

Abstract
Reactive microglia associated with the β-amyloid plaques in Alzheimer's disease (AD) brains initiate a sequence of inflammatory events integral to the disease process. We have observed that fibrillar β-amyloid peptides activate a tyrosine kinase-based signaling response in primary mouse microglia and the human monocytic cell line, THP-1, resulting in production of neurotoxic secretory products, proinflammatory cytokines, and reactive oxygen species. We report that most of the amyloid-induced tyrosine kinase activity was stimulated after activation of Src family members such as Lyn. However, transduction of the signaling response required for increased production of the cytokines TNFα and IL1-β was mediated by the nonreceptor tyrosine kinase, Syk. Additionally, β-amyloid stimulated an NFκB-dependent pathway in parallel that was required for cytokine production. Importantly, TNFα generated by the monocytes and microglia was responsible for the majority of the neuorotoxic activity secreted by these cells after β-amyloid stimulation but must act in concert with other factors elaborated by microglia to elicit neuronal death. Moreover, we observed that the neuronal loss was apoptotic in nature and involved increased neuronal expression of inducible nitric oxide synthase and subsequent peroxynitrite production. Selective inhibitors of inducible nitric oxide synthase effectively protected cells from toxicity associated with the microglial and monocytic secretory products. This study demonstrates a functional linkage between β-amyloid-dependent activation of microglia and several characteristic markers of neuronal death occurring in Alzheimer's disease brains.
Keywords: Alzheimer's disease, β-amyloid, microglia, THP-1 monocytes, signal transduction, tyrosine kinase, Lyn, Syk, inflammation, neurotoxicity, apoptosis, nitric oxide, nitrotyrosine, peroxynitrite, TNFα, cytokines
Alzheimer's disease (AD) is characterized by an accumulation of extracellular deposits of β-amyloid and abundant neurofibrillary tangles in the brain that is correlated with a progressive dementia and neuron loss (Berg et al., 1993; Braak and Braak, 1997). There is extensive and compelling evidence that amyloid deposition provokes a microglial-mediated inflammatory response that contributes significantly to the cell loss and cognitive decline that is characteristic of this disease (Akiyama et al., 2000). Significantly, both epidemiological and clinical trial data have demonstrated the value of anti-inflammatory therapies for lowering the incidence, slowing the progression, and reducing the symptomatic severity of AD (McGeer and Rogers, 1992; Rogers et al., 1993; Rich et al., 1995; McGeer et al., 1996; Aisen, 1997; Stewart et al., 1997; Mackenzie and Munoz, 1998).
It is now well documented that fibrillar forms of β-amyloid serve as an inflammatory stimulus for microglial lineage cells, and the signal transduction cascades mediating the effects of the amyloid peptides have been identified (Del Bo et al., 1995; Giulian et al., 1995;Klegeris et al., 1997; Lorton, 1997; McDonald et al., 1997, 1998;Bianca et al., 1999; Combs et al., 1999, 2000; Yates et al., 2000). Indeed, abundant reactive microglia and astrocytes surround the β-amyloid plaques in the AD brain (Itagaki et al., 1989; Miyazono et al., 1991; McGeer and Rogers, 1992; McGeer and McGeer, 1995; Cotman et al., 1996). Amyloid-dependent activation of microglia in vitro results in acquisition of a reactive phenotype with the production and secretion of proinflammatory products such as reactive oxygen species, cytokines, and neurotoxins. The identity of the neurotoxic agent(s) generated by reactive microglia has not been resolved (Banati et al., 1993; Giulian et al., 1995; Ii et al., 1996; Klegeris et al., 1997; Combs et al., 2000).
Despite the ambiguity of the effectors of neuron loss in the AD brain, recent data demonstrate that it is an apoptotic process, as evidenced by the presence of activated caspases and endonuclease cleaved DNA (Li et al., 1997; Selznick et al., 1999; Stadelmann et al., 1999). Furthermore, neurons in the AD brain display increased levels of markers of oxidative damage such as inducible nitric oxide synthase (iNOS) and the peroxynitrite marker, nitrotyrosine, suggesting that oxygen species may mediate neuronal apoptosis (Good et al., 1996;Vodovotz et al., 1996; Smith et al., 1997; Hensley et al., 1998). The proinflammatory cytokine, TNFα, is capable of inducing neuronal apoptosis in specific situations (Venters et al., 1999). It has been demonstrated in vitro that TNFα stimulation of neuronal cell lines leads to increased expression of inducible nitric oxide synthase and subsequent apoptosis (Ogura et al., 1997; Heneka et al., 1998). A number of recent in vitro studies have demonstrated that Aβ fibril stimulation increases microglial/monocytic TNFα production (Klegeris et al., 1997; Galimberti et al., 1999; Combs et al., 2000; Yates et al., 2000). Importantly, increased levels of TNFα have been reported in brains and plasma of AD patients (Fillit et al., 1991; Bruunsgaard et al., 1999; Tarkowski et al., 1999).
We report that fibrillar Aβ stimulation of mouse microglia and THP-1 monocytes results in a Syk kinase and NFκB-dependent production of TNFα that is responsible for increased iNOS expression, peroxynitrite production, and subsequent apoptosis in primary mouse neuronal cultures. These data establish a functional linkage between Aβ-stimulated microglial proinflammatory changes and the specific characteristics of neuron loss that occur in AD brains.
MATERIALS AND METHODS
Materials. The anti-MAP2 antibody was purchased from Sigma (St. Louis, MO). Anti-ERK2 and anti-c-fos antibodies were from Santa Cruz Biotechnologies (Santa Cruz, CA). The anti-phosphotyrosine antibody, 4G10, was from Upstate Biotechnology (Lake Placid, NY). Anti-phospho-CREB (Ser133) antibody was from New England Biolabs (Beverly, MA). Anti-active p65NLS (RelA) antibody and piceatannol were purchased from Boehringer Mannheim (Mannheim, Germany). Anti-phospho-IκBα antibody was from New England Biolabs. Anti-IκBα antibody and PP1 were purchased from Calbiochem (La Jolla, CA). Anti-active p38 and active-MAP kinase antibodies were from Promega (Madison, WI). Anti-nitrotyrosine antibody and NFkB SN-50 cell-permeable inhibitory peptide were both obtained from Biomol Research Laboratories (Plymouth Meeting, PA). Anti-human TNFα/IL-1β antibodies, mouse recombinant TNFα, and anti-mouse TNFα antibody were all purchased from R&D Systems (Minneapolis, MN). The anti-iNOS antibody as well as the specific iNOS inhibitors, 1400W.2HCl and AMT.HCl, and the neuronal nitric oxide synthase (nNOS) inhibitor Vinyl-l-NIO were purchased from Alexis Biochemicals (San Diego, CA). Affinity-purified horseradish peroxidase-conjugated goat anti-mouse and goat anti-rabbit antibodies were purchased from Amersham Pharmacia Biotech (Piscataway, NJ). FITC-conjugated goat anti-rabbit antibody was from Jackson Laboratories (Bar Harbor, ME). Peptides corresponding to amino acids 25–35 and 1–40 of human Aβ were purchased from Bachem (Philadelphia, PA). β-amyloid peptides were resuspended in sterile dH20. Fibrillar Aβ 1–40 and 25–35 peptides were prepared by reconstitution of the lyophilized peptides in sterile distilled water, followed by incubation for 1 week at 37°C. Neurobasal media and B27 supplements were purchased from Life Technologies (Rockville, MD).
Tissue culture. THP-1 cells are a monocytic cell line derived from peripheral blood of a human with acute monocytic leukemia and were purchased from the American Type Culture Collection (Manassas, VA). All experiments requiring THP-1 cells used undifferentiated cells. THP-1 cells were grown in RPMI-1640 (Whittaker Bioproducts, Walkersville, MD) supplemented with 10% heat-inactivated fetal calf serum, 5 × 10−5m 2-mercaptoethanol, 5 mmHEPES, and 2 μg/ml gentamicin in 5% CO2. Microglial and neuronal cultures were derived from postnatal day 1–2 and embryonic day (E) 16 mouse brain (C57BL/6J), respectively, as described previously (Combs et al., 1999, 2000). Neurons were grown in Neurobasal media (4.0 × 104 per 24-well tissue poly-l-lysine-coated tissue culture plate) with B27 supplement for 5 d in vitrobefore use.
