

Abstract
It is well known that there are individual differences in a sensitivity to analgesics. Several lines of evidence have suggested that the level of opioid-induced analgesia is dependent on the level of expression of the μ-opioid receptor (μ-OR). However, the molecular mechanisms underlying the diversity of the level of the opioid receptor and the opioid sensitivity among individuals remain to be elucidated. In the present study, we analyzed the opioid-receptor genes of CXBK recombinant-inbred mice, which show reduced sensitivity to opioids. Northern blotting, nucleotide sequencing, and in situhybridization histochemical analyses demonstrated that CXBK mice possessed μ-OR mRNA with a normal coding region but an abnormally long untranslated region (UTR). In addition, the μ-OR mRNA level in CXBK mice was less than in the control mice. Next, we produced littermate mice that had inherited two copies of the wild-type μ-OR gene, had inherited two copies of the CXBK μ-OR gene, and had inherited both copies of the μ-OR genes. In these mice, inheritance of the CXBK μ-OR gene was well correlated with less μ-OR mRNA and reduced opioid effects on nociception and locomotor activity. We conclude that the CXBK μ-OR gene is responsible for the CXBK phenotypes. Because UTR differences are known to affect the level of the corresponding mRNA and protein and because UTRs are more divergent among individuals than coding regions, the present findings suggest that opioid sensitivity may vary, depending on different μ-OR levels attributable to divergent UTR of μ-OR mRNA.
Keywords: CXBK mouse, μ-opioid receptor, UTR, individual difference, analgesia, morphine
The CXBK mouse strain is a recombinant-inbred strain derived by full-sib mating from a cross between C57BL/6By and BALB/cBy mice (Bailey, 1971). This strain has been used as a μ-opioid receptor (μ-OR)-deficient strain because CXBK mice have a low level of μ-agonist binding sites (Moskowitz and Goodman, 1985) and display a reduced sensitivity to morphine and the κ-agonist U-50488 (Ikeda et al., 1999). For example, an important role of opioids in the analgesia induced by electroacupuncture and stress has been suggested because a lower level of electroacupuncture analgesia (Peets and Pomeranz, 1978) and lower levels of analgesia after defeat (Miczek et al., 1982) or swimming (Marek et al., 1988) are observed in CXBK mice, respectively. However, there is no molecular biological evidence that shows a deficiency of the μ-OR gene in CXBK mice. Furthermore, we have demonstrated recently that CXBK mice display an apparently different phenotype from that of μ-OR knock-out (KO) mice. This suggests that CXBK mice do not completely lack μ-OR (Ikeda et al., 1999).
The main target of morphine is μ-OR, and this is considered to be one of the most important molecules in opioid-induced analgesia. Mice that lack the μ-OR gene (μ-OR-KO) are insensitive to morphine (Matthes et al., 1996; Sora et al., 1997b; Tian et al., 1997; Loh et al., 1998) and are less sensitive to δ- and κ-agonists (Sora et al., 1997a,1999; Matthes et al., 1998) despite a normal expression of δ- and κ-opioid receptors in the brain (Kitchen et al., 1997; Sora et al., 1997b). Heterozygous mice with only one μ-OR allele have 50% less μ-OR protein than wild-type mice and show a reduced sensitivity to morphine (Sora et al., 1997b; Loh et al., 1998). This suggests that the amount of μ-OR affects the sensitivity to opioid analgesics. Furthermore, polymorphisms in the μ-OR gene have been found recently in humans, and the relationships between these polymorphisms and a vulnerability to drug abuse and dependence have been investigated (Bergen et al., 1997; Berrettini et al., 1997; Bond et al., 1998). The findings suggest that diversity of the μ-OR gene may contribute to interindividual differences in a sensitivity to opioids.
Because the opioid sensitivity of CXBK mice is similar to that of μ-OR-KO heterozygous mice (Ikeda et al., 1999), we focused on the μ-OR gene as a candidate gene that could be responsible for the CXBK phenotypes. In the present study, we demonstrate that CXBK mice possess abnormally long μ-OR mRNA and that the CXBK μ-OR gene is correlated with a reduced μ-OR mRNA level and opioid sensitivity.
MATERIALS AND METHODS
Animals. The mice were housed in aluminum cages with littermates of the same sex (up to five per cage) in an environment maintained at 23 ± 1°C, a relative humidity of 50 ± 5%, and with a 12 hr light/dark cycle (lights on 7:00 A.M. to 7:00 P.M.). The mice had access to a standard commercial laboratory diet ad libitum (NMF; Oriental Yeast Co. Ltd., Tokyo, Japan) and water. CXBK mice were originally purchased from The Jackson Laboratory (Bar Harbor, ME). C57BL/6CrSlc (B6) and BALB/cCrSlc (BALB/c) mice were purchased from Japan SLC, Inc. (Shizuoka, Japan). The experimental procedures and housing conditions were approved by the Institutional Animal Care and Use Committee. All of the animals were cared for and treated humanely, in accordance with the animal experimentation guidelines of our institution.
Northern blot analyses. mRNAs were separately prepared from the brain of each naive adult male mouse (Messenger RNA Isolation kit; Stratagene, La Jolla, CA). RNA size markers were purchased from Novagen (Madison, WI). The RNAs were electrophoresed on 1% agarose gel containing formaldehyde and transferred to a nitrocellulose membrane (PROTRAN; Schleicher & Schuell, Dassel, Germany) or a nylon membrane (Hybond-N+; Amersham Pharmacia Biotech, Uppsala, Sweden). The probes for μ-, δ-, and κ-opioid receptor mRNAs were prepared by PCR withPfu DNA polymerase (Stratagene), pSPORμ, pSPORδ, and pSPORκ (Ikeda et al., 1995b, 1997) as the templates, respectively. The common pair of primers for fragments corresponding to the transmembrane V–VII regions of the receptors (∼100 amino acids) were 5′-CT(C/G)ATCATC(A/T)(C/T)(G/T)GT(C/G)TG(C/T)TA-3′ (sense primer) and 5′-GCGGATCCTTGAAGTT(C/T)TC(A/G)TCCAG-3′ (antisense primer). The hybridization was performed at 60°C for ∼20 hr in a hybridization solution (ExpressHyb Hybridization Solution; Clontech, Palo Alto, CA) with [32P]-labeled probe (2 × 106 cpm/ml). The blots were washed at 42°C in 0.1× SSC (150 mm NaCl and 25 mm sodium citrate) containing 0.1% SDS. Autoradiography was performed and analyzed by using BAS-5000 Imaging Analyzer (Fujifilm, Tokyo, Japan). The values of photostimulated luminescence (PSL), which are proportional to the radioactivity in arbitrary measured areas (Amemiya et al., 1987), were compared in quantitative analyses. The membranes were dehybridized in 0.1× SSC solution containing 0.1% SDS at 100°C for 10 min. Expressions of μ-, δ-, and κ-OR mRNAs were analyzed using the same membranes.
