

Abstract
Nitric oxide (NO) is a potent neuromodulator in the CNS and PNS. At the frog neuromuscular junction (nmj), exogenous application of NO reduces neurotransmitter release, and NO synthases (NOSs), the enzymes producing NO, are present at this synapse. This work aimed at studying the molecular mechanisms by which NO modulates synaptic efficacy at the nmj using electrophysiological recordings and Ca2+-imaging techniques. Bath application of the NO donors S-nitroso-N-acetylpenicillamine (SNAP) and sodium nitroprusside decreased end plate potential (EPP) amplitude as well as the frequency of miniature EPPs but not their amplitude. Ca2+ responses elicited in presynaptic terminals by single action potentials were unaffected by NO, but responses evoked by a short train of stimuli were increased. Tonic endogenous production of NO was observed as suggested by the increase in EPP amplitude by bath application of the NO scavenger hemoglobin and the neuronal NOS inhibitor 3-bromo-7-nitroindazole sodium salt. A soluble guanylate cyclase inhibitor, 6-anilino-5,8-quinolinedione (LY-83583), increased EPP amplitude and occluded the effects of the NO donor, suggesting that NO acts via a cGMP-dependent mechanism. High-frequency-induced depression was reduced in the presence of the NO scavenger but not by LY-83583. However, adenosine-induced depression was significantly reduced after bath perfusion of SNAP and in the presence of LY-83583. Our results indicate that NO regulates transmitter release and adenosine-induced depression via a cGMP-dependent mechanism that occurs after Ca2+ entry and that high-frequency-induced synaptic depression is regulated by NO in a cGMP-independent manner.
Keywords: nitric oxide, guanylate cyclase, adenosine, transmitter release, synaptic depression, calcium, perisynaptic Schwann cells
Neurotransmitter release is a highly organized and regulated process that provides a large degree of plasticity and adaptability (Illes, 1986; Wu and Saggau, 1997). It is modulated by a large number of second messengers each acting on specific elements involved in the transmitter release machinery. A very important second messenger known to regulate neurotransmitter release is nitric oxide (NO) (Brenman and Bredt, 1997).
NO is membrane permeable, and various forms of its synthesizing enzyme, the NO synthase (NOS), are found in neuronal and non-neuronal cells (Schmidt and Walter, 1994). In addition, most NOSs are activity dependent because of their Ca2+ dependency (Bredt and Snyder, 1992). In the CNS, there is evidence suggesting that NO might be implicated in synaptic plasticity phenomena such as long-term potentiation and depression in which it is thought to act as a retrograde messenger from postsynaptic neurons modulating surrounding presynaptic terminals (Izumi and Zorumski, 1997; Lev-Ram et al., 1997;Malen and Chapman, 1997; Calabresi et al., 1999). The major mechanism of action of NO is the activation of a soluble guanylate cyclase that in turn produces cGMP, causing a potentiation of protein kinase G (PKG) (Schmidt et al., 1993). Alternatively, NO has been shown to mediate post-translational modifications of proteins such as ADP-ribosylation (Duman et al., 1993), fatty acylation (Hess et al., 1993), andS-nitrosylation (Lipton et al., 1993). These modifications may prevent normal interactions between proteins involved in the synaptic vesicle–presynaptic membrane specific interactions occurring during exocytosis (Meffert et al., 1994, 1996).
There is also compelling evidence in support of the possibility that NO is an important modulator of synaptic transmission at the neuromuscular junction (nmj). First, exogenously applied NO reduces neurotransmitter release in immature (Wang et al., 1995) and mature (Lindgren and Laird, 1994) frog nmjs. Second, NO reduces the sensitivity to neurotransmitters of perisynaptic Schwann cells (PSCs), glial cells at the frog nmj (Descarries et al., 1998). Third, NOSs are found in skeletal muscle fibers (Silvagno et al., 1996; Okuda et al., 1997) where they are concentrated at the muscle end plate (Kusner and Kaminski, 1996). In addition, a form of neuronal NOS has also been found in PSCs (Descarries et al., 1998).
Although NO appears as a potent regulator of synaptic transmission at the mature nmj, there is no evidence yet whether endogenous NO is involved in the regulation of synaptic transmission, and little is known about the molecular mechanisms regulated by NO at the nmj. Thus, this work aimed to determine the mechanism of action of NO in its regulation of synaptic transmission and to test its involvement in high-frequency- and adenosine-induced depressions at the amphibian nmj.
Here, we report that there is a tonic production of NO at the frog nmj and that it reduces transmitter release via a cGMP-dependent and Ca2+-independent mechanism. We also present evidence that endogenous NO partially regulates adenosine-induced depression by a cGMP-dependent mechanism and high-frequency-induced depression by a cGMP-independent mechanism.
MATERIALS AND METHODS
Experiments were performed on nmjs of cutaneus pectoris muscles of Rana pipiens frogs. Frogs were double pithed, and muscles with their innervation were dissected and put into a recording chamber with the bottom filled with SylGard silicon elastomer (Dow Corning). Unless stated otherwise, all experiments were performed using normal frog Ringer's solutions (120 mm NaCl, 2 mm KCl, 1.8 mmCaCl2, 1 mmNaHCO3, and 15 mm HEPES). The pH was adjusted to 7.20 with NaOH (5N). All experiments were performed at room temperature (21–23°C).
Ca2+ imaging of nerve terminals. Frogs were double pithed and were partially submerged in a 5 mmMgCl2 Ringer's solution (no Ca2+ added) in a dissection dish. A small cut was made in the skin near the shoulder, and the pectoralis proprius nerve was cleared from the surrounding connective tissue, keeping the blood vessels intact. The nerve was then cut and rinsed with the Mg2+ Ringer's solution to prevent the cut end of the axons from collapsing. The nerve was then put on the animal skin, and crystals of Ca2+-green-1 dextran (molecular weight, 3000; Molecular Probes, Eugene, OR), a fluorescent Ca2+ indicator, were put next to the cut end of the nerve. The loading was performed in obscurity at room temperature for ∼10 hr to allow the indicator to be transported to the nerve terminals. We have shown that overnight treatments such as this one do not affect synaptic transmission and synapse–glia interactions (Jahromi et al., 1992; Robitaille et al., 1997, 1999).
A Bio-Rad (Hercules, CA) MRC 600 confocal microscope mounted on an Olympus BH2 upright microscope was used to collect images. The 488 nm excitation line of an argon ion laser was attenuated to 1% of maximal intensity using neutral density filters, and emitted fluorescence was filtered with a long-pass filter at 515 nm. A water-immersion lens was used (Olympus 40×; 0.75 numerical aperture).
Two types of experiments were performed. First, for single pulses and for short trains of stimulation (100 Hz; 100 msec), the line scan mode of the confocal microscope was used to monitor changes in fluorescence with a greater temporal resolution (Robitaille et al., 1999). The line scan mode permits successive readings at 2 msec intervals of a single line (0.22 μm thick) that was manually positioned over the center of a nerve terminal branch observed at zoom factor 4. Files of series of 512 lines were collected and analyzed off-line. Second, for longer stimulations (100 Hz; 7 sec), images (192 × 128 pixels) of nerve terminals were collected at intervals of 645 msec. Fluorescence emitted by the nerve terminals was measured, and changes in fluorescence were expressed as: %ΔF/F = (F−Frest)/Frest× 100.
All experiments were performed using suprathreshold stimulation, and muscle contractions were blocked using α-bungarotoxin (1.12 μm; Calbiochem, La Jolla, CA), an irreversible antagonist of nicotinic acetylcholine receptors (Dryden et al., 1974). Only one nerve terminal was monitored for each preparation.
