

Abstract
Hypoxic-ischemic injury to the periventricular cerebral white matter [periventricular leukomalacia (PVL)] results in cerebral palsy and is the leading cause of brain injury in premature infants. The principal feature of PVL is a chronic disturbance of myelination and suggests that oligodendrocyte (OL) lineage progression is disrupted by ischemic injury. We determined the OL lineage stages at risk for injury during the developmental window of vulnerability for PVL (23–32 weeks, postconceptional age). In 26 normal control autopsy human brains, OL lineage progression was defined in parietal white matter, a region of predilection for PVL. Three successive OL stages, the late OL progenitor, the immature OL, and the mature OL, were characterized between 18 and 41 weeks with anti-NG2 proteoglycan, O4, O1, and anti-myelin basic protein (anti-MBP) antibodies. NG2+O4+ late OL progenitors were the predominant stage throughout the latter half of gestation. Between 18 and 27 weeks, O4+O1+ immature OLs were a minor population (9.9 ± 2.1% of total OLs; n = 9). Between 28 and 41 weeks, an increase in immature OLs to 30.9 ± 2.1% of total OLs (n = 9) was accompanied by a progressive increase in MBP+ myelin sheaths that were restricted to the periventricular white matter. The developmental window of high risk for PVL thus precedes the onset of myelination and identifies the late OL progenitor as the major potential target. Moreover, the decline in incidence of PVL at ∼32 weeks coincides with the onset of myelination in the periventricular white matter and suggests that the risk for PVL is related to the presence of late OL progenitors in the periventricular white matter.
Keywords: development, cell lineage, cerebral white matter, cerebral cortex, myelination, O4 antibody, O1 antibody, NG2, myelin basic protein, immunohistochemistry, neurofilament protein, microglia, periventricular leukomalacia, prematurity
Periventricular leukomalacia (PVL) is a common lesion of the periventricular cerebral white matter that has its peak incidence in the premature infant and underlies the subsequent development of cerebral palsy and cognitive impairment in survivors of the newborn critical care nursery (Volpe, 2000). The risk for PVL is high during a well defined period in human brain development (23-32 weeks, postconceptional age). Two major factors related to cerebrovascular immaturity predispose the developing periventricular white matter to injury from hypoxia-ischemia (Volpe, 1998). These are the presence in periventricular white matter of arterial end and border zones (Takashima and Tanaka, 1978; Nakamura et al., 1994) and a propensity for the sick premature neonate to exhibit a pressure-passive circulation related to a disturbance of cerebral autoregulation (Pyrds, 1991; Menke et al., 1997).
The major pathological feature of PVL is a chronic disturbance of myelination and suggests that oligodendrocyte (OL) progenitors are a target of ischemic injury in PVL (Back and Volpe, 1997). The susceptibility of the rat OL lineage to oxidative stress, a well established sequela of ischemia and reperfusion (Traystman et al., 1991), is a maturation-dependent phenomenon, and late OL progenitorsin vitro are markedly more susceptible than mature OLs to free radical-mediated injury (Back et al., 1998; Fern and Moller, 2000). However, the susceptibility of human OL progenitors to hypoxia-ischemia has been difficult to evaluate directly, and there is no animal model that reproduces the myelination disturbances of PVL. The objectives of the present study were to determine the human OL lineage stages at risk for injury during the developmental window of vulnerability for PVL and to determine whether the risk for PVL might correlate with the presence of more immature stages in the cerebral white matter.
The human OL lineage has been characterized mainly in first trimester spinal cord (for review, see Hardy, 1997), and there are no studies from the human cerebrum that are relevant to the critical period of vulnerability for PVL. During rodent white matter developmentin vivo, OL progenitors express the platelet-derived growth factor-α receptor and the NG2 proteoglycan (Levine et al., 1993; Nishiyama et al., 1996, 1999; Reynolds and Hardy, 1997). Late OL progenitors (pre-OLs) are identified by labeling with the O4 antibody and for NG2 but not with the O1 monoclonal antibody (Warrington and Pfeiffer, 1992; Reynolds and Hardy, 1997). Next in succession are immature OLs that label with both the O4 and O1 monoclonal antibodies but not with myelin basic protein (Warrington and Pfeiffer, 1992). The mature OL is characterized by expression of myelin-associated markers that include myelin basic protein (MBP) (Sternberger et al., 1978).
We report here that the developmental window of high risk for PVL coincides with a period in human cerebral white matter development during which one major population of late OL progenitors predominates and identifies them as a potential target for injury in PVL. The decline in incidence of PVL at ∼32 weeks coincides with the onset of myelination in the periventricular white matter and suggests a developmental explanation for PVL that is related to the timing of appearance of late OL progenitors in the periventricular white matter.
MATERIALS AND METHODS
Study population. Cases without significant brain pathology were prospectively collected from the pediatric autopsy populations of the Departments of Pathology at Oregon Health Sciences University (Portland, OR), Brigham and Women's Hospital (Boston, MA), Children's Hospital (Boston, MA), and the National Institutes of Health Developmental Brain and Tissue Bank at the University of Miami (Miami, FL; provided under the direction of Dr. Carol Petito). “Normalcy” was defined as cases without gross brain pathology and was confirmed by histological data with reference to agonal-metabolic derangements, pertinent clinicopathologic data, and the cause of death. Postmortem intervals of <24 hr were accepted for 24 of the 26 cases (one case each at 26 and 30 hr). Cases were processed for standard histological stains [hematoxylin and eosin (H+E) and H+E plus Luxol fast blue] to permit evaluation of tissue sections by standard histopathological criteria.
Tissue preparation and sectioning. Tissue was immersed at the time of collection in ice-cold 4% paraformaldehyde in 0.1m phosphate buffer and stored at 2–4°C. Immunocytochemical labeling with the O4 and O1 mAbs was unaffected by fixation in excess of 18 months. However, fixation for 6–18 hr yielded optimal immunocytochemical labeling for NG2. Tissue blocks were adhered to a metal stage, chilled on ice, and embedded in 1% low-gelling temperature agarose (A-4018; Sigma, St. Louis, MO). Free-floating sections (50 μm) were cut on a Leica VTS-1000 vibrating microtome in ice-cold PBS.