In situ hybridization. The probe for μ-OR mRNA was a 45-mer oligonucleotide complementary to a part of a μ-OR cDNA sequence, including the initial methionine codon (Ikeda et al., 1996). The oligonucleotide was labeled with [33P]dATP using terminal deoxyribonucleotidyl transferase (TaKaRa, Kyoto, Japan) and purified using a Sephadex G-25 Spin Column (Boehringer Mannheim, Indianapolis, IN). The specific activity of the probe was 5 × 108 dpm/μg. In situhybridization histochemistry was performed as described previously (Ikeda et al., 1998). Briefly, horizontal and sagittal sections of adult male B6 and CXBK mouse brains were placed on slides and fixed with 4% paraformaldehyde/0.1 m sodium PBS. The sections were hybridized in a hybridization solution containing 5 × 103 dpm/μl probe for 16 hr at 42°C. The slides were washed three times in 0.1× SSC–0.1% Sarkosyl at 55°C for 40 min, dehydrated, and analyzed by using BAS-5000 Imaging Analyzer (Fujifilm). Values of PSL were compared by quantitative analyses. Afterward, the slides were exposed to Hyperfilm-β-max (Amersham Pharmacia Biotech) for 2 weeks to obtain x-ray film images.
Nucleotide sequencing. The CXBK and B6 mouse brain cDNAs were synthesized with the corresponding mRNAs as the templates (1st Strand cDNA Synthesis kit; Clontech). Genomic DNAs were prepared from mouse tail or liver. DNA fragments were amplified by PCR with Pfu DNA polymerase. The PCR primers for μ-OR cDNA were 5′-GCGCCTCCGTGTACTTCTAA-3′ (sense primer) and 5′-GATGGCAGCCTCTAAGTTTA-3′ (antisense primer). The nucleotide sequence of the PCR product was analyzed with the PCR primers and other primers as follows: 5′-AACCATGGACAGCAGCGCCG-3′, 5′-GCCACTAGCACGCTGCCCTT-3′, 5′-CAGTGGATCGAACTAACCACCAGCT-3′, and 5′-GGATTTTGCTCAGAATGGTGGCATG-3′ (Kaufman et al., 1995). The PCR primers for the μ-OR genes (5′-flanking region to the translation starting site) were 5′-AATGCATTCTTGCTCCTCAAGGATC-3′ (sense primer) and 5′-TCCCTGGGCCGGCGCTGCTGTCCAT-3′ (antisense primer). The nucleotide sequence of the PCR product was analyzed with the PCR primers and other primers as follows: 5′-AGTGGGGGCACATGAAACAGGCTTC-3′, 5′-GAGGGTTATTAATGTTGTCCTTTAC-3′, and 5′-GTTGTTACAAAGAAACTTAGAGTCT-3′ (Liang et al., 1995). The nucleotide sequencing was conducted by using PRISM 310 genetic analyzer (Applied Biosystems, Foster City, CA).
Behavioral tests. Naive adult (6–15 weeks old) mice were used in all the experiments. Each mouse was tested in the daytime (not earlier than 8:00 A.M. and not later than 5:00 P.M.). After the mouse was weighed, the tail-flick, open-field, and hot-plate tests were performed (in that sequence) to examine the basal reactivities and activity. Morphine hydrochloride (10 mg/ml) was purchased from Takeda Chemical Industries Ltd. (Osaka, Japan). (1S-trans-)-3,4-dichloro-N-methyl-N-(2-[1-pyrrolidinyl]cyclohexyl)benzeneacetamide hydrochloride [(−)-U-50488] (Research Biochemicals, Natick, MA) was dissolved in distilled water, and the stock solution was stored at −20°C until used. Each drug solution was diluted to 1 mg/ml with sterilized saline (0.9% NaCl) on each experimental day. The drug solution was injected intraperitoneally to the mouse at a dose of 10 ml/kg. The tail-flick, open-field, and hot-plate tests were performed 10, 15, and 20 min after the injection, respectively. The tail-flick test was performed according to the method of D'Amour and Smith (1941) with a slight modification (Ikeda et al., 1999). The cutoff time was 15 sec. The hot-plate test was performed according to the method of Woolfe and MacDonald (1944) with a slight modification (Ikeda et al., 1999). The temperature of the metal plate was adjusted to 52.0 ± 0.2°C. The latency, from the test start to the first jumping, was measured, and the cutoff time was 300 sec. The open-field test was performed as described previously (Ikeda et al., 1995a). The horizontal and vertical locomotions of the mouse were measured for 300 sec. In the present study, because the various kinds of locomotion were well correlated, the walking distance was used as the mouse locomotion. An ANOVA and Scheffe's F post hoc test were used to statistically analyze the between group data, with p < 0.05 accepted as statistically significant.
RESULTS
Abnormal μ-OR mRNA in CXBK mice
To investigate the expression of OR mRNAs in CXBK mice, we conducted Northern blot analyses (Fig.1A,B). The CXBK mice had a large-sized (14.5 kb) μ-OR mRNA in their brain, whereas the progenitor strain of mice, B6 mice, had 12 kb μ-OR mRNA. The heterozygotes between B6 and CXBK mice had both mRNAs, although the signal for the 14.5 kb mRNA was faint. The other progenitor stain of mice, BALB/c mice, had only 12 kb μ-OR mRNA. The signal intensity for μ-OR mRNA in CXBK mice was reduced to ∼60% of the intensity in B6 and BALB/c mice, when equal amounts of brain mRNAs were electrophoresed and analyzed. Although the size of δ-OR mRNA was the same in all strains, the signal intensity for δ-OR mRNA in CXBK mice was higher than that in B6 and BALB/c mice. The size of κ-OR mRNA and the signal intensity for the mRNA in all strains were not significantly different. The size difference in μ-OR mRNA suggests that the μ-OR gene in CXBK mice may be different from that of the progenitor strain mice.