Electrophysiology. The release of neurotransmitter was evoked by stimulating the motor nerve with single stimuli at a frequency of 0.2 Hz. Muscle contractions were blocked usingd-tubocurarine chloride (4.87 μm; Sigma, St. Louis, MO), a competitive antagonist of nicotinic acetylcholine receptors (Almon and Appel, 1976). Intracellular recordings of end plate potentials (EPPs) were performed using glass microelectrodes (10–15 MΩ) filled with KCl (2–3 m). Experiments were performed on muscle fibers with a membrane potential more negative than −70 mV and were discarded when it depolarized by >10 mV. For analysis of spontaneous activity, recordings were performed in normal Ringer's solution without d-tubocurarine chloride. In a few experiments in which both EPPs and miniature EPPs (MEPPs) were recorded during the same experiment, muscle contractions were prevented by the use of a low-[Ca2+] Ringer's solution (0.54 mm CaCl2 and 3.5 mm MgCl2). Similar results were obtained in both ionic conditions. Synaptic responses were recorded using an amplifier from Warner Instruments Corporation (gain of 10×) and then further amplified (100×) and filtered by a low-pass four-pole Bessel filter at 2 kHz (Warner Instruments Corporation). Data were acquired using a Digidata 1200 board controlled by the software Tomahacq (created by Mr. T. A. Goldthrope, University of Toronto) that was also used for data analysis. Only one muscle fiber was monitored on each preparation.
Drugs. Stock solutions ofS-nitroso-N-acetylpenicillamine (SNAP; Calbiochem), 6-anilino-5,8-quinolinedione (LY-83583; Calbiochem), and 3-bromo-7-nitroindazole sodium salt (3Br7NiNa; Calbiochem) were prepared in DMSO (Sigma) at 50, 40, and 250 mm,respectively. SNAP and 3Br7NiNa solutions were kept in the dark. Additional dilutions of SNAP (50–100 μm), LY-83583 (40 μm), and 3Br7NiNa (100 μm) in physiological solutions were prepared just before use.
Stock solutions of adenosine (Research Biochemicals, Natick, MA), 8-bromo-cGMP salt (8-Br-cGMP; Calbiochem), and sodium nitroprusside (SNP; Calbiochem) were diluted in water at 10, 10, and 50 mm, respectively. Additional dilutions of adenosine (10 μm), 8-Br-cGMP (100 μm), and SNP (50 μm) were made in physiological solutions just before use. Physiological solutions containing hemoglobin (30 μm) were prepared the day of the experiments.
Drugs were applied continuously with bath perfusion (2 ml/min) at room temperature.
Statistical analysis. All results are expressed as the mean ± SEM. In most experiments, two sets of data obtained from the same nmj were compared using a Student's paired t test. Otherwise, Student's t test was used to compare two sets of data obtained from different nmjs, and an ANOVA was used when three sets were compared.
RESULTS
The mechanisms of action of NO at the frog nmj were first investigated using electrophysiological and Ca2+-imaging techniques. The involvement of endogenous NO in the production of synaptic depression induced by high-frequency stimulation and adenosine was then determined.
Exogenous NO reduces transmitter release
It was reported previously that SNP, an NO donor, reduced neurotransmitter release at the frog nmj (Lindgren and Laird, 1994). Here we show that SNAP, another NO donor (Ignarro et al., 1981; Southam and Garthwaite, 1991), produced similar effects. Indeed, bath application of SNAP (50 μm) decreased EPP amplitude by 36 ± 3% (Fig.1A) (3.7 ± 1.0 mV in control vs 2.3 ± 0.6 mV in SNAP; p < 0.01, Student's one-tail paired t test; n = 10). The effects began after 5 min of perfusion with SNAP and were complete in 20 min. When tested, no reversal of the effects of the NO donor was detected for up to 90 min after its removal from the perfusion (data not shown).
Fig. 1.

NO reduces neurotransmitter release at the frog neuromuscular junction. A, Time course of changes in EPP amplitude (millivolts) before, during, and after bath application of SNAP (50 μm). The horizontal barrepresents the period of exposure to SNAP. The effects of SNAP started after 5 min and were complete by 20 min. There was no recovery, even after removal of SNAP from the perfusion. Similar results were obtained in 10 experiments. B, Left, Histogram of the mean ± SEM (n = 5) of MEPP frequency in control (open bar) and after a 30 min exposure to SNAP (filled bar). Right, Histogram of the mean ± SEM of MEPP amplitude in control (open bar) and after a 30 min exposure to SNAP (filled bar). Note that SNAP significantly reduced MEPP frequency, whereas it had no effect on MEPP amplitude (*p < 0.05, Student's one-tail paired t test).
To determine whether the effects of NO were presynaptic or postsynaptic, MEPP frequency and amplitude were measured in control and after a 30 min exposure to SNAP. Changes in MEPP frequency would be an indication of presynaptic changes in the probability of neurotransmitter release, whereas a change in MEPP amplitude and/or time course would indicate postsynaptic changes. In the presence of SNAP (100 μm), MEPP frequency was reduced by 39 ± 7% (5.27 ± 2.48 Hz in control vs 3.32 ± 1.81 Hz in SNAP;p < 0.05, Student's one-tail paired ttest; n = 5), whereas MEPP amplitude did not change significantly (567 ± 124 μV in control vs 667 ± 161 μV in SNAP; p > 0.05, Student's two-tail pairedt test; n = 4) (Fig. 1B). The reduction in MEPP frequency combined with the lack of effect on MEPP amplitude is consistent with presynaptic effects of NO. Thus, the reduction in EPP amplitude by NO was caused by a decrease in the probability of transmitter release.
The reduction in MEPP frequency is somewhat surprising because Lindgren and Laird (1994) reported that SNP had no effect on MEPP frequency at frog sartorius nmjs. This difference could be explained by the different nature of the NO donors used or by the different properties of the two synapses. This was tested by monitoring the effects of SNP on the cutaneus pectoris nmj. Similar to SNAP, SNP (50 μm) not only reduced EPP amplitude by 60 ± 12% (6.8 ± 1.0 mV in control vs 2.5 ± 0.5 mV in SNP;p < 0.05, Student's one-tail paired ttest; n = 3) but also reduced MEPP frequency by 44 ± 14% (4.09 ± 0.54 Hz in control vs 2.38 ± 0.65 Hz in SNP; p < 0.05, Student's one-tail paired ttest; n = 4) and did not change MEPP amplitude (456 ± 63 μV in control vs 487 ± 83 μV in SNP;p > 0.05, Student's two-tail paired ttest; n = 4). The fact that both NO donors had the same effects at the cutaneus pectoris nmj but somewhat different effects at the sartorius nmj (Lindgren and Laird, 1994) indicates that NO effects may vary according to the properties of the synapses.
Is endogenous NO produced in a tonic way?
To test whether NO was tonically produced at the frog nmj, synaptic transmission was monitored in the presence of hemoglobin, an NO scavenger (Murad et al., 1978). If there were a tonic production of NO at this synapse, the presence of an NO scavenger should cause an increase in EPP amplitude. As shown in Figure2A, bath application of hemoglobin (30 μm) caused a 28 ± 6% increase in EPP amplitude that raised from 5.2 ± 0.6 mV (control) to 6.7 ± 0.8 mV (hemoglobin; p < 0.01, Student's one-tail paired t test; n = 5).
Fig. 2.

NO chelation and inhibition of NOS potentiate synaptic transmission. A, Changes in EPP amplitude before and during bath application of the NO scavenger hemoglobin (30 μm). The horizontal bar represents the period of exposure to hemoglobin. Note that EPP amplitude increased in the presence of hemoglobin. Similar results were obtained in five experiments. B, Changes in EPP amplitude before and during bath application of the neuronal NOS inhibitor 3Br7NiNa (100 μm). The horizontal bar represents the period of exposure to 3Br7NiNa. EPP amplitude was increased in the presence of the NOS inhibitor. Similar results were obtained in five experiments.
