

 Celastrol is a natural bioactive compound extracted from the medicinal plant Tripterygium wilfordii Hook F.
Celastrol is a natural bioactive compound extracted from the medicinal plant Tripterygium wilfordii Hook F.
Abstract
Celastrol is a natural bioactive compound extracted from the medicinal plant Tripterygium wilfordii Hook F. It exhibits immunosuppressive, anti-inflammatory, and antioxidant activities. Cisplatin is a commonly used chemotherapeutic drug in the treatment of a wide range of tumors. Although very effective therapeutically, it can cause nephrotoxicity leading to dose reduction or discontinuation of treatment. This study aims to clarify the therapeutic potential of celastrol in cisplatin-induced nephrotoxicity. The possible protective effects of celastrol pretreatment against cisplatin-induced oxidative stress and genotoxicity were investigated. A rat kidney epithelial cell line NRK-52E was pretreated with the desired concentrations of celastrol (200 nM, 100 nM, and 50 nM) for 24 h. The cells were treated with 50 μM cisplatin for a further 24 h to see whether cisplatin caused the same or less toxicity compared to the vehicle control group. Alkaline comet assay was performed for genotoxicity assessment. Genotoxicity evaluation revealed that celastrol caused a statistically significant reduction in DNA damage. Oxidative stress parameters were evaluated by measuring the glutathione (GSH) and protein carbonyl (PC) levels and also by measuring the enzyme activities of glutathione peroxidase (GPx), glutathione reductase (GR), catalase (CAT) and superoxide dismutase (SOD) enzymes. Celastrol pretreatment increased the GSH content of the cells and ameliorated the protein carbonylation level. Likewise, celastrol pretreatment improved the GR and CAT activities. However, no significant difference was observed in GPx and SOD activities. In the light of these findings, celastrol treatment could be a therapeutic option to reduce cisplatin-induced nephrotoxicity. Further studies are needed for the clarification of its therapeutic potential.
Introduction
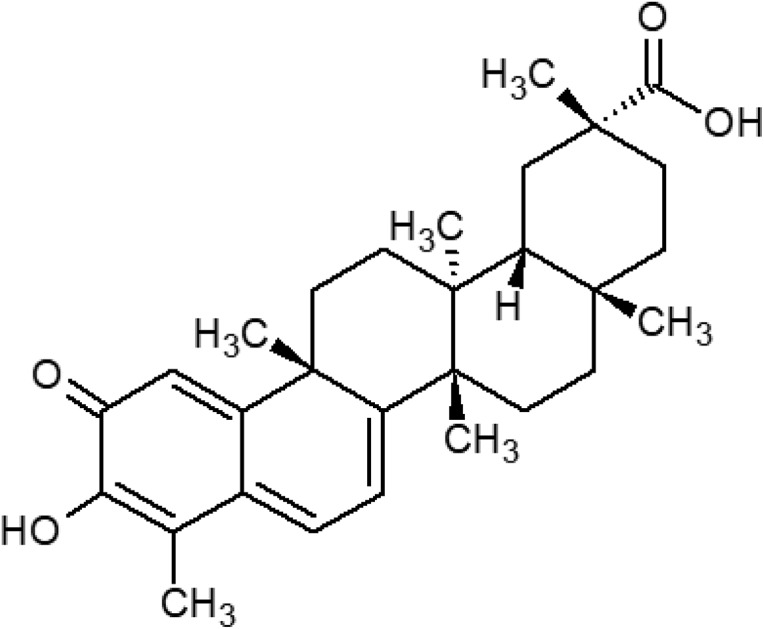
Celastrol is a natural bioactive compound and is extracted from the medicinal plant Tripterygium wilfordii Hook F. which is also known as Thunder God Vine. Celastrol exhibits immunosuppressive, anti-inflammatory, and antioxidant activities; therefore the plant has been used for the treatment of joint pain and fever in Chinese medicine for quite a long time.1,2 The chemical structure of celastrol is a pentacyclic triterpenoid which is presented in Fig. 1. In recent years, many studies have been carried out on the antioxidant activity of celastrol. These in vitro and in vivo studies have demonstrated that celastrol reduces reactive oxygen species (ROS) production, inhibits nitric oxide generation, increases antioxidant enzyme activity, and enhances glutathione (GSH) levels.3,4
Fig. 1. The molecular structure of celastrol.

