

Abstract
Fast IPSCs in the brain are predominantly caused by presynaptic release of GABA that activates GABAAreceptor (GABAAR) channels. The IPSCs are shaped by the gating and desensitization properties of postsynaptic GABAARs. Specifically, fast desensitization has been suggested to decrease IPSC amplitude and to increase IPSC duration by slowing deactivation; however, the mechanisms underlying desensitization, deactivation, and their coupling are poorly understood. Consistent with this suggestion, α1β3γ2L GABAARs desensitize with a prominent fast phase and deactivate slowly, whereas α1β3δ GABAARs desensitize without a fast phase and deactivate rapidly. Using the concentration-jump technique applied to excised patches, we studied GABAARs containing chimeras or exchange mutants between δ and γ2L subunits to gain insight into the structural bases for fast desensitization and its coupling to deactivation. We demonstrated that the N terminus and two adjacent residues (V233, Y234) in the first transmembrane domain (TM1) of the δ subunit were both required to abolish fast desensitization. Additionally, these residues in TM1 of the γ2L subunit (Y235, F236) were critical for desensitized states to prolong deactivation after removal of GABA, because mutations resulted in accelerated deactivation despite unaltered desensitization time course. Interestingly, control of desensitization and deactivation was independent of the identity (γ2L or δ subunit sequence) of TM2, indicating that structures related to the putative channel gate may play a less direct role in desensitization than previously suggested.
Keywords: GABAA receptor, desensitization, deactivation, rapid kinetics, concentration-jump, recombinant
Fast synaptic inhibition in the brain is predominantly attributable to release of GABA, which activates GABAA receptor (GABAAR) channels. GABAARs are members of a superfamily of ligand-gated ion channels (Ortells and Lunt, 1995), and seven different mammalian subunit families (α, β, γ, δ, ε, π, θ) and their subtypes (α1–6, β1–3, γ1–3) have been reported (Macdonald and Olsen, 1994; Davies et al., 1997; Hedblom and Kirkness, 1997; Bonnert et al., 1999). Multiple GABAAR isoforms, the majority of which are αβγ and αβδ heteromers (McKernan and Whiting, 1996), are composed of different combinations of five subunits (Nayeem et al., 1994) that together form a transmembrane chloride ion channel.
GABA concentration at central synaptic clefts has been estimated to reach 500–1000 μm rapidly (<1 msec) (Maconochie et al., 1994; Jones and Westbrook, 1995) before decaying within milliseconds because of diffusion and presynaptic terminal reuptake (Clements, 1996). The current relaxation time of IPSCs after clearance of GABA (deactivation), however, is substantially longer than predicted by the relatively low affinity and short burst durations of GABAARs (Macdonald et al., 1989; Fisher and Macdonald, 1997). Because the time course of IPSCs can be reproduced reasonably by native and recombinant GABAARs in excised membrane patches with 1–10 msec pulses of saturating GABA concentrations (Jones and Westbrook, 1995; Tia et al., 1996; Galarreta and Hestrin, 1997; Haas and Macdonald, 1999), its long duration relative to the brief GABA transient is likely attributable to intrinsic channel kinetics that do not depend on (although they may be modified by) a neuronal milieu.
Continued application of GABA to GABAARs leads to a decline in current, termed desensitization, that occurs with fast (∼10 msec), intermediate (∼150 msec), and slow (∼1500 msec) rates (Celentano and Wong, 1994; Jones and Westbrook, 1995; Dominguez-Perrot et al., 1996; Haas and Macdonald, 1999). Desensitization is generally considered a negative feedback mechanism that reduces the peak of IPSCs, but it has been suggested recently that desensitization may, in fact, enhance GABAergic transmission by prolonging IPSCs. Jones and Westbrook (1995) suggested that high-affinity, long-lived desensitized states delayed unbinding of GABA, thus allowing additional late openings to occur before unbinding and slowing deactivation. This coupling of fast desensitization and deactivation provides a mechanism that overcomes the kinetic limitations of low-affinity and low-efficacy synaptic receptors.
Not all GABAARs, however, have the same rates of desensitization and deactivation. We recently characterized the distinct desensitization and deactivation kinetics of α1β3γ2L and α1β3δ GABAARs (Haas and Macdonald, 1999). Receptors containing the γ2L subunit showed prominent fast desensitization accompanied by prolonged deactivation. In contrast, δ subunit-containing receptors lacked fast desensitization and deactivated rapidly, despite having a fourfold higher GABA sensitivity. To explore the structural bases for the coupling of fast desensitization and deactivation, we transiently coexpressed chimeras between the δ and γ2L subunits and exchange mutations in these subunits with α1 and β3 subunits in mouse L929 and human embryonic kidney (HEK) 293T fibroblasts and recorded macroscopic GABAAR currents evoked from excised outside-out patches using the concentration-jump technique.
MATERIALS AND METHODS
Construction of GABAAR chimeras and mutations. The chimeras were constructed by using restriction fragments (at engineered sites) or by a PCR-based overlap extension method or by site-directed mutagenesis in existing chimeras. The transition point of subunit amino acid sequence is listed for each chimera, as the N-terminal parent subunit with the last amino acid of that segment, followed by the C-terminal parent subunit with the first amino acid of that segment: δ-γ M1e, δG232-γY235; δ-γ M1pre-iso, δY234-γT237; δ-γ M1p, δP241-γC244; δ-γ M1i, δI255-γN258; δ-γ M2e, δR282-γK285; γ-δ M1e, γG234-δV233; and γ-δ M1i, γI257-δS256. Numbering refers to the mature peptide. Point mutations were made using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Oligonucleotide primers were synthesized by the University of Michigan DNA synthesis core facility (Ann Arbor, MI). The fidelity of the amino acid sequences surrounding the splice sites was confirmed by sequencing of the final constructs.
Expression of recombinant GABAARs.The cDNAs encoding rat α1, β3, γ2L, and δ GABAAR subunit subtypes, as well as the chimeras and mutant subunits, were individually subcloned into the plasmid expression vector pCMVNeo. Mouse L929 fibroblasts (American Type Culture Collection, Rockville, MD) and HEK293T cells (a gift from P. Connely, COR Therapeutics, San Francisco, CA) were maintained in DMEM, supplemented with 10% horse serum or 10% fetal bovine serum, respectively, at 37°C in 5% CO2 and 95% air. Cells were transfected with 4–8 μg of each subunit plasmid along with 1–2 μg of either EGFP plasmid for expression of the marker green fluorescent protein (Clontech, Palo Alto, CA,) or pHOOK (Invitrogen, Carlsbad, CA) for immunomagnetic bead separation (Greenfield et al., 1997), using a modified calcium phosphate coprecipitation technique as previously described (Angelotti et al., 1993). The next day, cells were replated, and recordings were made 18–30 hr later. Expression of receptors composed of αβ subunits can be distinguished from expression of receptors composed of αβγ or αβδ subunits by their smaller single-channel conductance (Angelotti et al., 1993; Fisher and Macdonald, 1997). Preliminary single-channel recordings were obtained for all mutant and chimeric receptor channels, and based on their single channel conductance, each mutant and chimera construct tested was shown to combine with α1 and β3 subtypes to produce functional receptor channels (data not shown).
Electrophysiology. Patch-clamp recordings were performed on outside-out membrane patches excised from transfected fibroblasts bathed in an external solution consisting of (in mm): NaCl 142; KCl or CsCl 8; MgCl2 6; CaCl2 1; HEPES 10; and glucose 10, pH 7.4, 325 mOsm. Glass microelectrodes were formed from thick-walled borosilicate glass (World Precision Instruments, Pittsburgh, PA) with a Flaming–Brown electrode puller (Sutter Instruments, San Rafael, CA), fire-polished, then coated with Q-dope (GC Electronics, Rockford, IL). Patch electrodes had resistances of 4–14 MΩ when filled with an internal solution consisting of (in mm): KCl or CsCl 153; MgCl21; MgATP 2; HEPES 10; and EGTA 5, pH 7.3, 300 mOsm. This combination of internal and external solutions produced a chloride equilibrium potential of ∼0 mV. Outside-out membrane patches were usually voltage-clamped at −50 to −75 mV using an EPC-7 (List, Darmstadt, Germany) or an Axon 200A amplifier (Axon Instruments, Foster City, CA). No voltage-dependent changes in kinetics were detected between −20 and −80 mV.