Cell stimulation. THP-1 cells and microglia were stimulated as described previously (Combs et al., 1999, 2000). Briefly, monocytes and microglia were removed from their respective media and transferred to serum-free RPMI for suspension stimulation or to Neurobasal media for adherent stimulation. Adherent stimulation was performed by plating the cells onto Aβ peptides bound to the surface of the dish (48 pmol/mm2). Bound fibrillar peptides were prepared as described previously (McDonald et al., 1997). Briefly, tissue culture wells were coated with nitrocellulose, and peptides were added to the coated wells and allowed to dry, immobilizing the peptides. The use of immobilized peptides prevented their subsequent collection in the conditioned medium and transfer to the neuronal cultures, avoiding any confounds arising from the action of amyloid fibrils on neurons. THP-1 cells and microglia (2.0 × 104 cells) were added to wells containing the bound peptides in 48-well tissue culture dishes in 0.5 ml of Neurobasal media for 48 hr. The conditioned media from these cells was clarified by centrifugation and added to neuronal cultures for 72 hr with vehicle (DMSO) or selected reagents. To determine the involvement of TNFα for the neuronal death, conditioned media was incubated with anti-human or anti-mouse (5 μg/ml media) TNFα-neutralizing antibodies for 15 min, 25°C before addition to neurons for 72 hr. The participation of IL-1β was evaluated similarly using an anti-human IL-1β antibody. Additionally, recombinant mouse TNFα (100 ng/ml) was added to neurons directly or into conditioned media with and without anti-human TNFα neutralizing antibody for 72 hr. The selective NOS inhibitors, 1400W.2HCl (iNOS, 5 and 10 μm), AMT.HCl (iNOS, 10 μm) and Vinyl-l-NIO (nNOS, 20 μm) were also added to neurons in the absence or presence of conditioned media. Neurons were fixed, stained, and counted after staining of the cultures using a mouse anti-MAP2 antibody. A counting grid was placed over the wells to count neuron and astrocyte numbers from eight identical fields for each condition. The average number of neurons and astrocytes (±SEM) was calculated for each condition. Each experiment was performed in duplicate and repeated three to four times.
Western blotting. Cells were lysed in 200 μl of ice-cold RIPA buffer [1% Triton, 0.1% SDS, 0.5% deoxycholate, 20 mm Tris, pH 7.4, 150 mmNaCl, 10 mm NaF, 1 mmNa3VO4, 1 mm EDTA, 1 mm EGTA, 0.2 mm phenylmethylsulfonyl fluoride (PMSF)], and insoluble material was removed by centrifugation at 10,000 ×g at 4°C for 10 min. Protein concentrations were quantitated by the method of Bradford (Bradford, 1976). Proteins were resolved by 7.5–9% SDS-PAGE, and the resulting Western blots were incubated with primary antibodies overnight at 4°C followed by incubation in horseradish peroxidase-conjugated secondary antibodies (1:3000, 1 hr, 25°C). Antibody binding was detected via enhanced chemiluminescence (Pierce, Rockford, IL). Blots were reprobed by stripping with 0.2N NaOH and shaking vigorously for 10 min at 25°C (Suck and Krupinska, 1996).
Immunoprecipitation. To perform immunoprecipitations, cells were lysed in 20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.5% NP-40, 1 mmEDTA, 1 mm EGTA, 0.2 mmPMSF. Cell lysates were incubated with immunoprecipitating antibody, anti-p65NLS, or anti-nitrotyrosine (1 μg antibody per milligram protein lysate), and Protein A-agarose for 2 hr at 4°C. The immunoprecipitates were washed three times in lysis buffer and then resolved by 7.5% SDS-PAGE and Western-blotted as described.
TNFα reporter assays. Luciferase reporter constructs for the human TNFα gene were transfected into THP-1 cells using DEAE-dextran together with a β-galactosidase reporter construct to control for transfection efficiency as described previously (Combs et al., 2000). The cells were transfected, and 48 hr later they were stimulated for 0–8 hr in serum-free RPMI media in the presence or absence of drugs/SN-50 peptide and fibrillar Aβ25–35 (60 μm) or Aβ1–40 (60 μm). The cells were lysed, and luciferase activity was measured and normalized to β-galactosidase activity. All assays were performed in duplicate in three separate experiments.
Immunocytochemistry. For immunocytochemistry, cells were fixed in 4% paraformaldehyde for 30 min. at 37°C. To count the numbers of surviving cells, neurons were stained with a mouse anti-MAP2 antibody (1:500). Immunoreactivity was visualized using 3,3′-diaminobenzidine tetrahydrochloride (DAB) (Vector Laboratories, Burlingame, CA). Immunodetection of iNOS and nitrotyrosine was performed using anti-iNOS (1:2000) and anti-nitrotyrosine (1:1000) antibodies and visualized using FITC-conjugated goat anti-rabbit antibody (1:500).
TUNEL protocol. After 72 hr treatment in the absence or presence of conditioned media, neurons were fixed in 4% paraformaldehyde for 30 min at 37°C and then DNAse-treated for 15 min. The terminal transferase reaction was then performed for 1 hr at 37°C to allow incorporation of biotinylated dUTP. Biotinylated dUTP was visualized using the Vector Elite ABC reagent kit according to manufacturer's protocol using DAB as the chromagen.
DNA ladder. After 48 hr treatment in the absence or presence of conditioned media, neurons were lysed in 5 mmTris-HCl, pH 7.4, 0.5% Triton X-100, 20 mm EDTA, 50ug/ml RNAse A, 0.2 mm PMSF, rocked at 4°C for 30 min, and spun (10 min, 16,000 rpm, 4°C). Endonuclease-cleaved DNA was phenol/chloroform-extracted and ethanol-precipitated from the supernatants and resolved by 1.8% agarose gel electrophoresis. Gels were stained with ethidium bromide for photography.
Statistical analysis. All experiments were performed in duplicate or triplicate a minimum of three to four times. Mean values (±SEM) for each experiment were determined, and values statistically different from controls were calculated using one-way ANOVA. The Tukey-Kramer multiple comparisons post-test was used to determinep values.
RESULTS
Aβ stimulation of THP-1 cells leads to activation of proinflammatory transcription factors
We have previously described tyrosine kinase-based signaling pathways activated by stimulation of microglia and THP-1 monocytes with full-length fibrillar Aβ1–40, Aβ1–42, or its active domain, Aβ25–35 (McDonald et al., 1997, 1998; Combs et al., 1999). We and others (Yates et al., 2000) have documented that primary microglia and the THP-1 monocytes respond similarly after exposure to all fibrillar amyloid peptides. Because of this consistent similarity in responsiveness, we initially performed experiments using the THP-1 cell line and Aβ25–25 fibrils and subsequently confirmed that these responses are also elicited by the full-length Aβ1–40 peptide and are observed in primary microglial cultures. Stimulation of the cells with Aβ fibrils resulted in increased cellular protein phosphotyrosine levels resulting from activation of several tyrosine kinases, including the Src family members Lyn, Syk, focal adhesion kinase, and PYK2 (Fig. 1) (McDonald et al., 1997; Combs et al., 1999). Tyrosine kinase activation lead to the activation of both the ERK MAP kinase and the p38 MAP kinase pathways (Fig. 1) (McDonald et al., 1998; Combs et al., 1999). We investigated the Aβ-dependent phosphorylation and activation of transcription factors involved in the upregulation of proinflammatory genes in these cells. Aβ stimulation resulted in the phosphorylation of CREB at the regulatory Ser133, reflecting activation of its transcriptional activity (Fig. 1) (McDonald et al., 1998). Stimulation with Aβ also resulted in increased expression of c-fos, indicating AP-1 transactivation, consistent with previous reports (Yates et al., 2000). Importantly, Aβ stimulation resulted in activation of the proinflammatory transcription factor, NFκB. Exposure of the cells to Aβ fibrils resulted in the phosphorylation of IκB followed by its proteolytic degradation (Karin, 1999). In parallel, increased levels of the active form of the Rel A (p65) subunit of the NFκB dimer were detected.
Fig. 1.

