

Abstract
The clb gene cluster encodes the biosynthesis of metabolites known as precolibactins and colibactins. The clb pathway is found in gut commensal E. coli, and clb metabolites are thought to initiate colorectal cancer via DNA cross-linking. Here we report confirmation of the structural assignment of the complex clb product precolibactin 886 via a biomimetic synthetic pathway. We show that a α-ketoimine linear precursor undergoes spontaneous cyclization to precolibactin 886 upon HPLC purification. Studies of this α-ketoimine and the related α-dicarbonyl revealed that these compounds are unexpectedly susceptible to nucleophilic cleavage under mildly basic conditions. This cleavage pathway forms other known clb metabolites or biosynthetic intermediates and explains the difficulties in isolating fully mature biosynthetic products. This cleavage also accounts for a recently identified colibactin–adenine adduct. The colibactin peptidase ClbP deacylates synthetic precolibactin 886 to form a non-genotoxic pyridone, suggesting precolibactin 886 lies off-path of the major biosynthetic route.
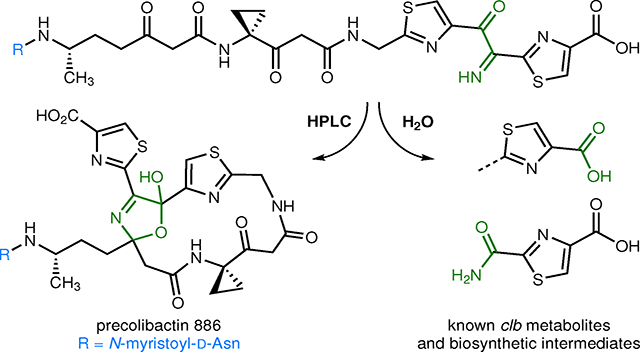
Graphical Abstract
In a seminal study, Oswald and co-workers discovered that strains of commensal E. coli containing a biosynthetic gene cluster – referred to as clb or pks – induce DNA damage in eukaryotic cells.1 Subsequent, the presence of clb+ E. coli was shown to be epidemiologically correlated with colorectal cancer (CRC) in humans,2,3 and literature now suggests these bacteria are tumorigenic.4–9 Therefore, elucidating the structures and mechanism of action of clb metabolites may help to understand if and how these metabolites contribute to the formation of CRC.
Precolibactin 886 (1, Fig. 1a)10 is one of the most complex isolated metabolites derived from the clb pathway; it accounts for all but three (clbL, clbO, and clbQ) biosynthetic enzymes in the gene cluster.10,11 The C37 and C38 atoms of precolibactin 886 (1) derive from an α-aminomalonate building block which originates biosynthetically from serine.10,12,13 Bacteria deficient in the genes responsible for the biosynthesis and transfer of this polyketide extender unit (clbDEFG and an adenylation (A) domain of clbH-A1) are not genotoxic, indicating that the α-aminomalonate building block is required for cytotoxicity.1,12,13
Fig. 1.

Overview of (pre)colibactin reactivity, mechanism of genotoxicity, and the synthetic strategy pursued herein. a, Structure of precolibactin 886 (1) and the putative biosynthetic precursor 2. The α-ketoimine 2 served as a synthetic precursor to precolibactin 886 (1). b, Genetic studies established that linear intermediates resembling 3 are off-loaded from the clb biosynthetic assembly line. In wild-type strains, these products undergo ClbP deacylation and transformation to the genotoxic imines 4. In mutant strains lacking functional ClbP, they transform to non-genotoxic pyridones, such as precolibactins A–C (6–8). The adenine adduct 5 was derived from DNA that had been exposed to clb+ E. coli cultures. c, Retrosynthetic analysis of the α-ketoimine 2.