GABA was applied to outside-out membrane patches using a rapid application system consisting of a double-barreled theta tube (Frederick Haer, Brunswick, ME) connected either to a piezoelectric translator (Burleigh Instruments, Fishers, NY) or to a Warner Perfusion Fast-Step (Warner Instrument Corporation, Hamden, CT). The fluid interface generated between control external recording solution and GABA-containing external solution was driven rapidly across the patch. The solution exchange time was monitored at the end of each recording by blowing out the patch and stepping a dilute external solution across the open electrode tip to measure a liquid junction current. The 10–90% rise times for solution exchange were consistently ∼400 μsec or less with either apparatus. All experiments were performed at room temperature (22–23°C).
For whole-cell experiments, GABA was applied to cells using multibarrel square glass attached to the Warner Perfusion Fast-Step (Warner Instrument Corporation). This enabled rapid solution changes, with maximal current rise times of <10 msec. Peak GABAR currents evoked by GABA at multiple concentrations were fitted to a sigmoidal function using a four parameter logistic equation (sigmoidal concentration–response) with a variable slope. The equation used to fit the concentration–response relationship was:
where I was the peak current at a given GABA concentration, and Imax was the maximal peak current.
Analysis of rapid application currents. Outside-out patch data were low-pass filtered at 2 or 3 kHz, digitized at 10 kHz, and analyzed using the pClamp8 software suite (Axon Instruments) and Origin 4.1 (Microcal, Northampton, MA). Multiple (3–50) GABA-elicited responses were acquired for each patch at 30–60 sec intervals and averaged to form ensemble currents for analysis. The desensitization or deactivation time courses of ensemble GABAAR currents were fit using the Levenberg–Marquardt least squares method with one or two component exponential functions of the form Σanτn +C, where n is the best number of exponential components, a is the relative amplitude of the component, τ is the time constant, and C is a constant term to account for residual current (incomplete desensitization). A second component was accepted only if it significantly improved the fit compared with a single exponential function, as determined by anF test on the sum of squared residuals. Three component fits were not considered for the application durations used in this study. The “fast” component was defined as the faster exponential function when the desensitization time course was fitted best by two exponential functions. If one exponential function was sufficient, the fast phase contribution was assigned as zero. However, to avoid misclassification, three patches containing the δ(VYΔYF) mutation were considered to have fast desensitization despite a single exponential fit because the τfast was <10 msec. Fast desensitization was quantified as the relative contribution of the fast exponential to the peak current, that is,a1/(a1+ a2 + C), wherea1 anda2 are the amplitude of the fast and slow exponential components respectively, and C is the constant term. The fitted peak current was given by the denominator. Because the % τfast values represent a mean of all patches for each isoform, small values (<10%) were obtained for isoforms in which most, but not all, patches showed no fast desensitization (zero fast phase), not because a small fast component was found in each patch. In none of these cases were the values significantly different from zero. For example, only 1 of 11 αβδ patches desensitized biphasically with 26% τfast, yielding a mean % τfast of 2.39% (Table1). The extent of desensitization was measured as (fitted peak current − fitted steady-state current)/(fitted peak current). For comparison of deactivation time courses, a weighted summation of the fast and slow decay components (af * τf +as * τs) was used, where τf and τswere the fast and slow decay time constants, andaf and aswere the relative initial proportion fast and slow, respectively. Numerical data were expressed as mean ± SEM. Statistical significance, unless otherwise stated, was p < 0.05, using unpaired two-tailed Student's t test (with a Welch's correction for unequal variances when necessary) or ANOVA as appropriate.
Table 1.
Summary of desensitization and deactivation kinetics
| Construct | % τfast | τfast(msec) | # fast/total | Extent (%) | τdeact. (msec) |
|---|---|---|---|---|---|
| αβδ | 2.4 ± 2.4 | – | 1 /11 | 12.4 ± 4.8 | 54.7 ± 4.8 |
| δ-γM2e | 6.1 ± 4.5 | – | 1 /10 | 17.8 ± 6.5 | 45.4 ± 11.3 |
| δ-γM1i | 9.6 ± 3.8 | – | 6 /26 | 33.1 ± 5.2* | 54.6 ± 8.1 |
| δ-γM1p | 8.2 ± 8.2 | – | 1 /6 | 14.7 ± 11.9 | 10.3 ± 2.2* |
| δ-γM1pre-iso | 3.5 ± 3.5 | – | 1 /14 | 15.8 ± 5.8 | 27.4 ± 4.9* |
| δ-γM1e | 40.8 ± 6.9 | 11.2 ± 2.8 | 13 /14 | 69.3 ± 7.5 | 107.5 ± 14.7* |
| αβγ | 41.3 ± 3.9 | 6.0 ± 0.7 | 13 /13 | 67.4 ± 3.8 | 171.8 ± 20.4 |
| δ(M2S) | 9.0 ± 4.8 | – | 3 /12 | 26.3 ± 8.4 | 61.7 ± 12.0 |
| γ(M2S) | 45.4 ± 2.2 | 7.9 ± 1.3 | 9 /9 | 69.6 ± 3.0 | 137.8 ± 24.6 |
| γ2L(YΔV) | 45.2 ± 7.8 | 6.9 ± 1.4 | 7 /7 | 68.1 ± 6.3 | 79.3 ± 21.8 |
| γ2L(FΔY) | 34.2 ± 5.2 | 8.0 ± 1.7 | 8 /8 | 57.7 ± 6.7 | 86.6 ± 18.7 |
| γ2L(YFΔVY) | 40.5 ± 5.9 | 9.0 ± 1.1 | 17 /19 | 67.5 ± 4.4 | 39.7 ± 4.7* |
| α(YFΔVY) | 18.5 ± 5.7* | 11.6 ± 1.7 | 6 /9 | 64.6 ± 3.6 | 56.5 ± 8.3 |
| β(YFΔVY) | 30.4 ± 4.8 | 7.8 ± 0.9 | 12 /14 | 61.5 ± 4.4 | 99.2 ± 12.5* |
| δ(VYΔYF) | 35.3 ± 3.6 | 9.0 ± 1.1 | 13 /21 | 38.4 ± 6.3* | 76.5 ± 6.0 |
| γ-δ M1e | 40.1 ± 9.2 | 11.8 ± 1.9 | 6 /7 | 56.2 ± 10.3 | 68.4 ± 11.4 |
| γ-δ M1i | 40.6 ± 5.7 | 10.5 ± 1.9 | 8 /8 | 66.8 ± 7.3 | 186.0 ± 29.0 |
% is the relative contribution of fast desensitization (see Results). τfast is shown only for constructs that had fast desensitization.
fast/total is the number of patches having fast desensitization/total patches studied. Extent (%) refers to the extent of current loss over the 400 msec GABA pulse (see Materials and Methods). τdeact is the weighted deactivation rate (see Materials and Methods). Asterisks indicate significant differences from both αβδ and αβγ isoforms. Data are mean ± SEM.
RESULTS
γ2L and δ subunits confer distinct desensitization and deactivation kinetics
To evaluate the kinetics of fast desensitization, 400 msec pulses of GABA (1 mm) were applied to excised outside-out membrane patches containing recombinant rat GABAAR isoforms using a rapid solution exchange protocol (see Materials and Methods). There were no kinetic differences observed between currents recorded from mouse (L929) and human (HEK293T) fibroblast expression systems, and thus the results were pooled. The current loss during the GABA pulse, attributed to receptor desensitization, was fitted with a single or double exponential function (see Materials and Methods). For α1β3γ2L (hereafter αβγ) receptors, rapid and extensive fast desensitization was evident followed by a small slower phase of desensitization (Fig.1A). The desensitization time course was fitted best with the sum of two exponential functions with fast (τfast < 10 msec) and slow (τslow ∼150 msec) time constants. The fast desensitizing component typically accounted for >40% of the current amplitude. In contrast, for α1β3δ (hereafter αβδ) receptors, only minimal desensitization occurred during the GABA application (Fig. 1B). Many αβδ receptor currents did not desensitize at all during the 400 msec pulse. When current loss was observed, it was fitted best with a single, small-amplitude exponential function with a slow rate (τslow ∼45 msec). Although αβδ receptors tended to have smaller peak currents than αβγ receptors (Haas and Macdonald, 1999; this study), there was no correlation between peak amplitude and desensitization rate among patches of the same isoform. The traces from Figure 1, A and B,were rescaled and overlaid to illustrate the different desensitization rates (Fig. 1C). Because of the presence of an additional slow phase of desensitization for both of these isoforms (τ ∼1500 msec) (Haas and Macdonald, 1999), the currents did not reach steady state during the 400 msec GABA application; however, the fast phase of desensitization could be resolved readily.