The anticancer drug cisplatin is commonly used in the treatment of a wide range of tumors such as in head and neck and bladder cancers. Although it is a very potent and effective chemotherapeutic drug, it causes some adverse effects which can range from mild to severe toxicities including ototoxicity, nephrotoxicity, neurotoxicity, and haematological toxicity.5,6 It has been known for a long time that cisplatin is nephrotoxic to humans and the main dose limiting adverse effect in cisplatin treatment is nephrotoxicity. In fact, nephrotoxicity was seen in 20% of the patients who underwent cisplatin therapy.7,8 The major excretion route for cisplatin is the kidneys. The drug is selectively transported by OCT2 (organic cation transporter 2) and the cisplatin concentration in proximal tubular cells is five times higher than that in blood. This mostly explains why the kidneys are the target of cisplatin toxicity.9,10
The major anticancer activity mechanism of cisplatin is DNA-adduct formation. Cisplatin–DNA adducts can inhibit cellular processes such as transcription, translation and DNA repair.11 Because of DNA damage, DNA replication and cell division are inhibited and the cells with severe and irretrievable DNA damage eventually die.12,13 The genotoxic effect of cisplatin, which is dose and time dependent, may lead to the formation of unrelated tumours years after the cessation of cisplatin therapy.14 Besides genotoxicity, several studies have shown that one of the suggested mechanisms of cisplatin-induced nephrotoxicity is oxidative stress.6,15–17
This study aims to clarify the potential of celastrol in the treatment of cancer. For this purpose, the possible protective effects of celastrol pretreatment against cisplatin-induced genotoxicity and oxidative stress were investigated in a rat kidney epithelial cell line (NRK-52E).
Materials and methods
Chemicals
Dulbecco's Modified Eagle's Medium (DMEM), fetal bovine serum (FBS), penicillin–streptomycin solution, phosphate-buffered saline (PBS) and trypsin–EDTA solution were obtained from Gibco Invitrogen Corp. (UK). HPLC grade celastrol (≥98%) and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (MO, USA). Unless otherwise stated, all other chemicals were of analytical grade and purchased from Sigma-Aldrich (MO, USA).
Dose selection
For decision making purposes, appropriate concentrations of celastrol and cisplatin exposure levels were determined based on published studies in terms of in vitro antioxidant and cytotoxic properties of these compounds, respectively.3,18,19 Several doses of celastrol and cisplatin were tested with the lactate dehydrogenase (LDH) activity test and trypan blue exclusion tests in our pilot prestudies (data not shown) as described in our previous publications.20,21 In order to test the protective effect of celastrol, the doses selected were 200 nM, 100 nM, and 50 nM prior to 50 μM cisplatin treatment which was determined based on the cytotoxicity test results, and all of the determined dose groups had more than 70% viability.
Experimental procedure
For in vitro experiments, celastrol was dissolved in 100% DMSO and diluted with medium to the desired concentrations as described in Table 1 (200 nM, 100 nM, and 50 nM). The vehicle control cells received an equal amount of DMSO (0.5%). The cells were cultured for 24 h for proper attachment before any drug treatment was applied. After the cells were attached in subconfluent matter, the cells were then pretreated with a medium containing the desired concentration of celastrol for 24 h to study the protective effect of celastrol. After 24 h, the cells were treated with 50 μM cisplatin prepared with fresh medium for a further 24 h to see whether cisplatin caused the same or less toxicity compared to the vehicle control group.
Table 1. Treatment groups of the experimental procedure.
| Celastrol pretreatment (nM) | 50 μM cisplatin | |
| Group I (control group) | – | – |
| Group II | – | + |
| Group III | 50 | + |
| Group IV | 100 | + |
| Group V | 200 | + |
Cell culture
Rat kidney epithelial cell line NRK-52E which was purchased from the American Type Culture Collection (ATCC, VA, USA) was grown in DMEM medium supplemented with 10% FBS and 1% penicillin–streptomycin. The cells were maintained at 37 °C in a humidified incubator containing 5% CO2. The medium was changed once every 2–3 days.
Genotoxicity assay
The alkaline comet assay method under in vitro conditions was used in this study and is also described in detail in our previous work.20 For the alkaline comet assay, cells were seeded into a six well plate at a density of 5 × 104 cells per well and the cells were finally suspended at 106 cells per mL for the genotoxicity assessment. The DNA damage was assessed from the percentage of DNA in the tail (TI %) and for this purpose one hundred cells were assessed per slide using a Comet assay IV image analysis system (Perceptive Instruments, UK). The slides were scored using one slide reader, blindly. 25 μM and 50 μM H2O2 was used as the positive control in the alkaline comet assay.
Oxidative stress assays
The imbalance between the free radicals and the antioxidant system components is defined as oxidative stress.22–24 For the purpose of investigating the antioxidant effect of celastrol pretreatment against oxidative stress on NRK-52E cells, the following assays were conducted. The NRK-52E cells were cultured in 75 cm2 flasks at a density of 7.5 × 105 and after the cell treatment with celastrol and cisplatin was completed, the cells were suspended in ice-cold PBS and homogenized by sonication and centrifuged at 15 000g for 10 min at 4 °C. The supernatant was used for antioxidant enzyme activity assays.
Glutathione (GSH) level
GSH is an important antioxidant protein which decreases quantitatively under oxidative stress conditions and increases to prevent the damage caused by free radicals in the cell.25 The GSH content of NRK-52E cells was determined using an enzyme-linked immunosorbent assay (ELISA) kit (Bioassay Technology Laboratory, China) based on the biotin double antibody sandwich technology. The GSH content of NRK-52E cells was measured at 450 nm using a microplate reader, according to the manufacturer's procedure.
