

Abstract
Recently, the cannabinoid (CB) receptor agonist anandamide (AEA) has been shown to excite perivascular terminals of primary sensory neurons via activation of the vanilloid receptor-1 (VR-1). To determine whether AEA stimulates central terminals of these neurons, via VR-1 activation, we studied the release of calcitonin gene-related peptide (CGRP)- and substance P (SP)-like immunoreactivities (LI) from slices of rat dorsal spinal cord. Mobilization of Ca2+ in rat dorsal root ganglion (DRG) neurons in culture was also studied. AEA (0.1–10 μm) increased the outflow of CGRP-LI and SP-LI from slices of the rat dorsal spinal cord in a Ca2+-dependent manner and increased [Ca2+]i in capsaicin-sensitive cultured DRG neurons. Both effects of AEA were abolished by capsaicin pretreatment and by the VR-1 antagonist capsazepine but not affected by the CB receptor antagonists AM281 or AM630. Both neuropeptide release and Ca2+ mobilization induced by electrical field stimulation (EFS) were inhibited by a low concentration of AEA (10 nm). Inhibition by AEA of EFS-induced responses was reversed by AM281 and AM630, but was not affected by capsazepine. Results indicate that stimulation of VR-1 with high concentrations of AEA excites central terminals of capsaicin-sensitive DRG neurons, thus causing neuropeptide release in the dorsal spinal cord. This novel activity opposes the CB receptor-mediated inhibitory action of low concentrations AEA. However, only if large amounts of endogenous AEA could be produced at the level of the dorsal spinal cord, they may not inhibit, but rather activate, nociceptive sensory neurons.
Keywords: anandamide (AEA), calcium, calcitonin gene-related peptide (CGRP), capsaicin, sensory neurons, substance P, vanilloid receptor-1 (VR-1)
Anandamide (AEA) is an arachidonate derivative that stimulates cannabinoid (CB) receptors (Devane et al., 1992). CB1 and CB2 receptor subtypes have been implicated in multiple biological actions, including inhibition of nociceptive responses (Hohmann et al., 1995; Tsou et al., 1996). A subset of neurons from dorsal root ganglia (DRG) with C or Aδ fibers are characterized for their unique sensitivity to the neurotoxin capsaicin (Holzer, 1991; Szallasi and Blumberg, 1999), which excites them by activating a nonselective cation channel [vanilloid receptor-1 (VR-1)] (Caterina et al., 1997). Capsaicin releases the peptide neurotransmitters calcitonin gene-related peptide (CGRP) and substance P (SP) from central and peripheral endings of these neurons. The release of SP and CGRP in the dorsal spinal cord has been associated with nociceptive transmission, whereas neuropeptide release in peripheral tissues causes neurogenic inflammatory responses (Otsuka and Yoshioka, 1993; Geppetti and Holzer, 1996). A number of mediators acting on inhibitory prejunctional receptors limit the sensory and local inflammatory actions of primary sensory neurons. The antihyperalgesic and anti-inflammatory actions of AEA are attributable, in part, to the activation of inhibitory CB1 receptors on central and peripheral endings of capsaicin-sensitive primary sensory neurons (Richardson et al., 1998a,b).
In contrast to the inhibitory action, recently, it has been reported that elevated concentrations of AEA excite peripheral terminals of capsaicin-sensitive primary sensory neurons via CB receptor-independent mechanisms (Zygmunt et al., 1999). Because the VR-1 antagonist capsazepine (Walpole et al., 1994) selectively abolished both AEA-induced release of CGRP in rodent peripheral arteries and AEA-induced activation of VR-1 transfected inXenopus oocytes and HEK293 cells, the proposal was advanced that elevated concentrations of AEA excites capsaicin-sensitive primary sensory neurons via VR-1 activation (Zygmunt et al., 1999). In addition, chemical similarities between AEA and certain ligands of VR-1 (Di Marzo et al., 1998; Beltramo and Piomelli, 1999; Melck et al., 1999) and the observation that AEA behaves as a full agonist at the VR-1 (Smart et al., 2000) support this hypothesis.
The discovery that AEA stimulates VR-1 has been obtained in heterologous systems (Xenopus oocytes and HEK293 cells;Zygmunt et al., 1999; Smart et al., 2000) expressing the VR-1 and in preparations (isolated rodent arteries) containing peripheral terminals of primary sensory neurons (Zygmunt et al., 1999). AEA is produced in endothelial cells, macrophages, and other peripheral cells (Devane et al., 1992). However, AEA is also produced by CNS neurons (Di Marzo et al., 1994), and VR-1 is highly expressed on central terminals of capsaicin-sensitive primary sensory neurons (Szallasi et al., 1994,1995; Caterina et al., 1997). The aim of this study was to investigate whether AEA could excite central endings of primary sensory neurons via the activation of VR-1. To examine this hypothesis, the ability of AEA to release CGRP and SP from slices of rat dorsal spinal cord was studied in vitro. In addition, the ability of AEA to mobilize Ca2+ in rat DRG neurons in culture was also examined. Results support the hypothesis that rat DRG neurons and their central terminals are stimulated by elevated concentration of AEA through its ability to activate VR-1.
MATERIALS AND METHODS
Animals and tissues. Male Sprague Dawley rats (Charles River, Varese, Italy) were used in all of the experiments. All experiments complied with the national guidelines and were approved by the regional ethical committee.