Fig. 1.
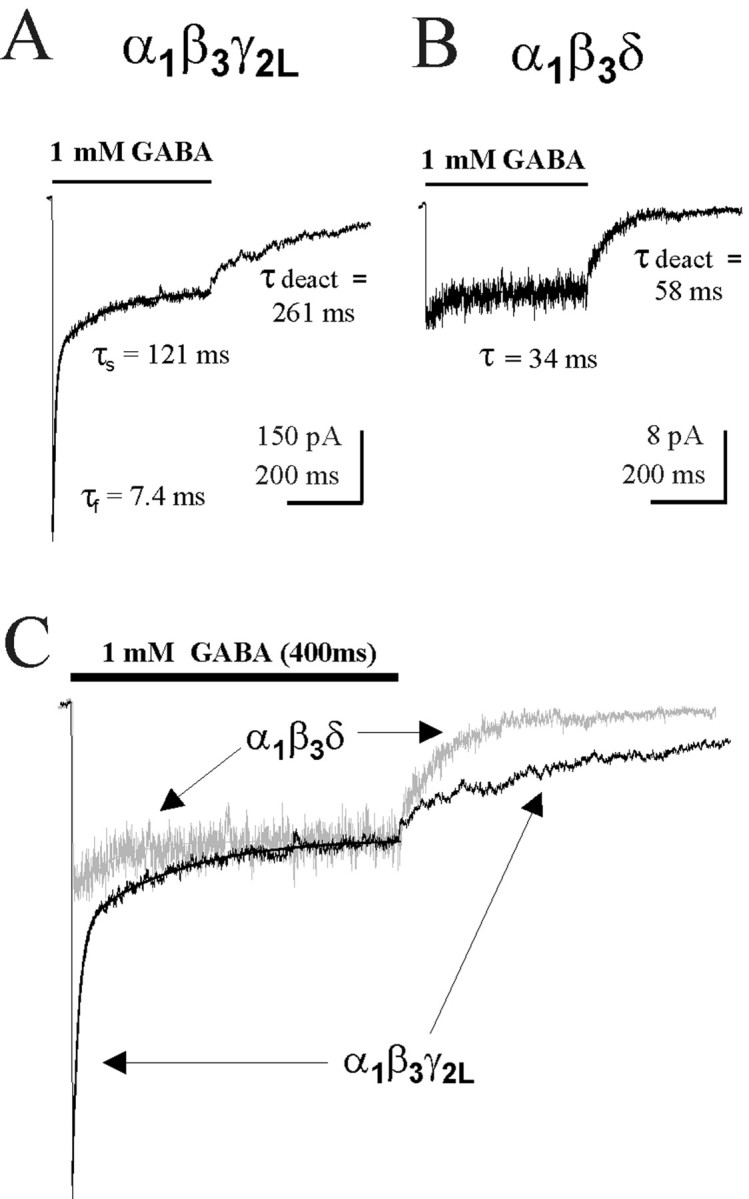
Macroscopic kinetics of αβδ and αβγ GABAAR isoforms. A, Rapid biphasic desensitization and prolonged deactivation were typical for αβγ receptors in response to a 400 msec pulse of 1 mm GABA. In this and all subsequent figures, outside-out patches excised from acutely transfected fibroblasts (mouse L929 and HEK293T) were exposed to GABA using the concentration-jump technique (see Materials and Methods). Patches were generally clamped at −50 to −75 mV. The traces shown are the average of multiple GABA-evoked currents from a single patch. Desensitization was described by the sum of fast (τfast) and slow (τslow) exponential functions. Deactivation was fit with one or two exponential functions, and the weighted deactivation τdeact is shown.B, Representative patch containing αβδ receptors showed minimal desensitization during a 400 msec pulse of GABA. This current was described by a single exponential decay function, τ = 34 msec. Deactivation of this isoform (τdeact = 54.7 ± 4.8 msec) was rapid compared with the α1β3γ2L isoform (τdeact = 171.8 ± 20.4 msec).C, Overlay of rescaled traces from A andB to emphasize the differences in fast desensitization (p < 0.0001) and deactivation (p < 0.0001) between these isoforms. The αβδ current was colored gray for clarity. Thecurved lines in each trace are the fitted exponential functions describing the desensitization time course.
The current relaxation during GABA washout (deactivation) also differed substantially between these isoforms. Consistent with our previous study (Haas and Macdonald, 1999), αβγ receptors deactivated slowly (τdeact = 171.8 ± 20.4 msec), whereas αβδ receptors deactivated rapidly (τdeact = 54.7 ± 4.8 msec) (Fig.1A,B). The rescaled and overlaid traces also illustrate the different deactivation rates (Fig. 1C). Although GABA affinity can influence the time course of deactivation, the pattern observed for these isoforms was opposite of that expected based on EC50 alone, because αβδ receptors have a threefold to fourfold lower GABA EC50(Saxena and Macdonald, 1996) but deactivate rapidly compared with αβγ receptors. Thus, minimal desensitization and rapid deactivation were characteristic of δ subunit-containing receptors, whereas pronounced fast desensitization and prolonged deactivation were characteristic of γ2L subunit-containing receptors.
Desensitization and deactivation of α1β3δ-γ2L chimera receptors
We generated δ and γ2L subunit chimeras to explore the molecular basis for desensitization and its coupling to deactivation (Fig. 2, left). During individual synaptic events, the duration of GABA in the cleft is brief, and fast desensitization is the only form of desensitization involved in shaping the synaptic current (Jones and Westbrook, 1995; Haas and Macdonald, 1999). Therefore we were interested in evaluating specifically the relative contribution of fast desensitization during a 400 msec pulse of saturating (1 mm) GABA (Fig. 2,middle). Extent (percentage of current loss) and weighted rates have been typically used to characterize desensitization, but these measures can obscure changes specific to the physiologically relevant fast phase. The fast phase of desensitization was defined here as the faster exponential function when a double exponential function was required to fit the time course of desensitization, usually (>95%) corresponding to a τfast < 15 msec (see Materials and Methods). If a single exponential function was sufficient to fit the current decay (usually τ ∼30–200 msec), the fast component amplitude was assigned as zero. Currents fitted with a single exponential time course usually exhibited <25% current loss during the 400 msec pulse. We summarized the desensitization patterns as the mean relative proportion of the fast exponential component (%τfast; see Materials and Methods) (Fig. 2,right, hatched bars). The 400 msec pulse length also allowed deactivation to be assessed, because the extent of desensitization was never complete with GABA applications of this duration. The deactivation time course was variable but could always be fitted with a single or double exponential function. To facilitate comparison among isoforms, a weighted deactivation rate (τdeact) was determined (Fig. 2, right, solid bars; see Materials and Methods).
Fig. 2.

Desensitization of GABAARs containing δ-γ2L chimeras. Desensitization and deactivation kinetics of GABAARs containing δ, γ2L, or δ-γ2L chimera subunits coexpressed with α1 and β3 subunits. Theleft column shows schematics of chimeras generated between the δ subunit (gray) and the γ2L subunit (white). N terminus is to the left (N-term), and boxes (1–4) indicate the four putative transmembrane domains. Representative normalized responses to a 400 msec pulse of 1 mm GABA are shown for each isoform in the center column. Desensitization was quantified as the relative contribution of a fast phase, %τfast (hatched bars, see Materials and Methods). Deactivation was fit with single or double exponential functions, and for comparison the weighted sum, τdeact, is shown (solid bars). Note the different scales used for %τfast and τdeact in the right column. The number of patches for each isoform is indicated in parentheses next to the sample currents. To facilitate comparison, shaded vertical bars indicate the mean (center of bar) and SEM range (thickness of bar) for wild-type αβδ and αβγ values of %τfast and τdeact. Theasterisk indicates significant difference from both αβδ (p < 0.01) and αβγ (p < 0.05) receptors. Horizontal calibration: 400 msec; vertical calibration: 6 pA for δ, 2 pA for M2e, 68 pA for M1e, and 340 pA for γ2L.