Protein carbonyl (PC) level
High levels of protein carbonyl groups have been observed under oxidative stress conditions. Thus, the measurement of the protein carbonylation level can be used as an oxidative stress biomarker.26,27 The protein carbonylation level of NRK-52E cells was determined using the enzyme-linked immunosorbent assay (ELISA) kit (Bioassay Technology Laboratory, China) based on the biotin double antibody sandwich technology. The PC level of NRK-52E cells was measured at 450 nm using a microplate reader, according to the manufacturer's procedure.
Determination of antioxidant enzyme activities
Glutathione peroxidase (GPx) and glutathione reductase (GR) activities were measured using an EnzyChrom™ Glutathione Peroxidase Assay Kit and an EnzyChrom™ Glutathione Reductase Assay Kit (BioAssay systems, USA) according to the manufacturer's protocols. CAT and SOD activities were assayed with the CAT100 Catalase assay kit (Sigma-Aldrich, USA) and 19160-SOD determination kit (Sigma-Aldrich, USA) following the manufacturer's protocols.
Although GSH can reduce the free radicals in an enzyme-free environment, GPx is an enzyme which helps the reaction proceed faster. GPx also catalyzes hydrogen peroxide (H2O2) transformation to water. The GPx activity in this study was evaluated by measuring NADPH consumption spectrophotometrically at 340 nm for 4 min, which is proportional to the GPx activity in the sample.28 Briefly, 90 μl of the working reagent was added to 10 μl of the sample/control and mixed. The substrate solution was pipetted into sample and control wells, and then the plate was immediately read at 340 nm.
GR is an enzyme which maintains the cellular GSH pool by reducing the oxidized form of GSH to its antioxidant state. The GR activity assay is based on the measurement of the absorbance change caused by the reduction in DTNB [5,50-dithiobis(2-nitrobenzoic acid)] at 412 nm. DTNB reacts with reduced GSH and forms a yellow product. The rate of the change in the optical density is directly proportional to the GSH concentration in the sample.29 Briefly, 80 μl of the working reagent was added to 20 μl of each sample and mixed. The plate was read at 412 nm at the end of 10 and 30 minutes of incubation.
H2O2, which is highly harmful to cells, is a by-product of various oxidase and superoxide dismutase reactions. CAT, a peroxisomal antioxidant enzyme, provides protection against oxidative damage to cells by catalyzing the reaction of H2O2 transformation to water and oxygen.22 In this study, the CAT activity was measured using spectrophotometry. The measurement was based on the decomposition rate of H2O2 for 5 minutes, which is proportional to the CAT activity in the sample.30 The absorbance was measured at 520 nm using a Shimadzu UV 1800 spectrophotometer (Shimadzu, Japan) in accordance with the kit protocol. Briefly, the samples of the protein were diluted to 2 mg ml–1. Assay buffer and colorimetric assay substrate solution were added to the samples and mixed well. At the end of 1–5 minutes of incubation, a stop solution was added and mixed. A color reagent was added into the mixture. After 15 minutes of incubation at room temperature, the plate was read at 520 nm.
Lastly, superoxide is a free radical produced via oxygen metabolism and if not eliminated it may cause oxidative stress and thus cellular damage. The antioxidant enzyme SOD catalyzes the dismutation of the superoxide radical into molecular oxygen or H2O2 to protect the cell. The SOD activity assay is based on the spectrometric measurements of the reduction in Dojindo's water-soluble tetrazolium salt (WST-1) by the superoxide anion at 450 nm, resulting in a colorful water-soluble formazan dye. SOD eliminates the superoxide anion; therefore, the IC50 value can be determined by a colorimetric method.31 For the determination of SOD activity, 200 μl of working solution and 20 μl of enzyme working solution were added into 20 μl of the sample and mixed. After incubation at 37 °C for 20 minutes, the plate was read at 450 nm using a microplate reader.
Statistical analysis
All of the assays were performed in triplicate for three separate experiments. The results were given as the mean ± SD and group mean comparisons were done by the one-way ANOVA test followed by the Tukey's test using SPSS software (SPSS Inc, USA).
Results
Effects of celastrol pretreatment on cisplatin-induced genotoxicity in NRK-52E cells
Appropriate doses for alkaline comet assay were determined for assessing DNA damage according to the cytotoxicity test results. The cell viability was found to be higher than 70% with the trypan blue exclusion test in dose groups when checked prior to the alkaline comet assay. Alkaline comet assay results are summarized in Fig. 2. According to our results 50 μM cisplatin treatment (Group II) caused statistically increased DNA damage (p < 0.05) compared to the negative control (Group I) in NRK-52E cells. In order to understand the protective effects of celastrol, cisplatin-exposed cells were pretreated with celastrol with concentrations of 50, 100 and 200 nM. Both 100 nM and 200 nM celastrol pretreatments caused statistically significant reduction (p < 0.05) in DNA damage compared to the solely cisplatin induced group (Group II).