Fig. 1.
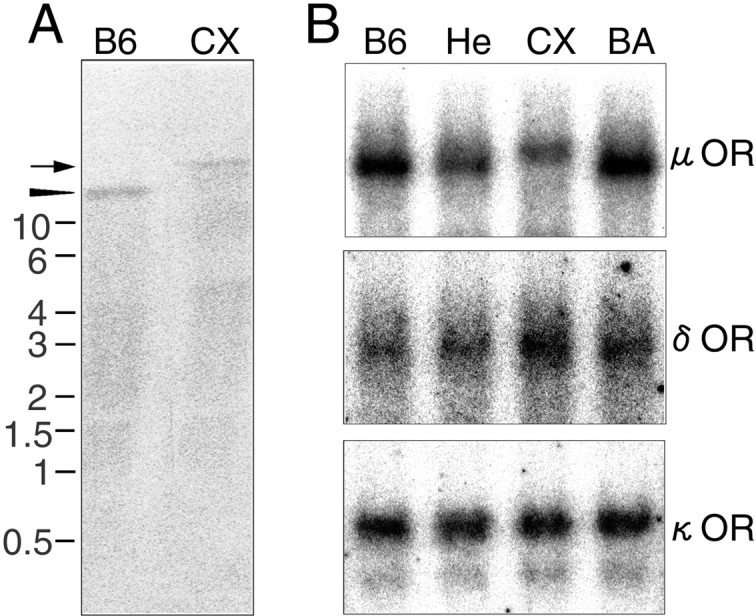
Northern blot analyses of CXBK mouse brain mRNAs.A, mRNAs (1.5 μg) of B6 and CXBK (CX) mouse brains were hybridized with cDNA probes for μ-OR mRNA. The size of the detected band in the CX lane was estimated at 14.5 kb (arrow), whereas that in the B6 lane was at 12 kb (arrowhead). B, mRNAs (4 μg) of B6, heterozygote (He) between B6 and CXBK, CXBK (CX), and BALB/c (BA) mouse brains were analyzed with cDNA probes for μ-, δ-, and κ-ORs. The sizes of μ-OR probe-positive bands in the B6 and BA lanes were estimated at 12 kb and that in the CX lane was at 14.5 kb. The ratios of the signal intensities for the μ-OR probe in the He, CX, andBA lanes to the intensity in the B6 lanewere 0.75, 0.6, and 1.1, respectively. The ratios of the signal intensity for the δ-OR probe in the He,CX, and BA lanes to the intensity in theB6 lane were 1.1, 1.7, and 1.4, respectively. The signal intensities for the κ-OR probe in all lanes were even. Thenumbers next to the photographs indicate the RNA size in kilobases.
Distribution of μ-OR mRNA in CXBK mice
Next, by using in situ hybridization histochemistry, we compared the expression of the μ-OR mRNA in the CXBK mouse brains with that in the B6 mouse brains (Fig.2). In the CXBK mouse brain, the μ-OR mRNA was expressed in a variety of brain regions in a similar manner to the B6 mouse brain. However, the signal intensity for the mRNA in the CXBK mouse brain was significantly lower (∼70% of that in the B6 mouse brain), which was consistent with the results of the Northern blot analyses. Similar results were obtained using sagittal sections of the B6 and CXBK mouse brains (data not shown). These results suggest that the expression level of the μ-OR mRNA was homogeneously lower in the CXBK mouse brains.
Fig. 2.
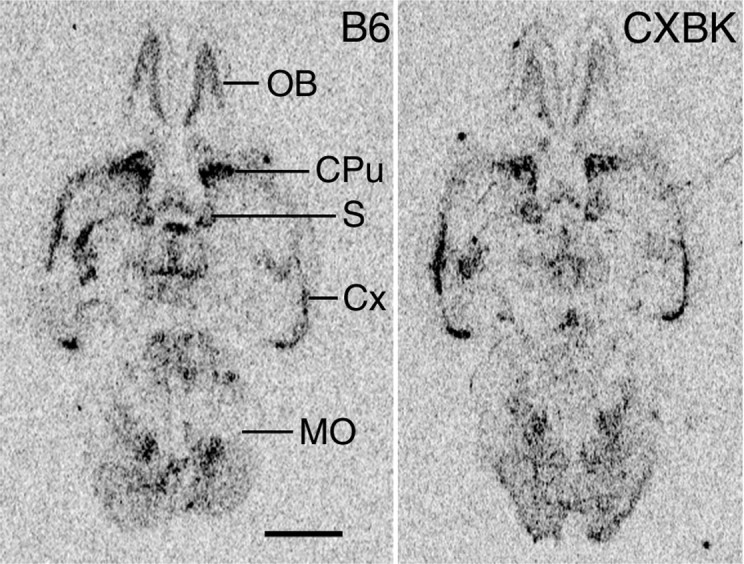
In situ hybridization showing the distribution of μ-OR mRNA in the B6 and CXBK mouse brains. Positive images made from an x-ray film are displayed. The signal intensity for the μ-OR mRNA in the CXBK mouse brain was lower than that in the B6 mouse brain (p < 0.005; n = 6 for each group; Student's t test). CPu, Caudate putamen;Cx, cerebral cortex; MO, medulla oblongata; OB, olfactory bulb; S, septum. Scale bar, 2 mm.
A nucleotide difference between B6 and CXBK μ-OR genes
A part (2184 bases; GenBank accession number AB047546) of the μ-OR mRNA, including the entire coding region, was compared in B6 and CXBK mice (Fig. 3). The sequence of the coding region (1197 bases) of the μ-OR mRNA in CXBK mice was identical to that in the B6 mice, indicating that the μ-OR protein structure is normal, but the untranslated region (UTR) of the μ-OR mRNA is abnormally long in CXBK mice. A sequence difference was not apparent in the examined 3′-UTR (726 bases), and there was only a single nucleotide sequence difference in the examined 5′-UTR (214 bases). This indicated that the difference in the size of the μ-OR mRNA between the B6 and the CXBK mice would be in the unexamined UTR of the μ-OR mRNA. We also compared a 5′-flanking region (1107 base pairs; GenBank accession number AB047547) with the translation starting site in the B6 and CXBK μ-OR genes. A sequence difference between them was not detected except that corresponding to the difference in the 5′-UTR. It was unlikely that the single nucleotide sequence difference caused whole CXBK phenotypes, because BALB/c mice possessed the same nucleotide sequence in this region as CXBK mice (data not shown). However, the nucleotide difference made it possible to distinguish the CXBK-derived μ-OR gene from the B6-derived μ-OR gene in the following experiments.