The δ and γ2L subunits share ∼35% overall sequence identity, which is much greater within the putative transmembrane domains. Therefore, splice sites for the first two δ-γ chimeras (M2e and M1e) were generated near transmembrane domains to minimize the potential for nonspecific structural perturbations in the resulting protein [see Fig. 2, left; αβδ receptors (gray) and αβγ receptors (white)]. The M1e and M2e chimeras were designed to isolate three major topological domains between δ and γ2L subunits: the extracellular N terminus; the first two transmembrane domains (TM1 and TM2), the latter of which contains residues lining the channel pore (Xu and Akabas, 1996), and their cytoplasmic loop; and the last two transmembrane domains (TM3 and TM4) with the large cytoplasmic loop connecting them and the small extracellular loop between TM2 and TM3. Currents from αβδ and αβγ receptors are shown for comparison with the chimeras (Fig. 2, middle). Currents from receptors containing the δ-γ M2e chimera, which has δ subunit sequence from the N terminus through the extracellular end of TM2, were similar to those of αβδ receptors (%τfast = 2.4 ± 2.4%; τdeact = 54.7 ± 4.8 msec; n = 11), having no fast desensitization and rapid deactivation (M2e, %τfast = 6.1 ± 4.5%, τdeact = 45.4 ± 11.3 msec;n = 10) (Fig. 2, right; Table 1). Thus, δ subunit sequence in TM3, TM4, and the cytoplasmic loop were not required for the δ subunit to abolish fast desensitization. In contrast, the pronounced fast desensitization (τfast = 11.2 ± 2.8 msec; %τfast = 40.8 ± 6.9%; n= 14) of receptors containing the δ-γ M1e chimera was similar to that of αβγ receptors (τfast = 6.0 ± 0.7 msec; %τfast = 41.3 + 3.9%;n = 13), although the rate of deactivation was somewhat faster (τdeact = 107.5 ± 14.7 msec vs τdeact = 171.8 ± 20.4 msec) (Fig. 2,right; Table 1). This chimera had δ subunit sequence in the entire N terminus, yet it rapidly desensitized, indicating that the N terminus of the δ subunit, potentially related to agonist binding, was insufficient to abolish fast desensitization. The results obtained using these chimeric subunits suggested that structures important for fast desensitization resided within the first two transmembrane domains.
There is extensive evidence from mutation studies for a role of TM2 in desensitization and gating in GABAARs (Im et al., 1995; Chang et al., 1996; Dalziel et al., 2000), nicotinic acetylcholine receptors (nAChRs) (Revah et al., 1991; Filatov and White, 1995; Labarca et al., 1995), and 5-HT3 receptors (Yakel et al., 1993). However, those experiments did not resolve fast phases of desensitization because of relatively slow perfusion systems, and because the implicated residues from those GABAAR studies were identical in γ2L and δ subunits, they could not be responsible for the distinct differences in desensitization between receptors containing the two subunits. The chimera strategy relies on sequence differences, and therefore, cannot determine the importance of conserved residues. However, given the results of using the first two chimeras and the strong suggestion in the literature that TM2 is involved in desensitization and gating, we mutated all four nonidentical sites in each subunit (Fig.3A) to the corresponding amino acids in the other subunit to create “TM2-swap” subunits (γ2L(M2S) and δ(M2S)) (Fig. 3B, left). Surprisingly, neither the desensitization nor the deactivation kinetics was sensitive to changes in TM2 sequence. The properties of receptors containing δ(M2S) (%τfast = 9.0 ± 4.8%; τdeact = 61.7 ± 12.0 msec;n = 12) and γ2L(M2S) (τfast = 7.9 ± 1.3 msec; %τfast = 45.4 ± 2.2%; τdeact = 137.8 ± 24.6 msec;n = 9) were similar to those containing wild-type δ and γ2L subunits, respectively (Fig. 3B, middle andright; Table 1).
Fig. 3.

Desensitization and deactivation differences are not specified by TM2. A, Desensitization and deactivation kinetics of receptors containing TM2 “swapped” subunits. The four divergent residues of TM2 shown in (B) have been exchanged in each subunit as indicated in the schematics. Neither desensitization nor deactivation was altered by this exchange for either subunit. For comparison,shaded vertical bars represent the range of values (mean ± SEM) obtained for wild-type αβδ and αβγ receptors. Horizontal calibration: 400 msec; vertical calibration: 10 pA for δ(M2S), 285 pA for γ2L(M2S). B, Amino acid sequence alignment of the second transmembrane domains of the δ and γ2L subunits. N-terminal end (intracellular side) is to theleft. The δ sequence is A259 through R282; the γ2L sequence is A261 through R284. Identical residues shared by these subunits are shaded, including the 9′ leucine (see Results).
Three subsequent chimeras with progressively less δ subunit sequence were generated, M1i, M1p, and M1pre-iso (Fig.4B, left) to further specify desensitization domains. A TM1 sequence alignment indicating the splice locations is shown in Figure 4A. Although TM2 sequence did not appear to control desensitization or deactivation, several reports suggested that the nearby TM1–TM2 cytoplasmic linker might be important. Cysteine scanning experiments suggested the channel gate to be very close to the cytoplasm in nAChRs (Wilson and Karlin, 1998), and we previously speculated that charge differences in the TM1–TM2 linker (NKD in the γ2L subunit, SQA in the δ subunit) might play a role in GABAAR gating (Fisher and Macdonald, 1997). Also, Lynch et al. (1997) showed that an alanine substitution in the linker increased desensitization of GlyRs. The M1i chimera contained δ subunit sequence through the intracellular end of TM1, whereas the TM1–TM2 linker and beyond were from the γ2L subunit. No fast desensitization, however, was observed with receptors containing this chimera (%τfast = 9.6 ± 3.8%; n = 26), limiting the important structures to those N-terminal to and including TM1. The extent of desensitization was greater (p < 0.01) than that of αβδ receptors (33.1 ± 5.2% compared with 12.4 ± 4.8%). Although it is possible that TM2 or the linker is involved in slower phases of desensitization, we did not observe such effects on extent of desensitization in the two TM2-swapped subunits or in two additional chimeras (see below) that also contained γ2L subunit sequence in these domains.
Fig. 4.

Isolation of TM1 residues that modulate desensitization. A, The left column shows additional δ-γ2L chimeras used to determine the role of TM1 in desensitization. The indicated chimeras (γ2L sequence iswhite, δ subunit sequence is gray) were expressed with α1 and β3 subunits. Representative traces are shown in middle column. Bar graphs are as in Figure 2. Data from the rapidly desensitizing M1e chimera (from Fig. 2) is shown again for comparison. The asterisk indicates significant difference from both αβδ (p < 0.01) and αβγ (p < 0.05) receptors. Horizontal calibration: 400 msec; vertical calibration: 4.5 pA for M1i, 22 pA for M1p, 35 pA for M1pre-iso, and 68 pA for M1e.B, Amino acid sequence alignment of the first transmembrane domain of the δ and γ2L subunits. N-terminal end (extracellular side) is to the left. The δ subunit sequence is R229 through I255; the γ2L subunit sequence is R231 through I257. Identical residues shared by these subunits areshaded. The lines witharrows indicate chimera splice positions where δ subunit sequence ended and γ2L subunit sequence began, corresponding to the four chimeras shown in (A). Theasterisks indicate the only residues that differ between the M1e and M1pre-iso chimeras.
The M1p chimera contained δ subunit sequence from the N terminus to the proline in the middle of TM1, and the M1pre-iso chimera contained δ subunit sequence in the N-terminus and two adjacent residues in TM1. Both chimeras abolished fast desensitization (M1p, %τfast = 8.2 ± 8.2%, n= 6; M1pre-iso, %τfast = 3.5 ± 3.5%,n = 14) (Fig. 4A, middleand right; Table 1). These data indicated that although most of TM1 was not involved in modulating desensitization, two TM1 residues adjacent to the N terminus (V and Y; Fig. 4B, asterisks) were required for the δ subunit to abolish fast desensitization. Consistent with the role of desensitization in shaping deactivation patterns, the deactivation of receptors containing the M1i, M1p, and M1pre-iso chimeras were at least as fast as αβδ receptors (M1I, τdeact = 54.6 ± 8.1 msec; M1p, τdeact = 10.3 ± 2.2 msec; M1pre-iso, τdeact = 27.4 ± 4.9 msec). The striking difference in fast desensitization between receptors containing the δ-γ M1e (shown again in Fig. 4A for comparison) and δ-γ M1pre-iso chimeras prompted us to generate exchange mutations based on the residues that differed between these constructs.