β-Amyloid fibrils stimulate activation of NFκB and multiple MAP kinase pathways in THP-1 cells. THP-1 cells were stimulated in serum-free RPMI with fibrillar Aβ25–35 (60 μm) for increasing times (0–60 min). Cell lysates were resolved by 7.5% SDS-PAGE and Western-blotted with selected antibodies. The p65 (RelA) subunit of NFκB was immunoprecipitated from the stimulated cell lysates and resolved by 7.5% SDS-PAGE and Western-blotted. ERK2 levels were evaluated in parallel as a protein loading control. The antibodies used were 4G10 (anti-phosphotyrosine), anti-phospho-ERK, anti-phospho-p38, anti-phospho-IκBα, anti-IκBα, anti-p65NLS, anti-c-fos, anti-phospho-CREB, and anti-ERK2.
Regulation of transcription factor activation through Lyn- and Syk-linked signaling pathways
Nonreceptor tyrosine kinases such as the Src family members Lyn and Syk kinase serve as membrane proximal signaling elements in the Aβ-dependent inflammatory response (McDonald et al., 1997; Combs et al., 1999). To determine whether transcription factor activation relied on these tyrosine kinase activities, we preincubated the THP-1 cells with the Src family-specific inhibitor PP1 (5 μm) as well as the Syk-selective inhibitor piceatannol (10 μm) for 30 min before a 60 min stimulation with fibrillar 60 μmAβ1–40 (Oliver et al., 1994; Hanke et al., 1996). As we have already observed, PP1 treatment effectively inhibited the Aβ-dependent increase in protein phosphotyrosine levels and activation of ERK and p38 MAP kinases (Fig. 2) (Combs et al., 1999). Accordingly, this resulted in a subsequent inhibition of the phosphorylation of CREB and partially prevented the increase in c-fos protein levels (Fig. 2). As demonstrated previously, pretreatment with piceatannol had little effect on decreasing protein phosphotyrosine levels or ERK and p38 MAP kinase activation (Fig. 2) (Combs et al., 1999) or the Aβ-dependent CREB phosphorylation (Fig. 2). However, piceatannol preincubation of THP-1 cells was able to completely eliminate the Aβ-dependent increase in c-fos expression (Fig. 2). Interestingly, pretreatment with either drug had no effect on inhibiting activation of NFκB as determined by decrease in IκB protein levels (Fig. 2) (Chen et al., 1996). These data demonstrate that Lyn and Syk each mediate a subset of the specific downstream signaling events elicited by Aβ treatment of the cells.
Fig. 2.

Regulation of transcription factor activation through Lyn- and Syk-linked signaling pathways. THP-1 cells were treated in serum-free RPMI with vehicle (DMSO) or 5 μmPP1 (Src inhibitor) or 10 μm piceatannol (Syk inhibitor) for 30 min before stimulation with fibrillar Aβ1–40 (60 μm) for 60 min. Cell lysates were resolved by 7.5% SDS-PAGE and Western-blotted with selected antibodies. The antibodies used were 4G10 (anti-phosphotyrosine), anti-phospho-ERK, anti-phospho-p38, anti-IκBα, anti-c-fos, anti-phospho-CREB, and anti-ERK2.
Aβ-dependent increase in TNFα expression is dependent on Syk and NFκB activity
Aβ fibrils stimulate an increase in monocytic/microglial proinflammatory cytokine production in vitro (Klegeris et al., 1997; Lorton, 1997; Meda et al., 1999; Combs et al., 2000; Yates et al., 2000). Aβ fibrils stimulated increased promoter activity for TNFα in transiently transfected THP-1 cells using a luciferase reporter linked to the human promoter elements of the TNFα gene (Fig.3A). We linked the Aβ-dependent increase in TNFα production to the proximal signaling events by incubating TNFα–luciferase reporter-transfected THP-1 cells with Aβ1–40 and Aβ25–35 fibrils for 5 hr in the presence or absence of piceatannol, PP1, or the NFκB inhibitory peptide, SN-50. TNFα promoter activity required activation of NFκB and Syk but not Src family kinases (Fig. 3B) (Lin et al., 1995). We also examined whether an Aβ-dependent increase in IL-1β production was regulated by activation of the proximal tyrosine kinase activities. Nontransfected THP-1 cells were stimulated with 60 μm Aβ25–35 fibrils for 5 hr in the presence or absence of PP1 and piceatannol, and expression of IL-1β was examined by Western blot of cell lysates. As was seen with TNFα, the Aβ-dependent increase in proIL-1β levels required activation of Syk but not Src family kinases (Fig. 3C). As reported previously, Aβ-stimulated levels of mature IL-1β were very low in these cells (Lorton et al., 1996).
Fig. 3.

β-Amyloid-induced TNFα and IL1-β gene expression require Syk activity. THP-1 cells were transiently transfected with a TNFα reporter construct and assayed for promoter activity 48 hr later. The cells were cotransfected with a β-galactosidase reporter construct to control for transfection efficiency. A, During the last 8 hr, cells were incubated in serum-free RPMI alone (black bars) or stimulated for increasing times (0–8 hr) with fibrillar Aβ25–35 (60 μm) (gray bars). B, To determine whether proximal tyrosine kinase and NFκB activities were required for TNFα promoter activity, the cells were incubated with drug/vehicle only (black bars) or with fibrillar Aβ25–35 (60 μm) (light gray bars) or Aβ1–40 (60 μm) (dark gray bars) for the last 5 hr in the presence or absence of PP1 (5 μm), piceatannol (10 μm), or 100 μg/ml SN-50 peptide. The data shown represent the average (±SEM) of three independent experiments. Unpaired ANOVA was performed with Tukey-Kramer post-comparison to evaluate statistical significance (**p < 0.001). C, To determine whether proximal tyrosine activities were required for β-amyloid-dependent IL1-β expression, the cells were incubated with drug/vehicle only or fibrillar Aβ25–35 (60 μm) for 5 hr in the presence or absence of PP1 (5 μm) or piceatannol (10 μm). Cell lyates were resolved by 9% SDS-PAGE and Western-blotted using anti-IL-1β antibody.
Conditioned media from Aβ-stimulated monocytes induces neuronal apoptosis
Microglia and monocytes secrete neurotoxic factors on stimulation with Aβ-fibrils (Combs et al., 1999, 2000). However, the nature of the neurotoxic factors has not been clearly defined (Banati et al., 1993; Giulian et al., 1995; Ii et al., 1996; Klegeris et al., 1997; Combs et al., 1999, 2000). We have investigated the method of neuronal death that occurs after stimulation with conditioned media from Aβ-stimulated THP-1 cells. Several studies have documented the presence of markers of neuronal apoptosis in AD brains as evidenced by increased immunoreactivity for active caspases and endonuclease-cleaved DNA (Li et al., 1997; Selznick et al., 1999;; Stadelmann et al., 1999). To verify that the method of microglial/monocytic-dependent death in our in vitro system was duplicating disease-related phenomena, we treated cortical neuron cultures for 48 hr using conditioned media from THP-1 cells stimulated for 48 hr with Aβ1–40 fibrils. To assess whether neurons were dying apoptotically, cultures were fixed and TdT-mediated dUTP nick end labeling (TUNEL) was performed to visualize the endonuclease-cleaved DNA. Conditioned media treatment resulted in a clear increase in numbers of TUNEL-labeled neuronal nuclei compared with control cultures (Fig.4A,B), and the DNA obtained from the neuronal cultures displayed a characteristic DNA ladder reflecting DNA fragmentation (Fig.4C).
Fig. 4.
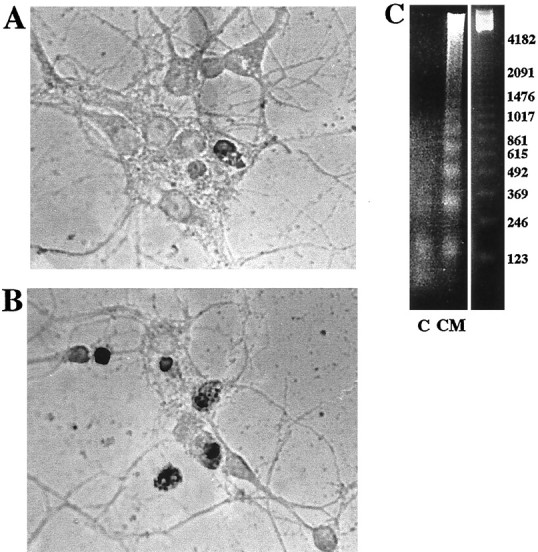
Proinflammatory products in conditioned media from Aβ-stimulated THP-1 cells induce neuronal apoptosis. Conditioned Neurobasal media from THP-1 monocytes stimulated for 48 hr without (A, control) or with (B) immobilized Aβ1–40 was applied to mouse cortical neuron cultures (E16, 5 d in vitro) for 48 hr. Neurons were fixed in 4% paraformaldehyde, and endonuclease-cleaved DNA was visualized by TUNEL staining. C, Nuclear extracts were collected from the same conditions, control (C) and conditioned media (CM)-treated neurons after 48 hr treatment, and fragmented DNA was visualized after agarose electrophoresis.