Fig. 2. Alkaline comet assay results of NRK-52E cells pretreated with celastrol (*p < 0.05, compared to Group I; #p < 0.05, compared to Group II). SD: standard deviation.

Effects of cisplatin and celastrol on PC and GSH levels in NRK-52E cells
The carbonylated protein level dramatically increased in Group II (the 50 μM cisplatin exposure group). Celastrol pretreatment significantly reduced the PC level in Group III compared to that in Group II (Fig. 3).
Fig. 3. Effects of celastrol pretreatment on (a) the PC level and (b) the GSH level of NRK-52E cells (*p < 0.05, compared to Group I; #p < 0.05, compared to Group II). (a) PC: protein carbonyl, (b) GSH: glutathione and SD: standard deviation.

The GSH content of the cells significantly decreased after 50 μM cisplatin exposure. When compared to Group II, celastrol pretreatment significantly improved the reduced GSH level at the studied celastrol concentrations and Group IV (100 nM celastrol) was the most effective group in this respect, as shown by the increase in the GSH level of the cells (Fig. 3).
Effects of cisplatin and celastrol on antioxidant enzyme activities in NRK-52E cells
The CAT activity decreased after 50 μM cisplatin exposure. As shown in Fig. 4, celastrol treatment before cisplatin exposure significantly attenuated the decreased CAT activity in Groups III and IV (50 nM and 100 nM celastrol concentrations) compared to Group II (only cisplatin treated cells). 50 μM cisplatin treatment caused a significant reduction in the GR activity. Celastrol pretreatment increased the reduced GR activity at all studied celastrol concentrations compared to Group II. However, GPx and SOD enzyme activities did not show any significant change in either Group II or the celastrol pretreatment groups.
Fig. 4. Effects of celastrol pretreatment on (a) CAT, (b) GR (c) GPx and (d) SOD levels of NRK-52E cells (*p < 0.05, compared to Group I; #p < 0.05, compared to Group II). CAT: catalase, GPx: glutathione peroxidase, GR: glutathione reductase, SOD: superoxide dismutase, and SD: standard deviation.

Discussion
Celastrol which is found in the extract of the Chinese medicinal plant T. wilfordii Hook F. has started to draw attention due to its possible therapeutic effects.32–36 Previous studies have shown that celastrol has an antioxidant activity37,38 and a cytoprotective effect.39 On the other hand, it has been stated that celastrol has anticancer effects inhibiting the growth of tumour cells at higher doses than those selected in this study.40,41 Apart from these effects, it was demonstrated that celastrol has an ameliorative effect on mitochondrial dysfunction in different cell lines.42–44 Besides determining the mitochondrial ameliorative function of celastrol, it has also been demonstrated under in vitro conditions that celastrol pretreatment attenuated cisplatin-induced nephrotoxicity by alleviating apoptosis at a 50 nM celastrol concentration on mouse renal tubule and human proximal tubule epithelial cell lines. These protective effects were also determined at a dose of 1 mg kg–1 celastrol on mice under in vivo conditions. The mechanism of this effect is demonstrated to be through antagonizing NF-κB-mediated inflammation and also by improving mitochondrial functions.44 In addition, it has been shown that celastrol post-treatment led to a recovery in acetaminophen-induced toxicity on HepG2 cells.45
It has been stated that celastrol exhibits anticancer effects by inhibiting the growth of tumor cells.40,41 Similar to our results, it has been stated that celastrol shows a protective effect against γ irradiation-induced oxidative stress and DNA damage.38 Besides its protective effect on DNA, celastrol has been shown to sensitize cancer cells to ionizing radiation and DNA-cross-linking agents at higher doses than those selected in this study.46–48 Considering the studied positive effects of celastrol, it has been thought that celastrol can be used to alleviate the adverse effects of antineoplastic drugs and sensitize tumor cells to anticancer agents in combination or supportive therapies. Although there are data on the use of celastrol in the treatment of cancer, we determined its protective effects against cisplatin-induced nephrotoxicity. These dual roles may increase its potential for clinical usage. However, there is a need for further studies and clinical data in order to clarify the possible effects of celastrol in cancer patients. Celastrol–anticancer drug interactions and its selectivity on damaged kidney cells are also important parameters to study and clarify.
Cisplatin is a widely used drug in cancer chemotherapy and it is a very effective treatment against a broad spectrum of tumours.49 While showing anticancer activity, cisplatin also affects kidney cells adversely, causing nephrotoxicity and this can lead to dose reduction or discontinuation of treatment.50–53 One of the important underlying mechanisms of cisplatin-induced nephrotoxicity has been shown to be DNA damage.54,55 The DNA damage inhibits DNA replication and thus cell division is inhibited, which eventually leads to apoptosis. The genotoxicity of cisplatin can be a major cause of treatment discontinuation.54 Our study is the first in vitro study focusing on the protective effect of celastrol on NRK-52E cells against cisplatin-induced nephrotoxicity with respect to oxidative stress and DNA damage. We found that celastrol led to a statistically significant reduction in DNA damage which was induced by cisplatin. These results show us that celastrol can be an effective herbal treatment against cisplatin-induced genotoxicity during cancer therapy.