Point mutations in TM1
The nondesensitizing receptors containing the δ-γ M1pre-iso chimera differed from receptors with the rapidly desensitizing δ-γ M1e chimera by only two TM1 residues, having a VY sequence in the δ subunit and a YF sequence in the γ2L subunit (Fig. 4B, asterisks). Interestingly, the γ2L subunit YF sequence is conserved among all known α, β, γ, and π GABAAR subunits across species and is similar in the human ε subunit (YV); it also differs in the ρ subunits, which form nondesensitizing homomers and contain an FF sequence in that TM1 location (Amin and Weiss, 1994). Thus, we predicted that the VY sequence in the homologous region of the δ subunit was responsible for its capacity to abolish fast desensitization. We introduced these δ subunit VY residues singly (γ2L(YΔV) and γ2L(FΔY)) and together (γ2L(YFΔVY)) into the γ2L subunit to determine whether they were sufficient to attenuate fast desensitization. However, despite their implication as the key residues from the chimera results, the desensitization of receptors containing these mutated γ2L subunits were indistinguishable from receptors containing the wild-type γ2L subunit (γ2L(YΔV), τfast = 6.9 ± 1.4 msec; %τfast = 45.2 ± 7.8%, n= 7; γ2L(FΔY), τfast = 8.0 ± 1.7 msec, %τfast = 34.2 ± 5.2%, n= 8; γ2L(YFΔVY), τfast = 9.0 ± 1.1 msec, %τfast = 40.5 ± 5.9%, n= 19) (Fig. 5, Table 1).
Fig. 5.

VY sequence in TM1 is not sufficient to abolish fast desensitization. The effect of mutations in TM1 of the γ2L subunit on desensitization and deactivation kinetics was determined. The YF pair was mutated, singly and together, in the γ2L subunit to the corresponding VY residues of the δ subunit, and expressed with α1 and β3 subunits. The middle column illustrates representative currents. Bar graphs are as in Figure 2. The rate and relative contribution of fast desensitization was not altered by any of the mutations compared to wild-type γ2L subunit-containing receptors. However, the weighted deactivation was significantly accelerated by both the single mutations and the double mutation in TM1. Theasterisk indicates significant difference from both αβδ (p < 0.05) and αβγ (p < 0.01) receptors. Horizontal calibration: 400 msec; vertical calibration: 180 pA for γ2L(YΔV), 53 pA for γ2L(FΔY), and 72 pA for γ2L(YFΔVY).
In summary, replacement of the entire N terminus of the γ2L subunit with δ subunit sequence (Fig. 2, δ-γ M1e chimera) had no effect on fast desensitization, and replacement of the YF sequence in TM1 of the γ2L subunit with the corresponding VY sequence of the δ subunit also failed to affect desensitization (Fig. 5, γ2L(YFΔVY)). Fast desensitization was abolished only when both of these exchanges were made in the γ2L subunit (Fig. 4A, δ-γ M1pre-iso). Thus, both the N terminus and the adjacent VY residues in TM1 of the δ subunit appeared to be necessary to abolish fast desensitization. Interestingly, deactivation was substantially accelerated when either or both of the YF pair was mutated to VY in the γ2L subunit, with the fastest deactivation observed in the double mutant (Fig. 5, 39.7 ± 4.7 msec, n = 19, compared with 171.8 ± 20.4 msec, n = 13, with the wild-type αβγ receptor). It appeared that prolonged deactivation had been functionally uncoupled from fast desensitization; whereas mutation of the YF sequence did not interfere with desensitization, it prevented desensitized states from contributing to deactivation.
It is possible that pentamer assembly and/or stoichiometry differed whether a δ or γ2L subunit was present. Recent studies suggested that N-terminal residues in α, β, and γ subunits were important for intersubunit contacts related to assembly (Klausberger et al., 2000; Taylor et al., 2000). Although subunit sequences important for the assembly of αβδ receptors have not yet been determined, it was possible that chimeras with δ subunit sequence in the N terminus assembled differently, in terms of position or stoichiometry, than the γ2L subunit mutants that contain γ2L subunit N terminus sequence. For example, if the effect of the δ subunit on desensitization was in fact mediated by the VY residues but was specific to position within the pentamer, the VY mutation in the γ2L subunit might not alter desensitization. The YF amino acid pair is conserved in all α, β, and γ subunits and predicted to lie near the outer vestibule of the channel. To address the issue of position-specific effects, we studied α1β3γ2L receptors containing YF to VY mutations in either α or β subunits rather than in the γ2L subunit. Together with the γ2L(YFΔVY) mutation, these mutations allowed us to evaluate the impact of VY residues in TM1 at every subunit position in the GABAAR pentamer. Neither subunit mutation abolished rapid desensitization (Fig.6, middle andright; Table 1), although its relative contribution was reduced in the α1(YFΔVY) receptors (%τfast = 18.5 ± 5.7%,n = 9) compared with wild type (%τfast = 41.3 ± 3.9%,n = 13). These results were inconsistent with a simple position-specific effect of the δ subunit VY sequence and provided further evidence for the requirement of both the TM1 VY residues and the δ N terminus to effectively abolish fast desensitization. However, both subunit mutations appeared to accelerate deactivation compared with αβγ receptors, suggesting that the uncoupling effect observed with the γ2L(YFΔVY) mutation might generalize to other subunits in the pentamer (α1(YFΔVY), τdeact = 56.5 ± 8.3 msec; β3(YFΔVY), τdeact = 99.2 ± 12.5 msec).
Fig. 6.

The effect of YF to VY mutation in the α1 or β3 subunits of αβγ receptors. α1(YFΔVY) was coexpressed with β3 and γ2L, and β3(YFΔVY) was coexpressed with α1 and γ2L. The middle column illustrates representative currents. Bar graphs are as in Figure 2. Theasterisks indicates significant difference (p < 0.05) from both αβδ and αβγ receptors. Horizontal calibration: 400 msec; vertical calibration: 150 pA for α1(YFΔVY), and 116 pA for β3(YFΔVY).
If the δ subunit required the combination of N-terminal sequence and adjacent VY residues in TM1 to abolish fast desensitization, the converse exchanges into the δ subunit (introducing γ2L subunit sequence) should “unmask” a rapid phase of desensitization. Accordingly, when the VY to YF exchange was made in the δ subunit, a fast component of desensitization (τfast = 9.0 ± 1.1 msec) was recorded in 13 of 21 patches (Fig. 7A, δ(VYΔYF)). When present, the mean contribution of this fast phase was similar to that observed with receptors containing the γ2L subunit (%τfast = 35.3 ± 3.6%,n = 10, compared with %τfast = 41.3 ± 3.9% for αβγ receptors). Although many patches did not exhibit a fast phase, the mean rate and extent of desensitization was significantly greater than observed with wild-type δ subunit (p < 0.05). The reason for this variability in occurrence of fast desensitization was unclear. Although mutation of the YF residues in the γ2L subunit accelerated deactivation, introduction of the YF pair into the δ subunit did not significantly prolong deactivation (τdeact = 76.5 ± 6.0 msec, compared with αβδ, τdeact = 54.7 ± 4.8 msec). Moreover, the deactivation rate was not significantly different in patches having fast desensitization compared with those that did not, suggesting that desensitized states were not coupled to prolonged deactivation in the mutant.
Fig. 7.

γ2L TM1 is critical for desensitization-deactivation coupling. A, Introducing γ2L subunit sequence of extracellular TM1 or the N terminus into the δ subunit increased fast desensitization (γ-δ M1e, δ(VYΔYF)). However, prolonged deactivation was only evident when both the N terminus and TM1 contained γ2L subunit sequence (γ-δ M1i). Note that receptors containing the δ(VYΔYF) subunit were divided into two groups: 13 of 21 that had fast desensitization, and eight that did not. The values for %τfast were calculated from the former group. The %τfast of all 21 patches was 21.8 ± 4.4%. Splice junctions of the chimeras are the same as the δ-γ M1e and M1i chimeras (Fig.4A). Horizontal calibration: 400 msec; vertical calibration: 53 pA for δ(VYΔYF), 11 pA for γ-δ M1e, and 17 pA for γ-δ M1i. B, Representative deactivation currents were normalized to the current amplitude at the offset of GABA application, and overlaid to illustrate the differences in rate. Wild-type δ and γ2L subunit-containing receptor currents were colored gray. Note that only receptors containing the γ-δ M1i chimera had both fast desensitization and prolonged deactivation resembling that of αβγ receptors. Scale bar, 100 msec.