Fig. 3.
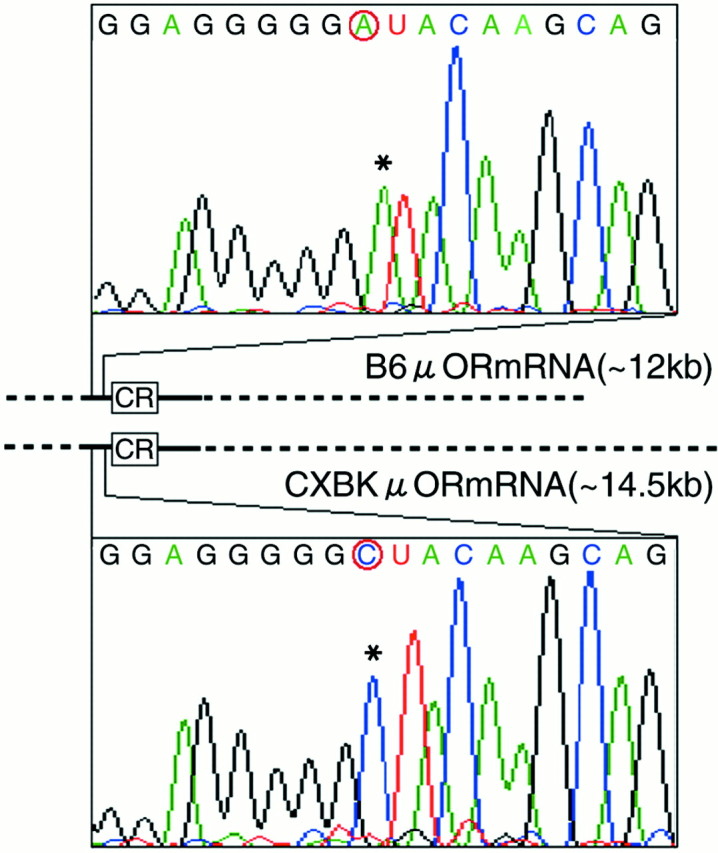
The nucleotide sequence differences in the μ-OR mRNAs in the B6 and CXBK mouse brains. The μ-opioid receptor mRNAs in the B6 and CXBK mouse brains are illustrated. The rectangles with CR denote the coding regions of the μ-OR mRNAs. Thesolid and broken lines denote the untranslated regions. The nucleotide sequences of the solid line were examined (214 base 5′-UTR and 726 base 3′-UTR). Chromatograms of nucleotide sequences of the regions containing a nucleotide sequence difference are shown. The difference was located at the 202 base upstream site from the translation starting site.Asterisks indicate the nucleotide sequence difference between the B6 and CXBK μ-OR mRNAs.
Mice inheriting two copies of the CXBK μ-OR gene (CXμ)
To understand the correlation between the CXBK μ-OR gene and the CXBK phenotypes, we prepared littermates by mating heterozygotes between B6 and CXBK mice (Fig.4A). These littermates were as follows: mice inheriting two copies of the B6 μ-OR gene (B6μ), mice inheriting the CXBK μ-OR gene (CXμ), and mice inheriting one copy of the B6 μ-OR gene and one copy of the CXBK μ-OR gene (Heμ). First, using these littermates, we conducted Northern blot analyses to clarify whether the differences in the size and amount of OR mRNAs in the CXBK mouse brains were attributable to the CXBK μ-OR gene (Fig. 4B). The sizes of the μ-OR mRNAs in B6μ and CXμ mice were estimated to be the same as the B6 and the CXBK mice, respectively. The signal intensities for the μ- and δ-OR mRNAs in CXμ mice were low and high, respectively, when compared with the signal intensities in B6 μ mice. Heμ mice possessed both of these μ-OR mRNAs in a similar manner to the heterozygotes between B6 and CXBK mice. These results suggest that the CXBK μ-OR gene caused the differences in the size and expression levels of the OR mRNAs in the CXBK mice.
Fig. 4.

Littermate mice inheriting two copies of the B6 μ-OR gene (B6μ), mice inheriting one B6 μ-OR gene and one CXBK μ-OR gene (Heμ), and mice inheriting two copies of the CXBK μ-OR gene (CXμ). A, Pedigree indicating relations of B6, CXBK, He, B6μ, CXμ, and Heμ mice. Littermate mice were prepared by mating of heterozygotes (He) between B6 and CXBK (CX) mice. B, Northern blot analyses of littermate mouse brain mRNAs with cDNA probes for μ- and δ-ORs. mRNAs (1.3 μg) in the brains of male littermate mice were analyzed. The sizes of the μ-OR probe-positive bands in theB6μ and CXμ lanes were estimated at 12 and 14.5 kb, respectively. The ratios of the signal intensity for the μ-OR probe in the Heμ andCXμ lanes to the signal intensity in the B6μ lane were 0.7 and 0.5, respectively. The ratios of the signal intensity for the δ-OR probe in the Heμ and CXμlanes to the signal intensity in theB6μ lane were 1.2 and 1.9, respectively.