Neuronal apoptosis produced by stimulation with conditioned media from Aβ-stimulated monocytes/microglia occurs in a TNFα/iNOS-dependent fashion
Activation of microglia or monocytes results in increased secretion of a number of proinflammatory products. We tested whether TNFα secreted by the monocytes/microglia was responsible for the apoptosis that we had observed. TNFα has a well described ability to synergize with other stimuli, resulting in apoptosis of a number of cell types (Fiers, 1991; Natoli et al., 1998). Moreover, TNFα-dependent neuronal apoptosis has been linked to the increased expression and activity of iNOS (Ogura et al., 1997; Heneka et al., 1998; Chung et al., 1999). Importantly, it has been reported that levels of TNFα and neuronal iNOS immunoreactivity are increased in the AD brain (Fillit et al., 1991; Vodovotz et al., 1996; Bruunsgaard et al., 1999; Tarkowski et al., 1999). We investigated whether conditioned media from Aβ-stimulated monocytes and microglia induced neuronal apoptosis in a manner requiring TNFα and iNOS activity. To determine whether TNFα was required for neuronal apoptosis, conditioned media from Aβ1–40 and Aβ25–35 stimulated microglia and monocytes was preincubated with neutralizing antibodies to mouse or human TNFα, respectively, before addition to neuronal cultures. Antibody preincubations completely attenuated the neuron loss caused by the conditioned media (Fig. 5). We determined that neutralizing antibodies for IL-1β did not improve neuron survival in the presence of conditioned media (Fig. 5). Importantly, addition of mouse TNFα to THP-1 cell-conditioned media containing anti-human-specific TNFα neutralizing antibody restored the ability of the media to elicit neuron death (Fig. 5). These data demonstrate that TNFα is responsible for much of the neuronal death observed in these cultures. However, direct administration of recombinant mouse TNFα to neurons had no effect on neuron survival (Fig. 5). These findings demonstrate that additional factors present in the conditioned media act in concert with TNFα to elicit apoptosis, consistent with the well described requirement of TNFα to act synergistically with other agents to signal an apoptotic response (Venters et al., 1999, 2000).
Fig. 5.

Conditioned media from Aβ-stimulated monocytes produces TNFα-dependent-neuronal apoptosis. Purified cultures of mouse cortical neurons (E16, 5 d in vitro) were incubated for 72 hr in Neurobasal media from unstimulated THP-1 cells or conditioned Neurobasal media (CM) obtained from THP-1 cells stimulated (48 hr) via surface-immobilized Aβ25–35 or Aβ1–40 fibrils (48 pmol/mm2). The incubations also included, as indicated, anti-human TNFα antibody (5 μg/ml), mouse TNFα (100 ng/ml), and anti-human IL-1β antibody (5 μg/ml). The neurons were fixed and stained for neuron-specific MAP2 protein, and surviving neurons were counted. Neurons from four fields/condition were counted in duplicate wells and averaged ± SEM. The mean values shown (±SEM) are representative of four independent experiments. Unpaired ANOVA was performed with Tukey-Kramer post-comparison to evaluate statistical significance (*p < 0.001).
We determined the involvement of neuronal iNOS activity in the conditioned media-dependent neuron death by adding iNOS-selective inhibitors to the media at the time of addition to the neuron cultures (Tracey et al., 1995; Garvey et al., 1997). The iNOS-selective inhibitors, AMT.HCl and 1400W.2HCl, blocked neuron death associated with treatment using either microglial or monocytic Aβ1–40 and Aβ25–35-stimulated conditioned media (Fig.6A,B). To determine whether nNOS activity also contributed to the neuronal death caused by conditioned media, the nNOS-specific inhibitor, Vinyl-l-NIO, was added to neurons at the time of addition of conditioned media (Babu and Griffith, 1998). In contrast to the results obtained with iNOS-selective inhibitors, Vinyl-l-NIO had no ability to ameliorate conditioned media-dependent death.
Fig. 6.

Conditioned media from Aβ-stimulated monocytes and microglia produces TNFα/iNOS-dependent-neuronal apoptosis. Purified cultures of mouse cortical neurons (E16, 5 din vitro) were treated for 72 hr in Neurobasal media from unstimulated microglia (B) or THP-1 cells (A) or conditioned Neurobasal media (CM) obtained from microglia (B) or THP-1 cells (A) stimulated (48 hr) via surface-immobilized Aβ25–35 or Aβ1–40 fibrils. The incubations also included, as indicated, 10 μm AMT.HCl, 5 μm 1400W.2HCl, 20 μm Vinyl-l-NIO, and anti-mouse TNFα antibody (5 μg/ml). To terminate experiments, the neurons were fixed and stained for neuron- specific MAP2 protein, and surviving neurons were counted. Neurons from four fields/condition were counted in duplicate wells and averaged ± SEM. The mean values shown (±SEM) are representative of four independent experiments. Unpaired ANOVA was performed with Tukey-Kramer post-comparison to evaluate statistical significance (*p < 0.001).
TNFα stimulates the increased expression of iNOS in neuronal cell types (Ogura et al., 1997; Heneka et al., 1998; Chung et al., 1999). The requirement of iNOS activity for neuronal apoptosis in our system correlated well with increased neuronal iNOS expression in cultures treated with conditioned media. Neurons treated with Aβ1–40-stimulated THP-1-conditioned media displayed increased iNOS immunoreactivity (Fig.7A,B). Addition of the anti-human TNFα antibody to the conditioned media prevented the increase in iNOS immunoreactivity after 72 hr of stimulation (Fig. 7C). Similarly, addition of mouse TNFα alone was able to induce an increase in iNOS immunoreactivity (Fig.7D). The TNFα-dependent increase in conditioned media-stimulated iNOS expression was verified by Western blot analysis of lysates from the treated neurons (Fig. 7E).
Fig. 7.

Aβ-stimulated conditioned media-dependent neuronal apoptosis involves increased iNOS expression. Mouse cortical neuron cultures (E16, 5 d in vitro) were treated for 72 hr in the absence (control) or presence of conditioned Neurobasal media (CM) obtained from THP-1 cells stimulated 48 hr with surface-immobilized Aβ1–40 fibrils with and without 5 μg/ml anti-human TNFα antibody or 100 ng/ml mouse TNFα. A–D, After treatment, neuronal cultures were fixed in 4% paraformaldehyde and stained using an anti-iNOS antibody. Antibody binding was visualized with goat anti-rabbit FITC-conjugated secondary antibody. E, Neuronal cultures were also collected after stimulation, and lysates were resolved by 7.5% SDS-PAGE and Western-blotted using the anti-iNOS antibody. Blots were stripped and reprobed using anti-ERK2 antibody to confirm equal protein loading.
Conditioned media-treated neurons display increased nitrotyrosine levels indicative of peroxynitrite production
One of the consequences of increased inducible nitric oxide synthase expression is the elevation of intracellular nitric oxide levels. Increased concentrations of nitric oxide favor the rapid reaction with superoxide anion to produce the strongly oxidizing agent peroxynitrite (Ischiropoulos et al., 1992a,b). In addition to the well known ability of peroxynitrite to mediate oxidative damage to sulfhydryl groups and lipid peroxidation, it also reacts with Cu, Zn, Mn, and Fe superoxide dismutase, enabling it to nitrate protein tyrosine residues (Beckman et al., 1992; Ischiropoulos et al., 1992a,b). Although the consequences of protein tyrosine nitration are not entirely clear, protein nitrotyrosine levels can be used as an indirect measure of peroxynitrite production (Beckman et al., 1992; Ischiropoulos et al., 1992a,b). Importantly, neurons in AD brains display increased nitrotyrosine immunoreactivity, indicative of increased peroxynitrite production (Good et al., 1996;Vodovotz et al., 1996). We asked whether our in vitroparadigm leading to increased neuronal iNOS expression would replicate AD-associated phenomenon and produce increased nitrotyrosine immunoreactivity. We found that neurons treated with Aβ-stimulated conditioned media from microglia and THP-1 cells displayed increased nitrotyrosine immunoreactivity relative to untreated control cultures (Fig.8A–D). The basal level of nitrotyrosine observed in these experiments is a consequence of elevated iNOS levels arising from in vitroculture conditions. Interestingly, addition of the iNOS-selective inhibitor 1400W.2HCl to conditioned media-treated cultures was able to completely eliminate the nitrotyrosine immunoreactivity (Fig.8E,F). Immunoprecipitation of nitrotyrosine-containing proteins from conditioned media-treated and control cultures revealed a selective increase in nitration of several high molecular weight species (Fig. 8G).