It has been known that cisplatin nephrotoxicity can be associated with oxidative stress involving an increase in the production of ROS, a decrease in antioxidant enzyme activity and depletion of GSH levels.56 There are many studies which focus on the amelioration of or protection from cisplatin-induced nephrotoxicity by antioxidant mechanisms.57–60 However, these therapies have not been approved yet and new studies to find the optimal agent are certainly needed. There are also comprehensive studies particularly for finding new therapeutic antioxidant substances of herbal origin which focus on protecting from cisplatin-induced nephrotoxicity.53,61
As an early marker of oxidative stress, protein carbonylation is commonly considered convenient.62 It has been reported that cisplatin increases the carbonylated protein level in cells.15 According to our results, after cisplatin exposure, carbonylation of proteins increased significantly. Moreover, it was seen that celastrol treatment at a 50 nM concentration before cisplatin exposure (Group III) decreased the carbonylated protein level in these cells. These results are in parallel with a study in which celastrol has been demonstrated to alleviate the carbonylated protein level in NRK-52E cells.42
In the antioxidant defense system (Fig. 5), GSH, which is synthesized and found in the cells of the body, plays crucially important roles. Under normal cellular conditions, GSH is found in its reduced form. When oxidative stress occurs, GSH gets oxidized and transforms into its oxidized form (GSSG). This helps maintain the cellular redox balance.63,64 There are many studies showing that cisplatin decreases the GSH content of the cells.65–68 According to our results, it was found that cisplatin leads to the depletion of the GSH content. It has also been stated in other studies that celastrol can enhance the GSH level.18,69 Our study revealed that celastrol pretreatment increases the GSH content of the cells at a 50 nM concentration.
Fig. 5. Schematic illustration of antioxidant systems in the cells. GPx: glutathione peroxidase, GR: glutathione reductase, SOD: superoxide dismutase, and CAT: catalase.

SOD, GPx, CAT, and GR are antioxidant enzymes which are essential for protection from oxidative stress in cells (Fig. 5). SOD turns the superoxide radicals into H2O2. H2O2 is one of the most important reactive oxygen species which is converted to H2O and O2 by GPx and CAT enzymes. During this reaction, GPx uses the reduced form of GSH and turns it into its oxidized form.70 GR catalyzes the reduction of the oxidized form and this enzyme is responsible for the supplementation of reduced GSH which manages the oxidant/antioxidant balance in healthy tissues.71 There are studies reporting that cisplatin decreases the antioxidant enzyme activity65,72,73 whilst celastrol has been demonstrated to increase the antioxidant enzyme activity.45,69,74
In the present study, it was seen that cisplatin exposure did not change GPx and SOD activities whereas GR and CAT activities decreased significantly with cisplatin exposure. Celastrol pretreatment increased the GR activity at all studied celastrol concentrations and increased the CAT activity at 100 nM and 200 nM celastrol concentrations (Groups IV and V). However, GPx and SOD enzyme activities did not significantly change with celastrol pretreatment.
Based on these results it is possible to say that celastrol pretreatment has a positive impact on the antioxidant enzyme activity and can be used prior to cisplatin treatment in order to mitigate the unintended effects of cisplatin. Nevertheless, celastrol's protective effect has also been studied when administered simultaneously with cisplatin treatment.44
Conclusion
It was seen that celastrol pretreatment provided significant DNA protection from cisplatin-induced genotoxicity in NRK-52E cells. Celastrol pretreatment also increased the GSH content of the cells and ameliorated the protein carbonylation level, enabled by the antioxidant properties of celastrol. Likewise, celastrol pretreatment improved the GR and CAT activities as shown in Fig. 6. In the light of these findings, celastrol pretreatment could be a therapeutic option to reduce cisplatin-induced nephrotoxicity. However, further investigation using other kidney cell lines and animal models and studies on humans are required.
Fig. 6. Possible protective mechanisms of celastrol against cisplatin-induced nephrotoxicity.

Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgments
This study was supported by the Scientific Research Project Coordination Unit of Istanbul University. Project number: BEK-2017-26079.