Reduced sensitivity to opioids of CXμ mice
Second, using these littermates, we investigated whether the CXBK μ-OR gene is associated with reduction of morphine effects in CXBK mice. We conducted tail-flick and hot-plate tests for morphine-induced analgesia (Fig.5A,B) and an open-field test for morphine-induced hyperactivity (Fig.5C). These mice responded to the heat stimuli with similar latencies and showed similar spontaneous activity when they were not given morphine. However, after intraperitoneal administration of 10 mg/kg morphine, CXμ mice responded to heat stimuli with a significantly shorter latency than the littermates in both analgesic tests, indicating that the CXμ mice showed lower morphine-induced analgesia. In the open-field test, B6μ and Heμ mice walked similar distances before and after morphine administration, indicating that the decrease in locomotor activity attributable to habituation was counterbalanced by morphine-induced hyperactivity in these mice. In contrast, CXμ mice walked significantly shorter distances after morphine administration than they did before morphine administration (p < 0.001; paired t test), indicating that morphine failed to counterbalance the inhibiting effects of habituation on the locomotion of CXμ mice. These results suggested that the reduced effects of morphine on nociception and locomotion in CXBK mice were correlated with the CXBK μ-OR gene. Third, using these littermates, we investigated whether the CXBK μ-OR gene caused reduced analgesic effects of (−)-U-50488, a selective κ-agonist, in CXBK mice. In a tail-flick test, CXμ mice responded to the heat stimulus with a shorter latency than the littermates after intraperitoneal administration of 10 mg/kg (−)-U-50488 (Fig.5D). This result suggest that the reduction of (−)-U-50488-induced analgesia in the CXBK mice was also associated with the CXBK μ-OR gene. These three correlations between the CXBK μ-OR gene and the CXBK phenotypes suggest that the CXBK μ-OR gene contributed to the CXBK phenotypes.
Fig. 5.

Reduced sensitivity to morphine and (−)-U-50488 of CXμ mice. The effects of morphine (10 mg/kg, i.p.) on nociception and locomotion in adult littermate B6μ, Heμ, and CXμ mice were investigated in tail-flick (A), hot-plate (B), and open-field (C) tests (n = 10 for each group). There was a significant difference in tail-flick latency between the B6μ and CXμ mice (p < 0.05; repeated-measure ANOVA). In the hot-plate and open-field tests, there were significant interactions between the morphine effect and μ-OR gene-type effect (p < 0.05; repeated-measure ANOVA) when CXμ and B6μ mice were compared. D, Analgesia induced by (−)-U-50488 (10 mg/kg, i.p.) in adult littermate B6μ, Heμ, and CXμ mice was investigated in a tail-flick test (n= 6 for each group). There was significant interaction between the (−)-U-50488 effect and the μ-OR gene-type effect (p < 0.05; repeated-measure ANOVA) when CXμ and B6μ mice were compared. The white andstriped bars represent the data before and after drug injections, respectively. All values are means ± SEM.
DISCUSSION
The gene responsible for CXBK phenotypes
When a novel phenotype appears during establishment of a recombinant-inbred strain, the phenotype is considered to be caused not by a single gene but by a combination of more than two genes (Bailey, 1981). However, in the present study, we found that CXBK and CXμ mice possessed an abnormal-sized μ-OR mRNA, whereas both the two progenitor mice and B6μ mice possessed a normal-sized μ-OR mRNA. This indicated that the CXBK μ-OR gene is different from the μ-OR gene of either of the progenitor mice. Furthermore, CXμ mice displayed the CXBK phenotypes, a reduced μ-OR mRNA level, and a reduced sensitivity to opioids. These findings suggest that the altered μ-OR gene in CXBK mice is responsible for the CXBK phenotypes. Because the μ-OR gene in CXBK mice seems to be different from either of the μ-OR genes in the progenitor strains, it may be appropriate to classify the CXBK mouse strain into the mutant strains. Our present findings could provide essential bases for the previous studies using CXBK mice as μ-OR-deficient mice (Peets and Pomeranz, 1978; Miczek et al., 1982; Marek et al., 1988). Further investigation of the sequence differences between CXBK and wild-type mice would reveal the molecular mechanisms underlying the reduced μ-OR mRNA level and the reduced sensitivity to opioids in CXBK mice.
OR mRNA levels in CXBK mice
In the present study, we demonstrate a molecular mechanism that may underlie the reduced level of μ-OR protein in CXBK mice. We found that the amount of μ-OR mRNA in the CXBK mouse brains was reduced to 60% of the normal μ-OR mRNA amount in the control B6 and BALB/c mouse brains. These results suggest that the μ-OR protein level was reduced because of the reduced amount of μ-OR mRNA in CXBK mice.Duttaroy et al. (1999), by using a RNase protection assay, have shown that the levels of μ-OR mRNAs are similar in CXBK and control mice. This is inconsistent with our present results. This apparent discrepancy could be attributable to differences in the detection methods, because intact mRNAs can be selectively detected in Northern blot analyses, whereas partially degraded mRNAs are included in the experimental data in RNase protection assays. Therefore, we conclude that the amount of intact μ-OR mRNA is reduced in CXBK mice. In addition, we observed that the amount of intact δ-OR mRNA was increased in CXBK mice, a finding that is consistent with a previous report showing an increase of δ-OR mRNA levels in several brain regions (Kest et al., 1998). Because the amounts of δ- and κ-OR mRNAs were reported to be unchanged in μ-OR-KO mice (Kitchen et al., 1997; Sora et al., 1997b), there might be a specific mechanism for elevating the amount of δ-OR mRNA in CXBK mice. Further investigations of the mechanism in CXBK mice may reveal interactions among the expression of μ-, δ-, and κ-OR mRNAs.
Interindividual differences in analgesia
Pain perception differs among individuals (Dellemijn, 1999; Mogil, 1999; Uhl et al., 1999), and the differences can be attributed to inherited factors as well as environmental factors. Several gene alterations associated with pathological pain perception have been identified by nucleotide sequencing or suggested by linkage analyses. These include mutations, in the tRNALeu(UUR) (Goto et al., 1990) or P/Q-type Ca2+ channel α1-subunit (Joutel et al., 1993; Ophoff et al., 1996) gene with familial migraine (Peroutka, 1998), mutations in the neurotrophic tyrosine kinase receptor type 1 (NTRK1) gene with congenital insensitivity to pain (Indo et al., 1996) and a region including the NTRK2 gene with hereditary sensory neuropathy type I (Nicholson et al., 1996). In addition to the genetic studies on pathological pain perception, differences in nonpathological pain perception have been studied by using interstrain differences in mice. For example, quantitative trait locus (QTL) analyses for basal nociceptive sensitivity and morphine-induced analgesia of the recombinant inbred strains between C57BL/6 (B6) and DBA/2, showed that the μ-OR (Belknap et al., 1995), δ-OR (Mogil et al., 1997), and serotonin-1B receptor (Mogil, 1999) genes were associated with the traits. Similar analyses revealed that three loci, including the μ-OR gene locus, were associated with morphine preference (Berrettini et al., 1994). However, nucleotide sequence differences in these genes have not yet been identified by the QTL methods. In the present study, by using a different approach of focusing on the OR genes, we found that the μ-OR gene difference was associated with a reduced level of μ-OR mRNA and a reduced sensitivity to opioids in CXBK mice. Considering the present findings together with the previous reports that show a reduced sensitivity to opioids in the heterozygous μ-OR-KO mice with 50% μ-OR mRNA (Sora et al., 1997b; Loh et al., 1998), we propose that the low amount of μ-OR mRNA causes the reduced sensitivity to opioids. Interindividual differences in opioid analgesia may be partly attributable to divergent μ-OR mRNA levels because of μ-OR gene differences.