This result suggests that there was a tonic production of NO at the frog nmj maintaining the synapse in a depressed state. If this were the case, blocking NO synthase activity should also increase transmitter release. This was tested by perfusing the neuronal NO synthase inhibitor 3Br7NiNa (Chapman et al., 1995) while monitoring its effects on EPP amplitude. As shown in Figure 2B, bath application of a small concentration of 3Br7NiNa (100 μm) (Wegener et al., 2000) increased EPP amplitude by 47 ± 18% (4.5 ± 0.7 mV in control vs 6.5 ± 1.0 mV in 3Br7NiNa; p < 0.05, Student's one-tail paired t test; n = 5). When tested, hemoglobin (30 μm) had no additional effect when perfused after 3Br7NiNa effects were complete (data not shown). These results indicate that there was a tonic production of NO at the frog nmj.
Is NO affecting Ca2+ entry in nerve terminals?
Knowing that neurotransmitter release is closely regulated by the intracellular Ca2+ concentration in nerve terminals (Katz and Miledi, 1967; Adler et al., 1991; Zucker, 1993), we tested whether NO could modulate the Ca2+concentration in nerve terminals, either by reducing Ca2+ entry triggered by action potentials and/or by affecting the resting level of Ca2+ in nerve terminals. Ca2+-green-1 dextran was backfilled into nerve terminals, and changes in the fluorescence of living terminals were monitored using confocal microscopy. Figure3A, top, illustrates a confocal image of nerve terminal branches loaded with Ca2+-green-1 dextran seen in false colors, and Figure 3A, bottom, shows a Ca2+ response induced by a brief train of stimuli (100 Hz; 100 msec) obtained using the line scan mode.
Fig. 3.

Effects of NO on stimulation-evoked Ca2+ responses in nerve terminals. Ca2+ responses were obtained using the line scan mode of the confocal microscope and monitored over a nerve terminal backfilled with Ca2+-green-1 dextran.A, Top, Gray scale confocal image of a branch of nerve terminal loaded with Ca2+-green-1 dextran. The line indicates the position of the line used to perform the line scan measurements. Bottom, Gray scale color image of the changes in fluorescence elicited by a brief train of stimuli (100 Hz; 100 msec) observed using the line scan mode of the confocal microscope. Black indicates a low level of Ca2+; white is a high level. Thetop of the image is time 0 msec; thebottom is time 1024 msec (512 lines at 2 msec intervals). Note the elevation in fluorescence induced by the stimulation (vertical bar on right).B, Time course of Ca2+ responses evoked by a single action potential in control (solid line) and after a 30 min exposure to SNAP (100 μm; dotted line). The arrowindicates the time of stimulation of the motor nerve. The peak and the duration of Ca2+ responses were unchanged in four experiments. C, Time course of Ca2+responses evoked by stimulation of the motor nerve (100 Hz; 100 msec) in control (solid line) and after a 30 min exposure to SNAP (50 μm; dotted line). Thehorizontal bar indicates the period of nerve stimulation. NO significantly raised the amplitude of Ca2+ responses in 10 experiments. D, Time course of Ca2+ responses evoked by prolonged stimulation of the motor nerve (100 Hz; 7 sec) in control (solid line) and after a 30 min exposure to SNAP (50 μm;dotted line). The horizontal barindicates the period of nerve stimulation. In 10 experiments, Ca2+ responses were unchanged in the presence of SNAP.
The effects of NO on Ca2+ responses evoked by single pulses were first monitored. As shown in Figure3B, Ca2+ responses were unchanged in the presence of SNAP (100 μm; 30 min exposure). The average maxima of Ca2+responses were 13.5 ± 1% ΔF/F in control and 13.5 ± 1% ΔF/F in the presence of NO (p > 0.05, Student's one-tail pairedt test; n = 4). The area under the curve of Ca2+ responses was also unchanged by SNAP (1867 ± 768% ΔF/F × msec in control vs 2053 ± 803% ΔF/F × msec in SNAP; p > 0.05, Student's one-tail pairedt test; n = 4), indicating that NO did not change the duration of Ca2+ responses evoked by single action potentials in nerve terminals. Therefore, the effects of NO on EPP amplitude cannot be explained by a reduction of Ca2+ entry that would change the global level of Ca2+ in nerve terminals.
We next tested the effects of NO on Ca2+entry during a train of stimuli at high frequency (100 Hz; 100 msec). Surprisingly, the amplitude of Ca2+responses was significantly higher in the presence of the NO donor (SNAP, 50 μm; 30 min exposure) than in control; the average maximum relative change in fluorescence was 160 ± 22% ΔF/F in control and 184 ± 27% ΔF/F in the presence of SNAP (Fig.3C) (p < 0.05, Student's one-tail t test; n = 10).
Hence, instead of reducing Ca2+ responses that would explain the NO-induced reduction in transmitter release, NO caused an increase in Ca2+ responses that would predict an elevation in neurotransmitter release. A reduction in Ca2+ entry should have been detected because we have reported a large reduction in the amplitude of Ca2+ responses in ionic conditions identical to those required to mimic the reduction in transmitter release observed with the NO donors [see Robitaille et al. (1999), their Fig. 5].
We then tested whether more prolonged stimulation would reveal a more pronounced effect of NO on the Ca2+responses. However, with stimulations at 100 Hz for 7 sec, there was no increase in the amplitude of Ca2+responses in the presence of SNAP (50 μm) (Fig.3D) (213 ± 23% ΔF/F in control vs 222 ± 17% ΔF/F in SNAP;p > 0.05, Student's one-tail ttest; n = 10). Also, the area under the curve of Ca2+ responses evoked by this type of stimulation with SNAP was not significantly different from control (5220 ± 579% ΔF/F × msec in control vs 6633 ± 657% ΔF/F × msec in SNAP; p > 0.05, Student's one-tailt test; n = 10). As a whole, these results indicate that NO effects on transmitter release cannot be explained by a global reduction in Ca2+ entry in the nerve terminal.
Is NO affecting the resting level of Ca2+ in nerve terminals?
We questioned whether NO would decrease the resting Ca2+ concentration in nerve terminals because MEPP frequency is sensitive to the resting level of Ca2+ in nerve terminals (Erulkar and Rahamimoff, 1978) and because MEPP frequency was reduced in the presence of NO. We monitored the resting fluorescence of living nerve terminals in 11 experiments in control and during bath application of SNAP (50–100 μm). There was no change in the fluorescence of nerve terminals in the presence of the NO donor where the baseline fluorescence was 29 ± 5 pixel values in control and 31 ± 6 pixel values after 30 min of perfusion with SNAP (50–100 μm) (p > 0.05, Student's one-tail paired t test; n = 11). It is unlikely that the lack of effect was caused by a lack of sensitivity because we reported, in similar conditions, small significant reduction in resting levels of Ca2+ as a consequence of a reduced Ca2+ gradient created by chelating extracellular Ca2+ with EGTA (Robitaille et al., 1999). Therefore, the effects of the NO donors on MEPP frequency cannot be explained by a reduction in the level of resting Ca2+ of nerve terminals. However, because only bulk Ca2+ was monitored, local differences in Ca2+ concentration near active zones cannot be excluded.
Do guanylate cyclase–cGMP-dependent mechanisms modulate transmitter release?
The main mode of action of NO that has been reported is the activation of a soluble guanylate cyclase and the production of cGMP (Schmidt et al., 1993). First, we considered whether a functional cGMP pathway was present and whether its activation could mimic the effects of NO. We monitored the effects of 8-Br-cGMP, a cell-permeable cGMP analog (Meyer and Miller, 1974), on synaptic transmission. Similar to SNAP, 8-Br-cGMP (100 μm) decreased EPP amplitude by 26 ± 4% (Fig.4A; 4.80 ± 0.77 mV in control vs 3.63 ± 0.25 mV in 8-Br-cGMP; p< 0.01, Student's one-tail paired t test;n = 6).
Fig. 4.