Fig. 8.

Aβ-stimulated conditioned media-dependent neuronal apoptosis is characterized by increased nitrotyrosine expression. Mouse cortical neuron cultures (E16, 5 d in vitro) were treated for 72 hr in the absence (c,control) or presence of conditioned Neurobasal media (CM) obtained from THP-1 cells or microglia-stimulated (48 hr) with surface-immobilized Aβ1–40 fibrils. Neuronal cultures were incubated in Neurobasal media from unstimulated THP-1 cells (A) or microglia (B), conditioned Neurobasal media from Aβ-stimulated THP-1 cells (C) or microglia (D), 10 μm 1400W.2HCl only (F), and Aβ-stimulated THP-1 cell (E) conditioned Neurobasal media + 10 μm 1400W.2HCl. After treatment, neuronal cultures were fixed in 4% paraformaldehyde and stained using an anti-nitrotyrosine antibody. Antibody binding was visualized with goat anti-rabbit FITC-conjugated secondary antibody. G, Neuronal cultures were also lysed after stimulation, and nitrotyrosine-containing proteins were immunoprecipitated and resolved by 7.5% SDS-PAGE and Western-blotted using the anti-nitrotyrosine antibody.
DISCUSSION
Our previous reports have characterized tyrosine kinase-based inflammatory signaling pathways activated in microglia and THP-1 monocytes with Aβ fibrils, resulting in production of proinfammatory cytokines, secretion of superoxide anions, and generation of neurotoxic products (McDonald et al., 1997, 1998; Combs et al., 1999, 2000). Numerous other reports have documented similar findings from microglial lineage cells after Aβ stimulation (Del Bo et al., 1995; Giulian et al., 1995; Lorton, 1996, 1997; Klegeris et al., 1997; Meda et al., 1999; Yates et al., 2000). We also demonstrated that treatment of monocytes with specific enzymatic inhibitors that target enzymes activated in the Aβ response were sufficient to prevent production of neurotoxins (Combs et al., 1999). Treatment of microglia and monocytes with activating ligands for the nuclear receptor PPARγ were also capable of preventing the Aβ-dependent production of neurotoxic factors (Combs et al., 2000). This protection is likely afforded through the ability of PPARγ to prevent NFκB and AP-1 dependent proinflammatory transcriptional events (Lemberger et al., 1996;Ricote et al., 1998). The present report verifies that the Aβ-stimulation of monocytes results in the activation of transcription factors involved in proinflammatory gene expression, such as NFκB and AP-1, and links this response to the previous activation of tyrosine kinase-dependent signaling events. Moreover, we have identified TNFα as the principal neurotoxic agent resulting from the proinflammatory transcriptional changes. Finally, we demonstrate that the mechanism of TNFα-mediated neuronal death is iNOS-dependent apoptosis. These data provide a mechanistic explanation of how fibrillar Aβ stimulation of microglia results in production of proinflammatory products and ultimately the oxidative damage-associated neuronal apoptosis observed in AD brains (Fig.9).
Fig. 9.

Aβ-stimulated microglial and monocytic proinflammatory products cause TNFα/iNOS-dependent neuronal apoptosis. Binding of Aβ fibrils to microglia initiates a tyrosine kinase-dependent signaling response involving Src family members and Syk as membrane proximal signaling elements. Src kinases, such as Lyn, mediate the activation of several tyrosine kinase activities associated with adhesion and phagocytosis of Aβ fibrils. In parallel, Syk kinase activity specifically regulates increased cytokine production in response to Aβ stimulation. A separate cytokine regulatory pathway using the proinflammatory transcription factor NFκB is also activated on Aβ stimulation to operate independent of Src and Syk activation. Aβ-stimulated microglia secrete TNFα plus an additional secretory factor(s) to produce neuronal apoptosis. The apoptosis requires iNOS activity and correlates with increased expression of iNOS and peroxynitrite.
Increased expression of proinflammatory cytokines by microglial lineage cells is a well described phenomenon that occurs in response to numerous activating stimuli. We as well as others (Klegeris et al., 1997; Lorton, 1997; Combs et al., 2000; Yates et al., 2000) have reported the increased expression of TNFα and IL-1β in microglial lineage cells in response to Aβ fibril stimulation. Interestingly, Aβ-dependent production of cytokines does not require the activity of Src-related kinases, although most stimulated tyrosine kinase activity is linked to Src family activation (Combs et al., 1999). Moreover, the Aβ-dependent increase in p38 and ERK MAP kinase activities as well as the subsequent phosphorylation of CREB and increase in c-fos expression required Src family member activation. In contrast, the Syk-selective inhibitor piceatannol was not effective at preventing the Aβ-dependent increase in protein phosphotyrosine levels, MAP kinase activities, or CREB phosphorylation, but dramatically prevented increased c-fos expression. It is critical to note that we have used a piceatannol concentration (10 μm) lower than that reported in our previous report (Combs et al., 1999). The lower drug concentration was required to maintain specificity of enzyme inhibition. Most importantly, Syk, but not Src, inhibition resulted in inhibition of the Aβ-dependent increase in production of IL-1β and TNFα promoter activitity. This is in agreement with our previous observation that Src kinase inhibitors were not effective at preventing the Aβ-dependent respiratory burst in these cells (Combs et al., 1999). These data suggest a bifurcation of Aβ-mediated signaling in microglia in which increased cytokine production involves a mechanistically distinct pathway from other parallel signaling cascades.
Importantly, inhibition of active NFκB nuclear translocation using the SN-50 peptide prevented the Aβ-stimulated increase of TNFα promoter activity. Interestingly, neither Src family nor Syk kinase inhibition prevented IκB degradation. These data suggest that the Aβ-mediated activation of the NFκB pathway relies on yet another class of membrane proximal signaling elements and demonstrates the functional redundancies used by these cells for regulating proinflammatory gene expression.
A major question unanswered by our previous studies was the identity of the neurotoxic factor(s) in our culture system as well as the characterization of the neuronal death. We now identify TNFα as a critical element required for iNOS-dependent apoptosis. TNFα actions observed here are transduced through binding to TNF receptor I because human TNFα binds selectively to TNF receptor I and not TNF receptor II (Fiers, 1991). There is an extensive and conflicting literature concerning the ability of TNFα to induce both pro-apoptotic (Sipe et al., 1996; Ogura et al., 1997; Heneka et al., 1998; Chung et al., 1999; Sortino et al., 1999; Venters et al., 1999,2000) and anti-apoptotic (Cheng et al., 1994; Houzen et al., 1997;Mattson et al., 1997; Shinpo et al., 1999; Sullivan et al., 1999;Tamatani et al., 1999; Yu et al., 1999) responses in neuronal cells. Importantly, the pro-apoptotic action of TNFα on primary neurons typically requires its specific presentation to neurons as part of an inflammatory milieu such as that derived from glial cells (Gelbard et al., 1993; Chao and Hu, 1994; Viviani et al., 1998;Downen et al., 1999; Venters et al., 1999). Similarly, the differential actions of TNFα on cellular survival has been rigorously examined in a number of cell types (Rath and Aggarwal, 1999; Sethi and Hotamisligil, 1999; Wallach et al., 1999; Smythe and Johnstone, 2000). It is now clear that divergent signaling pathways downstream of TNF receptor I lead to either cell survival or death (Natoli et al., 1998). In many cell types, the default TNFα signaling pathway is anti-apoptotic unless additional stimuli such as other cytokines or RNA/protein synthesis inhibitors are also applied (Fiers, 1991; Natoli et al., 1998; Xu et al., 1998; Jones et al., 2000). We have arrived at similar conclusions in interpreting the outcome of our experiments demonstrating that the apoptosis-inducing action of the conditioned media on neurons required both TNFα and an additional factor(s) that acts synergistically to promote cell death. TNFα alone was without effect on neuron survival. Indeed, recent data favoring this hypothesis demonstrated that the TNFα signaling pathway cross-talks with pathways used by the insulin-like growth factor-1 (IGF-1) receptor to result in neuronal death. These findings dramatically illustrate that TNFα can act not only as a neurotrophic factor but also as a “silencer of survival signaling” (Loddick and Rothwell, 1999;Venters et al., 1999, 2000).