Primary antibodies. The O4 and O1 mouse monoclonal antibodies were isolated and purified by ammonium sulfate fractionation from the media of cultured hybridoma cells (Bansal et al., 1989) that were the generous gift of Dr. Steven Pfeiffer (University of Connecticut Health Center, Farmington, CT). Before dilution, the concentration of purified O4 was 11.3 mg/ml, and that of O1 was 3.8 mg/ml. The panaxonal neurofilament marker SMI 312 and the anti-myelin basic protein antibody SMI 99 were mouse monoclonal antibodies from Sternberger Monoclonals (Lutherville, MD). The Ricinus communis lectin microglial marker (biotinylated agglutinin RCA120, L-2641) and the mouse monoclonal antibody neuronal marker anti-β-tubulin isotype III (T-8660) were from Sigma. A rabbit anti-bovine glial fibrillary acidic protein (GFAP) antibody (Z-0334) was from Dako (Carpinteria, CA). Unless otherwise noted, all immunohistochemical procedures used a 1 hr incubation in blocking buffer (5% NGS in PBS), overnight incubation at 2–4°C with primary antibodies, and a 2 hr incubation at room temperature (RT; 20°C) with secondary antibodies, and all washes were 10 min in a minimum of 5 ml of PBS at RT. Antibodies were diluted in PBS with 3% NGS.
Immunofluorescence histochemistry for O4 or O1 antibodies.For single immunofluorescent labeling, the O4 or O1 mAb (1:1000) was visualized with a μ-chain-specific fluorescein-conjugated IgM secondary antibody (1:100; FI-2020; Vector Laboratories, Burlingame, CA). For double immunofluorescent labeling with O4 and O1, a biotinylated O4 (bO4) antibody was synthesized (Research Genetics, Huntsville, AL), because both primary antibodies are of the same class (IgM). Tissue sections were first labeled overnight with O1 (1:250), followed by incubation for 2 hr at RT in fluorescein-conjugated IgM secondary antibody (1:100). Sections were next incubated overnight with bO4 antibody (1:100), followed by incubation with rhodamine red X-conjugated streptavidin (1:400 in PBS; 016-290-084; Jackson ImmunoResearch, West Grove, PA). For double immunofluorescent labeling with O4 or O1 (1:1000) and SMI 312 (1:1000), anti-β-tubulin isotype III (1:100), anti-GFAP (1:1000), or biotinylated agglutinin RCA120 (1:100), both primary antibodies were coincubated with tissue sections. SMI 312 and anti-β-tubulin isotype III were visualized with Fc fragment-specific rhodamine red X-conjugated anti-mouse IgG (1:200; 115-295-071; Jackson ImmunoResearch). Anti-GFAP was visualized with anti-rabbit IgG conjugated to Texas Red (TI-1000; Vector Laboratories). Biotinylated agglutinin RCA120 was visualized with rhodamine red X-conjugated streptavidin (1:400 in PBS). Nonspecific labeling with O1 mAb, when noted, was related to a prolonged postmortem interval or a high titer of O1. It was markedly decreased both in magnitude and distribution as the concentration of O1 was reduced from 800 to 4 μg/ml, whereas OL-specific labeling with O1 was unchanged. Specific labeling with O1 was achieved at a concentration of 1–2 μg/ml.
Immunofluorescence double labeling of O4 or O1 antibodies and NG2. After tissue sections were labeled with O4 or O1 (1:1000), they were washed, incubated for 5 min in ice-cold 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, and immediately washed three times in PBS at RT. After a 2 hr incubation with μ-chain-specific fluorescein-conjugated IgM (1:100), all subsequent incubations were done in the dark. Tissue sections were next incubated overnight at 2–4°C with a rabbit polyclonal antibody raised against NG2 (Ong and Levine, 1999) (1:1000 in PBS with 3% NGS and 0.1% Triton X-100). After incubation with biotinylated goat anti-rabbit IgG (1:200; 111-065-046; Jackson ImmunoResearch), sections were incubated for 2 hr with rhodamine red X-conjugated streptavidin (1:400). Immunocytochemical controls, processed under conditions identical to those described above, revealed no cross-reactivity between O4 or O1 and the Fc fragment-specific IgG secondary antibody. With omission of both primary antibodies, there was no detectable nonspecific labeling with either secondary antibody. It should be noted that the 5 min incubation in 4% paraformaldehyde was required to prevent the solubilization of the O4–antigen complex by 0.1% Triton X-100 that was necessary for optimal visualization of NG2.
Quantitative morphometric analysis of O4- and O1-labeled cells. Cell counts were obtained from a minimum of three sets of 50-μm-thick adjacent sections that were alternately stained with O4 or O1. Absolute counts were not determined because of the uneven thickness inherent in vibrating microtome-cut sections (Guillery and Herrup, 1997). The nucleus was selected as the smallest countable object and was visualized by immunofluorescent counterstain with Hoechst 33324. Cells were systematically counted in similar sectors of adjacent sections that included superficial, mid and deep cerebral white matter, and the germinal matrix. Cell profiles that contained a nucleus were counted with a 20× objective equipped with a counting grid that was mounted on a Nikon Eclipse TE 300 inverted fluorescent microscope. The total O4-labeled cells counted varied with the gestational age of the case and ranged from 433 to 2867. In 14 cases, at least 1000 total cells were counted. Immature OLs were calculated as the percentage of total (i.e., O4-labeled) cells that labeled with O1.
Because the nucleus was selected as the smallest countable object, a difference in the size of the nucleus for an O4- or an O1-labeled cell would introduce a bias into the relative cell counts. To estimate whether the nucleus differed in size for cells labeled with O4 versus O1, the average nuclear area was determined for O4- or O1-labeled cell profiles from three adjacent sections from a case at 18 and at 35 weeks. We predicted that if nuclear size varied with the state of OL differentiation, that this difference would be amplified by comparing a case at midgestation (i.e., 18 weeks) with one close to term. Nuclei, labeled with Hoechst 33324, were randomly but systematically sampled from a sector that included superficial, mid, and deep cerebral white matter. Double-labeled profiles were visualized with a 40× oil immersion objective (Nikon Plan Fluor; numerical aperture, 1.30) on a laser-scanning confocal microscope (Noran Instruments) equipped with argon ion and UV lasers coupled to an inverted microscope (Nikon Diaphot 200). Nuclei were visualized by excitation with 360 nm (λ) light using a UV bandpass filter, and fluorescence emission was detected at >470 nm (λ). The beam was attenuated using a 25 nm dichroic confocal aperture. For all measurements, the laser settings that included brightness, contrast, and exposure time (100 nsec) were held constant. Frame-averaged confocal images (128 frames/image) were digitalized at 512 × 480 pixels using microcomputer-based imaging software (Noran OZ with Intervision). For analysis of nuclear area, regions of interest were selected by image threshold analysis. Mean nuclear area was calculated from a minimum of 40 cellular profiles for all four groups sampled.
At 18 weeks, there was no significant difference (paired Student'st test) between the size of the nucleus of an O4+ cell (39.0 ± 8.3 μm2; n= 112 profiles) and that of an O1+ cell (37.5 ± 8.3 μm2; n = 40 profiles). At 35 weeks, there was also no difference between the size of the nucleus of an O4+ cell (54.5 ± 10.9 μm2; n = 164 profiles) and that of an O1+ cell (54.8 ± 10.6 μm2; n = 93 profiles). The size of the nucleus was significantly less (unpaired Student'st test) at 18 weeks than at 35 weeks for an O4+ cell (p < 0.0001) or for an O1+ cell (p < 0.0001). We concluded that at any given gestational age, there was no difference in the size of the nucleus of a pre-OL or an immature OL.