NO activates the soluble guanylate cyclase–cGMP-dependent pathway. A, Changes in EPP amplitude before and during bath application of 8-Br-cGMP (100 μm), a cell-permeable cGMP analog. The horizontal bar represents the period of exposure to the cGMP analog. Similar results were obtained in six experiments. B, Changes in EPP amplitude before and during bath application of LY-83583 (40 μm), a soluble guanylate cyclase antagonist. Thehorizontal bar represents the period of exposure to LY-83583. Note the rise in EPP amplitude in the presence of the soluble guanylate cyclase antagonist. Similar results were obtained in 14 experiments. C, Left, Histogram of the mean ± SEM of MEPP frequency in control (open bar) and after a 30 min exposure to LY-83583 (filled bar) (*p < 0.05; Student's one-tail pairedt test). Right, Histogram of the mean ± SEM of MEPP amplitude in control (open bar) and after a 30 min exposure to LY-83583 (filled bar). In six experiments, LY-83583 raised MEPP frequency and had no effects on MEPP amplitude. D, Changes in EPP amplitude in control and during bath application of LY-83583 and of LY-83583 simultaneously with SNAP. The horizontal barsrepresent exposure to the different drugs. In six experiments, LY-83583 (40 μm) increased EPP amplitude, and subsequent application of SNAP (100 μm) had no effect.
Because the presumed target of NO is a soluble guanylate cyclase, we used LY-83583, an inhibitor of that enzyme (Mülsch et al., 1988), and monitored its effects on synaptic transmission. As shown in Figure4B, bath application of LY-83583 (40 μm) increased EPP amplitude by 44 ± 9% (4.34 ± 0.75 mV in control vs 5.76 ± 0.84 mV in LY-83583;p < 0.001, Student's one-tail paired ttest; n = 14). As shown in Figure 4C, the effects of LY-83583 were presynaptic because MEPP frequency increased from 3.83 ± 0.79 Hz in control up to 7.26 ± 1.71 Hz in the presence LY-83583 (40 μm; p < 0.01, Student's one-tail paired t test; n = 6), whereas MEPP amplitude did not change (62 ± 24 μV in control vs 631 ± 138 μV in LY-83583; p > 0.05, Student's one-tail paired t test; n = 6).
These results indicate that there is a guanylate cyclase–cGMP pathway effective at the frog nmj. Similar to NO effects, activating that pathway reduced transmitter release, whereas blocking it increased transmitter release similar to the blockade of NOS activity with 3Br7NiNa. Also, these results strongly suggest that there is a tonic activity of that pathway at the frog nmj.
Is NO activating guanylate cyclase–cGMP-dependent pathways?
If the effects of NO on transmitter release were mediated by the activation of a soluble guanylate cyclase, the presence of LY-83583 should occlude the effects of SNAP. LY-83583 (40 μm) was first perfused for 30 min to reach a stable and maximal effect on EPP amplitude, and SNAP (100 μm) was then perfused along with LY-83583 (40 μm). As shown in Figure4D, the presence of LY-83583 prevented the effects of SNAP while EPP amplitude was reduced only by 4 ± 3% (4.12 ± 0.46 mV in LY-83583 vs 3.95 ± 0.44 mV in LY-83583 and SNAP;p > 0.05, Student's one-tail paired ttest; n = 6). Therefore, these results indicate that a guanylate cyclase–cGMP pathway mediated the effects of NO on EPP amplitude.
Does NO affect high-frequency-induced depression?
Because exogenous application of NO caused a depression of synaptic transmission and because NO is endogenously produced at the frog nmj, we considered whether NO would be involved in the depression induced by high-frequency (>10 Hz) stimulation of the motor nerve (Meriney and Grinnell, 1991; Robitaille, 1998).
We first tested the effects of the NO chelator hemoglobin on high-frequency-induced depression. If NO was indeed implicated in that form of depression, the amount of synaptic depression would be reduced in the presence of the NO scavenger. As shown in Figure5, A and B, the presence of hemoglobin (30 μm) in the perfusion reduced the amount of depression from 63 ± 6% (control) down to 49 ± 9% (p < 0.05, Student's one-tail paired t test; n = 6). Therefore, these results suggest the existence of an endogenous NO production that modulates the high-frequency-induced depression at the frog nmj.
Fig. 5.

Regulation of frequency-induced synaptic depression by endogenous NO. A, Changes in EPP amplitude expressed as a percentage of control before, during, and after high-frequency stimulation (10 Hz; horizontal bar) of the motor nerve in the absence (Ctrl) and 30 min after bath perfusion of hemoglobin (30 μm;Hemo). Similar results were obtained in six experiments.B, Histogram showing the average amount of depression in control (63 ± 6%; open bar) and with hemoglobin (49 ± 9%; filled bar). In six experiments, hemoglobin significantly reduced the proportion of synaptic depression (*p < 0.05, Student's one-tail pairedt test). C, Changes in EPP amplitude expressed as a percentage of control before, during, and after high-frequency stimulation (10 Hz; horizontal bar) of the motor nerve in the absence (Ctrl) and 30 min after bath perfusion of LY-83583 (40 μm;LY-83583). Similar results were obtained in five experiments. D, Histogram showing the average amount of depression in control (40 ± 11%; open bar) and with LY-83583 (52 ± 12%; filled bar). In five experiments, LY-83583 significantly raised the proportion of synaptic depression (*p < 0.05, Student's one-tail pairedt test).
Because the guanylate cyclase inhibitor mimicked the effects of the NO scavenger, one would expect that it should have effects on synaptic depression similar to those of the NO scavenger. To test whether the modulation of high-frequency-induced depression by NO was mediated via a cGMP-dependent mechanism, we tested the effects of LY-83583 on this form of depression. The presence of LY-83583 in the perfusion should mimic the effects of hemoglobin if NO was modulating this synaptic depression via the activation of a soluble guanylate cyclase. However, as shown in Figure 5, C and D, there was no reduction in the amount of depression in the presence of LY-83583. In fact, the amount of depression was significantly higher in all experiments; it increased from 40 ± 11% in control up to 52 ± 12% in the presence of LY-83583 (40 μm) (p < 0.01, Student's one-tail pairedt test; n = 5). This increase in depression may be attributed to a larger level of transmitter release produced by the guanylate cyclase inhibitor. Indeed, depression is known to be more pronounced when the level of transmitter release is high (Zucker, 1989); an increase of 30% in transmitter release results in an increase in depression by ∼15% (D. Papas and R. Robitaille, unpublished observations). Hence, these results indicate that high-frequency depression is modulated by NO via a guanylate cyclase-independent mechanism.
Is NO implicated in adenosine-induced depression?
Another form of synaptic depression at the nmj is mediated by adenosine (Silinsky, 1984) and develops slowly at moderate rates of stimulation (∼2 Hz) (Redman and Silinsky, 1994). Interestingly, as seen in NO-induced depression of transmitter release, it was shown recently that the adenosine-dependent depression does not affect Ca2+ entry or resting [Ca2+] in the nerve terminal (Robitaille et al., 1999). Moreover, adenosine was also shown to regulate the level of depression elicited by high-frequency stimulation (Meriney and Grinnell, 1991), and there is evidence that adenosine stimulates NO production in endothelial cells (Li et al., 1995).
The level of synaptic depression induced by adenosine was first determined. In control experiments in which adenosine (10 μm) was applied alone without any other treatment, a reduction of 53 ± 3% in transmitter release was observed (5.77 ± 0.79 mV in control vs 2.78 ± 0.45 mV in adenosine;p < 0.001, Student's one-tail paired ttest; n = 10). This is consistent with values reported previously in the literature (Silinsky, 1984; Redman and Silinsky, 1994; Robitaille et al., 1999). If NO-dependent mechanisms were involved in the adenosine-induced depression, their activation would occlude the effects of subsequent application of adenosine on transmitter release. As shown in Figure6A, bath application of adenosine (10 μm) after a 30 min perfusion with the NO donor SNAP (50 μm) still significantly reduced transmitter release (p < 0.01, Student's one-tail paired t test; n = 6), whereas EPP amplitude was reduced by 42 ± 4% (2.13 ± 0.49 mV in control vs 1.20 ± 0.29 mV in adenosine). However, the adenosine-induced depression after bath application of SNAP was significantly smaller than the one observed when adenosine was applied alone (Fig. 6C) (p < 0.05, ANOVA), suggesting that part of the adenosine-induced depression was occluded by the presence of the NO donor.