CGRP- and SP-like immunoreactivities release studies. Rats (250–300 gm) were terminally anesthetized and decapitated. The spinal cord was removed, and thick slices (∼0.4 mm) from the dorsal part of the cervical and lumbar enlargements were prepared at 4°C using a tissue slicer (McIlwain Tissue Chopper). Slices (∼100 mg) were placed in 2 ml chambers and superfused at 0.4 ml/min with a Krebs' solution of the following composition (in mm): NaCl 119, NaHCO3 25, KH2PO4 1.2, MgSO4 1.5, CaCl2 2.5, KCl 4.7, and d-glucose 11. To the basic Krebs' solution, the following agents were added: 0.1% bovine serum albumin (BSA), 1 μm phosphoramidon, and 1 μm captopril (to minimize peptide degradation), maintained at 37°C and gassed with 95% O2 and 5% CO2. After a 90 min stabilization period, 10 min fractions were collected into acetic acid (final solution, 2 N). Two prestimuli samples were taken at 10 min intervals followed by a third set of samples during stimulation. A final poststimulus 10 min sample was also collected. At the end of the experiment, tissues were blotted and weighed. Fractions were freeze-dried, reconstituted with assay buffer, and analyzed by enzyme immunoassays for CGRP-and SP-like immunoreactivities (LI) according to the methods reported previously (Frobert et al., 1999; Ricciardolo et al., 2000). The detection limits of the assays were 5 pg/ml for CGRP and 2 pg/ml for SP. The level of release of CGRP-LI and SP-LI were calculated by subtracting the mean prestimulus value from those values obtained during and after stimulation. The results are expressed as femtomoles of peptide per gram of tissue per 20 min. The highest concentration of AEA (10 μm), methanandamide (META) (1 μm), palmitoylethanolandamide (PEA) (1 μm), capsaicin (1 μm), capsazepine (10 μm), and AM281 and AM630 (both 10 μm) did not show any significant cross-reactivity with CGRP and SP antisera. The initial electrical field stimulation (EFS) (10 Hz, 100 mA/cm2, 1 msec pulse duration, 10 sec train every 20 sec for 5 min) was delivered by a Grass Instruments S88 stimulator for 5 min, and the perfusate was collected during this 5 min and the following 15 min (two 10 min fractions, S1). A second EFS (for 5 min) was delivered after a 60 min interval, and the perfusate was also collected for 20 min (S2). Treatment with drugs or their vehicles were performed during the second EFS (S2).
Primary culture. Rats (1–3 d old) were terminally anesthetized and decapitated. DRG were removed one by one from all spinal segments and rapidly placed in cold PBS before being transferred to collagenase–dispase (1 mg/ml dissolved in Ca2+–Mg2+-free PBS) for 35 min at 37°C. Enrichment of the fraction of nociceptive neurons was obtained following the methods reported previously (Gilabert and McNaughton, 1997). After the enzymatic treatment, ganglia were rinsed three times with Ca2+–Mg2+-free PBS and then placed in 2 ml of cold DMEM supplemented with 10% fetal bovine serum (FBS) (heat inactivated), 2 mml-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. The ganglia were then dissociated into single cells by several passages through a series of syringe needles (23 gauge down to 25 gauge). Finally, the complex of medium and ganglia cells were sieved through a 40 μm filter to remove debris and topped up with 8 ml of DMEM and centrifuged (200 × g for 5 min). The final cell pellet was resuspended in DMEM [supplemented with 100 ng/ml mouse nerve growth factor (mouse NGF-7S) and 2.5 μmcytosine-β-d-arabinofuranoside free base (AraC)]. Cells were plated on poly-l-lysine (8.3 μm)- and laminin (5 μm)-coated 25 mm glass coverslips and kept for 5–8 d at 37°C in a humidified incubator gassed with 5% CO2 and air. Cells were fed on the second day (and subsequent alternate days) with DMEM (with 1% FBS instead of 10% FBS).
Ca2+ fluorescence measurements.For the intracellular Ca2+([Ca2+]i) fluorescence measurements, plated neurons (5–8 d old) were loaded with fura-2 AM (3 μm) in Ca2+ buffer solution of the following composition (in mm): CaCl21.4, KCl 5.4, MgSO4 0.4, NaCl 135,d-glucose 5, and HEPES 10 with BSA (0.1%), pH 7.4, for 40 min at 37°C. The plated neurons were then washed twice with the Ca2+ buffer solution and transferred to a chamber on the stage of Nikon eclipse TE300 microscope. Fura-2 AM was excited at 340 and 380 nm to indicate relative [Ca2+]i changes by theF340/F380ratio recorded with a dynamic image analysis system (Laboratory Automation 2.0; RCS, Florence, Italy).
After transferring the plated neurons to the chamber, neurons were allowed (at least 10 min) to attain a stable fluorescence before beginning the experiment. AEA (1–30 μm), capsaicin (0.001–1 μm), META (10 μm), PEA (10 μm), or their respective vehicles were added to the chamber. In some experiments, to desensitize the neurons 60 min before the beginning of the Ca2+ fluorescence experiments, plated neurons were preexposed to capsaicin (10 μm) for 60 min. In a separate set of experiments, during the Ca2+ fluorescence, the cells were excited with electrical stimulation by means of two platinum bands (each covering a quarter of the radius) placed into the chamber at 180° from each other. Electrical stimulation (10 Hz, 40 mA/cm2, 1 msec pulse duration, for 10 sec) was delivered twice with a resting period of 20 min between each stimulation. A calibration curve was performed using buffer containing fura-2 AM and determinant concentrations of free Ca2+ (Kudo et al., 1986). This curve was then used to convert the data obtained fromF340/F380ratio to [Ca2+]i(nanomolar).
Materials. Drugs and reagents were obtained from the indicated companies: AEA and META (Alexis, Vinci, Italy); mouse NGF-7S and collagenase–dispase (Roche Diagnostics, Monza, Italy); DMEM, heat-inactivated FBS, l-glutamine (200 mm), penicillin–streptomycin (10,000 units to 10 mg), and Ca2+–Mg2+-free PBS (Life Technologies, San Giuliano Milanese, Italy); fura-2 AM (Societa' Italiana Chimici, Rome, Italy); AM281 [1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-4-morpholinyl-1H-pyrazole-3-carboxamide], AM630 [6-iodo-2-methyl-1-[2-(4-morpholinyl) ethyl]-1H-indol-3-yl] (4-methoxyphenyl) methanone], and PEA (Tocris Cookson, Bristol, UK); BSA, captopril, AraC, capsaicin, capsazepine, EDTA, HEPES, ionomycin, laminin, phosphoramidon, poly-l-lysine, tetrodotoxin, and other reagents (Sigma, Milan, Italy). The stock concentration of AEA (10 mm), AM281 (10 mm), AM630 (10 mm), capsaicin (10 mm), capsazepine (10 mm), and META (10 mm) were prepared in 100% ethanol. Fura-2 AM and ionomycin were dissolved in DMSO. All other drugs were dissolved in distilled water. The appropriate dilutions were then made in Krebs' buffer solution.