TM1 of the γ2L subtype is critical for desensitization–deactivation coupling
To investigate further the structures involved in fast desensitization and its coupling to deactivation, we generated two “reverse” chimeras, splicing N-terminal γ2L subunit sequence to δ subunit sequence (Fig. 7A, left). Whereas the initial set of δ-γ chimeras was designed to confer a δ subunit-specific property (minimal desensitization) to a γ2L subunit, these γ-δ chimeras were generated to confer properties specific to the γ2L subunit (fast desensitization and prolonged deactivation) to a δ subunit. We hypothesized that replacement of the δ subunit N terminus by the γ2L subunit N terminus, like the VY to YF exchange in δ, would confer fast desensitization. However, deactivation should remain rapid (as seen with wild-type δ subunit-containing receptors) because the VY sequence would be present in TM1. We confirmed this hypothesis, because the fast desensitization of receptors containing the γ-δ M1e chimera resembled αβγ receptor desensitization (%τfast = 40.1 ± 9.2%,n = 7, compared with αβγ, %τfast = 41.3 ± 3.9%), whereas the deactivation was rapid and resembled that of αβδ receptors (τdeact = 68.4 + 11.4 msec, compared with αβδ, τdeact = 54.7 ± 4.8 msec) (Fig. 7A, middle and right; Table 1). Because we were still interested in whether additional γ2L subunit sequence in TM1 could restore the coupling of fast desensitization and prolonged deactivation, we tested the γ-δ M1i chimera that extends γ2L subunit sequence into TM1, including the critical YF sites (Fig.7A, left). Consistent with the role of TM1 in desensitization–deactivation coupling, the kinetics of both desensitization and deactivation (Fig. 7A, middleand right) of receptors containing this chimera were indistinguishable from wild-type αβγ receptors (%τfast = 40.6 ± 5.7%; τdeact = 186.0 ± 29.0 msec;n = 8). The deactivation currents are expanded and normalized in Figure 7B to illustrate more clearly the differences in deactivation rate. Rapid deactivation was observed with receptors containing the minimally desensitizing δ subunit. Receptors containing the γ-δ M1e chimera and the γ2L(YFΔVY) mutant subunit exhibited similarly rapid deactivation despite fast desensitization. Prolonged deactivation resembling αβγ currents was only observed with receptors containing the γ-δ M1i chimera.
To determine whether other properties of the GABAR were affected by the apparent uncoupling of desensitization and deactivation, we generated GABA concentration–response curves and performed paired pulse analysis on α1β3γ2L(YFΔVY)receptor currents for comparison with those from wild-type α1β3γ2L receptors. Figure8A illustrates a small right-shift in the GABA concentration–response curves (α1β3γ2L EC50 = 7.8 μm,n = 7; α1β3γ2L(YFΔVY)EC50 = 11.0 μm,n = 7), obtained using whole-cell recording and fast GABA application (maximal current rise times were <10 msec). Although changes in both binding and gating steps can influence EC50, these data suggested that a large change inkoff was not responsible for the rapid deactivation of this mutant receptor. Paired 5–10 msec pulses of 1 mm GABA, separated by 10–800 msec intervals were applied to excised outside-out patches to examine recovery from desensitization. When compared with α1β3γ2L receptors (Fig.8B), the α1β3γ2L(YFΔVY)mutation resulted in substantial reduction of paired pulse depression. Note the fourfold different time scales in Figure 8, B andC, to emphasize the lack of severe paired pulse depression for even 10 msec interpulse intervals for the α1β3γ2L(YFΔVY)mutation.
Fig. 8.

GABA concentration–response relationship and paired pulse analysis for αβγ and αβγ(YFΔVY) isoforms. A, Whole-cell concentration–response curves were generated for αβγ and αβγ(YFΔVY) isoforms using rapid perfusion. Currents were normalized to the maximal peak current for each cell. The relation was fit with a sigmoidal curve (see Materials and Methods). Each curve was obtained from the mean responses of seven cells. B, Brief (5–10 msec) pulses of 1 mmGABA were applied to excised outside-out patches at various intervals to investigate paired pulse depression of αβγ GABARs. Note the different horizontal time scales in B andC. Intervals in B were 100, 200, 400, and 800 msec. Similar results were obtained in three other patches. C, Same paradigm as in B, except shorter interpulse intervals were used to emphasize the decrease in paired pulse depression observed for αβγ(YFΔVY) GABARs. Intervals inC were 10, 25, 50, 100, and 200 msec. Similar results were obtained in three other patches.
DISCUSSION
Mechanism of desensitization
Desensitization is an intrinsic property of many ligand-gated ion channels (for review, see Jones and Westbrook, 1996). Fast desensitization of GABAARs is most relevant for brief synaptic events during which the fastest microscopic rate constants dominate channel behavior regarding visits to open, closed, or desensitized states. The fast (τfast ∼10 msec) phase of desensitization, easily resolved with our experimental protocol, is assumed to represent rapid entry into a “fast” desensitized kinetic state. Even when patches are used in concentration-jump experiments, our models indicated that fast desensitization truncates the peak current despite fast activation (<1 msec) (Haas and Macdonald, 1999). However, the resulting errors would not alter our interpretations, because such a truncation would cause an underestimation of the already prominent fast contribution.
Several studies have implicated TM2 residues in the modulation of desensitization in nAChRs, GABAARs, and 5-HT3Rs (Revah et al., 1991; Yakel et al., 1993;Filatov and White, 1995; Im et al., 1995; Labarca et al., 1995;Chang et al., 1996; Dalziel et al., 2000). It seemed, therefore, that desensitization was modulated or at least affected by structures at or near the channel gate. The structural events surrounding the transduction of agonist binding at the extracellular N terminus to conformational changes at the channel gate, however, remain the object of much speculation. In fact, the physical nature of the gate is not currently agreed on. Electron micrograph data obtained from nAChRs of Torpedo electric organ indicated a region of high density near the center of the pore-lining TM2 of each subunit attributed to the 9′ leucine of TM2 (Unwin, 1995). However, such a gate location is incompatible with evidence from the accessibility of engineered cysteines in TM2 of both nAChRs and GABAARs. These studies constrained the location of the gate, or at least the narrowest portion of the pore, to the cytoplasmic end of the channel (Xu and Akabas, 1996; Wilson and Karlin, 1998). However, in the γ2L and δ subunits the 9′ leucine and the 10 flanking TM2 residues are conserved, and because these subunits confer dramatically different desensitization kinetics, there must be other domains that govern desensitization. Our data clearly indicated that the channel-lining residues of the δ subunit are not the basis for its abolishing fast desensitization. Although this finding does not eliminate the possibility of TM2 involvement in occluding the channel during desensitization, it does suggest that this domain could play only a passive role in the process. The N terminus and extracellular end of TM1 can modulate fast desensitization independent of the identity (γ2L vs δ subunit sequence) of TM2. This was evident in the results obtained with several chimeras (for example, Fig. 4A,compare δ-γ M1e and M1pre-iso), and in particular, with the TM2 swap subunits (Fig. 3A). It is worth noting, however, that none of the aforementioned TM2 mutation studies resolved the fast phase of desensitization. Thus, it is possible that slower forms of desensitization were actually affected that we cannot detect with relatively short pulses. Conserved residues may in fact control slow desensitization, which has a similar time constant in receptors containing γ2L or δ subunits (∼1500 msec). Concentration-jump experiments on receptors containing 9′ leucine mutations may clarify this issue.
