

Abstract
Rocker (gene symbol rkr), a new neurological mutant phenotype, was found in descendents of a chemically mutagenized male mouse. Mutant mice display an ataxic, unstable gait accompanied by an intention tremor, typical of cerebellar dysfunction. These mice are fertile and appear to have a normal life span. Segregation analysis reveals rocker to be an autosomal recessive trait. The overall cytoarchitecture of the young adult brain appears normal, including its gross cerebellar morphology. Golgi-Cox staining, however, reveals dendritic abnormalities in the mature cerebellar cortex characterized by a reduction of branching in the Purkinje cell dendritic arbor and a “weeping willow” appearance of the secondary branches. Using simple sequence length polymorphism markers, the rocker locus was mapped to mouse chromosome 8 within 2 centimorgans of the calcium channel α1a subunit (Cacna1a, formerly known as tottering) locus. Complementation tests with the leaner mutant allele (Cacna1ala) produced mutant animals, thus identifying rocker as a new allele ofCacna1a (Cacna1arkr). Sequence analysis of the cDNA revealed rocker to be a point mutation resulting in an amino acid exchange: T1310K between transmembrane regions 5 and 6 in the third homologous domain. Important distinctions between rocker and the previously characterized alleles of this locus include the absence of aberrant tyrosine hydroxylase expression in Purkinje cells and the separation of the absence seizures (spike/wave type discharges) from the paroxysmal dyskinesia phenotype. Overall these findings point to an important dissociation between the seizure phenotypes and the abnormalities in catecholamine metabolism, and they emphasize the value of allelic series in the study of gene function.
Keywords: P/Q-type calcium channel, tottering, dendrite maintenance, cerebellar catecholamine metabolism, mouse mutant, Ca2+ channel α subunit, cerebellar Purkinje cells, gene mapping
In neurons, voltage-dependent Ca2+ channels (VDCCs) are involved in diverse functions, including excitability (Llinas, 1988), neurotransmitter release (Wheeler et al., 1994; Dunlap et al., 1995;Scholz and Miller, 1995), and regulation of gene expression (Bading et al., 1993). In maturing neurons, VDCCs and calcium entry have been implicated in neuronal migration (Komuro and Rakic, 1992), neurite outgrowth (Kater and Mills, 1991; Moorman and Hume, 1993; Manivannan and Terakawa, 1994), axon and dendrite extension (Cohan et al., 1987;McCobb et al., 1989), and establishment of early synaptic connections (Llinas and Sugimori, 1979; Mills and Kater, 1990; Vigers and Pfenninger, 1991; Komuro and Rakic, 1992; Johnson and Byerly, 1993;Spitzer, 1994).
VDCCs in mammalian neurons have been classified into five groups: L, N, P/Q, R, and T, on the basis of their electrophysiological and pharmacological properties. They are multisubunit complexes, the expression and targeting of which require the assembly of the pore-forming subunit α1 with β, α2/δ, and γ subunits. The calcium channel α1a subunit gene (Cacna1a) encodes the pore-forming protein of P/Q-type channels. It is diffusely localized in brain, with high levels of expression in cerebellar granule and Purkinje cells (Stea et al., 1994; Tanaka et al., 1995; Westenbroek et al., 1995). Mutations of Cacna1a have been shown to cause human neurological diseases such as familial hemiplegic migraine, episodic ataxia-2 (Ophoff et al., 1996), and spinocerebellar ataxia 6 (Zhuchenko et al., 1997). In mice, mutations in the gene are found in the neurological mutants tottering(Cacna1atg, formerly tg),leaner (Cacna1atg-la, formerlyla and tgla) (Fletcher et al., 1996; Doyle et al., 1997), and rolling mouse Nagoya(Cacna1atg-rol, formerly roland tgrol) (Mori et al., 2000) and are expected in the other known mutant allele, tottering-3J(Cacna1atg-3J, formerlytg3J) (Green et al., 1988). Mutations in the auxiliary subunits also result in neurological dysfunction in mice. The β4 subunit is defective in lethargic (Burgess et al., 1997), and γ2 is defective in stargazer (Letts et al., 1998).
The Cacna1a mutants, although clearly allelic, exhibit distinct phenotypes (Tsuji and Meier, 1971) ranging from the mildertottering (Green and Sidman, 1962) and rolling(Oda, 1981) alleles to the more severe leaner (Sidman et al., 1965; Meier and MacPike, 1971) and tottering-3J (Green et al., 1988). Phenotypic features that may be absent in some alleles or may vary in onset and severity include ataxia, paroxysmal dyskinesia, absence seizures (Meier and MacPike, 1971), electrocorticographic “spike-and-wave” activity pattern similar to that observed in human petit mal epilepsy (Noebels and Sidman, 1979), reduced current density (Dove et al., 1998; Wakamori et al., 1998; Mori et al., 2000; Qian and Noebels, 2000), cerebellar cell death (Herrup and Wilczynski, 1982), increased adrenergic terminals from locus coeruleus (LC) neurons (Levitt and Noebels, 1981), increased tyrosine hydroxylase (TH) expression in Purkinje cells (Hess and Wilson, 1991;Austin et al., 1992; Sawada et al., 1999), and ectopic dendritic spines and axonal torpedoes on Purkinje cells (Rhyu et al., 1999a,b) (Table1). Here we describe a newCacna1a mutant allele, rocker(Cacna1arkr), that presents a fifth distinct phenotype in this allelomorphic series.
Table 1.
Phenotypes of Cacna1a alleles
| tottering | leaner | rolling | tottering-3J | rocker | tg/la | tg/rol | rkr/la | |
|---|---|---|---|---|---|---|---|---|
| Viability | Normal | Reduced | Normal | Reduced | Normal | Normal | Normal | Normal |
| Age of onset of ataxia | P21–28 | P8–10 | P10–14 | P12–14 | P21–28 | P15–17 | P10–14 | P15–17 |
| Ataxia | + (1) | +++ (1) | ++ (2) | +++ (3) | + | ++ (1) | ++ (2) | ++ |
| Paroxysmal dyskinesia | + (4–6) | – | – | ? | – | + (1) | – | ? |
| Absence seizures | + (4–6) | + (7) | – ? (8) | + ? (3) | + | + (7) | ? | + ? |
| Increased LC innervation | + (9) | ? | + (10) | ? | ? | ? | ? | – ? |
| Cerebellar cell death | – (11, 12) | + (9) | + (11, 13)/– (14, 15) | ? | – | – (12) | ? | – ? |
| Purkinje cell TH expression | + (16, 17) | + (16, 17) | + (18) | ? | – | + (17) | ? | + |
| Purkinje cell axonal torpedoes | + (19) | + (19) | + (20) | ? | – | + (19) | ? | – |
| Purkinje cell ectopic spines | + (19) | + (19) | + (20) | ? | – | + (19) | ? | + |
| Purkinje cell dendritic weeping | – | – | ? | ? | + | ? | ? | – |
| Purkinje cell current density | 60% of control (21) | 40% of control (21, 22) | 60% of control (15) | ? | ? | ? | ? | ? |
+++, Strong phenotype; ++, moderate phenotype; +, mild phenotype; –, no overt phenotype; ?, needs further study.
1, Meier and MacPike (1971); 2, Oda (1973); 3, Green et al. (1988); 4,Green and Sidman (1962); 5, Sidman et al. (1965); 6, Noebels and Sidman (1979); 7, Noebels (1984); 8, Oda (1981); 9, Levitt and Noebels (1981); 10, Muramoto et al. (1982); 11, Nakane (1976); 12, Isaacs and Abbott (1995); 13, Nishimura (1975); 14, Mukaiyama and Mizuno (1976); 15, Mori et al. (2000); 16, Hess and Wilson (1991); 17, Austin et al. (1992); 18, Muramoto et al. (1981); 19, Rhyu et al. (1999a); 20, Rhyu et al. (1999b); 21, Wakamori et al. (1998); 22, Dove et al. (1998).