The TNFα-dependent neuronal apoptosis observed in our experiments revealed a dependence on iNOS activity. This requirement for iNOS activity correlated well with increased iNOS expression after treatment of cortical neurons with Aβ-stimulated conditioned media. More importantly, the increase in iNOS expression was directly dependent on TNFα stimulation. Neuronal cells expressed low basal levels of iNOS as a consequence of cell culture conditions. However, conditioned media and TNFα treatment both stimulated an increase in iNOS protein levels. The mechanism of iNOS-dependent death involves the production of nitric oxide. Although nitric oxide is a relatively weak oxidizing agent, it rapidly reacts with superoxide anion to form the strong oxidizing and protein nitrating agent peroxynitrite (Beckman et al., 1992; Ischiropoulos et al., 1992a,b). Peroxynitrite-dependent apoptosis is a well described phenomenon that occurs in the presence of excess intracellular nitric oxide concentrations (Troy et al., 1996;Estevez et al., 1998; Heneka et al., 1999). The increase in protein nitrotyrosine above the basal in vitro levels observed in the neuronal cultures after treatment with Aβ-stimulated conditioned media from microglia and monocytes is evidence of increased formation of peroxynitrite (Beckman et al., 1992; Keller et al., 1998). This change correlated well with the increase in iNOS expression and was inhibited with specific iNOS inhibitors. These data strongly suggest that the mechanism of apoptosis signaled by conditioned media involves peroxynitrite-mediated oxidative damage.
The present study represents a continuation of our efforts to characterize our in vitro system modeling the microglial-dependent inflammatory changes occurring in AD brains. In this report we have described a bifurcation of Aβ-stimulated signaling events in monocytes. Significantly, this behavior allows for development of pathway-specific therapeutic approaches affecting selective microglial phenotypic changes in response to Aβ-stimulation. We have also shown that increased TNFα production after Aβ-stimulation leads to iNOS-dependent neuronal apoptosis. Microglial-mediated neuron death is not likely to be the sole mechanism of neuronal loss in AD. However, it is encouraging that several markers of “at risk” neurons in AD brains are duplicated in our culture system, suggesting that it is accurately modeling disease events. Elucidation of the microglial-dependent inflammatory changes occurring in AD will continue to offer molecular targets for therapeutic intervention.
Footnotes
This work was supported by grants from National Institutes of Health (AG08012 and AG16740) to G.L. and the Blachett Hooker Rockefeller Foundation. C.C. was supported by a training grant from National Institutes of Health (HD0710422). The TNFα construct was a generous gift from Dr. Andre Nel. We thank Dr. Michael Heneka for his comments on this manuscript and Dr. Mark Smith for useful discussion.
Correspondence should be addressed to Dr. Gary Landreth, Alzheimer Research Laboratory, E504, Case Western Reserve University School of Medicine, 10900 Euclid Avenue, Cleveland, OH 44106. E-mail:gel2@po.cwru.edu.
Dr. Combs's present address: Department of Pharmacology, Physiology and Therapeutics, University of North Dakota School of Medicine, Grand Forks, ND 58203.
REFERENCES
- 1.Aisen PS. Inflammation and Alzheimer's disease: mechanisms and therapeutic strategies. Gerontology. 1997;43:143–149. doi: 10.1159/000213842. [DOI] [PubMed] [Google Scholar]
- 2.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babu B, Griffith O. N5-(1-Imino-3-butenyl)-L-ornithine. A neuronal isoform selective mechanism-based inactivator of nitric oxide synthase. J Biol Chem. 1998;273:8882–8889. doi: 10.1074/jbc.273.15.8882. [DOI] [PubMed] [Google Scholar]
- 4.Banati R, Gehrmann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. Glia. 1993;7:111–118. doi: 10.1002/glia.440070117. [DOI] [PubMed] [Google Scholar]
- 5.Beckman J, Ischiropoulos H, Zhu L, van der Woerd M, Smith C, Chen J, Harrison J, Martin J, Tsai M. Kinetics of superoxide dismutase- and iron-catalyzed nitration of phenolics by peroxynitrite. Arch Biochem Biophys. 1992;298:438–445. doi: 10.1016/0003-9861(92)90432-v. [DOI] [PubMed] [Google Scholar]
- 6.Berg L, McKeel DW, Miller J, Baty J, Morris J. Neuropathological indexes of Alzheimer's disease in demented and nondemented persons aged 80 years and older. Arch Neurol. 1993;50:349–358. doi: 10.1001/archneur.1993.00540040011008. [DOI] [PubMed] [Google Scholar]
- 7.Bianca VD, Dusi S, Bianchini E, Dal Pra I, Rossi F. beta-amyloid activates the O•2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer's disease. J Biol Chem. 1999;274:15493–15499. doi: 10.1074/jbc.274.22.15493. [DOI] [PubMed] [Google Scholar]
- 8.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- 9.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 10.Bruunsgaard H, Andersen-Ranberg K, Jeune B, Pedersen AN, Skinhoj P, Pedersen BK. A high plasma concentration of TNF-alpha is associated with dementia in centenarians. J Gerontol A Biol Sci Med Sci. 1999;54:M357–364. doi: 10.1093/gerona/54.7.m357. [DOI] [PubMed] [Google Scholar]
- 11.Chao CC, Hu S. Tumor necrosis factor-alpha potentiates glutamate neurotoxicity in human fetal brain cell cultures. Dev Neurosci. 1994;16:172–179. doi: 10.1159/000112104. [DOI] [PubMed] [Google Scholar]
- 12.Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 13.Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- 14.Chung KC, Park JH, Kim CH, Ahn YS. Tumor necrosis factor-alpha and phorbol 12-myristate 13-acetate differentially modulate cytotoxic effect of nitric oxide generated by serum deprivation in neuronal PC12 cells. J Neurochem. 1999;72:1482–1488. doi: 10.1046/j.1471-4159.1999.721482.x. [DOI] [PubMed] [Google Scholar]
- 15.Combs CK, Johnson DE, Cannady SB, Lehman TM, Landreth GE. Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of β-amyloid and prion proteins. J Neurosci. 1999;19:928–939. doi: 10.1523/JNEUROSCI.19-03-00928.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer's disease: inhibition of β-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARγ agonists. J Neurosci. 2000;20:558–567. doi: 10.1523/JNEUROSCI.20-02-00558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cotman CW, Tenner AJ, Cummings BJ. β-amyloid converts an acute phase injury response to chronic injury responses. Neurobiol Aging. 1996;17:723–731. doi: 10.1016/0197-4580(96)00117-0. [DOI] [PubMed] [Google Scholar]
- 18.Del Bo R, Angeretti N, Lucca E, De Simoni MG, Forloni G. Reciprocal control of inflammatory cytokines, IL-1 and IL-6, and beta-amyloid production in cultures. Neurosci Lett. 1995;188:70–74. doi: 10.1016/0304-3940(95)11384-9. [DOI] [PubMed] [Google Scholar]
- 19.Downen M, Amaral TD, Hua LL, Zhao ML, Lee SC. Neuronal death in cytokine-activated primary human brain cell culture: role of tumor necrosis factor-alpha. Glia. 1999;28:114–127. [PubMed] [Google Scholar]
- 20.Estevez AG, Spear N, Manuel SM, Barbeito L, Radi R, Beckman JS. Role of endogenous nitric oxide and peroxynitrite formation in the survival and death of motor neurons in culture. Prog Brain Res. 1998;118:269–280. doi: 10.1016/s0079-6123(08)63214-8. [DOI] [PubMed] [Google Scholar]
- 21.Fiers W. Tumor necrosis factor. Characterization at the molecular, cellular and in vivo level. FEBS Lett. 1991;285:199–212. doi: 10.1016/0014-5793(91)80803-b. [DOI] [PubMed] [Google Scholar]