Fig. 6.

Involvement of NO in adenosine-induced depression.A, Changes in EPP amplitude expressed as a percentage of control amplitude before, during, and after bath perfusion with adenosine (10 μm; Ado; horizontal bar) after an application of SNAP (50 μm) for 30 min. The gray horizontal bar represents the mean ± 1 SEM obtained when adenosine was applied alone, with no previous treatment. Similar results were obtained in six experiments.B, Changes in EPP amplitude expressed as a percentage of control amplitude before, during, and after bath perfusion of adenosine (10 μm; Ado; horizontal bar) together with LY-83583 (40 μm) after an application of LY-83583 for 30 min. The gray horizontal bar represents the mean ± 1 SEM obtained when adenosine was applied alone, with no previous treatment. C, Histogram showing the average of adenosine-induced depression expressed as a percentage of control EPP amplitude and obtained in the three conditions tested. The amount of adenosine (Ado) depression was significantly reduced when the NO-dependent mechanisms were activated (Ado after SNAP) or when the cGMP-dependent cascade was blocked (Ado with LY-83583) (*p < 0.05, ANOVA test).
We next investigated whether guanylate cyclase-dependent mechanisms regulate adenosine-induced depression. This was tested by monitoring the effects of adenosine after bath application of LY-83583 (40 μm). As shown in Figure 6B, adenosine significantly reduced EPP amplitude in the presence of LY-83583 by 42 ± 2% (6.37 ± 1.73 mV in control vs 3.63 ± 0.95 mV in adenosine and LY-83583; p < 0.05, Student's one-tail paired t test; n = 4). However, this reduction in transmitter release was significantly smaller in the presence of the guanylate cyclase inhibitor than when adenosine was applied alone (Fig. 6C) (p < 0.05, ANOVA). These results suggest that adenosine-dependent depression was partially regulated by NO via guanylate cyclase-dependent mechanisms.
DISCUSSION
Here we report that NO reduces the amount of neurotransmitter released via a cGMP-dependent pathway and that a tonic production of NO occurs at this synapse. This regulation cannot be accounted for by a global reduction in Ca2+ entry in nerve terminals. High-frequency-induced depression is partially modulated by NO possibly via cGMP-independent mechanisms, whereas adenosine-induced depression is also modulated by NO but via cGMP-dependent mechanisms. Hence, NO is an important endogenous regulator of synaptic efficacy at the adult amphibian nmj.
NO reduces synaptic efficacy at the amphibian synapse
The reduction in synaptic transmission by NO was consequent to a reduction in neurotransmitter release because the NO donors SNAP and SNP reduced EPP amplitude and MEPP frequency without affecting MEPP amplitude and time course. This is consistent with the data on cultured immature frog nmj (Wang et al., 1995). However, Lindgren and Laird (1994) reported that SNP reduced transmitter release without affecting MEPP frequency at a mature nmj of the sartorius muscle. An interesting explanation for the difference between their results and those reported here might be related to the properties of the nmjs in which the nmjs of the sartorius muscle are weaker (i.e., release less neurotransmitter per nerve terminal) than are those of the cutaneus pectoris (Grinnell and Herrera, 1980).
Our results strongly suggest that the effects observed in our study are related to NO-dependent mechanisms. Indeed, different NO donors produced identical effects on the same preparation, suggesting that the effects were not caused by other by-products produced by the different donors. In addition, drugs that prevented NO effects or the activation of the target of NO (i.e., NO scavenger, NOS inhibitor, and the soluble guanylate cyclase inhibitor) increased transmitter release. Finally, the effects of hemoglobin were occluded by the action of the NOS inhibitor, whereas inhibition of guanylate cyclase prevented NO-induced depression during SNAP application.
Lindgren and Laird (1994) showed that a brief exposure to SNP depressed synaptic transmission for as long as 60 min. We also observed long-lasting effects on synaptic transmission even after SNAP removal from the perfusion. This persistent effect of NO is not consistent with our finding that hemoglobin, 3Br7NiNa, and LY-83583 increased EPP amplitude or with the fact that hemoglobin also increased EPP amplitude in the sartorius nmj (Lindgren and Laird, 1994). Interestingly, in cultured nmjs (Wang et al., 1995), the long-lasting NO effects are induced after exposure for a period of 20 min, suggesting that a concentration and/or duration range of NO exposures is required to produce the prolonged effects. Also, it is likely that the release of endogenous NO differs from a bath with NO donors in terms of concentration and duration.
NO regulation of Ca2+ entry in nerve terminals
The NO donor did not affect Ca2+responses evoked by single action potentials but increased the responses induced by brief trains of stimuli, suggesting that NO modulates a frequency-dependent Ca2+mechanism. Potential targets of NO might be the sarcoplasmic Ca2+/ATPase pump that is known to beS-nitrosylated by NO (Ishii et al., 1998) and the IP3 receptor known to be phosphorylated by PKGs (Haug et al., 1999). Neurotransmitter release at the frog nmj is controlled by these two Ca2+ regulatory mechanisms (Castonguay and Robitaille, 2001). Also, there is evidence that NO facilitates N-type Ca2+channel activation via a cGMP–PKG pathway (Hirooka et al., 2000). These channels are clustered at active zones of the frog nmj, regulating transmitter release (Robitaille et al., 1990; Cohen et al., 1991). Regardless of the mechanisms regulating Ca2+ entry, NO reduction of transmitter release at the amphibian nmj cannot be explained by a reduction in Ca2+ entry, suggesting that a regulation occurs after that step (Gray et al., 1999). Our results further suggest that the reduction in transmitter release by NO at a higher frequency of stimulation is underestimated because of the potentiation of Ca2+ entry by NO in those conditions.
Guanylate cyclase-dependent and -independent NO regulation of transmitter release
The activation of a soluble guanylate cyclase leading to the production of cGMP seems to be the main mechanism by which NO modulates transmitter release during low activity. This mechanism also seems to be involved in the regulation of transmitter release at the immature nmj (Wang et al., 1995). At the frog nmj, NO-induced production of cGMP could activate cGMP-dependent protein kinase (PKG). This possibility is supported by the evidence that PKG is present together with NOS at the rat nmj (Chao et al., 1997) and that there is a PKG-dependent regulation of transmitter release at a number of synapses (Gray et al., 1999; Yawo, 1999) including the frog nmj (Branisteanu et al., 1988).
NO regulation of high-frequency- and adenosine-induced synaptic depression
Our results indicate that depression of transmitter release induced by repetitive stimulation at high frequency is modulated by NO as suggested by the reduction in the amount of depression in the presence of the NO scavenger. This is the first direct evidence that the production and release of NO, as shown by the partial occlusion with an NO scavenger, are directly involved in the modulation of synaptic depression at the nmj.
Because transmitter release is similarly sensitive to the NO scavenger and the guanylate cyclase inhibitor, the effect of the latter on synaptic depression should have been also similar. However, the guanylate cyclase inhibitor did not reduce depression but rather increased it. Hence, this suggests that the NO regulation of synaptic depression occurs in a guanylate cyclase-independent manner. The increase in depression may be caused by the fact that depression is larger when transmitter release is increased (Zucker, 1989) (Pappas and Robitaille, unpublished observations). However, a guanylate cyclase regulation of depression, unrelated to NO, cannot be excluded. In agreement with the hypothesis that synaptic depression is caused by a reduced availability of the vesicular pool (Zucker, 1989), NO could induce post-translational changes of synaptic proteins involved in the SNAP/soluble NSF attachment protein receptor complex (Meffert et al., 1994, 1996), leading to a regulation of the exocytosis–endocytosis cycle.