UTR differences and interindividual differences
The stability, localization, and translation of mRNAs are known to be affected by the UTRs (Decker and Parker, 1995). It has been demonstrated in the nervous system that the 3′-UTR of the Ca2+ channel α1BmRNA mediates calcium-dependent stabilization of the mRNA (Brook et al., 1992) and that the 3′-UTR of the growth-associated protein of 43 kDa mRNA is required for the stabilization of the mRNA in response to treatment with phorbol esters (Fu et al., 1992). In addition, recent studies have shown that alteration of UTR is related to several diseases. For example, expanded CTG repeat in the 3′-UTR of myotonic dystrophy protein kinase mRNA is responsible for myotonic dystrophy (Verkerk et al., 1991; Mahadevan et al., 1992; Gecz et al., 1996). The triplet repeats in the 5′-UTRs of fragile X mental retardation-1 (FMR1) (Gu et al., 1996) and FMR2 (Tsai et al., 1997;Schorge et al., 1999) mRNAs are associated with fragile X syndrome. The present study, which demonstrates a reduced level of μ-OR mRNA and a reduced sensitivity to opioids attributable to an abnormal UTR in CXBK mice, provides a novel aspect of the importance of the UTR. Because UTRs show little evolutionary conservation (Levitt, 1991), the resulting diversity of UTRs might be the molecular mechanisms for various interindividual differences. Analyses of UTRs could lead to identification of the gene responsible for diseases and individual differences and might contribute in the future to custom-made medical treatment.
Footnotes
This research was supported by research grants from the Cooperative Research Program of the RIKEN Brain Science Institute and the Ministry of Education, Science, Sports, and Culture of Japan. We thank Dr. Raymond Kado, Dr. Tsuyoshi Koide, Dr. Nobuhiko Kojima, and Sheldon J. Moss for critical reading and discussion. We also thank Naomi Mihira, Yoshitaka Miyazaki, Tsutomu Oowada, and Yoshimasa Yamada for technical assistance.
Correspondence should be addressed to Kazutaka Ikeda, Department of Psychopharmacology, Tokyo Institute of Psychiatry, 2-1-8 Kamikitazawa, Setagaya, Tokyo 156-8585, Japan. E-mail: ikedak@prit.go.jp.
REFERENCES
- 1.Amemiya Y, Wakabayashi K, Tanaka H, Ueno Y, Miyahara J. Laser-stimulated luminescence used to measure x-ray diffraction of a contracting striated muscle. Science. 1987;237:164–168. doi: 10.1126/science.3496662. [DOI] [PubMed] [Google Scholar]
- 2.Bailey D. Recombinant inbred strains and bilineal congenic strains. In: Foster H, Small J, Fox J, editors. The mouse in biomedical research. Academic; New York: 1981. pp. 223–239. [Google Scholar]
- 3.Bailey DW. Cumulative effect or independent effect? Transplantation. 1971;11:419–422. doi: 10.1097/00007890-197104000-00014. [DOI] [PubMed] [Google Scholar]
- 4.Belknap JK, Mogil JS, Helms ML, Richards SP, O'Toole LA, Bergeson SE, Buck KJ. Localization to chromosome 10 of a locus influencing morphine analgesia in crosses derived from C57BL/6 and DBA/2 strains. Life Sci. 1995;57:PL117–PL124. doi: 10.1016/0024-3205(95)02040-p. [DOI] [PubMed] [Google Scholar]
- 5.Bergen AW, Kokoszka J, Peterson R, Long JC, Virkkunen M, Linnoila M, Goldman D. Mu opioid receptor gene variants: lack of association with alcohol dependence. Mol Psychiatry. 1997;2:490–494. doi: 10.1038/sj.mp.4000331. [DOI] [PubMed] [Google Scholar]
- 6.Berrettini WH, Ferraro TN, Alexander RC, Buchberg AM, Vogel WH. Quantitative trait loci mapping of three loci controlling morphine preference using inbred mouse strains. Nat Genet. 1994;7:54–58. doi: 10.1038/ng0594-54. [DOI] [PubMed] [Google Scholar]
- 7.Berrettini WH, Hoehe MR, Ferraro TN, DeMaria PA, Gottheil E. Human mu opioid receptor gene polymorphisms and vulnerability to substance abuse. Addiction Biology. 1997;2:303–308. doi: 10.1080/13556219772598. [DOI] [PubMed] [Google Scholar]
- 8.Bond C, LaForge KS, Tian M, Melia D, Zhang S, Borg L, Gong J, Schluger J, Strong JA, Leal SM, Tischfield JA, Kreek MJ, Yu L. Single-nucleotide polymorphism in the human mu opioid receptor gene alters beta-endorphin binding and activity: possible implications for opiate addiction. Proc Natl Acad Sci USA. 1998;95:9608–9613. doi: 10.1073/pnas.95.16.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, Sohn R, Zemelman B, Snell RG, Rundle SA, Crow S, Davies J, Shelbourne P, Buxton J, Jones C, Juvonen V, Johnson K, Harper PS, Shaw DJ, Housman DE. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;69:385. doi: 10.1016/0092-8674(92)90418-c. [DOI] [PubMed] [Google Scholar]
- 10.D'Amour F, Smith D. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
- 11.Decker CJ, Parker R. Diversity of cytoplasmic functions for the 3′ untranslated region of eukaryotic transcripts. Curr Opin Cell Biol. 1995;7:386–392. doi: 10.1016/0955-0674(95)80094-8. [DOI] [PubMed] [Google Scholar]
- 12.Dellemijn P. Are opioids effective in relieving neuropathic pain? Pain. 1999;80:453–462. doi: 10.1016/S0304-3959(98)00256-5. [DOI] [PubMed] [Google Scholar]
