

Abstract
At nondepressed Aplysia sensory to motor synapses, serotonin (5-HT) facilitates transmitter release primarily through a protein kinase A pathway. In contrast, at depressedAplysia sensory to motor synapses, 5-HT facilitates transmitter release primarily through a protein kinase C (PKC)-dependent pathway. It is known that only two phorbol ester-activated PKC isoforms, the Ca2+-dependent PKC Apl I and the Ca2+-independent PKC Apl II, exist in the Aplysia nervous system. For the first time, we have now been able to functionally determine which isoform of PKC is involved in a particular form of plasticity. We microinjected cultured sensorimotor pairs of neurons with various PKC constructs tagged with the enhanced green fluorescent protein as a reporter for successful plasmid expression. Our results demonstrate that short-term facilitation of depressed synapses is mediated by PKC Apl II. Dominant-negative PKC Apl II, but not dominant-negative PKC Apl I, disrupted the normal kinetics of 5-HT-induced facilitation by completely blocking its rapid onset. This effect was specific to depressed synapses, because dominant-negative PKC Apl II did not inhibit 5-HT-mediated facilitation of nondepressed synapses. Our results suggest that not only different signal transduction pathways but also different isoforms of a specific cascade may mediate physiological responses according to the state of a synapse.
Keywords: Aplysia, protein kinase C, memory, synaptic facilitation, serotonin, plasmids
In Aplysia, the effect of the facilitating neurotransmitter serotonin (5-HT) on sensorimotor (SM) synapses is mediated through multiple kinase pathways that become involved at different times according to the state of the neurons (Ghirardi et al., 1992; Klein, 1995; Byrne and Kandel, 1996). At nondepressed synapses, spike broadening can contribute to transmitter release. In this case, activation of PKA is critical for 5-HT-induced facilitation (Ghirardi et al., 1992). At depressed Aplysiasynapses, pools of releasable neurotransmitter vesicles are depleted (Gingrich and Byrne, 1985; Bailey and Chen, 1988; Zhao and Klein, 2000) (but see Eliot et al., 1994), and spike broadening is not effective at enhancing secretion (Hochner et al., 1986). In this case, activation of protein kinase C becomes critical for 5-HT-induced facilitation (Braha et al., 1990; Sacktor and Schwartz, 1990; Ghirardi et al., 1992).
Protein kinase Cs are a large family of serine–threonine kinases with a ubiquitous role in cellular responses (Nishizuka, 1988). Twelve PKC isoforms have been identified in mammals, and each of them may act in specific cell transduction pathways (Mochly-Rosen and Gordon, 1998). Signaling pathways through PKCs regulate many aspects of neuronal function and development, including ion channels, neurotransmitter receptors, cytoskeletal dynamics, and both short-term and long-term plasticity (Tanaka and Nishizuka, 1994). Most strikingly, PKCs appear to be ubiquitously involved in the modulation of neurotransmitter release (Malenka et al., 1986; Shapira et al., 1987; Ghirardi et al., 1992; Sánchez-Prieto et al., 1996).
Only two phorbol ester-activated PKCs exist in the Aplysianervous system (Sossin et al., 1993). These two isoforms, the Ca2+-dependent PKC Apl I and the Ca2+-independent PKC Apl II (Kruger et al., 1991), show distinct patterns of activation. Purified PKC Apl I is activated at lower concentrations of phosphatidylserine (PS) than PKC Apl II, and this occurs because of the distinct C2 domains of the kinases (Sossin et al., 1996b; Pepio and Sossin, 1998; Pepio et al., 1998). Consistent with these properties of purified PKCs, PKC Apl I is translocated to membranes rapidly by physiological activators, whereas persistent presence of transmitter is required to translocate PKC Apl II (Sossin and Schwartz, 1992; Sossin et al., 1994, 1996a). Thus, it was suggested that short-term events mediated by PKC, such as the synaptic facilitation of depressed synapses, would be mediated by PKC Apl I, whereas events that require longer incubations with neuromodulators, such as intermediate-term facilitation (Ghirardi et al., 1995), or those that are seen with phorbol ester, such as facilitation of naive synapses (Sugita et al., 1997a) and increases in excitability (Sugita et al., 1997b; Manseau et al., 1998), could be mediated by PKC Apl II (Sossin and Schwartz, 1992).
In this study, we used electrophysiological techniques to directly examine the functional role of PKC Apl I and PKC Apl II in short-term facilitation (STF) of depressed SM synapses. The identification of distinct functions for these kinases was limited until now by a lack of selective pharmacological inhibitors. Here, we transfectedAplysia neurons with various enhanced green fluorescent protein (EGFP)-PKC hybrid constructs. We found that overexpressing a mutant form of PKC Apl II, but not PKC Apl I, specifically inhibited 5-HT-induced recovery of depressed SM synapses, which strongly suggests that this form of plasticity is mediated by PKC Apl II.
MATERIALS AND METHODS
Plasmid construction
Wild-type EGFP-PKCs. Full-length clones of PKC Apl I and PKC Apl II were present in pBluescript SK (Invitrogen, Carlsbad, CA) and baculovirus vectors (Sossin et al., 1996b). Initially, hemagglutinin (HA) tags were attached to the 5′ end of the PKCs. PKC Apl I was excised from pBluescript SK withSacI, cut back with Klenow (Promega, Madison, WI) to make a blunt end, and inserted into the SmaI site of the HA vector (gift from J. Ngsee, University of Ottawa, Ottawa, Canada). PKC Apl II was excised from pBluescript SK withEcoRI, filled in with Klenow, and inserted into theStuI site of the HA vector. These constructs were then excised from the HA vector with KpnI and SacI and inserted into pNEX-3 (gift from B. Kaang, Seoul National University, Seoul, South Korea) cut with the same enzymes. The HA-PKC Apl I was cut with SpeI, filled in with Klenow to create a blunt end, excised with XhoI, and then inserted into the baculovirus transfer vector BB4 (Invitrogen), which had been cut withEcoRI, filled in to create a blunt end, and then cut withXhoI. Similarly, HA-PKC Apl II was excised usingXhoI/EcoRI and inserted into BB4 cut with the same enzymes. EGFP-NI (Clontech, Palo Alto, CA) was amplified by PCR (primers in Table 1) and inserted into the 5′ end of PKC Apl I and PKC Apl II using XhoI. TheXhoI site was then cut and filled in to create the correct reading frame. The EGFP-PKCs were then excised with SacI and inserted into BB4 cut with SacI. All clonings were confirmed by sequencing over insertion sites.
Table 1.
List of primers for PCRs
| Construct name | Primers | Cloning sites | Introduced sites |
|---|---|---|---|
| Apl I K349-R | O5 5′-GAGCTTCACAGTAGACATCGG | BsmI | BstBI |
| I3 5′-GCAATTCGAATCTTGAAGAAGGACG | BstEII | ||
| I5 5′-GCAATTCGAATCTTGAAGAAGGACG | |||
| O3 5′-CAATGCCTTGAGTATGGAGG | |||
| Apl II K432-R | O5 5′-AGTGAGTCCCATGAGTCCCC | NheI | MluI |
| I3 5′-AACGCGTATAGCATATACTTCATCTGTCC | SstI | ||
| I5 5′-TACGCGTTTTGAAAAGGATGTG | |||
| O3 5′-TGAACATGAGATCTCCTCC | |||
| Apl II T706-E | O5 5′-CAGAGGACCCTGTCCTGGAACCGGTGGATCCGG | Eco0109I | AgeI |
| NotI | |||
| O3 T3 primer | |||
| Apl II S725-E | O5 5′-TCCCCGCGGCTTCGAATTTGCCAATCCAGACTACG | SacII | BstB |
| O3 T3 primer | |||
| EGFP–PKCs | O5 5′-CAGAGCTGGTTTAGTGAACCG | XhoI | |
| O3 5′-CCCTCGAGGCTTGTACAGCTCGTCCATG | |||
| PKC Apl I C2 | O5 5′-GGGAATTCGAGAAGAACGTGCCCT | KpnI | |
| O3 5′-GGGGTACCTTAGCTCTCTGTGATGTCA | EcoRI | ||
| PKC Apl II C2 | O5 5′-GGGAATTCGCTTCAATGTCGCGGAGG |
KpnI
EcoRI |
|
| O3 5′-GGGGTACCTTACACTTTCTCCTTTTGTGGAC |
Dominant-negative PKCs. Lysine to arginine (K-R) mutations were generated with a two-step mutagenic procedure by PCR. First-round PCR used the PKC Apl I or PKC Apl II cDNA in pBluescript SK (Invitrogen) as a template and either an outside 5′ primer (O5) and an inside 3′ primer (I3) or an inside 5′ primer (I5) and an outside 3′ primer (O3) (for primers, see Table 1). The products from the first-round synthesis were combined and used as the template for second-round synthesis using O5 and O3. The resultant product was cut with appropriate enzymes (Table 1) and inserted into PKCs in pBluescript SK. A new site was formed by the mutagenesis (Table 1) and was used to confirm the cloning. A HA tag was then added to the 5′ end of PKC Apl I and PKC Apl II as described above for the wild-type PKCs. Subsequent conversion of the conserved phosphorylated sites to glutamic acid was done as described for PKC Apl I (Nakhost et al., 1999) or by taking advantage of pre-existing restriction sites to mutagenize the sites for PKC Apl II with PCR (Table 1). Clones were inserted into either the BB4 vector or pNEX-3 as described above for wild-type PKCs. EGFP was inserted at the 5′ ends of the PKCs as described above for wild-type PKCs in pNEX-3, and then EGFP-PKCs were inserted into BB4 as described above. The clones were sequenced over the entire amplified region when PCR was used to confirm that no additional changes were made.