Precolibactin 886 (1) was isolated by Qian, Moore, and co-workers from the fermentation broth of a clbP/clbQ double mutant.10 Recently, a derivative of precolibactin 886 (1) bearing a terminal oxazole was disclosed.14 Deactivation of the pathway-dedicated serine protease ClbP15,16 has been widely used to facilitate the isolation of clb metabolites. This modification leads to the persistence of a terminal N-myristoyl-D-Asn residue (blue in 1)17–19 and accumulation of the more stable precolibactins. Deactivation of the type-II thioesterase ClbQ,20 which is required for genotoxicity,1 increased the production of precolibactin 886 (1) 22-fold by reducing the release of advanced intermediates from the biosynthetic assembly line.10 Despite the use of this strategically-optimized double mutant, only 2.8 mg of precolibactin 886 (1) were obtained from a 1000-L fermentation.10 Qian and Moore proposed that the 15-atom macrocycle and 5-hydroxy-3-oxazoline ring (green in 1) found in precolibactin 886 (1) arises from the enzymatic oxidation and cyclization of a linear α-ketoamine precursor. This proposal is supported by literature demonstrating the addition of α-ketoimines to ketones.21
In wild-type strains possessing functional ClbP, N-deacylation initiates an alternative reaction pathway that ultimately forms unsaturated imines resembling 4 (Fig. 1b).11,22 Synthetic derivatives of 4 alkylated linearized plasmid DNA by nucleotide addition to the cyclopropane and activated DNA damage response pathways in human cells.22,23 A dimer of 4 formed stable DNA cross-links, and a derivative possessing a gem-dimethyl group in place of the cyclopropane did not damage DNA, providing robust support for DNA alkylation by cyclopropane opening.16 Further supporting this model, the adenine adduct 5 was subsequently identified in clb+ E. coli cultures treated with exogenous DNA.24 This product was later established as an N3-substituted adenine adduct, which could also be detected in human cells and in colonic epithelial cells of a mouse model when treated with clb+ E. coli.25 These studies suggest a basis for clb+ E. coli genotoxicity and explain earlier determinations that functional ClbP is required for cytopathic effects.1,15–19
Here we report a biomimetic synthesis of precolibactin 886 (1) and the surprising discovery that the C36–C37 bond in 2 is unstable toward nucleophilic cleavage. This degradation generates other known clb metabolites and biosynthetic precursors, and explains the difficulties in isolating advanced clb metabolites. We show that ClbP deacylation of precolibactin 886 (1) forms a non-genotoxic pyridone, suggesting it lies off of the major colibactin biosynthetic pathway.
Results
Total synthesis of the linear precursor 2
Our retrosynthetic analysis is shown in Fig. 1c. As the α-ketoimine motif was found to be unstable, it was introduced in the final step. We envisioned accessing 2 by double oxidation of the amino alcohol 9. The latter was readily deconstructed to the β-ketothioesters 10 and 1126 and the azido alcohol 12.
The synthesis of the azido alcohol 12 is shown in Fig. 2. The thiazole 13 is accessible in one step and 74% yield from commercial reagents.26 Semi-reduction of the ester (di-iso-butylaluminum hydride, DIBAL-H) provided the aldehyde 14 (88%). Alternatively, removal of the carbamate (hydrochloric acid) followed by condensation with benzophenone imine generated the imine 15 (85%, two steps). This approach exploits the pseudo-symmetry within the thiazoles and greatly expedited material throughput. Silver-catalyzed coupling27,28 of 14 and 15, followed by hydrolysis, then provided the amino alcohol 16 (2:1 mixture of diastereomers, stereochemistry not assigned). Wong diazo transfer29,30 to 16 using imidazole sulfonyl azide31 in methanol yielded the azido alcohol 17 with concomitant transesterification of the ester (56% over three steps). Treatment of the diazo transfer product 17 with hydrochloric acid then formed the primary ammonium ion 12. Coupling with the β-ketothioester 11 (silver trifluoroacetate, triethylamine) generated the β-ketoamide 18 (85%, two steps).26 Saponification of the ester (lithium hydroxide, 87%) followed by removal of the tert-butyl carbamate (hydrochloric acid) produced the carboxylic acid 19. A second silver-mediated fragment coupling between 19 and the β-ketothioester 10 provided the linear intermediate 20 (78%, two steps). Staudinger reduction of 20 (trimethylphosphine) proceeded smoothly to generate the target amino alcohol 9 (>99%).
Fig. 2.

Synthesis of the azide 20 and the amino alcohol 9. Our synthetic approach exploits the pseudo-symmetry within the linked thiazole residues to access this fragment from the single synthetic intermediate 13. The linear precursors 9 and 20 are assembled by sequential N-acylation reactions, using the β-ketothioesters 10 and 11 as acylating agents.