Statistical analysis. Results are expressed as mean ± SEM. Statistical analysis was performed by means of the Student'st test or ANOVA and the Dunnett's test when required. The SEs of proportions and comparisons between proportions of responding neurons were performed with the methods reported by Glantz (1992) with the Primer of Biostatistics package. Ifp < 0.05, the results were considered significant.
RESULTS
CGRP-LI and SP-LI release
We examined the effect of AEA on the release of CGRP-LI and SP-LI on superfused slices of rat dorsal spinal cord, in which these peptides are confined to the central projections of DRG neurons. AEA (0.01–10 μm) produced a concentration-related increase in CGRP-LI outflow (Fig.1), threshold concentration of AEA being 100 nm. Similar results were obtained when the outflow of SP-LI was studied (n = 5; data not shown). Two other cannabinoid agonists, META (1 μm) and PEA (1 μm), also increased CGRP-LI outflow from slices of rat dorsal spinal cord, whereas the vehicle of AEA, PEA, and META (ethanol 0.1%) was inactive (Fig. 1). The increase in CGRP-LI (Fig.2A) and SP-LI (Fig.2B) outflow evoked by AEA (1 μm) was abolished in experiments performed with a Ca2+-free medium, containing 1 mm EDTA or in tissues pretreated with capsaicin (10 μm for 60 min before AEA administration). The VR-1 antagonist capsazepine (10 μm) also abolished the increase in CGRP-LI and SP-LI outflow evoked by AEA (1 μm) (Fig.2A,B). The increase in CGRP-LI outflow induced by 1 μm PEA (154 ± 28 fmol/gm for 20 min; n = 5) and 1 μm MEA (183 ± 34 fmol/gm for 20 min;n = 5) was markedly inhibited by capsazepine (10 μm) by 64% (55 ± 12 fmol/gm for 20 min;n = 4; p < 0.05) and by 73% (50 ± 14 fmol/gm for 20 min; n = 4; p < 0.05), respectively. In contrast, the increase in CGRP-LI (Fig.2A) and SP-LI (n = 6; data not shown) outflow evoked by AEA (1 μm) was unaffected by pretreatment with the CB receptor antagonists AM281 (10 μm) and AM630 (10 μm). For pretreatment with capsaicin or in the presence of capsazepine, AM281 and AM630 had no significant effect on basal CGRP-LI or SP-LI outflow (n = 4–6; data not shown). Previous experiments (data not shown) indicated that basal outflow of both CGRP-LI and SP-LI declined progressively from 0 to 60 min of incubation and remained constant and very close to the detection limit of the assays after 60 min. Thus, basal outflow detected after 90 min of incubation seems to reflect nonspecific immunoreactivity rather than release or leakage of neuropeptides. This could explain why the various treatments did not show any significant effect on basal outflow of both CGRP-LI and SP-LI.
Fig. 1.

Outflow of CGRP-LI from slices of the cervical and lumbar enlargements of the rat dorsal spinal cord. Effect of AEA (μm), PEA (1 μm), and META (1 μm) or vehicle (VEH). Each entry is the mean ± SEM of at least five experiments. *p < 0.05 versus vehicle controls.
Fig. 2.
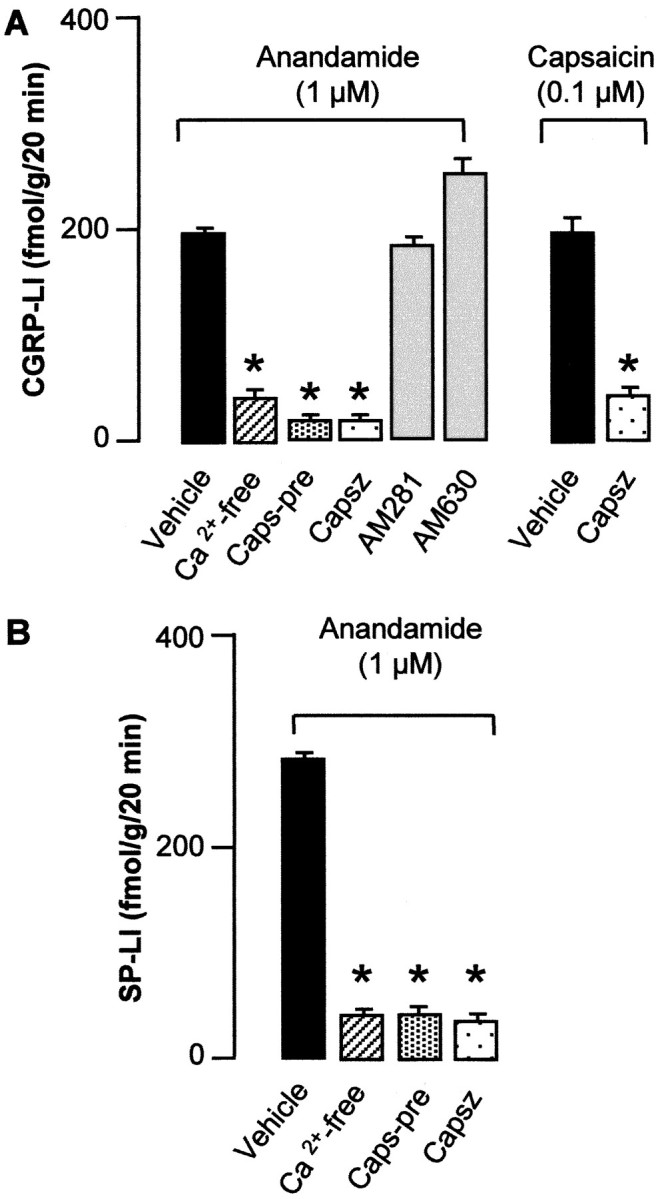
Outflow of CGRP-LI (A) and SP-LI (B) above baseline from slices of the cervical and lumbar enlargements of the rat dorsal spinal cord induced by anandamide or capsaicin. Effect of a Ca2+-free medium and 1 mm EDTA, pretreatment with capsaicin (Caps-pre; 10 μm for 60 min, 60 min before AEA), capsazepine (Capsz; 10 μm), and the CB receptor antagonists AM281 (10 μm) and AM630 (10 μm). Each entry is the mean ± SEM of at least five experiments. *p < 0.05 versus vehicle controls.