MATERIALS AND METHODS
Animal husbandry. Ataxic animals were discovered at the Children's Hospital (Boston, MA) in the fourth generation of a male mouse mutagenized with ethylnitrosourea (ENU) at Kansas State University (Bode, 1984; Bode et al., 1987). The original ENU-mutagenized male was of mixed genetic background. To assist in the genetic analysis, the rocker gene was backcrossed onto the C57BL/6J strain five times by mating homozygousrocker animals with C57BL/6J animals (obtained from The Jackson Laboratory, Bar Harbor, ME). The mutant gene passed through both males and females and continued to be crossed onto the C57BL/6J strain. Generation of mutants was achieved through the mating of homozygous rocker males with heterozygous females, as well as through heterozygous intercrosses. All animals were maintained in the Case Western Reserve University Medical School Animal Resource Center, a facility fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. The mice were provided a commercial diet and water ad libitum under conventional conditions with controlled temperature, humidity, and lighting.
cDNA cloning and sequence analysis. cDNAs encoding the α 1a subunit were isolated through reverse transcriptase (RT)-PCR using the random primers DNA labeling system (Life Technologies, Grand Island, NY) and total RNA. Eighteen PCR primers were designed according to the published sequence data (Fletcher et al., 1996) such that the entire 6495 bp reported sequence was covered. cDNA RT-PCR products were sequenced using an automated sequencer (ABI prism model 377; Applied Biosystems, Foster City, CA). Sequence was also obtained manually using an Amplicycle Sequencing Kit (PerkinElmer Life Sciences, Norwalk, CT) with α-32P incorporation according to the manufacturer's instructions. Sequence reactions were electrophoresed through a 6% acrylamide denaturing gel and exposed overnight at −70°C on X-OMAT AR film (Eastman Kodak, Rochester, NY). Sequence information was obtained separately from four mutants and four background control mice.
Histology. After being deeply anesthetized with Avertin (0.02 cc/gm), animals were perfused through the heart with 4% paraformaldehyde in 0.1 m sodium phosphate buffer (PB), pH 7.4. The brain was dissected free of the skull case and stored overnight in fresh fix. The following day, the tissue was dehydrated through graded alcohols, infiltrated with paraffin (Paraplast Plus; Fisher, Pittsburgh, PA), bisected on the midline, and embedded in the sagittal plane. A full set of serial 10 μm sections was collected for each half cerebellum counted. Using standard histological methods, every 20th section was stained with 0.2% cresyl violet and examined at 400×. Purkinje cells were counted by previously described profile-based counting methods (Herrup and Sunter, 1987). Purkinje cells were identified by their large size and position in the Purkinje cell layer. No obvious Purkinje-like cells were present in the molecular layer; Purkinje cells in the internal granule cell layer might have escaped detection because they would have been mistaken for Golgi II cells. All identified Purkinje cells that had a portion of their nucleus in the section were counted. The nucleus-positive Purkinje cell counts were graphed as a function of distance from the midline. The areas under the curves are proportional to the total number of Purkinje cells in the cerebellum. This estimate was corrected for split-cell counting errors by the method of Hendry (1976).
Immunohistochemistry. Animals were anesthetized deeply with Avertin and perfused intracardially with 4% paraformaldehyde in PB. The brains were immediately removed from the cranium and fixed for an additional 4 hr at 4°C. The brains were then cryoprotected by sinking in 18% (w/v) sucrose in PBS at 4°C overnight. Most samples were embedded in OCT, frozen in powdered dry ice for 5 min, and then allowed to equilibrate to the cutting temperature of the cryostat (−20°C). Cryostat sections (10 μm) were cut and allowed to air dry on gelatin-coated slides.
For single-label immunofluorescence, sections were first rinsed in 1× PBS for 5 min, followed by a 1 hr wash at room temperature in PBS containing 0.5% Tween 20 and 5% normal goat serum (NGS). All antibodies were diluted in PBS containing 0.5% Tween 20 and 5% normal goat serum (see Table 2); sections were coverslipped with parafilm and incubated overnight at 4°C in a humid chamber. The sections were rinsed for 20 min in PBS, incubated in goat anti-mouse IgG (or goat anti-rabbit IgG; 1:200 dilution) for 1.5 hr, rinsed for 20 min in PBS, and then coverslipped using a glycerol/PBS mix at 1:1 ratio. Secondary antibodies were Cy3-conjugated goat anti-mouse IgG and Cy2-conjugated goat anti-rat IgG (Jackson ImmunoResearch, West Grove, PA). Control sections were incubated without primary antibody and used to determine the level of nonspecific staining. A Leitz DMRB microscope was used to view the immunofluorescent sections using rhodamine or fluorescein filters.
Table 2.
Primary antibodies used in current study
| Antibody | Dilution | Supplier |
|---|---|---|
| Mouse-anti calbindin | 1:1000 | Sigma |
| Mouse-anti-NeuN | 1:100 | Richard Mullen (Mullen et al., 1992) |
| Mouse-anti-NF160 | 1:40 | Sigma |
| Rabbit-anti-TrkB | 1:1000 | Santa Cruz |
| Mouse-anti-Map2 | 1:200 | Sigma |
| Mouse-anti-MBP | 1:500 | Boehringer Mannheim |
| Rabbit-anti-GFAP | 1:250 | Accurate |
| Mouse-anti-SV2 | 1:100 | Kathleen M. Buckley (Buckley and Kelly, 1985) |
Tissue processed for immunohistochemistry with the anti-tyrosine hydroxylase antibody was frozen under powdered dry ice, and 30-μm-thick sections were cut into PBS with a sliding microtome. Some sections were stored at −20°C in an antifreeze solution [10% (w/v), polyvinylpyrrolidone 30% (w/v) sucrose, 10% (w/v) ethylene glycol, and 50 mm sodium phosphate, pH 7.4]. Before staining, sections were rinsed in 50 mm Tris, pH 7.4, 1.5% NaCl (TBS), and permeabilized by incubation in 0.3% Triton X-100 in TBS for 20–30 min at room temperature with gentle agitation. Sections were then blocked by incubation in 5% NGS in TBS for 30 min at room temperature. The monoclonal antibody against TH was diluted in 1% NGS in TBS 1:10,000 (Pel-Freez Biologicals, Browndeer, WI), and sections were incubated overnight at 4°C. Sections were then processed with the Vectastain Elite avidin–biotinylated peroxidase reagents (Vector Laboratories, Burlingame, CA). Sections were incubated for 1 hr with biotinylated goat anti-mouse secondary antibody (1:200) in 1% NGS in TBS at room temperature, washed for 10 min in 1% NGS in TBS, and then washed three times for 7 min each in TBS alone. Then they were incubated with avidin–biotinyl peroxidase complex (1:50 dilution of each reagent in TBS) for 1 hr at room temperature. After being washed three times for 10 min each in TBS, sections were developed using 0.5 μg/ml diaminobenzidine and 0.1% H2O2 in TBS. Sections were rinsed, mounted on glass slides, dehydrated in graded ethanol solutions, cleared with xylene, and coverslipped.
Golgi-Cox. Animals were asphyxiated by carbon dioxide, and their brains were removed and immersed in fixative (10 mg/ml potassium dichromate, 10 mg/ml mercuric chloride, and 4.5 mg/ml potassium chromate; precipitate was cleared with 0.1N HCl). Brains were stored in the dark, undisturbed for 6–8 weeks. The tissue was dehydrated first in equal parts of acetone and alcohol for 24 hr at room temperature. This solution was replaced with ether alcohol (anhydrous ether/ethyl alcohol, 1:1) for 24 hr at room temperature. The brains were then infiltrated with low-viscosity nitrocellulose (LVN) celloidin/parlodion (Fisher Scientific, Pittsburg, PA) at concentrations of 5% (2 d), 10% (1 d), and 12% (5 d). Brains were embedded in fresh 12% LVN and hardened with chloroform vapors overnight. The blocks were stored in 70% ethanol until being cut with a sliding microtome. Sections (100 μm) were hydrated in distilled water for 15 min and developed in 5% sodium sulfite for 20 min. Sections were then dehydrated in 95% ethanol for 1 hr, cleared in terpineol overnight, mounted on slides, and coverslipped with Permount.