Unlike high-frequency-induced depression, adenosine-induced depression appears modulated by NO in a guanylate cyclase-dependent manner as suggested by the partial occlusion of adenosine effects by an NO donor and the guanylate cyclase inhibitor. Interestingly, Hirsh et al. (1990)reported that the adenosine depression was only partially occluded by the presence of cAMP analogs. Hence, one possibility might be that part of the effects of adenosine is mediated by an NO-dependent activation of the guanylate cyclase–cGMP cascade. Alternatively, NO may regulate adenosine-induced depression by acting on the cAMP-dependent system perhaps via the action of cGMP on phosphodiesterase activity (Doerner and Alger, 1988).
Hence, our results suggest that there are two NO-dependent functional mechanisms at the frog nmj, each acting within a certain frequency range: the regulation of NO at a low level of transmitter release appears to occur via guanylate cyclase-dependent mechanisms, whereas NO regulation at a higher level of transmitter release appears to occur via cGMP-independent mechanisms. Interestingly, both PKG (cGMP)-dependent and -independent NO regulations of glutamate release have been reported for rat hippocampal nerve terminals; the former is associated with a low level of NO production, and the latter is associated with a high level of NO (Sequeira et al., 1999).
Model of NO regulation of synaptic efficacy at the nmj
Our results indicate the presence of guanylate cyclase-dependent and -independent regulation of transmitter release in a frequency-dependent manner, and the use of the NO chelator hemoglobin and the NOS inhibitor 3Br7NiNa indicates that NO is produced tonically. In addition, there is evidence that an NOS is located in the muscle fiber and is concentrated at the end plate region (Brenman et al., 1995) and in PSCs, glial cells at the nmj (Descarries et al., 1998). The muscular NO seems critical for the consolidation of the synapse (Wang et al., 1995). On the basis of this knowledge, we propose the following model of NO modulation of synaptic efficacy at the nmj in which the tonic production of NO would originate from the muscle fibers to serve as a feedback signal for synapse maintenance (Fig.7A). The tonic NO production would also reduce the sensitivity of PSCs to neurotransmitters because NO application reduced the Ca2+ responses elicited in PSCs by transmitter substances (Descarries et al., 1998). In addition, this tonic production of NO by the muscle fibers would modulate transmitter release at a low level of activity. It is unlikely, in our experimental conditions, that muscle fibers produced NO in an activity-dependent manner because muscle activity was quite low owing to the partial blockade of postsynaptic receptors whereas the PSCs were fully activated because they are unaffected by nicotinic antagonists (Jahromi et al., 1992; Robitaille et al., 1997). Hence, although a tonic glial NO production cannot be excluded, we propose that the activity-induced, Ca2+-dependent NO production originates from the PSCs (Fig. 7B) because their intracellular Ca2+ elevation is frequency dependent (Jahromi et al., 1992; Robitaille, 1995; Bourque and Robitaille, 1998). That rise in Ca2+would then activate neuronal NOS present in PSCs. This glial NO production could mediate the PSC-mediated modulation of synaptic depression at the amphibian nmj (Robitaille, 1998; Castonguay et al., 2001).
Fig. 7.
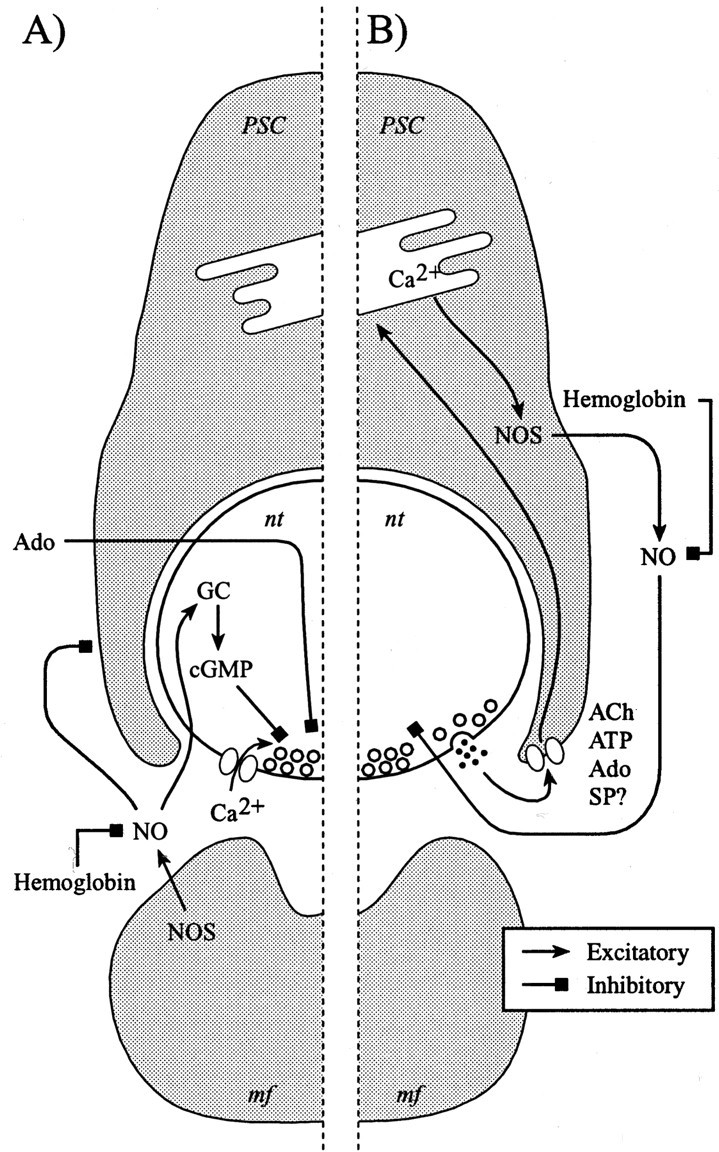
Model of differential frequency-dependent regulation of transmitter release by NO. The diagram of a cross section of a frog nmj depicts a PSC covering the nerve terminal facing a muscle fiber. A, Model of the tonic action of NO at the amphibian nmj, where we propose that the tonic production of NO originates from the muscle fibers. This NO modulates transmitter release via a cGMP-dependent pathway, whereas it may keep the sensitivity of PSCs for various neurotransmitters in a reduced state. The adenosine-induced depression is also regulated by the cGMP-dependent pathway that is consistent with the fact that this form of depression occurs during a low level of transmitter release.B, Proposed model of the activity-dependent production of NO by the PSCs. This would occur as a consequence of the activation of the PSCs during prolonged and repetitive stimulation that would trigger the release of Ca2+ from internal stores and the activation of the neuronal type of NOS present in PSCs. This NO would then be modulating the release of neurotransmitter in a cGMP-independent manner, perhaps via direct protein modification that would alter the availability of the synaptic vesicles for exocytosis or endocytosis. GC, Guanylate cyclase;mf, muscle fiber; nt, nerve terminal;SP, substance P.
Footnotes
This work was supported by Medical Research Council of Canada Grant MT14137, by awards from the EJLB Research Foundation and The Alfred P. Sloan Foundation, and by a team grant from Fonds pour la Formation de Chercheurs et de l'aide à la Recherche to R.R. S.T. was supported by a studentship from the Medical Research Council of Canada, and R.R. was a Junior II Scholar from the Fonds de la Recherche en Santé du Québec and a Medical Research Council scientist.
Correspondence should be addressed to Dr. Richard Robitaille, Département de physiologie, P.O. Box 6128, station centre-ville, Montreal, Quebec, Canada H3C 3J7. E-mail:richard.robitaille@umontreal.ca.