References
- Allison A. C., Cacabelos R., Lombardi V. R. M., Álvarez X. A., Vigo C. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2001;25:1341–1357. doi: 10.1016/s0278-5846(01)00192-0. [DOI] [PubMed] [Google Scholar]
- Venkatesha S. H., Dudics S., Astry B., Moudgil K. D. Pathog. Dis. 2016;74:1–12. doi: 10.1093/femspd/ftw059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Zhao H., Lobo N., Guo X., Gentleman S. M., Ma D. J. Alzheimer's Dis. 2014;41:835–844. doi: 10.3233/JAD-131799. [DOI] [PubMed] [Google Scholar]
- Li C., Li J., Fan J., Meng L., Cao L. Chin. J. Cell. Mol. Immunol. 2017;33:1328–1334. [PubMed] [Google Scholar]
- El-Awady E.-S. E., Moustafa Y. M., Abo-Elmatty D. M., Radwan A. Eur. J. Pharmacol. 2011;650:335–341. doi: 10.1016/j.ejphar.2010.09.085. [DOI] [PubMed] [Google Scholar]
- Yao X., Panichpisal K., Kurtzman N., Nugent K. Am. J. Med. Sci. 2007;334:115–124. doi: 10.1097/MAJ.0b013e31812dfe1e. [DOI] [PubMed] [Google Scholar]
- Peres L. A. B., da Cunha Júnior A. D. J. Bras. Nefrol. 2013;35:332–340. doi: 10.5935/0101-2800.20130052. [DOI] [PubMed] [Google Scholar]
- Goldstein R. S., Mayor G. H. Life Sci. 1983;32:685–690. doi: 10.1016/0024-3205(83)90299-0. [DOI] [PubMed] [Google Scholar]
- Ciarimboli G., Ludwig T., Lang D., Pavenstädt H., Koepsell H., Piechota H.-J., Haier J., Jaehde U., Zisowsky J., Schlatter E. Am. J. Pathol. 2005;167:1477–1484. doi: 10.1016/S0002-9440(10)61234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann M., Burkhardt G., Kohler H. Nephrol., Dial., Transplant. 1997;12:2478–2480. doi: 10.1093/ndt/12.12.2478. [DOI] [PubMed] [Google Scholar]
- Wozniak K., Czechowska A., Blasiak J. Chem.-Biol. Interact. 2004;147:309–318. doi: 10.1016/j.cbi.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Jamieson E. R., Lippard S. J. Chem. Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- Pabla N., Dong Z. Kidney Int. 2008;73:994–1007. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- Brozovic G., Orsolic N., Knezevic F., Horvat Knezevic A., Benkovic V., Sakic K., Borojevic N., Dikic D. J. Appl. Genet. 2011;52:355–361. doi: 10.1007/s13353-011-0046-0. [DOI] [PubMed] [Google Scholar]
- Santos N. A. G., Catão C. S., Martins N. M., Curti C., Bianchi M. L. P., Santos A. C. Arch. Toxicol. 2007;81:495–504. doi: 10.1007/s00204-006-0173-2. [DOI] [PubMed] [Google Scholar]
- Somani S. M., Husain K., Whitworth C., Trammell G. L., Malafa M., Rybak L. P. Pharmacol. Toxicol. 2008;86:234–241. doi: 10.1034/j.1600-0773.2000.d01-41.x. [DOI] [PubMed] [Google Scholar]
- Gumulec J., Balvan J., Sztalmachova M., Raudenska M., Dvorakova V., Knopfova L., Polanska H., Hudcova K., Ruttkay-Nedecky B., Babula P., Adam V., Kızek R., Stiborova M., Masarık M. Int. J. Oncol. 2014;44:923–933. doi: 10.3892/ijo.2013.2223. [DOI] [PubMed] [Google Scholar]
- Deng Y.-N., Shi J., Liu J., Qu Q.-M. Neurochem. Int. 2013;63:1–9. doi: 10.1016/j.neuint.2013.04.005. [DOI] [PubMed] [Google Scholar]
- He Y., Zhu Q., Chen M., Huang Q., Wang W., Li Q., Huang Y., Di W. Oncotarget. 2016;7:70803–70821. doi: 10.18632/oncotarget.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcagli E., Alpertunga B., Fenga C., Berktas M., Tsitsimpikou C., Wilks M. F., Tsatsakis A. M. Food Chem. Toxicol. 2016;89:1–7. doi: 10.1016/j.fct.2015.12.027. [DOI] [PubMed] [Google Scholar]
- Demirel G., Alpertunga B., Ozden S., Pezzuto J. M. Pharm. Biol. 2015;53:1302–1310. doi: 10.3109/13880209.2014.976714. [DOI] [PubMed] [Google Scholar]
- Auten R. L., Davis J. M. Pediatr. Res. 2009;66:121–127. doi: 10.1203/PDR.0b013e3181a9eafb. [DOI] [PubMed] [Google Scholar]
- Koch K., Havermann S., Büchter C., Wätjen W. Sci. World J. 2014;2014:1–15. [Google Scholar]
- Burton G. J., Jauniaux E. Best Pract. Res., Clin. Endocrinol. Metab. 2011;25:287–299. doi: 10.1016/j.bpobgyn.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lushchak V. I. J. Amino Acids. 2012;2012:1–26. doi: 10.1155/2012/736837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle-Donne I., Rossi R., Giustarini D., Milzani A., Colombo R. Clin. Chim. Acta. 2003;329:23–38. doi: 10.1016/s0009-8981(03)00003-2. [DOI] [PubMed] [Google Scholar]