C2 domain constructs. The C2 domains were amplified with PCR (Table 1) and inserted into EGFP-C1 (Clontech) using KpnI and EcoRI. The boundaries for the C2 domains were identical to those used in earlier glutathione S-transferase and MBP-C2 domain constructs (Sossin et al., 1996b; Pepio and Sossin, 1998; Pepio et al., 1998). EGFP-PKC Apl I C2 and EGFP-PKC Apl II C2 were excised from the Clontech vector with NheI andKpnI and inserted into pNEX-3 at XhoI andKpnI and into BB4 with NheI andKpnI. The clones were sequenced over the entire amplified region when PCR was used to confirm that no changes were made.
Expression in baculovirus
Recombination of the transfer vectors into baculovirus and generation of high-titer baculovirus stocks were performed as described previously (Sossin et al., 1996b). To characterize the activity of PKCs, Sf9 or Sf21 cells were infected at a multiplicity of infection of 5 for 3 d. Extracts of Sf9 or Sf21 cells were made as described previously and assayed directly after dilution or after initial purification over a DEAE column (Sossin et al., 1996b). Separation into supernatant and pellet fractions and translocation of C2 domains was as described previously (Sossin et al., 1996b).
PKC kinase assay
The reaction mixture (30 μl) contained 50 mmTris-HCl, pH 7.5, 10 mm MgCl2, 5 mm EGTA, and 2 μm Aε-pep (LNRRRGSMRRRVHQVNGH) in the presence or absence of 50 μg/ml dioleol phosphatidylserine (Avanti Polar Lipids, Alabaster, AL) and 20 nm 12-O-tetradecanoyl-phorbol-13-acetate (TPA). Aε-pep is a synthetic peptide based on the pseudosubstrate peptide of PKC Apl II, which is phosphorylated well by both PKC Apl I and PKC Apl II (Sossin and Schwartz, 1992; Sossin et al., 1993). After addition of 10 μl of purified PKCs, diluted to remain in the linear range of the assay (Sossin and Schwartz, 1992), the reaction was started with 10 μl of [γ-32P]ATP (1 μCi; 50 μm final concentration; New England Nuclear, Boston, MA). After 30 min at 20°C, 40 μl of the 50 μl reaction mixture was spotted onto a Whatman phosphocellulose paper disk, which was washed in 100 ml of 2% (w/v) ATP. The disks were then rinsed four times for 5 min with 0.425% (v/v) phosphoric acid, and radioactivity was counted by scintillation. Each value is the average of duplicate assays. The mixed micelle assay to determine dependence on PS was performed similarly but with 2 mol percent dioctylglycerol, 0.3% Triton X-100, and varying levels of PS.
Cell culture preparation
The isolation of sensory and motoneurons from Aplysiawas as described by Manseau et al. (1998). Briefly, adult animals were anesthetized by injection of 60–100 ml of isotonic MgCl2 solution. Pleuropedal and abdominal ganglia were removed, desheathed, and digested in 1% protease–artificial seawater. Tail sensory neurons (SNs) from the ventrocaudal cluster of pleural ganglia and siphon (LFS) motoneurons from the abdominal ganglion were mechanically dissociated and transferred to separate dishes (Falcon #1008; Becton Dickinson Canada Inc., Mississauga, Canada) containing a mixture of L-15 (modified forAplysia) (Schacher and Proshansky, 1983), hemolymph (7%), and bovine serum albumin (0.01%) (Klein, 1995). This procedure allowed easy manipulation of the neurons for many days.
Microinjection of the plasmid vectors
On day 1 after isolation, sensory neurons (usually round and devoid of any processes) were plated and allowed to adhere in a final dish (Falcon #3001) containing only L-15. Microinjections of plasmid solutions (2% fast green and ∼0.2 μm DNA in distilled water) were done with backfilled glass pipettes (∼5 MΩ) using a pico-injector (PLI-100; Medical Systems, Greenvale, NY). After impalement, sensory neurons were rapidly filled by delivering short air puffs (50–150 psi) until the cell soma became uniformly green.
Fluorescence microscopy
Injected sensory neurons were visualized at 4× with a Nikon fluorescence microscope (Optiphot-2) equipped with a BM510 filter (Nikon, Tokyo, Japan). The excitation light source was a 100 W high-pressure mercury lamp. Depending on the plasmid, the success rate of expression ranged from 50 to 90%; smaller constructs, such as the EGFP-C2 domain fusion protein, were expressed more easily than full-length EGFP-PKCs. Expression was usually stable 24 hr after the injection and could be maintained for >1 week.
Preparation of neuronal pairs
An arbitrary scale of fluorescence (from 0 to 5) was established to evaluate the labeling of each sensory neuron. Sensory neurons that were positive for plasmid expression (3–5 on the scale) were individually paired with motoneurons kept aside until then, in a hemolymph-enriched medium. After the pairing, neurons were still easily identifiable because of their distinct natural pigmentation (Klein, 1995) and the fast green injection in sensory neurons. Electrophysiological recordings started 2 d after pairing to allow the formation of new synaptic contacts (mostly soma to soma) and a full maturation of the PKC transduction pathway involved in short-term plasticity (Sun and Schacher, 1996). Fluorescence of the sensory neurons was monitored once again just before recording. Neuronal pairs showing no more fluorescence at this point were excluded from the final analysis. Therefore, the overall sequence was as follows: day 1, dissection of ganglia plus isolation of sensory neurons and motoneurons; day 2, injection of sensory neurons with plasmids; day 3, pairing of motoneurons with fluorescent sensory neurons; day 4, rest; day 5, electrophysiological recordings.
Electrophysiology
All recordings were done in L-15 at room temperature (21–24°C) using Axoclamp-2A and Axoprobe-1A amplifiers (Axon Instruments, Foster City, CA) in the current-clamp configuration (bridge mode). The major criteria for selection of healthy neurons was a stable membrane potential for both SNs and motoneurons, a holding current (at −50 mV) that did not exceed 0.3 pA for the SN, and, most importantly, the presence of a synapse. When these criteria were met, the experiment was started. The neuronal pair was kept in the study if at least two postsynaptic potentials were clearly visible and if both SNs and motoneurons were still capable of producing action potentials at the end of the experiment. The resting potential of sensory neurons was not measured until the end of the experiment to prevent the generation of unwanted spontaneous spikes.
Changes in synaptic transmission
In experiments on short-term facilitation of depressed synapses, a hyperpolarizing current was passed (10–15 MΩ glass pipette filled with 2 m KAc) to prevent spike generation during neuron impalement. The motoneuron was impaled first, and its membrane potential was stabilized at −80 mV. The sensory neuron was then impaled, and its membrane potential was stabilized at −50 mV. Short intracellular pulses were delivered, and once the threshold for action potential was reached, the stimulation intensity and interval was kept constant through the experiment. The series of EPSPs were recorded in the motoneuron. 5-HT (10 μm final concentration) was added directly to the bath near the cells and mixed gently. The amount of facilitation was calculated as the percent change between normalized EPSPs 41–43 (after 5-HT) and EPSPs 38–40 (before 5-HT). In experiments on short-term facilitation of rested synapses, we used extracellular stimulation (Manseau et al., 1998). A single depolarizing stimulus was applied to the sensory neuron, and the initial EPSP amplitude was recorded. At 2 min, 5-HT was applied to the bath (10 μm final concentration), and a second EPSP was recorded 3 min later in the presence of 5-HT. Data were acquired and analyzed digitally using Clampex 7 and a modified version of pClamp (Axon Instruments), provided by Dr. M. V. Storozhuk (A. A. Bogomolety Institute of Physiology, Kiev, Ukraine). Experiments comparing the effects of the various constructs were done in parallel as much as possible. Data are expressed as mean ± SEM.