RESULTS
Clinicopathologic information
The parietal white matter at the level of the trigone was analyzed in 26 human brains, ranging in age from 18 to 41 postconceptional weeks. The mean postmortem interval of autopsy cases was 19 ± 7 hr. This study involved analysis of cases at the following postconceptional ages in weeks: 18 (n = 1), 20 (n = 3), 21 (n = 1), 22 (n = 3), 24 (n = 2), 26 (n = 1), 27 (n = 2), 28 (n = 1), 30 (n = 2), 31 (n = 1), 34 (n = 1), 35 (n = 2), 36 (n = 1), 37 (n = 1), 40 (n = 3), and 41 (n = 1). The brains were free of PVL or other major malformative or acquired lesions. The causes of death were as follows: therapeutic abortion (n = 7), stillborn (n = 1), extreme prematurity (n = 1), congenital heart disease (n = 3), major noncerebral congenital malformations (n = 5), respiratory distress (n = 5), fetal distress at delivery (n= 2), sepsis (n = 1), and undetermined (n = 1).
Numerous O4- and NG2-labeled cells populate the human cerebrum by midgestation
In a series of 13 cases between 18 and 27 weeks, we first examined the distribution of cells that labeled with the O4 antibody. At 18 weeks, numerous O4-labeled somata were present in both the parietal white matter and the cortical mantle. The distribution of O4-labeled somata in the white matter was nonuniform (Fig.1A). They were more numerous in the superficial (Fig. 1B) and mid (Fig.1C) cerebral white matter, and fewer somata were present in the deep white matter (Fig. 1D) or the germinal matrix (Fig. 1E). Many O4-labeled somata were also present in the cortical mantle as early as 18 weeks (Fig.2A). These cells appeared less differentiated than those in the white matter and were associated with a fine meshwork of narrow processes.
Fig. 1.
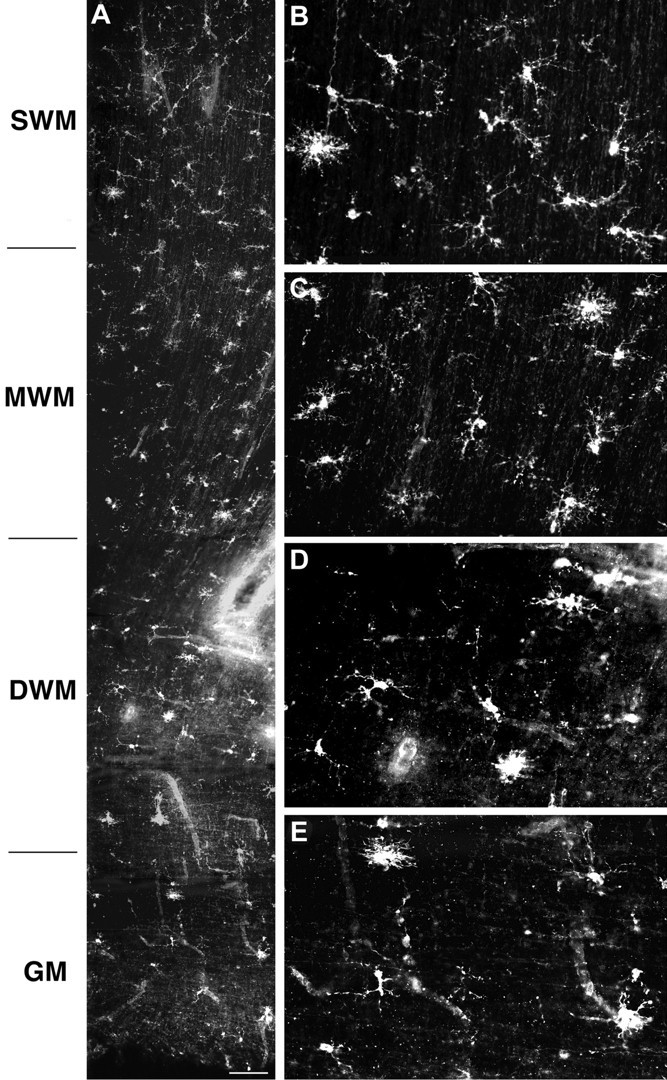
Distribution of O4 mAb-labeled cells in the cerebral white matter at 18 weeks. A, Low-power photomontage showing the regional distribution of cells in the superficial (SWM), mid (MWM), and deep (DWM) cerebral white matter and the germinal matrix (GM). B–E, Detail of the morphology and distribution of cells shown in A in theSWM (B), MWM(C), DWM(D), and GM(E). Scale bar: A, 110 μm.
Fig. 2.
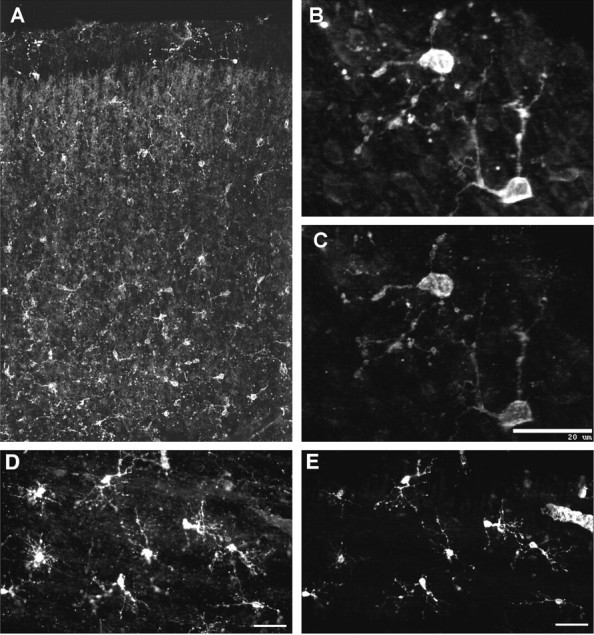
A, Low-power photomontage shows the regional distribution of O4 mAb-labeled cells in the cortical mantle from a case at 18 weeks. B–E, Confocal laser digital images demonstrate that OL precursors in the cortical mantle (B, C) and in the cerebral white matter (D, E) labeled for both the O4 antibody (B, D) and NG2 (C, E). Note that the cells in the cortical mantle appear morphologically less mature compared with those in the white matter. Scale bars: B, C, 20 μm; D, E, 25 μm.