Genetic mapping. To localize the rocker mutation to a specific mouse chromosomal region, we began genetic mapping using differences in simple sequence length polymorphisms (SSLP). We selected MIT SSLP markers that amplified PCR products of different sizes in the C3H/HeJ and C57BL/6J strains. We began by selecting markers that allowed us to scan the entire mouse genome at a resolution of 20 centimorgans (cM). Homozygous rocker mutants on a C57BL/6J genetic background were crossed to C3H/HeJ mice producing (C57BL/6JxC3H/HeJ rkr/+) F1s. These were then backcrossed to homozygous C57BL/6J-rkr/rkr animals. To test for linkage to different chromosomal regions, we used a pooled PCR method (Pacek et al., 1993). Pools of DNA aliquots were made such that each pool consisted of equal amounts of DNA from each of 30 ataxic (rkr/rkr) or normally behaving (+/rkr) animals. The neurological phenotype of each mouse was determined by observation of abnormal gait at 3–4 weeks of age. Tail biopsies were taken and digested with 0.4 μg/μl proteinase K. Digests were spun at 13,000 × g for 5 min at room temperature. Either PCR was performed directly on diluted proteinase K supernatant (1:50) from individual animals, or DNA was extracted using phenol/chloroform (1:1) followed by ethanol precipitation. Genotyping was done on pooled samples containing equal amounts of DNA (1 μg per animal) from each mouse (30 animals total). Samples from ataxic (rkr/rkr) animals were pooled separately from samples of normally behaving (+/rkr) backcross littermates. Aliquots of the pools were then assayed by PCR for each of the selected SSLP loci. Primers for these loci were obtained from Research Genetics. PCR amplification was done on an MJ Research (Watertown, MA) thermal cycler in a total volume of 50 μl containing 100 ng of mouse genomic DNA. Amplification conditions were as follows: denaturation at 94°C for 1 min, followed by 30–35 cycles of (1) 95°C for 30 sec, (2) 55°C for 30 sec, and (3) 72°C for 30 sec. In this paradigm, if a given SSLP is unlinked torocker, the ataxic and wild-type pools should each amplify two bands (C3H/HeJ and C57BL/6J) of intensity ratio 1:3. By contrast, if the SSLP locus is linked to rocker, then the ataxic pool should predominantly amplify the C57BL/6J band, whereas the wild-type pool should amplify the two bands in a ratio closer to 1:1. An example of each result is shown in Figure 5A.
Fig. 5.

Mapping of rocker phenotype to mouse chromosome 8. The second generation of backcrossed animals was separated into affected and wild-type groups based on the observed ataxic phenotype. DNAs from animals of known phenotype were mixed to create known ratios of B6 and C3H DNA as controls for the PCR amplification. Duplicate lanes from the affected pool (rkr pool) and wild-type pool (wt pool) were amplified with a number of MIT markers that recognize SSLPs in the mouse genome (two examples are shown in A).A, The first set of primers, D8Mit95, amplifies a 158 bp product from the B6 strain and a 154 bp product from the C3H strain. The second set of primers, D8Mit162, amplifies a 149 bp B6 band and a 131 bp C3H band. Therocker pool shows strong bands from the B6 and a much weaker band amplified from the C3H. The wild-type pool shows a more equal amplification of both the B6 and C3H PCR products. In each experiment, a control lane of B6 alone shows a single band, and a second control lane shows a heterozygote animal (F1, a known 1:1 ratio of B6/C3H) producing both the B6 and C3H PCR products. The negative control (no DNA lane) showed no nonspecific amplification. The estimated size of PCR products was determined using a 123 bp DNA marker (L).B, MIT markers on mouse chromosome 8 used to localize the candidate region in individual animals.
Electrocorticographic recordings. Silver wire electrodes (0.005 inch diameter) soldered to a microminiature connector were implanted bilaterally into the subdural space over the frontal and parietal cortex of anesthetized mice several days before recording (Noebels and Sidman, 1979). Cortical activity was recorded using a digital electroencephalograph (TECA) from mutants and controls moving freely in the test cage for prolonged periods. Seizure behavior was observed directly and annotated on all recordings.
RESULTS
The neurological mouse mutant was initially observed in descendents of an ENU-mutagenized male mouse. The ataxic phenotype proved to be heritable as a Mendelian autosomal recessive trait and was given the provisional gene name rocker (gene symbolrkr). Rocker mutants display an ataxic gait initiated by an action tremor at 3–4 weeks of age, suggesting a possible cerebellar dysfunction. Affected animals are first recognizable by a splayed stance of their hindlimbs and their clumsy, uncoordinated behavior, especially on uneven surfaces. Despite the motor problems, they still exhibit exploratory behavior and are capable of quick movements. The rocker mutant mouse is able to swim and manages to keep its head above the surface. No paroxysmal dyskinesia phenotype has been observed in the rockerhomozygotes. At weaning age, there may be a slight decrease in size of the affected animals compared with their normal littermates, but the homozygous mutants of both sexes are fertile and appear to have a normal life span.
On initial analysis of the rocker brain [postnatal day (P) 60–120], the cytoarchitecture appeared normal. Given the spectrum of the behavioral symptoms, we extended our examination of the cerebellum. We found a normal cerebellar morphology and cytoarchitecture inrocker homozygotes (Fig.1B) compared with wild type (Fig. 1A). Measurements of the molecular layer width and total area revealed no significant difference between affected and nonaffected littermates. We performed Purkinje cell counts to investigate whether there was any loss of Purkinje neurons in 1-year-old animals (Fig. 2). No significant difference was seen between the number of Purkinje cells inrocker versus wild-type cerebella. To investigate the possible regional difference, our cell counts were also analyzed by folia. No Purkinje cell decrease was detected in any of the individual folia or in the anterior portion of the cerebellum (data not shown). To extend this structural analysis, rocker brains were immunostained with antibodies against a number of different neuronal and non-neuronal antigens. No abnormal staining pattern was observed inrocker cerebellum compared with wild-type animals using antibodies against either neuronal antigens (NeuN, NF160, and TrkB) or non-neuronal antigens (MBP, GFAP, and SV2). These antibodies are summarized in Table 2. Consistent with the normal cell counts, calbindin immunohistochemistry showed no gaps in the Purkinje cell monolayer.
Fig. 1.
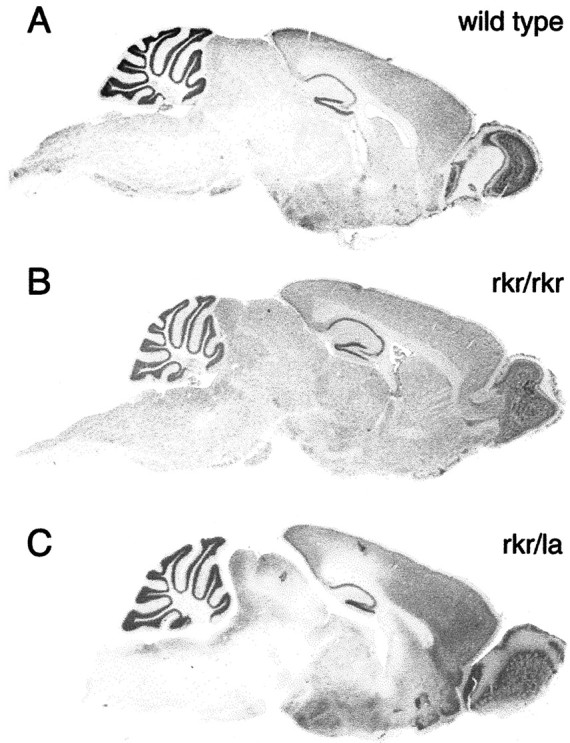
Sagittal sections of wild-type, rocker (rkr/rkr), and compound heterozygote (rkr/la)mice stained with 0.2% cresyl violet. Rocker mutants show normal cytoarchitecture of whole mouse brains. Comparison of wild type (A) with homozygote rocker(B) or compound heterozygote (C) revealed no significant difference in cerebellar morphology.
Fig. 2.

Adult rocker mutants show no loss of cerebellar Purkinje cells. The total number of Purkinje cells is graphed as a function of the distance from the midline.Rocker mutants >1 year of age (black line with diamonds and black dashed line with triangles) showed no significant difference from the age-matched wild-type control (gray line with circles). Gaps at the midline reflect sections that could not be counted because of a technical artifact of sectioning.
In contrast to the near absence of phenotype in the immunocytochemical studies, Golgi-Cox staining revealed dendritic structural abnormalities in the cerebellar Purkinje cells. During normal aging, the wild-type controls in our study underwent a slight reduction in the branching complexity of the Purkinje cell dendritic arbor (Fig.3A,B). By contrast, age-matched rocker animals aged ≥1 year displayed a Purkinje cell arbor that was significantly reduced in dendritic mass (Fig. 3C,D). In addition to the overall reduction in the size and complexity of the dendritic arbor, we observed what appeared to be an abnormal secondary outgrowth of the tips of the distal dendrites (Fig. 3D, arrow). The ends of the secondary branches appeared to extend, often turning and growing back toward the cell body. This resulted in a bent or serpentine-like appearance at the ends of the dendrites, and these extensions appeared to have grown without the usual branching that is found during normal dendritic development. The shafts of main Purkinje dendrites also showed a thickening in the upper molecular layer. We examined the hippocampus and the cerebral cortex and found that in cresyl violet-stained sections as well as Golgi impregnations, both areas were indistinguishable from wild-type animals (Figs.1A,B,4A,B). The dendritic changes illustrated in the Purkinje cells are atrophic in nature because the Purkinje cell dendrites in rocker animals examined at 6 months of age exhibit a normal appearance (data not shown).