- 22.Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, Lawlor B, Wolf-Klein G. Elevated circulating tumor necrosis factor levels in Alzheimer's disease. Neurosci Lett. 1991;129:318–320. doi: 10.1016/0304-3940(91)90490-k. [DOI] [PubMed] [Google Scholar]
- 23.Galimberti D, Baron P, Meda L, Prat E, Scarpini E, Delgado R, Catania A, Lipton JM, Scarlato G. Alpha-MSH peptides inhibit production of nitric oxide and tumor necrosis factor-alpha by microglial cells activated with beta-amyloid and interferon gamma. Biochem Biophys Res Commun. 1999;263:251–256. doi: 10.1006/bbrc.1999.1276. [DOI] [PubMed] [Google Scholar]
- 24.Garvey E, Oplinger J, Furfine E, Kiff R, Laszlo F, Whittle B, Knowles R. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J Biol Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- 25.Gelbard HH, Dzenko KA, DiLoreto D, del Cerro C, del Cerro M, Epstein LG. Neurotoxic effects of tumor necrosis factor alpha in primary human neuronal cultures are mediated by activation of the glutamate AMPA receptor subtype: implications for AIDS neuropathogenesis. Dev Neurosci. 1993;15:417–422. doi: 10.1159/000111367. [DOI] [PubMed] [Google Scholar]
- 26.Giulian D, Haverkamp LJ, Li J, Karshin WL, Yu J, Tom D, Li X, Kirkpatrick JB. Senile plaques stimulate microglia to release a neurotoxin found in Alzheimer brain. Neurochem Int. 1995;27:119–137. doi: 10.1016/0197-0186(95)00067-i. [DOI] [PubMed] [Google Scholar]
- 27.Good PF, Werner P, Hsu A, Olanow CW, Perl DP. Evidence of neuronal oxidative damage in Alzheimer's disease. Am J Pathol. 1996;149:21–28. [PMC free article] [PubMed] [Google Scholar]
- 28.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 29.Heneka MT, Loschmann PA, Gleichmann M, Weller M, Schulz JB, Wullner U, Klockgether T. Induction of nitric oxide synthase and nitric oxide-mediated apoptosis in neuronal PC12 cells after stimulation with tumor necrosis factor-alpha/lipopolysaccharide. J Neurochem. 1998;71:88–94. doi: 10.1046/j.1471-4159.1998.71010088.x. [DOI] [PubMed] [Google Scholar]
- 30.Heneka MT, Feinstein DL, Galea E, Gleichmann M, Wullner U, Klockgether T. Peroxisome proliferator-activated receptor gamma agonists protect cerebellar granule cells from cytokine-induced apoptotic cell death by inhibition of inducible nitric oxide synthase. J Neuroimmunol. 1999;100:156–168. doi: 10.1016/s0165-5728(99)00192-7. [DOI] [PubMed] [Google Scholar]
- 31.Hensley K, Maidt ML, Yu Z, Sang H, Markesbery WR, Floyd RA. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J Neurosci. 1998;18:8126–8132. doi: 10.1523/JNEUROSCI.18-20-08126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Houzen H, Kikuchi S, Kanno M, Shinpo K, Tashiro K. Tumor necrosis factor enhancement of transient outward potassium currents in cultured rat cortical neurons. J Neurosci Res. 1997;50:990–999. doi: 10.1002/(SICI)1097-4547(19971215)50:6<990::AID-JNR9>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 33.Ii M, Sunamoto M, Ohnishi K, Ichimori Y. β-amyloid protein-dependent nitric oxide production from microglial cells and neurotoxicity. Brain Res. 1996;720:93–100. doi: 10.1016/0006-8993(96)00156-4. [DOI] [PubMed] [Google Scholar]
- 34.Ischiropoulos H, Zhu L, Beckman J. Peroxynitrite formation from macrophage-derived nitric oxide. Arch Biochem Biophys. 1992a;298:446–451. doi: 10.1016/0003-9861(92)90433-w. [DOI] [PubMed] [Google Scholar]
- 35.Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin J, Smith C, Beckman J. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys. 1992b;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- 36.Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–182. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 37.Jones BE, Lo CR, Liu H, Srinivasan A, Streetz K, Valentino KL, Czaja MJ. Hepatocytes sensitized to tumor necrosis factor-alpha cytotoxicity undergo apoptosis through caspase-dependent and caspase-independent pathways. J Biol Chem. 2000;275:705–712. doi: 10.1074/jbc.275.1.705. [DOI] [PubMed] [Google Scholar]
- 38.Karin M. The beginning of the end: IkappaB kinase (IKK) and NF-kappaB activation. J Biol Chem. 1999;274:27339–27342. doi: 10.1074/jbc.274.39.27339. [DOI] [PubMed] [Google Scholar]
- 39.Keller JN, Kindy MS, Holtsberg FW, St Clair DK, Yen HC, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klegeris A, Walker DG, McGeer PL. Interaction of Alzheimer beta-amyloid peptide with the human monocytic cell line THP-1 results in a protein kinase C-dependent secretion of tumor necrosis factor-alpha. Brain Res. 1997;747:114–121. doi: 10.1016/s0006-8993(96)01229-2. [DOI] [PubMed] [Google Scholar]
- 41.Lemberger T, Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: a nuclear receptor signaling pathway in lipid physiology. Annu Rev Cell Dev Biol. 1996;12:335–363. doi: 10.1146/annurev.cellbio.12.1.335. [DOI] [PubMed] [Google Scholar]
- 42.Li WP, Chan WY, Lai HW, Yew DT. Terminal dUTP nick end labeling (TUNEL) positive cells in the different regions of the brain in normal aging and Alzheimer patients. J Mol Neurosci. 1997;8:75–82. doi: 10.1007/BF02736774. [DOI] [PubMed] [Google Scholar]
- 43.Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem. 1995;270:14255–14258. doi: 10.1074/jbc.270.24.14255. [DOI] [PubMed] [Google Scholar]
- 44.Loddick SA, Rothwell NJ. Mechanisms of tumor necrosis factor alpha action on neurodegeneration interaction with insulin-like growth factor-1. Proc Natl Acad Sci USA. 1999;96:9449–9451. doi: 10.1073/pnas.96.17.9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lorton D. beta-Amyloid-induced IL-1 beta release from an activated human monocyte cell line is calcium- and G-protein-dependent. Mech Ageing Dev. 1997;94:199–211. doi: 10.1016/s0047-6374(96)01847-7. [DOI] [PubMed] [Google Scholar]
- 46.Lorton D, Kocsis JM, King L, Madden K, Brunden KR. beta-Amyloid induces increased release of interleukin-1 beta from lipopolysaccharide-activated human monocytes. J Neuroimmunol. 1996;67:21–29. doi: 10.1016/0165-5728(96)00030-6. [DOI] [PubMed] [Google Scholar]
- 47.Mackenzie IR, Munoz DG. Nonsteroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology. 1998;50:986–990. doi: 10.1212/wnl.50.4.986. [DOI] [PubMed] [Google Scholar]
- 48.Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K. Activation of NF-kappaB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res. 1997;49:681–697. doi: 10.1002/(SICI)1097-4547(19970915)49:6<681::AID-JNR3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 49.McDonald DR, Brunden KR, Landreth GE. Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci. 1997;17:2284–2294. doi: 10.1523/JNEUROSCI.17-07-02284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McDonald D, Bamberger M, Combs C, Landreth G. β-amyloid fibril activate parallel mitogen-activated protein kinase pathways in microglia and THP-1 monocytes. J Neurosci. 1998;18:4451–4460. doi: 10.1523/JNEUROSCI.18-12-04451.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Rev. 1995;21:195–218. doi: 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- 52.McGeer PL, Rogers J. Anti-inflammatory agents as a therapeutic approach to Alzheimer's disease. Neurology. 1992;42:447–449. doi: 10.1212/wnl.42.2.447. [DOI] [PubMed] [Google Scholar]
- 53.McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 1996;47:425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- 54.Meda L, Baron P, Prat E, Scarpini E, Scarlato G, Cassatella MA, Rossi F. Proinflammatory profile of cytokine production by human monocytes and murine microglia stimulated with beta-amyloid[25–35]. J Neuroimmunol. 1999;93:45–52. doi: 10.1016/s0165-5728(98)00188-x. [DOI] [PubMed] [Google Scholar]