Oxidation and cyclization studies
We then probed the oxidation of 9 to the α-ketoimine 2 (Fig. 3a). However, the reaction was difficult to monitor by thin-layered chromatography (TLC) or liquid chromatography/mass spectrometry (LC/MS), and all attempts to isolate the product derived from two-fold oxidation of 9 were unsuccessful. We surmised that the α-ketoimine 2, if formed, was hydrating or hydrolyzing on attempted isolation.32,33 To circumvent this, we studied the oxidation of 9 by NMR spectroscopy. The addition of 4.0 equiv of 2-iodoxybenzoic acid (IBX) to a solution of 9 in methyl sulfoxide-d6 at 23 °C resulted in consumption of the amino alcohol 9 within 30 min. A sharp singlet that we attributed to the N-H resonance of the α-ketoimine emerged at δ 12.02 ppm. However, this resonance under-integrated relative to the C20 methyl group, suggesting transformation of the starting material to other unidentified products.
Fig. 3.

Synthesis of precolibactin 886 (1) and reactivity of the α-ketoimine and α-dicarbonyl residues. a, Two-fold oxidation of the 1,2-aminoalcohol 9 generates the α-ketoimine 2. This intermediate is unstable toward hydrolysis to the α-diketone 22. We were unable to effect the cyclization of 2 under a broad range of acidic, basic, or thermal conditions. However, we found that the α-ketoimine 2 converted to precolibactin 886 (1) upon HPLC purification. Oxidation of the azidoalcohol 20 with excess IBX also formed the α-ketoimine 2, presumably by loss of dinitrogen from an α-ketoazide intermediate (not shown). b, Synthesis of the α-ketoimine 25 and the α-dicarbonyl 26. The α-ketoimine 25 partially hydrolyzed to the α-dicarbonyl 26 upon exposure to water during work-up. Alternatively, this intermediate could be deliberately transformed to 26 by exposure to silica gel. c, Oxidation of the N-(tert-butoxycarbonyl)-1,2-amino alcohol 27. Oxidation with excess DMP generated the N-(tert-butoxycarbonyl) aminal 29. Alternatively, oxidation with 0.9 equiv of DMP formed the α-aminoketone 28. The α-aminoketone 28 transformed spontaneously to the hemiaminal 29 on standing.
Nonetheless, we attempted to promote macrocyclization of the α-ketoimine 2 under thermal, basic, or acidic conditions. However, in each instance we were unable to detect (by 1H NMR spectroscopy) any signals that could be attributed to precolibactin 886 (1). Under most conditions, we observed hydrolysis to the α-diketone 22, as evidenced by a gradual disappearance of the N–H resonance and the appearance of a new signal at 185.8 ppm in the 13C NMR spectrum (assigned as the C37 carbonyl). The hydrolysis was faster under acidic conditions, as expected. Surprisingly, under basic conditions precolibactin B (7) was formed by an unforeseen hydrolytic cleavage of the C36–C37 bond, followed by cyclodehydration.26 Attempts to promote the cyclization thermally led to decomposition.
In light of these difficulties, we hypothesized that in the isolation studies10 cyclization to precolibactin 886 (1) occurred during HPLC (high pressure liquid chromatography) analysis and purification. Such reactivity would be undetectable because all reported fermentations of clb+ E. coli have been analyzed (and purified) by HPLC, to our knowledge. To test this, we directly injected solutions of the α-ketoimine 2 onto a semipreparative HPLC system. Under these conditions cyclization did occur, and from 10 mg of 2 we obtained ~0.3 mg of precolibactin 886 (1, ~3%). Spectroscopic data for synthetic precolibactin 886 (1) were in full agreement with natural material10 (See Supplementary Table 1 and Supplementary Fig. 2). The C36 stereocenter was formed as a ~1:1.9 mixture of diastereomers in the ring closure (stereochemistry not assigned). The dr of natural precolibactin 886 (1), which also formed non-stereoselectively, was not reported.10 Importantly, H32 are magnetically-equivalent in the linear precursor 2 and appear as an apparent doublet (δ 4.52, J = 6.0 Hz). Formation of the macrocycle breaks this degeneracy leading to a doublets of doublets resonating at δ 4.27 (J = 17.2, 5.0 Hz) and δ 4.79 (J = 17.2, 7.1 Hz). We did not observe cyclization of the amino alcohol 9 or the monooxidation product 21, suggesting the imine is formed prior to macrocyclization in the bacterial extracts. Finally, precolibactin 886 (1) could also be accessed in comparable yield from the azido alcohol 20. Oxidation of 20 with 2-iodoxybenzoic acid (IBX) formed the α-ketoimine 2 directly, presumably via loss of dinitrogen from an α-azidoketone intermediate.30 The generation of precolibactin 886 (1) from the azido alcohol 20 provides further support for the intermediacy of the α-ketoimine 2.