NG2-labeled cells were visualized in four cases between 18 and 26 weeks of gestation. In all cases, numerous NG2-labeled cells were present in the white matter and cortical mantle. The morphology and distribution of these cells were very similar to that visualized with the O4 antibody. Immunocytochemical double-labeling studies with the O4 and anti-NG2 antibodies demonstrated that the entire population of NG2-labeled cells in the cortical mantle (Fig. 2C) and in the white matter (Fig. 2E) also labeled with O4 (Fig.2B,D).
The O4+NG2+ cells were morphologically diverse (Fig.3). Some cells were characterized by an asymmetric bipolar morphology (Fig. 3A). The asymmetry derived from an initial short process that branched into two major processes. Other bipolar cells were typically elongated or fusiform in shape with a similar degree of branching from each pole (Fig.3B). Another population of asymmetric simple multipolar cells had several processes that were elaborated from one pole of the soma (Fig. 3C). Other simple multipolar cells had more uniformly distributed processes (Fig. 3D). Both types of bipolar cells were most numerous in the superficial white matter and were less common in the mid and deep white matter.
Fig. 3.

Representative confocal laser microscopic digital three-dimensional reconstructions of NG2-positive cells in the cerebral white matter. Representative examples of the variety of morphological subtypes of pre-OLs identified in the white matter are shown.A, An asymmetric bipolar cell. B, A symmetric bipolar cell. C, An asymmetric multipolar cell. D, Simple and complex multipolar cells. Scale bars: A–D, 25 μm.
To confirm that the NG2+O4+ cells were distinct from other neural cells in fetal human brain, we stained sections with antibodies specific for neurons, microglia, and astrocytes. Neuronal somata and processes identified by β-tubulin isotype III differed in morphology and distribution from that of O4+ somata in the cortical mantle (Fig.4A) and white matter (Fig. 4B). Astrocytes identified by glial fibrillary acidic protein also did not overlap with O4+ somata (data not shown). We examined two markers that label microglia in the developing human brain (Streit et al., 1999). The LN-3 antibody, reactive against HLA-DR antigen, detected reactive microglia at sites of cerebral necrosis in a case at term gestation but failed to detect most of the resident population of ramified and/or resting microglia (data not shown). However, the R. communis lectin RCA120 detected numerous ramified microglia in the cerebral white matter and cortical mantle in this same case and five additional cases at 18, 20, 31, 35, and 40 weeks of gestation. Although the RCA120-labeled microglia had a morphology and distribution similar to that of the bipolar and simple multipolar cells visualized with the O4 antibody, we found no overlap in the distribution of cells labeled by these two markers (Fig.4C).
Fig. 4.
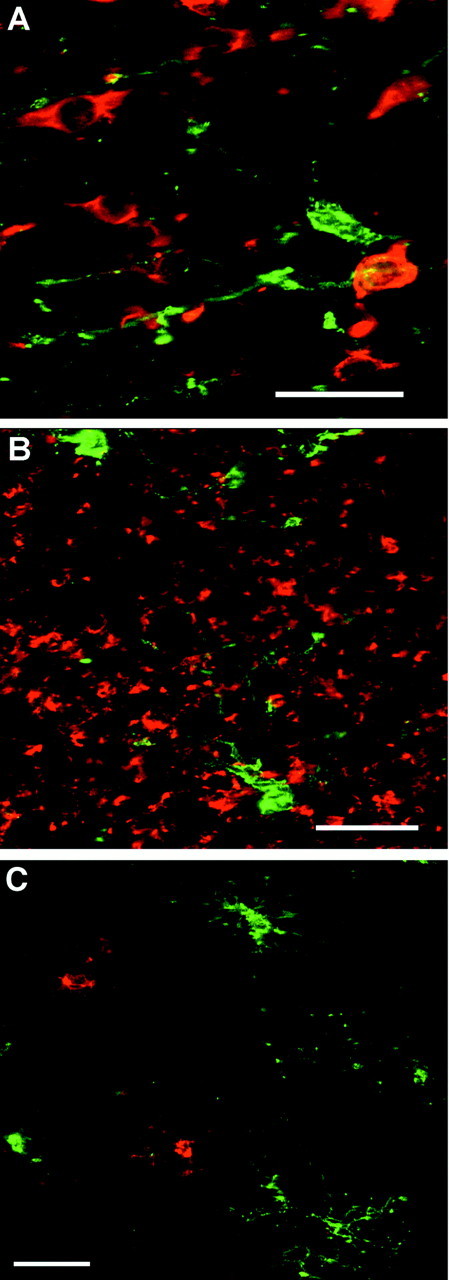
Representative confocal laser microscopic digital images demonstrate the specificity of the O4 antibody for OL precursors. A, B, O4-labeled cells (green) differ in morphology and distribution from neuronal somata (red) in the cortical mantle (A) and from neuronal processes (red) in the superficial white matter (B) that were visualized with anti-β-tubulin isotype III. C, Microglia labeled with the R. communis lectin (red) have a similar morphology but a distinctly different distribution from that of O4-labeled cells (green) in the superficial white matter. Scale bars: A, 20 μm; B, C, 25 μm.
Few O1-labeled cells are present in the human cerebrum between 18 and 27 weeks
In all 13 cases examined between 18 and 27 weeks, numerous cells were visualized with the O4 antibody (Fig.5A), but few O1-labeled cells were visualized in the white matter (Fig. 5B), and none was visualized in the cerebral cortex. All of the O1-labeled cells displayed a complex multipolar morphology characterized by a round soma that elaborated numerous extensively branched processes.
Fig. 5.
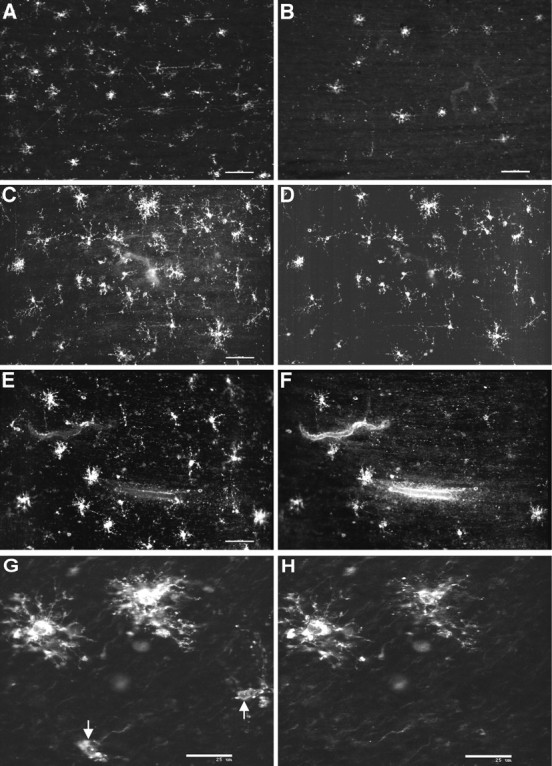
Pre-OLs are more abundant than immature OLs in human parietal white matter at midgestation. Representative examples of the morphology and distribution of pre-OLs and immature OLs from cases between 18 and 22 weeks of gestation in human parietal white matter are shown. A, B, Cells labeled with O4 (A) were more numerous than those labeled with O1 (B). C, D, The morphology and distribution of cells labeled with the native O4 antibody (C) and a bO4 antibody (D) were quite similar. E, F, Immunofluorescent double labeling with the bO4 (E) and O1 (F) antibodies in the mid cerebral white matter demonstrates that bO4 and O1 labeled a minor population of immature OLs with a complex multipolar morphology. G, H, Confocal laser microscopic digital images are shown of the morphology of complex multipolar immature OLs that labeled with both the bO4 (G) and the O1 (H) antibodies. Note the less differentiated-appearing pre-OLs that labeled with bO4 but not O1 (arrows). Scale bars: A, B, 100 μm; C–F, 60 μm; G, H, 25 μm.