Fig. 4.
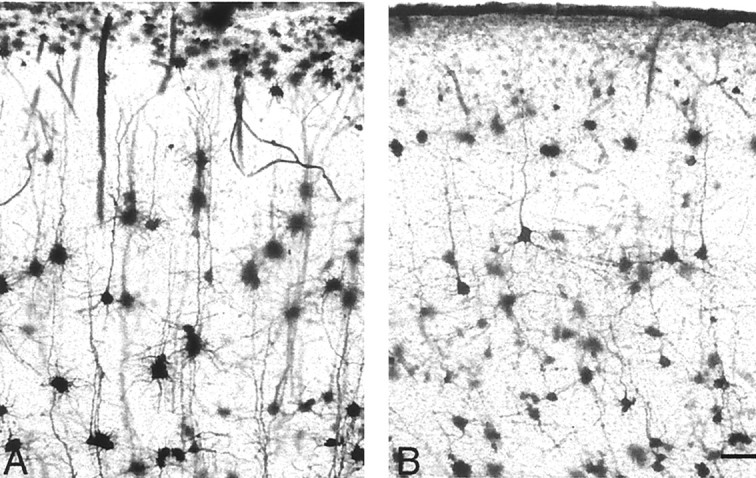
Golgi preparations were used to examine other areas of the brain known to express the calcium channel α 1a, such as the cerebral cortex. No distinguishable differences are evident between wild-type (A) and rocker(B) mutants. Scale bar, 50 μm.
Fig. 3.
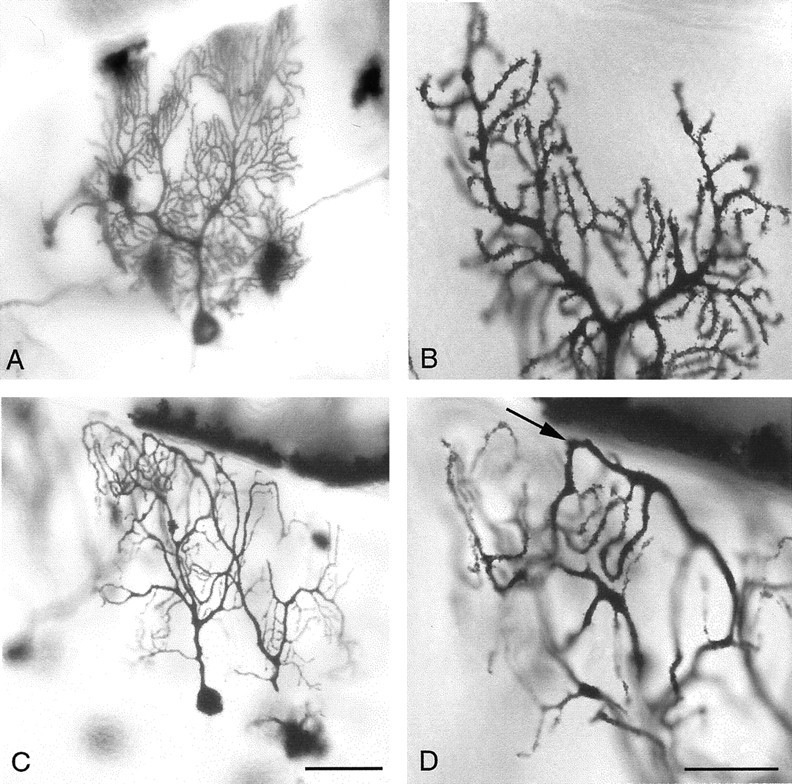
Golgi impregnation of individual cerebellar Purkinje cells shows dendritic abnormalities in the adultrocker mutants. A, B, Branching in the wild-type Purkinje cell usually appears very dense, with an increasing number of branching points near the distal tips.C, D, Purkinje cells in older rockermutants show a decrease in branching and have downturned distal ends of their dendritic tree (D, arrow). Scale bars: A, C, 50 μm; B,D, 25 μm.
Heritable changes in descendents of ENU-mutagenized males are usually assumed to result from a point mutation. The offspring of the male that produced the rocker mutation included a new myotonia allele named Clcn1adr-K (Neumann and Weber, 1989;Gronemeier et al., 1994). We eliminated this additional mutation as the source of the ataxic phenotype by complementation studies that showed no allelism between Clcn1adr-K androcker. Furthermore, sequence analysis of rockermuscle RT-PCR products confirmed that rocker was a different mutation because it did not have the single amino acid change, I553T, observed in the chloride channel mutant,Clcn1adr-K (data not shown).
To localize the rocker mutation to a specific mouse chromosomal region, we began genetic mapping using differences in SSLP. Substantial linkage disequilibrium was found in the amplification products of the D8Mit162 primer pair (Fig.5A). Analysis of other markers in this region confirmed this observation, and thus we were able to localize rocker to mouse chromosome 8. An expanded series of MIT markers was then analyzed in DNA from 107 individual progeny (Fig.5B). This allowed us to narrow the location ofrocker to a 2 cM region between D8Mit162 andD8Mit45. Because of its location and mutant phenotypes,Cacna1a emerged as a significant candidate gene (Fig.5B). To test for complementation, mating pairs were set up between heterozygous leaner animals (Cacna1a+/la) (The Jackson Laboratory, Bar Harbor, ME) and heterozygous rocker animals. Theleaner mutation is maintained at The Jackson Laboratory in repulsion with the tightly linked semidominant mutationoligosyndactylism (Os) (Sidman et al., 1965). Heterozygous Os induces fusion of the second and third digit on all four paws; homozygous Os/Os is lethal early in embryogenesis. Therefore, only the progeny of Os+/+la× Os+/+la with normal paws are homozygous forleaner. In a cross of heterozygous rocker and heterozygous leaner, 50% of the pups would be expected to have Os, and 50% would have normal paws. Of the normal-pawed animals, half would be expected to have an affected phenotype if there were no complementation. Of 65 progeny examined from this mating, 16 ataxic animals were identified, all with normal paws. The mating of heterozygous rocker and heterozygousleaner mice led to the production of offspring, one-fourth of whom were ataxic. This is consistent with the assumption that therocker and leaner mutations are allelic.
The rocker mutation was assumed to be a small missense mutation consistent with its appearance in an ENU mutagenesis experiment. The sequence change was identified by automated sequencing of the mouse α1a subunit cDNA from the rocker mouse brain. This analysis revealed several single-nucleotide polymorphisms that distinguish C57BL/6J and C3H/HeJ (the current and original background strains, respectively). Only one of these was unique torocker, a C-to-A change at nucleotide residue 3929 (Fig.6A). Wild-type animals from both parental strains (C57BL/6J and C3H/H3J) that were used in the outcross have C at position 3929. Both types of splice variation, α1A-a and α1A-b, were found at the three different sites (Bourinet et al., 1999) in the isolated cDNA fragments, revealing no specific link between particular splice variants and C3929A substitution. Together with the failure of genetic complementation, this sequence change firmly establishes rocker as a new Cacna1aallele, Cacna1arkr.
Fig. 6.

Determination of rocker mutation.A, The sequence alteration in the rockermutant. Rocker contains a cytosine (C) to adenosine (A) change at nucleotide position 3929 that results in a threonine (T) to lysine (K) alteration at amino acid position 1310. B, Proposed transmembrane topography of the α 1A subunit and positions oftottering (tg), rolling (rol), leaner (la), and rocker (rkr) (indicated by arrows).
This mutation predicts a nonconservative amino acid substitution, changing threonine (T) to lysine (K) at position 1310. T1310 is nine amino acids into the extracellular, pore-forming loop that connects segments 5 and 6 of repeat III (Fig.6A,B). This amino acid change results in the uncharacteristic arrangement of a charged polar side chain located on the extracellular membrane near the region that is thought to form the ion pore that passes calcium ions through the cell membrane.