After confirming the formation of precolibactin 886 (1) during HPLC purification, we attempted to form precolibactin 886 (1) deliberately by evaluating the cyclization of the α-ketoimine 2 in aqueous methanol or aqueous acetonitrile. While we were able to observe precolibactin 886 (1) by HPLC/MS analysis, we did not observe an increase in isolated yield after incubating for 24 h before purification. As a result, we conclude that a small percentage of the fleetingly stable α-ketoimine 2 cyclizes to form macrocylic precolibactin 886 (1) during LC/MS analysis/HPLC purification and that this isolate is an artifact of these analytical methods.
On the chemical instability of the α-ketoimine 2
The low yield of precolibactin 886 (1) and the unexpected generation of precolibactin B (7) from 9 motivated us to study the reactivity of the model α-ketoimine 25 and its hydrolysis product 26 in detail (Fig. 3b and Table 1). The amino alcohol 23 (see Supplementary Fig. 1 for synthesis) was efficiently oxidized to the α-ketoimine 25 by treatment with excess Swern reagent. 1H NMR analysis indicated that the sample was >95% pure (nominally 93% yield). The imine N–H resonance appeared at δ 12.07 ppm in methyl sulfoxide-d6, in good agreement with the fully elongated α-ketoimine 2 (δ 12.02 ppm). As expected, the imine readily hydrolyzed. For example, the α-diketone 26 began to form if the α-ketoimine 25 was briefly exposed to aqueous conditions on work-up. Alternatively, the addition of silica gel to solutions of the α-ketoimine 25 formed the α-diketone 26 (38% from 23). The α-diketone 26 was independently synthesized by Wong diazo transfer29,30 to 23 using imidazole sulfonyl azide31 (96%), followed by oxidation with the Dess–Martin periodinane34 (DMP, 2.5 equiv, 54%). The C37 carbon atom of 26 was observed at δ 184.6 in methyl sulfoxide-d6, in good agreement with the fully elongated α-diketone 22 (δ 185.8).
Table 1.
Nucleophilic cleavage of the α-ketoimine 25 and the α-diketone 26.
 | |||
|---|---|---|---|
| entry | substrate | nucleophile | product(s) (yield)a |
| 1 | 25 | methanolb | 30a (42%) + 30d (~42%)e |
| 2 | 25 | waterc | 30b (51%) |
| 3 | 25 | pyrrolidined | 30c (36%) |
| 4 | 26 | methanolb | 30a (45%) + 30e (41%) |
| 5 | 26 | waterc | 30b (49%) + 30f (25%) |
| 6 | 26 | pyrrolidined | 30c (41%) |
Isolated yield after purification by flash-column chromatography.
25 or 26 (1 equiv), NaHCO3 (9.0 equiv), methanol, 23 °C, 48 h (for 25 and 26).
25 or 26 (1 equiv), tetrahydrofuran–saturated aqueous sodium bicarbonate solution (1:1, v/v), 23 °C, 48 h (for 25), 72 h (for 26).
25 or 26 (1 equiv), pyrrolidine (14 equiv), dichloromethane, 23 °C, 48 h (for 25), 72 h (for 26).
Estimated by HPLC analysis of the product mixture.
Studies of the oxidation of the N-(tert-butoxycarbonyl)-1,2-amino alcohol 27 provided unexpected results (Fig. 3c). Oxidation with an excess of DMP, under Swern conditions, or using sulfur trioxide–pyridine complex formed the product of two-fold oxidation, the hemiaminal 29. Attempted oxidation of 27 using 0.9 equiv of DMP, followed by immediate purification by flash-column chromatography, generated the expected α-amino ketone 28, but this spontaneously oxidized to 29 after prolonged exposure to air. These data suggest intermediates such as 28 undergo facile oxidation and support a pathway for the spontaneous generation of the α-ketoimine in the precolibactin 886 precursor 2 from the α-aminoketone substrate expected to be formed based on biosynthetic logic.