REFERENCES
- 1.Adler EM, Augustine GJ, Duffy SN, Charlton MP. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci. 1991;11:1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almon RR, Appel SH. Cholinergic sites in skeletal muscle. II. Interaction of an agonist and two antagonists with the acetylcholine site. Biochemistry. 1976;15:3667–3671. doi: 10.1021/bi00662a004. [DOI] [PubMed] [Google Scholar]
- 3.Bourque MJ, Robitaille R. Endogenous peptidergic modulation of perisynaptic Schwann cells at the frog neuromuscular junction. J Physiol (Lond) 1998;512:197–209. doi: 10.1111/j.1469-7793.1998.197bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Branisteanu DD, Popescu LM, Branisteanu DD, Haulica ID. Cyclic GMP and protein kinase G inhibit the quantal transmitter release induced by protein kinase C. Brain Res. 1988;464:263–266. doi: 10.1016/0169-328x(88)90034-4. [DOI] [PubMed] [Google Scholar]
- 5.Bredt DS, Snyder SH. Nitric oxide, a novel neuronal messenger. Neuron. 1992;8:3–11. doi: 10.1016/0896-6273(92)90104-l. [DOI] [PubMed] [Google Scholar]
- 6.Brenman JE, Bredt DS. Synaptic signaling by nitric oxide. Curr Opin Neurobiol. 1997;7:374–378. doi: 10.1016/s0959-4388(97)80065-7. [DOI] [PubMed] [Google Scholar]
- 7.Brenman JE, Chao DS, Xia H, Aldape K, Bredt DS. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743–752. doi: 10.1016/0092-8674(95)90471-9. [DOI] [PubMed] [Google Scholar]
- 8.Calabresi P, Gubellini P, Centonze D, Sancesario G, Morello M, Giorgi M, Pisani A, Bernardi G. A critical role of the nitric oxide/cGMP pathway in corticostriatal long-term depression. J Neurosci. 1999;19:2489–2499. doi: 10.1523/JNEUROSCI.19-07-02489.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castonguay A, Robitaille R (2001) Differential regulation of transmitter release by presynaptic and glial Ca2+ internal stores at the neuromuscular synapse. J Neurosci, in press. [DOI] [PMC free article] [PubMed]
- 10.Castonguay A, Levesque S, Robitaille R (2001) Dynamic contributions of glial cells to synaptic function. Prog Brain Res, in press. [DOI] [PubMed]
- 11.Chao DS, Silvagno F, Xia H, Cornwell TL, Lincoln TM, Bredt DS. Nitric oxide synthase and cyclic GMP-dependent protein kinase concentrated at the neuromuscular endplate. Neuroscience. 1997;76:665–672. doi: 10.1016/s0306-4522(96)00367-3. [DOI] [PubMed] [Google Scholar]
- 12.Chapman V, Buritova J, Honore P, Besson JM. 7-Nitro-indazole, a selective inhibitor of neuronal nitric oxide synthase reduces formalin evoked c-Fos expression in dorsal horn neurons of the rat spinal cord. Brain Res. 1995;697:258–261. doi: 10.1016/0006-8993(95)00973-t. [DOI] [PubMed] [Google Scholar]
- 13.Cohen MW, Jones OT, Angelides KJ. Distribution of Ca2+ channels on the frog nerve terminals revealed by fluorescent ω-conotoxin. J Neurosci. 1991;11:1032–1039. doi: 10.1523/JNEUROSCI.11-04-01032.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Descarries LM, Cai S, Robitaille R. Localization and characterization of nitric oxide synthase at the frog neuromuscular junction. J Neurocytol. 1998;27:829–840. doi: 10.1023/a:1006907531778. [DOI] [PubMed] [Google Scholar]
- 15.Doerner D, Alger BE. Cyclic GMP depresses hippocampal Ca2+ current through a mechanism independent of cGMP-dependent protein kinase. Neuron. 1988;1:693–699. doi: 10.1016/0896-6273(88)90168-7. [DOI] [PubMed] [Google Scholar]
- 16.Dryden WF, Harvey AL, Marshall IG. Pharmacological studies on the bungarotoxins. Eur J Pharmacol. 1974;26:256–267. doi: 10.1016/0014-2999(74)90235-0. [DOI] [PubMed] [Google Scholar]
- 17.Duman RS, Terwilliger RZ, Nestler EJ. Alterations in nitric oxide-stimulated endogenous ADP-ribosylation associated with long-term potentiation in rat hippocampus. J Neurochem. 1993;61:1542–1545. doi: 10.1111/j.1471-4159.1993.tb13652.x. [DOI] [PubMed] [Google Scholar]
- 18.Erulkar SD, Rahamimoff R. The role of calcium ions in tetanic and post-tetanic increase of miniature end-plate potential frequency. J Physiol (Lond) 1978;278:501–511. doi: 10.1113/jphysiol.1978.sp012320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gray DB, Polo-Parada L, Pilar GR, Eang P, Metzger RR, Klann E, Meriney SD. A nitric oxide/cyclic GMP-dependent protein kinase pathway alters transmitter release and inhibition by somatostatin at a site downstream of calcium entry. J Neurochem. 1999;72:1981–1990. doi: 10.1046/j.1471-4159.1999.0721981.x. [DOI] [PubMed] [Google Scholar]
- 20.Grinnell AD, Herrera AA. Physiological regulation of synaptic effectiveness at frog neuromuscular junctions. J Physiol (Lond) 1980;307:301–317. doi: 10.1113/jphysiol.1980.sp013436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haug LS, Jensen V, Hvalby O, Walaas SI, Ostvold AC. Phosphorylation of the inositol 1,4,5-trisphosphate receptor by cyclic nucleotide-dependent kinases in vitro and in rat cerebellar slices in situ. J Biol Chem. 1999;274:7467–7473. doi: 10.1074/jbc.274.11.7467. [DOI] [PubMed] [Google Scholar]
- 22.Hess DT, Patterson SI, Smith DS, Skene JHP. Neuronal growth cone collapse and inhibition of protein fatty acylation by nitric oxide. Nature. 1993;366:562–565. doi: 10.1038/366562a0. [DOI] [PubMed] [Google Scholar]
- 23.Hirooka K, Kourennyi DE, Barnes S. Calcium channel activation facilitated by nitric oxide in retinal ganglion cells. J Neurophysiol. 2000;83:198–206. doi: 10.1152/jn.2000.83.1.198. [DOI] [PubMed] [Google Scholar]
- 24.Hirsh JK, Silinsky EM, Solsona CS. The role of cyclic AMP and its protein kinase in mediating acetylcholine release and the action of adenosine at frog motor nerve endings. Br J Pharmacol. 1990;101:311–318. doi: 10.1111/j.1476-5381.1990.tb12707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ignarro LJ, Lippton H, Edwards JC, Baricos WH, Hyman AL, Kadowitz PJ, Gruetter CA. Mechanism of vascular smooth muscle relaxation by organic nitrates, nitrites, nitroprusside and nitric oxide: evidence for the involvement of S-nitrosothiols as active intermediates. J Pharmacol Exp Ther. 1981;218:739–749. [PubMed] [Google Scholar]
- 26.Illes P. Mechanisms of receptor-mediated modulation of transmitter release in noradrenergic, cholinergic and sensory neurones. Neuroscience. 1986;17:909–928. doi: 10.1016/0306-4522(86)90071-0. [DOI] [PubMed] [Google Scholar]
- 27.Ishii T, Sunami O, Saitoh N, Nishio H, Takeuchi T, Hata F. Inhibition of skeletal muscle sarcoplasmic reticulum Ca2+-ATPase by nitric oxide. FEBS Lett. 1998;440:218–222. doi: 10.1016/s0014-5793(98)01460-4. [DOI] [PubMed] [Google Scholar]
- 28.Izumi Y, Zorumski CF. Involvement of nitric oxide in low glucose-mediated inhibition of hippocampal long-term potentiation. Synapse. 1997;25:258–262. doi: 10.1002/(SICI)1098-2396(199703)25:3<258::AID-SYN4>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 29.Jahromi BS, Robitaille R, Charlton MP. Transmitter release increases intracellular calcium in perisynaptic Schwann cells in situ. Neuron. 1992;8:1069–1077. doi: 10.1016/0896-6273(92)90128-z. [DOI] [PubMed] [Google Scholar]