The identification of rocker as a Cacna1a allele prompted us to reexamine several phenotypes of the new mutation. Because of previous reports of axonal swellings in the Purkinje cells of Cacna1a mutants (Rhyu et al., 1999a,b), we carefully examined the rocker mice. No swellings of the Purkinje cell axons were observed in the internal granule cell layer of either the wild-type or the rocker mutants (Fig.7A,C). The shafts of the Purkinje cell dendrite in totteringanimals have been shown to bear ectopic spines. Despite the unusual structural features of the rocker dendrite, no ectopic spines were observed on either the primary or the distal dendritic branches (Figs. 7D, 3D,respectively).
Fig. 7.
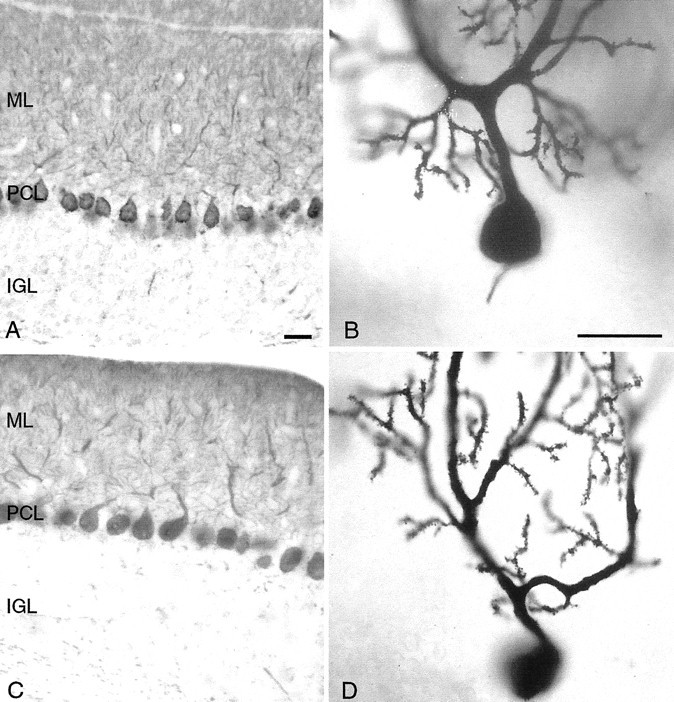
Calbindin immunohistochemistry staining in adult wild-type (A, B) and rocker (C, D) cerebellum reveals no obvious structural abnormalities. No axonal swellings of the Purkinje cell axons are observed in the internal granule cell layer of either the wild-type orrocker mutants. No gaps in the Purkinje cell layer (PCL) are evident, and no decrease appears in the size of the molecular (ML) or internal granule (IGL) cell layers. The structure of the Purkinje cell dendritic arbor also appears normal. No ectopic spines are observed on proximal dendrites of either the wild-type or rockeranimals. Scale bars: shown in A for A andC, 25 μm; shown in B forB and D, 25 μm.
Previous studies of tottering and leaner mutant mice have shown abnormalities in the temporal regulation of TH expression in posterior vermal cerebellar Purkinje cells. Wild-type animals show a transient Purkinje cell expression of TH that disappears by P40 (Fig.8A,B). By contrast, in tottering and leaner mutants, this transient expression persists into adulthood. We examined normal and rocker animals at 3 months of age and at P40. To our surprise, we observed no Purkinje cell expression of TH at these ages in the rocker mutant (Fig. 8C,D). The lack of persistence of TH staining marks a significant variance betweenrocker and the previously studied alleles at theCacna1a locus.
Fig. 8.
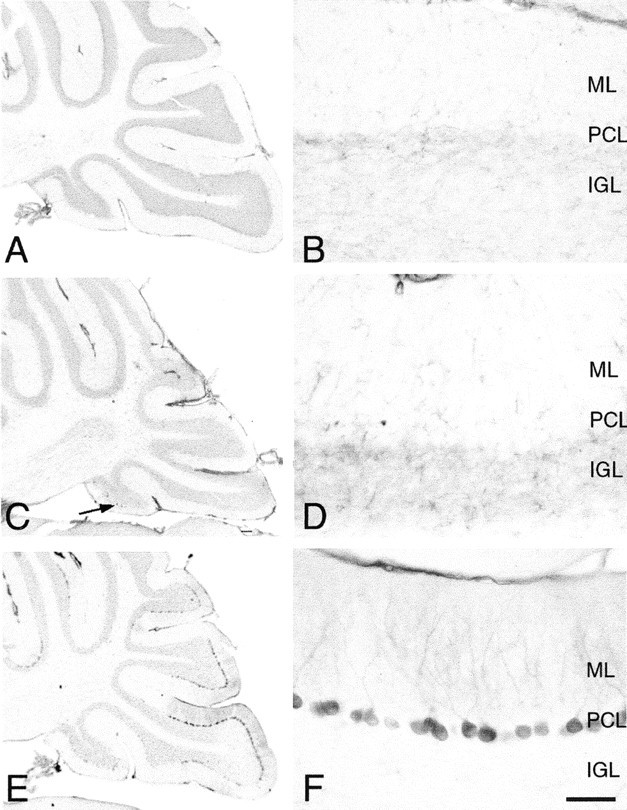
Immunostaining for tyrosine hydroxylase in the cerebellum of wild-type (A, B), rocker(C, D), and compound heterozygote (rkr/la) (E, F) adult animals reveal rocker to be unique among the α 1a mutants examined. A–D, Except for the occasional Purkinje cell TH staining (C, arrow), both wild type and rocker show the expected downregulation of TH expression. E, F, Large subpopulations of Purkinje cells show persistent TH expression in the compound heterozygote cerebellum. Scale bar: shown in F forB, D, and F, 50 μm.PCL, Purkinje cell layer; ML, molecular cell layer; IGL, internal granule cell layer.
Another irregularity associated with tottering mice is the presence of a characteristic pattern of EEG abnormalities that coincide with absence seizures. To determine whether rocker mice had the spike-and-wave patterns, we performed chronic monitoring of electroencephalograph activity from adult rockerhomozygotes. These studies revealed the spontaneous appearance of bilaterally synchronous and symmetrical 6–7 Hz spike-wave discharges while the mouse was awake. These episodes occurred at a mean frequency range of 41–65 episodes per hour and a mean burst duration of 1–1.7 sec (Fig. 9). Behavioral arrest and, on occasion, myoclonic movements of the vibrissae or neck always accompanied spike-wave episodes. Normal motor behavior resumed immediately on termination of the cortical discharge. The general morphology and temporal parameters of the cortical burst activity and behavioral arrest seizures are essentially identical to those recorded from tottering mutants.
Fig. 9.

Electrocorticographic activity recorded from awake adult rocker mutant. Traces from left and right hemispheres show bilateral, spontaneous cortical six to seven spike-wave synchronous discharge during behavioral arrest seizure. Electroencephalograph activity reverts to normal, low-amplitude, high-frequency rhythmic patterns immediately after the behavioral seizure episode. Calibration: 200 μV, 1 sec.
The Cacna1a gene product forms a membrane pore without multimerization (unlike the entire family of potassium channels that function as tetramers). The rocker/leaner compound heterozygote (Cacna1arkr/la)offers an unusual opportunity to study the effects of co-expression of two different mutant channels in a single neuronal membrane. Depending on the phenotype, the compound heterozygote might be expected to show either additive effects or dominance of one or the other allele. The severity of the ataxia appeared to be intermediate between that of theleaner and rocker homozygotes (as is reported for the tottering/leaner compound heterozygote). Thus the ataxia phenotype is additive. By contrast, the leaner allele is dominant over rocker in the expression of the dystonic movement phenotype. Although rocker does not have dyskinesia, both the compound heterozygote (Cacna1arkr/la) and leanermouse do. Rocker is dominant over leaner with respect to the phenotype of cell loss because no decrement in Purkinje cell counts is observed in the cerebellar cortex of the compound heterozygote (Fig. 1C) (cell counts not shown). In the phenotype of TH persistence, leaner is dominant overrocker; posterior Purkinje cells of the compound heterozygote retained significant TH levels in their cytoplasm well into adulthood (Fig. 8E,F). Finally, Golgi analysis of the compound heterozygote revealed ectopic spines appearing on secondary dendrites. This is aleaner-like phenotype, illustrating dominance of theleaner allele. No axonal swellings were observed (data not shown).