We then studied the reactivity of the α-ketomine 25 and the α-diketone 26 in detail (Table 1). Unexpectedly, 25 and 26 readily underwent cleavage of the C36–C37 bond [precolibactin 886 (1) numbering]. For example, dissolution of 25 in methanol followed by the addition of sodium bicarbonate resulted in formation of the ester 30a (42%) and the carboxamide 30d. Bond cleavage even occurred in the absence of base, albeit more slowly: the half-life of the α-ketoimine 25 in neutral, distilled methanol-d4 was ~12 h at 24 °C (1H NMR analysis). Alternatively, the α-ketoimine 25 was transformed to the carboxylic acid 30b by treatment with saturated aqueous sodium bicarbonate solution in tetrahydrofuran (51%), and the amide 30c was formed by treatment with excess pyrrolidine (36%). In the case of entries 2 and 3 the products derived from the right-hand thiazole were not readily identified. The α-diketone 26 underwent parallel transformations forming the methyl esters 30a and 30e, the carboxylic acids 30b and 30f, and the amide 30c. The remainder of the mass balance in these reactions is accounted for by the formation of unidentified decomposition products.
In light of this, we re-examined the products resulting from oxidation of the amino alcohol 9 by LC/MS and tandem MS. As expected, we detected hydrolysis of the α-ketoimine 2 to its corresponding α-diketone 22 (Fig. 4a, Supplementary Fig. 4). We also detected hydrolytic cleavage of the C36–C37 bond to generate 32, and, as reported in an earlier study,26 double ring closure of 32 to provide precolibactin B (7). The addition of L-cysteine resulted in generation of 33, the linear precursor to precolibactin A (6, Fig. 1b). The structures of 31–33 and precolibactin B (7) were confirmed by tandem MS and LC/MS co-injection with the bacterial extracts derived from a clb+ ΔclbP E. coli culture (Supplementary Figs. 3, 5–7). We also detected competitive hydrolysis of the C23–C24 bond to generate the acylasparagine derivative 31, which has previously been detected in clb cultures.19 The formation of these products from the α-ketoimine 2 under mild conditions suggests that this intermediate is decomposing during HPLC purification and provides an explanation for the low overall yield of precolibactin 886 (1).
Fig. 4.

The α-ketoimine 2 undergoes transformation to known clb metabolites or biosynthetic intermediates under mild conditions, while ClbP deacylation of precolibactin 886 (1) forms the non-genotoxic pyridone 35. a, Cleavage reactions of the α-ketoimine 2. Hydrolysis of 2 generates the α-dicarbonyl 22. Alternatively, hydrolytic cleavage of the C23–C24 or C36–C37 bonds in 2 forms the carboxylic acids 31 and 32, respectively. The cleavage product 32 was found to convert to precolibactin B (7). The addition of L-cysteine to 2 results in cleavage of the C36–C37 bond and generation of the N-acyl cysteine derivative 33. b, ClbP deacylation of synthetic precolibactin 886 (1) generates the amine 34. The amine 34 was found to convert to the pyridone 35, suggesting that precolibactin 886 (1) lies off of the major biosynthetic pathway c, Time course analysis of the conversion of synthetic precolibactin 886 (1) to the deacylation product 34 and the pyridone 35 when exposed to E. coli expressing or lacking functional ClbP. The conversion of precolibactin 886 (1) was only detected in the presence of a ClbP-expressing strain (n = 1).
To establish precolibactin 886 (1) as a potential substrate of the promiscuous colibactin peptidase ClbP, we added 1 to cultures of E. coli expressing functional ClbP (pPEB018)19 and a control strain lacking ClbP (pBAD18) (Fig. 4b). The expected deacylation product 34 was formed in the ClbP expressing strain but it was undetectable in the ClbP-deficient strain. The concentration of 34 increased in a time-dependent fashion. However, we determined that the macrocycle 34 was unstable under the cultivation conditions. As it was produced, 34 underwent conversion to the pyridone 35. The structure of 35 formed in this experiment was confirmed by LC/MS co-injection and tandem MS with a synthetic standard (Supplementary Fig. 8). The pyridone 35 derives from C36–C37 bond cleavage and double cyclodehydration.