Capsaicin (0.1 μm) increased significantly the outflow of CGRP-LI from slices of rat dorsal spinal cord in a manner quantitatively similar to AEA (1 μm). This effect of capsaicin was significantly attenuated by capsazepine (10 μm) (Fig. 2A). EFS (10 Hz, 100 mA/cm2, 1 msec pulse duration, 10 sec train every 20 sec for 5 min) induced a significant increase in CGRP-LI outflow above baseline (S1, 92 ± 12 fmol/gm for 20 min;n = 7) that was practically abolished by tetrodotoxin (0.3 μm) (S2, 12 ± 6 fmol/gm for 20 min;n = 4; p < 0.01). The effect of EFS was reproducible 60 min after the first stimulation (S2, 76 ± 9 fmol/gm for 10 min; n = 7; S2/S1 ratio, 0.82 ± 0.04). EFS-induced increase in CGRP-LI outflow was significantly inhibited by a low concentration of AEA (10 nm) (S2/S1, 0.24 ± 0.03; n = 4; p < 0.05) but not by AEA vehicle (S2/S1, 0.73 ± 0.08;n = 4). The inhibitory effect of AEA on EFS-induced increase in CGRP-LI outflow was reversed by AM281 (10 μm) (S2/S1, 0.74 ± 0.07;n = 4). In contrast, capsazepine (10 μm) did not affect EFS-induced increase in CGRP-LI outflow (S2/S1, 0.76 ± 0.06; n = 4).
Ca2+ mobilization experiments
The effect of AEA on mobilization of Ca2+ and its modulation was examined in cultured DRG neurons obtained from newborn rats. Under the present experimental conditions, baseline [Ca2+]i was 93 ± 3 nm (n = 196), and 91% (174 of 196) of the tested cells responded to capsaicin (1 μm) with an increase in [Ca2+]i that was 621 ± 121 nm. AEA (1–30 μm) evoked a concentration-dependent and prompt increase in [Ca2+]i that reached a maximum within 20–30 sec (Fig.3A,B), whereas the decay of the response varied with the concentration of the stimulus used. Threshold concentrations of AEA to elicit a visible increase in [Ca2+]i was 1 μm. At the maximum concentration used, AEA (30 μm) evoked a peak in [Ca2+]i transients (462 ± 89 nm;n = 18) that declined but did not return to baseline levels after 10 min (data not shown). In a series of experiments, cells that responded to AEA (1 μm) were 52% (44 of 84) of those that responded to capsaicin (1 μm, 10 min after AEA) (Fig. 4B). In no instance did AEA (1 μm) increase [Ca2+]i in cells that did not respond to capsaicin (1 μm). All plated cells responded to KCl (30 mm) with a peak in [Ca2+]itransient of 650 ± 121 nm(n = 30). Exposure to AEA (1 μm) did not affect the magnitude of the response to capsaicin (1 μm) (n= 29; data not shown). Furthermore, all of the neurons that responded to 30 μm AEA responded also to 1 μm capsaicin. However, the response to capsaicin was 49% lower (311 ± 86 nm;n = 16) than the response obtained in neurons pretreated with the vehicle of AEA (609 ± 143 nm;n = 12; p < 0,05), thus suggesting that AEA caused a certain degree of desensitization.
Fig. 3.
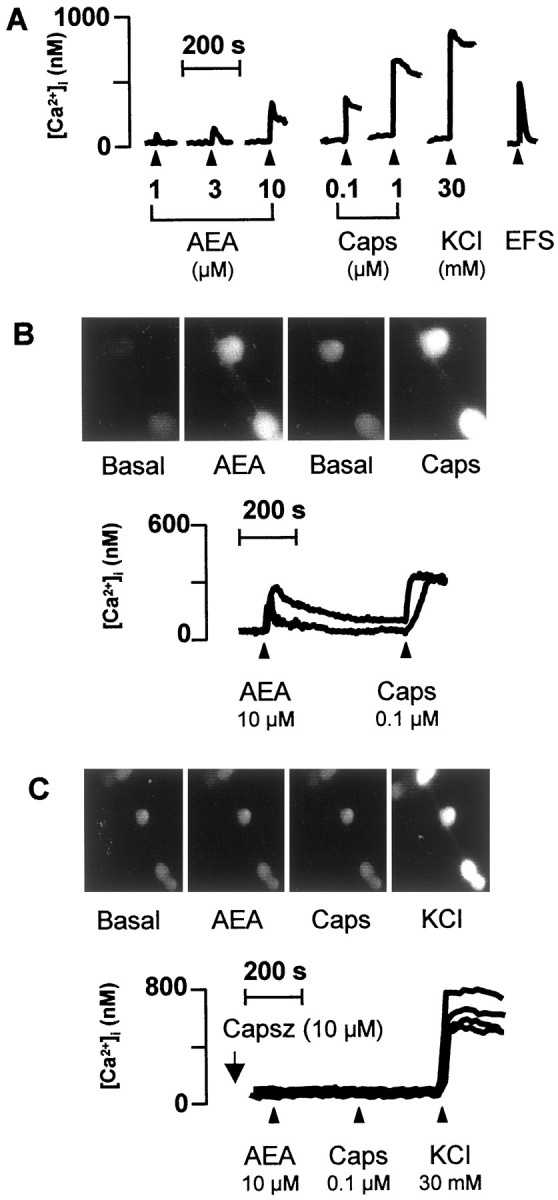
A, Ca2+mobilization induced by AEA, capsaicin (Caps), KCl, and EFS (10 Hz, 40 mA/cm2, 1 msec pulse duration, for 10 sec) (administered at the arrowhead) in cultured newborn rat DRG neurons. B, Neurons responding to anandamide responded also to capsaicin. C, The presence of capsazepine (Capsz) abolished the response to anandamide and capsaicin. Each line shows [Ca2+]i measured in the soma of single neuron.