There is some evidence suggesting that structures in or near the agonist-binding site or sites can modulate desensitization. For example, not all agonists at the AMPA-type glutamate receptor induce desensitization. It has been proposed that ligand-induced structural changes in the extracellular domains initiate desensitization (Stern-Bach et al., 1998), which may account for agonist-specific desensitization. Also, point mutations in the agonist-binding site can abolish AMPA receptor desensitization (Stern-Bach et al., 1998). Electron micrograph images of Torpedo nAChRs in (presumably) the desensitized state revealed significant movement of the N-terminal extracellular domain around the level of the ACh-binding pockets (Unwin, 1995). However, the chimera containing the entire δ subunit N terminus (δ-γ M1e chimera, Fig. 2) failed to abolish fast desensitization, providing compelling evidence against the δ subunit acting purely through an effect on GABA binding.
It appeared, however, that the N terminus was interacting with TM1 to modulate desensitization. Involvement of residues in the extracellular end of TM1 raised the possibility of a conformational change near the outer mouth of the channel that could be prevented by the VY residues. This effect could only occur, however, in the structural context of the δ subunit N terminus, which may serve to anchor the position in TM1; introducing the VY into other subunits did not reproduce the effect of the δ subunit. TM1 may be interleaved with TM2 segments toward the extracellular end of the channel, based on cysteine-scanning data (Akabas et al., 1994; Akabas and Karlin, 1995), which could serve to couple conformational changes in TM1 to TM2. An alternative interpretation is that fast desensitization develops in or near the gate. The δ subunit N terminus might abolish entry into this state via conformational changes that are transduced through TM1 to the gate region. Interestingly, the involvement of extracellular domains and a smaller “pre-M1” domain in desensitization was recently reported in NMDA receptors (Krupp et al., 1998; Villarroel et al., 1998), raising the possibility that evolutionarily divergent ligand-gated channels share similar desensitization mechanisms.
Desensitization–deactivation coupling
The role of desensitized states in prolonging deactivation of native and recombinant GABAARs is supported by the work of several independent laboratories (Jones and Westbrook, 1995; Tia et al., 1997; Dominguez-Perot et al., 1996; Haas and Macdonald, 1999), yet the structural basis for this phenomenon is unclear. Although the current decay after removal of agonist (deactivation) ultimately reflects GABA unbinding, dissociation is itself sensitive to conformational changes in the receptor associated with gating and desensitization (see Colquhoun, 1998, for a commentary on the “binding-gating” problem). Although this logic is somewhat model-dependent, there is experimental evidence in support of the more general idea that open and desensitized states do not allow agonist dissociation (Jones and Westbrook, 1995; Chang and Weiss, 1999; Haas and Macdonald, 1999). The trapping of agonist by desensitization could be a primarily local structural consequence of ligand binding, or alternatively, the manifestation of desensitization, that is occlusion of the channel pore, might indirectly alter the binding site to prevent dissociation. Our data are more consistent with the latter interpretation, because TM1 residues that are presumably distant from extracellular GABA-binding regions were critical for desensitized states to prolong deactivation. Initial evidence for the critical role of TM1 came from mutations of the VY residues in the γ2L subunit that increased the rate of deactivation without affecting fast desensitization. However, more compelling evidence came from the “reverse” (γ-δ) chimeras. Rapid desensitization, but not prolonged deactivation, was conferred to a δ subunit by replacement of the N terminus with γ2L subunit sequence. Restoring the functional coupling of fast desensitization and prolonged deactivation was only accomplished when γ2L subunit sequence extended into TM1. If desensitized states can trap agonist on the receptors, then these conformations associated with the channel pore must somehow effect remote structural changes in the ligand-binding domains. TM1 is positioned in such a way that it might allow the propagation of conformational changes not only from the N terminus to the gate, but also from the gate back to the N terminus. Interestingly, as with fast desensitization, deactivation is controlled by N-terminal structures acting with or through TM1, whereas structures near the gate (TM2) appeared to play a less direct role.
From a kinetic standpoint, several possibilities exist that might explain our observation of fast deactivation in spite of the presence of fast desensitization for certain chimeras and mutant subunits. We evaluated some of these possibilities for receptors containing the γ2L(YFΔVY) subunit. The simplest one is that the microscopic unbinding rate has increased dramatically, which would favor GABA dissociation instead of reopening after recovery from desensitized states (assuming desensitized states are connected to liganded closed states). However, this parameter change predicts a large increase in GABA EC50, which was not observed (Fig. 8A). Another possibility is that recovery from desensitization occurs on a much slower time scale, so that late channel openings would not contribute to the deactivation during the time we examined after removal of GABA. This can be ruled out because it predicts severe paired pulse depression, which was not observed (Fig. 8, compare B, C). In fact, less paired pulse depression was observed for this mutation, which may indicate increased recovery from desensitization. Rapid recovery from desensitization would also accelerate deactivation, because it is the duration of sorties to desensitized states that delay the unbinding of GABA and allow the late openings that prolong deactivation. However, simply increasing the rate of exit from fast desensitization predicts a decrease in the rate and extent of macroscopic desensitization that was not observed (Fig. 5). Because reopening after recovery from desensitization depends on the relative rates toward unbinding versus rates toward open or pre-open states, decreasing the latter rates would favor unbinding. Detailed single-channel analysis is necessary to evaluate changes in gating related to decreased entry into various open states or decreased burst/cluster duration. Finally, it is possible that the observed uncoupling of fast desensitization and prolonged deactivation is because the mutations allow dissociation of GABA to occur directly from desensitized conformations.
Footnotes
This work was supported by National Institutes of Health Grant R01-NS33300 (R.L.M.), and National Institute on Drug Abuse Training Fellowships T32-DA07281-03 (K.F.H., M.T.B.). We thank Hyun Chung and Fang Sun for generating the chimeric and mutant constructs.
Correspondence should be addressed to Dr. Robert L. Macdonald, Department of Neurology, University of Michigan, 1103 East Huron, Ann Arbor, MI 48104-1687. E-mail: rlmacd@umich.edu.
REFERENCES
- 1.Akabas MH, Karlin A. Identification of acetylcholine receptor channel-lining residues in the M1 segment of the α-subunit. Biochemistry. 1995;34:12496–12500. doi: 10.1021/bi00039a002. [DOI] [PubMed] [Google Scholar]
- 2.Akabas MH, Kaufmann C, Archdeacon P, Karlin A. Identification of acetylcholine receptor channel-lining residues in the entire M2 segment of the α subunit. Neuron. 1994;13:919–927. doi: 10.1016/0896-6273(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 3.Amin J, Weiss DS. Homomeric ρ1 GABA channels: activation properties and domains. Receptors Channels. 1994;2:227–236. [PubMed] [Google Scholar]
- 4.Angelotti TP, Uhler MD, Macdonald RL. Assembly of GABAA receptor subunits: analysis of transient single-cell expression utilizing a fluorescent substrate/marker gene technique. J Neurosci. 1993;13:1418–1428. doi: 10.1523/JNEUROSCI.13-04-01418.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonnert TP, McKernan RM, Farrar S, Le Bourdelles B, Heavens RP, Smith DW, Hewson L, Rigby MR, Sirinathsinghji DJS, Brown N, Wafford KA, Whiting PJ. θ, a novel γ-aminobutyric acid type A receptor subunit. Proc Natl Acad Sci USA. 1999;96:9891–9896. doi: 10.1073/pnas.96.17.9891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Celentano JJ, Wong RKS. Multiphasic desensitization of the GABAA receptor in outside-out patches. Biophys J. 1994;66:1039–1050. doi: 10.1016/S0006-3495(94)80885-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang Y, Weiss DS. Channel opening locks agonist onto the GABAC receptor. Nat Neurosci. 1999;2:219–225. doi: 10.1038/6313. [DOI] [PubMed] [Google Scholar]