Discussion
The prevailing model for colibactin biosynthesis and genotoxicity involves off-loading of fully linear products (see 3, Fig. 1b) from the biosynthetic assembly line. In wild-type strains these intermediates are deacylated in the periplasm by ClbP and transform, via spontaneous cyclization reactions, to genotoxic unsaturated imines (see 4, Fig. 1b). In mutant strains lacking functional ClbP, artificial cyclization reactions prevail resulting in the generation of non-genotoxic pyridone-containing compounds (e.g., 6–8, Fig. 1b).7,22 The studies reported herein allow us to expand this model to accommodate the structure of precolibactin 886 (1, Fig. 5a). Our data suggest a biosynthetic pathway for 1 involving off-loading of the linear α-amino ketone 36 from the assembly line. It has previously been suggested that 36 is oxidized by the enzyme ClbK;10 however, our studies of the amino alcohol 27 (Fig. 3c) raise the possibility that oxidation occurs spontaneously. Irrespective of how it is formed, the resulting α-ketoimine 2 then partitions among several reaction pathways. We speculate that in wild-type or mutant strains hydrolysis of the α-ketoimine to the α-diketone 22 is rapid and predominates. In mutant strains lacking functional ClbP, the hydrolysis product 22 degrades by cleavage of the C23–C24 bond and/or cleavage of the C36–C37 bond. The latter pathway generates metabolites such as precolibactin A (6)26,35 and precolibactin B (7)36 that have previously been detected in ΔclbP cultures. In wild-type strains, deacylation of 22 followed by cyclocondensation generates unsaturated imines 4 structurally analogous to those formed from simpler clb metabolites. With the difficulties we encountered in inducing cyclization of the linear α-ketoimine 2 to precolibactin 886 (1), and the very low yield of the latter from prior isolation studies,10 we suggest that macrocyclization is a minor pathway. This is also consistent with our observation that ClbP cleavage of 1 leads to the non-genotoxic pyridone breakdown product 35 (Fig. 4b) as the major detectable product in cell culture.
Fig. 5.

Biosynthetic and chemical reactivity model based on the studies reported herein. a, Our data suggest that the α-ketoamine 36 is off-loaded from the biosynthetic assembly line. This product undergoes oxidation, either spontaneously or by the enzyme ClbK. The α-ketoimine 2 that is formed then partitions among several pathways. Hydrolytic cleavage of the C23–C24 or C36–C37 bonds forms the carboxylic acids 31 or 32, respectively (see Fig. 4a). Alternatively, macrocyclization (upon HPLC analysis or purification) generates precolibactin 886 (1). Hydrolysis of the imine forms the α-dicarbonyl 22. This intermediate is also susceptible to hydrolytic cleavage, to provide the carboxylic acids 31 or 32, respectively (see Fig. 4a). ClbP deacylation of the α-dicarbonyl in wild-type strains may generate unsaturated imines resembling 4 (Fig. 1b). c, Potential mechanism for hydrolytic cleavage of the α-ketoimine 2 or the α-dicarbonyl 22. Hydration of the α-ketoimine 2 generates the hemiaminal 37. Isomerization to the hydroxyepoxide 38, followed by rearrangement, forms the enol ester 39. Addition of water to the C36 carbonyl of 39, followed by collapse of the tetrahedral intermediate, provides the carboxylic acid 32 and the aminoenol 40. Oxidation of 40 generates the thiazole 30d.
