Hebert AS, et al (2018). Conserved lipid and small-molecule modulation of COQ8 reveals regulation of the ancient kinase-like UbiB family. Cell Chem Biol , 154–165.e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheeff ED, Bourne PE. (2005). Structural evolution of the protein kinase-like superfamily. PLoS Comput Biol , e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner B, Westerburg H, Forne I, Imhof A, Neupert W, Mokranjac D. (2012). Role of the AAA protease Yme1 in folding of proteins in the intermembrane space of mitochondria. Mol Biol Cell , 4335–4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shai N, Yifrach E, van Roermund CWT, Cohen N, Bibi C, Lodewijk IJ, Cavellini L, Meurisse J, Schuster R, Zada L, et al (2018). Systematic mapping of contact sites reveals tethers and a function for the peroxisome–mitochondria contact. Nat Commun , 1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinzel M, Tan T, Wendling P, Kalbacher H, Ozbalci C, Chelius X, Westermann B, Brugger B, Rapaport D, Dimmer KS. (2016). Mcp3 is a novel mitochondrial outer membrane protein that follows a unique IMP-dependent biogenesis pathway. EMBO Rep , 965–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinzel M, Zeitler A, Dimmer KS. (2018). Interaction network of the mitochondrial outer membrane protein Mcp3. FEBS Lett , 3210–3220. [DOI] [PubMed] [Google Scholar]
- Sogo LF, Yaffe MP. (1994). Regulation of mitochondrial morphology and inheritance by Mdm10p, a protein of the mitochondrial outer membrane. J Cell Biol , 1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud DA, Oeljeklaus S, Wiese S, Bohnert M, Lewandrowski U, Sickmann A, Guiard B, van der Laan M, Warscheid B, Wiedemann N. (2011). Composition and topology of the endoplasmic reticulum–mitochondria encounter structure. J Mol Biol , 743–750. [DOI] [PubMed] [Google Scholar]
- Tamura Y, Onguka O, Aiken Hobbs AE, Jensen RE, Iijima M, Claypool SM, Sesaki H. (2012a). Role for two conserved intermembrane space proteins, Ups1p and Up2p, in intra-mitochondrial phospholipid trafficking. J Biol Chem , 15205–15218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y, Onguka O, Itoh K, Endo T, Iijima M, Claypool SM, Sesaki H. (2012b). Phosphatidylethanolamine biosynthesis in mitochondria: phosphatidylserine (PS) trafficking is independent of a PS decarboxylase and intermembrane space proteins UPS1P and UPS2P. J Biol Chem , 43961–43971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan T, Ozbalci C, Brugger B, Rapaport D, Dimmer KS. (2013). Mcp1 and Mcp2, two novel proteins involved in mitochondrial lipid homeostasis. J Cell Sci , 3563–3574. [DOI] [PubMed] [Google Scholar]
- Tatsuta T. (2017). Quantitative analysis of glycerophospholipids in mitochondria by mass spectrometry. Methods Mol Biol , 79–103. [DOI] [PubMed] [Google Scholar]
- Thomas BJ, Rothstein R. (1989). Elevated recombination rates in transcriptionally active DNA. Cell , 619–630. [DOI] [PubMed] [Google Scholar]
- Trotter PJ, Pedretti J, Voelker DR. (1993). Phosphatidylserine decarboxylase from Saccharomyces cerevisiae. Isolation of mutants, cloning of the gene, and creation of a null allele. J Biol Chem , 21416–21424. [PubMed] [Google Scholar]
- van den Brink-van der Laan E, Killian JA, de Kruijff B. (2004). Nonbilayer lipids affect peripheral and integral membrane proteins via changes in the lateral pressure profile. Biochim Biophys Acta , 275–288. [DOI] [PubMed] [Google Scholar]
- Van Heusden GP, Nebohacova M, Overbeeke TL, Steensma HY. (1998). The Saccharomyces cerevisiae TGL2 gene encodes a protein with lipolytic activity and can complement an Escherichia coli diacylglycerol kinase disruptant. Yeast , 225–232. [DOI] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW. (2008). Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol , 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukotic M, Nolte H, Konig T, Saita S, Ananjew M, Kruger M, Tatsuta T, Langer T. (2017). Acylglycerol kinase mutated in Sengers syndrome is a subunit of the TIM22 protein translocase in mitochondria. Mol Cell , 471–483.e477. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Alberti-Segui C, Rebischung C, Philippsen P. (1997). Heterologous HIS3 marker and GFP reporter modules for PCR-targeting in Saccharomyces cerevisiae. Yeast , 1065–1075. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pohlmann R, Philippsen P. (1994). New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast , 1793–1808. [DOI] [PubMed] [Google Scholar]
- Waegemann K, Popov-Celeketic D, Neupert W, Azem A, Mokranjac D. (2015). Cooperation of TOM and TIM23 complexes during translocation of proteins into mitochondria. J Mol Biol , 1075–1084. [DOI] [PubMed] [Google Scholar]
- Waggoner DW, Johnson LB, Mann PC, Morris V, Guastella J, Bajjalieh SM. (2004). MuLK, a eukaryotic multi-substrate lipid kinase. J Biol Chem , 38228–38235. [DOI] [PubMed] [Google Scholar]
- Walther DM, Papic D, Bos MP, Tommassen J, Rapaport D. (2009). Signals in bacterial beta-barrel proteins are functional in eukaryotic cells for targeting to and assembly in mitochondria. Proc Natl Acad Sci USA , 2531–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckbecker D, Longen S, Riemer J, Herrmann JM. (2012). Atp23 biogenesis reveals a chaperone-like folding activity of Mia40 in the IMS of mitochondria. EMBO J , 4348–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B, Neupert W. (2000). Mitochondria-targeted green fluorescent proteins: convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast , 1421–1427. [DOI] [PubMed] [Google Scholar]
- Wollweber F, von der Malsburg K, van der Laan M. (2017). Mitochondrial contact site and cristae organizing system: a central player in membrane shaping and crosstalk. Biochim Biophys Acta , 1481–1489. [DOI] [PubMed] [Google Scholar]
- Xie LX, Hsieh EJ, Watanabe S, Allan CM, Chen JY, Tran UC, Clarke CF. (2011). Expression of the human atypical kinase ADCK3 rescues coenzyme Q biosynthesis and phosphorylation of Coq polypeptides in yeast coq8 mutants. Biochim Biophys Acta , 348–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.