DISCUSSION
In this study, we identified the mouse mutant rocker(rkr) as a new allele of the calcium channel α1A subunit gene, Cacna1arkr, using mapping results, the failure of complementation with leaner animals, and sequencing to identify a point mutation that occurs only in ataxic animals. Rocker is the fifth mutant mouse Cacna1aallele in an allelomorphic series in which each mutation presents a distinct phenotypic syndrome.
Comparison with other alleles of mouse Cacna1a
Overall, the phenotype of this new mutation bears the greatest similarity to the original tottering mutation.Rocker mutant mice display an ataxic gait initiated by an action tremor. The age of onset of the rocker behavioral abnormalities (P21–28) is approximately the same astottering. Analysis of the overall cytoarchitecture of the adult rocker brain (P60–120) reveals no gross cytological abnormalities. We observed no reduction in the size of any of the three layers of cerebellar cortex of the rocker cerebellum, and we found no evidence of cell death (pyknotic cell bodies) in the molecular or internal granule cell layers of rocker mutants. In 1-year-old animals, there is no detectable decrease in the number ofrocker Purkinje cells (Fig. 2). This plus the absence of major alterations in cerebellar morphology (Fig. 1B) also identify rocker as similar to the totteringbrain, as opposed to leaner or rolling in which significant cell loss is reported (Muramoto et al., 1981; Herrup and Wilczynski, 1982). Furthermore, electroencephalographic recordings reveal frequent, spontaneous seizures when generalized, bilateral spike-and-wave discharges are observed, similar to those found intottering. The point mutation identified inrocker also appears similar to tottering in its structural location on the channel. Both mutations are extracellular and located near the pore-forming region of the channel (Fig.6B).
Although our initial observations suggest a close similarity to thetottering phenotype, further examination ofrocker reveals interesting and informative differences. In other Cacna1a alleles, an abnormal persistence of TH gene expression into adulthood has been reported in posterior/medial cerebellar Purkinje cells (Muramoto et al., 1981; Hess and Wilson, 1991; Austin et al., 1992; Sawada et al., 1999). In rocker,however, TH expression is downregulated on a time course identical to wild type. Although an occasional Purkinje cell was positive inrocker (Fig. 8C, arrow), similar observations have been reported in wild-type animals (Austin et al., 1992; Fujii et al., 1994; Abbott et al., 1996; Sawada et al., 1999). Previous explanations for the persistence of TH expression took note of the known effect of calcium on the regulation of immediate early gene expression (Ghosh et al., 1994) and the responsiveness of the TH promoter to c-fos, calcium, and neuronal activity (Gizang-Ginsberg and Ziff, 1990; Kilbourne et al., 1992; Sawada et al., 1999). The existence of TH-positive Purkinje cells in other ataxic mutant mice has suggested that abnormal expression of TH in Purkinje cells may not be specific to the Cacna1a allelic group (Muramoto et al., 1981; Sawada et al., 1999) and instead may be indicative of a developmental arrest or a more general predictor of neuronal dysfunction caused by alterations in cellular Ca2+ currents. One possibility is that a certain magnitude of change (or absolute concentration) of Ca2+ must be achieved to initiate the downregulation of TH. Electrophysiological examination of the calcium flux in rocker Purkinje cells might help evaluate the validity of this hypothesis. The lack of TH expression inrocker, even in the presence of absence seizures, allows us to rule out the spike-and-wave discharges as sufficient to result in the sustained TH expression, and vice versa. Furthermore, unlike thetottering phenotype, rocker animals display no evidence of motor seizures. These important clinical distinctions will allow rocker to serve as a useful model to assist in the determination of the different molecular or cellular mechanisms responsible for each of these phenotypes.
Golgi-Cox staining reveals dendritic abnormalities in the agedrocker cerebellar cortex. Our analysis of adultrocker animals (>1 year of age) reveals a reduction of branching in the Purkinje cell dendritic arbor and a weeping willow appearance of the secondary branches. Age-matched wild-type controls show no similar reduction in the Purkinje cell dendritic arbor. Our examination of younger rocker animals revealed that the Purkinje cell initially develops a fully branched dendritic arbor, which then regresses. Although Golgi-Cox analysis has been done for the other alleles at the Cacna1a locus, aged animals have not been examined. Other mouse mutants, such as weaver andreeler, have displayed cerebellar Purkinje cells with a weeping dendritic phenotype, as have irradiation- or toxin-induced agranular models. These insults were applied during cerebellar development, however, and have been attributed to a disruption in the afferent input and the formation, rather than the maintenance, of the dendritic tree.
Unlike the other known alleles, rocker mutants do not have ectopic Purkinje cell spines (Figs. 3D, 7D). Recent analysis by Rhyu and colleagues (1999a,b) has shown ectopic spines on the secondary branches of Purkinje cells in thetottering, rolling, and leaner animals. Although we see a dramatic reduction in the number of tertiary branches in the adult rocker, no aberrant sprouting of spines is seen on the primary dendrites. The structural changes seen in totteringand leaner and the absence of ectopic spines inrocker suggest that the calcium channel defect inrocker may be sufficient to sustain initial dendritic development but may be less efficient in the processes needed to maintain dendritic structure. Whether this is a direct effect of Ca2+ changes on the biochemistry of dendritic maintenance or a secondary effect of altered synaptic function remains to be determined.
The compound heterozygote (Cacna1arkr/la)
Additional insights into the function of the Cacna1agene can be found in the phenotype of the rocker/leanercompound heterozygote (Cacna1arkr/la). The severity of the motor phenotype of the compound heterozygote is intermediate between the rocker and leaner phenotype and is first observed at P15–20. Although previous studies inleaner animals have shown a continuous cell loss of both Purkinje and granule cells in the cerebellum beginning at about P30, we observed no such loss in compound heterozygotes (data not shown) (Herrup and Wilczynski, 1982; Heckroth and Abbott, 1994).
The rocker/leaner compound heterozygous mice maintained the expression of TH in the cerebellar Purkinje cells although heterozygousleaner/wild-type mice have a wild-type phenotype (Fig.8E,F). This suggests that the net disruption in either rocker or heterozygousleaner animals is inadequate to produce a persistent TH phenotype, whereas the combination of leaner androcker is sufficient. The dominance of leanerover rocker, but wild type over leaner suggests a threshold model in which a certain absolute level of Ca2+ physiology disruption is required to block the regression of TH staining. Leaner channels in the presence of wild type are sufficiently normal to trigger a regression but replace the wild-type channel with a rocker form, and there is enough imbalance to block regression. Preliminary studies of other phenotypes in the compound heterozygous mice also produce differences in dominance of the alleles. The presence of ectopic spines, as in leaner animals, and the lack of axonal swellings, as in rocker animals, suggest that further examination of the compound heterozygote will yield insights into allelic interactions. It would also be of interest to examine these phenotypes in a rocker/tottering compound heterozygote.
The contribution of VDCCs to neuronal function in the intact animal is clearly complex. The many mutants that disrupt these channels not only illustrate the importance of the VDCCs but also emphasize the value of the allelic series of mutations in a channel gene. Structural analysis of the different mutations will lead to a better understanding of the different mechanisms responsible for the diversity of resulting phenotypes. Identification of this allelic series comes at an opportune time to complement the rapid advances being made in the biophysics of voltage-gated calcium channels. Mutations that have different phenotypes will allow us to separate the molecular and cellular pathways responsible for these phenotypes. Rocker is of increased value not for its similarity to the other alleles in this series, but for the distinct differences it introduces.Rocker is the first allele to present absence seizures without the intermittent movement disorder. It is also the first to have no sustained expression of TH in the Purkinje cells, and it is the only allele to have no ectopic spines. Finally, it is the first reported Cacna1a allele with structural abnormalities in the mature Purkinje cell dendrite.
Multiple systems in the CNS develop abnormalities in both the murine and human hereditary diseases at this locus. Previous studies that compared properties of the native and recombinant mutant channels suggest that single tottering mutations are directly responsible for the neuropathic phenotypes of reduction in current density and deviations in gating behavior that lead to neuronal death and cerebellar atrophy (Wakamori et al., 1998; Qian and Noebels, 2000). The phenotype of rocker mice emphasizes how many of these traits are affected differently by the various alleles.
Footnotes
This work was supported by National Institutes of Health Grant NS20591 (K.H.), Medical Research Council Grant MT-15507 (P.E.N.), and National Institutes of Health Grant MH61092 (J.L.N.) We thank Caleb Davis for technical assistance and extend special thanks to Dr. Aravinda Chakravarti for his gracious persistence in obtaining cDNA sequence from the entire rocker gene.