- 30.Katz B, Miledi R. A study of synaptic transmission in the absence of nerve impulses. J Physiol (Lond) 1967;192:407–436. doi: 10.1113/jphysiol.1967.sp008307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kusner LL, Kaminski HJ. Nitric oxide synthase is concentrated at the skeletal muscle endplate. Brain Res. 1996;730:238–242. doi: 10.1016/0006-8993(96)00675-0. [DOI] [PubMed] [Google Scholar]
- 32.Lev-Ram V, Jiang T, Wood J, Lawrence DS, Tsien RY. Synergies and coincidence requirements between NO, cGMP, and Ca2+ in the induction of cerebellar long-term depression. Neuron. 1997;18:1025–1038. doi: 10.1016/s0896-6273(00)80340-2. [DOI] [PubMed] [Google Scholar]
- 33.Li J-M, Fenton RA, Cutler BS, Dobson JG., Jr Adenosine enhances nitric oxide production by vascular endothelial cells. Am J Physiol. 1995;269:C519–C523. doi: 10.1152/ajpcell.1995.269.2.C519. [DOI] [PubMed] [Google Scholar]
- 34.Lindgren CA, Laird MV. Nitroprusside inhibits neurotransmitter release at the frog neuromuscular junction. NeuroReport. 1994;5:2205–2208. doi: 10.1097/00001756-199410270-00054. [DOI] [PubMed] [Google Scholar]
- 35.Lipton SA, Choi Y-B, Pan Z-H, Lei SZ, Chen H-SV, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 36.Malen PL, Chapman PF. Nitric oxide facilitates long-term potentiation, but not long-term depression. J Neurosci. 1997;17:2645–2651. doi: 10.1523/JNEUROSCI.17-07-02645.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meffert MK, Premack BA, Chulman H. Nitric oxide stimulates Ca2+-independent synaptic vesicle release. Neuron. 1994;12:1235–1244. doi: 10.1016/0896-6273(94)90440-5. [DOI] [PubMed] [Google Scholar]
- 38.Meffert MK, Calakos NC, Scheller RH, Schulman H. Nitric oxide modulates synaptic vesicle docking/fusion reactions. Neuron. 1996;16:1229–1236. doi: 10.1016/s0896-6273(00)80149-x. [DOI] [PubMed] [Google Scholar]
- 39.Meriney SD, Grinnell AD. Endogenous adenosine modulates stimulation-induced depression at the frog neuromuscular junction. J Physiol (Lond) 1991;443:441–455. doi: 10.1113/jphysiol.1991.sp018843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyer RB, Jr, Miller JP. Analogs of cyclic AMP and cyclic GMP: general methods of synthesis and the relationship of structure to enzymatic activity. Life Sci. 1974;14:1019–1040. doi: 10.1016/0024-3205(74)90228-8. [DOI] [PubMed] [Google Scholar]
- 41.Mülsch A, Busse R, Liebau S, Förstermann U. LY 83583 interferes with the release of endothelium-derived relaxing factor and inhibits soluble guanylate cyclase. J Pharmacol Exp Ther. 1988;247:283–288. [PubMed] [Google Scholar]
- 42.Murad F, Mittal CK, Arnold WP, Katsuki S, Kimura H. Guanylate cyclase: activation by azide, nitro compounds, nitric oxide and hydroxyl radical and inhibition by hemoglobin and myoglobin. Adv Cyclic Nucleotide Res. 1978;9:145–158. [PubMed] [Google Scholar]
- 43.Okuda S, Kanda F, Kawahara Y, Chihara K. Regulation of inducible nitric oxide synthase expression in L6 rat skeletal muscle cells. Am J Physiol. 1997;272:C35–C40. doi: 10.1152/ajpcell.1997.272.1.C35. [DOI] [PubMed] [Google Scholar]
- 44.Redman RS, Silinsky EM. ATP released together with acetylcholine as the mediator of neuromuscular depression at the frog nerve endings. J Physiol (Lond) 1994;477:117–127. doi: 10.1113/jphysiol.1994.sp020176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robitaille R. Purinergic receptors and their activation by endogenous purines at perisynaptic glial cells of the frog neuromuscular junction. J Neurosci. 1995;15:7121–7131. doi: 10.1523/JNEUROSCI.15-11-07121.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robitaille R. Modulation of synaptic efficacy and synaptic depression by glial cells at the frog neuromuscular junction. Neuron. 1998;21:847–855. doi: 10.1016/s0896-6273(00)80600-5. [DOI] [PubMed] [Google Scholar]
- 47.Robitaille R, Adler EM, Charlton MP. Strategic location of calcium channels at transmitter release sites of frog neuromuscular synapses. Neuron. 1990;5:773–779. doi: 10.1016/0896-6273(90)90336-e. [DOI] [PubMed] [Google Scholar]
- 48.Robitaille R, Jahromi BS, Charlton MP. Muscarinic Ca2+ responses resistant to muscarinic antagonists at perisynaptic Schwann cells of the frog neuromuscular junction. J Physiol (Lond) 1997;504:337–347. doi: 10.1111/j.1469-7793.1997.337be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robitaille R, Thomas S, Charlton MP. Effects of adenosine on Ca2+ entry in the nerve terminal of the frog neuromuscular junction. Can J Physiol Pharmacol. 1999;77:707–714. [PubMed] [Google Scholar]
- 50.Schmidt HH, Walter U. NO at work. Cell. 1994;78:919–925. doi: 10.1016/0092-8674(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 51.Schmidt HH, Lohmann SM, Walter U. The nitric oxide and cGMP signal transduction system: regulation and mechanism of action. Biochim Biophys Acta. 1993;1178:153–175. doi: 10.1016/0167-4889(93)90006-b. [DOI] [PubMed] [Google Scholar]
- 52.Sequeira SM, Carvalho AP, Carvalho CM. Both protein kinase G dependent and independent mechanisms are involved in the modulation of glutamate release by nitric oxide in rat hippocampal nerve terminals. Neurosci Lett. 1999;261:29–32. doi: 10.1016/s0304-3940(98)01002-7. [DOI] [PubMed] [Google Scholar]
- 53.Silinsky EM. On the mechanism by which adenosine receptor activation inhibits the release of acetylcholine from motor nerve endings. J Physiol (Lond) 1984;346:243–256. doi: 10.1113/jphysiol.1984.sp015019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Silvagno F, Xia H, Bredt DS. Neuronal nitric oxide synthase-μ, an alternatively spliced isoform expressed in differentiated skeletal muscle. J Biol Chem. 1996;271:11201–11208. doi: 10.1074/jbc.271.19.11204. [DOI] [PubMed] [Google Scholar]
- 55.Southam E, Garthwaite J. Comparative effects of some nitric oxide donors on cyclic GMP levels in rat cerebellar slices. Neurosci Lett. 1991;130:107–111. doi: 10.1016/0304-3940(91)90239-p. [DOI] [PubMed] [Google Scholar]
- 56.Wang T, Xie Z, Lu B. Nitric oxide mediates activity-dependent synaptic suppression at developing neuromuscular synapses. Nature. 1995;374:262–266. doi: 10.1038/374262a0. [DOI] [PubMed] [Google Scholar]
- 57.Wegener G, Volke V, Rosenberg R. Endogenous nitric oxide decreases hippocampal levels of serotonin and dopamine in vivo. Br J Pharmacol. 2000;130:575–580. doi: 10.1038/sj.bjp.0703349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- 59.Yawo H. Involvement of cGMP-dependent protein kinase in adrenergic potentiation of transmitter release from the Calyx-type presynaptic terminal. J Neurosci. 1999;19:5293–5300. doi: 10.1523/JNEUROSCI.19-13-05293.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zucker R. Short term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–32. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]
- 61.Zucker RS. Calcium and transmitter release. J Physiol (Paris) 1993;87:25–36. doi: 10.1016/0928-4257(93)90021-k. [DOI] [PubMed] [Google Scholar]