- Suzuki Y. J., Carini M., Butterfield D. A. Antioxid. Redox Signaling. 2010;12:323–325. doi: 10.1089/ars.2009.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedaghatfard F., Razavi S. A., Hedayati M., Razmi N., Rahnama A. Eur. Online J. Nat. Soc. Sci. 2016;5:15–21. [Google Scholar]
- Mannervik B. Curr. Protoc. Toxicol. 1999:7.2.1–7.2.4. [Google Scholar]
- Shangari N. and O'Brien P. J., in Current Protocols in Toxicology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2006, vol. 27, p. 7.7.1–7.7.16. [DOI] [PubMed] [Google Scholar]
- Peskin A. V., Winterbourn C. C. Clin. Chim. Acta. 2000;293:157–166. doi: 10.1016/s0009-8981(99)00246-6. [DOI] [PubMed] [Google Scholar]
- Tang M., Cao X., Zhang K., Li Y., Zheng Q., Li G., He Q., Li S., Xu G., Zhang K. Cell Death Dis. 2018;9:601. doi: 10.1038/s41419-018-0666-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boridy S., Le P. U., Petrecca K., Maysinger D. Cell Death Dis. 2014;5:e1216–e1216. doi: 10.1038/cddis.2014.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhill C. Nat. Rev. Endocrinol. 2018;14:626–626. doi: 10.1038/s41574-018-0093-2. [DOI] [PubMed] [Google Scholar]
- Kuchta K., Xiang Y., Huang S., Tang Y., Peng X., Wang X., Zhu Y., Li J., Xu J., Lin Z., Pan T. Prostate Cancer Prostatic Dis. 2017;20:156–164. doi: 10.1038/pcan.2016.61. [DOI] [PubMed] [Google Scholar]
- Kashyap D., Sharma A., Tuli H. S., Sak K., Mukherjee T., Bishayee A. Crit. Rev. Oncol. Hematol. 2018;128:70–81. doi: 10.1016/j.critrevonc.2018.05.019. [DOI] [PubMed] [Google Scholar]
- Choi B.-S., Kim H., Lee H. J., Sapkota K., Park S. E., Kim S., Kim S.-J. Neurochem. Res. 2014;39:84–96. doi: 10.1007/s11064-013-1193-y. [DOI] [PubMed] [Google Scholar]
- Han X., Tan Y., Fang Y., Li F. Exp. Ther. Med. 2018;16:685–694. doi: 10.3892/etm.2018.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide S. D., Bosman J. D., Mbadugha B. N. A., Kawahara T. L. A., Matsumoto G., Kim S., Gu W., Devlin J. P., Silverman R. B., Morimoto R. I. J. Biol. Chem. 2004;279:56053–56060. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- Chen G., Zhang X., Zhao M., Wang Y., Cheng X., Wang D., Xu Y., Du Z., Yu X. BMC Cancer. 2011;11:170. doi: 10.1186/1471-2407-11-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava S., Jeengar M. K., Reddy V. S., Reddy G. B., Naidu V. G. M. Exp. Mol. Pathol. 2015;98:313–327. doi: 10.1016/j.yexmp.2015.03.031. [DOI] [PubMed] [Google Scholar]
- Bakar M., Sarmidi M., Kai C., Huri H., Yaakob H., Bakar M. H. A., Sarmidi M. R., Kai C. K., Huri H. Z., Yaakob H. Int. J. Mol. Sci. 2014;15:22227–22257. doi: 10.3390/ijms151222227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu Bakar M. H., Tan J. S. Biomed. Pharmacother. 2017;93:903–912. doi: 10.1016/j.biopha.2017.07.021. [DOI] [PubMed] [Google Scholar]
- Yu X., Meng X., Xu M., Zhang X., Zhang Y., Ding G., Huang S., Zhang A., Jia Z. EBioMedicine. 2018;36:266–280. doi: 10.1016/j.ebiom.2018.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jannuzzi A., Kara M., Alpertunga B. Hum. Exp. Toxicol. 2018;37:742–751. doi: 10.1177/0960327117734622. [DOI] [PubMed] [Google Scholar]
- Lo Iacono M., Monica V., Vavalà T., Gisabella M., Saviozzi S., Bracco E., Novello S., Papotti M., Scagliotti G. V. Int. J. Cancer. 2015;136:2598–2609. doi: 10.1002/ijc.29302. [DOI] [PubMed] [Google Scholar]
- Wang G.-Z., Liu Y.-Q., Cheng X., Zhou G.-B. Cancer Sci. 2015;106:902–908. doi: 10.1111/cas.12679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y., Desano J. T., Meng Y., Ji Q., Lawrence T. S., Xu L. Int. J. Radiat. Oncol., Biol., Phys. 2009;74(4):1217–1225. doi: 10.1016/j.ijrobp.2009.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kart A., Cigremis Y., Karaman M., Ozen H. Exp. Toxicol. Pathol. 2010;62:45–52. doi: 10.1016/j.etp.2009.02.066. [DOI] [PubMed] [Google Scholar]