Fig. 4.

The effect of pretreatment with capsazepine (10 μm) or its vehicle on Ca2+mobilization (A) induced by increasing concentrations of anandamide or capsaicin in cultured newborn rat DRG neurons. B, The number of neurons (as a percentage of those responding to capsaicin 1 μm) responding to increasing concentrations of anandamide or capsaicin in the presence of capsazepine or its vehicle. Each entry is the mean ± SEM of at least 27 cells. *p < 0.05 versus respective vehicle.
In cells (n = 63) exposed to capsaicin (10 μm for 60 min), addition of AEA (30 μm) or capsaicin (1 μm) failed to produce any significant increase in [Ca2+]i (data not shown). In the presence of capsazepine (10 μm), the magnitude of the increase in [Ca2+]i and the number of cells excited by increasing concentrations of either AEA or capsaicin were markedly reduced (Figs. 3C,4A,B). The increase in [Ca2+]i evoked by AEA (1 μm) (375 ± 97 nm;n = 21) was not affected by AM281 (10 μm) (412 ± 67 nm; n = 22) and AM630 (10 μm) (338 ± 81 nm;n = 18). EFS (10 Hz, 40 mA/cm2, 1 msec pulse duration, for 10 sec) caused a TTX-sensitive (0.3 μm) (n = 14; data not shown) increase in [Ca2+]i that was reproducible at 20 min intervals (Fig.5A,B). Capsazepine (10 μm) did not affect the increase in [Ca2+]iproduced by EFS, an effect that, however, was reduced significantly by AEA (10 nm) in a manner reversible by AM281 (10 μm) (Fig. 5). Capsazepine, AM281, and AM630 (all 10 μm) did not affect baseline [Ca2+]i(n = 54; data not shown).
Fig. 5.
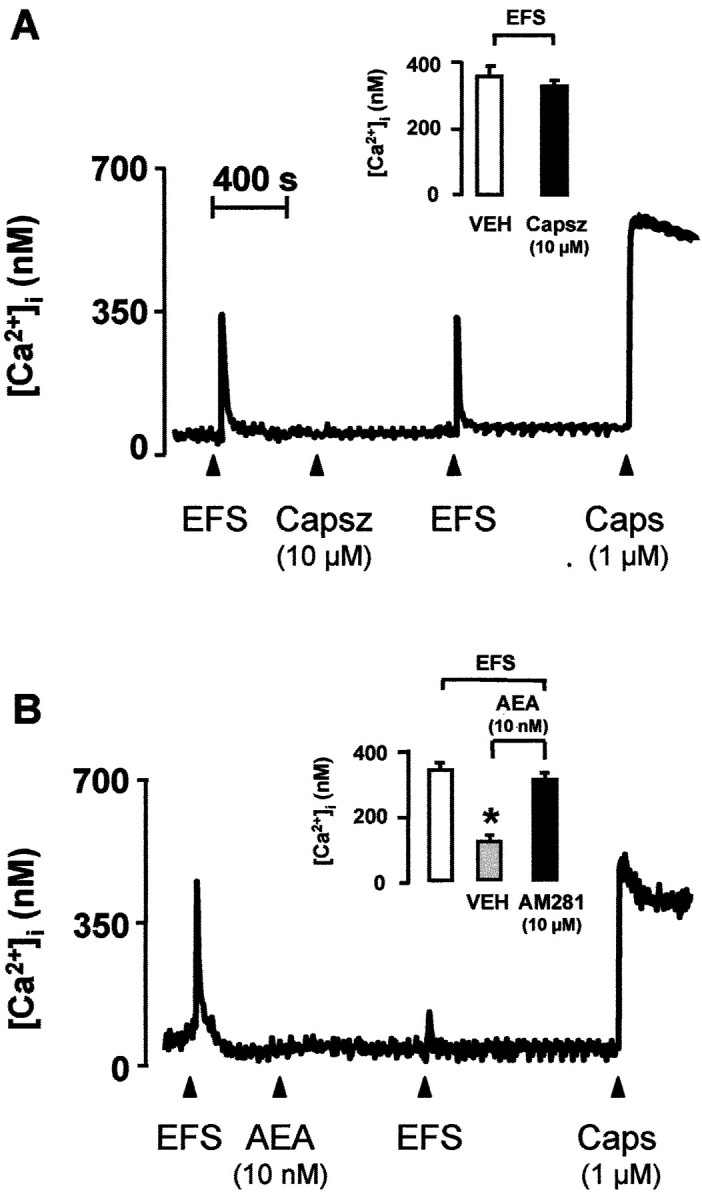
Ca2+ mobilization induced by EFS (10 Hz, 40 mA/cm2, 1 msec pulse duration, for 10 sec) and capsaicin (Caps) (administered at thearrowhead) in cultured newborn rat DRG neurons.A, Capsazepine (Capsz) did not affect the response to EFS. B, In this case, a low concentration of AEA reduced the response to EFS in a manner reversible by the CB receptor antagonist AM281. Each line shows [Ca2+]i measured in the soma of single neuron. Each column is the mean ± SEM of at least 42 cells. *p < 0.01 versus vehicle (VEH).