We next compared the distribution of cells visualized with the O1 antibody with that of cells labeled with a bO4 antibody (see Materials and Methods). We first determined the effect of biotinylation of O4 on the specificity of the antibody. Double-labeling studies with the bO4 (Fig. 5D) and native O4 (Fig. 5C) antibodies demonstrated a complete overlap in the staining obtained with the two antibodies. The O1-labeled cells (Fig.5F) were a small subset of the cells that labeled with bO4 antibody (Fig. 5E). All of the O1-labeled cells also labeled with the bO4 antibody. The double-labeled cells displayed a complex multipolar morphology (Fig. 5G,H), whereas the cells that labeled with only the bO4 antibody had a more simple appearance (Fig. 5G, arrows). Double-labeling studies with O1 and for NG2 revealed an occasional NG2-positive cell that stained weakly for O1, but most of these cells did not stain for O1 (data not shown; see also Fig. 7C,D).
Fig. 7.

Double immunofluorescent-labeling studies from a case at 32 weeks. Pre-OLs are a morphologically and immunohistochemically distinct population of OL precursors that persist in human cerebral white matter during the expansion in the immature OL population. Pre-OLs were distinguished by diverse morphologies similar to those observed earlier in development. A, B, Representative examples of pre-OLs in the superficial white matter that strongly labeled with O4 (A) and for NG2 (B) are shown. C, Note inC that the NG2-positive pre-OLs (arrowheads) appear morphologically less mature compared with an O1-positive immature OL (D) with a complex multipolar morphology (arrow). Scale bars:A–D, 20 μm.
O4+O1+ immature OLs increase at ∼30 weeks
Approximately 30 weeks marked the onset of an increase in the number of O4+ cells that displayed a complex multipolar morphology (Fig. 6A,B). These cells were most extensive in the deep and mid cerebral white matter, were sparsely localized to the superficial white matter, and were not detected in the cerebral cortex. In 12 cases between 30 and 41 weeks, the O4 and O1 antibodies identified a similar population of complex multipolar cells. Fluorescent double-labeling studies with the bO4 and O1 antibodies confirmed that these cells were a single population of O4+O1+ immature OLs (Fig. 6C,D). Interspersed among the immature OLs were numerous O4+O1− pre-OLs (Fig.6C,D, arrows). The pre-OLs appeared morphologically less mature than did the immature OLs and were most numerous in the superficial white matter.
Fig. 6.

Pre-OLs persist as the expansion in the immature OL population occurs at ∼30 weeks. Representative confocal laser microscopic digital images from a case at 31 weeks demonstrate the striking change in the morphology of OL precursors that occurs with the expansion of the immature OL population in the parietal white matter.A, B, A low-power image (A) and detail (B) demonstrate the marked increase in complex multipolar cells visualized here in the mid cerebral white matter with the O4 antibody. C, D, Double immunofluorescent-labeling studies with a biotinylated O4 antibody (C) and the O1 antibody (D) revealed that many of the O4-labeled cells were O4+O1− pre-OLs (arrows) that were interspersed among the O4+O1+ immature OLs. Scale bars: A, B, 100 μm; C, D, 20 μm.
In two cases at 32 and 37 weeks, numerous cells were identified that labeled with O4 and for NG2. In general, cells that appeared morphologically less mature labeled with similar intensity for both antibodies (Fig. 7A,B). However, complex multipolar cells that labeled with O1 (Fig.7D, arrow) or with O4 (data not shown) labeled weakly for NG2. The less mature-appearing cells labeled strongly for NG2 but did not label with O1 (Fig. 7C, arrowheads).
Distribution of MBP between 18 and 40 weeks
The distribution of MBP, visualized by SMI 99 immunoreactivity, was determined in the parietal white matter in 15 cases between 18 and 40 weeks. In seven cases examined between 18 and 27 weeks, no MBP labeling was detected (Fig.8A). In eight cases examined between 30 and 40 weeks, MBP labeling was first detected at ∼30 weeks and was restricted to the periventricular white matter, which included the optic radiation (Fig. 8B). By 40 weeks, MBP labeling was much more extensively visualized in the periventricular white matter (Fig. 8C) and was often present in more superficial regions of the white matter (data not shown).
Fig. 8.

The temporal progression of myelinogenesis in human parietal periventricular white matter between 18 and 40 weeks was assessed by immunohistochemical localization of MBP. Representative examples of the distribution of MBP are shown at 20 weeks (A), 30 weeks (B), and 40 weeks (C). The asterisk is adjacent to the ependymal surface of the lateral ventricle. Scale bar:A–C, 100 μm.
Pre-OLs predominate throughout the high-risk period for PVL
To identify the major OL lineage stage in the parietal white matter when the risk of PVL is high (i.e., 23-32 weeks), we determined the relative percentage of pre-OLs and immature OLs present in 18 individual cases that ranged in age from 18 to 41 weeks. To quantify these two OL stages, the nucleus was selected as the smallest countable object. We confirmed that at any given gestational age there was no difference in the size of the nucleus of a pre-OL or an immature OL (see Materials and Methods). Figure 9shows the temporal progression of the percentage of immature OLs between 18 and 41 weeks. Between 18 and 27 weeks (n = 9), the O1-labeled cells comprised 9.9 ± 2.1% of total cells (mean ± SE; one-way ANOVA). At ∼28–30 weeks, a progressive increase in the number of O1-labeled cells was observed. Between 28 and 41 weeks (n = 9), the O1-labeled cells comprised 30.9 ± 2.1% of total cells (mean ± SE; one-way ANOVA). There was thus approximately a threefold increase in the percentage of O1-positive cells observed at 28–41 weeks when compared with 18–27 weeks (p < 0.0001).
Fig. 9.