Drugs and solutions
The following were used: protease type IX and 5-HT (Sigma, St. Louis, MO), TPA (Sigma), and PS (Avanti Polar Lipids).
RESULTS
Properties of the various Aplysia PKC constructs used for functional studies
A schematic diagram of the various plasmids that were made for the functional study of specific PKC isoforms is shown in Figure1. Three types of constructs were made for each isoform: full-length wild-type PKC, mutant PKC, and a deletion mutant encoding only the C2 domain of PKC. Each type of construct was tagged at its N-terminal with the EGFP. The various constructs were expressed in Sf9 or Sf21 cells using baculovirus to test their biochemical properties. All kinases were expressed at the expected molecular weights (Fig.2A,B). Addition of EGFP did not change the specific activity of the PKCs (Fig.2C), and the EGFP-tagged kinases retained their distinct activation by PS (Fig. 2D). These results suggest that addition of EGFP does not affect kinase activity. To make possible dominant-negative constructs, PKCs were inactivated by a K-R mutation in the catalytic site. These constructs would be expected to compete with endogenous PKCs for binding to PKC binding proteins and/or substrates but will not lead to phosphorylation of PKC substrates, and thus these constructs would block the action of endogenous PKCs. This substitution rendered the constructs insoluble, as shown by their disappearance from the supernatant fraction (Fig.3, K-R). Maturation of PKC requires two autophosphorylations in the catalytic domain; these are needed for PKC stability and function, and nonphosphorylated PKC can be trapped in the particulate fraction (Newton, 1995). To obviate this possible problem, we converted the autophosphorylation sites in PKC Apl I and PKC Apl II to glutamic acid, which mimics phosphorylation. We have shown previously that, for PKC Apl I, conversion of the two sites to glutamic acid does not affect the specific activity or the requirements for activation of the kinase (Nakhost et al., 1999) (data not shown). Conversion of the autophosphorylation sites to glutamic acid partially restored solubility of the kinases [Fig. 3, PKC (K-R, T-E, S-E), EGFP-PKC (K-R, T-E, S-E)]. As expected, kinases with K-R mutations have no kinase activity, and conversion of the phosphorylation sites to glutamic acid did not restore kinase activity (Fig. 2C). Throughout the rest of the paper, we will refer to the EGFP-PKC (K-R, T-E, S-E) constructs as “mutant” or “dominant-negative” PKC constructs.
Fig. 1.

Schematic diagram showing the major EGFP-PKC fusion constructs prepared for this study. Note that the C2 domain of PKC Apl II is on the N-terminal side. The general architecture of PKCs is conserved. A filled box represents the pseudosubstrate region. C1 binds DAG and phorbol esters; C2 binds to RACKs and mediates Ca2+ sensitivity in PKC Apl I; C3 and C4 are the catalytic domains. An example of a successful sensory neuron transfection with an EGFP-PKC fusion construct is shown (for details, see Materials and Methods). The distribution of fluorescence in the sensory neuron is cytoplasmic. The motoneuron was not injected and shows no fluorescence. The picture was taken at 20× magnification.
Fig. 2.

Specific activities of the various constructs. Sf21 cells were fractionated 3 d after infection with baculovirus encoding either PKC Apl I, PKC Apl II, EGFP-HA-PKC Apl I; EGFP-HA-PKC Apl II, EGFP-HA-PKC Apl I (K-R), EGFP-HA-PKC Apl II (K-R), EGFP-HA-PKC Apl I (K-R, T-E, S-E), or EGFP-HA-PKC Apl II (K-R, T-E, S-E). Supernatant fractions were diluted and assayed for maximal activity (stimulated by saturating amounts of PS-TPA) or assayed using the mixed micelle assay with 1% dioctylglycerol and increasing levels of PS (Sossin et al., 1996a). A,B, A fraction of the supernatant was used for Western blotting with an antibody to PKC Apl I (A) or to PKC Apl II (B) to determine the relative amounts of the kinases present. C, There were no differences in activity between wild-type PKCs and EGFP-PKCs. Specific activity was calculated as activity per amount of PKC (determined by Western blotting). There was no detectable activity after mutating the catalytic lysine to arginine, either before or after converting the autophosphorylation sites to glutamic acid and adding EGFP.D, Mixed micelle assays of wild-type PKCs and EGFP-PKCs demonstrate that addition of EGFP does not change the distinct PS requirements between PKC Apl I and PKC Apl II. Error bars indicate SEM; n = 3.
Fig. 3.
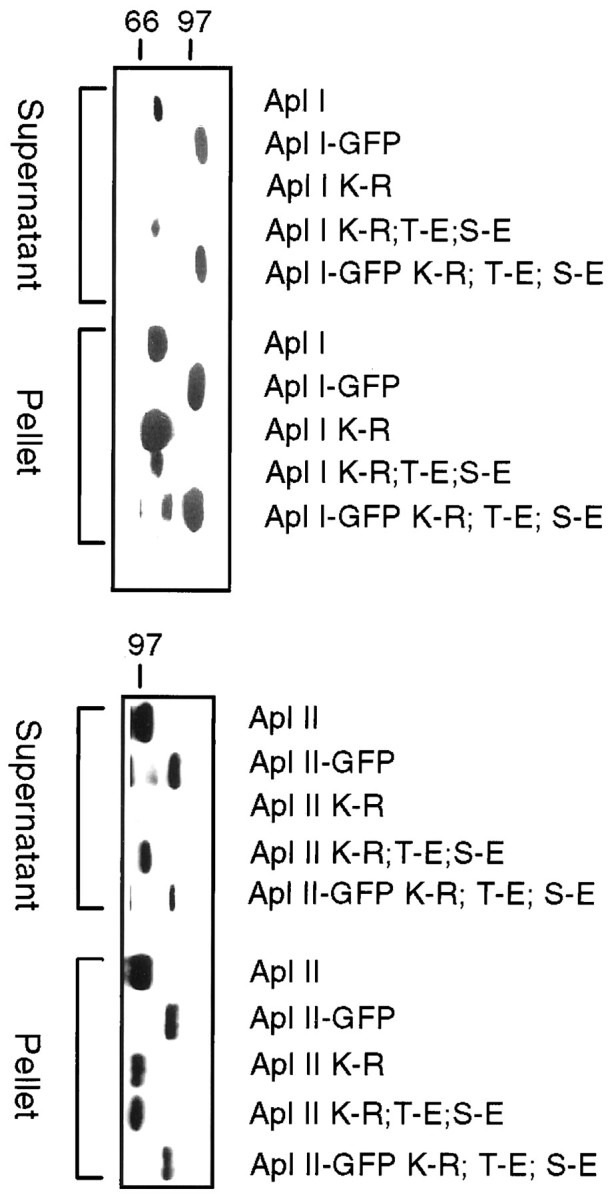
Solubility of PKC expressed in Sf9 cells with a baculovirus system. Sf9 cells were fractionated 3 d after infection with the baculovirus construct shown. The cells were lysed, separated into supernatant and pellet, and separated on SDS-PAGE gels. Approximately threefold more of the supernatant fraction was loaded than the pellet. Mutating the catalytic lysine (K-R) causes loss of soluble PKC. This is partially restored by converting phosphorylation sites to glutamic acid in PKC Apl I (K-R, T-E, S-E), EGFP-PKC Apl I (K-R, T-E, S-E), PKC Apl II (K-R, T-E, S-E), and EGFP-PKC Apl II (K-R, T-E, S-E).