DISCUSSION
In this study, we have shown that micromolar concentrations of AEA cause release of CGRP-LI and SP-LI from central terminals of capsaicin-sensitive primary sensory neurons. This conclusion derives from the following observations. First, the ability of AEA to increase sensory neuropeptide outflow from slices of rat dorsal spinal cord was abolished by capsaicin pretreatment. It is known that exposure to elevated concentrations of capsaicin causes desensitization and cell death of a specific subpopulation of primary sensory neurons (Holzer, 1991; Szallasi and Blumberg, 1999), uniquely sensitive to the dual excitatory–neurotoxic action of this drug. Although the existence of SP-containing neuronal cell bodies has been documented in the spinal cord, this observation strongly points to central terminals of capsaicin-sensitive neurons as the sole source of the increase in CGRP-LI and SP-LI outflow induced by AEA. Second, the AEA-induced increase in both CGRP-LI and SP-LI outflow was virtually abolished in experiments performed in Ca2+-free conditions. We also found that elevated concentrations of AEA mobilized Ca2+ exclusively in those cultured DRG neurons that responded to capsaicin. This latter finding is in agreement with the recent observation that AEA mobilizes Ca2+ in a proportion of capsaicin-sensitive rat trigeminal neurons in culture (Szoke et al., 2000). An interesting aspect of our findings is that similar threshold concentrations of AEA produced both neuropeptide release and mobilization of Ca2+. The conclusion that AEA promotes a neurosecretory response from central terminals of capsaicin-sensitive DRG neurons is consistent with a recent report (Zygmunt et al., 1999) that elevated concentrations of AEA release CGRP from peripheral endings of capsaicin-sensitive neurons in the rat hepatic and mesenteric arteries and guinea pig basilar artery.
The most interesting observation of the present study, however, relates to the molecular mechanism responsible for the excitatory action of AEA on central terminals of primary sensory neurons. This result derives from parallel findings obtained in release and Ca2+ mobilization experiments. CB receptors via their ability to reduce adenylyl cyclase activity (Howlett, 1995) exert an inhibitory role on target cells. Thus, the possibility that AEA stimulates neuropeptide release via CB receptors directly on sensory nerve terminals seems unlikely. This and the additional hypothesis that CB receptors control sensory neuropeptide release in the dorsal spinal cord, by prejunctionally reducing the release of an inhibitory transmitter(s), liberated from adjacent nerve fibers, seems to be excluded because the CB receptor antagonists did not affect AEA-induced CGRP-LI or SP-LI release. In contrast with these negative findings, we found that CB receptor antagonists reversed the AEA-induced inhibition of EFS-induced sensory neuropeptide release. This observation confirms and extends previous findings showing that AEA, via CB1 receptor activation, reduces both high K+- and capsaicin-induced release of CGRP from dorsal spinal cord (Richardson et al., 1998). The fact that AM281 and AM630 reversed the inhibition induced by AEA of EFS-induced [Ca2+]i transient in cultured DRG neurons strongly supports the view that inhibitory CB receptors are expressed on the neuronal cell bodies and possibly on their central endings, as suggested by previous morphological evidence (Hohmann and Herkenham, 1999a,b).
Data obtained with the CB receptor antagonists and with capsazepine, a drug that inhibits capsaicin action on sensory nerves (Walpole et al., 1994), point to the role of VR-1 as the molecular structure that mediates the excitatory action of AEA on central terminals of DRG neurons. Furthermore, capsazepine abolished AEA-induced release of both CGRP-LI and SP-LI from the dorsal spinal cord. Although the concentration of capsazepine used was rather high (10 μm), selectivity was demonstrated by the fact that it abolished capsaicin-induced neuropeptide release but did not affect CGRP-LI release induced by EFS. Similarly, in the Ca2+ assay in cultured DRG neurons, capsazepine shifted to the right the concentration–response curves to capsaicin and to AEA without affecting the moderate increase in [Ca2+]i evoked by EFS.
The present results show that AEA-related compounds, such as META and PEA, apparently share the same properties of AEA on VR-1. Although a previous study showed that META, but not PEA, activates VR-1 (Zygmunt et al., 1999), another paper indicated that PEA is an efficient, although less potent agonist than AEA, to activate the human VR-1 in transfected HEK293 cells (Smart et al., 2000). VR-1 is closely related to the family of channels activated by transient receptor potential (TRP) (Caterina et al., 1997). Certain TRP channels are stimulated by micromolar concentrations of lipid derivatives, such as arachidonic acid and diacylglycerol (Chyb et al., 1999). The similarity between the chemical structure of these compounds and the observation that META and PEA, although apparently less efficacious than AEA, caused a capsazepine-sensitive CGRP-LI release suggests that they may also activate VR-1 in the dorsal spinal cord. Together, the present and previous observations (Zygmunt et al., 1999; Smart et al., 2000) suggest the stimulating hypothesis that additional lipidic molecules may exist, which, showing affinity for VR-1 higher than that of AEA, might be better candidates as endogenous ligand for VR-1.
The present findings confirm previous results showing that, in peripheral terminals of capsaicin-sensitive primary sensory neurons (Zygmunt et al., 1999; Smart et al., 2000) AEA has an excitatory role via VR-1 activation and extends this observation to the central terminals of these neurons. These results may have important pathophysiological implications. Apart from certain xenobiotics, such as capsaicin and resinferatoxin (Szallasi and Blumberg, 1999), VR-1 has been recognized to mediate influx of cations and excite a subset of primary sensory neurons in response to noxious heat (>43°C) (Szolcsanyi, 1977; Caterina et al., 1997) and possibly protons (pH <6) (Bevan and Geppetti, 1994; Tominaga et al., 1998). The critical role of VR-1 in thermal hyperalgesia has been confirmed by findings obtained in VR-1 knock-out mice (Caterina et al., 2000; Davis et al., 2000). Heat is an obvious stimulus to activate VR-1 in cutaneous afferents and under specific circumstances in visceral afferents. Likewise, a low pH may be encountered in a variety of inflammatory conditions in peripheral tissues, including tissue injury, ischemia, asthma exacerbations, acid back diffusion in the gastric wall, and other conditions (Bevan and Geppetti, 1994; Holzer, 1998; Hunt et al., 2000). Less obvious is, however, the role of heat and low pH in the activation of VR-1 on the central terminals of DRG neurons.