The facile C36–C37 bond cleavage we have observed has several important implications for colibactins and precolibactins First the adenine adduct 5 (Fig. 1b) was characterized in the digestion mixtures of DNA that had been cross-linked by clb+ E. coli.24,25 While it is conceivable that 5 derives from potentially more abundant monoalkylation products, the facile cleavage of the α-dicarbonyl residue we have documented raises the possibility that 5 is generated by degradation of a cross-linked product. While genetic studies indicate that every gene in the clb cluster is required for cytopathic effects,1 implicating the fully elaborated colibactin as the causative genotoxic agent, cleavage of the C36–C37 bond provides an explanation for the difficulties encountered in identifying this molecule (or the corresponding precolibactin). The remaining biosynthetically unaccounted clb enzymes ClbL and ClbO, which are required for genotoxicity, append substituents to the terminal thiazole of advanced clb metabolites. C36–C37 bond cleavage would effectively remove these substituents from advanced precolibactins, rendering isolation of the corresponding precolibactin or colibactin products impossible. A possible mechanism for the hydrolytic cleavage of the C36–C37 bond in the α-ketoimine 2 is shown in Fig. 5b. Formation of the hemiaminal 37, followed by rearrangement would generate the enolester 39. Hydrolysis would generate the carboxylic acid 32 and liberate the enol 40. Oxidation of the enol 40 would generate the observed carboxamide 30d. This mechanism is reminiscent of thiazolium ion reactivity,37 but employs a thiazole rather than a thiazolium ion as the nascent leaving group. Related pathways for the cleavage of 1,2-dicarbonyl compounds by thiazolium ions have been proposed.38
Conclusion
In sum, we have reported the first total synthesis of precolibactin 886 (1), one of the most advanced clb products. Our studies confirm the structural assignment of this unusual metabolite, and suggest it may be an artifact deriving from HPLC analysis and purification of the bacterial extracts. Significantly, our data have revealed the unexpected electrophilic reactivity of the α-ketoimine of precolibactin 886 (1); the mild cleavage of the C36–C37 bond we have observed provides an explanation for the difficulties in isolating advanced clb metabolites and accounts for the structures of recently isolated colibactin–adenine adducts. Finally, our synthetic studies suggest the unusual α-ketoimine may arise by spontaneous oxidation of an unstable α-amino ketone. This work defines the unexpected reactivities of advanced clb metabolites, provides a path for characterizing the structures of DNA crosslinked products, and explains the underlying cellular and chemical degradation pathways of (pre)colibactins.
Supplementary Material
Acknowledgement
We thank Professor David Spiegel for helpful discussions regarding the mechanism of hydrolytic cleavage of 2. Financial support from the National Institutes of Health (R01GM110506 to S.B.H., 1DP2-CA186575 to J.M.C., R01CA215553 to S.B.H. and J.M.C.), the Chemistry Biology Interface Training Program (T32GM067543 to K.M.W.), the Charles H. Revson foundation (postdoctoral fellowship to A.R.H.) and Yale University is gratefully acknowledged.
Footnotes
Competing interest. The authors declare no competing interests.
Data availability statement. All relevant data are available from the authors, and/or are included with the manuscript and Supplementary Information.
References
- 1.Nougayrède J-P et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 313, 848–851 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Arthur JC et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buc E et al. High prevalence of mucosa-associated e. Coli producing cyclomodulin and genotoxin in colon cancer. PLoS One 8, e56964 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trautman EP & Crawford JM Linking biosynthetic gene clusters to their metabolites via pathway-targeted molecular networking. Curr. Top. Med. Chem 16, 1–12 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balskus EP Colibactin: Understanding an elusive gut bacterial genotoxin. Nat. Prod. Rep 32, 1534–1540 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Taieb F, Petit C, Nougayrede JP & Oswald E The enterobacterial genotoxins: Cytolethal distending toxin and colibactin. EcoSal Plus 7, doi: 10.1128/ecosalplus.ESP-0008-2016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Healy AR & Herzon SB Molecular basis of gut microbiome-associated colorectal cancer: A synthetic perspective. J. Am. Chem. Soc 139, 14817–14824 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faïs T, Delmas J, Barnich N, Bonnet R & Dalmasso G Colibactin: More than a new bacterial toxin. Toxins 10, 151 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dejea CM et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 359, 592 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li ZR et al. Divergent biosynthesis yields a cytotoxic aminomalonate-containing precolibactin. Nat. Chem. Biol 12, 773–775 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trautman EP, Healy AR, Shine EE, Herzon SB & Crawford JM Domain-targeted metabolomics delineates the heterocycle assembly steps of colibactin biosynthesis. J. Am. Chem. Soc 139, 4195–4201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brachmann AO