Facilitation of depressed synapses is blocked by mutant PKC Apl II
Both wild-type and mutant EGFP-PKCs were successfully expressed in sensory neurons. The level of expression was evaluated using an arbitrary scale of fluorescence (from 0 to 5). Only the sensory neurons that were positive for plasmid expression (3–5 on the scale) were individually paired with motoneurons. The EGFP-PKCs were excluded from the nucleus of sensory neurons and restricted to the cytoplasm (Fig. 1). To study the function of specific PKC isoforms inAplysia, sensory neurons expressing EGFP or EGFP-PKC constructs were tested for the plasticity of their synaptic connections with motoneurons. In a preliminary control experiment, we found that GFP expression by itself had no effect on the kinetics of depression and facilitation of SM pairs (Table 2). Thus, using EGFP-expressing pairs as a control, we examined the effects of mutant PKCs in sensory neurons. In mutant PKC Apl I- and mutant PKC Apl II-transfected pairs, repeated stimulation induced a profound homosynaptic depression that was indistinguishable from controls (Fig.4A, Table3). However, although 5-HT produced a robust facilitation in the EGFP and mutant PKC Apl I groups, this effect was greatly reduced in SM pairs expressing the PKC Apl II mutant, suggesting that it acted as a dominant-negative construct (Fig. 4). Although the time course of facilitation was rapid (peak within 40 sec) in mutant PKC Apl I and control groups, facilitation in mutant PKC Apl II pairs was reduced and developed gradually; it became significant after 1.5 min. The difference between mutant PKC Apl II and mutant PKC Apl I was not because of differences in expression, because neither the arbitrary scale of fluorescence (measured on a scale of 1–5; PKC Apl I mutant, 3.4 ± 0.16,n = 10; PKC Apl II mutant, 3.33 ± 0.17,n = 9) nor quantitation of imaged neurons (PKC Apl I mutant, 40 ± 10, intensity per pixel, n = 3; PKC Apl II mutant, 35 ± 10, n = 3) suggested any difference in the level of expression of these proteins. The effect of mutant PKC Apl II was dependent on the state of the synapse. Mutant PKC Apl II had no effect on the facilitation by 5-HT at nondepressed synapses (Fig. 4D).
Table 2.
Effect of GFP expression on the kinetics of depression and facilitation of SM pairs
| n | Initial EPSP (mV) | Depression: EPSPs 9–11 (% of initial) | Facilitation: EPSPs 12–14 (% of initial) | |
|---|---|---|---|---|
| GFP | 12 | 4.8 ± 2.8 | 18 ± 5% | 125 ± 24% |
| Noninjected | 6 | 6.3 ± 1.9 | 27 ± 3% | 94 ± 16% |
| ttests | NS | NS | NS |
At moderately depressed synapses, GFP expression had no effect on synaptic properties. Mean initial postsynaptic potential, depression (EPSPs 9–11), and facilitation (EPSPs 12–14) of sensorimotor synapses were comparable in the GFP-positive and noninjected groups. In this control experiment, 5-HT was applied between EPSPs 11 and 12 (interstimulus interval, 30 sec).
Fig. 4.

Short-term facilitation of depressed synapses is blocked by a dominant-negative mutant of PKC Apl II (Apl II-m). A, Sensory to motor transmission was depressed by a series of 40 repeated intracellular stimuli (interstimulus interval, 20 sec), and 5-HT (10 μm) was applied to induce synaptic facilitation. In this experiment, 5-HT facilitated control EGFP-expressing synapses to 96 ± 27 of their initial EPSP amplitude (n = 12). The expression of mutant forms of either PKC Apl I (n = 10) or PKC Apl II (n = 9) did not change the slope of depression, but mutant PKC Apl II dramatically reduced the effect of 5-HT relative to the control group (EGFP). B, Traces from two different synapses expressing EGFP and the mutant form of PKC Apl II are shown.C, Comparing the amount of facilitation [percent change between normalized EPSPs 41–43 (after 5-HT) and EPSPs 38–40 (before 5-HT)] between the three groups reveals a significant inhibition by the mutant form of PKC Apl II (p < 0.01; unpaired one-tailed t test with Welch's correction).D, Facilitation of rested SM synapses is unaffected by overexpression of mutant form (Apl II-m) of PKC Apl II. An initial EPSP was induced by single extracellular stimulation to the sensory neuron. After 5-HT (10 μm), a second EPSP was recorded. The interstimulus interval between the two EPSPs was 5 min. The EPSP amplitude was normalized to the initial control value (for details, see Materials and Methods). Facilitation was determined by comparing the percent change between the two normalized EPSPs [EPSP 2 (after 5-HT) − EPSP 1 (before 5-HT); EGFP,n = 4; mutant Apl II, n = 5]. No significant differences were observed (unpaired one-tailedt test with Welch's correction).
Table 3.
Comparison of intrinsic and synaptic properties of sensory neurons expressing EGFP or various EGFP-PKC constructs
| Apl I-m | Apl I-w | Apl II-m | Apl II-w | Apl I-C2 | Apl II-C2 | EGFP | ANOVA | |
|---|---|---|---|---|---|---|---|---|
| Ihold(pA) | −0.08 ± 0.03 | −0.15 ± 0.03** | −0.20 ± 0.10 | −0.08 ± 0.03 | −0.11 ± 0.03 | −0.03 ± 0.01 | −0.05 ± 0.01 | * |
| Rin(MΩ) | 111.7 ± 15.7 | 136.4 ± 23.4 | 92.8 ± 7.8 | 109.5 ± 7.8 | 133.8 ± 14.8 | 140.8 ± 11.8* | 107.1 ± 10.8 | NS |
| Vr(mV) | −29.8 ± 4.0 | −31.4 ± 1.7 | −35.0 ± 2.3 | −32.3 ± 1.8 | −36.3 ± 0.9* | −34.6 ± 1.5 | −31.4 ± 1.9 | NS |
| Initial EPSP (mV) | 3.8 ± 1.1 | 4.6 ± 2.1 | 3.0 ± 0.9* | 13.9 ± 5.1 | 9.3 ± 3.5 | 6.9 ± 2.2 | 7.5 ± 2.3 | NS |
| EPSPs 6–10 (% of initial) | 33 ± 3.8 | 23 ± 5.3 | 32 ± 5.0 | 26 ± 3.5 | 40 ± 8.4 | 40 ± 9.0 | 35 ± 5.6 | NS |
| EPSPs 36–40 (% of initial) | 7.9 ± 2.2 | 4.5 ± 1.2 | 6.5 ± 2.2 | 10.1 ± 2.4 | 11.3 ± 3.3 | 12.5 ± 4.2 | 7.7 ± 2.9 | NS |
The holding current (Ihold) is the current needed to hold the SN at −50 mV before the first stimulus. Resting potential (Vr) and input resistance (Rin) were taken at the end of each experiment. The values for Vr are therefore underestimated, because at this point, 5-HT was present in the bathing solution and the SNs were often spontaneously active when hyperpolarization was removed. EPSPs 6–10, Early depression; EPSPs 36–40, late depression; ANOVA, one-way ANOVA. Significant differences between individual groups and the control EGFP group are indicated by asterisks (**p< 0.01, *p < 0.05; unpaired one-tailed ttest with Welch's correction).
Expression of the mutant constructs did not alter the shape and latency of the postsynaptic response or the intrinsic electrical properties of sensory neurons (Table 3). The frequency, speed, and pattern of neurite formation were also similar between the groups, and there was no evidence that pairs expressing one type of construct failed more often to form detectable chemical connections. The initial size of synaptic potentials from mutant PKC Apl II SM pairs was smaller than in controls (Table 3). However, it was not different from the initial EPSP of the mutant PKC Apl I group (Student's t test, NS), which had normal kinetics of facilitation. Thus, it seems unlikely that mutant PKC Apl II synapses did not respond to 5-HT simply because they had smaller synapses initially. Moreover, the mean amplitude of initial EPSPs from the EGFP and mutant PKC Apl II groups was similar in the experiment using nondepressed synapses, indicating that dominant-negative PKC Apl II by itself does not cause a decrease in the initial synaptic weight.
There was no evidence that the expression of the dominant-negative PKC Apl II affected the type of synaptic contact that was formed. Royer et al. (2000) recently suggested that the plasticity of the SM junction depends on the switching off and on of one subpopulation of synapses while a second population transmits stably and is less affected by modulatory agents. If dominant-negative PKC Apl II-transfected neurons only formed contacts of this latter type, this could explain the lack of facilitation. However, if this was the case, there would be no homosynaptic depression at these synapses. We have observed a normal depression rate (Fig. 4, Table 3).
Thus, these results demonstrate that expression of mutant PKC Apl II, but not similarly mutated PKC Apl I, specifically blocks 5-HT-mediated synaptic facilitation of depressed synapses without affecting most synaptic properties and without altering the mechanism underlying synaptic depression. Indeed, the actions of the mutant PKC Apl II were also dependent on the state of the synapse; mutant PKC Apl II blocked facilitation of the depressed synapse but had no effect on facilitation of nondepressed synapses.