VR-1 is abundantly present in central endings of primary sensory neurons in the dorsal part of the spinal cord and brainstem (Szallasi et al., 1995, Caterina et al., 1997). Its expression at this level may be solely the consequence of the transport process from the cell body that occurs, in a bidirectional way, to both central and peripheral processes of pseudounipolar DRG neurons. Should heat and low pH be the sole agents capable of stimulating VR-1, its expression on central projections of DRG neurons would, at best, result in occasional activation during severe spinal cord injury. However, the discovery of a putative class of chemical messengers that stimulate VR-1 (Zygmunt et al., 1999) and that are produced locally within the CNS (Di Marzo et al., 1994) suggests the hypothesis that pathophysiological processes occurring within the dorsal spinal cord or even at the peripheral levels may release AEA, or of an AEA-related molecule, in amounts sufficiently high to activate central terminals of DRG neurons expressing VR-1. Testing the biological effects of AEA or derivatives of 12- or 15-lipoxygenases in VR-1 knock-out mice (Caterina et al., 2000; Davis et al., 2000) or in animals pretreated with capsazepine may be of help to elucidate the novel role of putative capsaicin-like endogenous substances in the processing of nociceptive information via the activation of VR-1 at the spinal cord level.
Biological processes leading to release of AEA in the CNS are still poorly understood (Di Marzo et al., 1994), and precise information regarding the ability of neurons or other cells in the dorsal spinal cord to release elevated amounts of AEA is lacking. In addition, we must underline that endogenous AEA may potentially reach levels (nearly nanomolar) in peripheral tissues or in the spinal cord sufficient to reduce neuropeptide release and their nociceptive and inflammatory effects via the CB receptor-dependent inhibitory mechanism. In contrast, considering that nearly micromolar concentrations of AEA are required to activate the excitatory VR-1-dependent pathway, it seems unlikely that these exceedingly high concentrations of AEA are reached in peripheral tissue as well as in the spinal cord, even during pathophysiological conditions. In other terms, the possibility that endogenous AEA promotes neurogenic inflammatory responses or activates nociceptive pathways stimulating VR-1 must be supported by evidence showing that sufficiently high concentrations of this molecule are released. Recent data indicate that different lipoxygenase derivatives have similar potency, but greater efficacy than AEA in stimulating VR-1 (Hwang et al., 2000). Thus, these molecules may be more suitable candidates than AEA as the final mediators of a nociceptive and proinflammatory pathway that targets VR-1 on primary sensory neurons.
Footnotes
This study was supported in part by grants from Ministero dell'Università della Ricera Scientifica e Tecnologica, Rome and the European Respiratory Society.
M.T. and S.A. contributed equally to this work.
Correspondence should be addressed to Dr. Pierangelo Geppetti, Department of Experimental and Clinical Medicine, Headache Center, University of Ferrara, Via Fossato di Mortara 19, 44100 Ferrara, Italy. E-mail: p.geppetti@unife.it.
REFERENCES
- 1.Beltramo M, Piomelli D. Anandamide transport inhibition by the vanilloid agonist olvanil. Eur J Pharmacol. 1999;364:75–78. doi: 10.1016/s0014-2999(98)00821-8. [DOI] [PubMed] [Google Scholar]
- 2.Bevan S, Geppetti P. Protons: small stimulants of capsaicin-sensitive sensory nerves. Trends Neurosci. 1994;17:509–512. doi: 10.1016/0166-2236(94)90149-x. [DOI] [PubMed] [Google Scholar]
- 3.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 4.Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- 5.Chyb S, Raghu P, Hardie RC. Polyunsaturated fatty acids activate the Drosophila light-sensitive channels TRP and TRPL. Nature. 1999;397:255–259. doi: 10.1038/16703. [DOI] [PubMed] [Google Scholar]
- 6.Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- 7.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 8.Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- 9.Di Marzo V, Bisogno T, Melck D, Ross R, Brockie H, Stevenson L, Pertwee R, De Petrocellis L. Interactions between synthetic vanilloids and the endogenous cannabinoid system. FEBS Lett. 1998;436:449–454. doi: 10.1016/s0014-5793(98)01175-2. [DOI] [PubMed] [Google Scholar]
- 10.Frobert Y, Nevers MC, Amadesi S, Volland H, Brune P, Geppetti P, Grassi J, Creminon C. A sensitive sandwich enzyme immunoassay for calcitonin gene-related peptide (CGRP): characterization and application. Peptides. 1999;20:275–284. doi: 10.1016/s0196-9781(98)00172-7. [DOI] [PubMed] [Google Scholar]
- 11.Geppetti P, Holzer P. Neurogenic inflammation. CRC; Boca Raton, FL: 1996. [Google Scholar]
- 12.Gilabert R, McNaughton P. Enrichment of the fraction of nociceptive neurones in cultures of primary sensory neurones. J Neurosci Methods. 1997;71:191–198. doi: 10.1016/s0165-0270(96)00144-6. [DOI] [PubMed] [Google Scholar]
- 13.Glantz SA. Primer of bio-statistics. McGraw-Hill; New York: 1992. [Google Scholar]
- 14.Hohmann AG, Herkenham M. Localization of central cannabinoid CB1 receptor messenger RNA in neuronal subpopulations of rat dorsal root ganglia: a double-label in situ hybridization study. Neuroscience. 1999a;90:923–931. doi: 10.1016/s0306-4522(98)00524-7. [DOI] [PubMed] [Google Scholar]