- 8.Chang Y, Wang R, Barot S, Weiss DS. Stoichiometry of a recombinant GABAA receptor. J Neurosci. 1996;16:5415–5424. doi: 10.1523/JNEUROSCI.16-17-05415.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clements JD. Transmitter timecourse in the synaptic cleft: its role in central synaptic function. Trends Neurosci. 1996;19:163–171. doi: 10.1016/s0166-2236(96)10024-2. [DOI] [PubMed] [Google Scholar]
- 10.Colquhoun D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and the effects of mutating receptors. Br J Pharmacol. 1998;125:924–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dalziel JE, Cox GB, Gage PW, Birnir B. Mutating the highly conserved second membrane-spanning region 9′ leucine residue in the α1 or β1 subunit produces subunit-specific changes in the function of human α1β1 γ-aminobutyric acidA receptors. Mol Pharmacol. 2000;57:875–882. [PubMed] [Google Scholar]
- 12.Davies PA, Hanna MC, Hales TG, Kirkness EF. Insensitivity to anaesthetic agents conferred by a class of GABAA receptor subunit. Nature. 1997;385:820–823. doi: 10.1038/385820a0. [DOI] [PubMed] [Google Scholar]
- 13.Dominguez-Perrot C, Feltz P, Poulter MO. Recombinant GABAA receptor desensitization: the role of the gamma2 subunit and its physiological significance. J Physiol (Lond) 1996;497:145–159. doi: 10.1113/jphysiol.1996.sp021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filatov GN, White MW. The role of conserved leucines in the M2 domain of the acetylcholine receptor in channel gating. Mol Pharmacol. 1995;48:379–384. [PubMed] [Google Scholar]
- 15.Fisher J, Macdonald RL. Single channel properties of recombinant GABAA receptors containing γ2 of δ subtypes expressed with α1 and β3 subtypes in mouse L929 cells. J Physiol (Lond) 1997;505:283–297. doi: 10.1111/j.1469-7793.1997.283bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galarreta M, Hestrin S. Properties of GABAA receptors underlying inhibitory synaptic currents in neocortical pyramidal neurons. J Neurosci. 1997;17:7220–7227. doi: 10.1523/JNEUROSCI.17-19-07220.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenfield LJ, Jr, Sun F, Neelands TR, Burgard EC, Donnelly JL, Macdonald RL. Expression of functional GABAA receptors in the transfected L929 cells isolated by immunomagnetic bead separation. Neuropharmacology. 1997;36:63–73. doi: 10.1016/s0028-3908(96)00150-5. [DOI] [PubMed] [Google Scholar]
- 18.Haas KF, Macdonald RL. GABAA receptor subunit γ2 and δ subtypes confer unique kinetic properties on recombinant GABAA receptors in mouse fibroblasts. J Physiol (Lond) 1999;514:27–45. doi: 10.1111/j.1469-7793.1999.027af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hedblom E, Kirkness EF. A novel class of GABAA receptor subunit in tissues of the reproductive system. J Biol Chem. 1997;272:15346–15350. doi: 10.1074/jbc.272.24.15346. [DOI] [PubMed] [Google Scholar]
- 20.Im WB, Binder JA, Dillon GH, Pregenzer JF, Im HK, Altman RA. Acceleration of GABA-dependent desensitization by mutating threonine 266 to alanine of the alpha 6 subunit of rat GABAA receptors. Neurosci Lett. 1995;186:203–207. doi: 10.1016/0304-3940(95)11293-6. [DOI] [PubMed] [Google Scholar]
- 21.Jones MV, Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 22.Jones MV, Westbrook GL. The impact of receptor desensitization on fast synaptic transmission. Trends Neurosci. 1996;19:96–101. doi: 10.1016/s0166-2236(96)80037-3. [DOI] [PubMed] [Google Scholar]
- 23.Klausberger T, Fuchs K, Mayer B, Ehya N, Sieghart W. GABAA receptor assembly: identification and structure of γ2 sequences forming the intersubunit contacts with α1 and β3 subunits. J Biol Chem. 2000;275:8921–8928. doi: 10.1074/jbc.275.12.8921. [DOI] [PubMed] [Google Scholar]
- 24.Krupp JJ, Vissel B, Heinemann SF, Westbrook GL. N-terminal domains in the NR2 subunit control desensitization of NMDA receptors. Neuron. 1998;20:317–327. doi: 10.1016/s0896-6273(00)80459-6. [DOI] [PubMed] [Google Scholar]
- 25.Labarca C, Nowak MW, Zhang H, Tang L, Deshpande P, Lester HA. Channel gating governed symmetrically by conserved leucine residues in the M2 domain of nicotinic receptors. Nature. 1995;376:514–516. doi: 10.1038/376514a0. [DOI] [PubMed] [Google Scholar]
- 26.Lynch JW, Rajendra S, Pierce KD, Handford CA, Barry PH, Schofield PR. Identification of intracellular and extracellular domains mediating signal transduction in the inhibitory glycine receptor chloride channel. EMBO J. 1997;16:110–120. doi: 10.1093/emboj/16.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macdonald RL, Olsen RW. GABAA receptor channels. Annu Rev Neurosci. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- 28.Macdonald RL, Rogers CJ, Twyman RE. Kinetic properties of the GABAA receptor main conductance state of mouse spinal cord neurones in culture. J Physiol (Lond) 1989;410:479–499. doi: 10.1113/jphysiol.1989.sp017545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maconochie DJ, Zempel JM, Steinbach JH. How quickly can GABAA receptors open? Neuron. 1994;12:61–71. doi: 10.1016/0896-6273(94)90152-x. [DOI] [PubMed] [Google Scholar]
- 30.McKernan RM, Whiting PJ. Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci. 1996;19:139–143. doi: 10.1016/s0166-2236(96)80023-3. [DOI] [PubMed] [Google Scholar]
- 31.Nayeem N, Green TP, Martin IL, Barnard EA. Quaternary structure of the native GABAA receptor determined by electron microscopic image analysis. J Neurochem. 1994;62:815–818. doi: 10.1046/j.1471-4159.1994.62020815.x. [DOI] [PubMed] [Google Scholar]
- 32.Ortells MO, Lunt GG. Evolutionary history of the ligand-gated ion-channel superfamily of receptors. Trends Neurosci. 1995;18:121–127. doi: 10.1016/0166-2236(95)93887-4. [DOI] [PubMed] [Google Scholar]
- 33.Revah F, Bertrand D, Galzi JL, Devillers-Thiery A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux JP. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature. 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- 34.Saxena NC, Macdonald RL. Properties of putative cerebellar γ-aminobutyric acidA receptor isoforms. Mol Pharmacol. 1996;49:567–579. [PubMed] [Google Scholar]
- 35.Stern-Bach Y, Russo S, Neuman M, Rosenmund C. A point mutation in the glutamate binding site blocks desensitization in AMPA receptors. Neuron. 1998;21:907–918. doi: 10.1016/s0896-6273(00)80605-4. [DOI] [PubMed] [Google Scholar]
- 36.Taylor PM, Connolly CN, Kittler JT, Gorrie GH, Hosie A, Smart TG, Moss SJ. Identification of residues within GABAA receptor α subunits that mediate specific assembly with receptor β subunits. J Neurosci. 2000;20:1297–1306. doi: 10.1523/JNEUROSCI.20-04-01297.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tia S, Wang JF, Kotchabhakdi N, Vicini S. Developmental changes of inhibitory synaptic currents in cerebellar granule neurons: role of GABAA receptor alpha 6 subunit. J Neurosci. 1996;16:3630–3640. doi: 10.1523/JNEUROSCI.16-11-03630.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tia S, Wang JF, Kotchabhakdi N, Vicini S. Distinct deactivation and desensitization kinetics of recombinant GABAA receptors. Neuropharmacology. 1997;35:1375–1382. doi: 10.1016/s0028-3908(96)00018-4. [DOI] [PubMed] [Google Scholar]
- 39.Unwin N. Acetylcholine receptor channel imaged in the open state. Nature. 1995;373:37–43. doi: 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- 40.Villarroel A, Regalado MP, Lerma J. Glycine-independent NMDA receptor desensitization: localization of structural determinants. Neuron. 1998;20:329–339. doi: 10.1016/s0896-6273(00)80460-2. [DOI] [PubMed] [Google Scholar]
- 41.Wilson GG, Karlin A. The location of the gate in the acetylcholine receptor channel. Neuron. 1998;20:1269–1281. doi: 10.1016/s0896-6273(00)80506-1. [DOI] [PubMed] [Google Scholar]
- 42.Xu M, Akabas MH. Identification of channel-lining residues in the M2 membrane-spanning segment of the GABAA receptor α1 subunit. J Gen Physiol. 1996;107:195–205. doi: 10.1085/jgp.107.2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yakel JL, Lagrutta A, Adelman JP, North RA. Single amino acid substitution affects desensitization kinetics of the 5-hydroxytryptamine type 3 receptor expressed in Xenopus oocytes. Proc Natl Acad Sci USA. 1993;90:5030–5033. doi: 10.1073/pnas.90.11.5030. [DOI] [PMC free article] [PubMed] [Google Scholar]