Wild-type PKC expression also blocks the PKC-dependent facilitation
Because dominant-negative PKC and pharmacological PKC inhibitors block the facilitation of depressed synapses by 5-HT, it is possible that an increased level of wild-type PKC would conversely enhance this form of plasticity. We tested this possibility by transfecting EGFP-tagged wild-type forms of PKC Apl I or PKC Apl II constructs in SNs. Overexpression of wild-type PKC did not enhance facilitation. In fact, it was found that both isoforms significantly reduced the effect of 5-HT at depressed synapses (Fig. 5). This result is in contrast to the observation that dominant-negative constructs produce an isoform-selective block of STF. The effect of wild-type constructs also differed from that of mutants, in that the amplitude of facilitation was reduced but their kinetics remained unchanged.
Fig. 5.

Short-term facilitation of depressed synapses is decreased by overexpression of wild-type PKC Apl I (Apl I-w) (n = 11) and PKC Apl II (Apl II-w) (n = 12) (p < 0.05; unpaired one-tailedt test with Welch's correction). A, Notice that, in this case, the kinetics of facilitation is not affected. The control EGFP data (n = 12) shown in this figure are the same as in Figure 4 and are used for comparison.B, Comparison of the amount of facilitation [percent change between normalized EPSPs 41–43 (after 5-HT) and EPSPs 38–40 (before 5-HT)] between the three groups. C, Expression of wild-type PKC Apl II had no effect on the facilitation of nondepressed synapses (EGFP, n = 4; wild-type Apl II, n = 5; unpaired one-tailed ttest with Welch's correction).
Because of their different biochemical properties (active vs inactive) and considering their differential effect on the kinetics of STF, it is likely that the actions of wild-type and mutant PKC constructs are mediated through distinct mechanisms. Overexpression of wild-type PKC may increase the basal level of PKC activity in neurons, and this could lead to a homeostatic downregulation of PKC pathways. Activation of PKC may also lead to inhibition or activation of the PKA pathway (Sugita et al., 1997b). To determine whether overexpression of PKC affects PKA-mediated signal transduction, we examined the effect of 5-HT at rested synapses of neurons overexpressing the wild-type PKC Apl II. We found that the wild-type PKC Apl II construct had no effect on facilitation of rested synapses (Fig. 5C). This result suggests that overexpression of this isoform does not alter the PKA pathway or the PKA-coupled 5-HT receptors. Thus, like the mutant PKC Apl II, the effects of overexpression of the wild-type PKC Apl II are dependent on the state of the synapses, inhibiting facilitation by 5-HT at depressed synapses but not at nondepressed synapses.
Facilitation may not involve protein–protein interaction with the C2 domain of PKC
The C2 domain of PKC is involved in isoform-specific recognition and binding with membrane-anchoring proteins known as receptors for activated C kinases (RACKs) (Mochly-Rosen et al., 1991; Ron et al., 1994b; Johnson et al., 1996; Csukai et al., 1997). This binding may ensure the correct placement of PKC in isoform-specific macromolecular complexes directing the enzyme–substrate interaction (Mochly-Rosen and Gordon, 1998; Sim and Scott, 1999). Selective inhibition of C2-RACK interactions has been used successfully in other systems to block the activation of individual PKC isoforms (Ron and Mochly-Rosen, 1994a;Johnson et al., 1996; Yedovitzky et al., 1997). Therefore, we tested whether the C2 domains of PKC Apl I or PKC Apl II could block facilitation of depressed synapses. To make sure that our EGFP-C2 constructs were functional, we expressed them in Sf21 cells with baculovirus. We have reported previously that the C2 domain of PKC Apl I binds PS in a Ca2+-dependent manner, whereas the C2 domain of PKC Apl II does not (Pepio et al., 1998). Indeed, similar results were seen with the EGFP-tagged C2 domains (Fig.6A). After expression in sensory neurons, EGFP and EGFP-C2 domain constructs were uniformly distributed in the soma and in every compartment of the SN, including the nucleus (data not shown). However, their expression in sensory neurons had no effect on facilitation of depressed synapses (Fig.6B,C).
Fig. 6.

The C2 regions of Aplysia PKCs have no effect on facilitation. A, Properties of the C2 constructs. The distinct translocation of the two C2 domains is retained in the EGFP-C2 constructs; PKC Apl I binds to PS in a Ca2+-dependent manner, and PKC Apl II does not bind constitutively to PS or translocate in the presence of Ca2+. Sf21 cells were fractionated 3 d after infection with either PKC Apl I EGFP-C2 or PKC Apl II EGFP-C2. Supernatants were incubated with buffer, PS (40 μg/ml), or PS (40 μg/ml) and Ca2+ (500 μm).B, Short-term facilitation of depressed synapses is unaffected by overexpression of the C2 domains from either PKC Apl I (n = 12) or PKC Apl II (n = 13) (not significantly different; unpaired one-tailed t test with Welch's correction). Control EGFP (n = 12) is as in Figure 4. C, Summary of the amount of facilitation between the three groups.
DISCUSSION
Use of a dominant-negative mutant to study the function of PKC isoforms in Aplysia
Traditionally, signal transduction studies inAplysia have relied mostly on pharmacological inhibitors or activators of specific enzymes. Go6976 and Ro-32-0432 are pharmacological inhibitors that are reported to distinguish Ca2+-activated and Ca2+-independent PKCs in vertebrates, but they do not show selectivity for inhibition of PKC Apl I and PKC Apl II (data not shown). Pseudosubstrate-based inhibitors have been used as isoform-specific inhibitors, but the pseudosubstrates from PKC Apl I and PKC Apl II inhibit both enzymes (Sossin and Schwartz, 1992). More recently, it has become possible to transfect Aplysianeurons in intact ganglia (Kaang et al., 1992; Kaang, 1996; Chang et al., 2000) and in culture (Bailey et al., 1997). To determine the roles of the two PKC isoforms known inAplysia, we used mutants of PKC as isoform-specific inhibitors. A similar strategy has been used successfully in other systems (Brodie et al., 1999; Soh et al., 1999; Buchner, 2000) and inAplysia for determining the importance of kinases or transcription factors (Castellucci et al., 1980; Dash et al., 1990;Bartsch et al., 1998).
Effect of mutant PKC expression in SM neurons shows the involvement of PKC Apl II in 5-HT-induced synaptic recovery from depression
Dominant-negative PKC Apl II blocked the rapid onset of facilitation; however, after 1.5 min, there was no significant difference between the cells expressing dominant-negative PKC Apl II and controls. We believe that the dominant-negative PKC Apl II works by binding to a complex of proteins involved in transmitter release and thus blocking endogenous PKC from binding to this complex. However, the endogenous PKC is competing with the dominant-negative PKC for this site, and it will eventually succeed in phosphorylating the appropriate protein to increase transmitter release. If the half-life of phosphorylation is long enough, then over time enough protein will be phosphorylated to increase transmitter release; we believe this explains the transience of the dominant-negative inhibition. Consistent with this interpretation, we have been unable to block the PDBu-induced increase in excitability, which develops over tens of minutes (Sugita et al., 1997b; Manseau et al., 1998) using the dominant-negative PKCs (data not shown). The effect of the dominant-negative PKCs may be limited to actions of PKC that require protein–protein interactions to prelocalize PKC. These may be actions of PKC that must occur shortly after stimulation, as is the case for the role of PKC in invertebrate phototransduction (Tsunoda et al., 1997), in which prelocalization of PKC is essential.
Role of PKCs in regulating transmitter release
PKC regulates transmitter release in many systems (for review, see Vaughan et al., 1998). In some cases, PKC increases transmitter release through a phosphorylation-dependent modulation of Ca2+ channels (Swartz, 1993;Sánchez-Prieto et al., 1996; Hamid et al., 1999). InAplysia, modulation of Ca2+channels does not make a large contribution to synaptic plasticity (Klein, 1995). Other possible actions of PKC are to increase the pool of transmitter available for release (Smith et al., 1998; Stevens and Sullivan, 1998; Walaas, 1999; Zhao and Klein, 2000) and to facilitate a late stage in the release process (Chen et al., 1999; Yawo, 1999). Either of these possibilities is consistent with the role of PKC Apl II in Aplysia neurons. PKC Apl II is a homolog of the vertebrate PKCs ε and η. Interestingly PKC ε is enriched in presynaptic terminals (Tanaka and Nishizuka, 1994) and has been implicated in the regulation of transmitter release from synaptosomes (Prekeris et al., 1996), as well as in the regulation of exocytosis in several non-neuronal preparations (Ozawa et al., 1993; Turner et al., 1994; Hong et al., 1997). Thus, the role of this isoform of PKC in regulating exocytosis may be conserved. It will be interesting to see whether the regulation of transmitter release induced by PDBu is affected in the recently described ε knock-out mice (Khasar et al., 1999), because it is still seen in PKC γ knock-outs (Goda et al., 1996) and only partially decreased in the PKC β knock-out mouse (Weeber et al., 2000).