- 15.Hohmann AG, Herkenham M. Cannabinoid receptors undergo axonal flow in sensory nerves. Neuroscience. 1999b;92:1171–1175. doi: 10.1016/s0306-4522(99)00220-1. [DOI] [PubMed] [Google Scholar]
- 16.Hohmann AG, Martin WJ, Tsou K, Walker JM. Inhibition of noxious stimulus-evoked activity of spinal cord dorsal horn neurons by the cannabinoid WIN 55,212–2. Life Sci. 1995;56:2111–2118. doi: 10.1016/0024-3205(95)00196-d. [DOI] [PubMed] [Google Scholar]
- 17.Holzer P. Capsaicin: cellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacol Rev. 1991;43:143–201. [PubMed] [Google Scholar]
- 18.Holzer P. Neural emergency system in the stomach. Gastroenterology. 1998;114:823–839. doi: 10.1016/s0016-5085(98)70597-9. [DOI] [PubMed] [Google Scholar]
- 19.Howlett AC. Pharmacology of cannabinoid receptors. Annu Rev Pharmacol Toxicol. 1995;35:607–634. doi: 10.1146/annurev.pa.35.040195.003135. [DOI] [PubMed] [Google Scholar]
- 20.Hunt JF, Fang K, Malik R, Snyder A, Malhotra N, Platts-Mills TA, Gaston B. Endogenous airway acidification. Implications for asthma pathophysiology. Am J Respir Crit Care Med. 2000;161:694–699. doi: 10.1164/ajrccm.161.3.9911005. [DOI] [PubMed] [Google Scholar]
- 21.Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ, Jung J, Cho S, Min KH, Suh YG, Kim D, Oh U. Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc Natl Acad Sci USA. 2000;97:6155–6160. doi: 10.1073/pnas.97.11.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kudo Y, Ozaki K, Miyakawa A, Amano T, Ogura A. Monitoring of intracellular Ca2+ elevation in a single neural cell using a fluorescence microscope/video-camera system. Jpn J Pharmacol. 1986;41:345–351. doi: 10.1254/jjp.41.345. [DOI] [PubMed] [Google Scholar]
- 23.Melck D, Bisogno T, De Petrocellis L, Chuang H, Julius D, Bifulco M, Di Marzo V. Unsaturated long-chain N-acyl-vanillyl-amides (N-AVAMs): vanilloid receptor ligands that inhibit anandamide-facilitated transport and bind to CB1 cannabinoid receptors. Biochem Biophys Res Commun. 1999;262:275–284. doi: 10.1006/bbrc.1999.1105. [DOI] [PubMed] [Google Scholar]
- 24.Otsuka M, Yoshioka K. Neurotransmitter functions of mammalian tachykinins. Physiol Rev. 1993;73:229–308. doi: 10.1152/physrev.1993.73.2.229. [DOI] [PubMed] [Google Scholar]
- 25.Ricciardolo FL, Steinhoff M, Amadesi S, Guerrini R, Tognetto M, Trevisani MCreminon C, Bertrand C, Bunnett NW, Fabbri LM, Salvadori S, Geppetti P. Presence and bronchomotor activity of protease-activated receptor-2 in guinea pig airways. Am J Respir Crit Care Med. 2000;161:1672–1680. doi: 10.1164/ajrccm.161.5.9907133. [DOI] [PubMed] [Google Scholar]
- 26.Richardson JD, Aanonsen L, Hargreaves KM. Antihyperalgesic effects of spinal cannabinoids. Eur J Pharmacol. 1998a;345:145–153. doi: 10.1016/s0014-2999(97)01621-x. [DOI] [PubMed] [Google Scholar]
- 27.Richardson JD, Kilo S, Hargreaves KM. Cannabinoids reduce hyperalgesia and inflammation via interaction with peripheral CB1 receptors. Pain. 1998b;75:111–119. doi: 10.1016/S0304-3959(97)00213-3. [DOI] [PubMed] [Google Scholar]
- 28.Smart D, Gunthorpe MJ, Jerman JC, Nasir S, Gray J, Muir AI, Chambers JK, Randall AD, Davis JB. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1). Br J Pharmacol. 2000;129:227–230. doi: 10.1038/sj.bjp.0703050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szallasi A, Blumberg PM. Vanilloid (Capsaicin) receptors and mechanisms. Pharmacol Rev. 1999;51:159–212. [PubMed] [Google Scholar]
- 30.Szallasi A, Nilsson S, Hokfelt T, Lundberg JM. Visualizing vanilloid (capsaicin) receptors in pig spinal cord by [3H]resiniferatoxin autoradiography. Brain Res. 1994;655:237–240. doi: 10.1016/0006-8993(94)91619-5. [DOI] [PubMed] [Google Scholar]
- 31.Szallasi A, Nilsson S, Farkas-Szallasi T, Blumberg PM, Hokfelt T, Lundberg JM. Vanilloid (capsaicin) receptors in the rat: distribution in the brain, regional differences in the spinal cord, axonal transport to the periphery, and depletion by systemic vanilloid treatment. Brain Res. 1995;703:175–183. doi: 10.1016/0006-8993(95)01094-7. [DOI] [PubMed] [Google Scholar]
- 32.Szoke E, Balla Z, Csernoch L, Czeh G, Szolcsanyi J. Interacting effects of capsaicin and anandamide on intracellular calcium in sensory neurones. NeuroReport. 2000;11:1949–1952. doi: 10.1097/00001756-200006260-00028. [DOI] [PubMed] [Google Scholar]
- 33.Szolcsanyi J. A pharmacological approach to elucidation of the role of different nerve fibres and receptor endings in mediation of pain. J Physiol (Paris) 1977;73:251–259. [PubMed] [Google Scholar]
- 34.Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 35.Tsou K, Lowitz KA, Hohmann AG, Martin WJ, Hathaway CB, Bereiter DA, Walker JM. Suppression of noxious stimulus-evoked expression of Fos protein-like immunoreactivity in rat spinal cord by a selective cannabinoid agonist. Neuroscience. 1996;70:791–798. doi: 10.1016/s0306-4522(96)83015-6. [DOI] [PubMed] [Google Scholar]
- 36.Walpole CS, Bevan S, Bovermann G, Boelsterli JJ, Breckenridge R, Davies JW, Hughes GA, James I, Oberer L, Winter J, Wrigglesworth R. The discovery of capsazepine, the first competitive antagonist of the sensory neuron excitants capsaicin and resiniferatoxin. J Med Chem. 1994;37:1942–1954. doi: 10.1021/jm00039a006. [DOI] [PubMed] [Google Scholar]
- 37.Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, Julius D, Hogestatt ED. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]