- 13.Duttaroy A, Shen J, Shah S, Chen B, Sehba F, Carroll J, Yoburn BC. Opioid receptor upregulation in mu-opioid receptor deficient CXBK and outbred Swiss Webster mice. Life Sci. 1999;65:113–123. doi: 10.1016/s0024-3205(99)00228-3. [DOI] [PubMed] [Google Scholar]
- 14.Fu YH, Pizzuti A, Fenwick RG, Jr, King J, Rajnarayan S, Dunne PW, Dubel J, Nasser GA, Ashizawa T, de Jong P, Wieringa B, Korneluk R, Perryman MB, Epstein HF, Caskey CT. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–1258. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- 15.Gecz J, Gedeon AK, Sutherland GR, Mulley JC. Identification of the gene FMR2, associated with FRAXE mental retardation. Nat Genet. 1996;13:105–108. doi: 10.1038/ng0596-105. [DOI] [PubMed] [Google Scholar]
- 16.Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651–653. doi: 10.1038/348651a0. [DOI] [PubMed] [Google Scholar]
- 17.Gu Y, Shen Y, Gibbs RA, Nelson DL. Identification of FMR2, a novel gene associated with the FRAXE CCG repeat and CpG island. Nat Genet. 1996;13:109–113. doi: 10.1038/ng0596-109. [DOI] [PubMed] [Google Scholar]
- 18.Ikeda K, Araki K, Takayama C, Inoue Y, Yagi T, Aizawa S, Mishina M. Reduced spontaneous activity of mice defective in the epsilon 4 subunit of the NMDA receptor channel. Mol Brain Res. 1995a;33:61–71. doi: 10.1016/0169-328x(95)00107-4. [DOI] [PubMed] [Google Scholar]
- 19.Ikeda K, Kobayashi T, Ichikawa T, Usui H, Kumanishi T. Functional couplings of the delta- and the kappa-opioid receptors with the G-protein-activated K+ channel. Biochem Biophys Res Commun. 1995b;208:302–308. doi: 10.1006/bbrc.1995.1338. [DOI] [PubMed] [Google Scholar]
- 20.Ikeda K, Kobayashi T, Ichikawa T, Usui H, Abe S, Kumanishi T. Comparison of the three mouse G-protein-activated K+ (GIRK) channels and functional couplings of the opioid receptors with the GIRK1 channel. Ann NY Acad Sci. 1996;801:95–109. doi: 10.1111/j.1749-6632.1996.tb17434.x. [DOI] [PubMed] [Google Scholar]
- 21.Ikeda K, Kobayashi K, Kobayashi T, Ichikawa T, Kumanishi T, Kishida H, Yano R, Manabe T. Functional coupling of the nociceptin/ orphanin FQ receptor with the G-protein-activated K+ (GIRK) channel. Mol Brain Res. 1997;45:117–126. doi: 10.1016/s0169-328x(96)00252-5. [DOI] [PubMed] [Google Scholar]
- 22.Ikeda K, Watanabe M, Ichikawa T, Kobayashi T, Yano R, Kumanishi T. Distribution of prepro-nociceptin/orphanin FQ mRNA and its receptor mRNA in developing and adult mouse central nervous systems. J Comp Neurol. 1998;399:139–151. doi: 10.1002/(sici)1096-9861(19980914)399:1<139::aid-cne11>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 23.Ikeda K, Ichikawa T, Kobayashi T, Kumanishi T, Oike S, Yano R. Unique behavioural phenotypes of recombinant-inbred CXBK mice: partial deficiency of sensitivity to μ- and κ-agonists. Neurosci Res. 1999;34:149–155. doi: 10.1016/s0168-0102(99)00047-4. [DOI] [PubMed] [Google Scholar]
- 24.Indo Y, Tsuruta M, Hayashida Y, Karim MA, Ohta K, Kawano T, Mitsubuchi H, Tonoki H, Awaya Y, Matsuda I. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat Genet. 1996;13:485–488. doi: 10.1038/ng0896-485. [DOI] [PubMed] [Google Scholar]
- 25.Joutel A, Bousser MG, Biousse V, Labauge P, Chabriat H, Nibbio A, Maciazek J, Meyer B, Bach MA, Weissenbach J, Lathrop GM, Tournier-Lasserve E. A gene for familial hemiplegic migraine maps to chromosome 19. Nat Genet. 1993;5:40–45. doi: 10.1038/ng0993-40. [DOI] [PubMed] [Google Scholar]
- 26.Kaufman DL, Keith DE, Jr, Anton B, Tian J, Magendzo K, Newman D, Tran TH, Lee DS, Wen C, Xia YR, Lusis AJ, Evans CJ. Characterization of the murine mu opioid receptor gene. J Biol Chem. 1995;270:15877–15883. doi: 10.1074/jbc.270.26.15877. [DOI] [PubMed] [Google Scholar]
- 27.Kest B, Beczkowska I, Franklin SO, Lee CE, Mogil JS, Inturrisi CE. Differences in delta opioid receptor antinociception, binding, and mRNA levels between BALB/c and CXBK mice. Brain Res. 1998;805:131–137. doi: 10.1016/s0006-8993(98)00696-9. [DOI] [PubMed] [Google Scholar]
- 28.Kitchen I, Slowe SJ, Matthes HW, Kieffer B. Quantitative autoradiographic mapping of mu-, delta- and kappa-opioid receptors in knockout mice lacking the mu-opioid receptor gene. Brain Res. 1997;778:73–88. doi: 10.1016/s0006-8993(97)00988-8. [DOI] [PubMed] [Google Scholar]
- 29.Levitt RC. Polymorphisms in the transcribed 3′ untranslated region of eukaryotic genes. Genomics. 1991;11:484–489. doi: 10.1016/0888-7543(91)90168-e. [DOI] [PubMed] [Google Scholar]
- 30.Liang Y, Mestek A, Yu L, Carr LG. Cloning and characterization of the promoter region of the mouse mu opioid receptor gene. Brain Res. 1995;679:82–88. doi: 10.1016/0006-8993(95)00222-c. [DOI] [PubMed] [Google Scholar]
- 31.Loh HH, Liu HC, Cavalli A, Yang W, Chen YF, Wei LN. mu Opioid receptor knockout in mice: effects on ligand-induced analgesia and morphine lethality. Mol Brain Res. 1998;54:321–326. doi: 10.1016/s0169-328x(97)00353-7. [DOI] [PubMed] [Google Scholar]