Correspondence should be sent to Dr. Karl Herrup, Alzheimer Research Laboratory, Case Western Reserve University, E 504 School of Medicine, 10900 Euclid Avenue, Cleveland, OH 44106. E-mail:kxh26@po.cwru.edu.
REFERENCES
- 1.Abbott LC, Isaacs KR, Heckroth JA. Co-localization of tyrosine hydroxylase and zebrin II immunoreactivities in Purkinje cells of the mutant mice, tottering and tottering/leaner. Neuroscience. 1996;71:461–475. doi: 10.1016/0306-4522(95)00444-0. [DOI] [PubMed] [Google Scholar]
- 2.Austin MC, Schultzberg M, Abbott LC, Montpied P, Evers JR, Paul SM, Crawley JN. Expression of tyrosine hydroxylase in cerebellar Purkinje neurons of the mutant tottering and leaner mouse. Brain Res Mol Brain Res. 1992;15:227–240. doi: 10.1016/0169-328x(92)90113-p. [DOI] [PubMed] [Google Scholar]
- 3.Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 4.Bode VC. Ethylnitrosourea mutagenesis and the isolation of mutant alleles for specific genes located in the T region of mouse chromosome 17. Genetics. 1984;108:457–470. doi: 10.1093/genetics/108.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bode VC, Justice MJ, McDonald JD. Visible mutations induced by ethylnitrosourea mutagenesis. Mouse News Lett. 1987;77:131–132. [Google Scholar]
- 6.Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi GW, Nargeot J, Snutch TP. Splicing of alpha 1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat Neurosci. 1999;2:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- 7.Buckley K, Kelly RB. Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. J Cell Biol. 1985;100:1284–1294. doi: 10.1083/jcb.100.4.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burgess DL, Jones JM, Meisler MH, Noebels JL. Mutation of the Ca2+ channel beta subunit gene Cchb4 is associated with ataxia and seizures in the lethargic (lh) mouse. Cell. 1997;88:385–392. doi: 10.1016/s0092-8674(00)81877-2. [DOI] [PubMed] [Google Scholar]
- 9.Cohan CS, Connor JA, Kater SB. Electrically and chemically mediated increases in intracellular calcium in neuronal growth cones. J Neurosci. 1987;7:3588–3599. doi: 10.1523/JNEUROSCI.07-11-03588.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dove LS, Abbott LC, Griffith WH. Whole-cell and single-channel analysis of P-type calcium currents in cerebellar Purkinje cells of leaner mutant mice. J Neurosci. 1998;18:7687–7699. doi: 10.1523/JNEUROSCI.18-19-07687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doyle J, Ren X, Lennon G, Stubbs L. Mutations in the Cacnl1a4 calcium channel gene are associated with seizures, cerebellar degeneration, and ataxia in tottering and leaner mutant mice. Mamm Genome. 1997;8:113–120. doi: 10.1007/s003359900369. [DOI] [PubMed] [Google Scholar]
- 12.Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 13.Fletcher CF, Lutz CM, O'Sullivan TN, Shaughnessy JD, Jr, Hawkes R, Frankel WN, Copeland NG, Jenkins NA. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87:607–617. doi: 10.1016/s0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- 14.Fujii T, Sakai M, Nagatsu I. Immunohistochemical demonstration of expression of tyrosine hydroxylase in cerebellar Purkinje cells of the human and mouse. Neurosci Lett. 1994;165:161–163. doi: 10.1016/0304-3940(94)90734-x. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh A, Ginty DD, Bading H, Greenberg ME. Calcium regulation of gene expression in neuronal cells. J Neurobiol. 1994;25:294–303. doi: 10.1002/neu.480250309. [DOI] [PubMed] [Google Scholar]
- 16.Gizang-Ginsberg E, Ziff EB. Nerve growth factor regulates tyrosine hydroxylase gene transcription through a nucleoprotein complex that contains c-Fos. Genes Dev. 1990;4:477–491. doi: 10.1101/gad.4.4.477. [DOI] [PubMed] [Google Scholar]
- 17.Green MC, Sidman RL. Tottering–a neuromuscular mutation in the mouse. J Hered. 1962;53:233–237. doi: 10.1093/oxfordjournals.jhered.a107180. [DOI] [PubMed] [Google Scholar]
- 18.Green RC, Neumann PE, Davisson MT. Tottering-3J (tg 3J): A new epileptic mutant. Mouse News Lett. 1988;80:162. [Google Scholar]
- 19.Gronemeier M, Condie A, Prosser J, Steinmeyer K, Jentsch TJ, Jockusch H. Nonsense and missense mutations in the muscular chloride channel gene Clc-1 of myotonic mice. J Biol Chem. 1994;269:5963–5967. [PubMed] [Google Scholar]
- 20.Heckroth JA, Abbott LC. Purkinje cell loss from alternating sagittal zones in the cerebellum of leaner mutant mice. Brain Res. 1994;658:93–104. doi: 10.1016/s0006-8993(09)90014-2. [DOI] [PubMed] [Google Scholar]
- 21.Hendry IA. A method to correct adequately for the change in neuronal size when estimating neuronal numbers after nerve growth factor treatment. J Neurocytol. 1976;5:337–349. doi: 10.1007/BF01175119. [DOI] [PubMed] [Google Scholar]
- 22.Herrup K, Sunter K. Numerical matching during cerebellar development: quantitative analysis of granule cell death in staggerer mouse chimeras. J Neurosci. 1987;7:829–836. doi: 10.1523/JNEUROSCI.07-03-00829.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herrup K, Wilczynski SL. Cerebellar cell degeneration in the leaner mutant mouse. Neuroscience. 1982;7:2185–2196. doi: 10.1016/0306-4522(82)90129-4. [DOI] [PubMed] [Google Scholar]
- 24.Hess EJ, Wilson MC. Tottering and leaner mutations perturb transient developmental expression of tyrosine hydroxylase in embryologically distinct Purkinje cells. Neuron. 1991;6:123–132. doi: 10.1016/0896-6273(91)90127-l. [DOI] [PubMed] [Google Scholar]
- 25.Isaacs KR, Abbott LC. Cerebellar volume decreases in the tottering mouse are specific to the molecular layer. Brain Res Bull. 1995;36:309–314. doi: 10.1016/0361-9230(94)00207-h. [DOI] [PubMed] [Google Scholar]
- 26.Johnson BD, Byerly L. A cytoskeletal mechanism for Ca2+ channel metabolic dependence and inactivation by intracellular Ca2+. Neuron. 1993;10:797–804. doi: 10.1016/0896-6273(93)90196-x. [DOI] [PubMed] [Google Scholar]
- 27.Kater SB, Mills LR. Regulation of growth cone behavior by calcium. J Neurosci. 1991;11:891–899. doi: 10.1523/JNEUROSCI.11-04-00891.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kilbourne EJ, Nankova BB, Lewis EJ, McMahon A, Osaka H, Sabban DB, Sabban EL. Regulated expression of the tyrosine hydroxylase gene by membrane depolarization. Identification of the responsive element and possible second messengers. J Biol Chem. 1992;267:7563–7569. [PubMed] [Google Scholar]
- 29.Komuro H, Rakic P. Selective role of N-type calcium channels in neuronal migration. Science. 1992;257:806–809. doi: 10.1126/science.1323145. [DOI] [PubMed] [Google Scholar]
- 30.Letts VA, Felix R, Biddlecome GH, Arikkath J, Mahaffey CL, Valenzuela A, Bartlett FS, II, Mori Y, Campbell KP, Frankel WN. The mouse stargazer gene encodes a neuronal Ca2+-channel gamma subunit. Nat Genet. 1998;19:340–347. doi: 10.1038/1228. [DOI] [PubMed] [Google Scholar]
- 31.Levitt P, Noebels JL. Mutant mouse tottering: selective increase of locus ceruleus axons in a defined single-locus mutation. Proc Natl Acad Sci USA. 1981;78:4630–4634. doi: 10.1073/pnas.78.7.4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Llinas R, Sugimori M. Calcium conductances in Purkinje cell dendrites: their role in development and integration. Prog Brain Res. 1979;51:323–334. doi: 10.1016/S0079-6123(08)61312-6. [DOI] [PubMed] [Google Scholar]