Quantitative analysis of the timing of appearance of pre-OLs and immature OLs in human cerebral white matter between 18 and 41 weeks. Each time point represents an individual case in which the percentage of immature OLs was determined from the total OL somata that labeled with the O1 antibody. Because the O4 antibody was found to label the entire population of OL precursors at all ages examined, total OL precursors were determined from the number of cells that labeled with O4 (see Materials and Methods). Note that O1+ immature OLs are a minor population between 18 and 28 weeks. Approximately 28–30 weeks mark the onset of an expansion in the immature OL population.
DISCUSSION
Definition of the cellular and molecular events that regulate OL lineage progression in the human CNS is important for the understanding of developmental disorders of cerebral white matter (Back and Volpe, 1999). This study identified three successive stages of the human OL lineage between midgestation and term birth in human cerebral white matter (Fig. 10): the pre-OL, the immature OL, and the mature OL. The major features of the lineage progression were the following: (1) Pre-OLs were present as early as 18 weeks in both the cerebral white matter and cortex and were the major OL stage throughout the latter half of gestation. Pre-OLs labeled for NG2 and with the O4 antibody but not with the O1 antibody. (2) Between 18 and 27 weeks, immature OLs were a minor population, present only in the white matter, that labeled strongly with the O4 and O1 antibodies but very weakly for NG2. Immature OLs were further distinguished by a complex multipolar morphology, whereas pre-OLs were morphologically more diverse and appeared less differentiated. (3) At ∼30 weeks, immature OLs increased in number only in the white matter. The increase in immature OLs coincided with the restricted localization of MBP-positive myelin sheaths to the periventricular white matter. Thus, during the high-risk period for PVL (Fig. 10), pre-OLs comprised ∼90% of the total cells derived from the OL lineage.
Fig. 10.

Summary diagram of OL lineage progression in human cerebral white matter during the latter half of gestation. Note that the high-risk period for PVL coincides with the developmental epoch when the white matter is mostly populated by O4+O1− pre-OLs that also label with NG2. O4+O1+ immature OLs are a minor population until ∼30 weeks (dotted line) when they undergo a marked expansion. This expansion in the immature OL population is accompanied by the appearance in the periventricular white matter of MBP+ mature OLs. Hence the decline in incidence of PVL at ∼32 weeks coincides with OL maturation in the periventricular white matter.wks, Weeks.
The human OL lineage was defined with several OL-specific markers and by morphological criteria (Pfeiffer et al., 1993; Nishiyama et al., 1999). Despite the morphological diversity of human pre-OLs, their immunohistochemical phenotype identified them as oligodendroglial in origin and distinct from astroglia, resting or reactive microglia, and neuronal precursors. Restricted foci of O4-positive progenitors were detected in the ventral ventricular zone of the human spinal cord as early as 6 weeks (Hajihosseini et al., 1996). In agreement with most (Levine et al., 1993; Nishiyama et al., 1997; Reynolds and Hardy, 1997;Chang et al., 2000) but not all (Pouly et al., 1999) studies, our results in developing human brain also indicate that NG2 is specific to the OL lineage in vivo.
The timing of appearance of early human OL progenitors cannot be established from our study. Although early OL progenitors were cultured from fetal (Rivkin et al., 1995) and adult (Scolding et al., 1995; Roy et al., 1999) human brain, we did not detect NG2+O4− OL progenitors at midgestation or later in parietal white matter or at the foramen of Monro, a region with a well developed ventricular zone (data not shown). These previous studies may have isolated pre-OLs that reverted to an early progenitor phenotype in culture, as do rat pre-OLs in culture (Gard and Pfeiffer, 1993; Back et al., 1998). We also did not observe migratory streams of OL progenitors, but rather pre-OLs were diffusely distributed throughout the white matter by 18 weeks. In contrast to the rodent spinal cord (for review, see Hardy, 1997), in which the migration of OL progenitors occurs at midgestation, O4+ progenitors migrate from the human ventral ventricular zone of the cord considerably earlier between 6 and 8 weeks (Hajihosseini et al., 1996). NG2+ OL progenitors are abundant in the adult rat and human brain (Levine et al., 1993; Nishiyama et al., 1996, 1997; Reynolds and Hardy, 1997; Ong and Levine, 1999; Chang et al., 2000). In adult human brain, NG2+ progenitors appear to be oligodendroglial in origin and as abundant as mature OLs (Chang et al., 2000). The relationship of these progenitors to pre-OLs in fetal or adult (Armstrong et al., 1992;Wolswijk, 1998) human brain is presently unclear. Because adult NG2-positive progenitors would appear to be more abundant than we observed in developing human brain, it is possible that adult human progenitors may subserve more diverse functions than during development.
The role of the minor population of human immature OLs between 18 and 28 weeks is unclear. Although O1-positive cells at 18 weeks had no significant difference in nuclear area compared with that of O4-positive cells (see Materials and Methods), they were morphologically more complex. In contrast to the rat, in which O1 positivity correlates with a rapid commitment to myelination (Warrington and Pfeiffer, 1992), human immature OLs were detected at least 3 months before the onset of myelinogenesis. Myelinogenesis began at ∼30 weeks and was restricted to the periventricular white matter despite the presence of numerous immature OLs in more superficial white matter. Hence, factors extrinsic to the immature OL seem to influence the onset and progression of myelinogenesis.
The restricted localization of early myelinogenesis to the periventricular white matter appears to be developmentally specific. Although MBP-positive myelin sheaths were detected immunohistochemically by 30 weeks, sulfatide by thin layer chromatography (TLC) and MBP by SDS-PAGE were not detected in parietal white matter until after birth (Kinney et al., 1994). The appearance of microscopic myelin does not occur until at least after the first postnatal month, and myelin tubes were not detected until 11–13 postconceptional months (Brody et al., 1987; Kinney et al., 1989). Hence, immunohistochemistry appears to be more sensitive than TLC or SDS-PAGE to detect myelin constituents. Interestingly, recent studies of infants between 24 and 40 weeks after conception with diffusion tensor magnetic resonance imaging showed changes in water diffusion that correlate with the time course for parietal myelinogenesis determined here by immunohistochemistry. Thus, in central white matter, relative anisotropy, a measure of preferred directionality of water parallel to fiber tracts, increased markedly from 28 to 40 weeks and occurred in parallel with a decline in overall water diffusion, as measured by the apparent diffusion coefficient (Huppi et al., 1998). This combination of findings implies restriction of diffusion perpendicular to fiber tracts and could relate to the progressive increase in myelinogenesis that we observed between 30 and 40 weeks.
We found substantial differences in the temporal progression of myelinogenesis between rodents and man. Whereas numerous human immature OLs are present before birth, they do not increase in the rat until the first postnatal week (Reynolds and Hardy, 1997; Back et al., 1998). Human myelinogenesis begins early in the third trimester, but myelinogenesis does not begin in the rat forebrain until approximately the second postnatal week. The marked difference in timing of OL lineage progression between humans and rats may partly account for the failure to develop a suitable perinatal rat model of PVL. Our results underscore the importance of human developmental studies to identify temporal mismatches between lineage progression in humans and experimental animal models.