- 32.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O'Hoy K, Leblond S, Earle-Macdonald J, de Jong PJ, Wieringa B, Korneluk RG. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 33.Marek P, Yirmiya R, Liebeskind JC. Strain differences in the magnitude of swimming-induced analgesia in mice correlate with brain opiate receptor concentration. Brain Res. 1988;447:188–190. doi: 10.1016/0006-8993(88)90984-5. [DOI] [PubMed] [Google Scholar]
- 34.Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le Meur M, Dolle P, Tzavara E, Hanoune J, Roques BP, Kieffer BL. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- 35.Matthes HW, Smadja C, Valverde O, Vonesch JL, Foutz AS, Boudinot E, Denavit-Saubie M, Severini C, Negri L, Roques BP, Maldonado R, Kieffer BL. Activity of the delta-opioid receptor is partially reduced, whereas activity of the kappa-receptor is maintained in mice lacking the mu-receptor. J Neurosci. 1998;18:7285–7295. doi: 10.1523/JNEUROSCI.18-18-07285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miczek KA, Thompson ML, Shuster L. Opioid-like analgesia in defeated mice. Science. 1982;215:1520–1522. doi: 10.1126/science.7199758. [DOI] [PubMed] [Google Scholar]
- 37.Mogil JS. The genetic mediation of individual differences in sensitivity to pain and its inhibition. Proc Natl Acad Sci USA. 1999;96:7744–7751. doi: 10.1073/pnas.96.14.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mogil JS, Richards SP, O'Toole LA, Helms ML, Mitchell SR, Belknap JK. Genetic sensitivity to hot-plate nociception in DBA/2J and C57BL/6J inbred mouse strains: possible sex-specific mediation by delta2-opioid receptors. Pain. 1997;70:267–277. doi: 10.1016/s0304-3959(97)03333-2. [DOI] [PubMed] [Google Scholar]
- 39.Moskowitz AS, Goodman RR. Autoradiographic analysis of mu1, mu2, and delta opioid binding in the central nervous system of C57BL/6BY and CXBK (opioid receptor-deficient) mice. Brain Res. 1985;360:108–116. doi: 10.1016/0006-8993(85)91226-0. [DOI] [PubMed] [Google Scholar]
- 40.Nicholson GA, Dawkins JL, Blair IP, Kennerson ML, Gordon MJ, Cherryson AK, Nash J, Bananis T. The gene for hereditary sensory neuropathy type I (HSN-I) maps to chromosome 9q22.1-q22.3. Nat Genet. 1996;13:101–104. doi: 10.1038/ng0596-101. [DOI] [PubMed] [Google Scholar]
- 41.Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- 42.Peets JM, Pomeranz B. CXBK mice deficient in opiate receptors show poor electroacupuncture analgesia. Nature. 1978;273:675–676. doi: 10.1038/273675a0. [DOI] [PubMed] [Google Scholar]
- 43.Peroutka SJ. Genetic basis of migraine. Clin Neurosci. 1998;5:34–37. [PubMed] [Google Scholar]
- 44.Schorge S, Gupta S, Lin Z, McEnery MW, Lipscombe D. Calcium channel activation stabilizes a neuronal calcium channel mRNA. Nat Neurosci. 1999;2:785–790. doi: 10.1038/12153. [DOI] [PubMed] [Google Scholar]
- 45.Sora I, Funada M, Uhl GR. The mu-opioid receptor is necessary for [d-Pen2,d-Pen5]enkephalin-induced analgesia. Eur J Pharmacol. 1997a;324:R1–R2. doi: 10.1016/s0014-2999(97)10016-4. [DOI] [PubMed] [Google Scholar]
- 46.Sora I, Takahashi N, Funada M, Ujike H, Revay RS, Donovan DM, Miner LL, Uhl GR. Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc Natl Acad Sci USA. 1997b;94:1544–1549. doi: 10.1073/pnas.94.4.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sora I, Li XF, Funada M, Kinsey S, Uhl GR. Visceral chemical nociception in mice lacking mu-opioid receptors: effects of morphine, SNC80 and U-50,488. Eur J Pharmacol. 1999;366:R3–R5. doi: 10.1016/s0014-2999(98)00933-9. [DOI] [PubMed] [Google Scholar]
- 48.Tian M, Broxmeyer HE, Fan Y, Lai Z, Zhang S, Aronica S, Cooper S, Bigsby RM, Steinmetz R, Engle SJ, Mestek A, Pollock JD, Lehman MN, Jansen HT, Ying M, Stambrook PJ, Tischfield JA, Yu L. Altered hematopoiesis, behavior, and sexual function in mu opioid receptor-deficient mice. J Exp Med. 1997;185:1517–1522. doi: 10.1084/jem.185.8.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsai KC, Cansino VV, Kohn DT, Neve RL, Perrone-Bizzozero NI. Post-transcriptional regulation of the GAP-43 gene by specific sequences in the 3′ untranslated region of the mRNA. J Neurosci. 1997;17:1950–1958. doi: 10.1523/JNEUROSCI.17-06-01950.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uhl GR, Sora I, Wang Z. The mu opiate receptor as a candidate gene for pain: polymorphisms, variations in expression, nociception, and opiate responses. Proc Natl Acad Sci USA. 1999;96:7752–7755. doi: 10.1073/pnas.96.14.7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, Eussen BE, van Ommen GJB, Blonden LAJ, Riggins GJ, Chastain JL, Kunst CB, Galjaard H, Caskey CT, Nelson DL, Oostra BA, Warren ST. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 52.Woolfe G, Macdonald A. The evaluation of the analgesic action of pethidine hydrochloride (demerol). J Pharmacol Exp Ther. 1944;80:300–307. [Google Scholar]