Effect of wild-type PKC overexpression
In our experiments, facilitation of depressed SM synapses was blocked by overexpression of wild-type PKC Apl I and PKC Apl II. It is unlikely that these constructs are acting as dominant-negatives. First, there are no differences between the activity of the EGFP-tagged PKCs and wild-type PKCs (Fig. 2). Second, unlike the dominant-negatives, facilitation is observed immediately after the 5-HT pulse in wild-type PKC-expressing SM pairs, suggesting that there is no competition with endogenous kinases. A simple explanation for the effect of wild-type PKCs is a homeostatic downregulation of the pathway between 5-HT receptors and PKC activation, perhaps by phosphorylating and desensitizing 5-HT receptors coupled to PKC activation. Alternatively, there may be downregulation of steps between PKC activation and facilitation. There was no effect of EGFP-tagged PKC Apl II on the 5-HT-mediated facilitation at nondepressed synapses, suggesting that this homeostatic regulation did not affect 5-HT receptors linked to cAMP production.
PKC Apl II-mediated EPSP facilitation may not involve isotype-selective interaction of the C2 domain with other proteins
RACKs recognize the C2 domain of PKC in an isotype-selective manner (Mochly-Rosen and Gordon, 1998). No evidence was found in this study that would indicate that these interactions are important for the regulation of transmitter release. PKC also has protein–protein interactions that are not localized to the C2 domain, including interactions with the C1 domain, catalytic subunit, and C-terminal sequences (Mochly-Rosen and Gordon, 1998; Jaken and Parker, 2000). The interaction of PKC Apl II with actin filaments does not require the C2 domain and could be important for the regulation of transmitter release (Nakhost et al., 1998).
Reconciliation with the biochemical data
The dominant-negative effect of mutant PKC Apl II on the 5-HT-mediated reversal of synaptic depression is in apparent contradiction with evidence that a short 5-HT pulse only induces translocation of PKC Apl I (Sossin and Schwartz, 1992). If PKC Apl II is translocated only at the synapse, previous studies would not have been sensitive enough to detect this translocation. Indeed, PKC Apl II is abundant in growth cones (Nakhost et al., 1998), and translocation of PKC Apl II by phorbol esters in synaptosomes is enhanced compared with the cell body (Sossin and Schwartz, 1994). Alternatively, PKC may not need to be translocated if it is already prelocalized at important sites through anchoring proteins (Tsunoda et al., 1997). Thus, our data show quite clearly that translocation of PKCs may be a good starting point to examine activation of the kinase, but it is limited in terms of predicting the isoforms of PKC that will be involved in a specific physiological function, especially if that function is localized to a small region of the cell.
State dependence of PKC activation
The efficacy of information transfer to a postsynaptic target is dependent on the previous history of synaptic activity. Facilitation of rested and depressed synapses is mediated by different mechanisms, and previous studies have emphasized that different kinases become involved according to the state of the neuron (Ghirardi et al., 1992; Byrne and Kandel, 1996). Our results confirm this finding, because dominant-negative PKC Apl II blocked 5-HT facilitation of depressed synapses but not naive synapses. PKC may phosphorylate a protein that is only rate-limiting when synapses are depressed. However, phorbol esters can also increase transmitter release at naive synapses (Braha et al., 1990; Sugita et al., 1997a; our unpublished data). Another interesting possibility is that PKC Apl II activation could be dependent on the state of the neuron. 5-HT would only induce activation at depressed synapses. We may be able to test this hypothesis if we can measure EGFP-PKC translocation at synapses. It may even be possible for the same protein to be phosphorylated by PKA at rested synapses and by PKC at depressed synapses. For example, the state of the synapse has been shown recently to be an important factor in which kinase phosphorylates the AMPA receptor after long-term potentiation (Lee et al., 2000).
There may also be other state dependences that specifically involve the PKC Apl I isoform. Recovery from depression after high-frequency firing may involve a Ca2+-dependent replenishment of the releasable pool of vesicles (Gingrich and Byrne, 1985). PKC is required for the persistence of synaptic facilitation when serotonin is paired with activity (Sutton and Carew, 2000). Because PKC Apl I is regulated by Ca2+, it may play a role in the regulation of transmitter release in these states of the synapse.
Summary
We have demonstrated that the dominant-negative constructs of PKC can be used as isotype-specific PKC blockers in the nervous system ofAplysia. Our results show that PKC Apl II partially mediates short-term facilitation at the sensorimotor synapse. In the future, it will be interesting to use imaging to see whether the action of PKC involves isotype-specific translocation at Aplysia synapses. Dominant-negative PKCs can be tested on other possible downstream effects of PKC, such as growth cone extension, the regulation of ion channels, and increases in spontaneous release.
Footnotes
This work was funded in part by a Fonds pour la Formation de Chercheurs et l'Aide à la Recherche fellowship (F.M.), by Medical Research Council of Canada Grants MT-14142 (V.F.C.) and MT-12046, and by Natural Sciences and Engineering Research Council Grant 187018 (W.S.). W.S. is the recipient of a Chercheur-Boursier from the Fonds de la Recherche en Santé du Québec. We thank Drs. M. Klein and P. McPherson for their helpful comments regarding this manuscript and France Cartier for typing this manuscript.
Correspondence should be addressed to Vincent F. Castellucci, Centre de Recherche en Sciences Neurologiques, Département de Physiologie, Université de Montréal, Montréal, Canada H3C 3J7. E-mail: castellv@physio.umontreal.ca.
REFERENCES
- 1.Bailey CH, Chen M. Morphological basis of short-term habituation in Aplysia. J Neurosci. 1988;8:2452–2459. doi: 10.1523/JNEUROSCI.08-07-02452.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailey CH, Kaang BK, Chen M, Martin KC, Lim CS, Casadio A, Kandel ER. Mutation in the phosphorylation sites of MAP kinase blocks learning-related internalization of apCAM in Aplysia sensory neurons. Neuron. 1997;18:913–924. doi: 10.1016/s0896-6273(00)80331-1. [DOI] [PubMed] [Google Scholar]
- 3.Bartsch D, Casadio A, Karl KA, Serodio P, Kandel ER. CREB 1 encodes a nuclear activator, a repressor, and a cytoplasmic modulator that form a regulatory unit critical for long-term facilitation. Cell. 1998;95:211–223. doi: 10.1016/s0092-8674(00)81752-3. [DOI] [PubMed] [Google Scholar]
- 4.Braha O, Dale N, Hochner B, Klein M, Abrams TW, Kandel ER. Second messengers involved in the two processes of presynaptic facilitation that contribute to sensitization and dishabituation in Aplysia sensory neurons. Proc Natl Acad Sci USA. 1990;87:2040–2044. doi: 10.1073/pnas.87.5.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brodie CK, Bogi P, Acs P, Lazarovici P, Petrovics G, Anderson WB, Blumberg PM. Protein kinase Cε plays a role in neurite outgrowth in response to epidermal growth factor and nerve growth factor in PC12 cells. Cell Growth Differ. 1999;10:183–191. [PubMed] [Google Scholar]
- 6.Buchner K. The role of protein kinase C in the regulation of cell growth and in signalling to the cell nucleus. J Cancer Res Clin Oncol. 2000;126:1–11. doi: 10.1007/pl00008458. [DOI] [PubMed] [Google Scholar]