- 55.Miyazono M, Iwaki T, Kitamoto T, Kaneko Y, Doh-ura K, Tateishi J. A comparative immunohistochemical study of kuru and senile plaques with a special reference to glial reactions at various stages of amyloid plaque formation. Am J Pathol. 1991;139:589–598. [PMC free article] [PubMed] [Google Scholar]
- 56.Natoli G, Costanzo A, Guido F, Moretti F, Levrero M. Apoptotic, non-apoptotic, and anti-apoptotic pathways of tumor necrosis factor signalling. Biochem Pharmacol. 1998;56:915–920. doi: 10.1016/s0006-2952(98)00154-3. [DOI] [PubMed] [Google Scholar]
- 57.Ogura T, Tatemichi M, Esumi H. TNF-alpha mediates inducible nitric oxide synthase expression in human neuroblastoma cell line by cisplatin. Biochem Biophys Res Commun. 1997;233:788–791. doi: 10.1006/bbrc.1997.6558. [DOI] [PubMed] [Google Scholar]
- 58.Oliver JM, Burg DL, Wilson BS, McLaughlin JL, Geahler RL. Inhibition of mast cell FceR1-mediated signalling and effector function by the syk-selective inhibitor, piceatannol. J Biol Chem. 1994;269:29697–29703. [PubMed] [Google Scholar]
- 59.Rath PC, Aggarwal BB. TNF-induced signaling in apoptosis. J Clin Immunol. 1999;19:350–364. doi: 10.1023/a:1020546615229. [DOI] [PubMed] [Google Scholar]
- 60.Rich JB, Rasmusson DX, Folstein MF, Carson KA, Kawas C, Brandt J. Nonsteroidal anti-inflammatory drugs in Alzheimer's disease. Neurology. 1995;45:51–55. doi: 10.1212/wnl.45.1.51. [DOI] [PubMed] [Google Scholar]
- 61.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 62.Rogers J, Kirby LC, Hempelman SR, Berry DL, McGeer PL, Kaszniak AW, Zalinski J, Cofield M, Mansukhani L, Willson P, Kogan F. Clinical trial of indomethacin in Alzheimer's disease. Neurology. 1993;43:1609–1611. doi: 10.1212/wnl.43.8.1609. [DOI] [PubMed] [Google Scholar]
- 63.Selznick LA, Holtzman DM, Han BH, Gokden M, Srinivasan AN, Johnson EM, Jr, Roth KA. In situ immunodetection of neuronal caspase-3 activation in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:1020–1026. doi: 10.1097/00005072-199909000-00012. [DOI] [PubMed] [Google Scholar]
- 64.Sethi JK, Hotamisligil GS. The role of TNF alpha in adipocyte metabolism. Semin Cell Dev Biol. 1999;10:19–29. doi: 10.1006/scdb.1998.0273. [DOI] [PubMed] [Google Scholar]
- 65.Shinpo K, Kikuchi S, Moriwaka F, Tashiro K. Protective effects of the TNF-ceramide pathway against glutamate neurotoxicity on cultured mesencephalic neurons. Brain Res. 1999;819:170–173. doi: 10.1016/s0006-8993(98)01354-7. [DOI] [PubMed] [Google Scholar]
- 66.Sipe KJ, Srisawasdi D, Dantzer R, Kelley KW, Weyhenmeyer JA. An endogenous 55 kDa TNF receptor mediates cell death in a neural cell line. Brain Res Mol Brain Res. 1996;38:222–232. doi: 10.1016/0169-328x(95)00310-o. [DOI] [PubMed] [Google Scholar]
- 67.Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smythe MJ, Johnstone RW. Role of TNF in lymphocyte-mediated cytotoxicity. Microsc Res Tech. 2000;50:196–208. doi: 10.1002/1097-0029(20000801)50:3<196::AID-JEMT3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 69.Sortino MA, Condorelli F, Vancheri C, Canonico PL. Tumor necrosis factor-alpha induces apoptosis in immortalized hypothalamic neurons: involvement of ceramide-generating pathways. Endocrinology. 1999;140:4841–4849. doi: 10.1210/endo.140.10.7062. [DOI] [PubMed] [Google Scholar]
- 70.Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Bruck W, Jellinger K, Lassmann H. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer's disease. Evidence for apoptotic cell death. Am J Pathol. 1999;155:1459–1466. doi: 10.1016/S0002-9440(10)65460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- 72.Suck RWL, Krupinska K. Repeated probing of western blots obtained from coomasie brilliant blue-stained or unstained polyacrylamide gels. Biotechniques. 1996;21:418–422. doi: 10.2144/96213bm17. [DOI] [PubMed] [Google Scholar]
- 73.Sullivan PG, Bruce-Keller AJ, Rabchevsky AG, Christakos S, Clair DK, Mattson MP, Scheff SW. Exacerbation of damage and altered NF-kappaB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J Neurosci. 1999;19:6248–6256. doi: 10.1523/JNEUROSCI.19-15-06248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tamatani M, Che YH, Matsuzaki H, Ogawa S, Okado H, Miyake S, Mizuno T, Tohyama M. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J Biol Chem. 1999;274:8531–8538. doi: 10.1074/jbc.274.13.8531. [DOI] [PubMed] [Google Scholar]
- 75.Tarkowski E, Blennow K, Wallin A, Tarkowski A. Intracerebral production of tumor necrosis factor-alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J Clin Immunol. 1999;19:223–230. doi: 10.1023/a:1020568013953. [DOI] [PubMed] [Google Scholar]
- 76.Tracey W, Nakane M, Basha F, Carter G. In vivo pharmacological evaluation of two novel type II (inducible) nitric oxide synthase inhibitors. Can J Physiol Pharmacol. 1995;73:665–669. doi: 10.1139/y95-085. [DOI] [PubMed] [Google Scholar]
- 77.Troy CM, Derossi D, Prochiantz A, Greene LA, Shelanski ML. Downregulation of Cu/Zn superoxide dismutase leads to cell death via the nitric oxide-peroxynitrite pathway. J Neurosci. 1996;16:253–261. doi: 10.1523/JNEUROSCI.16-01-00253.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Venters HD, Tang Q, Liu Q, VanHoy RW, Dantzer R, Kelley KW. A new mechanism of neurodegeneration: a proinflammatory cytokine inhibits receptor signaling by a survival peptide. Proc Natl Acad Sci USA. 1999;96:9879–9884. doi: 10.1073/pnas.96.17.9879. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 79.Venters HD, Dantzer R, Kelley KW. A new concept in neurodegeneration: TNFalpha is a silencer of survival signals. Trends Neurosci. 2000;23:175–180. doi: 10.1016/s0166-2236(99)01533-7. [DOI] [PubMed] [Google Scholar]
- 80.Viviani B, Corsini E, Galli CL, Marinovich M. Glia increase degeneration of hippocampal neurons through release of tumor necrosis factor-alpha. Toxicol Appl Pharmacol. 1998;150:271–276. doi: 10.1006/taap.1998.8406. [DOI] [PubMed] [Google Scholar]
- 81.Vodovotz Y, Lucia MS, Flanders KC, Chesler L, Xie QW, Smith TW, Weidner J, Mumford R, Webber R, Nathan C, Roberts AB, Lippa CF, Sporn MB. Inducible nitric oxide synthase in tangle-bearing neurons of patients with Alzheimer's disease. J Exp Med. 1996;184:1425–1433. doi: 10.1084/jem.184.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol. 1999;17:331–367. doi: 10.1146/annurev.immunol.17.1.331. [DOI] [PubMed] [Google Scholar]
- 83.Xu Y, Bialik S, Jones B, Iimuro Y, Kitsis R, Srinivasan A, Brenner D, Czaja M. NF-κB inactivation converts a hepatocyte cell line TNF-α response from proliferation to apoptosis. Am J Physiol. 1998;275:C1058–C1066. doi: 10.1152/ajpcell.1998.275.4.C1058. [DOI] [PubMed] [Google Scholar]
- 84.Yates SL, Burgess LH, Kocsis-Angle J, Antal JM, Dority MD, Embury PB, Piotrkowski AM, Brunden KR. Amyloid beta and amylin fibrils induce increases in proinflammatory cytokine and chemokine production by THP-1 cells and murine microglia. J Neurochem. 2000;74:1017–1025. doi: 10.1046/j.1471-4159.2000.0741017.x. [DOI] [PubMed] [Google Scholar]
- 85.Yu Z, Zhou D, Bruce-Keller AJ, Kindy MS, Mattson MP. Lack of the p50 subunit of nuclear factor-kappaB increases the vulnerability of hippocampal neurons to excitotoxic injury. J Neurosci. 1999;19:8856–8865. doi: 10.1523/JNEUROSCI.19-20-08856.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]