et al. Colibactin biosynthesis and biological activity depend on the rare aminomalonyl polyketide precursor. Chem. Commun 51, 13138–13141 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Zha L, Wilson MR, Brotherton CA & Balskus EP Characterization of polyketide synthase machinery from the pks island facilitates isolation of a candidate precolibactin. ACS Chem. Biol 11, 1287–1295 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Li Z-R et al. Macrocyclic colibactin induces DNA double-strand breaks via copper-mediated oxidative cleavage. bioRxiv, 530204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dubois D et al. Clbp is a prototype of a peptidase subgroup involved in biosynthesis of nonribosomal peptides. J. Biol. Chem 286, 35562–35570 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cougnoux A et al. Analysis of structure-function relationships in the colibactin-maturating enzyme clbp. J. Mol. Biol 424, 203–214 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Brotherton CA & Balskus EP A prodrug resistance mechanism is involved in colibactin biosynthesis and cytotoxicity. J. Am. Chem. Soc 135, 3359–3362 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Bian X et al. In vivo evidence for a prodrug activation mechanism during colibactin maturation. Chembiochem 14, 1194–1197 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Vizcaino MI, Engel P, Trautman E & Crawford JM Comparative metabolomics and structural characterizations illuminate colibactin pathway-dependent small molecules. J. Am. Chem. Soc 136, 9244–9247 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guntaka NS, Healy AR, Crawford JM, Herzon SB & Bruner SD Structure and functional analysis of clbq, an unusual intermediate-releasing thioesterase from the colibactin biosynthetic pathway. ACS Chem Biol 12, 2598–2608 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patonay T & Hoffman RV Base-promoted reactions of α-azido ketones with aldehydes and ketones: A novel entry to α-azido-β-hydroxy ketones and 2,5-dihydro-5-hydroxyoxazoles. J. Org. Chem 60, 2368–2377 (1995). [Google Scholar]
- 22.Healy AR, Nikolayevskiy H, Patel JR, Crawford JM & Herzon SB A mechanistic model for colibactin-induced genotoxicity. J. Am. Chem. Soc 138, 15563–15570 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shine EE et al. Model colibactins exhibit human cell genotoxicity in the absence of host bacteria. ACS Chem. Biol 13, 3286–3293 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xue M, Shine E, Wang W, Crawford JM & Herzon SB Characterization of natural colibactin-nucleobase adducts by tandem mass spectrometry and isotopic labeling. Support for DNA alkylation by cyclopropane ring opening. Biochemistry 57, 6391–6394 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson MR et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 363, eaar7785 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Healy AR, Vizcaino MI, Crawford JM & Herzon SB Convergent and modular synthesis of candidate precolibactins. Structural revision of precolibactin a. J. Am. Chem. Soc 138, 5426–5432 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seashore-Ludlow B, Torssell S & Somfai P Addition of azomethine ylides to aldehydes: Mechanistic dichotomy of differentially substituted α-imino esters. Eur. J. Org. Chem 2010, 3927–3933 (2010). [Google Scholar]
- 28.Lou S, Ramirez A & Conlon DA Catalytic syn-selective direct aldol reactions of benzophenone glycine imine with aromatic, heteroaromatic and aliphatic aldehydes. Adv. Synth. Catal 357, 28–34 (2015). [Google Scholar]
- 29.Alper PB, Hung S-C & Wong C-H Metal catalyzed diazo transfer for the synthesis of azides from amines. Tetrahedron Lett. 37, 6029–6032 (1996). [Google Scholar]
- 30.Nyffeler PT, Liang C-H, Koeller KM & Wong C-H The chemistry of amine–azide interconversion: Catalytic diazotransfer and regioselective azide reduction. J. Am. Chem. Soc 124, 10773–10778 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Goddard-Borger ED & Stick RV An efficient, inexpensive, and shelf-stable diazotransfer reagent: Imidazole-1-sulfonyl azide hydrochloride. Org. Lett 9, 3797–3800 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Edwards OE & Purushothaman KK Some reactions of alicyclic α-azidoketones. Can. J. Chem 42, 712–716 (1964). [Google Scholar]
- 33.Faiz S et al. Synthesis and consecutive reactions of alpha-azido ketones: A review. Molecules 20, 14699–14745 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dess DB & Martin JC A useful 12-i-5 triacetoxyperiodinane (the dess-martin periodinane) for the selective oxidation of primary or secondary alcohols and a variety of related 12-i-5 species. J. Am. Chem. Soc 113, 7277–7287 (1991). [Google Scholar]
- 35.Vizcaino MI & Crawford JM The colibactin warhead crosslinks DNA. Nat. Chem 7, 411–417 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li ZR et al. Critical intermediates reveal new biosynthetic events in the enigmatic colibactin pathway. Chembiochem 16, 1715–1719 (2015). [DOI] [PubMed] [Google Scholar]
- 37.White MJ & Leeper FJ Kinetics of the thiazolium ion-catalyzed benzoin condensation. J. Org. Chem 66, 5124–5131 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Kim T & Spiegel DA The unique reactivity of n-phenacyl-derived thiazolium salts toward α-dicarbonyl compounds. Rejuvenation Res. 16, 43–50 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.