Clinical correlations
A central unresolved question regarding the pathogenesis of PVL is the cellular basis for the increased susceptibility of fetal cerebral white matter to injury as gestational age decreases (Back and Volpe, 1997). Rorke (1982) reported an apparent depletion of myelination glia, putative OL precursors, in cases with PVL, but this observation has not been reevaluated with OL-specific markers. One aim of our study was, thus, to identify the stages in the OL lineage that may be susceptible to injury at sites of PVL. Because impaired myelination is the principal pathological sequela of PVL, OL precursors are hypothesized to be a target cell of ischemia-reperfusion injury to the white matter (Kinney and Back, 1998). That OL precursors may be susceptible to injury in PVL is supported by recent in vitro studies that demonstrated that OL progenitors are significantly more susceptible than are mature OLs to death triggered by oxidative stress (Back et al., 1998; Fern and Moller, 2000). We demonstrated here that the parietal periventricular white matter, a region of high predilection for PVL, is populated mostly by pre-OLs between 24 and 32 weeks of gestation, the period when the incidence of PVL is highest (Fig. 10). Interestingly, the developmental window for PVL thus coincides with a period before the onset of myelination of periventricular white matter. This period is dominated by one major population of pre-OLs and identifies this OL stage as a potential target for hypoxic-ischemic injury in PVL. The decline in the incidence of PVL coincides with the onset of maturation of the periventricular white matter at ∼30 weeks. Hence, the differentiation of OL precursors to mature OLs may underlie the increased resistance of the periventricular white matter to injury and could account for the decline in the incidence of PVL as term gestation approaches.
Future studies
Further studies are needed to define better the biology of human pre-OLs in vivo. Although rodent pre-OLs are mitotically active, it remains unresolved, for example, whether human pre-OLs actively divide in vivo during development or adulthood and whether their proliferation is affected by brain injury (Wolswijk, 1998). Also unclear is whether human pre-OLs are directly killed by hypoxia-ischemia or whether cell death might be triggered by disruption of trophic support during a critical period in white matter development. To understand the cellular pathogenesis of PVL, studies are needed to determine whether late OL progenitors are depleted at sites of PVL and, if depleted, what mechanisms may regulate their potential to repopulate the white matter after injury.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS 01855 to S.A.B., NIH Grant P30 HD 33703, Oregon Child Health Research Center, Training Program Award for Pediatric Physician-Scientists to S.A.B., NIH–National Institute of Child Health and Human Development Grant P30 HD 18655 to J.J.V., a Grass Foundation Morison Fellowship and a Hearst Foundation Award to S.A.B., and an award from the Sudden Infant Death Syndrome Alliance to H.C.K. We gratefully acknowledge the support of Drs. Linda Wallen, Randall Nixon, and Geoffrey Murdoch at Oregon Health Sciences University and the NIH Developmental Brain and Tissue Bank at the University of Miami under the direction of Dr. Carol Petito (NO1-HD-8-3284) for their support in the acquisition of cases. We also thank Drs. Edward G. Jones, Gregory Popken, and Clifford Saper for guidance on the morphometric studies, Dr. Peter Rotwein for the generous use of his microscope, and Dr. Gary Sexton for generous assistance with statistical analysis. We are grateful to Drs. Julie Ellison, John Fiala, Jonathan Flax, Montey Gates, John Kornhauser, Jeffrey Macklis, Akiko Nishiyama, Steven Pfeiffer, Scott Pomeroy, and Wolfgang Streit for their helpful advice and discussions. We thank Luciana Rava and Richard Belliveau for their excellent technical assistance.
Correspondence should be addressed to Dr. Stephen A. Back, Department of Pediatrics, Hatfield Research Center, Room 516, Oregon Health Sciences University, 3181 Southwest Sam Jackson Park Road, Portland, OR 97201-3098. E-mail: Backs@ohsu.edu.
REFERENCES
- 1.Armstrong RC, Dorn HJ, Kufta CV, Friedman E, Dubois-Dalcq ME. Pre-oligodendrocytes from adult human CNS. J Neurosci. 1992;12:1538–1547. doi: 10.1523/JNEUROSCI.12-04-01538.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Back SA, Volpe JJ. Cellular and molecular pathogenesis of periventricular white matter injury. Ment Retard Dev Disabil Res Rev. 1997;3:96–107. [Google Scholar]
- 3.Back SA, Volpe JJ. Approaches to the study of diseases involving oligodendroglial death. In: Koliatsos V, Ratan RR, editors. Cell death and diseases of the nervous system. Humana; Totowa, NJ: 1999. pp. 401–428. [Google Scholar]
- 4.Back SA, Gan X-D, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bansal R, Warrington AE, Gard A, Ranscht B, Pfeiffer SE. Multiple and novel specificities of monoclonal antibodies O1, O4, and R-mAb used in the analysis of oligodendrocyte development. J Neurosci Res. 1989;24:548–557. doi: 10.1002/jnr.490240413. [DOI] [PubMed] [Google Scholar]
- 6.Brody BA, Kinney HC, Kloman AS, Gilles FH. Sequence of central nervous system myelination in human infancy. I. An autopsy study of myelination. J Neuropathol Exp Neurol. 1987;46:283–301. doi: 10.1097/00005072-198705000-00005. [DOI] [PubMed] [Google Scholar]
- 7.Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD. NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci. 2000;20:6404–6412. doi: 10.1523/JNEUROSCI.20-17-06404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fern R, Moller T. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci. 2000;20:34–42. doi: 10.1523/JNEUROSCI.20-01-00034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gard AL, Pfeiffer SE. Glial cell mitogens bFGF and PDGF differentially regulate development of O4+ GalC− oligodendrocyte progenitors. Dev Biol. 1993;159:618–630. doi: 10.1006/dbio.1993.1269. [DOI] [PubMed] [Google Scholar]