- 7.Byrne JH, Kandel ER. Presynaptic facilitation revisited: state and time dependence. J Neurosci. 1996;16:425–435. doi: 10.1523/JNEUROSCI.16-02-00425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castellucci VF, Kandel ER, Schwartz JH, Wilson FD, Nairn AC, Greengard P. Intracellular injection of the catalytic subunit of cyclic AMP-dependent protein kinase stimulates facilitation of transmitter release underlying behavioral sensitization in Aplysia. Proc Natl Acad Sci USA. 1980;77:7492–7496. doi: 10.1073/pnas.77.12.7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang DJ, Li XC, Lee YS, Kim HK, Kim US, Cho NJ, Lo X, Weiss KR, Kandel ER, Kaang BK. Activation of a heterologously expressed octopamine receptor coupled only to adenylyl cyclase produces all the features of presynaptic facilitation in Aplysia sensory neurons. Proc Natl Acad Sci USA. 2000;97:1829–1834. doi: 10.1073/pnas.97.4.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen YA, Duvvuri V, Schulman H, Scheller RH. Calmodulin and protein kinase C increase Ca2+-stimulated secretion by modulating membrane-attached exocytic machinery. J Biol Chem. 1999;274:26469–26476. doi: 10.1074/jbc.274.37.26469. [DOI] [PubMed] [Google Scholar]
- 11.Csukai M, Chen CH, De Matteis MA, Mochly-Rosen D. The coatomer protein β-COP, a selective binding protein (RACK) for protein kinase Cε. J Biol Chem. 1997;272:29200–29206. doi: 10.1074/jbc.272.46.29200. [DOI] [PubMed] [Google Scholar]
- 12.Dash PK, Hochner B, Kandel ER. Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature. 1990;345:718–721. doi: 10.1038/345718a0. [DOI] [PubMed] [Google Scholar]
- 13.Eliot LS, Kandel ER, Hawkins RD. Modulation of spontaneous transmitter release during depression and posttetanic potentiation of Aplysia sensory-motor neuron synapses isolated in culture. J Neurosci. 1994;14:3280–3292. doi: 10.1523/JNEUROSCI.14-05-03280.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghirardi M, Braha O, Hochner B, Montarolo PG, Kandel ER, Dale N. Roles of PKA and PKC in facilitation of evoked and spontaneous transmitter release at depressed and nondepressed synapses in Aplysia sensory neurons. Neuron. 1992;9:479–489. doi: 10.1016/0896-6273(92)90185-g. [DOI] [PubMed] [Google Scholar]
- 15.Ghirardi M, Montarolo PG, Kandel ER. A novel intermediate stage in the transition between short- and long-term facilitation in the sensory to motor neuron synapse of Aplysia. Neuron. 1995;14:413–420. doi: 10.1016/0896-6273(95)90297-x. [DOI] [PubMed] [Google Scholar]
- 16.Gingrich KJ, Byrne JH. Simulation of synaptic depression, posttetanic potentiation, and presynaptic facilitation of synaptic potentials from sensory neurons mediating gill-withdrawal reflex in Aplysia. J Neurophysiol. 1985;53:652–669. doi: 10.1152/jn.1985.53.3.652. [DOI] [PubMed] [Google Scholar]
- 17.Goda Y, Stevens CF, Tonegawa S. Phorbol ester effects at hippocampal synapses act independently of the γ isoform of PKC. Learn Mem. 1996;3:182–187. doi: 10.1101/lm.3.2-3.182. [DOI] [PubMed] [Google Scholar]
- 18.Hamid J, Nelson D, Spaetgens R, Dubel SJ, Snutch TP, Zamponi GW. Identification of an integration center for cross-talk between protein kinase C and G protein modulation of N-type calcium channels. J Biol Chem. 1999;274:6195–6202. doi: 10.1074/jbc.274.10.6195. [DOI] [PubMed] [Google Scholar]
- 19.Hochner B, Klein M, Schacher S, Kandel ER. Additional component in the cellular mechanism of presynaptic facilitation contributes to behavioral dishabituation in Aplysia. Proc Natl Acad Sci USA. 1986;83:8794–8798. doi: 10.1073/pnas.83.22.8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong DH, Forstner JF, Forstner GG. Protein kinase Cε is the likely mediator of mucin exocytosis in human colonic cell lines. Am J Physiol. 1997;272:G31–G37. doi: 10.1152/ajpgi.1997.272.1.G31. [DOI] [PubMed] [Google Scholar]
- 21.Jaken S, Parker PJ. Protein kinase C binding partners. Bioessays. 2000;22:245–254. doi: 10.1002/(SICI)1521-1878(200003)22:3<245::AID-BIES6>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 22.Johnson JA, Gray MO, Chen CH, Mochly-Rosen D. A protein kinase C translocation inhibitor as an isozyme-selective antagonist of cardiac function. J Biol Chem. 1996;271:24962–24966. doi: 10.1074/jbc.271.40.24962. [DOI] [PubMed] [Google Scholar]
- 23.Kaang BK. Parameters influencing ectopic gene expression in Aplysia neurons. Neurosci Lett. 1996;221:29–32. doi: 10.1016/s0304-3940(96)13279-1. [DOI] [PubMed] [Google Scholar]
- 24.Kaang BK, Pfaffinger PJ, Grant SG, Kandel ER, Furukawa Y. Overexpression of an Aplysia shaker K+ channel gene modifies the electrical properties and synaptic efficacy of identified Aplysia neurons. Proc Natl Acad Sci USA. 1992;89:1133–1137. doi: 10.1073/pnas.89.3.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase Cε mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klein M. Modulation of ion currents and regulation of transmitter release in short-term synaptic plasticity: the rise and fall of the action potential. Invert Neurosci. 1995;1:15–24. doi: 10.1007/BF02331828. [DOI] [PubMed] [Google Scholar]
- 27.Kruger KE, Sossin WS, Sacktor TC, Bergold PJ, Beushausen S, Schwartz JH. Cloning and characterization of Ca2+-dependent and Ca2+-independent PKCs expressed in Aplysia sensory cells. J Neurosci. 1991;11:2303–2313. doi: 10.1523/JNEUROSCI.11-08-02303.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 29.Malenka RC, Madison DV, Nicoll RA. Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature. 1986;321:175–177. doi: 10.1038/321175a0. [DOI] [PubMed] [Google Scholar]
- 30.Manseau F, Sossin WS, Castellucci VF. Long-term changes in excitability induced by protein kinase C activation in Aplysia sensory neurons. J Neurophysiol. 1998;79:1210–1218. doi: 10.1152/jn.1998.79.3.1210. [DOI] [PubMed] [Google Scholar]
- 31.Mochly-Rosen D, Gordon AS. Anchoring proteins for protein kinase C: a means for isozyme selectivity. FASEB J. 1998;12:35–42. [PubMed] [Google Scholar]
- 32.Mochly-Rosen D, Khaner H, Lopez J. Identification of intracellular receptor proteins for activated protein kinase C. Proc Natl Acad Sci USA. 1991;88:3997–4000. doi: 10.1073/pnas.88.9.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakhost A, Forscher P, Sossin WS. Binding of protein kinase C isoforms to actin in Aplysia. J Neurochem. 1998;71:1221–1231. doi: 10.1046/j.1471-4159.1998.71031221.x. [DOI] [PubMed] [Google Scholar]
- 34.Nakhost A, Dyer JR, Pepio AM, Fan X, Sossin WS. Protein kinase C phosphorylated at a conserved threonine is retained in the cytoplasm. J Biol Chem. 1999;274:28944–28949. doi: 10.1074/jbc.274.41.28944. [DOI] [PubMed] [Google Scholar]
- 35.Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 36.Nishizuka Y. The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature. 1988;334:661–665. doi: 10.1038/334661a0. [DOI] [PubMed] [Google Scholar]
- 37.Ozawa K, Szallasi Z, Kazanietz MG, Blumberg PM, Mischak H, Mushinski JF, Beaven MA. Ca2+-dependent and Ca2+-independent isozymes of protein kinase C mediate exocytosis in antigen-stimulated rat basophilic RBL-2H3 cells. Reconstitution of secretory responses with Ca2+ and purified isozymes in washed permeabilized cells. J Biol Chem. 1993;268:1749–1756. [PubMed] [Google Scholar]
- 38.Pepio AM, Sossin WS. The C2 domain of the Ca2+-independent protein kinase C Apl II inhibits phorbol ester binding to the C1 domain in a phosphatidic acid-sensitive manner. Biochemistry. 1998;37:1256–1263. doi: 10.1021/bi971841u. [DOI] [PubMed] [Google Scholar]
- 39.Pepio AM, Fan X, Sossin WS (1998) The role of C2 domains in Ca2+-activated and Ca2+-independent protein kinase Cs inAplysia. J Biol Chem [Erratum (1998) 273:22856] 273:19040–19048. [DOI] [PubMed]