- McKeage M. J. Drug Saf. 1995;13:228–244. doi: 10.2165/00002018-199513040-00003. [DOI] [PubMed] [Google Scholar]
- Mishra J., Mori K., Ma Q., Kelly C., Barasch J., Devarajan P. Am. J. Nephrol. 2004;24:307–315. doi: 10.1159/000078452. [DOI] [PubMed] [Google Scholar]
- Rabik C. A., Dolan M. E. Cancer Treat. Rev. 2007;33:9–23. doi: 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirino Y. I., Sánchez-González D. J., Martínez-Martínez C. M., Cruz C., Pedraza-Chaverri J. Toxicology. 2008;245:18–23. doi: 10.1016/j.tox.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Dasari S., Bernard Tchounwou P. Eur. J. Pharmacol. 2014;740:364–378. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S., Pabla N., Tang C., He L., Dong Z. Arch. Toxicol. 2015;89:2197–2205. doi: 10.1007/s00204-015-1633-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-González P. D., López-Hernández F. J., López-Novoa J. M., Morales A. I. Crit. Rev. Toxicol. 2011;41:803–821. doi: 10.3109/10408444.2011.602662. [DOI] [PubMed] [Google Scholar]
- Jung H. W., Chung Y. S., Kim Y. S., Park Y.-K. Exp. Mol. Med. 2007;39:715–721. doi: 10.1038/emm.2007.78. [DOI] [PubMed] [Google Scholar]
- Knight R. J., Collis M. G., Yates M. S., Bowmer C. J. Br. J. Pharmacol. 1991;104:1062–1068. doi: 10.1111/j.1476-5381.1991.tb12550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Jin H., Sun H., Zhang Z., Zheng J., Li S., Han S. Eur. J. Pharmacol. 2016;772:124–130. doi: 10.1016/j.ejphar.2015.12.042. [DOI] [PubMed] [Google Scholar]
- Saleh S., El-Demerdash E. Basic Clin. Pharmacol. Toxicol. 2005;97:91–97. doi: 10.1111/j.1742-7843.2005.pto_114.x. [DOI] [PubMed] [Google Scholar]
- Wu F., Li Y., Song H., Zhang Y., Zhang Y., Jiang M., Wang F., Mu Q., Zhang W., Li L., Tang D. Evidence-Based Complementary Altern. Med. 2016;2016:1–9. [Google Scholar]
- Sassa H., Takaishi Y., Terada H. Biochem. Biophys. Res. Commun. 1990;172:890–897. doi: 10.1016/0006-291x(90)90759-g. [DOI] [PubMed] [Google Scholar]
- Kerksick C., Willoughby D. J. Int. Soc. Sports Nutr. 2005;2:38. doi: 10.1186/1550-2783-2-2-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Raheem I. T., Abdel-Ghany A. A., Mohamed G. A. Biol. Pharm. Bull. 2009;32:61–67. doi: 10.1248/bpb.32.61. [DOI] [PubMed] [Google Scholar]
- Almaghrabi O. A. Saudi J. Biol. Sci. 2015;22:227–231. doi: 10.1016/j.sjbs.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badary O. A., Abdel-Maksoud S., Ahmed W. A., Owieda G. H. Life Sci. 2005;76:2125–2135. doi: 10.1016/j.lfs.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Durak İ., Özbek H., Karaayvaz M., Öztürk H. S. Drug Chem. Toxicol. 2002;25:1–8. doi: 10.1081/dct-100108468. [DOI] [PubMed] [Google Scholar]
- Santos N. A. G., Bezerra C. S. C., Martins N. M., Curti C., Bianchi M. L. P., Santos A. C. Cancer Chemother. Pharmacol. 2007;61:145–155. doi: 10.1007/s00280-007-0459-y. [DOI] [PubMed] [Google Scholar]
- Divya T., Dineshbabu V., Soumyakrishnan S., Sureshkumar A., Sudhandiran G. Chem.-Biol. Interact. 2016;246:52–62. doi: 10.1016/j.cbi.2016.01.006. [DOI] [PubMed] [Google Scholar]
- MatÉs J. M., Pérez-Gómez C., De Castro I. N. Clin. Biochem. 1999;32:595–603. doi: 10.1016/s0009-9120(99)00075-2. [DOI] [PubMed] [Google Scholar]
- Couto N., Wood J., Barber J. Free Radicals Biol. Med. 2016;95:27–42. doi: 10.1016/j.freeradbiomed.2016.02.028. [DOI] [PubMed] [Google Scholar]
- Sahu B. D., Kuncha M., Sindhura G. J., Sistla R. Phytomedicine. 2013;20:453–460. doi: 10.1016/j.phymed.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Valentovic M. A., Ball J. G., Mike Brown J., Terneus M. V., McQuade E., Van Meter S., Hedrick H. M., Roy A. A., Williams T. Toxicol. In Vitro. 2014;28:248–257. doi: 10.1016/j.tiv.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaker M. E., Ashamallah S. A., Houssen M. E. Chem.-Biol. Interact. 2014;210:26–33. doi: 10.1016/j.cbi.2013.12.007. [DOI] [PubMed] [Google Scholar]