- 10.Guillery R, Herrup K. Quantification without pontification: choosing a method for counting objects in sectioned tissue. J Comp Neurol. 1997;386:2–7. doi: 10.1002/(sici)1096-9861(19970915)386:1<2::aid-cne2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 11.Hajihosseini M, Tham T, Dubois-Dalcq M. Origin of oligodendrocytes within the human spinal cord. J Neurosci. 1996;16:7981–7994. doi: 10.1523/JNEUROSCI.16-24-07981.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hardy R. Dorsoventral patterning and oligodendroglial specification in the developing central nervous system. J Neurosci Res. 1997;50:139–145. doi: 10.1002/(SICI)1097-4547(19971015)50:2<139::AID-JNR3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 13.Huppi P, Maier S, Peled S, Zientara G, Barnes P, Jolesz P, Volpe J. Microstructural development of human newborn cerebral white matter assessed in vivo by diffusion tensor magnetic resonance imaging. Pediatr Res. 1998;44:584–590. doi: 10.1203/00006450-199810000-00019. [DOI] [PubMed] [Google Scholar]
- 14.Kinney HC, Back SA. Human oligodendroglial development: relationship to periventricular leukomalacia. Semin Pediatr Neurol. 1998;5:180–189. doi: 10.1016/s1071-9091(98)80033-8. [DOI] [PubMed] [Google Scholar]
- 15.Kinney HC, Brody BA, Kloman AS, Gilles FH. Sequence of central nervous system myelination in human infancy. II. Patterns of myelination in autopsied infants. J Neuropathol Exp Neurol. 1989;47:217–234. doi: 10.1097/00005072-198805000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Kinney HC, Karthigasan J, Borenshteyn N, Kirschner DA. Myelination in the developing human brain: biochemical correlates. Neurochem Res. 1994;19:983–996. doi: 10.1007/BF00968708. [DOI] [PubMed] [Google Scholar]
- 17.Levine JM, Stincone F, Lee YS. Development and differentiation of glial precursor cells in the rat cerebellum. Glia. 1993;7:307–321. doi: 10.1002/glia.440070406. [DOI] [PubMed] [Google Scholar]
- 18.Menke J, Michel E, Hillebrand S. Cross-spectral analysis of cerebral autoregulation dynamics in high risk preterm infants during the perinatal period. Pediatr Res. 1997;42:690–699. doi: 10.1203/00006450-199711000-00023. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura Y, Okudera T, Hashimoto T. Vascular architecture in white matter of neonates: its relationship to periventricular leukomalacia. J Neuropathol Exp Neurol. 1994;53:582–589. doi: 10.1097/00005072-199411000-00005. [DOI] [PubMed] [Google Scholar]
- 20.Nishiyama A, Lin X-H, Giese N, Heldin C, Stallcup W. Co-localization of NG2 proteoglycan and PDGF α receptor on O2A progenitor cells in the developing rat brain. J Neurosci Res. 1996;42:299–314. doi: 10.1002/(SICI)1097-4547(19960201)43:3<299::AID-JNR5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 21.Nishiyama A, Yu M, Drazba J, Tuohy V. Normal and reactive NG2+ glial cells are distinct from resting and activated microglia. J Neurosci Res. 1997;48:299–312. doi: 10.1002/(sici)1097-4547(19970515)48:4<299::aid-jnr2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 22.Nishiyama A, Chang A, Trapp B. NG2+ glial cells: a novel glial cell population in the adult brain. J Neuropathol Exp Neurol. 1999;58:1113–1124. doi: 10.1097/00005072-199911000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Ong WY, Levine JM. A light and electron microscopic study of NG2 chondroitin sulfate proteoglycan-positive oligodendrocyte precursor cells in the normal and kainate-lesioned hippocampus. Neuroscience. 1999;92:83–95. doi: 10.1016/s0306-4522(98)00751-9. [DOI] [PubMed] [Google Scholar]
- 24.Pfeiffer SE, Warrington AE, Bansal R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 1993;3:191–197. doi: 10.1016/0962-8924(93)90213-k. [DOI] [PubMed] [Google Scholar]
- 25.Pouly S, Becher B, Blain M, Antel JP. Expression of a homologue of rat NG2 on human microglia. Glia. 1999;27:259–268. doi: 10.1002/(sici)1098-1136(199909)27:3<259::aid-glia7>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 26.Pyrds O. Control of cerebral circulation in the high risk neonate. Ann Neurol. 1991;30:321–329. doi: 10.1002/ana.410300302. [DOI] [PubMed] [Google Scholar]
- 27.Reynolds R, Hardy R. Oligodendroglial progenitors labeled with the O4 antibody persist in the adult rat cerebral cortex in vivo. J Neurosci Res. 1997;47:455–470. doi: 10.1002/(sici)1097-4547(19970301)47:5<455::aid-jnr1>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 28.Rivkin MJ, Flax J, Mozell R, Osathanondh R, Volpe JJ, Villa-Komaroff L. Oligodendroglial development in human fetal cerebrum. Ann Neurol. 1995;38:92–101. doi: 10.1002/ana.410380116. [DOI] [PubMed] [Google Scholar]
- 29.Rorke LB. Pathology of perinatal brain injury, pp 45–63. Raven; New York: 1982. [Google Scholar]
- 30.Roy N, Wang S, Harrison-Restelli C, Benraiss A, Fraser R, Gravel M, Braun P, Goldman S. Identification, isolation, and promoter-defined separation of mitotic oligodendrocyte progenitor cells from the adult human subcortical white matter. J Neurosci. 1999;19:9986–9995. doi: 10.1523/JNEUROSCI.19-22-09986.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scolding N, Rayner P, Sussman J, Shaw C, Compston D. A proliferative adult human oligodendrocyte progenitor. NeuroReport. 1995;6:441–445. doi: 10.1097/00001756-199502000-00009. [DOI] [PubMed] [Google Scholar]
- 32.Sternberger N, Itoyama Y, Kies M, Webster HD. Immunocytochemical method to identify basic protein in myelin-forming oligodendrocytes of newborn rat CNS. J Neurocytol. 1978;7:251–263. doi: 10.1007/BF01217922. [DOI] [PubMed] [Google Scholar]
- 33.Streit W, Walter S, Pennel N. Reactive microgliosis. Prog Neurobiol. 1999;57:563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- 34.Takashima S, Tanaka K. Development of cerebrovascular architecture and its relationship to periventricular leukomalacia. Arch Neurol. 1978;35:11–16. doi: 10.1001/archneur.1978.00500250015003. [DOI] [PubMed] [Google Scholar]
- 35.Traystman RJ, Kirsch JR, Koehler RC. Oxygen radical mechanisms of brain injury following ischemia and reperfusion. Am J Physiol. 1991;71:1185–1195. doi: 10.1152/jappl.1991.71.4.1185. [DOI] [PubMed] [Google Scholar]
- 36.Volpe JJ. Brain injury in the premature infant: overview of clinical aspects, neuropathology, and pathogenesis. Semin Pediatr Neurol. 1998;5:135–151. doi: 10.1016/s1071-9091(98)80030-2. [DOI] [PubMed] [Google Scholar]
- 37.Volpe JJ. Neurology of the newborn. Saunders; Philadelphia: 2000. [Google Scholar]
- 38.Warrington AE, Pfeiffer SE (1992) Proliferation and differentiation of 04+ oligodendrocytes in postnatal rat cerebellum: analysis in unfixed tissue slices using anti-glycolipid antibodies. J Neurosci Res 338–353. [DOI] [PubMed]
- 39.Wolswijk G. Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci. 1998;18:601–609. doi: 10.1523/JNEUROSCI.18-02-00601.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]