- 40.Prekeris R, Mayhew MW, Cooper JB, Terrian DM. Identification and localization of an actin-binding motif that is unique to the ε isoform of protein kinase C and participates in the regulation of synaptic function. J Cell Biol. 1996;132:77–90. doi: 10.1083/jcb.132.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ron D, Mochly-Rosen D. Agonists and antagonists of protein kinase C function, derived from its binding proteins. J Biol Chem. 1994a;269:21395–21398. [PubMed] [Google Scholar]
- 42.Ron D, Chen CH, Caldwell J, Jamieson L, Orr E, Mochly-Rosen D. Cloning of an intracellular receptor for protein kinase C: a homolog of the β subunit of G proteins. Proc Natl Acad Sci USA. 1994b;91:839–843. doi: 10.1073/pnas.91.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Royer S, Coulson RL, Klein M. Switching off and on of synaptic sites at Aplysia sensorimotor synapses. J Neurosci. 2000;20:626–638. doi: 10.1523/JNEUROSCI.20-02-00626.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sacktor TC, Schwartz JH. Sensitizing stimuli cause translocation of protein kinase C in Aplysia sensory neurons. Proc Natl Acad Sci USA. 1990;87:2036–2039. doi: 10.1073/pnas.87.5.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sánchez-Prieto J, Budd DC, Herrero I, Vazquez E, Nicholls DG. Presynaptic receptors and the control of glutamate exocytosis. Trends Neurosci. 1996;19:235–239. doi: 10.1016/0166-2236(96)10031-x. [DOI] [PubMed] [Google Scholar]
- 46.Schacher S, Proshansky E. Neurite regeneration by Aplysia neurons in dissociated cell culture: modulation by Aplysia hemolymph and the presence of the initial axonal segment. J Neurosci. 1983;3:2403–2413. doi: 10.1523/JNEUROSCI.03-12-02403.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shapira R, Silberberg SD, Ginsburg S, Rahamimoff R. Activation of protein kinase C augments evoked transmitter release. Nature. 1987;325:58–60. doi: 10.1038/325058a0. [DOI] [PubMed] [Google Scholar]
- 48.Sim AT, Scott JD. Targeting of PKA, PKC, and protein phosphatases to cellular microdomains. Cell Calcium. 1999;26:209–217. doi: 10.1054/ceca.1999.0072. [DOI] [PubMed] [Google Scholar]
- 49.Smith C, Moser T, Xu T, Neher E. Cytosolic Ca2+ acts by two separate pathways to modulate the supply of release-competent vesicles in chromaffin cells. Neuron. 1998;20:1243–1253. doi: 10.1016/s0896-6273(00)80504-8. [DOI] [PubMed] [Google Scholar]
- 50.Soh JW, Lee EH, Prywes R, Weinstein IB. Novel roles of specific isoforms of protein kinase C in activation of the c-fos serum response element. Mol Cell Biol. 1999;19:1313–1324. doi: 10.1128/mcb.19.2.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sossin WS, Schwartz JH. Selective activation of Ca2+-activated PKCs in Aplysia neurons by 5-HT. J Neurosci. 1992;12:1160–1168. doi: 10.1523/JNEUROSCI.12-04-01160.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sossin WS, Schwartz JH. Translocation of protein kinase Cs in Aplysia neurons: evidence for complex regulation. Brain Res Mol Brain Res. 1994;24:210–218. doi: 10.1016/0169-328x(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 53.Sossin WS, Diaz-Arrastia R, Schwartz JH. Characterization of two isoforms of protein kinase C in the nervous system of Aplysia californica. J Biol Chem. 1993;268:5763–5768. [PubMed] [Google Scholar]
- 54.Sossin WS, Sacktor TC, Schwartz JH. Persistent activation of protein kinase C during the development of long-term facilitation in Aplysia. Learn Mem. 1994;1:189–202. [PubMed] [Google Scholar]
- 55.Sossin WS, Chen CS, Toker A. Stimulation of an insulin receptor activates and down-regulates the Ca2+-independent protein kinase C, Apl II, through a Wortmannin-sensitive signaling pathway in Aplysia. J Neurochem. 1996a;67:220–228. doi: 10.1046/j.1471-4159.1996.67010220.x. [DOI] [PubMed] [Google Scholar]
- 56.Sossin WS, Fan X, Saberi F. Expression and characterization of Aplysia protein kinase C: a negative regulatory role for the E region. J Neurosci. 1996b;16:10–18. doi: 10.1523/JNEUROSCI.16-01-00010.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stevens CF, Sullivan JM. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron. 1998;21:885–893. doi: 10.1016/s0896-6273(00)80603-0. [DOI] [PubMed] [Google Scholar]
- 58.Sugita S, Baxter DA, Byrne JH. Differential effects of 4-aminopyridine, serotonin, and phorbol esters on facilitation of sensorimotor connections in Aplysia. J Neurophysiol. 1997a;77:177–185. doi: 10.1152/jn.1997.77.1.177. [DOI] [PubMed] [Google Scholar]
- 59.Sugita S, Baxter DA, Byrne JH. Modulation of a cAMP/protein kinase A cascade by protein kinase C in sensory neurons of Aplysia. J Neurosci. 1997b;17:7237–7244. doi: 10.1523/JNEUROSCI.17-19-07237.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun ZY, Schacher S. Development of short-term heterosynaptic facilitation at Aplysia sensorimotor synapses in vitro is accompanied by changes in the functional expression of presynaptic serotonin receptors. J Neurophysiol. 1996;76:2250–2261. doi: 10.1152/jn.1996.76.4.2250. [DOI] [PubMed] [Google Scholar]
- 61.Sutton MA, Carew TJ. Parallel molecular pathways mediate expression of distinct forms of intermediate-term facilitation at tail sensory-motor synapses in Aplysia. Neuron. 2000;26:219–231. doi: 10.1016/s0896-6273(00)81152-6. [DOI] [PubMed] [Google Scholar]
- 62.Swartz KJ. Modulation of Ca2+ channels by protein kinase C in rat central and peripheral neurons: disruption of G protein-mediated inhibition. Neuron. 1993;11:305–320. doi: 10.1016/0896-6273(93)90186-u. [DOI] [PubMed] [Google Scholar]
- 63.Tanaka C, Nishizuka Y. The protein kinase C family for neuronal signaling. Annu Rev Neurosci. 1994;17:551–567. doi: 10.1146/annurev.ne.17.030194.003003. [DOI] [PubMed] [Google Scholar]
- 64.Tsunoda S, Sierralta J, Sun Y, Bodner R, Suzuki E, Becker A, Socolich M, Zuker CS. A multivalent PDZ-domain protein assembles signalling complexes in a G-protein-coupled cascade. Nature. 1997;388:243–249. doi: 10.1038/40805. [DOI] [PubMed] [Google Scholar]
- 65.Turner NA, Rumsby MG, Walker JH, McMorris FA, Ball SG, Vaughan PF. A role for protein kinase C subtypes α and ε in phorbol-ester-enhanced K+ and carbachol-evoked noradrenaline release from the human neuroblastoma SH-SY5Y. Biochem J. 1994;297:407–413. doi: 10.1042/bj2970407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vaughan PF, Walker JH, Peers C. The regulation of neurotransmitter secretion by protein kinase C. Mol Neurobiol. 1998;18:125–155. doi: 10.1007/BF02914269. [DOI] [PubMed] [Google Scholar]
- 67.Walaas SI. Regulation of calcium-dependent [3H]noradrenaline release from rat cerebrocortical synaptosomes by protein kinase C and modulation of the actin cytoskeleton. Neurochem Int. 1999;34:221–233. doi: 10.1016/s0197-0186(99)00007-8. [DOI] [PubMed] [Google Scholar]
- 68.Weeber EJ, Atkins CM, Selcher JC, Varga AW, Mirnikjoo B, Paylor R, Leitges M, Sweatt JD. A role for the β isoform of protein kinase C in fear conditioning. J Neurosci. 2000;20:5906–5914. doi: 10.1523/JNEUROSCI.20-16-05906.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yawo H. Protein kinase C potentiates transmitter release from the chick ciliary presynaptic terminal by increasing the exocytotic fusion probability. J Physiol (Lond) 1999;515:169–180. doi: 10.1111/j.1469-7793.1999.169ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yedovitzky M, Mochly-Rosen D, Johnson JA, Gray MO, Ron D, Abramovitch E, Cerasi E, Nesher R. Translocation inhibitors define specificity of protein kinase C isoenzymes in pancreatic β-cells. J Biol Chem. 1997;272:1417–1420. doi: 10.1074/jbc.272.3.1417. [DOI] [PubMed] [Google Scholar]
- 71.Zhao Y, Klein M. Modulation of release efficacy and of the RRP of transmitter in synaptic depression, facilitation by serotonin and post-tetanic potentiation at Aplysia sensorimotor synapses in culture. Soc Neurosci Abstr. 2000;26:1632. [Google Scholar]