- 33.Llinas RR. The intrinsic electrophysiological properties of mammalian neurons: insights into central nervous system function. Science. 1988;242:1654–1664. doi: 10.1126/science.3059497. [DOI] [PubMed] [Google Scholar]
- 34.Manivannan S, Terakawa S. Rapid sprouting of filopodia in nerve terminals of chromaffin cells, PC12 cells, and dorsal root neurons induced by electrical stimulation. J Neurosci. 1994;14:5917–5928. doi: 10.1523/JNEUROSCI.14-10-05917.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCobb DP, Best PM, Beam KG. Development alters the expression of calcium currents in chick limb motoneurons. Neuron. 1989;2:1633–1643. doi: 10.1016/0896-6273(89)90052-4. [DOI] [PubMed] [Google Scholar]
- 36.Meier H, MacPike AD. Three syndromes produced by two mutant genes in the mouse. Clinical, pathological, and ultrastructural bases of tottering, leaner, and heterozygous mice. J Hered. 1971;62:297–302. doi: 10.1093/oxfordjournals.jhered.a108176. [DOI] [PubMed] [Google Scholar]
- 37.Mills LR, Kater SB. Neuron-specific and state-specific differences in calcium homeostasis regulate the generation and degeneration of neuronal architecture. Neuron. 1990;4:149–163. doi: 10.1016/0896-6273(90)90451-k. [DOI] [PubMed] [Google Scholar]
- 38.Moorman SJ, Hume RI. Omega-conotoxin prevents myelin-evoked growth cone collapse in neonatal rat locus coeruleus neurons in vitro. J Neurosci. 1993;13:4727–4736. doi: 10.1523/JNEUROSCI.13-11-04727.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mori Y, Wakamori M, Oda S, Fletcher CF, Sekiguchi N, Mori E, Copeland NG, Jenkins NA, Matsushita K, Matsuyama Z, Imoto K. Reduced voltage sensitivity of activation of P/Q-type Ca2+ channels is associated with the ataxic mouse mutation rolling Nagoya (tgrol). J Neurosci. 2000;20:5654–5662. doi: 10.1523/JNEUROSCI.20-15-05654.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mukaiyama M, Mizuno K. Cerebellum of rolling mouse Nagoya. Saishin Igaku. 1976;31:233–236. [Google Scholar]
- 41.Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- 42.Muramoto O, Kanazawa I, Ando K. Neurotransmitter abnormality in Rolling mouse Nagoya, an ataxic mutant mouse. Brain Res. 1981;215:295–304. doi: 10.1016/0006-8993(81)90508-4. [DOI] [PubMed] [Google Scholar]
- 43.Muramoto O, Ando K, Kanazawa I. Central noradrenaline metabolism in cerebellar ataxic mice. Brain Res. 1982;237:387–395. doi: 10.1016/0006-8993(82)90450-4. [DOI] [PubMed] [Google Scholar]
- 44.Nakane K. Postnatal development of brain with congenital ataxia, rolling (rol) and tottering (tg). Teratology. 1976;14:248–249. [Google Scholar]
- 45.Neumann P, Weber T. A new allele of adr. Mouse News Lett. 1989;83:157. [Google Scholar]
- 46.Nishimura Y. The cerebellum of rolling mouse Nagoya. Adv Neurol Sci. 1975;19:670–672. [Google Scholar]
- 47.Noebels JL. Isolating single genes of the inherited epilepsies. Ann Neurol. 1984;16[Suppl]:S18–S21. doi: 10.1002/ana.410160705. [DOI] [PubMed] [Google Scholar]
- 48.Noebels JL, Sidman RL. Inherited epilepsy: spike-wave and focal motor seizures in the mutant mouse tottering. Science. 1979;204:1334–1336. doi: 10.1126/science.572084. [DOI] [PubMed] [Google Scholar]
- 49.Oda S. The observation of rolling mouse Nagoya (rol), a new neurological mutant, and its maintenance. Jikken Dobutsu. 1973;22:281–288. doi: 10.1538/expanim1957.22.4_281. [DOI] [PubMed] [Google Scholar]
- 50.Oda S. A new allele of the tottering locus, rolling mouse Nagoya, on chromosome no. 8 in the mouse. Jpn J Genet. 1981;56:295–299. [Google Scholar]
- 51.Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- 52.Pacek P, Sajantila A, Syvanen AC. Determination of allele frequencies at loci with length polymorphism by quantitative analysis of DNA amplified from pooled samples. PCR Methods Appl. 1993;2:313–317. doi: 10.1101/gr.2.4.313. [DOI] [PubMed] [Google Scholar]
- 53.Qian J, Noebels JL. Presynaptic Ca2+ influx at a mouse central synapse with Ca2+ channel subunit mutations. J Neurosci. 2000;20:163–170. doi: 10.1523/JNEUROSCI.20-01-00163.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rhyu IJ, Abbott LC, Walker DB, Sotelo C. An ultrastructural study of granule cell/Purkinje cell synapses in tottering (tg/tg), leaner (tg(la)/tg(la)) and compound heterozygous tottering/leaner (tg/tg(la)) mice. Neuroscience. 1999a;90:717–728. doi: 10.1016/s0306-4522(98)00518-1. [DOI] [PubMed] [Google Scholar]
- 55.Rhyu IJ, Oda S, Uhm CS, Kim H, Suh YS, Abbott LC. Morphologic investigation of rolling mouse Nagoya (tg(rol)/tg(rol)) cerebellar Purkinje cells: an ataxic mutant, revisited. Neurosci Lett. 1999b;266:49–52. doi: 10.1016/s0304-3940(99)00254-2. [DOI] [PubMed] [Google Scholar]
- 56.Sawada K, Komatsu S, Haga H, Sun XZ, Hisano S, Fukui Y. Abnormal expression of tyrosine hydroxylase immunoreactivity in cerebellar cortex of ataxic mutant mice. Brain Res. 1999;829:107–112. doi: 10.1016/s0006-8993(99)01347-5. [DOI] [PubMed] [Google Scholar]
- 57.Scholz KP, Miller RJ. Developmental changes in presynaptic calcium channels coupled to glutamate release in cultured rat hippocampal neurons. J Neurosci. 1995;15:4612–4617. doi: 10.1523/JNEUROSCI.15-06-04612.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sidman RL, Green MC, Appel SH. Catalog of the neurological mutants of the mouse. Harvard UP; Cambridge, MA: 1965. [DOI] [PubMed] [Google Scholar]
- 59.Spitzer NC. Development of voltage-dependent and ligand-gated channels in excitable membranes. Prog Brain Res. 1994;102:169–179. doi: 10.1016/S0079-6123(08)60538-5. [DOI] [PubMed] [Google Scholar]
- 60.Stea A, Tomlinson WJ, Soong TW, Bourinet E, Dubel SJ, Vincent SR, Snutch TP. Localization and functional properties of a rat brain alpha 1A calcium channel reflect similarities to neuronal Q- and P-type channels. Proc Natl Acad Sci USA. 1994;91:10576–10580. doi: 10.1073/pnas.91.22.10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tanaka O, Sakagami H, Kondo H. Localization of mRNAs of voltage-dependent Ca(2+)-channels: four subtypes of alpha 1- and beta-subunits in developing and mature rat brain. Brain Res Mol Brain Res. 1995;30:1–16. doi: 10.1016/0169-328x(94)00265-g. [DOI] [PubMed] [Google Scholar]
- 62.Tsuji S, Meier H. Evidence for allelism of leaner and tottering in the mouse. Genet Res. 1971;17:83–88. doi: 10.1017/s0016672300012040. [DOI] [PubMed] [Google Scholar]
- 63.Vigers AJ, Pfenninger KH. N-type and L-type calcium channels are present in nerve growth cones. Numbers increase on synaptogenesis. Brain Res Dev Brain Res. 1991;60:197–203. doi: 10.1016/0165-3806(91)90048-n. [DOI] [PubMed] [Google Scholar]
- 64.Wakamori M, Yamazaki K, Matsunodaira H, Teramoto T, Tanaka I, Niidome T, Sawada K, Nishizawa Y, Sekiguchi N, Mori E, Mori Y, Imoto K. Single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel. J Biol Chem. 1998;273:34857–34867. doi: 10.1074/jbc.273.52.34857. [DOI] [PubMed] [Google Scholar]
- 65.Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TV, Snutch TP, Catterall WA. Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J Neurosci. 1995;15:6403–6418. doi: 10.1523/JNEUROSCI.15-10-06403.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- 67.Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15:62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]